AcpJtbPoHapma
eawwieeN
oabJatg
MINIREVIEW
Molecular Basis of Mendelian Disorders among Jews
Joel Zlotogora,* Gideon Bach,† and Arnold Munnich‡
*Department of Community Genetics, Ministry of Health, Israel; †Department of Human Genetics,
Molecular Genetics and Metabolism 69, 169–180 (2000)doi:10.1006/mgme.2000.2969, available online at http://www.idealibrary.com on
Hadassah Medical Center, Hebrew University, Jerusalem, Israel; and ‡Departementr-Enfa
nd in
de Genetique, Hopital Necke
Received November 22, 1999, a
THE JEWISH PEOPLE
The Jewish people originate from the Middle East.fter the destruction of the First Temple, part of the
ommunity remained in Palestine while anotherart moved eastward founding the Babylonianewry, ancestors of the Iraqi and Iranian Jews. Withhe rise of the Greco–Roman empires, Oriental Jewsegan to move also westward as far as Spain andortugal, where a large Jewish community devel-ped in the Middle Ages. Sephardic Jews (from theebrew word for an area often identified with Spain)re descendants of the Jews who were forcibly ex-elled during the Inquisition in Spain and settledostly in the countries along the Mediterranean Sea
nd in the Netherlands and the New World.The major movement of Oriental Jews toward
astern Europe was in the Middle Ages to Francend Germany. Later, there were two importantaves of migrations of the Ashkenazi Jews (from aord meaning Germany) in central Europe: the first
n the 15th–16th century eastward to Bohemia andventually to Poland–Lithuania and then from thend of the 18th century back to the west (Germany,etherlands, England, United States).The Jewish communities differed in their cultural
utlook and way of life, in their spoken language,nd in their traditions. While differences existedetween the various regions where the Ashkenaziews were living, there are no true subgroups
mong them. On the other hand, most of the Orien-al and Sepharadic Jewish communities remainedeographically separated and developed as distinct169
nts Malades, Paris, France
revised form January 22, 2000
identities. Therefore, a more useful classification ofthe non-Ashkenazi Jews is one using their country/region of origin together with the community theycome from. While the non-Ashkenazi communitiesrepresented more than 90% of the Jews in the 12thcentury, because of the size expansion of Ashkena-zim, they represented only 10% of the world Jewryin 1930. Today, the estimated number of Jewsworldwide is 13–14 million, some 75% of whom areof Ashkenazi origin. In 1996 there were 4.6 millionJews living in Israel, 50% of whom were of Ash-kenazi origin (1).
Since consanguineous marriages are allowed inthe Jewish religion, they were common in all thecommunities, including the Ashkenazi Jews. In asurvey in Israel after the foundation of the State theconsanguinity rate varied from 1.4% among Ash-kenazi Jews to 28.7% among Jews from Iraq (2). Inthe past decades intercommunity marriages havebeen more and more frequent, and within each of thecommunities consanguineous marriages have beendeclining rapidly (T. Cohen, personal communica-tion).
The first observations that various genetic dis-eases are relatively frequent among Jews were madeamong Ashkenazi Jews probably because of the sizeof the community as well as the relative advance inmedicine in the countries in which they lived. Soonafter the foundation of the state of Israel many phy-sicians, in particular, the late Haim Sheba and the
late Richard Goodman, initiated studies on geneticdiseases among the various Jewish communities (3).Since then, the molecular bases of most of the dis-1096-7192/00 $35.00Copyright © 2000 by Academic Press
All rights of reproduction in any form reserved.

tGaSfJioL
1 CH, A
orders have been elucidated and many of the muta-tions identified (Table 1).
Some of the disorders, such as thalassemia, famil-ial Mediterannean fever, and G6PD deficiency,which are found with a relatively high frequencyamong some of the Jewish communities, are alsofrequent in the non-Jewish local population. How-ever, these are exceptions and in most cases the highfrequency of the genetic diseases is particular to theJews. Among the Jewish people, some genetic disor-ders are found with a relatively high frequency inseveral communities, but often the high frequency islimited to one single community.
ASHKENAZI JEWS
At the end of the 19th century and the beginningof the 20th century, there was an important Jewishemigration from central Eastern Europe mainly tothe American continent, South Africa, and Israel.Most of the Ashkenazi Jewish community remainingin Europe was decimated in the Holocaust, and afterthe war many of the survivors immigrated to Israel.Today, the largest Ashkenazi Jewish communitiesare in the United States and in Israel. Several ad-ditional large Ashkenazi communities are in Russia,Ukraine, Canada, United Kingdom, Argentina, Bra-zil, and South Africa.
Genetic disorders relatively frequent among Ash-kenazi Jews may be separated according to theirmolecular basis into two groups: those in which onemutation is found in more than 95% of the diseasealleles and those in which multiple mutations arepresent. Within the second group is a subgroup inwhich one of the multiple mutations is predominant.
Disorders in which One Mutation Is Found
Bloom syndrome and Fanconi anemia. Amongthe diseases in which a single major mutation ispresent, both Bloom syndrome and Fanconi anemiaare rare even among the Ashkenazim. In Bloomsyndrome the most prominent clinical symptoms arethe severe pre- and postnatal growth retardationand the unusually high rate of neoplasia. Most of thepatients reported in the world affected with Bloomsyndrome are Ashkenazi Jews, and among them aunique mutation, [BLM, 6-BP del/7-BP ins], is found(4,5). In population studies, the frequencies of het-erozygotes for this mutation were 1:107 and 1:231,
70 ZLOTOGORA, BA
respectively (6,7).
Familial dysautonomia is a disease due to a dis-turbance of autonomic and peripheral sensory func-
Nsm
tions and is the only other disorder present almostexclusively among Ashkenazi Jews. The gene wasmapped to 9q31–q33, and the strong linkage dis-equilibrium found among the Ashkenazi Jewish pa-tients seems to point to a founder mutation (8,9).
Familial hypercholesterolemia, a relatively com-mon genetic disorder in the general population (1:500), is found with a much higher prevalence amongSouth African Jews (1:67). Most of the South AfricanJewish community originated from East Europe,mainly Lithuania, and settled in South Africa at theend of the 19th century. The mutation [LDLR,G197del] probably originated from a founder since itis present not only in South African Jews but also inJews originating from Lithuania living in Israel(10).
Idiopathic torsion dystonia of early onset is un-usual since a unique recurrent 3-bp GAG deletion ofthe gene DYT1 is present in all the patients irre-spective of their origin. However, among the Ash-kenazi Jews more than 90% of the patients have asimilar haplotype pointing to a common origin,which was probably relatively recent (350 years)(11,12).
Disorders Caused by Multiple Mutations
Lysosomal storage diseases. Three lysosomalstorage diseases are relatively frequent among theAshkenazi Jews, namely, Tay–Sachs disease,Niemann–Pick-type disease type A, and Gaucherdisease, which are caused by multiple mutations.Each of these disorders is secondary to the deficiencyof a different key lysosomal enzyme in the catabo-lism of sphingolipids. Among Ashkenazi Jewish pa-tients affected with Tay–Sachs disease three majormutations in the a subunit of b-hexosaminidasewere identified (13). The two most frequent muta-tions are [HEXA, 4-BP ins, EX11] (73% of the mu-tant alleles) and [HEXA, IVS12 1 1G3 C] (17.6% ofhe mutant alleles). The third mutation, [HEXA,269S], which is relatively rare (3.5% of the mutantlleles), is responsible for the adult type of Tay–achs disease. In the adult type of Gaucher disease,
our mutations are found in most affected Ashkenaziewish patients but one, [GBA, N370S], is predom-nant and present in 85% of the patients (14). Thether mutations are rarer, [GBA, 84GG] (6%), [GBA,444P] (3.5%), and [GBA, IVS2, 11G3 A] (1%). For
ND MUNNICH
iemann–Pick disease type A, three mutations withimilar frequency account for more than 95% of theutant alleles found among Ashkenazi Jewish pa-
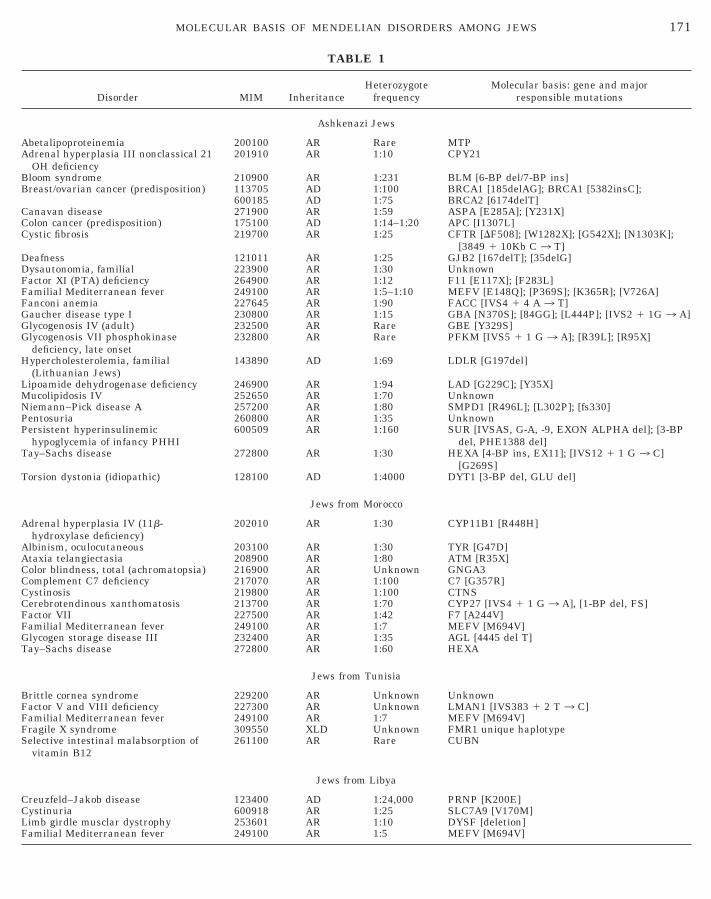
TABLE 1
Disorder MIM InheritanceHeterozygote
frequencyMolecular basis: gene and major
responsible mutations
Ashkenazi Jews
Abetalipoproteinemia 200100 AR Rare MTPAdrenal hyperplasia III nonclassical 21
OH deficiency201910 AR 1:10 CPY21
Bloom syndrome 210900 AR 1:231 BLM [6-BP del/7-BP ins]Breast/ovarian cancer (predisposition) 113705 AD 1:100 BRCA1 [185delAG]; BRCA1 [5382insC];
600185 AD 1:75 BRCA2 [6174delT]Canavan disease 271900 AR 1:59 ASPA [E285A]; [Y231X]Colon cancer (predisposition) 175100 AD 1:14–1:20 APC [I1307L]Cystic fibrosis 219700 AR 1:25 CFTR [DF508]; [W1282X]; [G542X]; [N1303K];
[3849 1 10Kb C 3 T]Deafness 121011 AR 1:25 GJB2 [167delT]; [35delG]Dysautonomia, familial 223900 AR 1:30 UnknownFactor XI (PTA) deficiency 264900 AR 1:12 F11 [E117X]; [F283L]Familial Mediterranean fever 249100 AR 1:5–1:10 MEFV [E148Q]; [P369S]; [K365R]; [V726A]Fanconi anemia 227645 AR 1:90 FACC [IVS4 1 4 A 3 T]Gaucher disease type I 230800 AR 1:15 GBA [N370S]; [84GG]; [L444P]; [IVS2 1 1G 3 A]Glycogenosis IV (adult) 232500 AR Rare GBE [Y329S]Glycogenosis VII phosphokinase
deficiency, late onset232800 AR Rare PFKM [IVS5 1 1 G 3 A]; [R39L]; [R95X]
Hypercholesterolemia, familial(Lithuanian Jews)
143890 AD 1:69 LDLR [G197del]
Lipoamide dehydrogenase deficiency 246900 AR 1:94 LAD [G229C]; [Y35X]Mucolipidosis IV 252650 AR 1:70 UnknownNiemann–Pick disease A 257200 AR 1:80 SMPD1 [R496L]; [L302P]; [fs330]Pentosuria 260800 AR 1:35 UnknownPersistent hyperinsulinemic
hypoglycemia of infancy PHHI600509 AR 1:160 SUR [IVSAS, G-A, -9, EXON ALPHA del]; [3-BP
del, PHE1388 del]Tay–Sachs disease 272800 AR 1:30 HEXA [4-BP ins, EX11]; [IVS12 1 1 G 3 C]
[G269S]Torsion dystonia (idiopathic) 128100 AD 1:4000 DYT1 [3-BP del, GLU del]
Jews from Morocco
Adrenal hyperplasia IV (11b-hydroxylase deficiency)
202010 AR 1:30 CYP11B1 [R448H]
Albinism, oculocutaneous 203100 AR 1:30 TYR [G47D]Ataxia telangiectasia 208900 AR 1:80 ATM [R35X]Color blindness, total (achromatopsia) 216900 AR Unknown GNGA3Complement C7 deficiency 217070 AR 1:100 C7 [G357R]Cystinosis 219800 AR 1:100 CTNSCerebrotendinous xanthomatosis 213700 AR 1:70 CYP27 [IVS4 1 1 G 3 A], [1-BP del, FS]Factor VII 227500 AR 1:42 F7 [A244V]Familial Mediterranean fever 249100 AR 1:7 MEFV [M694V]Glycogen storage disease III 232400 AR 1:35 AGL [4445 del T]Tay–Sachs disease 272800 AR 1:60 HEXA
Jews from Tunisia
Brittle cornea syndrome 229200 AR Unknown UnknownFactor V and VIII deficiency 227300 AR Unknown LMAN1 [IVS383 1 2 T 3 C]Familial Mediterranean fever 249100 AR 1:7 MEFV [M694V]Fragile X syndrome 309550 XLD Unknown FMR1 unique haplotypeSelective intestinal malabsorption of
vitamin B12261100 AR Rare CUBN
Jews from Libya
171MOLECULAR BASIS OF MENDELIAN DISORDERS AMONG JEWS
Creuzfeld–Jakob disease 123400 AD 1:24,000 PRNP [K200E]Cystinuria 600918 AR 1:25 SLC7A9 [V170M]Limb girdle musclar dystrophy 253601 AR 1:10 DYSF [deletion]Familial Mediterranean fever 249100 AR 1:5 MEFV [M694V]
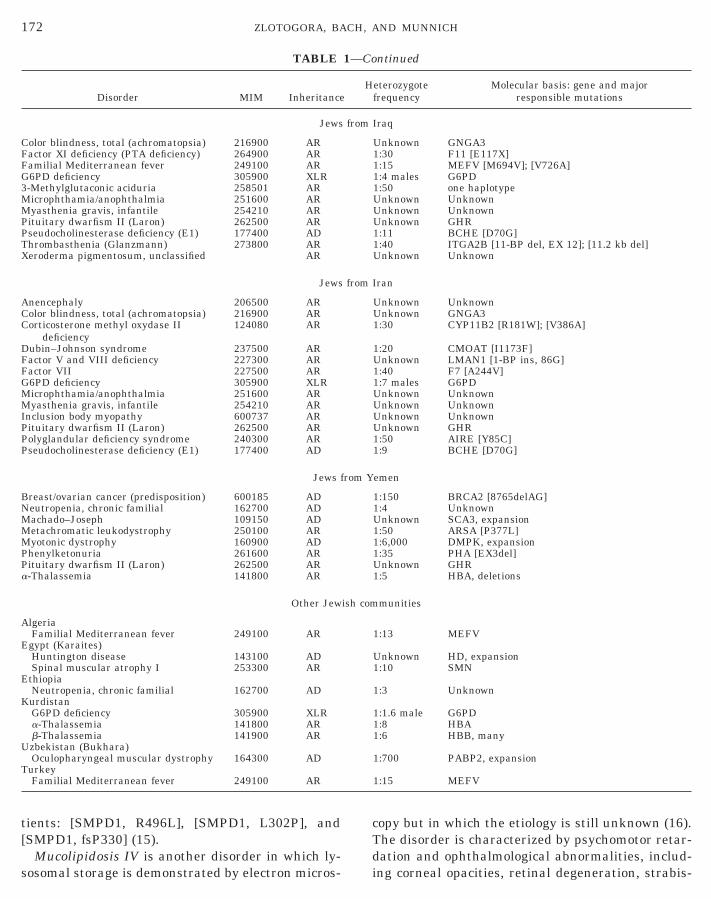
tients: [SMPD1, R496L], [SMPD1, L302P], and
TABLE
Disorder MIM Inheritance
Jews
Color blindness, total (achromatopsia) 216900 ARFactor XI deficiency (PTA deficiency) 264900 ARFamilial Mediterranean fever 249100 ARG6PD deficiency 305900 XLR3-Methylglutaconic aciduria 258501 ARMicrophthamia/anophthalmia 251600 ARMyasthenia gravis, infantile 254210 ARPituitary dwarfism II (Laron) 262500 ARPseudocholinesterase deficiency (E1) 177400 ADThrombasthenia (Glanzmann) 273800 ARXeroderma pigmentosum, unclassified AR
Jews
Anencephaly 206500 ARColor blindness, total (achromatopsia) 216900 ARCorticosterone methyl oxydase II
deficiency124080 AR
Dubin–Johnson syndrome 237500 ARFactor V and VIII deficiency 227300 ARFactor VII 227500 ARG6PD deficiency 305900 XLRMicrophthamia/anophthalmia 251600 ARMyasthenia gravis, infantile 254210 ARInclusion body myopathy 600737 ARPituitary dwarfism II (Laron) 262500 ARPolyglandular deficiency syndrome 240300 ARPseudocholinesterase deficiency (E1) 177400 AD
Jews
Breast/ovarian cancer (predisposition) 600185 ADNeutropenia, chronic familial 162700 ADMachado–Joseph 109150 ADMetachromatic leukodystrophy 250100 ARMyotonic dystrophy 160900 ADPhenylketonuria 261600 ARPituitary dwarfism II (Laron) 262500 ARa-Thalassemia 141800 AR
Other Jew
AlgeriaFamilial Mediterranean fever 249100 AR
Egypt (Karaites)Huntington disease 143100 ADSpinal muscular atrophy I 253300 AR
EthiopiaNeutropenia, chronic familial 162700 AD
KurdistanG6PD deficiency 305900 XLRa-Thalassemia 141800 ARb-Thalassemia 141900 AR
Uzbekistan (Bukhara)Oculopharyngeal muscular dystrophy 164300 AD
TurkeyFamilial Mediterranean fever 249100 AR
172 ZLOTOGORA, BA
[SMPD1, fsP330] (15).Mucolipidosis IV is another disorder in which ly-
sosomal storage is demonstrated by electron micros-
copy but in which the etiology is still unknown (16).
ntinued
eterozygotefrequency
Molecular basis: gene and majorresponsible mutations
Iraq
Unknown GNGA31:30 F11 [E117X]1:15 MEFV [M694V]; [V726A]1:4 males G6PD1:50 one haplotypeUnknown UnknownUnknown UnknownUnknown GHR1:11 BCHE [D70G]1:40 ITGA2B [11-BP del, EX 12]; [11.2 kb del]Unknown Unknown
Iran
Unknown UnknownUnknown GNGA31:30 CYP11B2 [R181W]; [V386A]
1:20 CMOAT [I1173F]Unknown LMAN1 [1-BP ins, 86G]1:40 F7 [A244V]1:7 males G6PDUnknown UnknownUnknown UnknownUnknown UnknownUnknown GHR1:50 AIRE [Y85C]1:9 BCHE [D70G]
emen
1:150 BRCA2 [8765delAG]1:4 UnknownUnknown SCA3, expansion1:50 ARSA [P377L]1:6,000 DMPK, expansion1:35 PHA [EX3del]Unknown GHR1:5 HBA, deletions
munities
1:13 MEFV
Unknown HD, expansion1:10 SMN
1:3 Unknown
1:1.6 male G6PD1:8 HBA1:6 HBB, many
1:700 PABP2, expansion
1:15 MEFV
ND MUNNICH
1—Co
H
from
from
from Y
ish com
CH, A
The disorder is characterized by psychomotor retar-dation and ophthalmological abnormalities, includ-ing corneal opacities, retinal degeneration, strabis-

ttEkiiU
JsFfiJTamct
ipfpfteegTwmem
JttkmW
G
ELIA
mus, and often myopia. The gene was mapped to19p13.2–13.3 by linkage analysis and it seems thatthere are at least two founder chromosomes (17).
Among the other disorders in which more thanone mutation is frequent among Ashkenazi Jews,three are relatively prevalent: Canavan disease, fac-tor XI deficiency, and lipoamide dehydrogenase de-ficiency.
Canavan disease is a rare degenerative disease ofhe brain due to the deficiency of the enzyme aspar-oacylase in which one major mutation, [ASPA,285A], accounts for 83% of the alleles among Ash-enazi Jews in the United States and all the allelesn Israel (18,19). Another mutation, [ASPA, Y231X],s frequent among Ashkenazi Jews living in thenited States (15% of the alleles).
Factor XI deficiency is frequent among Ashkenaziews because of two different mutations, each with aimilar frequency: [F11, E117X] (type II) and [F11,283L] (type III) (20). While type III mutation is
ound only among Ashkenazi Jews, type II mutations also found with a high frequency among Iraqiews and at low frequencies in other Jewish groups.he distribution of this mutation and its presence onsingle haplotype among the different Jewish com-unities suggest that it might have occurred in a
ommon founder from an ancestral Jewish popula-ion (21).
Lipoamide dehydrogenase deficiency is character-zed by recurrent attacks of vomiting, abdominalains, and encephalopathy with abnormal liverunction (22). The age of onset is variable and theatients who presented in the neonatal period suf-ered thereafter from neurological damage, whilehose who presented in childhood suffered from ex-rtional fatigue between the episodes but were oth-rwise asymptomatic. Two mutations in the LADene have been found among Ashkenazi patients.he [LAD, G229C] mutation is the most frequent,hile the [LAD, Y35X] is rare. The patients with theost severe clinical picture were compound het-
rozygotes, while those with later onset were ho-ozygotes for the [LAD, G229C] mutation.
Cystic fibrosis has a prevalence among Ashkenaziews almost similar to that in Caucasian popula-ions; however, the distribution of the different mu-ations in the CFTR gene is unique. Among Ash-
MOLECULAR BASIS OF MEND
enazi Jews, five mutations represent 97% of theutations found among the patients: [CFTR,1282X] (49%), [CFTR, DF508] (31%), [CFTR,
542X], [CFTR, N1303K] and [CFTR, 3849 1 10 kbC 3 T] (23,24).
Nonsyndromic deafness. Two frameshift muta-tions in the GBJ2 gene encoding for connesin 26 arefrequent among Ashkenazi Jews (25). One of thesemutations, [GJB2, 167delT], is present in approxi-mately 4% of the Ashkenazi Jews. Conservation ofthe haplotypes suggests that the allele has a singleorigin, while the other mutation, [GJB2, 35delG], isa hotspot for recurrent mutations.
Predisposition to breast and colon cancer. Thecharacterization of genes involved in the predisposi-tion to cancer in recent years led to the identificationof several mutations frequent among AshkenaziJews. Based on population screening among Ash-kenazi Jews, the carrier frequency of the mutationsin BRCA1 is estimated to be 0.9% for [BRCA1,185delAG] and 0.13% for [BRCA1, 5382insC], andthe carrier frequency of the mutation [BRCA2,6174delT] was estimated to be 0.9–1.5% (26,27). The[BRCA1, 185delAG] mutation is also found amongIraqi and Moroccan Jews at rates comparable tothose found in Ashkenazi Jews and probably have acommon origin since it is found on the same haplo-type as in Ashkenazi Jews (28). A twofold increasedlifetime risk for colorectal cancer among AshkenaziJews is conferred by a mutation in the APC gene.This mutation [APC, I1307K] creates a small hyper-mutable region in the gene and indirectly cancerpredisposition (29,30).
JEWS FROM NORTH AFRICA
In each of the countries of North Africa, Morocco,Algeria, Tunisia, and Libya an ancient Jewish com-munity existed for more than 2000 years. Severalwaves of Jewish immigrants joined these communi-ties, the most important being the one after theexpulsion from Spain and Portugal. The new emi-grants remained separated mostly from the localJewish communities; for instance, in Morocco therewere three different groups: Sephardim, Ashkena-zim, and Berber Jews. Most of the Jews of the dif-ferent North African countries left for Israel and/orFrance after the foundation of the state of Israel andwhen each of the respective countries achieved in-dependence.
Familial Mediterranean fever (FMF) is a common
173N DISORDERS AMONG JEWS
disorder in the Mediterannean region and it repre-sents the most frequent genetic disorder among theJews from North Africa (1:700). The highest fre-

1
fAtmmdJb
1 CH, A
quency of FMF is found among the Jews from Libya(1:200). One ancient mutation, [MEFV, M694V], re-sponsible for most Jewish cases from North Africa isalso present among the Iraqi Jews as well as non-Jewish patients (31). Another frequent mutation,[MEFV, E48Q], has been observed on several hap-lotypes in patients of various origins and therefore isprobably a recurrent mutation. However, amongJews from North Africa this mutation, [MEFV,E48Q], is found on a unique haplotype, suggestingtherefore a common founder in this community (32).The disease has been also reported among Ash-kenazi Jews; however, the high frequency of carriersas opposed to the incidence of the disease in thispopulation suggests a reduced penetrance (33).
Ataxia telangiectasia, glycogen storage diseasetype III, and cystinosis. These genetic disorders arerelatively frequent among Jews from different com-munities in North Africa, particularly MoroccanJews. The predominant clinical features of ataxiatelangiectasia are neurologic symptoms and telangi-ectasias that typically appear after the age of 2years. Variable immunodeficiency is also found inthese patients and may lead to recurrent upper re-spiratory and lung diseases. In addition, the pa-tients with ataxia telangiectasia have an increasedrisk for different types of cancer, in particular, lym-phomas as a result of chromosomal instability. Asingle mutation, [ATM, R35X], was found in almostall the Jewish patients from North Africa (34).
With glycogen storage disease type III, the patientsusually present in childhood with symptoms similarto those of glycogen storage disease type I; however,in some cases, there is a spontaneous improvementat puberty. A single mutation, [AGL, 4,455delT], hasbeen found in all North African Jewish patientsexamined (35,36).
With cystinosis, the clinical picture is dominatedby the symptomatology related to the renal Fanconisyndrome. The gene was recently cloned and molec-ular studies among Jews have not yet been reported(37,38).
Other genetic disorders are found with a rela-tively high frequency in specific Jewish communitiesfrom North Africa.
Deficiency of factor VII is a very rare autosomalrecessive disorder in the general population inwhich some of the affected individuals have a bleed-
74 ZLOTOGORA, BA
ing tendency. The disorder has been reported mostlyamong Jews from Morocco and from Iran and in bothcommunities the patients have the same mutation,
ac
[F7, A244V], with a common haplotype suggesting acommon founder (39).
Adrenal hyperplasia IV is also a relatively fre-quent among Jews from Morocco. It is a disorder inwhich the clinical symptomatology is variable, re-lated to the androgen and mineralocorticoid excesssecondary to the deficiency of the steroid 11b-hy-droxylase. Among the Moroccan Jewish patients asingle mutation, [CYP11B1, R448H], has been iden-tified in almost all the alleles and since most fami-lies originated from the Atlas mountains in Moroccoit is probable that the founder was a Berber Jew(40,41).
Creuzfeld–Jakob disease and fragile X syndrome.These disorders are relatively frequent among Jewsfrom Tunisia probably because of founder effects forboth disorders (42–44). A single mutation in theprion gene, [PRNP, K200E], was found in all theJewish patients from Tunisia and Libya withCreuzfeld–Jakob disease, and there is an unusuallyhigh incidence of a unique haplotype of the FMR1gene with CGG repeats devoid of AGG interruptionspredisposing to the expansion to the full mutationamong Tunisian Jews.
Cerebrotendinous xanthomatosis and Tay–Sachsdisease. Among Jews from Morocco both disordersare caused by multiple frequent mutations. In cere-brotendinous xanthomatosis juvenile cataracts areoften the presenting symptom, while tendon xantho-mas often develop later in the third and fourth de-cade. Most patients have low intelligence or arementally retarded and develop progressive spastic-ity with ataxia. The disease is prevalent only amongMoroccan Jews and among them two mutations arefrequent: [CYP27, IVS4 1 1G 3 A] and [CYP27,-BP del, FS] (45).Tay–Sachs disease of the infantile type is less
requent among Morrocan Jews than among theshkenazi Jews, but in both cases it is due to mul-
iple mutations mostly different between these com-unities. These observations suggest that a similarechanism for the relatively high frequency of the
isease in each community. Among the Moroccanewish patients seven different mutations haveeen identified, three of which were frequent (46).
JEWS FROM IRAQ AND IRAN
ND MUNNICH
The Jewish communities of Iraq and Iran aremong the oldest and trace their history back to 6thentury b.c.e., the time of the First Temple. The two

ELIA
communities have a common history in the pre-Islamic period and thereafter were mostly distinct.Part of the communities migrated and founded newcommunities, such as the “Baghdadis” who begansettling in India at the end of the 18th century andPersian Jews who came to Afghanistan fleeing theforced conversion in Mashad in the first half of the19th century.
After the legalization of immigration from Iraq in1950, most of the Iraqi Jews emigrated to Israel.Jewish immigration from Iran occurred mainly afterthe creation of the state of Israel and on the even ofthe Islamic Revolution to Israel as well as otherdestinations in particular the United States, wherea relatively large Iranian Jewish community is liv-ing.
Disorders Frequent in Jews from Iran and Iraq
Thalassemia and G6PD deficiency are frequentamong Jews from Iran, Iraq, and Kurdistan as wellas in the non-Jewish populations from these areasprobably because of an advantage of the carriersagainst malaria infections.
Pseudocholinesterase deficiency is a very commondisorder in Jews from Iran and Iraq (47). Individu-als with pseudocholinesterase deficiency arehealthy, but when they are treated with the musclerelaxant succinylcholine there is a prolonged paral-ysis instead of a short effect of the drug. This is dueto a mutation in the gene coding for the enzymebutyrylcholinesterase, which usually has a broaderspectrum than cholinesterase. The mutation[BCHE, D70G] is present with a high frequency inJews from Iran (7%) and Iraq (5%).
Laron dwarfism (pituitary dwarfism II) is associ-ated with defective growth hormone receptor. Two ofthe Iraqi Jewish patients studied were homozygousfor a deletion of a large portion of the extracellularhormone binding domain of the human growth hor-mone receptor (48,49).
Congenital familial myasthenia gravis is a disor-der in which the most common manifestations areptosis, limited facial expression with a myopathicappearance together with mandibular prognathism,malocclusion, and open bite (50). The molecular ba-sis of this rare disease has not been yet elucidated.
MOLECULAR BASIS OF MEND
Disorders Frequent Mainly among Iraqi Jews
3-Methylglutaconic aciduria was first describedamong Iraqi Jews and includes bilateral optic atro-
phy and neurological signs. The visual impairmentand decreased distance acuity do not seem to beprogressive (51). The patients present with neuro-logical signs related to the extrapyramidal system,and most have a low borderline intelligence up tomild mental retardation. All the patients excretelarge amounts of 3-methylglutaconic acid in theurine. Linkage disequilibrium was demonstrated inthe region 19q13.3; however, the gene has still notbeen isolated (52).
Xeroderma pigmentosum with relatively mildmanifestations is another disorder first reportedamong adults from the Iraqi Jewish community. Thedisease has not been well characterized and its fre-quency is unknown.
Glanzmann thrombasthenia is a disorder of plate-let function manifested by severe bleeding tendency.The disease may be caused by mutations in eitherglycoprotein IIb or IIIa genes. Among the Iraqi Jewsthe incidence is estimated to be 1:7700, and twomutations have been characterized (53,54). Themost frequent mutation is an 11-bp deletion in exon12 of the GPIIIa gene (85% of the alleles). Anothermutation also found in this community is an 11.2-kbdeletion between intron 9 and exon 14 leading to ashift in the reading frame and a stop codon (lessthan 8% of the alleles).
Disorders Frequent Mainly among Iranian Jews
Dubin–Johnson syndrome is a chronic disorderwith intermittent jaundice. The disease has beenreported to be relatively frequent among Jews fromIran but also in some cases from Iraq and Morocco(55). A common I1173F mutation in the canalicularmutispecific organic anion transporter gene was re-cently identified in all the of Iranian Jewish origin(56).
Inclusion body myopathy is a disease in whichmuscular weakness usually appears in the thirddecade as gait difficulties. The progression is grad-ual but the quadriceps stay strong even in advancedstage of the disorder, allowing the patients to standand walk until late in the course of the disease.While most known patients are Iranian Jews, a feworiginate from Afghanistan and Egypt. According tothe known migrations from the Iranian Jewish com-munity it is probable that all these patients have anIranian origin (57). The gene was mapped to chro-
175N DISORDERS AMONG JEWS
mosome 9p1–q1 but has not yet been identified (58).Corticosterone methyloxidase II deficiency may
present with a very wide range of severity in clinical

ontts
CH, A
symptoms, which may resemble Addison crisis tofailure to thrive or growth retardation. Some pa-tients are even apparently asymptomatic (59). Allthe Iranian Jewish patients examined were homozy-gous for two different mutations in CYP11B2: one,[CYP11B2, R181W], reduces 18-hydroxylase activitybut leaves 11b-hydroxylase intact; the other,[CYP11B2, V386A], causes a small reduction in theproduction of 18-hydrocorticosterone (60). Some ofthe healthy individuals from the patients’ familieswere found to be homozygous for one or the othermutation. Therefore, each mutation is insufficient toproduce a disease state.
Autoimmune polyendocrinopathy–candidiasis–ec-todermal dystrophy (APCED), or polyglandular de-ficiency syndrome, is relatively frequent among theIranian Jews and the Finns. The clinical picture isvariable and progressive but the cardinal featuresare hypoparathyroidism, adrenal insufficiency, andchronic mucocutaneous candidiasis. One majorunique mutation [AIRE, Y85C] was found in all theIranian Jewish patients (61,62).
JEWS FROM KURDISTAN
An isolated ancient Jewish community has beenliving in the mountains of Kurdistan and includesthree separate subgroups: the largest in NorthernIraq, the second largest in Iran, and the smallest ineastern Turkey. In 1950 almost all the communityimmigrated to Israel.
Thalassemia and G6PD deficiency are particu-larly frequent among Kurdish Jews, with an esti-mated carrier frequency of 15–20% for b-thalasse-mia and 1:7 males for G6PD deficiency. The G6PDMediterranean mutation seems to be the major mu-tation among Kurdish Jews, while for b-thalassemia13 mutations in the b-globin gene were identifiedamong the Kurdish Jews, 4 of which were charac-terized for the first time in this population: [HBB,TRP37FS], [HBB, C88A], [HBB, PRO36FS], and[HBB, 3-UNT, A–G, 16] (63). There was very littleverlap in the mutations found in the three commu-ities, further demonstrating the isolation of thehree communities even one from the other, in par-icular, because of the geographic conditions. Iteems that b-thalassemia was frequent among the
Kurdish Jews because of both multiple founder ef-
176 ZLOTOGORA, BA
fects and the selective advantage of the heterozy-gotes against malaria, which was prevalent in theregion.
JEWS FROM UZBESKISTAN
The Jews of Uzbekistan are divided into two cat-egories: the Ashkenazim who came to the regionfrom other parts of the former Soviet Union and theindigenous Bukharan community. Bukharan Jewryis an ancient community that claims descent to the5th-century exiles from Persia.
Oculopharyngeal muscular dystrophy is a raredisorder relatively frequent among Jews fromUzbekistan (1:700) and French-Canadian Jews. Thecharacteristic clinical symptoms are ptosis and dys-phagia, which appear late in life and are progres-sive. A (GCG) repeat of the poly(A)-binding protein 2gene is expanded in this disease (64). The expansionis between 8 and 13 repeats, and the compoundheterozygotes with an expansion on one allele and anormal allele with 7 repeats are more severely af-fected than heterozygotes with another type of nor-mal allele (65).
JEWS FROM YEMEN
The Jews of Yemen trace their origins back tobiblical times; the community probably arose fromthe settlement of Jewish traders and merchants.Several disorders are found with a relatively highfrequency in the community; in each of them theexistence of a single founder is probable.
Phenylketonuria is found with an incidence of1:5000 among Jews from Yemen. All the families ofthe affected patients originated from San’a, where acommon ancestor lived in the 18th century (66). Thepossibility that the increased incidence of the dis-ease in this isolated population is the result of afounder effect is supported by molecular studieswhich demonstrated a similar deletion in the gene ofall the patients (67).
Metachromatic leukodystrophy is a rare neurode-generative disease secondary to the deficiency of thelysosomal enzyme arylsulfatase A. The disease isfrequent among Jews originating from the isolate ofHabban (1:75 live births), a town in South Yemen inwhich a small Jewish community lived for centuriesisolated even from the larger Yemenite Jewish com-munity (68). The disease is also relatively frequentin Jews from Yemen, and all the Jewish patientsfrom the region are homozygous for a same muta-
ND MUNNICH
tion, [ARSA, P377L] (69). It is not known whetherthe founder lived in Yemen and emigrated to Hab-ban, where because of the structure of the popula-

ELIA
tion the disorder became frequent, or whether thegene was introduced in Yemen from Habban.
Two dominant disorders due to a triplet expansionare relatively frequent among Yemenite Jews:Machado–Joseph and myotonic dystrophy.
Machado–Joseph disease is one of the spinocere-bellar ataxias (SCA3) first reported to be frequentamong Portuguese immigrants. It is an autosomaldominant neurologic disorder caused by a (CAG)n
trinucleotide expansion in a gene located on 14q.Because of the high consanguinity rate among theYemenite Jews and the late onset of the disease,several patients homozygous for Machado–Josephdisease have been diagnosed in this community (70).
Myotonic dystrophy is a disorder the prevalence ofwhich among the Yemenite Jews is one of the high-est reported: 17/100,000, compared with an averageof 2.2–5.5/100,000 in Europeans (64). The existencein this community of a high frequency of (CTG)18
alleles (15%) and of the largest size range of (CTG)n
alleles seen in any of the populations studied isprobably the cause for the high prevalence of thedisease since these alleles represent the pool fromwhich the full mutations arise (71).
CONCLUSIONS
When a genetic disease is present in a single Jew-ish community, in most cases it is because of afounder effect. Examples of such disorders are phe-nylketonuria and metachromatic leukodystrophyamong Jews from Yemen, Bloom syndrome amongAshkenazi Jews, and ataxia telangiectasia amongJews from Morocco. However, there are several ex-ceptions and in some disorders more than one fre-quent mutation was responsible for the relativelyhigh frequency in a unique community, for instance,cerebrotendinous xanthomatosis among Jews fromMorocco or thrombasthenia among Jews from Iraq.The most demonstrative example of this group ofdisorders is Tay–Sachs disease, which is found witha relative high frequency in two distinct Jewish com-munities, the Ashkenazi and the Moroccan Jews. Ineach case the disease is caused by multiple muta-tions in the hexosaminidase gene, mutations whichare mostly different in each of the two communities.The existence of multiple mutations responsible fora relatively frequent disorder in a definite popula-
MOLECULAR BASIS OF MEND
tion has been reported in other population and itscauses are not clear. It seems, however, that it mightbe related to a selective advantage for carriers of the
mutations in the environment particular to the pop-ulation. The example of the Kurdish Jews andb-thalassemia is very illustrative since in the threeisolated communities in Kurdistan the disease wasfrequent because of multiple mutations, which evendiffered from one community to the other.
Among the disorders present in more than oneJewish community there are several examples inwhich there was a common ancient founder wholived before or at an early time after the dispersionof the Jews from the land of Israel. For instance,there are two examples of mutations common to theAshkenazi and Iraqi Jews pointing to ancient com-mon ancestor, one leading to the deficiency of factorXI gene and the other in the BRCA1 gene leading tobreast and ovarian cancer predisposition. In othercases the origin of the common ancestor was proba-bly more recent as, for instance, the mutation caus-ing hypercholesterolemia among Ashkenazi Jewsliving in Israel or in South Africa which originatesfrom a common ancestor in Lithuania.
The knowledge of the different diseases, which arefrequent among the different Jewish communities,is important as a tool for better clinical diagnoses.Many of those disorders are rare and therefore dif-ficult to diagnose. The knowledge of the patient’sorigin may be very useful since it may allow for adifferential diagnosis based also on the disordersknown to be relatively frequent in the community oforigin of the patient. With the rapid elucidation ofthe molecular basis of most of these diseases, thefinal diagnosis using molecular methods will be rel-atively easy.
REFERENCES
1. Statistical Abstracts Jerusalem, 1997.
2. Goldschmidt E, Ronen A, Ronen I. Changing marriage sys-tem in the Jewish communities in Israel. Ann Hum Genet24:191–204, 1960.
3. Goodman RM. Genetic Disorders among the Jewish People.Baltimore, MD: John Hopkins Univ. Press, 1979.
4. Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, CiocciS, Proytcheva M, German J. The Bloom’s syndrome geneproduct is homologous to RecQ helicases. Cell 83:655–666,1995.
5. German J, Bloom D, Passarge E, Fried K, Goodman RM,Katzenellenbogen I, Larson Z, Legum C, Levine S, Wahr-man J. Bloom’s syndrome. VI. The disorder in Israel and anestimation of the gene frequency in the Ashkenazim. Am J
177N DISORDERS AMONG JEWS
Hum Genet 29:553–562, 1977.
6. Eng LL, Desnick RJ, German J, Ellis NA. Carrier frequencyof the Bloom syndrome blmAsh mutation in the AshkenaziJewish population. Mol Genet Metab 64:286–290, 1998.

CH, A
7. Oddoux C, Clayton CM, Nelson HR, Oster H. Prevalence ofBloom syndrome heterozygotes among Ashkenazi Jews.Am J Hum Genet 64:1241–1243, 1999.
8. Blumenfeld A, Slaugenhaupt SA, Axelrod FB, Lucente DE,Maayan C, Liebert CB, Ozelius LJ, Trofatter JA, Haines JL,Breakefield XO, Gusella JF. Localization of the gene forfamilial dysautonomia on chromosome 9 and definition ofDNA markers for genetic diagnosis. Nature Genet 4:160–164, 1993.
9. Maayan C, Kaplan E, Shachar S, Peleg O, Gofrey S. Inci-dence of familial dysautonomia in Israel 1977–1981. ClinGenet 32:106–108, 1987.
10. Meiner V, Landsberger D, Berkman N, Reshef A, Segal P,Seftel HC, van der Westhuyzen DR, Jeenah MS, CoetzeeGA, Leitersdorf E. A common mutation causing familialhypercholesterolemia in Ashkenazi Jews. Am J Hum Genet49:443–449, 1991.
11. Risch N, de Leon D, Ozelius L, Kramer P, Almasy L, SingerB, Fahn S, Breakefield X, Breesman S. Genetic analysis ofidiopathic torsion dystonia in Ashkenazi Jews and theirrecent descent from a small founder population. NatureGenet 9:152–159, 1995.
12. Ozelius LJ, Hewett JW, Page CE, Bressman SB, KramerPL, Shalish C, deLeon D, Brin MF, Raymond D, Corey DP,Fahn S, Risch NJ, Buckler AJ, Guesella JF, Breakefield XO.The early onset torsion dystonia gene (DYT1) encodes anATP-binding protein. Nature Genet 17:40–48, 1997.
13. Paw BH, Tieu PT, Kaback MM, Lim J, Neufeld EF. Fre-quency of the three Hex A mutant allele among Jewish andnon-Jewish carriers identified in a Tay-Sachs screening pro-gram. Am J Hum Genet 47:698–705, 1990.
14. Beutler E, Gelbart T, Kuhl W, Zimran A, West C. Mutationsin Jewish patients with Gaucher disease. Blood 79:1662–1666, 1992.
15. Levran O, Desnick RJ, Schuchman EH. Identification andexpression of a common missense mutation (L302P) in theacid sphingomyelinase gene of Ashkenazi Jewish type ANiemann-Pick disease patients. Blood 80:2081–2087, 1992.
16. Amir N, Zlotogora J, Bach G. Mucolipidosis IV clinical spec-trum and natural history. Pediatrics 79:953–959, 1987.
17. Slaugenhaupt SA, Acierno JS, Helbling LA, Bove C, GoldinE, Bach G, Schiffmann R, Gusella JF. Mapping of the mu-colipidosis IV gene to chromosome 19p and definition offounder haplotypes. Am J Hum Genet 65:1999.
18. Elpeleg ON, Anikser Y, Barash V, Branski D, Shaag A. Thefrequency of the C854 mutation in the aspartoacylase genein Ashkenazi Jews in Israel. Am J Hum Genet 55:287–288,1994.
19. Kaul R, Gao P, Aloya M, Balamurugan K, Petrosky A,Michals K, Matalon R. Canavan disease: Mutations amongJewish and non-jewish patients. Am J Hum Genet 55:34–41, 1994.
20. Asakai R, Chung DW, Ratnoff OD, Davie EW. Factor XI(plasma thromboplastin antecedent) deficiency in AshkenaziJews is a bleeding disorder that can result from three typesof point mutations. Proc Natl Acad Sci USA 86:7667–7671,
178 ZLOTOGORA, BA
1989.21. Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI
deficiency in Ashkenazi Jews in Israel. N Engl J Med 325:153–158, 1991.
22. Shaag A, Saada A, Berger I, Mandel H, Joseph A, Feigen-baum A, Elpeleg ON. Molecular basis of lipoamide dehydro-genase deficiency in Ashkenazi Jews. Am J Med Genet 82:177–182, 1999.
23. Kerem E, Kalman YM, Yahav Y, Shoshani Z, Abeliovich D,Szeinberg A, Rivlin J, Blau H, Tal A, Ben-Tur L, Springer C,Augarten A, Godfrey S, Lerer I, Branski D, Friedman M,Kerem B. Highly variable incidence of cystic fibrosis anddifferent mutation distribution among different Jewish eth-nic groups in Israel. Hum Genet 96:193–197, 1995.
24. Abeliovich D, Quint A, Weinberg N, Lerer I, Verchezon G,Lerer I, Ekstein J, Rubinstein E. Cystic fibrosis heterozy-gote screening in the orthodox community of AshkenaziJews: The Dor Yesharim approach and heterozygote fre-quency. Eur J Hum Genet 4:338–341, 1996.
25. Struewing JP, Abeliovich D, Peretz T, Avishai N, KabackMM, Collins FS, Brody LC. The carrier frequency of theBRCA1 185delAG mutation is approximately 1 percent inAshkenazi Jewish individuals. Nature Genet 11:198–200,1995.
26. Morell RJ, Kim HJ, Hood LJ, et al. Mutations in the con-nexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 339:1500–1505, 1998.
27. Oddoux C, Struewing JP, Clayton CM, Neuhausen S, BrodyLC, Kaback M, Haas B, et al. The carrier frequency of the6174delT BRCA2 mutation in Ashkenazi Jewish individualsis approximatively 1%. Nature Genet 14:188–190, 1996.
28. Bar-Sade RB, Kruglikova A, Modan B, Gak E, Hirsh-Yechezkel G, Theodor L, Novikov I, Gershoni-Baruch R,Risel S, Papa MZ, Ben-Baruch G, Friedman E. The185delAG BRCA1 mutation originated before the dispersionof Jews in the Diaspora and is not limited to ashkenazim.Hum Mol Genet 7:801–805, 1998.
29. Laken SJ, Petersen GM, Gruber SB, Oddoux C, Osterer H,Giardiello FM, Hamilton SR, Hampel H, Markowitz A,Klimstra D, Jhanwar S, Winawer S, Offit K, Luce MC,Kinzler KW, Vogelstein B. Familial colorectal cancer in Ash-kenazim due to a hypermutable tact in APC. Nature Genet17:79–83, 1997.
30. Woodage T, King SM, Wacholder S, Hartage P, Strewing JP,McAdams M, Laken SJ, Tucker MA, Brody LC. The APCI1307 allele and cancer risk in a community-based study ofAshkenazi Jews. Nature Genet 20:62–65, 1998.
31. Sohar E, Gafni J, Pras M, Heller H. Familial Mediterraneanfever: A survey of 470 cases and review of the literature.Am J Med 43:227–253, 1967.
32. Bernot A, da Silva C, Petit JL, Cruaud C, Caloustian C,Castet V, Ahmed-Arab M, Dross C, Dupont M, Cattan D,Smaoui N, Dode C, Pecheux C, Nedelec B, Medaxian J,Rozenbaum M, Rosner I, Delpech M, Grateau G, Demaille J,Weissenbach J, Touitou I. Non-founder mutations in theMEFV gene establish this gene as the cause of familialMediterranean fever. Hum Mol Genet 7:1317–1325, 1998.
33. Aksentijevich I, Torosyany, Samuels J, Centola M, Pras E,
ND MUNNICH
Chae JJ, Oddoux C, Wood G, Azzaro MP, Palumbo G, Gius-tolisi R, Pras M, Ostrer H, Kastner DL. Mutation and hap-lotype studies of Familial Mediterranean Fever reveal newancestral relationships and evidence for a high carrier fre-

4
4
4
ELIA
quency with reduced penetrance in the Ashkenazi Jewishpopulation. Am J Hum Genet 64:949–962, 1999.
34. Gilad S, Bar-Shira A, Harnik R, Shkedy D, Ziv Y, KhosraviR, Brown K, Vanagaite L, Xu G, Frydman M, Lavin MF, HillD, Tagle DA, Shiloh Y. Ataxia telangiectasia: Founder effectamong North African Jews. Hum Mol Genet 5:2033–2037,1996.
35. Levin S, Moses SW, Chayot R, Jadoga N, Steinitz. Glycogenstorage disease in Israel: A clinical, biochemical and geneticstudy. Isr J Med Sci 3:397–410, 1967.
36. Parvari R, Moses S, Shen J, Hershkovitz E, Lerner A, ChenYT. A single base deletion in the 39 coding region of glycogendebranching enzyme is prevalent in glycogen storage dis-ease type IIIA in a population of North African Jewishpatients. Eur J Genet 5:266–270, 1997.
37. Gadoth N, Moses SW, Boichis H. Cystinosis in Israel.Harefuah 88:113–114, 1975.
38. Gribouval O, Broyer M, Bates GP, van’Hoff W, Antignac C.A novel gene encoding an integral membrane protein ismutated in nephropathic cystinosis. Nature Genet 18:319–324, 1998.
39. Tamary H, Fromovich Y, Shalmon L, Reich Z, Dym O, LanirN, Brenner B, Paz M, Luder AS, Blau O, Korostishevsky M,Zaizov R, Seligsohn U. Ala244val is a common probablyancient mutation causing factor VII deficiency in Moroccanand Iranian Jews. Thromb Haemostas 76:283–291, 1996.
40. Rosler A, Lieberman E, Cohen T. High frequency of congen-ital adrenal hyperplasia (11 b hydroxylase deficiency)among Jews from Morocco. Am J Med Genet 42:827–834,1992.
1. White PC, Dupont J, New MI, Leiberman, Hochberg Z,Rosler A. A mutation in CYP11B1 (Arg-448–His) associatedwith steroid 11 b-hydroxylase deficiency in Jews of Moroc-can origin. J Clin Invest 87:1664–1667, 1991.
2. Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, AvrahamiD, Scarlato G, Abramsky O, Prusiner SB, Gabizon R. Muta-tion of the prion protein in Libyan Jews with Creutzfeld–Jakob disease. N Engl J Med 324:1091–1097, 1991.
43. Kahana E, Alter M, Beraham J, Sofer D. Creutzfeld–Jakobdisease: Focus among Libyan Jews in Israel. Science 183:90–91, 1974.
4. Falik-Zakai TC, Shackak E, Yalon M, Borochowitz Z,Macphearson JN, Nelson DL, Eichler EE. Predisposition tothe fragile X syndrome in Jews of Tunisian descent is due tothe absence of AGG interruptions on a rare Mediterraneanhaplotype. Am J Hum Genet 60:103–112, 1997.
45. Leitersdorf E, Reshef A, Meiner V, Levitzki R, Schwartz SP,Dann EJ, Berkman N, Cali JJ, Klapholz L, Berginer VM.Frameshift and splice junction mutations in the sterol 27-hydroxylase gene cause cerebrotendinous xanthomatosis inJews of Moroccan origin. J Clin Invest 91:2488–2493, 1993.
46. Kaufman M, Grinshpun-Cohen J, Karpati M, Peleg L, Gold-man B, Akstein E, Adam A, Navon R. Tay Sachs disease andHEXA mutations among Morrocan Jews. Hum Mutat 10:295–300, 1997.
MOLECULAR BASIS OF MEND
47. Szeinberg A, Pipano S, Assa M, Medalie JH, Neufeld HN.High frequency of atypical pseudocholinesterase geneamong Iraqi and Iranian Jews. Clin Genet 3:123–127, 1972.
48. Adam A, Josefsberg Z, Pertzelan A, Zadik Z, Chemke JM,
Laron Z. Occurrence of four types of growth hormone relateddwarfism in Israeli communities. Eur J Pediatr 137:35–33,1981.
49. Pertzelan A, Adam A, Laron Z. Genetics aspects of pituitarydwarfism due to absence or biological inactivity of growthhormone. Isr J Med Sci 4:895–900, 1968.
50. Goldhammer Y, Blatt I, Sadeh M, Goodman RM. Congenitalmyasthenia associated with facial malformations in Iraqiand Iranian Jews: A new genetic syndrome. Brain 113:1291–1306, 1990.
51. Costeff H, Gadoth N, Apter N, Prialnic M, Savir H. A famil-ial syndrome of infantile optic atrophy, movement disorder,and spastic paraplegia. Neurology 39:595–597, 1989.
52. Nystuen A, Costeff H, Elpeleg ON, Apter N, Bonne-TamirBS, Mohrenweiser H, Haider N, Stone EM, Sheiffield VC.Iraqi-Jewish kindreds with optic atrophy plus (3-methyl-glutaconic aciduria type 3) demonstrate linkage disequilib-rium with the CTG repeat in the 39 untranslated region ofthe myotonic dystrophy protein kinase gene. Hum Mol Genet6:563–569, 1997.
53. Seligsohn U, Rososhansky S. A Glanzmann’s thrombasthe-nia cluster among Iraqi Jews in Israel. Tromb Haemostas52:230–231, 1984.
54. Rosenberg N, Yatuv R, Orion Y, Zivelin A, Pertz H, Selig-sohn U. Glanzmann thrombasthenia caused by an 11.2-kbdeletion in the glycoprotein IIIa (beta3) is a second mutationin Iraqi Jews that stemmed from a distinct founder. Blood15:3654–3662, 1997.
55. Seligsohn U, Rososhansky S. A Glanzmann’s thrombasthe-nia cluster among Iraqi Jews in Israel. Tromb Haemostas52:230–231, 1984.
56. Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing thequadriceps: A unique disorder in Iranian Jews. J Neurol Sci64:33–43, 1984.
57. Mor Cohen R, Zivelin A, Rosenberg N, Seligshon U. Identi-fication of a common Ile1173Phe mutation in the canalicularmutispecific organic anion transporter gene in patients withDubin Johnson of Iranian Jewish origin. Am J Hum Genet65:A110, 1999.
58. Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE,Seidman JG. Hereditary inclusion body myopathy maps tochromosome 9p1-q1. Hum Mol Genet 5:159–163, 1996.
59. Cohen T, Theodor R, Rosler A. Selective hypoaldosteronismin Iranian Jews: An autosomal recessive trait. Clin Genet11:25–50, 1977.
60. Pascoe L, Curnow KM, Slutsker L, Rosler A, White PC.Mutations in the human CYP11B2 (aldosterone synthetase)gene causing corticosterone methyloxidase II deficiency.Proc Nat Acad Sci USA 89:4996–5000, 1992.
61. Zlotogora J, Shapiro MS. Polyglandular autoimmune syn-drome in Iranian Jews. J Med Genet 29:834–836, 1992.
62. Bjorses P, Halonen M, Palvimo JJ, Kolmer M, Aaltonen J,Ellonen P, Perheentupa J, Ulmanen I, Peltonen L. Muta-tions in the AIRE gene: Effects on subcellular location andtransactivation function of the autoimmune polyendocri-
179N DISORDERS AMONG JEWS
nopathy-candidiasis-ectodermal dystrophy protein. Am JHum Genet 66:378–392, 2000.
63. Rund D, Filon D, Doewling CE, Rachmilewitz E, KazazianHH Jr, Oppenheim A. Diversity of molecular lesions causing

6
6
CH, A
beta thalassemia in Israel Jewish ethnic groups. In NewPerspectives in Genetic Markers and Diseases among theJewish People (Adam A, Bonne Tamir B, Eds.). Oxford:Oxford Univ. Press, 1991.
4. Blumen SC, Nisipeanu P, Sadeh M, Asherov A, Tome FM,Korczyn AD. Clinical features of oculophayngeal musculardystrophy among Bukhara Jews. Neuromusc Disord 3:575–577, 1993.
5. Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N,Tome FM, Lafreniere RG, Rommens JM, Uyama E, NohiraO, Blumen S, Korcyn AD, Heutink P, Mathieu J, DuranceauA, Codere F, Fardeau M, Rouleau GA. Short GCG expan-sions in the PABP2 gene cause oculopharyngeal musculardystrophy. Nature Genet 18:164–167, 1998.
66. Cohen BE, Bodonyi E, Szeinberg A. Phenylketonuria inJews. Lancet 1:344–345, 1961.
67. Avigad S, Cohen BE, Bauer S, Schwartz G, Frydman M,
180 ZLOTOGORA, BA
Woo SLC, Niny Y, Shiloh Y. A single origin of phenylketo-nuria in Yemenite Jews. Nature 344:168–170, 1990.
68. Zlotogora J, Bach G, Barak Y, Elian E. Metachromatic leu-kodystrophy in the Habbanite Jews: High frequency in a
genetic isolate and screening for heterozygotes. Am J HumGenet 32:663–669, 1980.
69. Zlotogora J, Bach G, Bosenberg C, Barak Y, von Figura K,Gieselmann V. Molecular basis of late infantile metachro-matic leukodystrophy in the Habbanite Jews. Hum Mutat5:137–143, 1995.
70. Lerer I, Merims D, Abeliovich D, Zlotogora J, Gadoth N.Machado Joseph disease: Correlation between the clinicalfeatures, the CAG repeat length and homozygosity for themutation. Eur J Hum Genet 4:3–7, 1996.
71. Mor-Cohen R, Magal N, Gadoth N, Shohat T, Shohat M.Correlation between the incidence of myotonic dystrophy indifferent groups in Israel and the number of CTG trinucle-otide repeats in the myotonin gene. Am J Med Genet 71:156–159, 1997.
72. Tishkoff SA, Goldman A, Calafell F, Speed WC, Deinard AS,Bonne-Tamir B, Kidd JR, Pakstis AJ, Jenkins T, Kidd KK. A
ND MUNNICH
global haplotype analysis of the myotonic dystrophy locus:Implications for the evolution of modern humans and for theorigin of myotonic dystrophy mutations. Am J Hum Genet62:1389–1402, 1998.
Recommended