
In: The Human Hypothalamus ISBN: 978-1-62081-806-0
Editor: Bertalan Dudás © 2013 Nova Science Publishers, Inc.
Chapter IX
HYPOTHALAMIC CONTROL OF APPETITE
AND ENERGY METABOLISM
Alex Reichenbach, Romana Stark and Zane B. Andrews
Introduction
The hypothalamus is a critical brain structure important for autonomic function, including
the regulation of energy metabolism, glucose metabolism, appetite and adiposity. This
hypothalamic control of energy metabolism is a highly complex system that involves the
central sensation, coordination and integration of peripherally derived nutrients and
hormones. These central hypothalamic processes are then translated into neurochemical code,
conveyed through the autonomic nervous system (ANS), to innervate peripheral target
tissues. In this manner, negative feedback from the periphery to the hypothalamus maintains
energy metabolism in equilibrium, ie energy intake is matched to energy expenditure. In
terms of energy metabolism, a disturbance in negative feedback from the periphery to the
central nervous system (CNS) can cause significant metabolic disease such as obesity and
diabetes. This chapter focuses on the hypothalamic control of appetite, with particular focus
on neuroendocrine feedback regulation of energy and glucose metabolism. Finally, this
chapter examines how dysfunctional hypothalamic circuits contribute to metabolic disease
such as obesity and diabetes.
Historical Overview
An important role for the hypothalamus in energy metabolism was first discovered over
100 years ago, when it was noted that hypothalamic injury, but not pituitary injury resulted in
marked obesity. Hetherington and Ranson demonstrated conclusively that this marked obesity
was due to electrolytic lesions of the hypothalamus (Hetherington and Ranson, 1940). In this
study, marked obesity developed from extensive bilateral damage (not unilateral damage) to
No part of this digital document may be reproduced, stored in a retrieval system or transmitted commercially in any form or by any means. The publisher has taken reasonable care in the preparation of this digital document, but makes no expressed or implied warranty of any kind and assumes no responsibility for any errors or omissions. No liability is assumed for incidental or consequential damages in connection with or arising out of information contained herein. This digital document is sold with the clear understanding that the publisher is not engaged in rendering legal, medical or any other professional services.

Alex Reichenbach, Romana Stark and B. Zane Andrews
248
the dorsomedial hypothalamic (DMH) and ventromedial hypothalamic (VMH) nuclei, the
arcuate nucleus (ARC), the fornix, the medial portion of the lateral hypothalamus and the
ventral premammillary nucleus. These authors did not define the VMH nucleus as a the key
hypothalamic site, but rather noted that “Symmetrical destruction of the ventral portion of this
area, including the nuclei and possibly other structures near the base, seems to be more
important than injury to the more dorsal structures for the production of maximum adiposity”
(Hetherington and Ranson, 1940). In 1942, Hetheringotn and Ranson described most
pronounced obesity with ‘bilateral anterior tuberal lesion’, which consist of lesions to three
quarters of the VMH, or ‘bilateral tubero-premammilary lesion’, which consist of lesions to
the caudal third of the VMH and premammillary area. The authors concluded that “Obesity
can be produced by fairly symmetrical lesions which destroy bilaterally: a) most of the VMH
together with some of the tissue immediately around them, especially on their lateral sides; 2)
the caudal ends of the VMH, the premammillary area, and a considerable part of the lateral
hypothalamic areas adjacent to it”. Brobeck et al validated these data in 1943, in which the
authors showed voracious hyperphagia due to lesions of the VMH (Brobeck et al., 1943).
These studies are the first to establish that the VMH functions normally as a “satiety center”
(Figure 1).
Although the presence of a hypothalamic “feeding center” had been previously
hypothesized by Clarke in 1939, this area wasn’t discovered until 1951 when Anand and
Brobeck showed that bilateral destruction of the lateral hypothalamus completely suppressed
spontaneous eating. Again, unilateral lesion to the lateral hypothalamus had no effect on
feeding behavior suggesting significant compensatory plasticity in the non-lesion side.
Furthermore, hyperphagia and obesity in rats with VMH lesions could be completely reversed
when the same rats had a second lateral hypothalamic lesion to the ‘feeding center’ (Anand
and Brobeck, 1951; Figure 1). This is the first evidence that hypothalamic neuronal circuits
controlling appetite act in a hierarchical manner, and suggests that the brain is wired to
prevent starvation, rather than prevent overeating. Despite the development of novel
molecular genetic technologies, these earlier observations still generally hold true. The VMH
‘satiety centre’ is a major site of feeding inhibition, through the direct actions of leptin on
leptin receptor-containing VMH neurons (Dhillon et al., 2006). The lateral hypothalamus
“feeding center” houses both appetite-stimulating orexin and melanin concentrating hormone
(MCH) neurons. Further, this nucleus plays a critical role in integrating and relaying
information from reward pathways in midbrain dopaminergic systems and homeostatic
appetite regulation via the melanocortin system.
The discovery of the melanocortin system was initially missed by early lesion studies and
is the missing piece of the hypothalamic puzzle in the control of appetite. The melanocortin
system comprises ARC neuropeptide Y (NPY)/agouti related peptide (AgRP) and
proopiomelanocortin (POMC) neurons and their projections to the paraventricular nucleus
(PVN). The critical role of these nuclei in the hypothalamic control of appetite will be
detailed in subsequent sections.
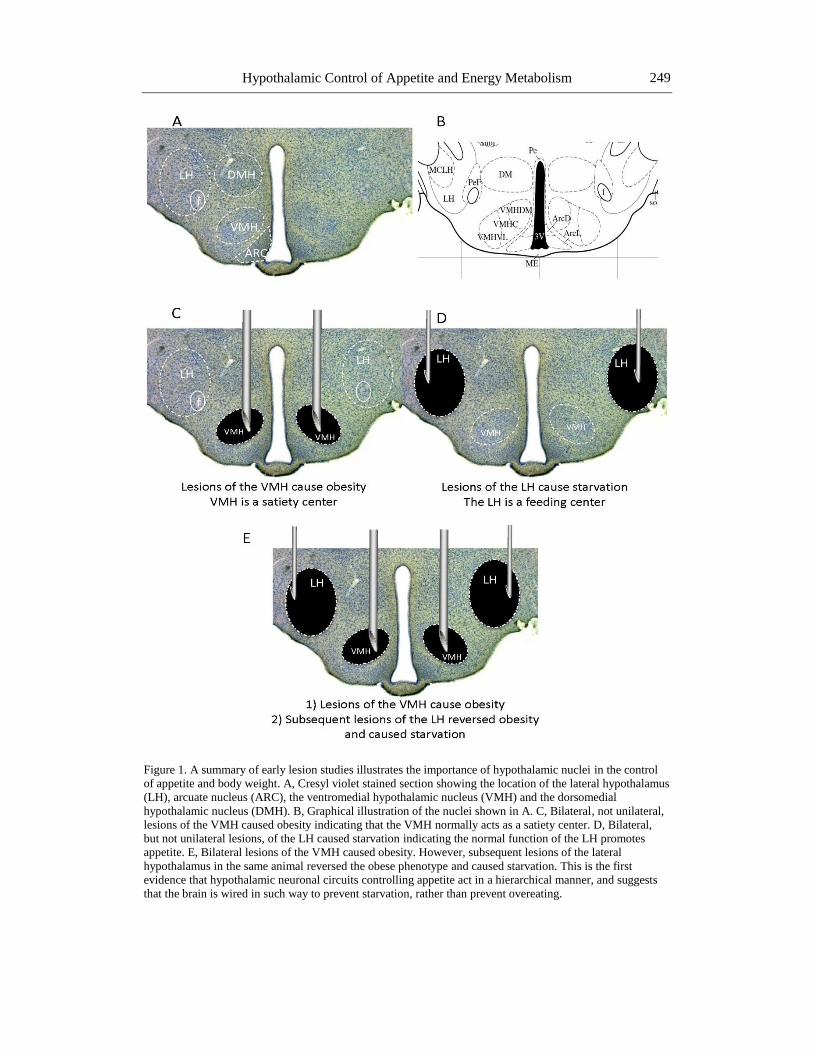
Hypothalamic Control of Appetite and Energy Metabolism 249
Figure 1. A summary of early lesion studies illustrates the importance of hypothalamic nuclei in the control
of appetite and body weight. A, Cresyl violet stained section showing the location of the lateral hypothalamus
(LH), arcuate nucleus (ARC), the ventromedial hypothalamic nucleus (VMH) and the dorsomedial
hypothalamic nucleus (DMH). B, Graphical illustration of the nuclei shown in A. C, Bilateral, not unilateral,
lesions of the VMH caused obesity indicating that the VMH normally acts as a satiety center. D, Bilateral,
but not unilateral lesions, of the LH caused starvation indicating the normal function of the LH promotes
appetite. E, Bilateral lesions of the VMH caused obesity. However, subsequent lesions of the lateral
hypothalamus in the same animal reversed the obese phenotype and caused starvation. This is the first
evidence that hypothalamic neuronal circuits controlling appetite act in a hierarchical manner, and suggests
that the brain is wired in such way to prevent starvation, rather than prevent overeating.

Alex Reichenbach, Romana Stark and B. Zane Andrews
250
Early studies suggested that humeral factors from peripheral tissues affect the
hypothalamic control of appetite. For example, denervation of the gastro-intestinal tract did
not inhibit feeding, suggesting that feeding can occur normally without sensation from the
stomach and intestine (Janowitz and Grossman, 1949). From early experiments researchers
realized that signals from the body must regulate the hypothalamic control of feeding. This
lead to Mayer’s proposal of the glucostatic theory in 1953, in which increased blood glucose
after a meal drives satiety (Mayer, 1953). In 1950, Kennedy proposed the lipostatic
hypothesis, in which feeding is regulated by “the concentration of certain metabolites, as yet
unspecified” (Kennedy, 1950). The nature of the lipostatic signal emanating from the adipose
tissue took another 44 years to be discovered. The discovery of leptin in adipose tissue in
1994 by Friedman and colleagues caused an explosion of interest in the hypothalamic
regulation of appetite and metabolism (Y. Zhang et al., 1994). However, the discovery of
leptin was based on pioneering work by Coleman and colleagues using parabiotic pairs of
mice (Coleman and Hummel, 1969). Parabiosis refers to a skin-to-skin anastomosis formed
by surgical joining two mice from the shoulder to the pelvic girdle. Within 3-4 days, after the
wound has healed, the parabiotic pairs share a common blood supply. Coleman hypothesized
that the obesity observed in mutant db/db mice was caused by a circulating factor and that this
factor would cause obesity in a normal mouse sharing a common parabiotic blood supply.
Although Coleman’s hypothesis was wrong, the results were nonetheless remarkable. The
normal mouse in this parabiotic pair died of starvation. Coleman discovered that the db/db
mouse produced a satiety factor so powerful that it drove the normal mouse to starvation
(Coleman and Hummel, 1969). A parabiotic pairing of the obese ob/ob mouse and a normal
mouse reduced weight gain and food intake in the ob/ob mouse, however the paired mice
lived for months until the end of the experiment (Coleman, 1973), suggesting the normal
mouse produced the same satiety factor as the db/db mouse but at insufficient amounts to
cause starvation. Parabiosis of the ob/ob mouse with the db/db mouse allowed Coleman to
make his profound conclusions. In this pairing, the ob/ob eventually starved to death after 20-
30 days while the db/db mouse gorged on food and gained weight at normal rates (Coleman,
1973). Coleman had demonstrated that the db/db mouse produced a satiety factor but did not
respond to it, whereas the ob/ob mouse responded to the factor but did not produce it (Figure
2). Further, lesion studies showed that the receptor for the satiety factor was found in the
VMH and ARC (Coleman and Hummel, 1970). The cloning of leptin and the leptin receptor
essentially verified all of Coleman’s predictions and highlighted a classic neuroendocrine
feedback loop that controls metabolism (Elmquist et al., 1998; Gautron and Elmquist, 2011).
Leptin, produced by the ob/ob gene in the adipose tissue, enters the blood and activates leptin
receptors, encoded by the db/db gene, in the hypothalamus to reduce food intake and activate
energy expenditure. A reduction in adipose tissue reduces leptin in the circulation and the
entire system is held in check; at a common set point. Many additional endocrine factors have
subsequently been discovered to regulate appetite and energy metabolism through feedback
mechanisms, including ghrelin, peptide YY (PYY) and glucagon like peptide 1. This chapter
will examine new developments in the hypothalamic control of appetite and energy
metabolism.
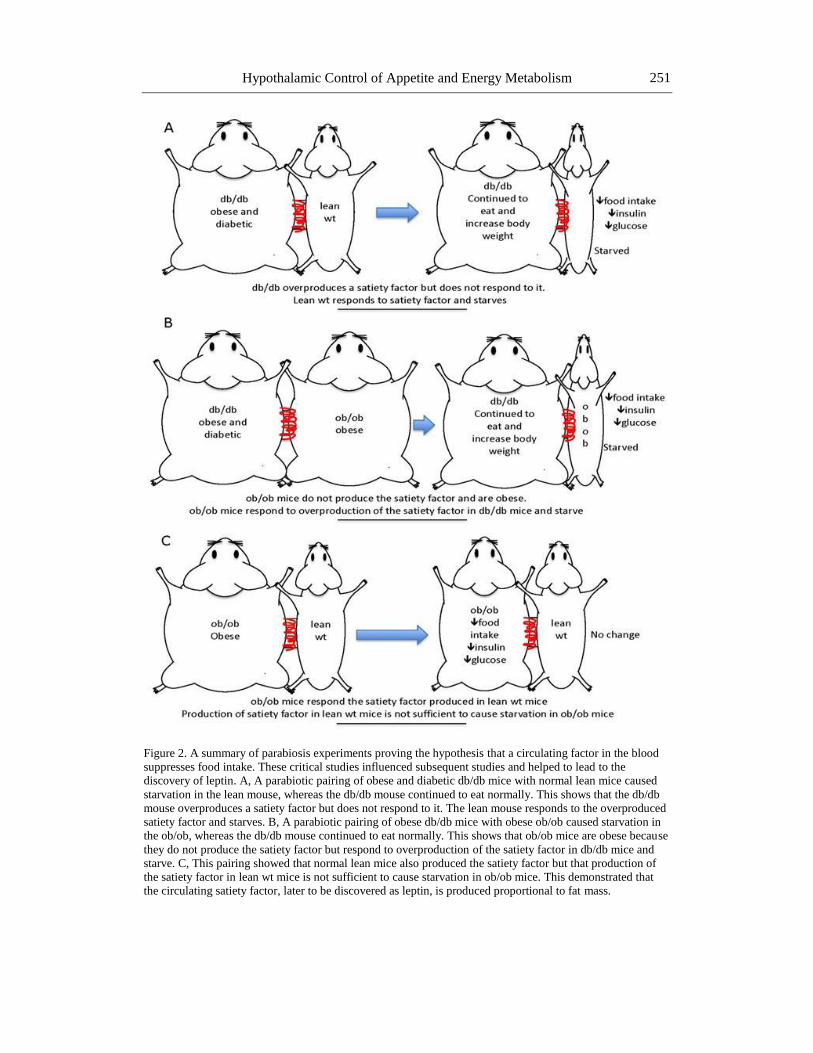
Hypothalamic Control of Appetite and Energy Metabolism 251
Figure 2. A summary of parabiosis experiments proving the hypothesis that a circulating factor in the blood
suppresses food intake. These critical studies influenced subsequent studies and helped to lead to the
discovery of leptin. A, A parabiotic pairing of obese and diabetic db/db mice with normal lean mice caused
starvation in the lean mouse, whereas the db/db mouse continued to eat normally. This shows that the db/db
mouse overproduces a satiety factor but does not respond to it. The lean mouse responds to the overproduced
satiety factor and starves. B, A parabiotic pairing of obese db/db mice with obese ob/ob caused starvation in
the ob/ob, whereas the db/db mouse continued to eat normally. This shows that ob/ob mice are obese because
they do not produce the satiety factor but respond to overproduction of the satiety factor in db/db mice and
starve. C, This pairing showed that normal lean mice also produced the satiety factor but that production of
the satiety factor in lean wt mice is not sufficient to cause starvation in ob/ob mice. This demonstrated that
the circulating satiety factor, later to be discovered as leptin, is produced proportional to fat mass.

Alex Reichenbach, Romana Stark and B. Zane Andrews
252
The Hypothalamic Nuclei Controlling Appetite
ARC
Although initial experiments by Hetherington and Anand showed that the VMH and
lateral hypothalamus are key appetite regulating centers in the hypothamalus, the ARC has
received the most attention over the last 15 years and as arguably the most critical
hypothalamic nucleus. There are two key appetite-regulating neuronal populations in the
ARC. NPY and AgRP are co-expressed in neurons of the ARC and are potent orexigenic
peptides, whereas the POMC precursor protein is cleaved into potent anorexigenic -
melanocyte-stimulating hormone (-MSH) and peptides (Figure 3). Because AgRP is only
expressed in the ARC nucleus, this chapter will refer to NPY/AgRP only as AgRP neurons.
AgRP and POMC neurons in the ARC are arguably considered “first-order” sensory neurons
in the control of food intake and receive, coordinate and respond to changes in metabolic
status. Both AgRP and POMC neurons project to the PVN, where the anorectic effects of -
MSH peptides are mediated by melanocortin 4 receptors (MC4R). NPY Y1 and Y5 receptors
in the PVN mediate the orexigenic effects of NPY, whereas AgRP antagonizes the effect of
-MSH on the MC4R (Figure 4). This system is collectively known as the melanocortin
system (Figure 3). A unique feature of the melanocortin system is the ability of AgRP
neurons to suppress POMC cell firing via inhibitory GABAergic inputs (Andrews et al.,
2008; Cowley et al., 2003). There is no evidence that POMC neurons feed back to inhibit
AgRP neuronal firing despite the expression of GABA in POMC neurons (Hentges et al.,
2004; Hentges et al., 2009). This anatomical arrangement of the melanocortin system
provides one simple mechanism through which the hypothalamus is geared to promote food
intake, rather than satiety. For instance, activation of AgRP neurons directly increases food
intake while simultaneously inhibiting satiety, analogous to driving a car, in which maximal
speed is achieved by releasing the brake and pressing the accelerator. From an evolutionary
standpoint, this melanocortin circuitry maintains a hunger stimulus during periods of food
scarcity and promotes food intake to ensure survival. However, in today’s energy-abundant
society, this circuitry contributes to the obesity epidemic by increasing hyperphagia.
The hypothalamic melanocortin circuits in the ARC and PVN have been extensive
studied, but it should be noted that POMC and AgRP neurons project to other regions of the
brain that are important for appetite regulation, including the medial preoptic area (MPA), the
nucleus of the solitary tract (NTS) and the parabrachial nucleus (PBN) (Broberger et al.,
1998; Elias et al., 1998). Understanding how these melanocortin circuits influence appetite
regulation in these distal target areas will be important future directions. Indeed, recent
research shows that GABAergic transmission in AgRP neurons innervating the PBN is
critical to prevent starvation (Wu et al., 2009; see below).
POMC Neurons Controlling Food Intake and Body Weight
The critical importance of POMC neurons in appetite and energy balance is highlighted
by elegant conditional gene ablation experiments. Ablation of POMC neurons in adulthood
produced an increase in food intake and body weight (Gropp et al., 2005). Because ablation of
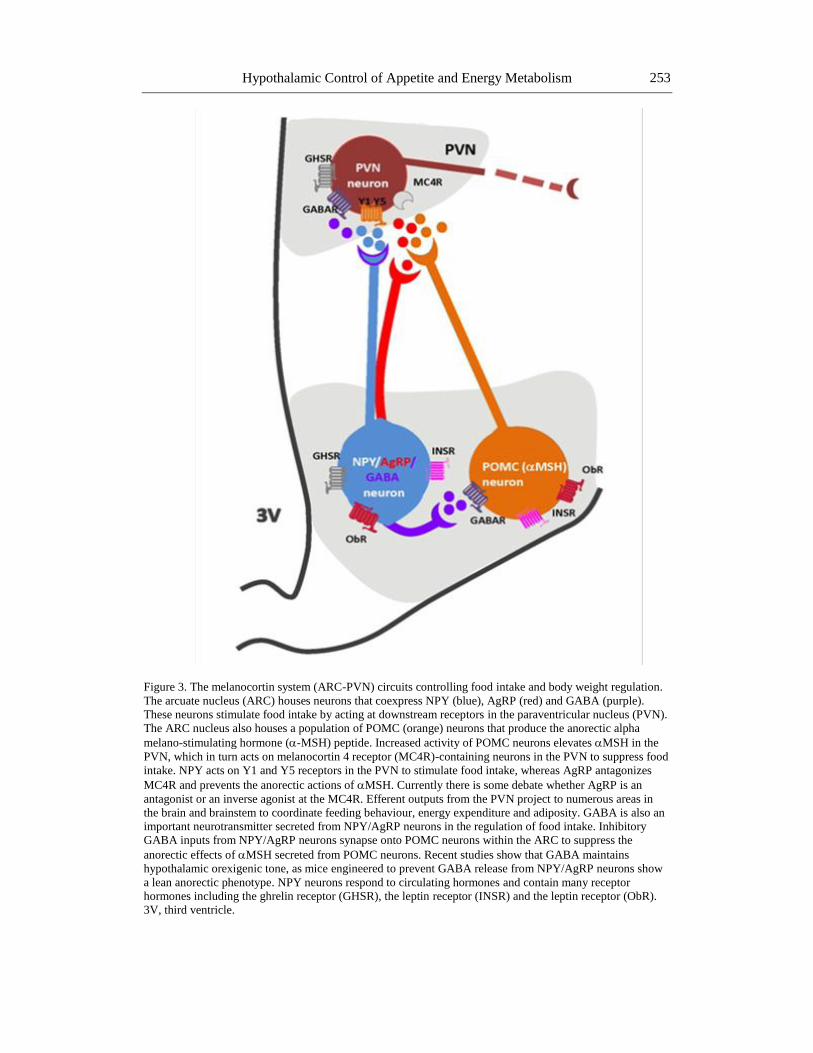
Hypothalamic Control of Appetite and Energy Metabolism 253
Figure 3. The melanocortin system (ARC-PVN) circuits controlling food intake and body weight regulation.
The arcuate nucleus (ARC) houses neurons that coexpress NPY (blue), AgRP (red) and GABA (purple).
These neurons stimulate food intake by acting at downstream receptors in the paraventricular nucleus (PVN).
The ARC nucleus also houses a population of POMC (orange) neurons that produce the anorectic alpha
melano-stimulating hormone (-MSH) peptide. Increased activity of POMC neurons elevates MSH in the
PVN, which in turn acts on melanocortin 4 receptor (MC4R)-containing neurons in the PVN to suppress food
intake. NPY acts on Y1 and Y5 receptors in the PVN to stimulate food intake, whereas AgRP antagonizes
MC4R and prevents the anorectic actions of MSH. Currently there is some debate whether AgRP is an
antagonist or an inverse agonist at the MC4R. Efferent outputs from the PVN project to numerous areas in
the brain and brainstem to coordinate feeding behaviour, energy expenditure and adiposity. GABA is also an
important neurotransmitter secreted from NPY/AgRP neurons in the regulation of food intake. Inhibitory
GABA inputs from NPY/AgRP neurons synapse onto POMC neurons within the ARC to suppress the
anorectic effects of MSH secreted from POMC neurons. Recent studies show that GABA maintains
hypothalamic orexigenic tone, as mice engineered to prevent GABA release from NPY/AgRP neurons show
a lean anorectic phenotype. NPY neurons respond to circulating hormones and contain many receptor
hormones including the ghrelin receptor (GHSR), the leptin receptor (INSR) and the leptin receptor (ObR).
3V, third ventricle.
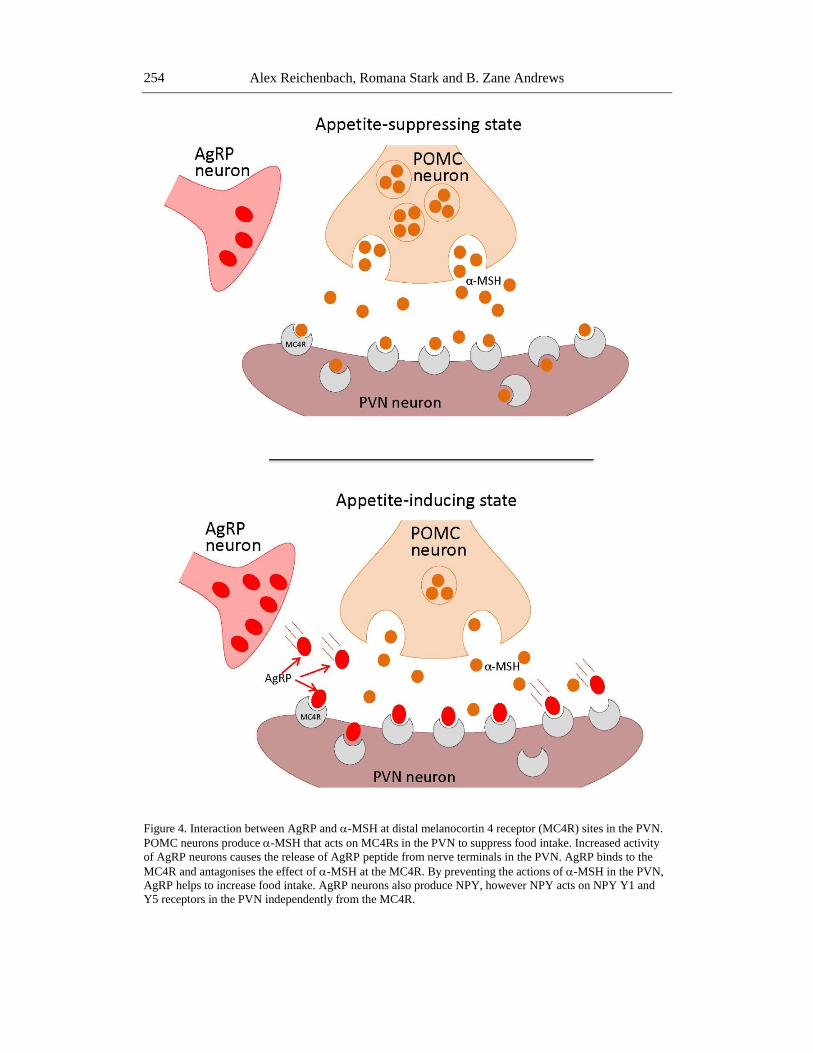
Alex Reichenbach, Romana Stark and B. Zane Andrews
254
Figure 4. Interaction between AgRP and -MSH at distal melanocortin 4 receptor (MC4R) sites in the PVN.
POMC neurons produce -MSH that acts on MC4Rs in the PVN to suppress food intake. Increased activity
of AgRP neurons causes the release of AgRP peptide from nerve terminals in the PVN. AgRP binds to the
MC4R and antagonises the effect of -MSH at the MC4R. By preventing the actions of -MSH in the PVN,
AgRP helps to increase food intake. AgRP neurons also produce NPY, however NPY acts on NPY Y1 and
Y5 receptors in the PVN independently from the MC4R.

Hypothalamic Control of Appetite and Energy Metabolism 255
POMC neurons leads to obesity, many recent studies have focused on the hormonal and
molecular mechanisms regulating POMC neuronal function and the physiological
consequence of perturbations to POMC neurons. Seminal studies show that POMC neurons
regulate the hypophagic actions of leptin signaling in the brain (Balthasar et al., 2004),
although not exclusively as VMH neurons are also important (Bingham et al., 2008; Dhillon
et al., 2006; see below). Activation of the leptin receptor on POMC neurons initiates
hypophagia through the janus kinase (JAK) - signal-transducer activator of transcription
(STAT) 3 pathway (Balthasar et al., 2004; Bates et al., 2003; Munzberg et al., 2003). Leptin
binds to its receptor and autophosphorylates JAK2 that then recruits and phosphorylates
STAT3 (Baumann et al., 1996). pSTAT3 dimerizes and enters the nucleus where is binds to
specific DNA elements in the POMC or AgRP promoter to activate POMC and repress AgRP
gene expression respectiviely (Bates et al., 2003; Figure 5). Indeed, nuclear pSTAT3
immunostaining is extensively used as a marker of leptin sensitive neurons in the brain. There
is a wealth of literature showing that leptin is a major regulator of POMC. Leptin-deficiency
or leptin receptor deficiency reduces POMC gene expression and leptin replacement to ob/ob
mice increases POMC (Korner et al., 1999; Mizuno et al., 1998). Moreover, fasting reduces
leptin levels and also reduces POMC gene expression (Korner et al., 1999; Mizuno et al.,
1998). After activating the STAT3 pathway, leptin induces suppressor of cytokine signaling 3
(SOCS3), which acts as a feedback inhibitor of leptin receptor signaling (Elias et al., 1999).
Dynamic control of the STAT3 pathway is required for ongoing leptin sensitivity in POMC
neurons. For example, the non-tyrosine phosphatase SHP2 helps to dephosphorylate the
STAT3 pathway and maintain leptin sensitivity, whereas dephosphorylation of pSTAT3 by
the protein tyrosine phosphatase PTPB1, reduces POMC leptin sensitivity (Banno et al.,
2010). However, POMC deletion of STAT3 results in only a slight increase in body weight
and food intake (Xu et al., 2007) suggesting other signaling pathways in POMC neurons are
involved in leptin’s anorectic action.
In addition to the STAT3 pathway, leptin can also utilize the phosphatidylinositol 3-
kinase (PI3K) pathway within POMC cells. PI3K phosphorylates the membrane lipidphosp-
hatidylinositol-4,5-bisphosphate (PIP2) after activation to form phosphatidylinositol-3,4,5-
trisphosphate (PIP3 ). The tumour suppressor PTEN (phosphatase and tensin homologue) is a
lipid phosphatase that dephosphorylates PIP3 to PIP2. Accumulation of PIP3 recruits several
kinases to the plasma membrane, which propagates further signaling cascades. Leptin directly
activates PI3K signaling in POMC neurons (Xu et al., 2005) and inhibiting the PI3K pathway
attenuates both the anorectic effects of insulin and leptin (Niswender et al., 2001; Niswender
and Schwartz, 2003). Genetic deletion of PI3K subunits in POMC neurons reduced acute
feeding responses to leptin and attenuated leptin-induced action potential firing in POMC
neurons (Hill et al., 2008). Despite preventing the acute actions of leptin, genetic deletion of
PI3K in POMC neurons had no effect on long-term body weight regulation.
When all the data is considered together, it is clear that leptin activates POMC neurons,
either via the STAT3 or the PI3K pathway, to regulate feeding and body weight. However,
gene deletion studies in POMC neurons show that other neurons and hypothalamic nuclei are
critical to reproduce the full metabolic phenotype seen in leptin-receptor deficient mice.
Because deletion of leptin receptors on POMC neurons does not fully recapitulate the obesity
seen in db/db leptin receptor-deficient mice, a recent study examined whether leptin-receptor
containing neurons regulate POMC neurons presynaptically. In this study, leptin receptors
Alex Reichenbach, Romana Stark and B. Zane Andrews
256
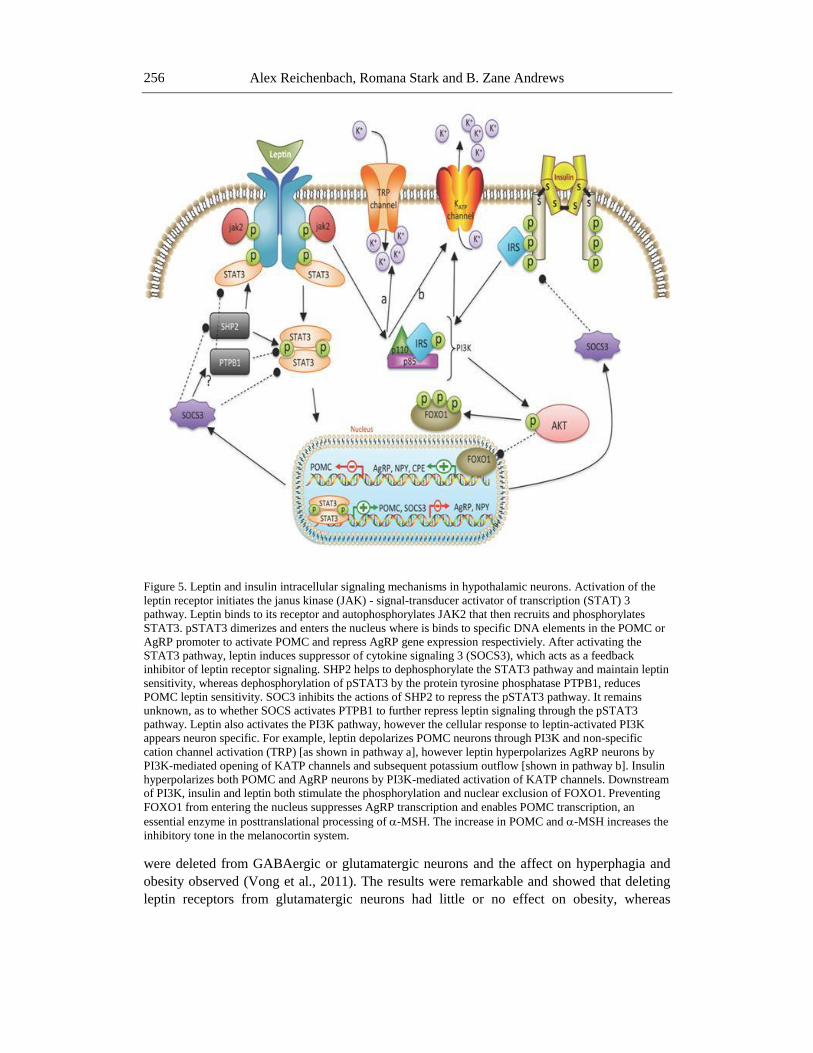
Figure 5. Leptin and insulin intracellular signaling mechanisms in hypothalamic neurons. Activation of the
leptin receptor initiates the janus kinase (JAK) - signal-transducer activator of transcription (STAT) 3
pathway. Leptin binds to its receptor and autophosphorylates JAK2 that then recruits and phosphorylates
STAT3. pSTAT3 dimerizes and enters the nucleus where is binds to specific DNA elements in the POMC or
AgRP promoter to activate POMC and repress AgRP gene expression respectiviely. After activating the
STAT3 pathway, leptin induces suppressor of cytokine signaling 3 (SOCS3), which acts as a feedback
inhibitor of leptin receptor signaling. SHP2 helps to dephosphorylate the STAT3 pathway and maintain leptin
sensitivity, whereas dephosphorylation of pSTAT3 by the protein tyrosine phosphatase PTPB1, reduces
POMC leptin sensitivity. SOC3 inhibits the actions of SHP2 to repress the pSTAT3 pathway. It remains
unknown, as to whether SOCS activates PTPB1 to further repress leptin signaling through the pSTAT3
pathway. Leptin also activates the PI3K pathway, however the cellular response to leptin-activated PI3K
appears neuron specific. For example, leptin depolarizes POMC neurons through PI3K and non-specific
cation channel activation (TRP) [as shown in pathway a], however leptin hyperpolarizes AgRP neurons by
PI3K-mediated opening of KATP channels and subsequent potassium outflow [shown in pathway b]. Insulin
hyperpolarizes both POMC and AgRP neurons by PI3K-mediated activation of KATP channels. Downstream
of PI3K, insulin and leptin both stimulate the phosphorylation and nuclear exclusion of FOXO1. Preventing
FOXO1 from entering the nucleus suppresses AgRP transcription and enables POMC transcription, an
essential enzyme in posttranslational processing of -MSH. The increase in POMC and -MSH increases the
inhibitory tone in the melanocortin system.
were deleted from GABAergic or glutamatergic neurons and the affect on hyperphagia and
obesity observed (Vong et al., 2011). The results were remarkable and showed that deleting
leptin receptors from glutamatergic neurons had little or no effect on obesity, whereas

Hypothalamic Control of Appetite and Energy Metabolism 257
deleting leptin receptors on GABAergic neurons caused marked obesity, similar to that seen
in leptin receptor deficient db/db mice. Further analysis showed that leptin suppressed
presynaptic GABAergic inhibitory inputs to POMC neurons, which subsequently disinhibited
POMC neuronal firing and increased satiety (Figure 6). These leptin receptor neurons that
synapse with POMC neurons are found in the ARC (majority of which are not AgRP
neurons), DMH and lateral hypothalamus. This study suggests that although leptin acts
directly on POMC neurons it also regulates POMC neurons indirectly by acting on upstream
presynaptic GABAergic, and not glutamatergic, neurons. Moreover, the genetic deletion of
leptin receptors from GABAergic neurons, rather than from POMC neurons, more closely
resembles the obese phenotype of leptin receptor deficient mice. Thus, presynaptic regulation
of POMC neuronal firing is essential to maintain normal energy metabolism.
In addition to leptin, insulin targets POMC neurons and influences food intake and
energy metabolism. POMC neurons express both insulin and leptin receptors, although a
recent study suggests that two distinct subpopulations of POMC neurons exist; those that
respond to insulin and those that respond to leptin (Williams et al., 2010). Indeed, insulin
directly activates PI3K signaling in POMC neurons and inhibiting PI3K blocks the anorectic
effects of insulin (Xu et al., 2005). However, recent studies have questioned the acute
anorectic role of insulin in rats (Jessen et al., 2010; Tups et al., 2010), or suggest that the
anorectic actions of insulin require simultaneous leptin receptor activation (Tups et al., 2010),
possibly due to enhanced intracellular signaling through STAT3 and PI3K pathway crosstalk
(Figure 5), as was recently described to regulate glucose homeostasis (Koch et al., 2010).
Moreover, genetic deletion of the insulin receptor in POMC neurons did not affect long term
body weight, glucose homeostasis or food intake (Konner et al., 2007), suggesting neurons
other than POMC are required for insulin’s ability to control energy homeostasis. Although
leptin and insulin both appear to have anorectic actions of food intake, only genetic deletion
of the leptin receptor on POMC neurons causes obesity (Balthasar et al., 2004; Hill et al.,
2010; Konner et al., 2007). Moreover, when both the insulin receptor and the leptin receptor
are deleted in POMC neurons, the obesity observed in the leptin receptor-deficient POMC
neurons is completely reversed. This suggests that insulin and leptin have different functions
in body weight regulation. Recent studies demonstrated that nicotine activates POMC
neurons and attenuates food intake, providing strong biological evidence that links cigarette
smoking to lower body weights (Mineur et al., 2011).
POMC Neurons Regulate Whole Body Glucose Homeostasis
POMC neurons not only affect energy metabolism by reducing food intake, but also play
a salient role in maintaining whole body glucose homeostasis. Studies show that impaired
glucose sensing in POMC neurons leads to peripheral glucose intolerance and insulin
resistance (Parton et al., 2007) via a brain-liver circuit involving hypothalamic neurons and
vagal autonomic innervation of liver (Yi et al., 2010). This effect involves the coordination
and integration of multiple hormonal inputs on POMC neurons. A recent elegant study
illustrated that both insulin and leptin receptor signaling in POMC neurons is required for
normal glucose homeostasis (Hill et al., 2010). In this study, single deletion of the insulin
receptor or the leptin receptor on POMC had no effect on glucose homeostasis, including
Alex Reichenbach, Romana Stark and B. Zane Andrews
258
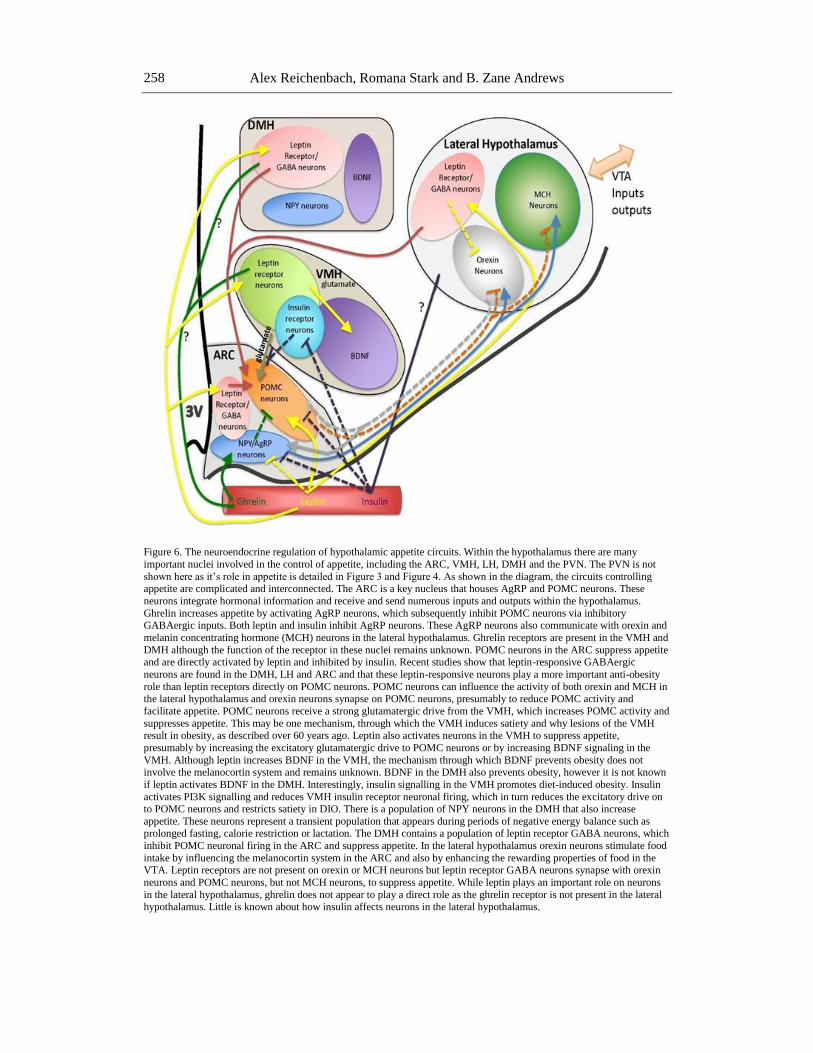
Figure 6. The neuroendocrine regulation of hypothalamic appetite circuits. Within the hypothalamus there are many
important nuclei involved in the control of appetite, including the ARC, VMH, LH, DMH and the PVN. The PVN is not
shown here as it’s role in appetite is detailed in Figure 3 and Figure 4. As shown in the diagram, the circuits controlling appetite are complicated and interconnected. The ARC is a key nucleus that houses AgRP and POMC neurons. These
neurons integrate hormonal information and receive and send numerous inputs and outputs within the hypothalamus.
Ghrelin increases appetite by activating AgRP neurons, which subsequently inhibit POMC neurons via inhibitory GABAergic inputs. Both leptin and insulin inhibit AgRP neurons. These AgRP neurons also communicate with orexin and
melanin concentrating hormone (MCH) neurons in the lateral hypothalamus. Ghrelin receptors are present in the VMH and
DMH although the function of the receptor in these nuclei remains unknown. POMC neurons in the ARC suppress appetite and are directly activated by leptin and inhibited by insulin. Recent studies show that leptin-responsive GABAergic
neurons are found in the DMH, LH and ARC and that these leptin-responsive neurons play a more important anti-obesity
role than leptin receptors directly on POMC neurons. POMC neurons can influence the activity of both orexin and MCH in the lateral hypothalamus and orexin neurons synapse on POMC neurons, presumably to reduce POMC activity and
facilitate appetite. POMC neurons receive a strong glutamatergic drive from the VMH, which increases POMC activity and
suppresses appetite. This may be one mechanism, through which the VMH induces satiety and why lesions of the VMH result in obesity, as described over 60 years ago. Leptin also activates neurons in the VMH to suppress appetite,
presumably by increasing the excitatory glutamatergic drive to POMC neurons or by increasing BDNF signaling in the
VMH. Although leptin increases BDNF in the VMH, the mechanism through which BDNF prevents obesity does not involve the melanocortin system and remains unknown. BDNF in the DMH also prevents obesity, however it is not known
if leptin activates BDNF in the DMH. Interestingly, insulin signalling in the VMH promotes diet-induced obesity. Insulin
activates PI3K signalling and reduces VMH insulin receptor neuronal firing, which in turn reduces the excitatory drive on to POMC neurons and restricts satiety in DIO. There is a population of NPY neurons in the DMH that also increase
appetite. These neurons represent a transient population that appears during periods of negative energy balance such as prolonged fasting, calorie restriction or lactation. The DMH contains a population of leptin receptor GABA neurons, which
inhibit POMC neuronal firing in the ARC and suppress appetite. In the lateral hypothalamus orexin neurons stimulate food
intake by influencing the melanocortin system in the ARC and also by enhancing the rewarding properties of food in the VTA. Leptin receptors are not present on orexin or MCH neurons but leptin receptor GABA neurons synapse with orexin
neurons and POMC neurons, but not MCH neurons, to suppress appetite. While leptin plays an important role on neurons
in the lateral hypothalamus, ghrelin does not appear to play a direct role as the ghrelin receptor is not present in the lateral hypothalamus. Little is known about how insulin affects neurons in the lateral hypothalamus.

Hypothalamic Control of Appetite and Energy Metabolism 259
plasma insulin concentration, hepatic glucose production, peripheral glucose disposal, glucose
infusion rate during euglycemic clamps, glucose tolerance tests and insulin tolerance tests, in
accord with a previously published paper (Konner et al., 2007). However, deletion of both
insulin and leptin receptors on POMC neurons caused insulin resistance and glucose
intolerance independent of changes in body weight. These results show a level of functional
redundancy in insulin and leptin actions on POMC neurons in terms of glucose homeostasis,
as deleting both insulin and leptin receptors prevents the compensatory change. Importantly,
these results highlight the need to examine how hormones and nutrients interact to affect not
just POMC function, but also other important neurons that control energy homeostasis. The
fact that impaired glucose tolerance occurs independent from changes in body weight and
food intake, highlights the complexity of signaling mechanisms within POMC neurons.
Although insulin and leptin receptors act together to maintain glucose homeostasis, the
downstream molecular mechanisms remain ambiguous. POMC neurons produce -MSH and
activate MC4R receptors in the PVN. Central -MSH injection can decrease basal insulin
release (Banno et al., 2007) or increase insulin-stimulated glucose uptake and production
(Heijboer et al., 2005; Obici et al., 2001). Further, MC4R antagonism failed to block
hyperinsulinemia-induced inhibition of hepatic glucose production (Obici et al., 2002a; Obici
et al., 2002b), but intracerebroventricular (icv) infusion of -MSH stimulates glucose
production via gluconeogenesis (Gutierrez-Juarez et al., 2004). These studies indicate -MSH
may act on non-MC4Rs in the hypothalamus to regulate glucose production. Future studies
should think beyond the traditional confines of the ARC-PVN melanocortin circuits to help
explain how POMC neurons control glucose homeostasis.
Agrp Neurons Regulate Food Intake and Body Weight
AgRP neurons in the ARC nucleus are critical for ghrelin-induced food intake as genetic
ablation of AgRP (Luquet et al, 2007) or double-knockout of NPY and AgRP prevents
ghrelin-induced food intake (Chen et al., 2004). Indeed, ghrelin induces feeding by robustly
stimulating NPY and AgRP neuronal activity as assessed by electrophysiology (Andrews et
al., 2008; Cowley et al., 2003) or fos immunoreactivity (Andrews et al., 2008; Cowley et al.,
2003; Hewson and Dickson, 2000; Wang et al., 2002) and gene expression (Chen et al., 2004;
Kamegai et al., 2000, 2001; Nakazato et al., 2001). Consistent with the effect of ghrelin on
NPY and AgRP neuronal activity, the GHSR1a is expressed on >90% of all NPY neurons in
the ARC (Willesen et al., 1999). However, the GHSR1a is only expressed on less than 8% of
POMC neurons (Willesen et al., 1999).
Despite the well-described effects of ghrelin on AgRP neurons in the ARC, neither NPY
nor AgRP single gene deletion (Erickson et al., 1996), nor NPY and AgRP double knockout
affected appetite and body weight (Qian et al., 2002). In order to rule out the development of
compensatory mechanisms that could potentially explain the lack of effect in knockout
models, two independent laboratories generated conditional AgRP neuronal ablation
techniques. Conditional ablation of AgRP neurons during adulthood in the ARC, using the
human diphtheria toxin targeted to the AgRP locus, results in a rapid reduction in food intake
and body weight (Gropp et al., 2005; Luquet et al., 2005). In addition, Luquet et al (Luquet et
al., 2005) showed that ablation of AgRP neurons during the early postnatal period did not

Alex Reichenbach, Romana Stark and B. Zane Andrews
260
result anorexia and weight loss. These results argue that reorganization of the hypothalamic
circuits during the developmental period can overcome AgRP neuronal ablation.
However, this conditional ablation approach is also not without technical limitations. For
example, ablation of the AgRP neurons with diphtheria toxin destroys the neuron and the
entire contents of the AgRP neuron, and as such the role of AgRP peptide is not tested, but
rather the AgRP neuron with all neurotransmitters and neuropeptides. Recent studies show
that GABA in AgRP neurons is the critical neurotransmitter affecting orexigenic pathways.
GABA Signaling in the ARC
GABA signaling in the brain influences appetite, and both AgRP neurons and POMC
neurons in the ARC contain GABA (Hentges et al., 2004; Hentges et al., 2009; Horvath et al.,
1997; Tong et al., 2008). Approximately 50-60% of all AgRP neurons express GABA
(Horvath et al., 1997; Luquet et al., 2005; Wu et al., 2008), which led researchers to
hypothesize that GABAergic neurotransmission in AgRP plays an important role in appetite.
Initial observations illustrated a melanocortin-dependent mechanism whereby inhibitory
GABAeric inputs from active AgRP neurons suppress POMC neuronal activity (Andrews et
al., 2008; Cowley et al., 2003). GABA co-localizes with AgRP neurons that innervate POMC
neurons in the ARC and distal target nuclei including the PVN (Horvath et al., 1997; Pu et al.,
1999). Ghrelin activation of AgRP neurons increases GABAergic inhibitory postsynaptic
currents and inhibitory synapses on POMC neurons (Andrews and Horvath, 2008; Andrews et
al., 2008). Increased GABAergic inhibitory inputs on POMC neurons elevates food intake by
lowering anorexigenic POMC neuronal activity. A recent study showed that deletion of
vesicular GABA transporter in AgRP neurons, which prevents the synaptic release of GABA,
removes the inhibitory tone onto postsynaptic POMC cells and produces a lean phenotype
that is resistant to diet-induced obesity (Tong et al., 2008). Further, these mice have an
attenuated hyperphagic response ghrelin (Tong et al., 2008). Although, GABA release from
AgRP neurons suppresses POMC activity, Vong et al (Vong et al., 2011) showed that only
30% of all presynaptic GABAergic POMC inputs came from AgRP neurons, the remaining
inputs came from non-AgRP GABA neurons in the ARC or from the DMH and lateral
hypothalamus.
GABAergic AgRP neurons also have melanocortin-independent effects on appetite.
Chronic blockade of the melanocortin pathway should lead to hyperphagia and obesity, as -
MSH cannot act on the MC4R to suppress food intake. However, ablation of AgRP neurons
still causes starvation even in mice with chronic blockade of the melanocortin pathway (Wu
et al., 2008). Wu et al discovered that GABA release from AgRP nerve terminals in the
parabrachical nucleus (PBN) in the hindbrain is essential to maintain appetite. Although
ablation of AgRP neurons caused starvation, this could be prevented by direct infusion of
GABAA agonists into the PBN. Moreover, infusion of the GABAA antagonist, bicuculline,
into the PBN inhibits feeding in a dose dependent manner and inactivation of GABA
biosynthesis in the ARC causes anorexia. These results show that sudden loss of AgRP
neurons prevents GABA signaling in the PBN and results in hyperactivity of a population of
PBN neurons. The synaptic output of these hyperactive PBN neurons must act as a brake on
an essential feeding circuit, which leads to starvation. Future studies are required to elucidate

Hypothalamic Control of Appetite and Energy Metabolism 261
the mechanisms underlying the function of the PBN in food intake. These studies clearly
show that GABA signaling in hypothalamic AgRP neurons regulates appetite.
Approximately 40% of POMC neurons in the ARC nucleus also co-express GABA
(Hentges et al., 2004; Hentges et al., 2009), although the direct effects of GABA signaling
from POMC neurons on appetite and energy metabolism remain enigmatic. Another 25% of
POMC neurons also express glutamate, raising the possibility that distinct subpopulations of
POMC neurons control physiologically distinct roles in energy metabolism. Because GABA
release from AgRP collateral projections in the ARC inhibit POMC neurons in the ARC, the
question remains; do POMC neurons use GABA to directly inhibit AgRP neurons and thus
create a complex short-loop feedback circuit in the ARC itself. Hentges et al (Hentges et al.,
2004) demonstrated that although ≈40% of POMC neurons co-express GABA in nerve
terminal regions, little or no POMC neurons co-express GABA or vesicular glutamate in the
ARC itself, suggesting that there is no GABAergic reciprocal feedback from POMC neurons
to AgRP neurons. These recent studies that focus on GABA in AgRP neurons herald a new
era in the central control of appetite. Although much of the work over the last 2 decades
focuses on neuropeptides such as NPY, AgRP and POMC, the future advances in the field
will elucidate how classic neurotransmitters such as GABA and glutamate affect neuronal
circuits controlling appetite. Indeed, the recent demonstration that leptin receptor deletion on
GABA, but not glutamatergic neurons (Vong et al., 2011), results in marked obesity
exemplifies this notion.
Evolutionary Considerations of the Melanocortin System
Even though the conditional ablation of AgRP and POMC neurons destroys the entire
neuron and not just the peptide of interest, these studies identify an intriguing evolutionary
adaptation. AgRP neuron-ablated mice without any intervention starved to the point of death,
whereas POMC neuron-ablated mice ‘only’ became obese (Gropp et al., 2005; Luquet et al.,
2005). These results imply a greater evolutionary selection pressure for AgRP cell survival,
via appropriate GABA transmission at innervation sites, compared to POMC cell survival in
the ARC. AgRP activity is a signal of negative energy balance in the brain that promotes food
intake to reestablish normal energy balance. On the other hand, POMC neurons respond to
signals of positive energy balance, such as glucose, insulin and leptin, and help to reduce food
intake and maintain normal energy balance. Given that our evolutionary history was almost
completely dominated by periods of negative energy balance, it is not surprising that AgRP
neurons developed different molecular mechanisms, compared to POMC neurons, to preserve
cell function and appetitive drive. POMC neuronal activity suppresses food intake and
therefore there is no adaptive evolutionary advantage to preserve POMC cell function.
Without AgRP neurons in the ARC neurons starvation would occur, whereas removing
POMC signaling ‘only’ causes obesity.
Hormonal Regulation of Agrp Neurons
Similar to POMC neurons, AgRP neurons receive hormonal input from the periphery
including insulin, leptin and ghrelin. Leptin receptors are found on AgRP neurons in the ARC

Alex Reichenbach, Romana Stark and B. Zane Andrews
262
(Draper et al., 2010) and leptin suppresses AgRP gene expression and transcription (Schwartz
et al., 1998) through nuclear exclusion of FOXO1 (Fukuda et al., 2008; Kitamura et al., 2006)
to restrict food intake. Leptin activates pSTAT3 and suppresses AgRP and NPY gene
expression (Xu et al., 2007). Leptin receptor activation of AgRP neurons plays an important
role in overall energy homeostasis as AgRP neurons lacking the leptin receptor displayed
adiposity and increased body weight relative to controls (van de Wall et al., 2008). Deleting
key intracellular signaling pathways had the same effect. Mice lacking STAT3 in AgRP were
mildly hyperphagic and unresponsive to leptin. Consistent with this result, constitutively
active STAT3 in AgRP neurons causes leanness and prevents high-fat diet-induced obesity
due to increased locomotor activity and subsequent energy expenditure (Mesaros et al., 2008).
No changes in food intake or gene expression were observed. Moreover, restoring leptin
receptors in the ARC nucleus increased locomotor activity (Coppari et al., 2005). These
studies show neuron-specific effects of leptin in the ARC. Leptin activates STAT3 signaling
in POMC neurons to suppress appetite and activates STAT3 in AgRP neurons to increase
energy expenditure. Collectively, these studies suggest that leptin-induced STAT3 signaling
in AgRP neurons provides an unappreciated level of tonic inhibition on AgRP and subsequent
food intake.
This idea is further supported by electrophysiological studies that show leptin
hyperpolarizes AgRP neurons by activating ATP-sensitive potassium channels to inhibit
action potential firing (Spanswick et al., 1997; van den Top et al., 2004). Further, leptin
application suppresses PI3K signaling in AgRP neurons but leptin withdrawal from the slice
preparation facilitates PI3K signaling in AgRP neurons, thereby mimicking low leptin
conditions that increase AgRP expression, such as fasting (Xu et al., 2005). Finally, a recent
study showed that leptin inhibits AgRP neuron firing through MAPK signaling and
subsequent L-calcium current inhibition (Wang et al., 2008), linking neuronal firing
properties to intracellular signal transduction.
Insulin is also an important regulator of AgRP neuronal function, as insulin inhibits
AgRP gene expression (Schwartz et al., 1992), restricts action potential firing via ATP-
dependent potassium channel (KATP)-induced hyperpolarization in AgRP neurons (Konner et
al., 2007) and suppresses food intake. The insulin receptor substrate is localized to AgRP
neurons (Pardini et al., 2006). The effects of insulin on AgRP neurons are mediated
predominantly through the phosphatidylinositol 3-OH-kinase (PI3K) (Xu et al., 2005). Brain-
specific deletion of insulin receptors causes hyperphagia and susceptibility to diet-induced
weight gain (Bruning et al., 2000), but despite the known effects of insulin on AgRP neurons,
deletion of the insulin receptor on AgRP neurons, or POMC neurons, did not affect food
intake or body weight (Konner et al., 2007). This illustrates that insulin receptor neurons,
other than, or in addition to AgRP and POMC neurons are required for the anorectic effect of
insulin on appetite and body weight.
Central insulin acts in the hypothalamus to robustly inhibit hepatic glucose production
(Obici et al., 2002) via an insulin receptor action in the hypothalamus (Obici, Feng et al.,
2002). This effect has been unequivocally ascribed to AgRP neurons as genetic deletion of
the insulin receptor on AgRP neurons, but not POMC neurons, fails to suppress hepatic
glucose production during euglycemic hyperinsulinemic clamp studies (Konner et al., 2007).
It is important to keep in mind that central NPY injection increases glucose production and
suppresses hepatic insulin sensitivity (Marks and Waite, 1997) and combining icv NPY
injections with hyperinsulinemic clamps partially blocks the inhibitory effect of peripheral

Hypothalamic Control of Appetite and Energy Metabolism 263
hyperinsulinemia on hepatic glucose production. Moreover, denervation of the hepatic
sympathetic nerves blocks the effect of central NPY on hepatic glucose production (van den
Hoek et al., 2008). Thus, the inhibitory effect of insulin on hepatic glucose production
requires an insulin-mediated suppression of AgRP neuronal activity and subsequent
sympathetic nerve activity.
VMH
Early studies from the 1940s identified the VMH as a critical nucleus regulating appetite.
Examination of the neuronal circuitry in the hypothalamus revealed that the VMH and ARC
nucleus are reciprocally connected. For example the VMH receives afferent projections from
the ARC (van den Hoek et al., 2008) and the ARC receives strong excitatory inputs from the
VMH (Sternson et al., 2005). The VMH contains MC4R and NPY Y1, Y2, Y5 receptors and
NPY infusions into the VMH increases feeding (Bouali et al., 1995; Harrold et al., 1999).
Sternson et al, used laser scanning photostimulation to show that POMC neurons received
strong excitatory input from the medial VMH, whereas AgRP neurons only received weak
inhibitory input from within the ARC. Fasting diminished the strength of the excitatory input
from the VMH to POMC neurons (Sternson et al., 2005). Further, the sensitivity of VMH
neurons to -MSH in food-deprived rats or rats pretreated with AgRP (Li and Davidowa,
2004) is suppressed, indicating that negative energy balance reduces the anorectic drive of
VMH neurons onto POMC neurons. A recent study showed that deleting glutamate synaptic
transmission from VMH neurons, increased long-term food intake and susceptibility to DIO
(Tong et al., 2007), suggesting that the major excitatory output from the VMH is to suppress
food intake, presumably acting on the POMC neurons, as described by Sternson et al
(Sternson et al., 2005).
Unlike the neurons in the ARC nucleus, little is known about the chemical phenotype of
VMH neurons that control appetite circuits. Nevertheless, brain-derived neurotrophic factor
(BDNF) is one key protein as it is abundantly expressed in the VMH. Genetic deletion of
BDNF or its receptor, TrkB, causes obesity in mice (Rios et al., 2001; Xu et al., 2003) and
they are two of only a few obesity candidate genes in humans (Ramachandrappa and Farooqi,
2011). Fasting reduces leptin levels and suppresses BDNF gene expression specifically in the
VMH, whereas leptin increases VMH BDNF gene expression, illustrating that BDNF is an
important regulatory step in leptin signaling in the VMH. Indeed, deleting BDNF in the VMH
and DMH produces hyperphagia and causes obesity (Unger et al., 2007). Importantly, the
melanocortin system does not mediate the anorectic effects of BDNF, as BDNF still
suppresses appetite and body weight in MC4R deficient mice (Xu et al., 2003). This indicates
that BDNF may influence appetite and body weight through other means, such as classic
neurotransmitter systems. Indeed, deleting glutamate release from VMH neurons results in
obesity and hyperphagia (Tong et al., 2007), support the idea that BDNF may regulate
glutamatergic neurotransmission in the VMH.
Leptin receptors are heavily expressed in the VMH (Elmquist et al., 1998), highlighting
that this nucleus is highly sensitive to changes in hormone signaling and metabolic state.
Leptin increases firing of VMH neurons and deleting the leptin receptor from VMH neurons,
using the steroidogenic factor 1 (SF-1) cre mouse, causes hyperphagia, reduces energy

Alex Reichenbach, Romana Stark and B. Zane Andrews
264
expenditure and predisposes mice to DIO (Bingham et al., 2008; Dhillon et al., 2006). These
mice are also hyperinsulinemic and glucose intolerant (Bingham et al., 2008; Dhillon et al.,
2006), highlighting an important role for leptin signaling in VMH neurons in glucose
homeostasis. SF-1 is a transcription factor exclusively expressed in the VMH and is required
for the development of this nucleus. The generation of an SF-1 cre mouse permits the ability
to knockout genes of interest only in the VMH to examine the function of this nucleus. These
results help to define the targets through which leptin regulates appetite and body weight. In
particular, leptin-receptor deficient db/db mice display severe obesity whereas leptin-receptor
deficient POMC mice only display mild obese, suggesting leptin targets neuronal
subpopulations other than POMC. Indeed, comparing the body weight phenotype of VMH
and POMC leptin-receptor deficient mice, closely approximates the obese phenotype seen in
leptin receptor db/db mice, suggesting these two circuits act independently but in parallel to
maintain body weight homeostasis. SOCS3 is an inhibitor of leptin receptor JAK-STAT
signaling and contributes to leptin resistance in DIO mice. Deletion of SOCS3 in VMH
neurons pronounces JAK-STAT signaling through increased pSTAT3 levels and enhances
sensitivity to peripherally injected leptin (Zhang et al., 2008). Furthermore, food intake was
reduced in either chow-fed or high fat fed mice but body weight was not different due to
compensatory reductions in energy expenditure. Despite no difference in body weight, mice
lacking SOC3 in VMH neurons had improved glucose homeostasis and protected from
hyperglycemia and hyperinsulinemia caused by DIO.
Because leptin activates PI3K signaling in POMC and inhibits PI3K signaling in AgRP
neurons (Xu et al., 2005), and the VMH regulates the anorectic effects of leptin, Xu et al,
deleted PI3K in VMH neurons (Xu et al., 2010) and examined the effects on energy
homeostasis. These mice were susceptible to DIO, had impaired energy expenditure and
showed a blunted response to leptin. Thus, these studies indicate that improving leptin
receptor signaling in the VMH improves glucose homeostasis and energy metabolism.
Although VMH neurons also express significant insulin receptor (Havrankova, Roth, and
Brownstein, 1978) and VMH neurons respond to insulin (Davidowa and Plagemann, 2001;
Spanswick et al., 2000), the physiological role of insulin on VMH neurons had not been
addressed until very recently. Klockener et al, (Klockener et al., 2011) showed that insulin
activates PI3K signaling in SF-1 neurons and reduces firing frequency in these cells by
activating KATP channels. Deletion of the insulin receptor on VMH neurons restricted
adiposity, leptin resistance and glucose intolerance associated with DIO. Intriguingly,
deletion of the insulin receptor on VMH neurons increased the firing of anorexigenic POMC
neurons in mice on a high fat diet. This result reveals that insulin-dependent PI3K signaling
in VMH neurons influences POMC neuronal firing and contributes to the development of
obesity, in contrast with the idea that the VMH is a ‘satiety center’. These results indicate that
the function of the VMH cannot be assessed by simple lesion studies and that the regulation
of appetite and energy metabolism in the VMH is more complex than originally appreciated.
Although both leptin and insulin utilize the PI3K pathway in VMH neurons, leptin increases
VMH cell firing whereas insulin inhibits VMH cell firing. This key difference presumably
underlies the contrasting body weight phenotypes after insulin or leptin receptor deletion on
VMH neurons.
Despite the well-described utility of SF-1 cre mice to probe questions around the function
of VMH neurons, little is known about the biological function of SF-1 itself. Elmquist and
colleagues recently deleted SF-1 from VMH neurons and discovered that this impaired energy

Hypothalamic Control of Appetite and Energy Metabolism 265
expenditure and increased susceptibility to DIO. Furthermore, these mice had reduced leptin
receptor expression leading to leptin resistance (Kim et al., 2011). Thus, SF-1 is a critical
transcription factor that programs the VMH to maintain energy homeostasis by regulating
leptin receptor expression.
The Lateral Hypothalamus
Early studies showed that the lateral hypothalamus was the hypothalamic feeding center,
because surgical lesions block feeding. Subsequent studies have identified two key neuronal
populations in the lateral hypothalamus that regulate appetite; Orexin and melanin
concentrating hormone (MCH) neurons. The orexin and MCH neurons are only found in the
lateral hypothalamus and both stimulate feeding after icv injection (Rossi et al., 1997; Sakurai
et al., 1998).
Orexin
The role of orexin on appetite has been questioned as the effects are relatively short
(Edwards et al., 1999) and ob/ob and db/db mice have reduced prepro-orexin gene expression
and peptide content (Stricker-Krongrad et al., 2002). Moreover, transgenic mice over-
expressing orexin are resistant to diet-induced obesity and maintain insulin sensitivity by
stimulating energy expenditure (Funato et al., 2009). Nevertheless, deletion of orexin causes
hypophagia (Hara et al., 2001), fasting increases neuronal activation in orexin neurons and
both fasting and hypoglycemia increase orexin mRNA (Diano et al., 2003; Sakurai et al.,
1998). There is a strong interaction between orexin neurons and the melanocortin system, as
orexin neurons synapse with AgRP and POMC neurons and NPY or AgRP neurons synapse
with orexin neurons (Dube et al., 1999; Horvath et al., 1999). This circuit regulates the
appetite-stimulating effects of orexin.
The regulation of orexin neurons by leptin is complex; for example, leptin inhibits
neuronal firing of orexin neurons and blocks neuronal activation of orexin caused by fasting
(Funato et al., 2009; Yamanaka et al., 2003). However, leptin stimulates preproorexin mRNA
(Funato et al., 2009; Tritos et al., 2001; Yamanaka et al., 2003). The key to understanding this
puzzle may lie in the leptin-sensitive neuronal circuitry in the lateral hypothalamus. Orexin
neurons do not express leptin receptors and leptin injection does not increase pSTAT3 in
orexin containing neurons (Leinninger et al., 2009b). However, leptin receptor neurons in the
lateral hypothalamus express GABA (Leinninger et al., 2009b) and directly synapse with
orexin neurons (Louis et al., 2010) and POMC neurons (Vong et al., 2011). Indeed, leptin
may be a key hormone to explain the different effects of orexin on energy homeostasis.
Although acute injection of orexin stimulates feeding, the long-term effect of orexin is to
promote activity, energy expenditure and decrease feeding (Funato et al., 2009; Tritos et al.,
2001; Yamanaka et al., 2003). The ability of leptin to stimulate orexin gene expression over
prolonged periods may drive increased energy expenditure whereas the acute effects of orexin
on food intake may require reduced leptin signaling, as seen during fasting (characterized by
increased orexin neuronal activity and low leptin). More recent studies show a primary role of
the orexin system in wakefulness (Funato et al., 2009; Yamanaka et al., 2003)

Alex Reichenbach, Romana Stark and B. Zane Andrews
266
Ghrelin is also thought to activate orexin neurons to increase food intake in a direct and
indirect manner via NPY neurons (Toshinai et al., 2003), however there is no evidence that
orexin neurons express the ghrelin receptor and little evidence that the receptor is present in
the lateral hypothalamus (Guan et al., 1997; Zigman et al., 2006). There is evidence that
ghrelin stimulates orexin-dependent feeding through the dopaminergic reward pathways in
the brain (Perello et al., 2010). Therefore, it seems likely that the orexin system in the lateral
hypothalamus is important for ghrelin-induced food intake via indirect mechanisms involving
the melanocortin system or the dopaminergic system. Future studies are required to elucidate
the exact mechanism of action.
Orexin affects glucose homeostasis as icv infusion increases plasma glucose
concentrations through an increase in hepatic glucose production that is blocked by hepatic
sympathetic denervation. Interestingly, orexin stimulates hepatic glucose production in the
same manner as NPY, suggesting that orexin neurons engage the melanocortin system to
affect plasma glucose concentrations. Direct injection of orexin into the VMH stimulates
glucose uptake in skeletal muscle through the sympathetic nervous (Shiuchi et al., 2009) and
overexpression of orexin restores glucose tolerance in DIO mice through an orexin-receptor 2
mediated action (Funato et al., 2009). These data collectively show that orexin plays
important roles in peripheral glucose homeostasis through the sympathetic nervous system.
MCH
MCH neurons stimulate food intake, as MCH and MCH-1 receptor mice knockout mice
are lean, hypophagic and more active than wild type controls (Marsh et al., 2002; Shimada et
al., 1998). In accordance, MCH overexpressing mice are overweight and more susceptible to
DIO (Ludwig et al., 2001). Classic obesity models such as ob/ob and db/db mice also show
increased MCH mRNA in the lateral hypothalamus and this can be reversed by leptin
treatment, suggesting MCH contributes to hyperphagia and weight gain (Qu et al., 1996;
Tritos et al., 2001). Similar to the orexin system, -MSH from POMC neurons in the
melanocortin system inhibits the activity of MCH neurons. MCH can antagonize the anorectic
effects of -MSH via indirect mechanisms since MCH does not prevent -MSH binding to
the MC4R or MC3R (Ludwig et al., 1998; Tritos et al., 1998). Despite the ability of leptin to
suppress MCH, these neurons do not contain leptin receptors (Leinninger et al., 2009a) and
unlike orexin neurons, do not receive inputs from leptin-receptor containing neurons in the
lateral hypothalamus (Louis et al., 2010). Thus, leptin receptor neurons, outside the lateral
hypothalamus, must regulate the robust effect of leptin to suppress MCH expression. Leptin-
induced activation of POMC neurons is the prime candidate for this mechanism. This still
requires further proof.
The role of MCH in glucose homeostasis is poorly defined. Although MCH
overexpression causes obesity and hyperglycemia (Ludwig et al., 2001), icv injection of
MCH had no effect on plasma glucose levels in wild type, MCH knockout or MCH1R
knockout mice (Yi et al., 2009). These results suggest that the hyperglycemia seen in MCH
overexpressing mice may be due to the adiposity rather than overexpression of MCH.
However, one recent paper showed that MCH neurons sense blood glucose levels and adjust
their output to maintain a euglycemic state in the periphery (Kong et al., 2010). The

Hypothalamic Control of Appetite and Energy Metabolism 267
interaction between MCH neurons and peripheral metabolic hormones, such as leptin and
insulin, in blood glucose control remains to be determined.
Recent studies also show that the lateral hypothalamus is also an important relay nucleus
connecting basic homeostatic functions with higher cognitive function. The lateral
hypothalamus integrates social, cognitive, rewarding and emotional aspects of palatable food,
which can override the homeostatic appetite systems. Consistent with these ideas, the lateral
hypothalamus neurons project to and receive inputs from, reward-associated brain regions,
such as the ventral tegmental area and the nucleus accumbens (Fadel and Deutch, 2002;
Leinninger et al., 2009a; Peyron et al., 1998).
These results described above highlight the importance of the lateral hypothalamus in
appetite regulation and energy homeostasis. Neurons in lateral hypothalamus, both MCH and
orexin, are directly wired into the ARC melanocortin system and dopaminergic reward
system. Therefore, the idea that the lateral hypothalamus is the ‘feeding center’ may be
misleading, as the orexigenic output may come from increased ARC AgRP neuronal activity,
suppressed POMC neuronal activity and elevated dopamine reward pathways associated with
palatable food intake. With the benefit of 60 years of research hindsight, the lateral
hypothalamic ‘feeding center’ should incorporate the ARC melanocortin and the dopamine
reward system.
Hypothalamic Synaptic Plasticity Regulates Food Intake
Synaptic plasticity is a term that describes changes in synaptic connections between two
cells. This plasticity manifests in many different ways such as the quantal release of
neurotransmitter at the synapse and the absolute number of synaptic contacts on a particular
cell. There are two main types of identifiable synapses at the electron microscopic level;
asymmetric excitatory glutamatergic synapses and symmetric inhibitory GABAergic
synapses. Synaptic plasticity is a term classically associated with memory formation and
hippocampal function but pioneering recent work shows that synaptic plasticity in
hypothalamic neurons plays an important role in the regulation of appetite and the
maintenance of energy balance.
The first evidence came from studies on leptin-deficient ob/ob mice. Pinto et al 2004
(Pinto et al., 2004), showed hyperphagic ob/ob mice had increased excitatory synapses and
decreased inhibitory synapses on NPY neurons, whereas POMC neurons showed reduced
excitatory synapses. This study shows that the synaptic organization of the melanocortin
system in ob/ob mice dramatically favors NPY activation and subsequent hyperphagia. When
leptin was administered systemically to ob/ob, synaptic input organization normalized to wild
type levels within 6 hours, several hours before an observable effect on feeding. The
discovery that leptin could modulate synaptic plasticity in the hypothalamus lead the same
laboratory to investigate whether this is a general phenomenon for all metabolic hormones
acting in the hypothalamus, or rather a leptin-specific effect on the melanocortin system.
Accordingly, the anorexigenic effects of estrogen caused an increase in excitatory synaptic
number on POMC neurons in a leptin-independent manner, as estrogen was still effective in
ob/ob and db/db mice (Gao et al., 2007). Moreover, the orexigenic hormone ghrelin can also
regulate synaptic plasticity on POMC neurons in the ARC nucleus. Ghrelin shifted the
synaptic profile of POMC neurons in the opposite direction caused by leptin, for example

Alex Reichenbach, Romana Stark and B. Zane Andrews
268
ghrelin decreased inhibitory inputs on POMC neurons thereby reducing satiety drive through
reduced activation of POMC neurons (Andrews et al., 2008; Pinto et al., 2004). This is a
particularly intriguingly observation as ghrelin does not act on POMC neurons and less than
<8% of POMC neurons contain the ghrelin receptor (Willesen et al., 1999). The ghrelin-
induced synaptic rearrangement on POMC neurons is due to GABAergic inhibitory inputs
directly from AgRP neurons (Andrews et al., 2008). Corticosterone also produced synaptic
rearrangement in AgRP and POMC neurons to favor increased food intake, i.e. increased
inhibitory synapses on POMC and increased excitatory synapses on AgRP neurons (Gyengesi
et al., 2010).
These studies clearly demonstrate that hormone-driven synaptic plasticity in the
melanocortin system influences feeding behavior (Figure 7). However, many questions
remain unanswered, for example do fuel metabolites, such as glucose and fatty acids, affect
synaptic plasticity in a similar manner to hormones. Considering that glucose and fatty acids
have direct effects on POMC and AgRP neuronal function and firing, and free access these
neurons in the ARC, it is highly likely that fuel metabolites affect synaptic plasticity. Indeed,
metabolic status also appears to mediate synaptic plasticity, perhaps offering the first clue that
fuel metabolites, such as glucose and fatty acids, may regulate synaptic plasticity. Diet-
induced obesity (DIO) reduces the total number of synapses on POMC neurons due to an
increase in gliosis and glial ensheathment around POMC neurons (Horvath et al., 2010).
DIO also affects NPY synaptic organization by reducing the number of excitatory
synapses. These data suggest that high fat diet has a profound influence on the synaptic input
organization of POMC and NPY neurons (Horvath et al., 2010).
The reactive gliosis that occurs after high fat diet appears to form a barrier between these
neurons and endocrine signals from the blood, implying hormonal signals cannot initiate the
appropriate synaptic rearrangement. The lateral hypothalamic orexin neurons are instrumental
to trigger arousal but are also important in food intake and energy metabolism. These neurons
exhibit an unusual synaptic input organization characterized by a large number of excitatory
synapses and minimal inhibitory inputs (Horvath and Gao, 2005). An overnight fast, which
elevates ghrelin and inhibits leptin, promotes the formation of more excitatory synapses and
synaptic currents onto orexin neurons. Leptin treatment during a fast or refeeding effectively
blocked the synaptic rearrangement on orexin neurons. This study demonstrates that synaptic
plasticity occurs in other regions of the hypothalamus, not only AgRP and POMC neurons. It
further emphasizes the role of metabolic status on synaptic reorganization and energy
metabolism.
While these studies indicate the importance of synaptic plasticity on hypothalamic
feeding circuits there are still many unanswered questioned. Most importantly, in these
studies described above, the authors are examining only axo-somatic synapses (axon terminal
synapses with target neuronal cell bodies), however the vast majority of synaptic input occurs
within the dendritic field. The technical difficulties currently preclude examining the synaptic
input organization at the level of the dendrite, however because changes in feeding behavior
parallel axo-somatic synaptic input, it is reasonable to expect axo-dendritic inputs do the
same. It remains to be determined what intracellular signaling mechanisms mediate changes
in synaptic input and how this occurs. However, chronic PI3K activation in POMC neurons
increases inhibitory synaptic input (Plum et al., 2006). Finally, because synapses are
protracting and retracting around neuronal cell bodies and dendrites, there is a large demand
Hypothalamic Control of Appetite and Energy Metabolism 269
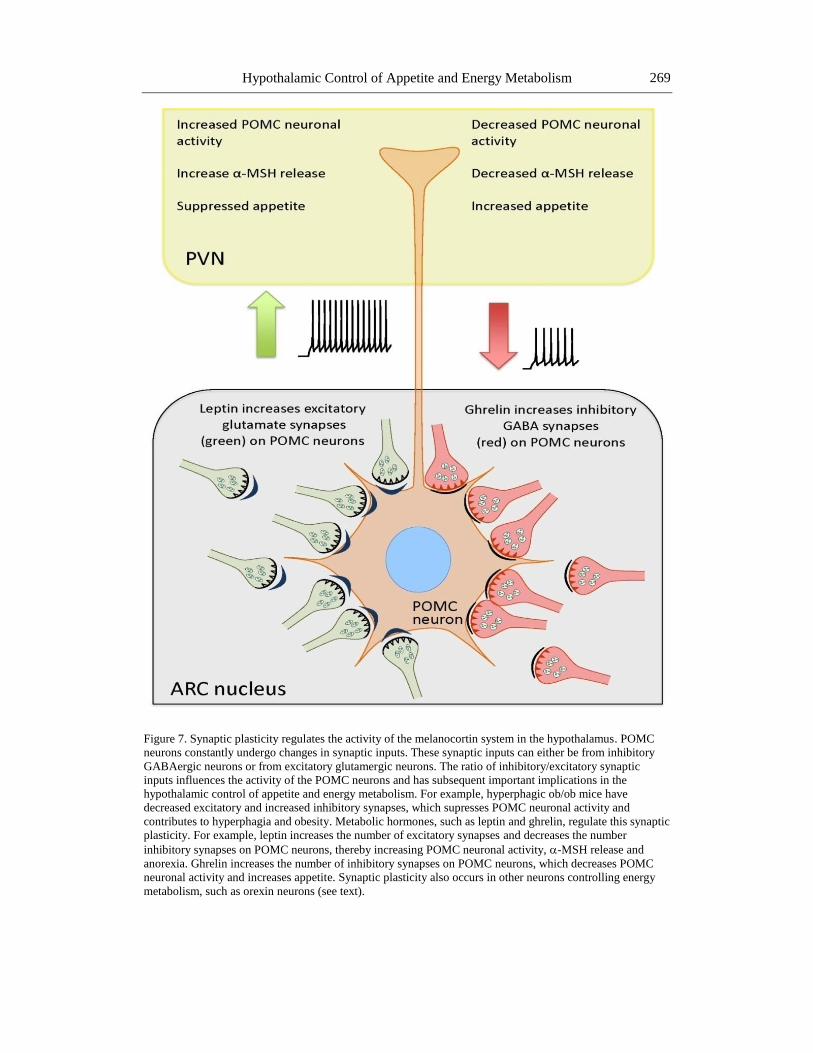
Figure 7. Synaptic plasticity regulates the activity of the melanocortin system in the hypothalamus. POMC
neurons constantly undergo changes in synaptic inputs. These synaptic inputs can either be from inhibitory
GABAergic neurons or from excitatory glutamergic neurons. The ratio of inhibitory/excitatory synaptic
inputs influences the activity of the POMC neurons and has subsequent important implications in the
hypothalamic control of appetite and energy metabolism. For example, hyperphagic ob/ob mice have
decreased excitatory and increased inhibitory synapses, which supresses POMC neuronal activity and
contributes to hyperphagia and obesity. Metabolic hormones, such as leptin and ghrelin, regulate this synaptic
plasticity. For example, leptin increases the number of excitatory synapses and decreases the number
inhibitory synapses on POMC neurons, thereby increasing POMC neuronal activity, -MSH release and
anorexia. Ghrelin increases the number of inhibitory synapses on POMC neurons, which decreases POMC
neuronal activity and increases appetite. Synaptic plasticity also occurs in other neurons controlling energy
metabolism, such as orexin neurons (see text).

Alex Reichenbach, Romana Stark and B. Zane Andrews
270
for local energy production. Indeed, enhanced synaptic activity increases mitochondrial
trafficking to the active area (Li et al., 2004) and mitochondrial biogenesis increases neuronal
plasticity (Lopez-Lluch et al., 2008). Therefore, determining how cellular bioenergetics status
affects synaptic plasticity will be the next important breakthrough in the field. Initial
observations show that impaired mitochondrial biogenesis in POMC neurons reduces
synaptic turnover (Andrews et al., 2008). In UCP2 knockout mice, ghrelin did not increase
inhibitory synaptic inputs on POMC and this attenuated ghrelin-induced food intake relative
control mice (Andrews et al., 2008).
Post-Translational Modifications
The melanocortin system is the most well described hypothalamic feeding circuit.
Important recent advances show that the POMC-derived peptide MSH undergoes extensive
post-translational modification before becoming the mature -MSH peptide, which is
responsible for activating the MC4R and inhibiting food intake. -MSH generated from
inappropriate POMC post-translational processing, such as -MSH1-12, does not suppress
food intake.
Prohormone convertase 1 (PC1) initially cleaves POMC into pro-ACTH and -lipo-
trophin (LPT) and then further cleaves pro-ACTH into ACTH1-39. LPT undergoes further
processing to form -MSH and -endorphin. Prohormone convertase 2 (PC2) cleaves
ACTH1-39 into ACTH1-17 and then mature -MSH is generated by the serial actions of
carboxypeptidase (CPE), peptidyl -amidating monooxygenase (PAM) and an unidentified n-
acetyltransferase (NAT) respectively. CPE removes the carboxy-terminal basic amino-acids
from ACTH1-17 to form -MSH1-14, which is then amidated by PAM to produce desacetyl--
MSH1-13. Mature acetyl--MSH is finally produced through the actions of NAT.
Consequentially, generating the anorectic -MSH peptide in the hypothalamus requires a
complex set of post-translational steps catalyzed by numerous enzymes (Figure 8).
Therefore, it is highly possible that any defect in the post-translational processing of -
MSH, could decrease hypothalamic -MSH and lead to hyperphagia and obesity. Indeed,
hyperinsulinemia in obese fa/fa mice is associated with a CPE mutation (Naggert et al., 1995)
and suppressed CPE activity (Fricker et al., 1996). Recent studies showed that deletion of
FOXO1 in POMC neurons increases CPE expression, which resulted in elevated -MSH in
the hypothalamus and reduced food intake. Moreover, CPE expression was decreased in DIO
and CPE overexpression in the hypothalamic arcuate nucleus also reduces food intake. These
studies clearly show that CPE activity in the hypothalamic arcuate nucleus regulates -MSH
peptide levels and maintains normal energy homeostasis. New evidence shows that another
carboxypeptidase (prolyl carboxypeptidase; PRCP) controls appetite by regulating the
degradation of -MSH. PRCP null mice are significantly leaner than controls on either a
chow or high fat diet. PRCP degrades active -MSH1-13 to inactive -MSH1-12, which has no
appetite-suppressing effect when injected centrally (Wallingford et al., 2009), and -MSH1-12
does not activate action potential firing in MC4R neurons in the PVN. Furthermore, a human
PRCP mutation is associated with the metabolic syndrome in males (McCarthy et al., 2003).
AgRP also undergoes post-translational modifications that affect antagonistic activity at the

Hypothalamic Control of Appetite and Energy Metabolism 271
MC4R. PC1 cleaves unprocessed full length AgRP and generates an AgRP83-132 peptide that
has greater antagonistic activity at the MC4R. Moreover, PC1 null mice have 3.3-fold more
unprocessed AgRP compared to controls (Creemers et al., 2006). PCs also play important
roles in proNPY processing (Brakch et al., 1997). The ability of PC1 to regulate POMC, NPY
and AgRP may have important implications for overall energy metabolism. Indeed, human
obesity is associated with a mutation in the PC1 gene (Ramachandrappa and Farooqi, 2011)
and PC activity is regulated by metabolic state, leptin and obesity. Future studies are required
to examine post-translational mechanisms regulating energy metabolism in the hypothalamus,
as targeting these processing steps may be useful to control obesity.
Figure 8. Post-translational modifications required to produce active acetyl -MSH in POMC neurons.
Prohormone convertase 1 (PC1) initially cleaves POMC into pro-ACTH and -lipotrophin (LPT) and then
further cleaves pro-ACTH into ACTH1-39. LPT undergoes further processing to form -MSH and -
endorphin. Prohormone convertase 2 (PC2) cleaves ACTH1-39 into ACTH1-17 and then mature -MSH is
generated by the serial actions of carboxypeptidase (CPE), peptidyl -amidating monooxygenase (PAM) and
an unidentified n-acetyltransferase (NAT) respectively. CPE removes the carboxy-terminal basic amino-acids
from ACTH1-17 to form -MSH1-14, which is then amidated by PAM to produce desacetyl--MSH1-13.
Mature acetyl--MSH1-13 is finally produced through the actions of NAT. -MSH1-13 is degraded to the
inactive -MSH1-12 by the actions of prolyl carboxypeptidase.

Alex Reichenbach, Romana Stark and B. Zane Andrews
272
Conclusion
The advent of novel molecular genetic techniques has dramatically aided our
understanding of hypothalamic systems regulating appetite and metabolism. Indeed, many of
the studies today are applying novel techniques to old questions, with a minor tweak. For
example, Hetherington and Brobeck described the important roles for the lateral
hypothalamus and the VMH in appetite and body weight. Today, scientists use sophisticated
molecular genetics to tweak these early discoveries, for example deleting leptin or insulin
receptor in these nuclei.
This chapter has systematically and in depth reviewed the current literature pertaining to
how the hypothalamus regulates appetite and energy metabolism. Within the hypothalamus
the ARC, VMH and LH are key nuclei regulating appetite and energy metabolism, however
there is a general desire in the field to develop a more general understanding of how the brain
regulates appetite and metabolism.
Indeed, new studies are beginning to focus on how the reward pathways regulate appetite
or how higher cognitive centers influence feeding behavior at multiple levels. These
approaches are likely to have significant impact on human health. Current technologies are
limited to examining the effect of single gene deletions in specific neurons, and while this is
an extremely useful tool to probe the role of single genes in neurons, it neglects to consider
the integrative function of multiple genes in a neuronal system.
Future challenges will elucidate how neuronal networks in the hypothalamus integrate
multiple hormonal and nutrient signaling molecules to regulate function. The next decade
should begin to focus on translating these basic discoveries into treatment strategies.
Acknowledgments
This work was supported by a Monash Fellowship, Monash University, Australia, an
Australia Research Council Future Fellowship and an NHMRC grant (NHMRC 1011274,
1026963, 1030037) to ZBA.
References
Anand, B. K., and Brobeck, J. R. (1951). Hypothalamic Control Of Food Intake In Rats And
Cats. Yale Journal Of Biology And Medicine, 24(2), 123-140.
Andrews, Z. B., and Horvath, T. L. (2008). Tasteless food reward. Neuron, 57(6), 806-808.
Andrews, Z. B., Liu, Z. W., Walllingford, N., Erion, D. M., Borok, E., Friedman, J. M., et al.
(2008). UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals.
Nature, 454(7206), 846-851.
Balthasar, N., Coppari, R., McMinn, J., Liu, S. M., Lee, C. E., Tang, V., et al. (2004). Leptin
receptor signaling in POMC neurons is required for normal body weight homeostasis.
Neuron, 42(6), 983-991.

Hypothalamic Control of Appetite and Energy Metabolism 273
Banno, R., Arima, H., Hayashi, M., Goto, M., Watanabe, M., Sato, I., et al. (2007). Central
administration of melanocortin agonist increased insulin sensitivity in diet-induced obese
rats. FEBS letters, 581(6), 1131-1136.
Banno, R., Zimmer, D., De Jonghe, B. C., Atienza, M., Rak, K., Yang, W., et al. (2010).
PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice.
J.Clin.Invest., 120(3), 720-734.
Bates, S. H., Stearns, W. H., Dundon, T. A., Schubert, M., Tso, A. W., Wang, Y., et al.
(2003). STAT3 signalling is required for leptin regulation of energy balance but not
reproduction. Nature, 421(6925), 856-859.
Baumann, H., Morella, K. K., White, D. W., Dembski, M., Bailon, P. S., Kim, H., et al.
(1996). The full-length leptin receptor has signaling capabilities of interleukin 6-type
cytokine receptors. Proc.Natl.Acad.Sci.U.S.A., 93(16), 8374-8378.
Bingham, N. C., Anderson, K. K., Reuter, A. L., Stallings, N. R., and Parker, K. L. (2008).
Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in
increased adiposity and a metabolic syndrome. Endocrinology, 149(5), 2138-2148.
Bouali, S. M., Fournier, A., St-Pierre, S., and Jolicoeur, F. B. (1995). Effects of NPY and
NPY2-36 on body temperature and food intake following administration into
hypothalamic nuclei. Brain Res.Bull., 36(2), 131-135.
Brakch, N., Rist, B., Beck-Sickinger, A. G., Goenaga, J., Wittek, R., Burger, E., et al. (1997).
Role of prohormone convertases in pro-neuropeptide Y processing: coexpression and in
vitro kinetic investigations. Biochemistry, 36(51), 16309-16320.
Brobeck, J. R., Tepperman, J., and Long, C. N. H. (1943). Experimental Hypothalamic
Hyperphagia In The Albino Rat. Yale Journal Of Biology And Medicine, 15(6), 831-853.
Broberger, C., De Lecea, L., Sutcliffe, J. G., and Hokfelt, T. (1998). Hypocretin/orexin- and
melanin-concentrating hormone-expressing cells form distinct populations in the rodent
lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein
systems. J.Comp.Neurol., 402(4), 460-474.
Bruning, J. C., Gautam, D., Burks, D. J., Gillette, J., Schubert, M., Orban, P. C., et al. (2000).
Role of brain insulin receptor in control of body weight and reproduction. Science,
289(5487), 2122-2125.
Chen, H. Y., Trumbauer, M. E., Chen, A. S., Weingarth, D. T., Adams, J. R., Frazier, E. G.,
et al. (2004). Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and
agouti-related protein. Endocrinology, 145(6), 2607-2612.
Coleman, D. L. (1973). Effects of parabiosis of obese with diabetes and normal mice.
Diabetologia, 9(4), 294-298.
Coleman, D. L., and Hummel, K. P. (1969). Effects of parabiosis of normal with genetically
diabetic mice. Am.J.Physiol., 217(5), 1298-1304.
Coleman, D. L., and Hummel, K. P. (1970). The effects of hypothalamics lesions in
genetically diabetic mice. Diabetologia, 6(3), 263-267.
Coppari, R., Ichinose, M., Lee, C. E., Pullen, A. E., Kenny, C. D., McGovern, R. A., et al.
(2005). The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on
glucose homeostasis and locomotor activity. Cell Metab., 1(1), 63-72.
Cowley, M. A., Smith, R. G., Diano, S., Tschop, M., Pronchuk, N., Grove, K. L., et al.
(2003). The distribution and mechanism of action of ghrelin in the CNS demonstrates a
novel hypothalamic circuit regulating energy homeostasis. Neuron, 37(4), 649-661.

Alex Reichenbach, Romana Stark and B. Zane Andrews
274
Creemers, J. W., Pritchard, L. E., Gyte, A., Le Rouzic, P., Meulemans, S., Wardlaw, S. L., et
al. (2006). Agouti-related protein is posttranslationally cleaved by proprotein convertase
1 to generate agouti-related protein (AGRP)83-132: interaction between AGRP83-132
and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology, 147(4),
1621-1631.
Davidowa, H., and Plagemann, A. (2001). Inhibition by insulin of hypothalamic VMN
neurons in rats overweight due to postnatal overfeeding. Neuroreport, 12(15), 3201-3204.
Dhillon, H., Zigman, J. M., Ye, C., Lee, C. E., McGovern, R. A., Tang, V., et al. (2006).
Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required
for normal body-weight homeostasis. Neuron, 49(2), 191-203.
Diano, S., Horvath, B., Urbanski, H. F., Sotonyi, P., and Horvath, T. L. (2003). Fasting
activates the nonhuman primate hypocretin (orexin) system and its postsynaptic targets.
Endocrinology, 144(9), 3774-3778.
Draper, S., Kirigiti, M., Glavas, M., Grayson, B., Chong, C. N., Jiang, B., et al. (2010).
Differential gene expression between neuropeptide Y expressing neurons of the
dorsomedial nucleus of the hypothalamus and the arcuate nucleus: microarray analysis
study. Brain Res., 1350, 139-150.
Dube, M. G., Kalra, S. P., and Kalra, P. S. (1999). Food intake elicited by central
administration of orexins/hypocretins: identification of hypothalamic sites of action.
Brain Res., 842(2), 473-477.
Edwards, C. M., Abusnana, S., Sunter, D., Murphy, K. G., Ghatei, M. A., and Bloom, S. R.
(1999). The effect of the orexins on food intake: comparison with neuropeptide Y,
melanin-concentrating hormone and galanin. J.Endocrinol., 160(3), R7-12.
Elias, C. F., Aschkenasi, C., Lee, C., Kelly, J., Ahima, R. S., Bjorbaek, C., et al. (1999).
Leptin differentially regulates NPY and POMC neurons projecting to the lateral
hypothalamic area. Neuron, 23(4), 775-786.
Elias, C. F., Saper, C. B., Maratos-Flier, E., Tritos, N. A., Lee, C., Kelly, J., et al. (1998).
Chemically defined projections linking the mediobasal hypothalamus and the lateral
hypothalamic area. J.Comp.Neurol., 402(4), 442-459.
Elmquist, J. K., Bjorbaek, C., Ahima, R. S., Flier, J. S., and Saper, C. B. (1998). Distributions
of leptin receptor mRNA isoforms in the rat brain. J.Comp.Neurol., 395(4), 535-547.
Erickson, J. C., Clegg, K. E., and Palmiter, R. D. (1996). Sensitivity to leptin and
susceptibility to seizures of mice lacking neuropeptide Y. Nature, 381(6581), 415-421.
Fadel, J., and Deutch, A. Y. (2002). Anatomical substrates of orexin-dopamine interactions:
lateral hypothalamic projections to the ventral tegmental area. Neuroscience, 111(2), 379-
387.
Fricker, L. D., Berman, Y. L., Leiter, E. H., and Devi, L. A. (1996). Carboxypeptidase E
activity is deficient in mice with the fat mutation. Effect on peptide processing.
J.Biol.Chem., 271(48), 30619-30624.
Fukuda, M., Jones, J. E., Olson, D., Hill, J., Lee, C. E., Gautron, L., et al. (2008). Monitoring
FoxO1 localization in chemically identified neurons. J.Neurosci., 28(50), 13640-13648.
Funato, H., Tsai, A. L., Willie, J. T., Kisanuki, Y., Williams, S. C., Sakurai, T., et al. (2009).
Enhanced orexin receptor-2 signaling prevents diet-induced obesity and improves leptin
sensitivity. Cell.Metab., 9(1), 64-76.

Hypothalamic Control of Appetite and Energy Metabolism 275
Gao, Q., Mezei, G., Nie, Y., Rao, Y., Choi, C. S., Bechmann, I., et al. (2007). Anorectic
estrogen mimics leptin's effect on the rewiring of melanocortin cells and Stat3 signaling
in obese animals. Nat.Med., 13(1), 89-94.
Gautron, L., and Elmquist, J. K. (2011). Sixteen years and counting: an update on leptin in
energy balance. J.Clin.Invest., 121(6), 2087-2093.
Gropp, E., Shanabrough, M., Borok, E., Xu, A. W., Janoschek, R., Buch, T., et al. (2005).
Agouti-related peptide-expressing neurons are mandatory for feeding. Nat.Neurosci.,
8(10), 1289-1291.
Guan, X. M., Yu, H., Palyha, O. C., McKee, K. K., Feighner, S. D., Sirinathsinghji, D. J., et
al. (1997). Distribution of mRNA encoding the growth hormone secretagogue receptor in
brain and peripheral tissues. Brain.Res.Mol.Brain.Res, 48(1), 23-29.
Gutierrez-Juarez, R., Obici, S., and Rossetti, L. (2004). Melanocortin-independent effects of
leptin on hepatic glucose fluxes. J.Biol.Chem., 279(48), 49704-49715.
Gyengesi, E., Liu, Z. W., D'Agostino, G., Gan, G., Horvath, T. L., Gao, X. B., et al. (2010).
Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons
in adult mice. Endocrinology, 151(11), 5395-5402.
Hara, J., Beuckmann, C. T., Nambu, T., Willie, J. T., Chemelli, R. M., Sinton, C. M., et al.
(2001). Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and
obesity. Neuron, 30(2), 345-354.
Harrold, J. A., Widdowson, P. S., and Williams, G. (1999). Altered energy balance causes
selective changes in melanocortin-4(MC4-R), but not melanocortin-3 (MC3-R), receptors
in specific hypothalamic regions: further evidence that activation of MC4-R is a
physiological inhibitor of feeding. Diabetes, 48(2), 267-271.
Havrankova, J., Roth, J., and Brownstein, M. (1978). Insulin receptors are widely distributed
in the central nervous system of the rat. Nature, 272(5656), 827-829.
Heijboer, A. C., van den Hoek, A. M., Pijl, H., Voshol, P. J., Havekes, L. M., Romijn, J. A.,
et al. (2005). Intracerebroventricular administration of melanotan II increases insulin
sensitivity of glucose disposal in mice. Diabetologia, 48(8), 1621-1626.
Hentges, S. T., Nishiyama, M., Overstreet, L. S., Stenzel-Poore, M., Williams, J. T., and Low,
M. J. (2004). GABA release from proopiomelanocortin neurons. J.Neurosci., 24(7),
1578-1583.
Hentges, S. T., Otero-Corchon, V., Pennock, R. L., King, C. M., and Low, M. J. (2009).
Proopiomelanocortin expression in both GABA and glutamate neurons. J.Neurosci.,
29(43), 13684-13690.
Hetherington, A. W., and Ranson, S. W. (1940). Hypothalamic Lesions And Adiposity In The
Rat. Anatomical Record, 1-4.
Hewson, A. K., and Dickson, S. L. (2000). Systemic administration of ghrelin induces Fos
and Egr-1 proteins in the hypothalamic arcuate nucleus of fasted and fed rats.
J.Neuroendocrinol., 12(11), 1047-1049.
Hill, J. W., Elias, C. F., Fukuda, M., Williams, K. W., Berglund, E. D., Holland, W. L., et al.
(2010). Direct insulin and leptin action on pro-opiomelanocortin neurons is required for
normal glucose homeostasis and fertility. Cell Metab., 11(4), 286-297.
Hill, J. W., Williams, K. W., Ye, C., Luo, J., Balthasar, N., Coppari, R., et al. (2008). Acute
effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in
mice. J.Clin.Invest., 118(5), 1796-1805.

Alex Reichenbach, Romana Stark and B. Zane Andrews
276
Horvath, T. L., Bechmann, I., Naftolin, F., Kalra, S. P., and Leranth, C. (1997). Heterogeneity
in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and
non-GABAergic subpopulations. Brain Res., 756(1-2), 283-286.
Horvath, T. L., Diano, S., and van den Pol, A. N. (1999). Synaptic interaction between
hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a
novel circuit implicated in metabolic and endocrine regulations. J.Neurosci., 19(3), 1072-
1087.
Horvath, T. L., and Gao, X. B. (2005). Input organization and plasticity of hypocretin
neurons: possible clues to obesity's association with insomnia. Cell.Metab., 1(4), 279-
286.
Horvath, T. L., Sarman, B., Garcia-Caceres, C., Enriori, P. J., Sotonyi, P., Shanabrough, M.,
et al. (2010). Synaptic input organization of the melanocortin system predicts diet-
induced hypothalamic reactive gliosis and obesity. Proc. Nat.Acad.Sci.U.S.A., 107(33),
14875-14880.
Janowitz, H. D., and Grossman, M. I. (1949). Hunger and appetite; some definitions and
concepts. Journal of Mt Sinai Hospital New York, 16(4), 231-240.
Jessen, L., Clegg, D. J., and Bouman, S. D. (2010). Evaluation of the lack of anorectic effect
of intracerebroventricular insulin in rats. Am.J.Physiol. Regulatory, integrative and
comparative physiology, 298(1), R43-50.
Kamegai, J., Tamura, H., Shimizu, T., Ishii, S., Sugihara, H., and Wakabayashi, I. (2000).
Central effect of ghrelin, an endogenous growth hormone secretagogue, on hypothalamic
peptide gene expression. Endocrinology, 141(12), 4797-4800.
Kamegai, J., Tamura, H., Shimizu, T., Ishii, S., Sugihara, H., and Wakabayashi, I. (2001).
Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and Agouti-
related protein mRNA levels and body weight in rats. Diabetes, 50(11), 2438-2443.
Kennedy, G. C. (1950). The hypothalamic control of food intake in rats. Proc.Royal
Soc.Biol.Sci., 137(889), 535-549.
Kim, K. W., Zhao, L., Donato, J., Jr., Kohno, D., Xu, Y., Elias, C. F., et al. (2011).
Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin
action in the ventral medial hypothalamic nucleus. Proc.Natl.Acad.Sci.U.S.A., 108(26),
10673-10678.
Kitamura, T., Feng, Y., Kitamura, Y. I., Chua, S. C., Jr., Xu, A. W., Barsh, G. S., et al.
(2006). Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food
intake. Nat.Med, 12(5), 534-540.
Klockener, T., Hess, S., Belgardt, B. F., Paeger, L., Verhagen, L. A., Husch, A., et al. (2011).
High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1
VMH neurons. Nat.Neurosci., 14(7), 911-918.
Koch, C., Augustine, R. A., Steger, J., Ganjam, G. K., Benzler, J., Pracht, C., et al. (2010).
Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic
insulin sensitivity. J.Neurosci. 30(48), 16180-16187.
Kong, D., Vong, L., Parton, L. E., Ye, C., Tong, Q., Hu, X., et al. (2010). Glucose stimulation
of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and
regulates peripheral glucose homeostasis. Cell.Metab., 12(5), 545-552.
Konner, A. C., Janoschek, R., Plum, L., Jordan, S. D., Rother, E., Ma, X., et al. (2007).
Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose
production. Cell.Metab., 5(6), 438-449.

Hypothalamic Control of Appetite and Energy Metabolism 277
Korner, J., Chua, S. C., Jr., Williams, J. A., Leibel, R. L., and Wardlaw, S. L. (1999).
Regulation of hypothalamic proopiomelanocortin by leptin in lean and obese rats.
Neuroendocrinology, 70(6), 377-383.
Leinninger, G. M., Jo, Y. H., Leshan, R. L., Louis, G. W., Yang, H., Barrera, J. G., et al.
(2009a). Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to
modulate the mesolimbic dopamine system and suppress feeding. Cell.Metab., 10(2), 89-
98.
Leinninger, G. M., Jo, Y. H., Leshan, R. L., Louis, G. W., Yang, H., Barrera, J. G., et al.
(2009b). Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to
modulate the mesolimbic dopamine system and suppress feeding. Cell.Metab., 10(2), 89-
98.
Li, Y. Z., and Davidowa, H. (2004). Food deprivation decreases responsiveness of
ventromedial hypothalamic neurons to melanocortins. J.Neurosci.Res., 77(4), 596-602.
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic
mitochondria in the morphogenesis and plasticity of spines and synapses. Cell, 119(6),
873-887.
Lopez-Lluch, G., Irusta, P. M., Navas, P., and de Cabo, R. (2008). Mitochondrial biogenesis
and healthy aging. Exp.Gerontol., 43(9), 813-819.
Louis, G. W., Leinninger, G. M., Rhodes, C. J., and Myers, M. G., Jr. (2010). Direct
innervation and modulation of orexin neurons by lateral hypothalamic LepRb neurons.
J.Neurosci., 30(34), 11278-11287.
Ludwig, D. S., Mountjoy, K. G., Tatro, J. B., Gillette, J. A., Frederich, R. C., Flier, J. S., et al.
(1998). Melanin-concentrating hormone: a functional melanocortin antagonist in the
hypothalamus. Am.J.Physiol, 274(4 Pt 1), E627-633.
Ludwig, D. S., Tritos, N. A., Mastaitis, J. W., Kulkarni, R., Kokkotou, E., Elmquist, J., et al.
(2001). Melanin-concentrating hormone overexpression in transgenic mice leads to
obesity and insulin resistance. J.Clin.Invest., 107(3), 379-386.
Luquet, S., Perez, F. A., Hnasko, T. S., and Palmiter, R. D. (2005). NPY/AgRP neurons are
essential for feeding in adult mice but can be ablated in neonates. Science, 310(5748),
683-685.
Luquet, S., Phillips, C. T., and Palmiter, R. D. (2007). NPY/AgRP neurons are not essential
for feeding responses to glucoprivation. Peptides, 28(2), 214-225.
Marks, J. L., and Waite, K. (1997). Intracerebroventricular neuropeptide Y acutely influences
glucose metabolism and insulin sensitivity in the rat. J.Neuroendocrinol., 9(2), 99-103.
Marsh, D. J., Weingarth, D. T., Novi, D. E., Chen, H. Y., Trumbauer, M. E., Chen, A. S., et
al. (2002). Melanin-concentrating hormone 1 receptor-deficient mice are lean,
hyperactive, and hyperphagic and have altered metabolism. Proc.Natl.Acad.Sci.U.S.A.,
99(5), 3240-3245.
Mayer, J. (1953). Glucostatic mechanism of regulation of food intake. N.Engl.J. Med., 249(1),
13-16.
McCarthy, J. J., Meyer, J., Moliterno, D. J., Newby, L. K., Rogers, W. J., and Topol, E. J.
(2003). Evidence for substantial effect modification by gender in a large-scale genetic
association study of the metabolic syndrome among coronary heart disease patients.
Hum.Genet., 114(1), 87-98.

Alex Reichenbach, Romana Stark and B. Zane Andrews
278
Mesaros, A., Koralov, S. B., Rother, E., Wunderlich, F. T., Ernst, M. B., Barsh, G. S., et al.
(2008). Activation of Stat3 signaling in AgRP neurons promotes locomotor activity.
Cell.Metab., 7(3), 236-248.
Mineur, Y. S., Abizaid, A., Rao, Y., Salas, R., DiLeone, R. J., Gundisch, D., et al. (2011).
Nicotine decreases food intake through activation of POMC neurons. Science, 332(6035),
1330-1332.
Mizuno, T. M., Kleopoulos, S. P., Bergen, H. T., Roberts, J. L., Priest, C. A., and Mobbs, C.
V. (1998). Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and
[corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes, 47(2), 294-
297.
Munzberg, H., Huo, L., Nillni, E. A., Hollenberg, A. N., and Bjorbaek, C. (2003). Role of
signal transducer and activator of transcription 3 in regulation of hypothalamic
proopiomelanocortin gene expression by leptin. Endocrinology, 144(5), 2121-2131.
Naggert, J. K., Fricker, L. D., Varlamov, O., Nishina, P. M., Rouille, Y., Steiner, D. F., et al.
(1995). Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E
mutation which reduces enzyme activity. Nat.Genet., 10(2), 135-142.
Nakazato, M., Murakami, N., Date, Y., Kojima, M., Matsuo, H., Kangawa, K., et al. (2001).
A role for ghrelin in the central regulation of feeding. Nature, 409(6817), 194-198.
Niswender, K. D., Morton, G. J., Stearns, W. H., Rhodes, C. J., Myers, M. G., Jr., and
Schwartz, M. W. (2001). Intracellular signalling. Key enzyme in leptin-induced anorexia.
Nature, 413(6858), 794-795.
Niswender, K. D., and Schwartz, M. W. (2003). Insulin and leptin revisited: adiposity signals
with overlapping physiological and intracellular signaling capabilities. [Review].
Frontiers in Neuroendocrinology, 24(1), 1-10.
Obici, S., Feng, Z., Karkanias, G., Baskin, D. G., and Rossetti, L. (2002a). Decreasing
hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats.
Nat.Neurosci., 5(6), 566-572.
Obici, S., Feng, Z., Tan, J., Liu, L., Karkanias, G., and Rossetti, L. (2001). Central
melanocortin receptors regulate insulin action. J.Clin.Invest., 108(7), 1079-1085.
Obici, S., Zhang, B. B., Karkanias, G., and Rossetti, L. (2002b). Hypothalamic insulin
signaling is required for inhibition of glucose production. Nat.Med., 8(12), 1376-1382.
Pardini, A. W., Nguyen, H. T., Figlewicz, D. P., Baskin, D. G., Williams, D. L., Kim, F., et
al. (2006). Distribution of insulin receptor substrate-2 in brain areas involved in energy
homeostasis. Brain Res., 1112(1), 169-178.
Parton, L. E., Ye, C. P., Coppari, R., Enriori, P. J., Choi, B., Zhang, C. Y., et al. (2007).
Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in
obesity. Nature, 449(7159), 228-232.
Perello, M., Sakata, I., Birnbaum, S., Chuang, J. C., Osborne-Lawrence, S., Rovinsky, S. A.,
et al. (2010). Ghrelin increases the rewarding value of high-fat diet in an orexin-
dependent manner. Biol.Psychiatry, 67(9), 880-886.
Peyron, C., Tighe, D. K., van den Pol, A. N., de Lecea, L., Heller, H. C., Sutcliffe, J. G., et al.
(1998). Neurons containing hypocretin (orexin) project to multiple neuronal systems.
J.Neurosci., 18(23), 9996-10015.
Pinto, S., Roseberry, A. G., Liu, H., Diano, S., Shanabrough, M., Cai, X., et al. (2004). Rapid
rewiring of arcuate nucleus feeding circuits by leptin. Science, 304(5667), 110-115.

Hypothalamic Control of Appetite and Energy Metabolism 279
Plum, L., Ma, X., Hampel, B., Balthasar, N., Coppari, R., Munzberg, H., et al. (2006).
Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to
diet-sensitive obesity. J.Clin.Invest., 116(7), 1886-1901.
Pu, S., Jain, M. R., Horvath, T. L., Diano, S., Kalra, P. S., and Kalra, S. P. (1999).
Interactions between neuropeptide Y and gamma-aminobutyric acid in stimulation of
feeding: a morphological and pharmacological analysis. Endocrinology, 140(2), 933-940.
Qian, S., Chen, H., Weingarth, D., Trumbauer, M. E., Novi, D. E., Guan, X., et al. (2002).
Neither agouti-related protein nor neuropeptide Y is critically required for the regulation
of energy homeostasis in mice. Mol.Cell Biol., 22(14), 5027-5035.
Qu, D., Ludwig, D. S., Gammeltoft, S., Piper, M., Pelleymounter, M. A., Cullen, M. J., et al.
(1996). A role for melanin-concentrating hormone in the central regulation of feeding
behaviour. Nature, 380(6571), 243-247.
Ramachandrappa, S., and Farooqi, I. S. (2011). Genetic approaches to understanding human
obesity. J.Clin.Invest., 121(6), 2080-2086.
Rios, M., Fan, G., Fekete, C., Kelly, J., Bates, B., Kuehn, R., et al. (2001). Conditional
deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and
hyperactivity. Mol.Endocrinol., 15(10), 1748-1757.
Rossi, M., Choi, S. J., O'Shea, D., Miyoshi, T., Ghatei, M. A., and Bloom, S. R. (1997).
Melanin-concentrating hormone acutely stimulates feeding, but chronic administration
has no effect on body weight. Endocrinology, 138(1), 351-355.
Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., et al. (1998).
Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-
coupled receptors that regulate feeding behavior. Cell, 92(4), 573-585.
Schwartz, M. W., Erickson, J. C., Baskin, D. G., and Palmiter, R. D. (1998). Effect of fasting
and leptin deficiency on hypothalamic neuropeptide Y gene transcription in vivo revealed
by expression of a lacZ reporter gene. Endocrinology, 139(5), 2629-2635.
Schwartz, M. W., Sipols, A. J., Marks, J. L., Sanacora, G., White, J. D., Scheurink, A., et al.
(1992). Inhibition of hypothalamic neuropeptide Y gene expression by insulin.
Endocrinology, 130(6), 3608-3616.
Shimada, M., Tritos, N. A., Lowell, B. B., Flier, J. S., and Maratos-Flier, E. (1998). Mice
lacking melanin-concentrating hormone are hypophagic and lean. Nature, 396(6712),
670-674.
Shiuchi, T., Haque, M. S., Okamoto, S., Inoue, T., Kageyama, H., Lee, S., et al. (2009).
Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle
via sympathetic nervous system. Cell.Metab., 10(6), 466-480.
Spanswick, D., Smith, M. A., Groppi, V. E., Logan, S. D., and Ashford, M. L. (1997). Leptin
inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels.
Nature, 390(6659), 521-525.
Spanswick, D., Smith, M. A., Mirshamsi, S., Routh, V. H., and Ashford, M. L. (2000). Insulin
activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats.
Nat.Neurosci., 3(8), 757-758.
Sternson, S. M., Shepherd, G. M., and Friedman, J. M. (2005). Topographic mapping of
VMH --> arcuate nucleus microcircuits and their reorganization by fasting.
Nature.Neurosci., 8(10), 1356-1363.

Alex Reichenbach, Romana Stark and B. Zane Andrews
280
Stricker-Krongrad, A., Richy, S., and Beck, B. (2002). Orexins/hypocretins in the ob/ob
mouse: hypothalamic gene expression, peptide content and metabolic effects.
Regul.Pept., 104(1-3), 11-20.
Tong, Q., Ye, C., McCrimmon, R. J., Dhillon, H., Choi, B., Kramer, M. D., et al. (2007).
Synaptic glutamate release by ventromedial hypothalamic neurons is part of the
neurocircuitry that prevents hypoglycemia. Cell.Metab., 5(5), 383-393.
Tong, Q., Ye, C. P., Jones, J. E., Elmquist, J. K., and Lowell, B. B. (2008). Synaptic release
of GABA by AgRP neurons is required for normal regulation of energy balance.
Nat.Neurosci. 11(9), 998-1000.
Toshinai, K., Date, Y., Murakami, N., Shimada, M., Mondal, M. S., Shimbara, T., et al.
(2003). Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology,
144(4), 1506-1512.
Tritos, N. A., Mastaitis, J. W., Kokkotou, E., and Maratos-Flier, E. (2001). Characterization
of melanin concentrating hormone and preproorexin expression in the murine
hypothalamus. Brain Res., 895(1-2), 160-166.
Tritos, N. A., Vicent, D., Gillette, J., Ludwig, D. S., Flier, E. S., and Maratos-Flier, E. (1998).
Functional interactions between melanin-concentrating hormone, neuropeptide Y, and
anorectic neuropeptides in the rat hypothalamus. Diabetes, 47(11), 1687-1692.
Tups, A., Anderson, G. M., Rizwan, M., Augustine, R. A., Chaussade, C., Shepherd, P. R., et
al. (2010). Both p110alpha and p110beta isoforms of phosphatidylinositol 3-OH-kinase
are required for insulin signalling in the hypothalamus. J.Neuroendocrinol., 22(6), 534-
542.
Unger, T. J., Calderon, G. A., Bradley, L. C., Sena-Esteves, M., and Rios, M. (2007).
Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult
mice results in hyperphagic behavior and obesity. J.Neurosci., 27(52), 14265-14274.
van de Wall, E., Leshan, R., Xu, A. W., Balthasar, N., Coppari, R., Liu, S. M., et al. (2008).
Collective and individual functions of leptin receptor modulated neurons controlling
metabolism and ingestion. Endocrinology, 149(4), 1773-1785.
van den Hoek, A. M., van Heijningen, C., Schroder-van der Elst, J. P., Ouwens, D. M.,
Havekes, L. M., Romijn, J. A., et al. (2008). Intracerebroventricular administration of
neuropeptide Y induces hepatic insulin resistance via sympathetic innervation. Diabetes,
57(9), 2304-2310.
van den Top, M., Lee, K., Whyment, A. D., Blanks, A. M., and Spanswick, D. (2004).
Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus.
Nat.Neurosci., 7(5), 493-494.
Vong, L., Ye, C., Yang, Z., Choi, B., Chua, S., Jr., and Lowell, B. B. (2011). Leptin action on
GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons.
Neuron, 71(1), 142-154.
Wallingford, N., Perroud, B., Gao, Q., Coppola, A., Gyengesi, E., Liu, Z. W., et al. (2009).
Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents.
J.Clin.Invest., 119(8), 2291-2303.
Wang, J. H., Wang, F., Yang, M. J., Yu, D. F., Wu, W. N., Liu, J., et al. (2008). Leptin
regulated calcium channels of neuropeptide Y and proopiomelanocortin neurons by
activation of different signal pathways. Neuroscience, 156(1), 89-98.

Hypothalamic Control of Appetite and Energy Metabolism 281
Wang, L., Saint-Pierre, D. H., and Tache, Y. (2002). Peripheral ghrelin selectively increases
Fos expression in neuropeptide Y - synthesizing neurons in mouse hypothalamic arcuate
nucleus. Neurosci.Lett, 325(1), 47-51.
Willesen, M. G., Kristensen, P., and Romer, J. (1999). Co-localization of growth hormone
secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat.
Neuroendocrinology, 70(5), 306-316.
Williams, K. W., Margatho, L. O., Lee, C. E., Choi, M., Lee, S., Scott, M. M., et al. (2010).
Segregation of acute leptin and insulin effects in distinct populations of arcuate
proopiomelanocortin neurons. J.Neurosci. 30(7), 2472-2479.
Wu, Q., Boyle, M. P., and Palmiter, R. D. (2009). Loss of GABAergic signaling by AgRP
neurons to the parabrachial nucleus leads to starvation. Cell, 137(7), 1225-1234.
Wu, Q., Howell, M. P., Cowley, M. A., and Palmiter, R. D. (2008). Starvation after AgRP
neuron ablation is independent of melanocortin signaling. Proc.Natl.Acad.Sci.U.S.A.,
105(7), 2687-2692.
Xu, A. W., Kaelin, C. B., Takeda, K., Akira, S., Schwartz, M. W., and Barsh, G. S. (2005).
PI3K integrates the action of insulin and leptin on hypothalamic neurons. J.Clin.Invest.,
115(4), 951-958.
Xu, A. W., Ste-Marie, L., Kaelin, C. B., and Barsh, G. S. (2007). Inactivation of signal
transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes
decreased pomc expression, mild obesity, and defects in compensatory refeeding.
Endocrinology, 148(1), 72-80.
Xu, B., Goulding, E. H., Zang, K., Cepoi, D., Cone, R. D., Jones, K. R., et al. (2003). Brain-
derived neurotrophic factor regulates energy balance downstream of melanocortin-4
receptor. Nat.Neurosci., 6(7), 736-742.
Xu, Y., Hill, J. W., Fukuda, M., Gautron, L., Sohn, J. W., Kim, K. W., et al. (2010). PI3K
signaling in the ventromedial hypothalamic nucleus is required for normal energy
homeostasis. Cell.Metab., 12(1), 88-95.
Yamanaka, A., Beuckmann, C. T., Willie, J. T., Hara, J., Tsujino, N., Mieda, M., et al. (2003).
Hypothalamic orexin neurons regulate arousal according to energy balance in mice.
Neuron, 38(5), 701-713.
Yi, C. X., la Fleur, S. E., Fliers, E., and Kalsbeek, A. (2010). The role of the autonomic
nervous liver innervation in the control of energy metabolism. Biochim.Biophys.Acta,
1802(4), 416-431.
Yi, C. X., Serlie, M. J., Ackermans, M. T., Foppen, E., Buijs, R. M., Sauerwein, H. P., et al.
(2009). A major role for perifornical orexin neurons in the control of glucose metabolism
in rats. Diabetes, 58(9), 1998-2005.
Zhang, R., Dhillon, H., Yin, H., Yoshimura, A., Lowell, B. B., Maratos-Flier, E., et al.
(2008). Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis
without affecting body weight. Endocrinology, 149(11), 5654-5661.
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994).
Positional cloning of the mouse obese gene and its human homologue. Nature,
372(6505), 425-432.
Zigman, J. M., Jones, J. E., Lee, C. E., Saper, C. B., and Elmquist, J. K. (2006). Expression of
ghrelin receptor mRNA in the rat and the mouse brain. J.Comp.Neurol., 494(3), 528-548.
Recommended

















