
Introduction
Autoimmune hemolytic anemia (AIHA) is an immunologic disorder in which antibodies are produced that target RBCs
shortening of red cell survival due to the products of an immune response
All require antigen-antibody reactions

Classification of Immune Hemolytic Anemia

Characteristics of Erythrocyte Autoantibodies
Warm AIHA caused by IgG autoantibodies that are optimally reactive at 37 C
CAD is typified by IgM autoantibodies that are optimally reactive at 4°C
PCH is a form of cold-reacting AIHA caused by IgG autoantibodies
mixed-type AIHA with both IgG warm and IgM cold autoantibodies

Warm-Reactive Autoantibodies
maximally reactive at 37°C
typically react with a patient’s own RBCs
IgG1 is the most common subclass
IgG1 and IgG3 activate C1 more readily, bind strongly to FcγRI, FcγRII, and FcγRIII, and increase RBC destruction in comparison with IgG2 and IgG4 subtypes

Cold-Reactive Autoantibodies
primarily associated with CAD and PCH PCH is the most common form in children Both are bind RBCs optimally at temperatures
below 37°C and usually below 31°C
Cold agglutinins are IgM autoantibodies that primarily cause extravascular hemolysis but can mediate intravascular hemolysis as well
PCH is IgG mediated and causes intravascular
hemolysis

pathogenesis of a cold-reactive autoantibody
bind host RBCs and activate complement
complement fixation by these antibodies occurs at 20°C to 25°C
Complement fixation cause RBC destruction via opsonization by macrophages

Cold Agglutinin Disease acute and often a result of Mycoplasma
pneumoniae infection
Other viruses:EBV,varicella and adenovirus
CAD typically occurs in the second or third week of illness
jaundice and pallor due to rapid onset of hemolysis
The hemolysis is self-limiting, and the degree of anemia is typically mild to moderate

Cold Agglutinin Disease (cont.)
cold autoantibody is an IgM with anti-I specificity associated with M. pneumoniae and anti-i specificity associated with EBV
high (≥256) cold agglutinin titers with postinfectious CAD
Treatment of postinfectious CAD is supportive and includes keeping the patient warm and using a blood warmer if the degree of anemia warrants RBC transfusion

Cold Agglutinin Disease (cont.)
chronic form of CAD is seen in elderly patients associated with lymphoma, chronic lymphocytic
anemia hemoglobinuria from intravascular hemolysis and
acrocyanosis of the ears, nose, fingers, and toes from autoagglutination of RBCs in the skin capillaries, particularly in cold weathe are seen
Autoagglutination can be enhanced by cooling the blood to 4°C and is reversed by warming to 37°C
Higher(≥1000) cold agglutinin titers

Paroxysmal Cold Hemoglobinuria
PCH occurs primarily in children as an acute transient condition following an upper respiratory or viral infection
detected by the Donath-Landsteiner test
PCH is the second most common form of AIHA in children

Clinical manifestation of PCH
In children, PCH classically presents within several weeks after a viral infection
sudden onset of hemoglobinuria, accompanied by fever, pallor, and jaundice
headache, abdominal pain, nausea, vomiting, or diarrhea
Hepatomegaly and splenomegaly were reported in up to 25% of cases

LABORATORY FINDING OF PCH
Reticulocytosis is characteristic, reticulocytopenia can be observed
PBS demonstrates RBC agglutination, polychromasia nucleated RBCs, and spherocytes
DAT is negative
Donath-Landsteiner test is positive
↑LDH,↑BUN,Cr,↑ unconjugated bilirubin, ↓haptoglobin

PRIMARY AIHA
incidence of about 1 per 75,000 to median age of diagnosis of 3.8 years typically after a recent infection Warm-reactive antibodies are IgG class and
account for the majority of AIHA cases in children IgM-mediated CAD is uncommon in children and
mostly affects adults and elderly persons PCH primarily presents in children, with the
median age at diagnosis being 5 years

PRIMARY AIHA
Clinical course:
1) Warm Ab:acute illness or intermittent remissions and relapses
2) Cold Ab:severe but acute self-limited illness requiring short-term supportive care with transfusion
improved mortality rate from 18% to 4% result of improvement in supportive care

In a largest cohort study of 265 pediatric patients with AIHA:
37% were diagnosed with Evans syndrome
4% had died
6% had no response or were in partial remission
90% were in complete remission while either receiving or not receiving therapy

Treatment of Acute AIHA
transfusion
Corticosteroid
IVIG
Plasma exchange
ACUTE AIHA TREATMENT

Transfusion Therapy
Mild anemia & no symptom:observation only Modrate to sever anemia or symptomatic:
transfusion therapy very severe anemia with hemoglobin levels˂6 :
RBC transfusion should be instituted as soon as possible
in warm, idiopathic AIHA, antibody is against blood group antigens that are present on most RBCs. no truly matched RBC units are possible, but transfusion can be safely performed

Corticosteroid Therapy
Mechanism to improve hemolysis in warm AIHA :
1-decrease in serum antibody concentration
2-decreasing sequestration in the spleen
Dosage:1-2 mg/kg q 6 h for 1-3 day
Then 2 mg/kg/day for children
1 mg/kg/day for adolescent:2-4 week
Then taper slowly in 3-12 mouth
overall clinical response is approximately 80%

Intravenous Immunoglobulin
Dose of 0.5-1 mg/kg/day ᵪ 5 day
overall clinical response is approximately 40%
not considered first-line treatment for AIHA in children
may be considered in a patient who is not responding to steroids

Therapeutic Plasma Exchange TPE in warm AIHA is considered a category III
intervention
TPE can significantly reduce antibody titers, in CAD(Ig M)
may be considered as a temporizing measure in a severely ill child who has a suboptimal response to RBC transfusion and may not have had time to respond to corticosteroid therapy

Treatment of Chronic or Refractory AIHA
Splenectomy and rituximab are the only second-line treatments with proven short-term efficacy
1) Rituximab : monoclonal antibody specific for the CD20 antigen complement-mediated cytotoxicity inhibition of B-cell proliferation induction of apoptosis 375 mg/m2 weekly for 1 to 6 weeks More than 90% of patients have a complete
response that lasted 7 to 28 months at least 1.5 gm/dL increase in Hb at a median of
12 days from the time the first dose of rituximab

Treatment of Chronic or Refractory AIHA(cont)
2)splenectomy: complete and partial response in approximately two
thirds of patients 50% of patients will remain in remission for years mortality and morbidity of laparoscopic splenectomy
for hematologic indications was 0.6% and 18% prior to surgery immunize with the polyvalent
polysaccharide vaccines against Streptococcus pneumoniae and Neisseria meningitides H. influenzae
penicillin prophylaxis twice a day for at least several years after surgery

Alternative Immunotherapeutic Agents
indicated in patients who do not respond to corticosteroids, rituximab, and splenectomy or who have contraindications to those therapies
Cyclophosphamide, cyclosporine, azathioprine 6-mercaptopurine and Campath-1H have been used to treat refractory AIHA
treatment should be continued for up to 6 months before it is considered to have failed

9 adult patient with severe refractory AIHA treted with cyclophosphamide 50 mg/kg/day for 4 days :
Six patients achieved complete remission with normal hemoglobin
three patients had partial remissions,as a hemoglobin level of at least 10 gm/dL without transfusion support
Patients became RBC transfusion–independent after a median of 19 days
High-dose cyclophosphamide was well tolerated common adverse effects included nausea,
vomiting, and transient myelosuppression

T-lymphocyte function inhibitor Cyclosporine, 6-mercaptopurine, and Cell-Cept
interfere with autoantibody synthesis cyclosporine has improved the course of AIHA
it is not routinely used because of the adverse effects
6-MP in 7 pediatric patients with AIHA was reported (five cases of primary AIHA and two cases of secondary AIHA) all responded
Cell-cept use in 3 adult all responded (Hb˃10) Campath-1H monoclonal antibodies improve
refractory disease but do not induce a prolonged remission

Hematopoietic Stem Cell Transplantation
36 patients with severe refractory autoimmune cytopenia, two underwent allogeneic HSCT and five patients autologous HSCT:
– two allogeneic HSCT achieved continuous remission
– five autologous HSCT, one died of treatment-related
causes, one died of progressive disease, two had no response, and one had a transient response
– this treatment option should be reserved for patients with severe life-threatening disease for whom all other therapies have failed

Treatment Cold-Reactive AIHA
In children, CAD typically occurs after an infection
In mild, compensated anemia not require
treatment or transfusions
If transfusion is indicated, a blood warmer need
In severe cases of cold, IgM-mediated AIHA, plasmapheresis can remove autoantibodies
treatment of PCH is also supportive with transfusion
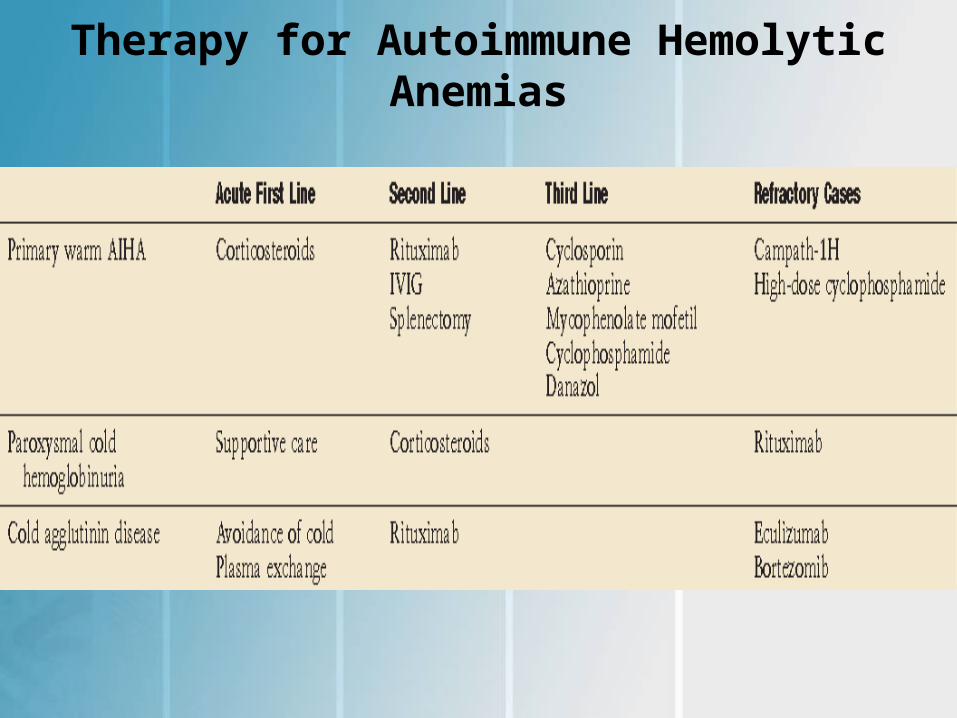
Therapy for Autoimmune Hemolytic Anemias

SECONDARY AUTOIMMUNE HEMOLYTIC ANEMIA
24% to 63% of all pediatric AIHA case
1) Evans Syndrome:– combination of ITP, AIHA, and/or autoimmune
neutropenia, or immune pancytopenia– Autoantibodies are directed at specific antigens on
RBCs, plt, or neutrophils but are not cross-reactive– 13% to 73% of published pediatric AIHA cases– Evans is a chronic disorder, by frequent exacerbations
and remissions– Neutropenia occurs in up to 55% of patients at
presentation

management of Evans syndrome :
treat symptomatic cytopenias including moderate to severe anemia, thrombocytopenia with bleeding, or prolonged and/or severe neutropenia (ANC˂ 500/μL)
first-line therapy is corticosteroids with or without IVIG steroids are useful in the acute setting, most patients
will require adjuvant or alternative therapy to sustain a remission
IVIG as a single agent has not been reported for treatment
Rituximab,an anti-CD20 monoclonal antibody, is an effective second-line

management of Evans syndrome(cont.)Splenectomy can achieve improvement of cytopenias,
but the response is often transient and relapse occurs in most cases 1 to 2 months later
splenectomy should be reserved for patients who do not respond to other second-line therapies
Cyclophosphamide has induced remissionAlemtuzumab (Campath-1H) is an IgG monoclonal AB
directed the CD52 antigen on T and B lymphocyte in the European Group study, of 22 patients who
underwent HSCT for refractory autoimmune cytopenias, 11 had Evans syndrome two autologous 9 allogeneic
- in autologous: one no response ,one relaps
- in allogeneic HSCT five continuous remission, one relapse, and three died of treatment-related causes

Acute Lymphoproliferative Syndrome ALPS, an inherited disorder of abnormal
lymphocyte survival caused by dysregulation of the Fas apoptotic pathway
excess of T-cell receptor αβ+CD3+CD4−CD8− T (double-negative T [DNT]) lymphocytes that accumulate in the lymph nodes, spleen, and peripheral blood
results in chronic lymphoproliferation, autoimmune disease, increased risk of malignancy

Acute Lymphoproliferative Syndrome (cont.)
clinical presentation : chronic lymphadenopathy, splenomegaly, multilineage cytopenias resulting from sequestration and autoimmune destruction, and an increased risk of B-cell lymphoma
60% to 70% have heterozygous germline mutations in FAS, autosomal-dominant fashion 10% of FAS mutations are acquired and no genetic defect in 30%
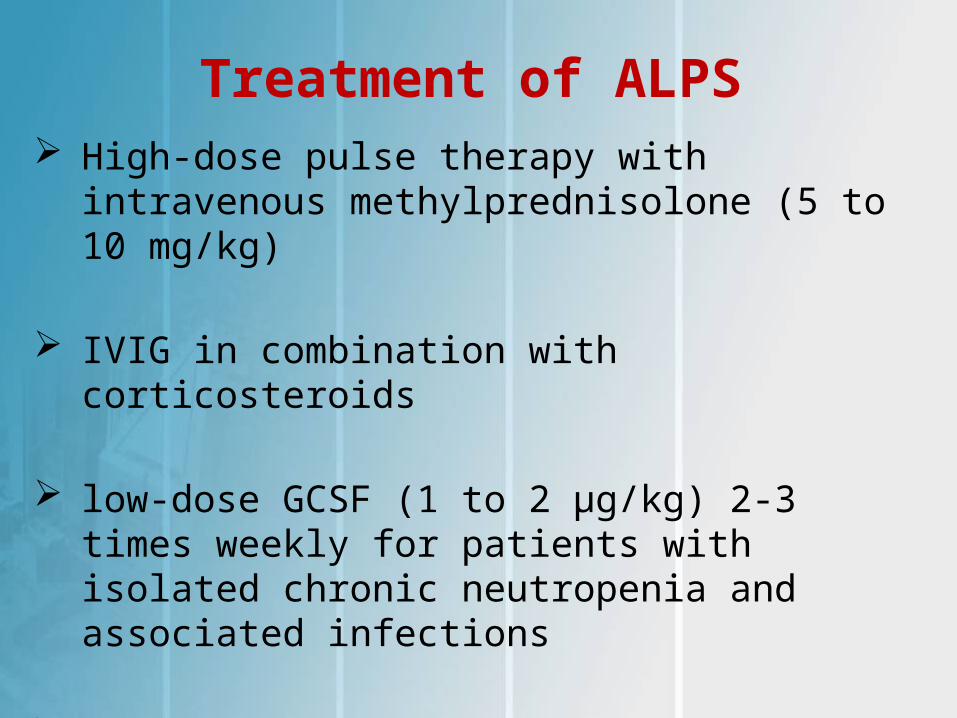
Treatment of ALPS High-dose pulse therapy with intravenous
methylprednisolone (5 to 10 mg/kg)
IVIG in combination with corticosteroids
low-dose GCSF (1 to 2 μg/kg) 2-3 times weekly for patients with isolated chronic neutropenia and associated infections
CellCept and sirolimus : ˃ 80% responses for their cytopenias

Other couse of secondary AIHA
Immunodeficiency: CVID, AIDS,WAS
Systemic Disorders: SLE, Sjögren, scleroderma
Malignancy: acute leukemia,lymphoma,MDS
Infections: EBV, CMV, M.pneumonia,varicella
Drug: Piperacillin, cefotetan, ceftriaxone

LOG
O
Recommended


















