
GEMS: THE GRID EMPOWERED MOLECULAR SIMULATOR
Antonio Laganà
Department of Chemistry, University of Perugia, Italy

THE PROJECT• Step 1 - SIMBEX (Simulator of Crossed Beam
Experiments) for atom diatom trajectory studies
• Step 2 - GEMS (Grid Empowered Molecular Simulator)
• Step 3 - Grid version of GEMS
• Step 4 - Some case studies
• Step 5 - The COMPCHEM Molecular Science Virtual Organization (VO)
• Step 6 - Next

1 - SIMBEX

SIMBEX: CROSSED BEAM EXPERIMENT of Perugia
MEASURABLES- Angular and time of flight product distributions
INFORMATION OBTAINABLE- Primary reaction products- Reaction mechanisms- Structure and life time of transient- Internal energy distribution of products- Key features of the potential
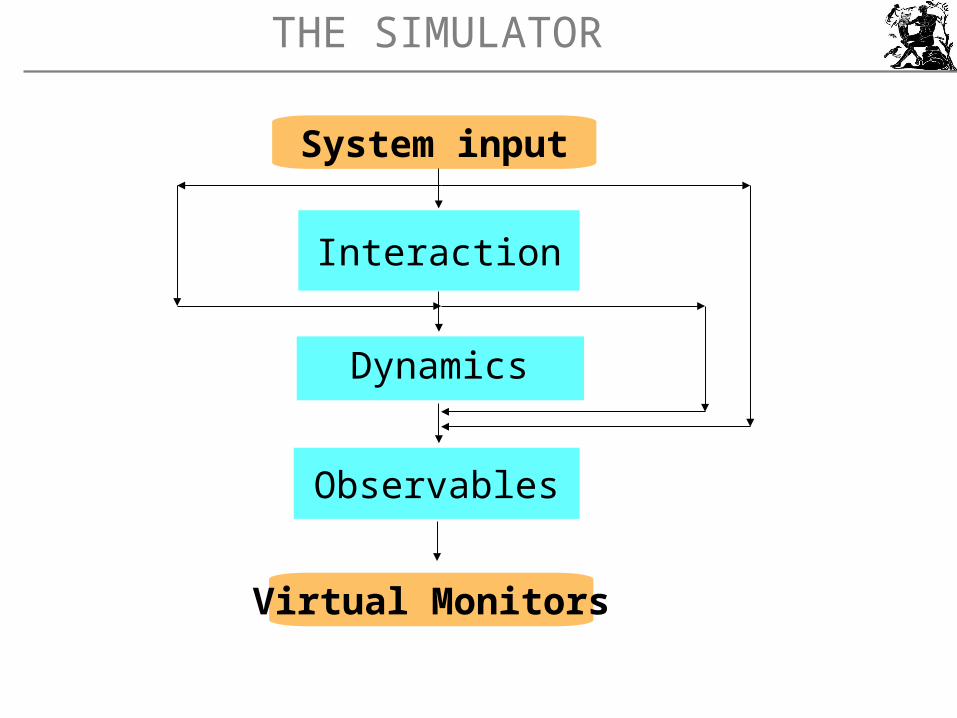
THE SIMULATOR
Interaction
Observables
Dynamics
Virtual Monitors
System input

The implemented INTERACTION module
INTERACTION
DYNAMICS
Is therea suitable
PES?
Import thePES parameters
NO
YES
START
CAVEATS
PES not needed in on the fly methods.
Seldomly a PES already exists
PESs can be semiempirical
Best if from a fit of ab initio values
Often PESs are of low accuracy

The implemented DYNAMICS module
DYNAMICS
OBSERVABLES
Are trajectory
calculationsaccepta-
ble?
NO
YES
TRAJ: application
using classical mechanics
calculations
CAVEATS
Implementation with trajectories
ABCtraj for atom diatom
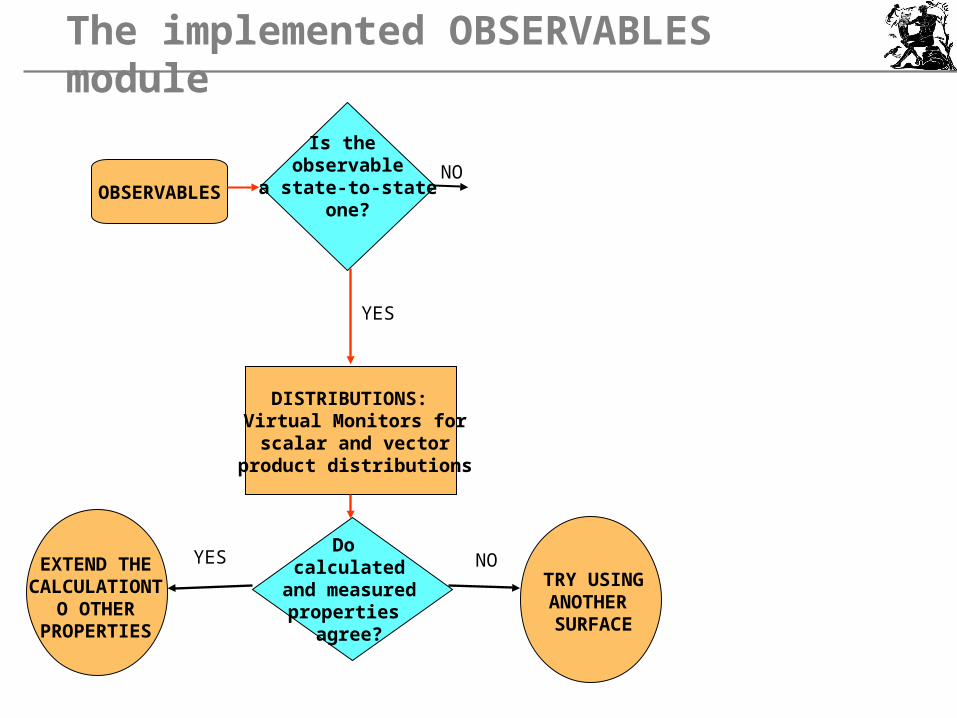
The implemented OBSERVABLES module
OBSERVABLESNO
YES
Is the observable
a state-to-stateone?
DISTRIBUTIONS: Virtual Monitors forscalar and vector
product distributions
Do calculated
and measuredproperties
agree?
EXTEND THECALCULATION
TO OTHERPROPERTIES
YES NOTRY USINGANOTHER SURFACE

PG
MI
PD
BO
BA
NA
RM
The prototype ChemGrid.it of grid.it
CILEA
UPV
UBCESCA
CINECA

High perfor-mance nets
GARR Fiber optics
Portals Security Communications
Resource Management MonitoringMiddleware
HP Components Problem Solving
Libraries Cost models
Program-Ming tools
Applications
Astrophysics Bioinformatics Earth observation
Geophysics Computational Chemistry

THE EGEE PRODUCTION GRID
• EGEE is a European project aimed at developing a European service grid infrastructure available to scientists.
• A prototype implementation of the Grid Molecular Simulator has been selected for the NA4 Activity of EGEE (Application Identification and Support)

THE EGEE PRODUCTION GRID

The GRIDified atom diatom TRAJ kernel
TRAJ
return
Iterate over initial conditionsthe integration of individualtrajectories (ABCTRAJ, etc.)
Define quantities of generaluse
Collect individual trajectory results

TRAJECTORY NATURAL CONCURRENCY
SEND “ready” status messageRECEIVE seedintegrate trajectoryupdate indicatorsSEND “ready” status messageGOTO RECEIVE
Worker:
DO traj_index =1, traj_number RECEIVE status message IF worker “ready” THEN generate seed SEND seed to worker ELSE GOTO RECEIVE endIF endDO
Master:

THE VIRTUAL MONI-TORS SHOWED THE PRODUCT ANGULAR DISTRIBUTIONS FOR THE VARIOUS CHANNELS
H+ICl→Cl + HI
H+ICl→H + ICl
H+ICl→HCl+I

Using history files to rationalize mechanisms
NEAR RECROSSING IN REACTIVE PROCESSES

2 – A GENERALIZATION OF SIMBEX TO GEMS: THE GRID EMPOWERED
MOLECULAR SIMULATOR

The molecular dynamics problem
twWHtwWt
i ,,ˆ,,
Electronic Schrödinger equation:
WwWEWwH nnnelec ;;ˆ
Nuclear Schrödinger equation:
tWt
itWH nnn ,,ˆ
Separation of electronic and nuclear motions

ELECTRONIC SCHRÖDINGER EQUATION
• Programs: often standard packages
• Methods - wavefunction quantum approaches (MRCI) - density functional theory (DFT)

• Classical
transform the Schrödinger equation into a set classical mechanics equations and integrate them in time
• Quantum - Integrate the equation in time for a given (or a set of
given or an average distribution) state(s) - Integrate the (stationary) equation in space for a given
energy and all energetically open states
NUCLEAR SCHRÖDINGER EQUATION
• Semiclassical
overimpose quantum effects of the associated wave to quantum mechanics outcomes

THE QUANTUM TREATMENT
Time dependent
{W} – set of position vectors of the nuclei or choices of center of mass coordinates like the already seen Jacobi Rτ and rτ vectors
HN - nuclear Hamiltonian
METHOD – integrate the first order time dependent equation using time as continuity variable and either collocating the system wavepacket on a grid (for R and r) or by expanding it on a basis set (for Θ)

Time independent
{W} – set of position vectors of the nuclei or choices of orthogonal coordinates of which one can act as continuity variable in going from one arrangement to another
HN - nuclear Hamiltonian
METHOD – segment the continuity variable in sectors and expand locally (in each sector) the wavefunction on the remaining (orthogonal) coordinates
THE QUANTUM TREATMENT

0
)()()(
1)(
2
tdtCTQTQ
Tk fNtrans
translational partition function
Qtrans(T) R
22
32
rotational partition function
oddjj
evenjjN
j
jTQ
exp123
exp126)(2
Flux-flux correlation function
)(tC f
By exact MCTDH or approximateSC-IVR calculations
FLUX CORRELATION FUNCTION FORMULATION OF THE RATE
COEFFICIENT

THE MCTDH METHOD• Diagonalisation of the thermal flux operator
defined onto a dividing surface to build a reduced Krylov subspace (iterative diagonalisation by consecutive application of the thermal flux operator on a trial wave function). The outcome is a set of eigenvalues and eigenstates of the thermal flux operator.
• Time propagation of the thermal flux eigenstates employing MCTDH.
• Calculation of observables: k(T), N(E).
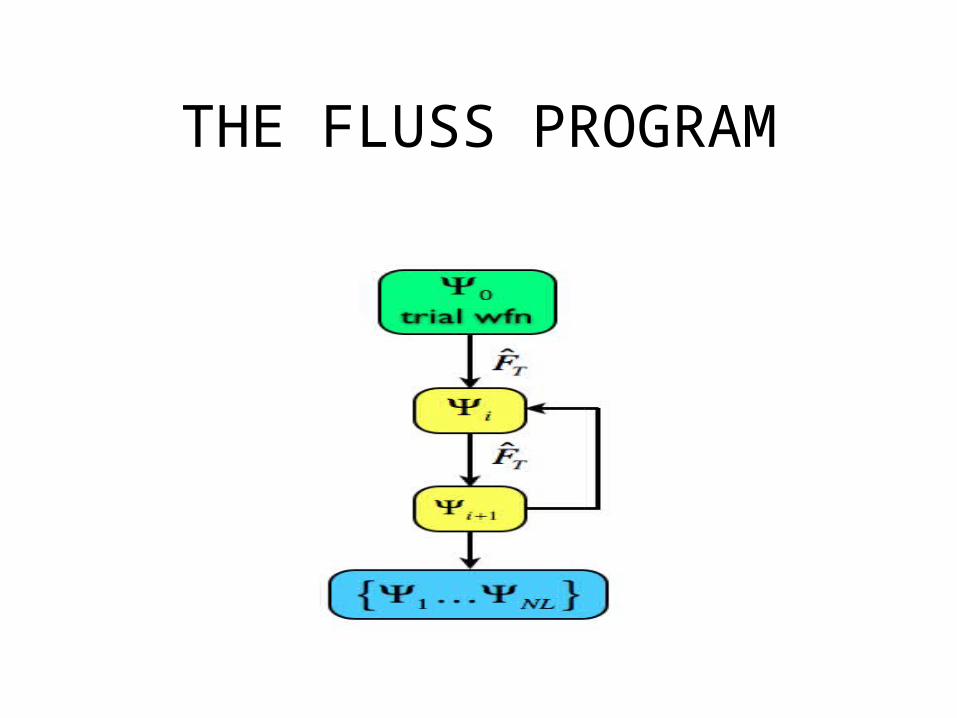
THE FLUSS PROGRAM

QDYN: the Quantum dynamics group in COMPCHEM (from COST Action D37)
• A COST Action to foster the constitution of a Molecular science community in the European Grid initiatives
• A working group (QDYN) to implement exact and approximate quantum methods
• Develop workflow and expert system tools for quantum chemical investigations
• Enhance collaborative research work in terms of service offer/request within quantum chemistry developers
• Foster the transfer of exact molecular treatments to industrial and commercial applications

MEMBERS OF QDYN
• A. LAGANA’, O. GERVASI (Perugia, Italy)
• G.G. BALINT KURTI (Bristol, UK)
• E. GARCIA (Vittoria, Spain)
• F. HUARTE (Barcelona, Spain)
• G. LENDVAY (Budapest, Hungary)
• G. NYMAN (Goteborg, Sweden)
• S. FARANTOS (Heraklion, Greece)
• M. LAUNAY (Rennes, France)

OTHER APPROACHES
• Reduced dimensionality quantum methods
• Classical, quasiclassical and molecular dynamics methods
• Semiclassical methods

3 – GRID IMPLEMENTATION

The extended INTERACTION module
INTERACTION
DYNAMICS
Is therea suitable Pes?
Are ab initiocalculationsavailable?
Are ab initiocalculations
feasible?
Import thePES routine
NO NO NO
YES YES YES
Take force fielddata and
procedures from relateddatabases
START
FITTING SUPSIM
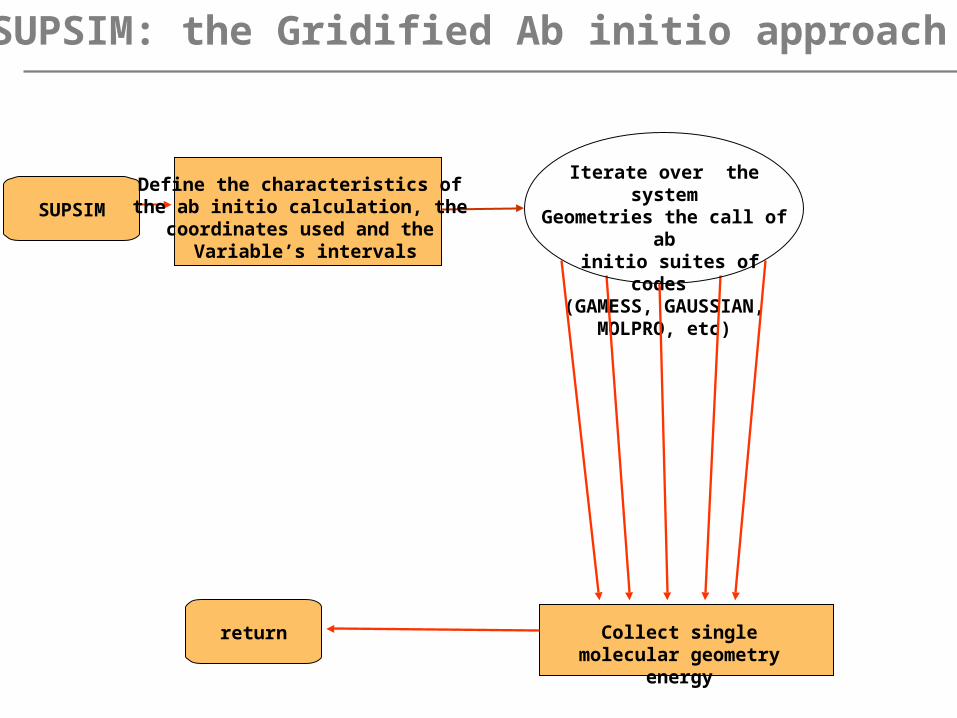
SUPSIM: the Gridified Ab initio approach
SUPSIM
return
Iterate over the systemGeometries the call of ab
initio suites of codes (GAMESS, GAUSSIAN,
MOLPRO, etc)
Define the characteristics of the ab initio calculation, the coordinates used and the
Variable’s intervals
Collect single molecular geometry energy

The FITTING portal
FITTING
Return
Are asym-ptotic values
accurate?
Are remai-ning valuesinaccurate?
Do ab initiovalues have the
proper sym-metry?
Enforce the propersymmetry
Application using fitting programs to
generate a PESroutine
Modify asym-ptotic values
NO NONO
Modify short andlong range values
YES YESYES
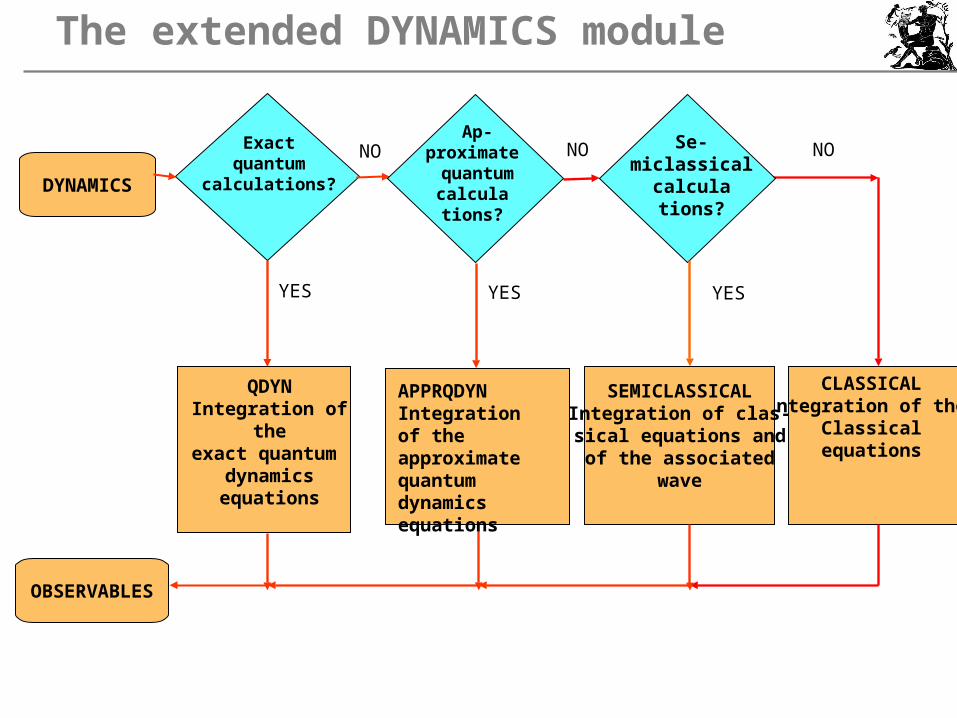
The extended DYNAMICS module
DYNAMICS
OBSERVABLES
Exact quantum
calculations?
NO NO
YES YES
CLASSICALIntegration of the
Classicalequations
APPRQDYNIntegration of the approximate quantum dynamicsequations
QDYNIntegration of theexact quantum
dynamics equations
SEMICLASSICALIntegration of clas-sical equations and
of the associatedwave
YES
NO Ap-
proximate quantumcalculations?
Se-miclassical
calculations?

The QDYN PROCEDURES
QUANTUMDYNAMICS
OBSERVABLES
Single Initial
quantum state?
Multiple initial
quantum states?
NO NO
YES YES
CRP: cumulative
reaction probabilities and TransitionState theory
TI: atom diatomS matrix
elements for a single energy
TD: atom diatom S matrix elements
for several energies
MCTDH: reactive flux over the
barrier
Statespecific
(summed overfinal states)
YES
Fully averaged

Gridified time dependent approaches
TD
return
•Iterate over initial conditions•the time propagation •(RWAVEPR, CYLHYP, etc.)
Define quantities of generaluse
•Collect single initial state•S matrix element
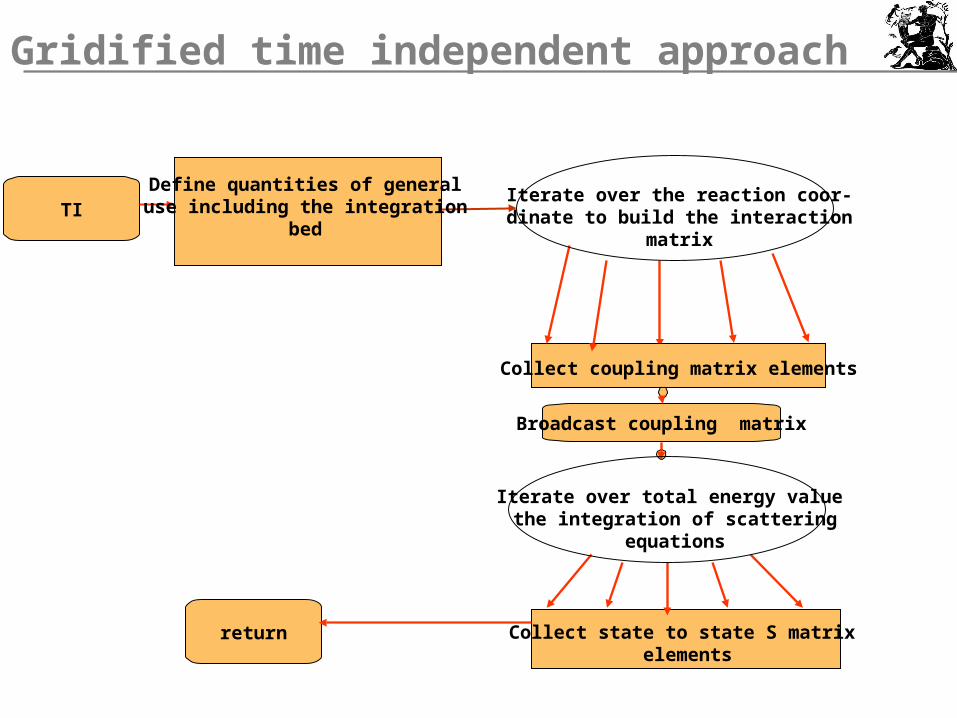
Gridified time independent approach
TI
return
Iterate over total energy value the integration of scattering
equations
Define quantities of generaluse including the integration
bed
Iterate over the reaction coor-dinate to build the interaction
matrix
Broadcast coupling matrix
Collect coupling matrix elements
Collect state to state S matrix elements

Gridified MCTDH method

The extended MEASURABLES module
OBSERVABLESNO NO
YES YES
Is the observable
a state-to-stateone?
Is theobservable
a state specificonee?
VM for thermal and thermodynamic pro-
perties including Molecular Virtual
Reality tools
CROSS: VM for statespecific cross sections,
rate constants and maps of
product intensity
DISTRIBUTIONS: VMfor scalar and vectorproduct distributions,
and state-to-state crosssections
Do calculated
and measuredproperties
agree?END
YES
INTERACTION
NO
Beam VM for Intensity in the
Lab frame

PROGRAMS BEING IMPLEMENTED ON THE GRID FOR PERSONAL USE
Perugia, ABC (also using PGRADE), RWAVEPR, CYLHYP, DL_POLY
Bristol, DIFFREALWAVE
Vittoria, RWAVEPR, VENUS
Vienna, COLUMBUS
Budapest, ABC, VENUS, RWAVEPR
Barcelona, MCTDH
Goteborg (On the fly Q-RBA?)
Heraklion, MODTINKER

4 – SOME CASE STUDIES

The N+N2 case study
)',()(),()( 12
412
4 vNSNvNSN gg

2NN : the LEPS potential energy surface
The collinear LEPS surface
Isoenergetic contour maps 1 eV spacing


Reactive state to state probabilities
0.146 eV
0.433 eV
0.717 eV
0.997 eV
E(v)
V=0
V=1
V=2
V=3
1.270 eV
1.543eV
V=4
V=5
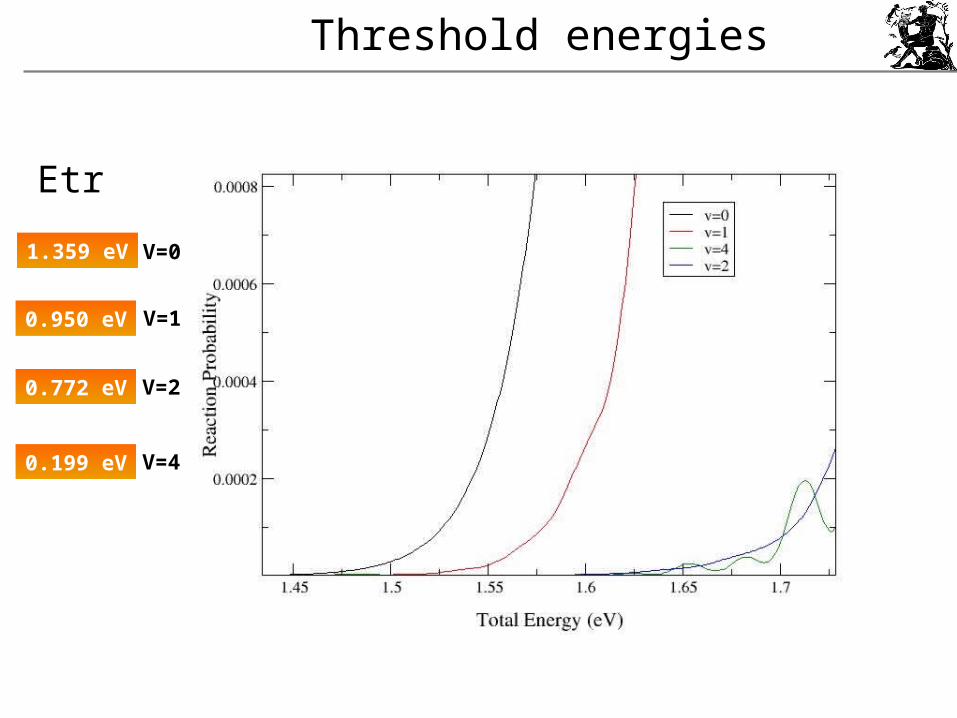
Threshold energies
1.359 eV
0.950 eV
0.772 eV
0.199 eV
Etr
V=0
V=1
V=2
V=4

2NN : the L3 potential energy surface
The bent L3 surface (125o transition state geometry)
Isoenergetic contour maps 1 eV spacing
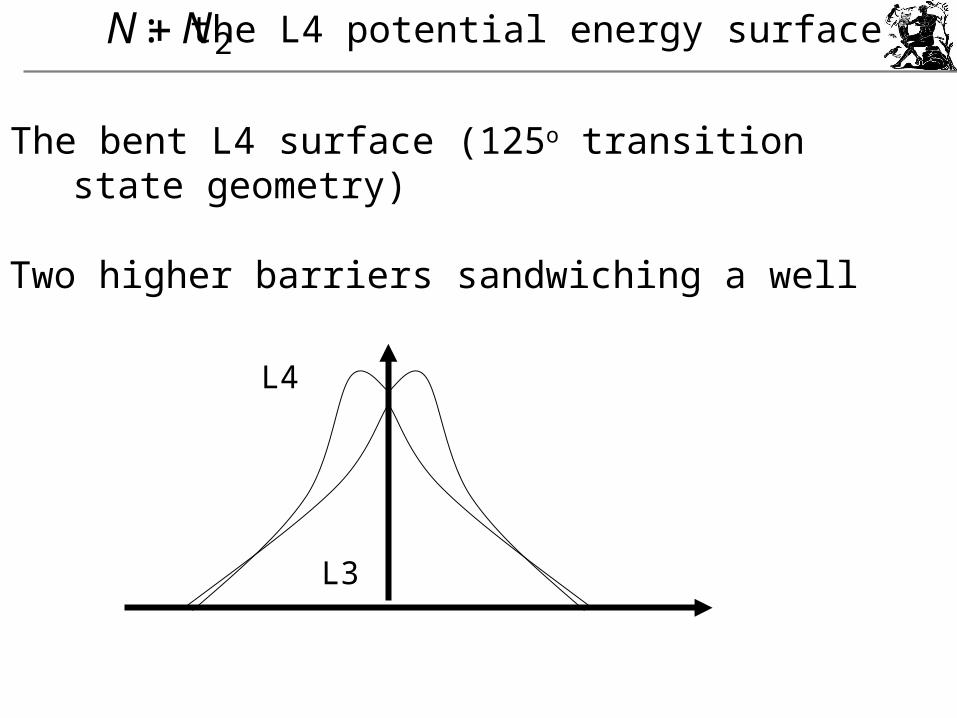
2NN : the L4 potential energy surface
The bent L4 surface (125o transition state geometry)
Two higher barriers sandwiching a well
L3
L4
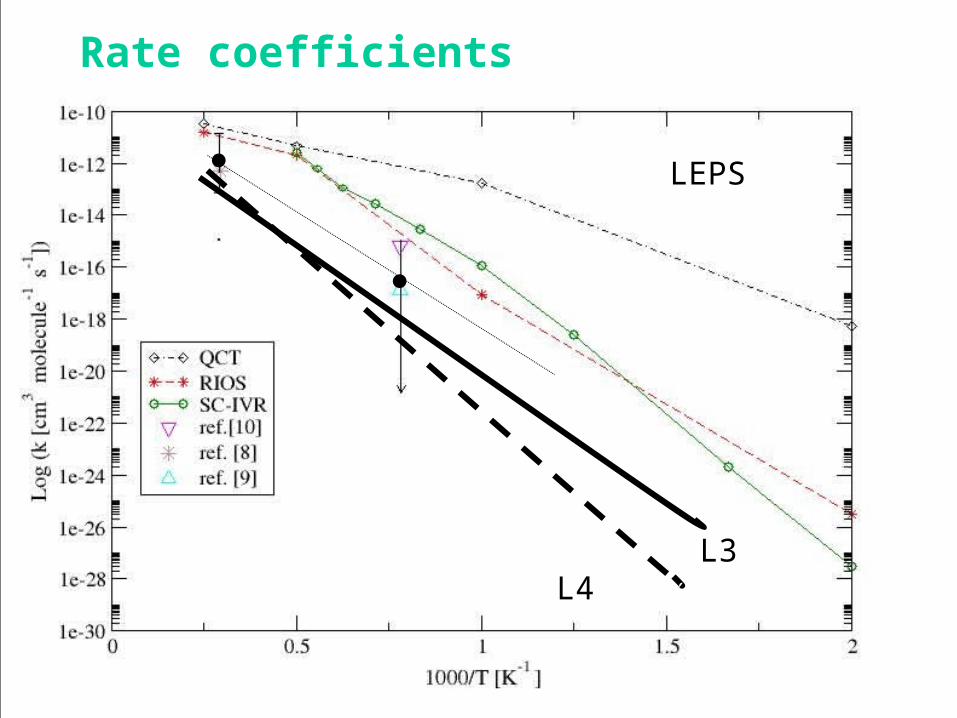
●
●
Rate coefficients
L3L4
LEPS

IONIC BIOLOGICAL CHANNELS
They are usually schematized as a sequence of:• Entrance gate• Bilayer pore• Selectivity filter
• Biological ionic channels play an important role in the control of ionic cellular concentrations and in synapses

ION FLOW THROUGH NANOTUBES
A nanotube model can be used to understand the ionic conductivity of cations (like Na+ or K+) through cellular membranes.
A life science application to the understanding of cellular micropores
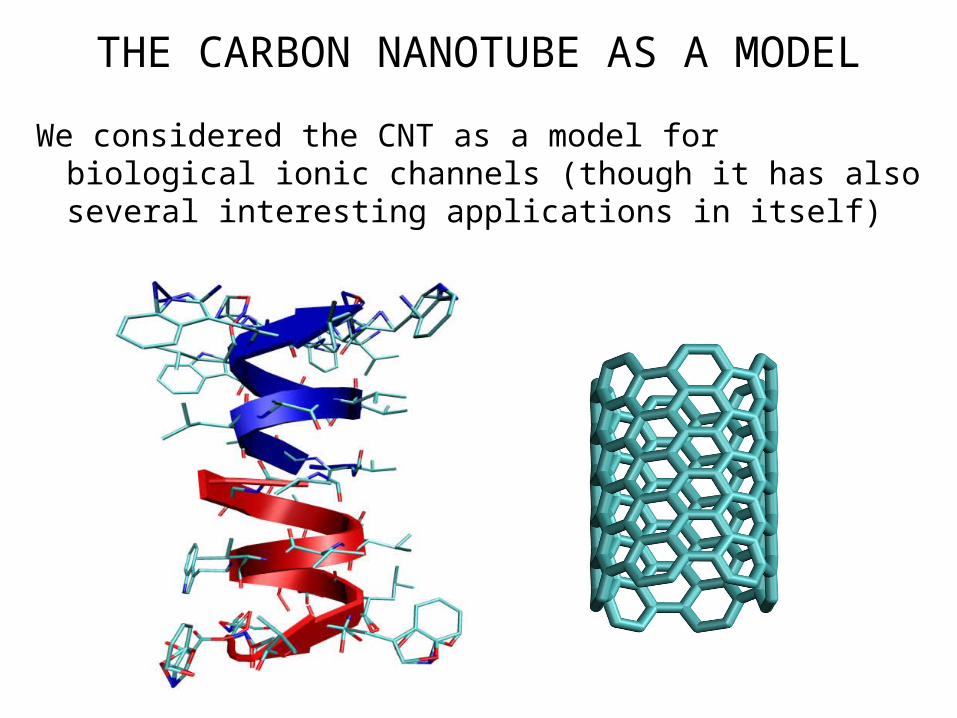
THE CARBON NANOTUBE AS A MODEL
We considered the CNT as a model for biological ionic channels (though it has also several interesting applications in itself)

MOLECULAR DYNAMICS

• H+/D+ ions flowing through a carbon nanotube
• A quantum scattering problem solved using a 3D time-dependent technique (the problem has been already solved using classical approaches)
• Implementation of a quantum scattering formalism based on polar cylindrical coordinates to single out resonances, interferences and tunneling
A quantum approach to ion flow in nanotubes

SCATTERING IN CYLINDRICAL SYMMETRY PROBLEMS
In the nanotube problem the symmetry is about cylindricalThe most suitable coordinates are the polar cylindrical ones (r,,z) The projection of the total angular momentum on z is a good quantum number

with k being the momentum along z.
iKθnK e)Rr
(ρJθ)ψ(r,
ikzeψ(z)
BASIS SET
R is the nanotube radius K is the angular momentum component on z
ρn is the nth zero of the Bessel function JK
The radial component is a Bessel function and the angular component is an imaginary exponential
The z component of the wavefunction is given by plane waves:

THE WAVEPACKET
- The initial (t=0) wavepacket is placed at one end of the nanotube- Its shape is that of an eigenfunction of the polar component of the Hamiltonian with a given component of the total angular momentum and a given radial excitation (that of the corresponding Bessel function)- Its z component is a Gaussian times a phase factor (corresponding to the linear momentum)
ikz2σ
)z(z
ee(z)2
20

-0.003
-0.002
-0.001
0.000
0.001
L=0
-0.003
-0.002
-0.001
0.000
L=5
-0.003
-0.002
-0.001
0.000
L=10
Out
goin
g F
lux
0 500 1000 1500 2000-0.003
-0.002
-0.001
0.000
L=30
Time (atomic units)
OUTGOING FLUX PLOTS: angular momentum
H+ - Elong=0.04 h Etransv=0.01 h
An increase of the value of the angular momentum quantum number slightly delays the flux (the increase of the centrifugal potential pushes the wavepacket closer to the nanotube walls).

Docking Proteina - Molecola piccola
Recettore: Adipocita Proteina che lega i lipidi
PDB code: 1LIC
Ligando: acido Esadecan sulfonico
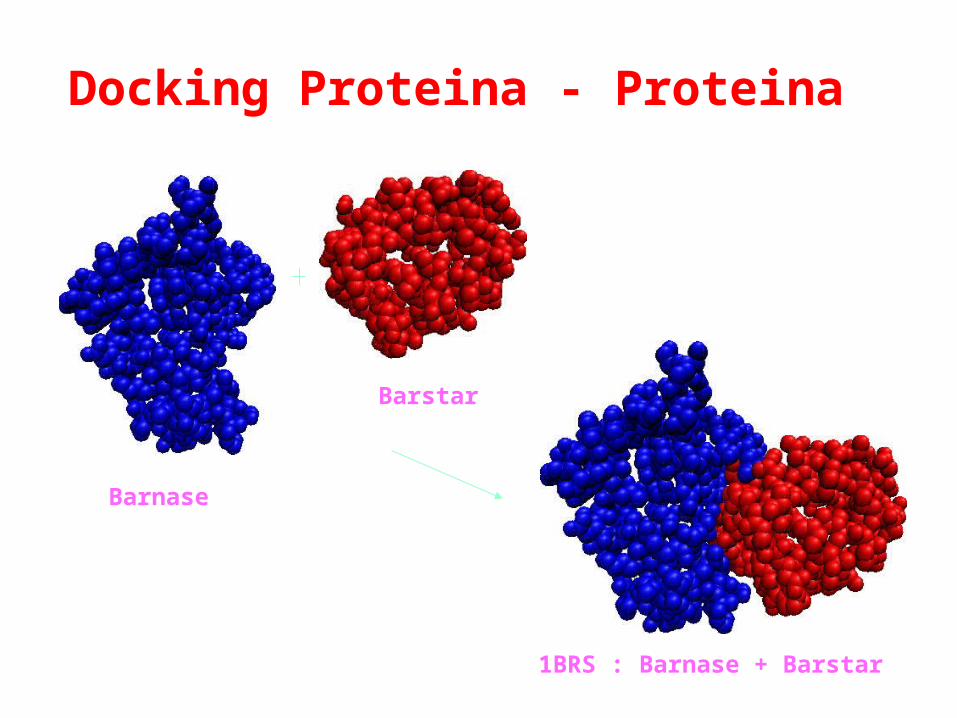
Barnase
Barstar
1BRS : Barnase + Barstar
Docking Proteina - Proteina

AIR POLLUTION SIMULATION
CPM10 Concentration from CHIMERE-aerosols

5 – THE COMPCHEM VIRTUAL ORGANIZATION

WHAT IS COMPCHEM
• COMPCHEM is a Virtual Organization (VO)
• VOs specialize a segment of the European Grid for specific purposes
• COMPCHEM is the VO of molecular and material sciences
• It is based, at present, on a subgrid of more than 8000 cpus (out of the 80000 of EGEE)

THE COMPCHEM APPROACH1. USER PASSIVE : Runs other’s programs ACTIVE: Implements at least one program for personal usage2. SW PROVIDER (from this level on one can earn credits) PASSIVE : Implements at least one program for other’s usage ACTIVE: Management at least one implemented program for cooperative usage 3. GRID DEPLOYER PASSIVE : Confers to the infrastructure at least a small cluster of processors ACTIVE: Contributes to deploy and manage the structure 4. STAKEHOLDER: Takes part to the development and the
management of the virtual organization• Further information at http://compchem.unipg.it

6 – FUTURE GUIDELINES

QUANTUM CHEMISTRY DATA STANDARDIZATION
• The Q5 data model and format was created for quantum chemistry (electronic structure) data by the WG 4 of D37
• Create D5 a data model for dynamics (in particular quantum dynamics)
• Extend the Q5 standard to D5

CREATE AND TEST WORKFLOWS
• Inter-job workflow
- Wrap the jobs
- Treat the jobs as objects
- Define composition rules and data links
• Intra-job workflows
- Define tools as for inter-job workflows via directives to be inserted inside the jobs

GETTING READY FOR EGI
• Broaden the molecular and material science user basis
• Introduce and gridify other suites of programs• Carry out massive calculations using the
gridified programs• Extend the usage of graphical interfaces and
virtual reality either to define initial conditions or to represent final observable properties
• Develop a credit system• Cluster COMPCHEM with other Grid VOs

ACKNOWLEDGEMENTS
• CDK group, Dept. Chemistry, Perugia (Crocchianti, Faginas, Pacifici, Skouteris, Costantini, Rampino, Manuali)
• HPC group, Dept. Math&Inf, Perugia (Gervasi, Tasso)
• Qdyn group, COST D37 (Garcia, Huarte, Lendvay, Nyman, Balint-Kurti, Farantos)
• Other groups of COST D37
Recommended

















