-
7/31/2019 Immuno . Lec 5
1/18
Antibody-Antigen Interaction
Sara Al-Zu'bi
Ziad Al-NasserThursday, 7/7/2011
5
30
-
7/31/2019 Immuno . Lec 5
2/18
1
The Dr. started the lecture by talking about the attendance and
seat numbers, I think all of us knowevery thing regarding that
:D
Dont look how many pages this lecture is, you will think its
long and boring before reading it!! Just lookat the info in it and
get the maximum benefit and good luck
we started talking about antigens, antibodies and
antigen-antibody
interaction, we talked in details about Immunoglobulins and the
structure of the antibody;
remember the specific parts, the Fab region (binds to the
antigen), Fc portion (binds to the cells or
complement), Hypervariable region, we talked also about antigens
and how they bind to the
antibodies, remember the forces that keeps antigens and
antibodies together (these forces are
electrical or non-covalent).
we talked about the benefit of the antigen-antibody interaction
to clear out infections; neutralize
viruses, binding to ligands; attachment proteins preventing
those from binding to receptors &
also we said that we can use this interaction for diagnostic
purposes; if we see antibodies, this is
an indication that we have been invaded by antigens so if you
determine the antibody type, then
you can by default determine the antigen type that has induced
the production of this antibody.
This -the substance that induce the production of antibodies by
itself- is called immunogen.
We also talked about cross-reacting antibodies: antibodies that
react with antigens that are not
responsible for their production, remember the significance of
that in explaining many
autoimmune diseases, drug allergy and giving you false positive
results in serology.
And dont forget the significance of the serological tests; to
have 100% specificity which means
zero false positive, 100% sensitivity means zero false negative
and not to have any problem.
We talked also about quantity and quality >> quality: the
test is either positive or negative,
quantity: measure the titer
Routinely, we do a lot of diagnostic tests in the lab in the
section of serology (or clinical
-
7/31/2019 Immuno . Lec 5
3/18
-
7/31/2019 Immuno . Lec 5
4/18
3
We also have a test called adsorption inhibition test as well
lots of applications related to that. (The Dr said
this sentence without any explanation just like that)
one of the most common tests that we do in our serology lab and
over the world of course where
simply we are using tagged secondary immunoglobulins, tagged
here is an enzyme which is
horseradish peroxidase enzyme), and you detect the enzyme
activity;
Enzyme + substrate enzyme + products;
** If the products are colored then its a positive test.
** The more the intense the color the more antibodies or
reaction you are going to have.
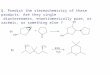
look at the picture and follow:
The antigen is known and adherent on the
bottom of the plastic plate.
We add the patients serum; if it has the
antibody that we are looking for; it is going to
bind to the antigen.
Incubate and wash; incubate: collect the
antibodies bind to the antigens and wash any
excess of the antibody.
Add secondary Immunoglobulins that are tagged with horseradish
peroxidase enzyme.
Incubate and wash
add the substrate so it will produce enzyme-substrate complex,
it will gives you back the
enzyme & products, products are colored and sometimes you
could add something to color the
products so,
- the color that develops means positive test that means the
patient has an antibodies against
the antigens,
- When no color is appeared, no enzyme activity, the antibodies
dont match the antigens so
they will be washed out so it will be negative.
So, to sum up:
** Agglutination inhibition test >> agglutination =
negative test, inhibition of agglutination = positive test.
** In case of pregnancy (positive test):
HCGH in urine antibodies interact with it no free antibodies no
agglutination when latex (HCG) added positive
** In case of NO pregnancy (negative test):
NO HCGH in urineNO antigen-antibody interaction free antibodies
agglutination when latex (HCG) added negative
-
7/31/2019 Immuno . Lec 5
5/18
4
** we do positive control as well at the same time here to be
sure, so ELISA has much higher specificity and
sensitivity compared to agglutination, precipitation or even the
complement fixation test, sensitive means that you
can detect very small amount of the antigen, so the chance of
giving false negative will be very very low.
Here, the substance that could be tagged is an immunofluorescent
material and we use asubstance called fluorscein isothiocyanate
(FITC),,, if you hit this substance with the source of UV
light, it will go into a higher state of energy and then it will
lose this energy in the form of light, so
it emits light. So,
- If you see light (means that it fluoresces) = positive
test.
- Iftheres no light = negative test.
In this test, we could have 2 variants: direct &
indirect.
Where we detect the antigen and the antibody is known and we
label the antibody with an
immunofluorescent material so you detect the antigen; we detect
tissue antigens
We have a slide and it contains an antigen that is unknown, I
want to know what the nature of
the antigen is, if Im suspecting, for example, x antigen so I
buy an antibody specific for that
antigen (x antigen).
So, to sum up:
** ELISA test >> color = positive test, no color =
negative test.
** In case of positive test:
Antigen is known antibodies interact with it when wash, the
antibodies still present secondary antibodies added
theyll interact with the primary onewhen wash, the complex wont
be washed add substrate enzyme activity
colored products.
** In case of negative test:
Antigen is known
no antigen-antibody interaction
when wash, the antibody will be washed out
secondaryantibodies added no interaction with the primary
onewhen wash, secondary antibodies also will be washed out
add substrate no enzyme activity no products or color.
-
7/31/2019 Immuno . Lec 5
6/18
5
Add the antibodies that are fluorescenated with the fluorescein
isothiocyanate and these
antibodies are known.
Incubate and wash.
Put it under the fluorescent microscope.
- if the antigen matches the antibodies, so it want to bind
& when you put it under the UV light
of the fluorescent microscope, the fluorescent material will
fluoresces and emits light so this is
the positive test.
- Ifthe antigen doesnt match the antibody, it wont bind so it
will be washed out so when we
put it under the UV light, no fluorescent material and no light
development,,, negative test.
-
- Used to detect the antibodies so the antigens have to be known
and its exactly like the ELISA test,but the difference is: instead
of using an enzyme on the secondary antibodies, we use the
fluorescein isothiocyanate.
-
-
The difference between the light and fluorescent
microscopes is the light that is used, UV in the
fluorescent and regular light in the light)
So, to sum up:
** Direct immunofluorescent test >> light = positive test,
no light = negative test.
** In case of positive test:
Antigen unknownmatching between antibody & antigen
fluorescenated antibodies interact with it exposure to UV
light emission of light positive
** In case of negative test:
Antigen unknown no matching between antibody & antigen no
antigen-antibody interaction exposure to UV light
no emission of light negative
-
7/31/2019 Immuno . Lec 5
7/18
6
- We have the antigen is known on the slide.
- Add the patients serum that contains antibodies which are
unknown.
- Incubate and wash so if it has attached it will stay.
- Then we add a secondary antibody with the fluorescenated
material on the top.
- incubate and wash- Put it under the fluorescent
microscope.
-
- - If the antigen matches the antibodies, then they will bind
and when we wash, they wont be
washed so the immunofluorescent material will fluoresces. This
is a positive test, that means that
the antibodies that you are looking for is matching that of the
antigen.
- - if the antigen doesnt match the antibodies so there will be
no binding and the antibodies will be
washed out and theres no immunofluorescent material so no
light,,, this is a negative test.
*** for example, if Im looking for anti-rubella antibodies, here
on the slide put rubella antigen
then anti-rubella antibodies are positive.
*** The indirect immunofluorescence is more sensitive (means
that its ability to detect smaller
amounts of the antigens or antibodies is better), because what
you need here is just one antibody
to bind to one antigenic determinant. Also, the intensity of
light is stronger and you could have so
many other antigens that the fluorescenated antibody could bind
to, they could bind to the
isotypes, allotypes and so on.
So, to sum up:
** Indirect immunofluorescent test >> light = positive
test, no light = negative test.
** In case of positive test:
Antigen is known antigen & antibodies match each other
primary antibodies interact with it when wash, the
antibodies still present secondary antibodies addedtheyll
interact with the primary onewhen wash, the complex
wont be washed exposure to UV light emission of light positive
test.
** In case of negative test:
Antigen is known no matching between the antigen and antibody no
antigen-antibody interaction when wash,the antibody will be washed
out secondary antibodies added no interaction with the primary
onewhen wash,
secondary antibodies also will be washed out exposure to UV
light no light.
-
7/31/2019 Immuno . Lec 5
8/18
7
We can use the immunofluorescence for the confirmation of
syphilis; the causative
agent is: treponema pallidum, the test called: fluorescent
treponema antibody
absorption test.
In the routine screening for syphilis, we use an antigen called
cardiolipin and the
antibodies react with cardiolipin [what cardiolipin has to do
with T.pallidum is like a
cross reaction in those patients, when they are infected with
syphilis organisms,
they developed immunoglobulins that react with cardiolipin]
BUT,,, this test is not that specific so we need to confirm the
test BY fluorescent
treponema antibody absorption test.
SO, we get the patients serum, we add some treponemes other than
T.pallidumto
absorb other antibodies that are not against T.pallidum, so the
patients serum will
be left only with antibodies supposed to react with the
T.pallidum, so we get
T.pallidumorganisms fixed into a slide and then we put the
patients serum on the
top then incubate & wash then add a secondary antibodies
with the fluorescenated
material then incubate & wash then we put it under the
fluorescent microscope so,
- If the patient is positive (have syphilis), then the primary
antibodies (in the serum)
are going to bind to the T.pallidum organisms(its shape like the
spring ( )) and
the fluorescenated antibodies bind to the primary antibodies so
it will fluoresces.
This is a positive test for syphilis that means that the patient
is having syphilis.
So, this is a confirmatory test
** We can do other tests here; like that >> you have
syphilis antigen of T.pallidum
adherent to RBCs for example and you can do microheme
agglutination and so on.
- Student: does the secondary antibody consider the primary
antibody
as an antigen?
- dr.: yes, the secondary antibody considers the primary
antibody as an
antigen and it will bind into those antigenic determinants that
we have
talked about; isotypes or allotypes or the carbohydrate antigen
and so
on. And many sites thats why its so sensitive.
-
7/31/2019 Immuno . Lec 5
9/18
8
Firstly, we willdiscuss flow cytometry then western immunoblot
technique
also, we can use immunofluorescence to determine the type of
cells, we can use a machine that
we have in the lab we call it: flow cytometer and the process is
called flow cytometry and you caneven sort these cells or separate
cells from each other through a process we call it FACS
(Fluorescenated Active Cell Sorter), you can separate the cell
that you want, you use that if you
want to determine the number of the CD4 cells, like in the AIDS
patients.
If I want for example to see how many CD4 cells I have, I want
even to separate those CD4 cells, we
identify different types of cells that we have in our body by a
specific marker present on their
surface; marker means specific antigen that is not shared by
others if that marker is there then we
call that CD4 cells or CD8 and so on
What do we do?!
We get the patients serum and we get the Buffy coat, which has
the WBCs, they contain T helper
cells plus other cells like CD8 (T cytotoxic), for example; the
CD4 has the CD4 antigen on the top
and the CD8 has the CD8 antigen on the top.
we start thinking of ELISA to detect antibodies against the
antigens of the HIV virus (indirect
process), BUT we could get 1-2% false positive SO we dont say
directly that this patient has AIDS
so we need to confirm that with another test (because if the
patient is diagnosed having AIDS then
hes died until proving otherwise so we have to be sure in making
diagnosis)..
** the test is called: and after we confirm that, the patient is
having
AIDS (HIV positive) then we start treatment and we start
monitoring that patient whether she or he
is improving or not we do that by counting the number of T
helper cells (CD4) because those
are the target cells of the HIV virus; since it binds to the
CD4+ cell antigen (this is the specifity)..
we have in our body around 1200 /l CD4 cells, usually if they go
below 200, the patient is going
into severe opportunistic type of infections, maybe 500 or less
start to have a trouble and so on.
We see how the patient is responding by calculating how many CD4
cells by a technique called
-
7/31/2019 Immuno . Lec 5
10/18
9
We add a fluorescenated antibodies against the CD4 (if I
want to detect CD8 cells, I will add a fluorescenated
antibodies against the CD8 antigens). So anti-CD4
fluorescenated antibodies bind to the CD4 cells.
In the flow cytometer; it has a very narrow tube that fitsthe
size of only one cell at a time, very small so these cells
start to pass; the CD8 or the CD4.
we have laser, it will hit the fluorescenated antibodies
(bound to cells) and every time it hits them, it will give
you a click -dot- (if theres no fluorescenated cells pass
then it will not click), So it will give you how many of
those cells have passed, also you could separate them
either to the right or to the left, so you can see how manydots
here from CD4 you have and how many dots of CD8
you do have.
>> For example, if you look into this patient (panel B),
the number of the dots of the CD4 is much
much less than here (panel A), that means this patient have
AIDS.
As you know, in making diagnosis of leukemias and lymphomas,
they detect certain markers on
lymphocytes (the abnormal one) or abnormal leukemic cells that
are not present on normal cells
and they do that exactly with the same mechanism to detect the
abnormal markers and herethese markers often are immature markers;
they supposed to be present in immature cells not in
the mature cells in the blood and so on.
We get the HIV virus and we do an electrophoresis to this virus
so it is going to be separated and all
the proteins of HIV virus are going to be separated in the gel
(the polyacrylamide gel) based on the
So, to sum up:
** Flow cytometry used to count number of cells and know their
types and separate them.
** We detect a specific cell marker like CD4, CD8, CD10.
** If you want to detect CD4:
take the serum add fluorescenated antibodies (anti-CD4) they
will bind to T helper cells use flow cytometer machine
has laser
when fluorescenated antibodies hit it, gives click
-
7/31/2019 Immuno . Lec 5
11/18
10
molecular weight or charges; the lower the molecular weight will
be fast as well the heavy will be
slow so now the proteins will be separated on the gel, then,
you get a nitrocellulose filter paper and you stick it in the
gel, then you do a transverse
electrophoresis from the gel into the nitrocellulose filter
paper so all the bands in the gel will betransferred into the
nitrocellulose filter paper so we get those filter papers and just
we cut them
into streps put them in blot and then we sell them to
labs!!!!!!!!!!
So nitrocellulose filter paper has all the HIV proteins adherent
into it, we dont see
them so now we want to see them or we want to see which antibody
is going to bind
to which protein on the nitrocellulose filter paper so We do an
ELISA for example.
Now, we take the patients serum and we put it in the blot so if
the patient has specific antibody,
its going to bind to these bands, then incubate for half an hour
and then we wash it out then we
add a secondary antibody with an enzyme like horseradish
peroxidase, incubate and then wash..
- the primary antibody is bound to the secondary antibody
Then you add the substrate and it will give a color so in the
nitrocellulose filter paper, you will see
colored bands,
- to make diagnosis of AIDS, we should show a band from every
group of antigens of the HIV
means that we have antigens on the surface of the virus, we have
the antigens for the reverse
transcriptase enzyme, we have antigens from the core of the
virus, you should have antigens
from each of those groups, if you see that then the patient is
going to be labeled as HIV
positive.
-
7/31/2019 Immuno . Lec 5
12/18
11
So this is how its going to look like
this is nitrocellulose, this is real test, you can see the
different proteins of
the HIV virus and the colors that develops so it shows all the
proteins, for
example if you look here into this patient -whos having just one
band-
and the negative control so one band here will not be enough to
make
the diagnosis so we tell the patient that we cant confirm, you
have to
come may be after one or 2 weeks or one month and we keep
monitoring
the patient tell we confirm either positive or negative.
this is a new technology where you can use a combination of
ELISA with immunofluorescence
together, if you want to detect more than one antigen at the
same time and we use this test if the
patient have hepatitis and we want to know which type of
hepatitis the patient has; A, B or C.
So we get a microbeads; bead is a glass sphere, very small
glass sphere and they are colored and each one of those can
give you a spectrum of color when you get it with the source
of UV light and you can bind an antigen on these spheres,
put antigens of hepatitis A, B or C and so on.,,
Add the patients serum on the top, if you have an infection
its going to bind to one of the beads
Extremely rare to have patients with hepatitis A or B or C
together,
we have never seen that but usually the infectious disease is
caused
by one organism; this is the rule of the thumb
So, to sum up:
** Western immunoblot technique used to detect antibodies!! To
confirm AIDS.** get patients serum (contain primary antibodies) add
it to nitrocellulose filter paper (have HIV antigens &
proteins) if
antibodies matches the antigens, they will interact add
secondary enzyme-tagged antibodiesDO ELISA TEST!!
-
7/31/2019 Immuno . Lec 5
13/18
12
So if the patient has one of them, A, B or C then its going to
bind the antibody
Add a secondary antibody that is fluorescenated
Hit it with the source of UV light so as you see here (the
picture)
After looking at the picture, the dr. said >> the color of
the 1st
,2nd
and the 3rd
and the differentcolors here for that of 1 so it means that if
that color for hepatitis A then the patient is going to
have only hepatitis A so the is designed to look for antibodies
thats feeling that theres so many
antigens at the same time and its very sensitive as you can see,
its an immunofluorescent
technique.
Now, we use hybridization techniques used to detect specific
sequence, the microarray test as
well. We now have the sequence of so many microorganisms and
then you can add antibodies into
that chip and determine the type of the infectious agent that we
are looking for.
Complement: complementary to antibody, it binds to see H2 part
of the heavy chain so we say
that the complement is fixed, this means that there will be no
more of them present.
** If I want look for the concentration of one of the complement
component of a patient lets say
C3 and its so low in that patient >> that means It could
be used up by antigen-antibody reaction or
genetically it could be deficient in that patient >> this
is the principle used up by the complement
fixation test.
The dr. invited us to visit the clinical
microbiology lab or the serology
section in the king Abdullah hospital
in order to get more details about
the tests used there.
So, to sum up:
** immunofluorescence microbeadbased immunoassay used to detect
more than an antigen at the same time.
** get the different antigens we want to detect put them in
different microbeadsadd pateints serum the antibody
that matches any of the antigens will bind to it add
fluorescenated antibodies expose to UV light each bead
fluoresces with different colors
-
7/31/2019 Immuno . Lec 5
14/18
-
7/31/2019 Immuno . Lec 5
15/18
14
Now, the dr. started with chapter 6. The pictures hes explaining
are in the book, I didnt put
them since the slides arent available yet!!
Going back to the antibody structure and the antibody diversity
which we talked about, do you
remember how many immunoglobulin specifities we -the humans-
have?! Around 1011 different
specifities and different shapes of antigens that can bind to!
From where do we get that number?
Also, we said around 1017-1018 different specifities of B-cells
we born with! How did you get into
that number? We said, if we are genetically diverse then we have
a higher chance to have both
diversities (in phenotype & genotype)
So, remember we said that antibodies come from B-cells and in
order to form an antibody, you
need genes that code for the polypeptide proteins of this
immunoglobulin. And also, we havegenes that will code to the area
that is so specific; the hypervariable region, the
complementary
determining region! Also, We need genes to code for the constant
region,,,
You will see that genes that will code for the variable regions
and the genes that will code for the
constant regions come from different areas, and the constant
regions, in the heavy chain and in
the light chain also they come from different regions!
How all of those come together to form the immunoglobulins and
how we calculated how many
different specifities we do have?
We will answer those questions in what we call
So, you have to know that proteins come out from genes; those
genes when they are expressed,
we call them EXONS and we have intervening sequences we call
them Introns, SO, the gene if it
was expressed, it go into transcription and then translation as
you know to end up with a protein.
So, the proteins that will develop are simply a reflection that
we have genes, for that, the different
polypeptides we get are related to the different genes that we
have.
So it starts happening in the Germline, at the DNA level
Variable (V), diversity (D), joining (J; the last 100 amino
acids in the variable region of the light
and heavy chains), leader (L), constant (C),, all will be
rearranged.
-
7/31/2019 Immuno . Lec 5
16/18
15
So, in the light and Heavy we have the variable area; V genes in
the light and V genes in the heavy!
And we have D genes in the heavy but not in the light.
What codes for the variable region of the light chain are V and
J segments and in the heavy
chain: V, D and J because the specific part of the Ig (the
variable region) comes from part of the
light and part of the heavy; Two polypeptide chains!
Now, we want to determine that specifity which comes from the
variable genes for the light
whether its kappa () or lambda () and the variable genes of the
heavy; the different isotypes
that you know; Mu (), Delta (), Gamma (), Epsilon () and Alpha
()!
And remember, we have diversity genes for the heavy and J genes
(joining) for both the light and
the heavy.
>> all Those will be rearranged and they will be
selective; 1V 1D 1J, you will have gene
rearrangement and then gene splicing at the level of the mRNA,
they come together from
different locations, they go into transcription, translation and
produce the polypeptide. You can
see it depends on how many different V genes I have, how many
different J genes I have or D
genes I have.
So V (D) J then recombination occurs at the level of the DNA,
transcription at the level of mRNA
and then translation and the polypeptide that will form the
Ig.
So, as an outcome of the proteins that will develop here will
form the heavy and light chain
polypeptides and those will come together to form the Ig; Ig
will assemble.>> This is all what we are going to talk about
for today and tomorrow. The chart below is summery
of whats explained before:
The introduction to understand this, one should know the
structure of Immunoglobulin, We have
heavy and light chains of the Immunoglobulin, we have variable
and constant regions, remember
on the light and the heavy, they are coded by different gene
segments; the more genes we have
the more the chance to have diversity.
germline heavy
and light chain
gene segments
V(D)J
recombination
transcription
translation
heavy and light
chain polypeptides
immunoglobulins
assembly
-
7/31/2019 Immuno . Lec 5
17/18
16
Genes will be rearranged during development, on B-cell
development and T-cell development for
the T-cell receptor, its almost the same. We have an L sequence
to start the process, then we have
J segment, D segment in the heavy in addition to V and C genes,
constant for the isotype classes
and subclasses that we have talked about before.
They will be next to each other by gene rearrangement and we
need enzymes to break and unite.
If you have those enzymes then the gene rearrangement is going
to take place and the
Immunoglobulins are going to develop, if genetically you are
missing these enzymes, then you are
going to be immunocompromised,, at the level of DNA; we have
RAG-1 and RAG-2 enzymes, and
they are so important for the production of those antibodies, if
they are missing you become
immuncompromised.
What really will add up to the development diversity is not only
the number of genes, the
rearrangement and rematching, but also we can see other things
that will add up into the
diversity, like for example; the somatic recombination that will
take place, possibilities, products
and then they assemble, V for binding (the variable region binds
to the antigen), C (the constant
part for the biological function; complement binding, T-cell
receptor binding or macrophages). So
you can understand that the most important part in the diversity
regarding the antigen binding is
the VARIABLE region not the constant.
So this is what I was talking about, here for an example; for an
Immunoglobulin, you must have a
light chain, which is either or never BOTH. And you require a
heavy chain class or subclass (1,
2, , 1, 2, 3, 4, or ). So what are the genes that are present in
those chains?! we start with
the DNA at the germline where we start with 5 and the L sequence
and then you have the variable
genes, we have 35 variable genes for (V1, V2 or V3); you need
just one gene of them and each
one gives you specifity as simple as that for the . And the same
thing for in which we have 30
different V genes, you dont have to remember the number of genes
in those just have an idea!
Then, we have J genes for joining; we said that the variable
region is formed from the products of
V and J, where the products of the J form the last 100 amino
acids.
So the specifity now for the will be determined by those genes
as you can see here, the V and theJ.
We have the C gene for the and we have one type for that
biological function. For the , we have
around 5 or 6 J genes, and also you can have more than 1
constant region (C1, C2, C3 and C4), so
we have more than 4 types for and those will add up to the
diversity as you know regarding to
the antigen binding, what really adds up to the antigen binding
as you know is the V and J only.
-
7/31/2019 Immuno . Lec 5
18/18
17
And then we have the heavy chain, the heavy here is the same
thing, we have around 50 V genes &
we have D genes, also will add up and those are around 25 genes
and then we have around 6 J
genes.
>>>> So the variable region, you must have one of
the V, one of the D and one of the J. you need
to have the constant regions of the heavy, which is one of the
(1, 2, , 1, 2, 3, 4, or ).
So the process starts here by the gene rearrangement,
this is a summary of what I was talking about (figure 6-1), you
have exons; expressed genes and
Introns; intervening in between and they are spliced out by
enzymes, so this gene is going to be
selected and then the Introns will be spliced out, so the J will
come very close, 1V and 1J, no more
than that, they will come together, V-J joining as you can
see.
For the heavy chain, they start with the V and the J, and then
the D will be added as you can see,
and you end up with V (D) J.both now go through the
transcription, so you have mRNA, and then into translation that
will code
for proteins of the V and J to form the variable region of the
light and then the variable region of
the heavy V (D) J.
This is more detailed of what I was talking (figures 6-2, 6-3)
(if you dont understand any thing, I
think that the dr. will repeat what hes said in the next lecture
so dont worry!!).
..
..
..
..