Indian Horticulture Journal 8(2/3): 45-51, April-September (2018)
ISSN: 2249-6823
©Indian Society of Advanced Horticulture
Research Paper
www.ihj.ind.in 544-18-1304-008
Comparative Transcriptome Sequencing of Cucumber Cultivars with Different Hypocotyl Length
Min Wang#1,2, Biao Jiang#1,2, Wenrui Liu1, Xiaoming He1, Qingwu Peng1, Zhaojun Liang1, Yu’e Lin*1
1Vegetable Research Institute, Guangdong Academy of Agricultural Sciences, Guangzhou - 510 640, China 2Guangdong Key Laboratory for New Technology Research of Vegetables, Guangzhou - 510 640, China
Received: 25 February 2018; Revised accepted: 30 May 2018
#These authors contributed equally to this work
*Corresponding author; e-mail: [email protected] A B S T R A C T
Cucumber (Cucumis sativus L.; 2n = 2x = 14), which belongs to the Cucurbitaceae, is one of the agriculturally and economically important vegetable crops around the world. It’s appropriate hypocotyl length is beneficial for establishing strong seedlings before transplanting. However, little knowledge is known about the molecular mechanism underlying hypocotyl elongation in the seedling traits. In our study, comparative analysis of hypocotyl transcriptome between long hypocotyl (B80) cultivar and short hypocotyl (JSH) cultivar would unravel novel genetic regulatory mechanisms involved in hypocotyl elongation. Using BGISEQ-500 platform, about 23.74 M reads per sample was averagely generated. The average mapping ratio with reference genome is 95.12% and the average mapping ratio with gene is 58.58%. A total of 20,861 genes were detected, among them, 1089 differentially expressed genes (DEGs) were detected between the two samples including 646 up-regulated genes and 443 down-expressed genes and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that the main activated genes in hypocotyl development were predominately involved in metabolic processes, biosynthesis of secondary metabolite pathways, catalytic activity, binding protein and other cellular components. In addition, the majority of related transcription factors (TFs) such as MYB, MYB related genes, and AP2-EREBP were identified in transcriptome. Multiple candidate cucumber genes in the hypocotyl development related pathways were also discovered. In all, these results offer specific genotype-dependent genes for genetic research of hypocotyl elongation in cucumber.
Key words: Cucumis sativus L, RNA-sequence, DEGs, Hypocotyl, Transcription factors Abbreviations: RNA seq: RNA sequencing DEGs: Differently expressed genes KEGG: Kyoto Encyclopedia of Genes and Genomes GO: Gene Ontology
ypocotyl, as an embryonic stem connecting
cotyledons and the primary root, has a relatively
simple architecture (Scheres et al. 1994). In Arabidopsis,
hypocotyl growth mainly relies on longitudinal cell
elongation after germination (Gendreau et al. 1997).
Cucumber (Cucumis sativus L.; 2n = 2x = 14), which
belongs to the Cucurbitaceae, is one of the agriculturally and
economically important vegetable crops around the world.
In general, cucumber is always cultivated by transplantation
way at seedling stage, thus, the seedling trait (hypocotyl
length, cotyledon size, the first female flower node) would
significantly influence plant development, flower bud
differentiation, and fruit development, finally impacting
plant production and quality (Ming et al. 2011).
Previous studies reported seedling related genes by
molecular genetic assay (Miao et al. 2012, Wang et al.
2016). At present, studies of hypocotyl length mainly focus
on the physiological (Zhou et al. 2010, Ming et al. 2011)
H
45
I
H
J

and genetic analysis under adversity stress. Several
researches demonstrated that is a quantitative trait controlled
by seveal genes and detected some QTLs (quantitative trait
locus) related to the trait (Jin et al. 2009, Zhang et al. 2011,
Miao et al. 2012, Wang et al. 2016). About 15 QTLs with
hypocotyl length were detected in low light environment
(Zhang et al. 2011). Two QTLs (Hl5.1 and Hl6.1) associated
with hypocotyl length was located at SSR23750-SSR00193
and SSR15818-SSR06003 of chromosome 5 and 6,
respectively (Miao et al. 2012). Combing recombinant
inbred lines population, we previously detected 10 QTLs of
hypocotyl length with the highest observed variation 15.1%
(Wang et al. 2016). A recent research isolated and
characterized a short hypocotyl gene (SH1), which regulated
low-dosage UVB-dependent hypocotyl elongation by
modulating UVR8 signaling pathway (Bo et al. 2016). The
appropriate hypocotyl length of cucumber is beneficial for
establishing strong seedlings before transplanting. During
seedling stage, cucumber with short hypocotyl shows
robuster and higher wind resistance than that with long
hypocotyl. Short cucumber hypocotyl has great application
value in the field cultivation and factory farming (Chen et
al. 2015).
RNA-Seq, combined with appropriate bioinformatics
tools, offers a pretty precise measurement of transcriptive
levels and their isoforms (Wang et al. 2010). And its high
accuracy and sensitivity makes suitable to study the whole
transcriptome (Jain et al. 2012). In addition, the RNA-Seq
allows identification of alternative splicing (AS) events,
novel transcripts and digital gene expression at the isoform
level in contrast to other methods such as microarray and tag
sequencing (Wang et al. 2010, Filichkin et al. 2010, Zhang
et al. 2010). In recent years, RNA-seq has been widely
applied to excavate transcriptomes of several plants under
different stress (Garg et al. 2014, Zhou et al. 2016, Song et
al. 2016). However, few studies under normal condition
were reported to analyze the transcriptive profiling between
different cultivars. Here, using deep RNA-Seq and digital
gene expression profile (DGE) analysis, we could rapidly
identify important genes expressed differently. And two
sequencing libraries prepared from long hypocotyl and short
hypocotyl samples were sequenced using a BGISEQ-500
platform. Overall, the research could provide a
comprehensive overview of transcriptional regulation and
complexity in hypocotyl elongation, which would offer
basic thesis for molecular mechanism of hypocotyl
elongation.
MATERIALS AND METHODS
Plant materials and growth conditions
Seeds of JSH short for Jin Shan (a South China type
cucumber variety) and B80 (North China type cucumber
variety) were germinated 48 hours at 37°C in a growth
chamber. Then we transplanted seedlings to soil-filled
containers and then incubated them in a greenhouse at 28°C and
60% relative humidity. Hypocotyl samples of three randomly
selected biological replicates were then collected from both
JSH and B80 plants (six samples in total). Plant tissues were
frozen in liquid nitrogen and stored at -80°C.
BGISEQ-500 library preparation and sequencing
A total of six samples (three replicates each of JSH and
B80 plants) were prepared for RNA extraction. High-quality
RNA was extracted from the leaves using Trizol reagent
(Invitrogen, USA) and then treated with DNase I
(Invitrogen). Total RNA was then purified and concentrated
using an RNeasy MinElute cleanup kit (Qiagen, Germany).
We prepared libraries using 2.5 μg total RNA of each
sample following the protocol described by Fehlmann et al.
(2016). Magnetic beads with Oligo (dT) were used to isolate
mRNA from total RNA. After mixing with fragmentation
buffer, mRNA was broken into short fragments. First-strand
cDNA was synthesized using random hexamer primers and
M-MuLV reverse transcriptase (RNase H). Second-strand
cDNA synthesis was then performed using DNA
polymerase I and RNase H. To select cDNA fragments 130-
160 bp in length, the PCR products were purified using an
AMPure XP system. Library quality was assessed on an
Agilent Bioanalyzer 2100 system.
Functional annotation and classification of the assembled
transcripts
Before assembly, adaptor sequences, empty reads,
low-quality sequences with an ‘N’ percentage over 10%
and sequences containing more than 50% bases with a Q-
value < 5 were removed using a custom Perl script. After
filtering, the remaining clean reads were used for
downstream bioinformatics analysis. Reads were mapped
to the Nipponbare reference genome using HISAT
software (Kim et al. 2015). Expression levels of each
gene were then calculated by quantifying the reads
according to the RPKM (reads per kilobase per million
reads) method (Li et al. 2011). Differential expression
analysis of the two conditions/groups was performed
using NOISeq, with different expression genes (DEGs)
identified according to the following criteria: fold change
≥ 2 and divergence probability ≥ 0.8.GO enrichment
analysis of DEGs was carried out using WEGO software
(Ye et al. 2006). To further understand DEG biological
functions, pathway enrichment analysis was performed
using the KEGG database (Kanehisa et al. 2008), the
major public pathway-related database.
Quantitative real-time PCR (qRT-PCR) validation of RNA-
Seq results
Three independent RNA samples per condition were
used to validate gene expression levels by qRT-PCR. Total
RNA was prepared from rice tissues using Trizol Reagent
(Life Tech). cDNA (20 µl) was synthesized from 1 µg of
RNA using a QuantiTect Reverse Transcription kit
(Qiagen). qRT-PCR amplifications were performed in 20 µl
reaction volumes containing 0.5 µl cDNA, 0.2 µM primer
mix and SYBR Premix Ex Taq (Takara) on an ABI PRISM
7900HT sequence detection system. The Actin gene was
used as an internal control. All primers used for qRT-PCR
were listed in (Table 1). Data was analyzed using the
relative quantification method (Livak and Schmittgen 2001).
Wang et al. 2018
Indian Horticulture Journal 8(2/3)
46

Table 1 Primers used in the assay of qRT-PCR Gens Forward primer Reverse Primer
Csa3G840450 TACGTCTTGCAGGACCAACT CTTGTGGACTTAGCCTCCCA
Csa7G060160 GCCTTCAAGCTGAGTCTGGT GAGTTCCAAGTACCGCAAGT
Csa4G629470 CAAGGCCTGGTTTCCCAATC CCAACCTTGCTGAGGGAGTA
Csa6G154500 CTCCATATCCTGCCGACCAT CATCATCGGCAAATGCAACG
Csa6G040640 GAAGCCACCATTGAAGCCAA TTGGTCCACGTGAAGGAACT
Csa6G505810 AGCCGATACATTGTCCCGA ACCAAACTCCTCCTCTGCTC
Csa2G237140 AGCCCACCAACAACTTTGAC TGGGTTGGTCTCACCAATGT
Csa7G378510 TGTTGCAATGGTTGATGGCA AGTGAAAGCATCCGCAATCC
Csa7G428260 CACTCAAAGGCACACAACCA TGCGCTTGTAATGTGTCTGG
Csa4G022900 ACAAGGCGTGTAACGAATGG TCGGTAACGGCTCTGAGTTT
Csa3G144140 GTAGGCGTACGTCTCTCCAA CTGACATTGGGAGGACCAGT
Csa2G094390 TGTCAATGCTCACTGGAGGT CGAATCTTTGGTGGTGCCAT
RESULTS
Phenotypic characteristic analysis
After seeding for one week, we observed the seedling
trait of JSH and B80. Results showed that the hypocotyl of
B80 is much longer than JSH (Fig 1A, B), which reached to
the significant different level (Fig 1C). In addition, the
cotyledon size between them also shows different. In detail,
the cotyledon length and width of JSH were 3.4 cm and 1.8
cm, respectively, while they were prominently larger in
contrast to B80 (Fig 1D).
Sequencing statistics
To further understand the molecular mechanism of
hypocotyl elongation in B80 plants, we carried out our
RNA-seq to explore detect gene expression. cDNA libraries
were prepared from leaves of cucumber seedlings to RNA-
Seq analysis on the platform of BGISEQ-500. Average
23.74 M reads were obtained in JSH and B80 seedling
leaves, respectively (Table 2). After compared with Chinese
Long genome for the references genome to map these reads,
the average mapping ratio with reference genome is 95.12%,
the average mapping ratio with gene is 58.58%. And a total
of 20,861 genes were detected.
Fig 1 Hypocotyl observation of JSH and B80
(A, B) Seedlings of JSH and B80 at cotyledon stage (C) Measurement of hypocotyl length (D) Measurement of cotyledon length and width Bar in (A, B) 3 cm Data is presented as the mean ± standard deviation (n = 9) *0.01≤P≤0.05; **P≤0.01; Student’s t test
Table 2 Summary of RNA-Seq results from hypocotyl tissues between JSH and B80
Sample Total raw
Reeds (Mb) Total clean reads
(Mb) Total clean bases (Gb)
Clean reads Q20 (%)
Clean reads Q30 (%)
Clean reads ratio (%)
B80-1 23.78 23.73 1.19 97.99 90.93 99.78 B80-2 23.75 23.69 1.18 97.67 89.87 99.76 B80-3 23.83 23.78 1.19 97.78 90.07 99.78 JSH-1 23.78 23.72 1.19 97.74 90.26 99.74 JSH-2 23.86 23.8 1.19 97.75 90.14 99.73 JSH-3 23.76 23.7 1.18 97.66 89.81 99.75
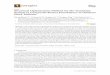
DEGs uncovered by RNA-Seq
Gene expression levels were quantified using RSEM
software (Li et al. 2011) and the FPKM (fragments per
kilobase of transcript per million mapped reads) method.
Putative differently expressed genes (DEGs) between B80
vs. JSH (B80_1 vs.JSH_1, B80_2 vs.JSH_2and B80_3
vs.JSH_3) were identified according to the following default
criteria: fold change ≥ 2 (B80/JSH) and divergence
probability ≥ 0.8. Results showed that a total of 646
transcripts demonstrated significant up- regulated, while 443
transcripts were predominantly down-regulated (Fig 2,
Table S1).
GO functional classification of DEGs
The GO standardized classification system for gene
function was used to analyze DEGs and understand the
molecular events involved in hypocotyl elongation. WebGO
tool was used to classify these detected significant DEGs
into three categories: “biological process,” “molecular
function”, and “cellular components” (Fig 3). In many cases,
a contig was associated with multiple functions. Under
biological process, the two largest subcategories were
metabolic process (375) and cellar process (278), which
followed by subcategories such as single-organism process,"
biological function, response to stimulus. Major
Comparative Transcriptome Sequencing of Cucumber Cultivars
47
subcategories under molecular function were “binding” and
“catalytic activity”. Finally, “membrane”, “cell”, and “cell
part”, followed by “organelle”, were the predominant
subcategories in the cellular component category in
hypocotyl.
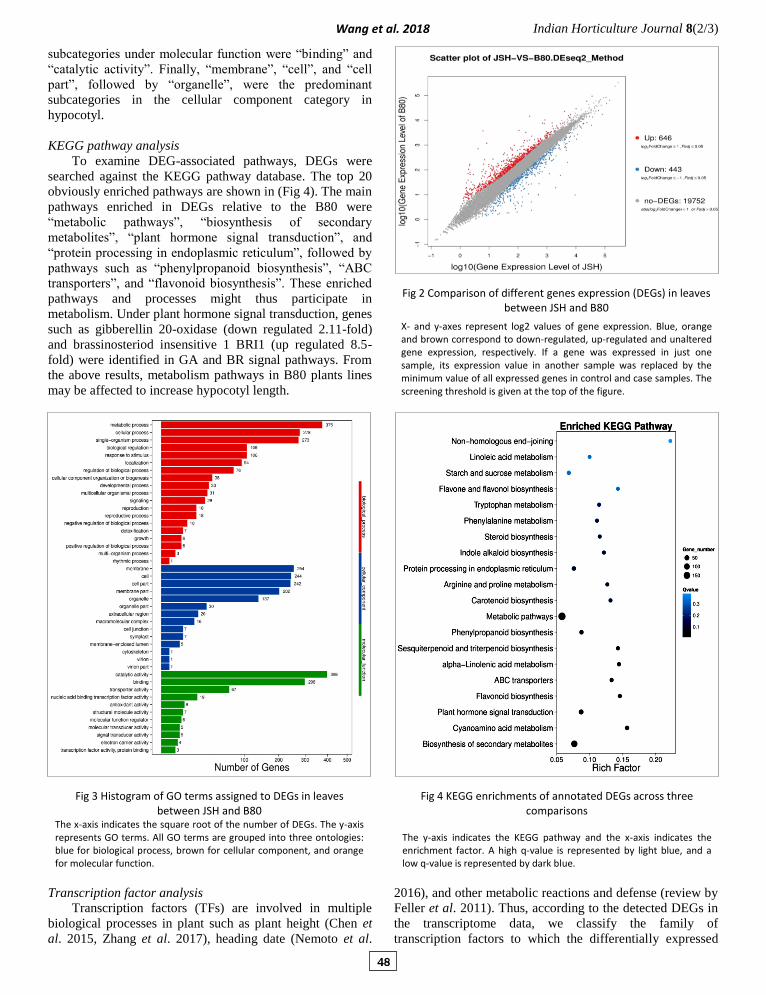
KEGG pathway analysis
To examine DEG-associated pathways, DEGs were
searched against the KEGG pathway database. The top 20
obviously enriched pathways are shown in (Fig 4). The main
pathways enriched in DEGs relative to the B80 were
“metabolic pathways”, “biosynthesis of secondary
metabolites”, “plant hormone signal transduction”, and
“protein processing in endoplasmic reticulum”, followed by
pathways such as “phenylpropanoid biosynthesis”, “ABC
transporters”, and “flavonoid biosynthesis”. These enriched
pathways and processes might thus participate in
metabolism. Under plant hormone signal transduction, genes
such as gibberellin 20-oxidase (down regulated 2.11-fold)
and brassinosteriod insensitive 1 BRI1 (up regulated 8.5-
fold) were identified in GA and BR signal pathways. From
the above results, metabolism pathways in B80 plants lines
may be affected to increase hypocotyl length.
Fig 2 Comparison of different genes expression (DEGs) in leaves between JSH and B80
X- and y-axes represent log2 values of gene expression. Blue, orange and brown correspond to down-regulated, up-regulated and unaltered gene expression, respectively. If a gene was expressed in just one sample, its expression value in another sample was replaced by the minimum value of all expressed genes in control and case samples. The screening threshold is given at the top of the figure.
Fig 3 Histogram of GO terms assigned to DEGs in leaves between JSH and B80
The x-axis indicates the square root of the number of DEGs. The y-axis represents GO terms. All GO terms are grouped into three ontologies: blue for biological process, brown for cellular component, and orange for molecular function.
Fig 4 KEGG enrichments of annotated DEGs across three comparisons
The y-axis indicates the KEGG pathway and the x-axis indicates the enrichment factor. A high q-value is represented by light blue, and a low q-value is represented by dark blue.
Transcription factor analysis
Transcription factors (TFs) are involved in multiple
biological processes in plant such as plant height (Chen et
al. 2015, Zhang et al. 2017), heading date (Nemoto et al.
2016), and other metabolic reactions and defense (review by
Feller et al. 2011). Thus, according to the detected DEGs in
the transcriptome data, we classify the family of
transcription factors to which the differentially expressed
Wang et al. 2018
Indian Horticulture Journal 8(2/3)
48

genes belong, and the results are shown in (Fig 5). In detail,
these TFs were classified into 59 families and the largest
group of differentially expressed TFs was MYB (215),
MYB-related (165), followed by the AP2-EREBP (136),
bHLH (122), NAC (83), WRKY (60) and others (Fig 5).
Among them, the representative TFs were listed in the
(Table S2). From the analysis, hypocotyl elongation might
be mainly attributed by MYB and MYB-related TFs.
Fig 5 DEGs classification on TF family
Verification of several genes among the DEGs
In order to confirm the accuracy of RNA-seq result, a
total of 12 genes were selected randomly based on
transcription upregulated and down regulated. These genes
encoded E3 ubiquitin-protein ligase (Csa3G840450),
BRASSINOSTEROID INSENSITIVE 1-associated receptor
kinase 1 (Csa7G060160), aminotransferase TAT2
(Csa4G629470), stem-specific protein TSJT1
(Csa6G154500), transcription factor MYB108-like
(Csa6G040640), auxin-induced protein (Csa6G505810),
salicylic acid-binding (Csa2G237140), LRR-repeat protein
(Csa7G378510), zinc finger protein WIP2-like
(Csa7G428260), gibberellin 20 oxidase (Csa4G022900), 3-
ketoacyl-CoA synthase (Csa3G144140), and sugar transport
protein (Csa2G094390). Expression quantities of the
selected genes using qRT-PCR were consistent with the
results obtained with RNA-Seq analysis, which means the
RNA-seq data were credible (Fig 6).
DISCUSSION
The study first represents the broad-scale gene
expression analysis of hypocotyl in cucumber to investigate
related genes expression profile changes in two materials
with differences in hypocotyl length. By this method, we
aimed to identify genes regulating hypocotyl length and
through these genes to explore what signal transduction
pathways and metabolism or physiological processes
involved in the hypocotyl development.
Fig 6 Quantitative real-time PCR (qRT-PCR) validation of several genes between JSH and B80 among DEGs from RNA-Seq results
Data is presented as the mean ± standard deviation (n = 9). *0.01≤P≤0.05; **P≤0.01; Student’s t test.
Multiple studies in plant could explore related genes
controlling the agricultural trait mainly by transcriptome
sequencing. For example, a study identified five cucumber
DEGs involved in gray mold combing the large-scale
transcriptome analysis during infection (Kong et al. 2015).
Heat shock proteins (HSPs) genes were reported to be
involved in the heat stress tolerance by an RNA-sequencing
approach of heat- and control-treated reproductive tissues in
rice (Gonzã et al. 2016). Genes of anthocyanin synthesis,
photosynthetic system reprogramming, cell wall remodeling,
and ascorbic acid (AsA) metabolism were involved in the
early nitrogen deficiency response in cucumber seedlings by
RNA-Seq-based transcriptome profiling (Zhao et al. 2015).
Similarly, combing the method, our results identified 1089
different expressed genes between JSH and B80, which
were largely enriched in the catalytic activity, binding,
metabolic process, cellular process, single-organism process,
membrane, and cell (Fig 3). These results indicate that
catalytic activity and metabolic process related genes play
essential roles in the growth and development of hypocotyl.
Gibberellin (GA) is essential for hypocotyl
development, which has been deeply studied in Arabidopsis
(Gomi et al. 2003, Alabadí et al. 2004, Stamm et al. 2017).
According to the KEGG analysis, we found that the
gibberellin receptor GID1 was prominently down-regulated
by 122.6-fold in B80 and the expression of gibberellin 20-
oxidase was also significantly decreased for 2.11-fold in
Comparative Transcriptome Sequencing of Cucumber Cultivars
49

B80, indicating the GA signaling transduction and
biosynthesis was affected in B80, resulting the different
hypocotyl length between JSH and B80.
MYB proteins are key factors in regulatory networks
controlling plant development, metabolism and responses to
biotic and abiotic stresses (review by Dubos et al. 2010).
Previous studies reported that MYB such as MYB46 and
MYB83 in Arabidopsis are regulators of the biosynthesis of
lignin, xylan and cellulose, involved in the secondary cell
wall thickening (Zhong et al. 2008, 2012, Hua et al. 2013).
In rice, OsMYB103L and MYB61 participated in the
secondary wall biosynthesis mediated by the GA pathway,
which can affect leaf shape, cellulose synthesis and
mechanical strength (Yafeng et al. 2015). OsMPH1, the
MYB-like transcription, factor regulates plant height and
improves grain yield in rice (Zhang et al. 2017). Our results
found that the MYB and MYB-related genes were mostly
enriched during these detected TFs (Fig 5) and the
expression of most MYBs was significantly increased in
B80 such as Csa6G040640 (up- regulated 5-fold),
Csa5G610490 (up-regulated 4.4-fold) and so on (Table S3).
In addition, we detected 12 genes expression level, which
was consistent with the transcriptome data, indicating these
expression changes of DEGs between JSH and B80 were
credible.
In conclusion, we hypothesized that higher expression
of MYBs in B80 might influence the synthesis of secondary
cell wall, resulting longer hypocotyl.
Acknowledgments
This work was supported by the Presidential foundation
of Guangdong academy of agricultural sciences (201813).
Conflicts of interest
This study is not related to any potentially competing
financial or other interest.
REFERENCES
Alabadí, D., J. Gil, J. M.A. Blázquez, and J. L. Garcia-Martinez. 2004. Gibberellins repress photomorphogenesis in darkness.
Plant Physiology 134(3): 1050.
Bo, K., H. Wang, Y. Pan, T. K. Behera, S. Pandey, C. Wen, Y. Wang, P. W. Simon, Y. Li, J. Chen, and Y. Weng. 2016.
Short hypocotyl 1 encodes a smarca3-like chromatin remodeling factor regulating elongation. Plant Physiology
172(2): 1273-1292.
Chen, J. 2015. Fine mapping of short hypocotyl locus in semi-wild Xishuangbanna cucumber. Plant and Animal Genome. (in
Chinese)
Chen, X., S. Lu, Y. Wang, X. Zhang, B. Lv, L. Luo, D. Xi, J. Shen, H. Ma, and F.Ming. 2015. OsNAC2 encoding a NAC
transcription factor that affects plant height through mediating the gibberellic acid pathway in rice. Plant Journal
82(2): 302-314.
Dubos, C., R.Stracke, E.Grotewold, B.Weisshaar, C. Martin, and L. Lepiniec. 2015. MYB transcription factors in
Arabidopsis. Trends Plant Science 15(10): 573-581.
Fehlmann, T., S. Reinheimer, C. Geng, X.Su, S.Drmanac, A. Alexeev, C. Zhang, C. Backes, N. Ludwig, M. Hart, D. An, Z.
Zhu, C. Xu, A. Chen, M.Ni, J.Liu, Y.Li, M.Poulter, Y.Li, C.Stähler, R.Drmanac, X.Xu, E.Meese, and A.Keller. 2016.
Cpas-based sequencing on the bgiseq-500 to explore small non-coding rnas. Clinical Epigenetics 8: 123.
Feller, A., K.Machemer, E.L.Braun, and E.Grotewold. 2011. Evolutionary and comparative analysis of MYB and bHLH
plant transcription factors. Plant Journal 66(1): 94-116.
Filichkin, S.A., H.D. Priest, S.A.Givan, R.Shen, D.W.Bryant, S.E.Fox, W.K.Wong, and T.C. Mockler. 2010. Genome-wide
mapping of alternative splicing in Arabidopsis thaliana. Genome Research 20(1): 45-58.
Garg, R. M.Verma, S.Agrawal, R.Shankar, M.Majee, and M. Jain. 2014. Deep transcriptome sequencing of wild halophyte
rice, Porteresia coarctata, provides novel insights into the salinity and submergence tolerance factors. DNA Research
21(1): 69-84.
Gendreau, E., J.Traas, T.Desnos, O.Grandjean, M.Caboche, and H.Höfte, 1997. Cellular basis of hypocotyl growth in
arabidopsis thaliana. Plant Physiology 114(1): 295-305.
Gomi, K., and Matsuoka, M. 2003. Gibberellin signalling pathway. Current Opinion in Plant Biology 6(5): 489-493.
Gonzã, l. N., L. Dreni, L. Lawas, M. Galbiati, M. L.Colombo, S. Heuer, K. S.V. Jagadish, and M.M. Kater .2016. Genome-
wide transcriptome analysis during anthesis reveals new insights into the molecular basis of heat stress responses in
tolerant and sensitive rice varieties. Plant Cell Physiology 57(1): 57-68.
Hua, C.W., N.Goué, M. N. Saidi, S.Legay, P.Sivadon, D.Goffner, J.Grima-Pettenati. 2013. Identification of novel
transcription factors regulating secondary cell wall formation in Arabidopsis. Front Plant Science 4(4): 189.
Jain, M. 2012. Next generation sequencing technologies for gene expression profiling in plants. Brief Funct Genomics 11(1):
63-70.
Jin, Z. A., L.T. Si, D.D. Li, and X. Gao. 2009. Genetic analysis of hypocotyls length of cucumber seedling under low light .
Jiangsu Agricultural Sciences 3: 158-161. (in Chinese)
Kanehisa, M., M.Araki, S.Goto, M. Hattori, M. Hirakawa, M.Itoh, T.Katayama, S.Kawashima, S.Okuda, T.Tokimatsu and Y.
Yamanishi. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Research 36(Database issue):
480-484.
Wang et al. 2018
Indian Horticulture Journal 8(2/3)
50

Kong, W., N.Chen, T.Liu, J. Zhu, J.Wang, X. He, and Y. Jin. 2015. Large-scale transcriptome analysis of cucumber and
botrytis cinerea during infection. Plos One 10(11): e0142221.
Li, B. and C.N. Dewey. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference
genome. BMC Bioinformatics 12(1): 323.
Livak K J, T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta
Delta C(T)) Method. Methods 25(2001): 402-408.
Miao, H., X.F Gu, S. P.Zhang, Z.H.Zhang, S.W.Huang, Y.Wang, and Z.Y. Fang. 2012. Mapping QTLs for seedling-
associated traits in cucumber. Acta Horticulturae Sinica 39(5): 879-887. (in Chinese)
Ming, C. H., F. L.Jiang, H. U.H, X. C.Zhou, F. H.Zhan, and Zhen, W. U. 2011. Effects of different leggy extent seedlings on
cucumber growth, yield and quality. China Vegetables 4: 29-34. (in Chinese)
Nemoto, Y., Y.Nonoue, M.Yano, and T. Izawa. 2016. Hd1, a CONSTANS ortholog in rice, functions as an Ehd1 repressor
through interaction with monocot-specific CCT-domain protein Ghd7. Plant Journal 86(3): 221-233.
Scheres B, H.Wolkenfelt, V.Willemsen, M.Terlouw, E.Lawson, C. Dean, and P. Weisbeek. 1994. Embryonic origin of the
Arabidopsis primary root and root meristem initials. Development 120(9): 2475-2487.
Song, L., S. Prince, B. Valliyodan, T. Joshi, J.V. Maldonado dos Santos, J. Wang, L. Lin, J.Wan, Y. Wang, D. Xu and
Nguyen H. T. 2016. Genome-wide transcriptome analysis of soybean primary root under varying water-deficit
conditions. BMC Genomics 17(1): 57.
Stamm, P., A.T. Topham, N. K. Mukhtar, M.D.Jackson, D.F.Tomé, J.L.Beynon, G.W. Bassel 2017. The transcription factor
ATHB5 affects GA-mediated plasticity in hypocotyl cell growth during seed germination. Plant Physiology 173(1):
907.
Wang, M., S.Liu, S.Zhang, H.Miao, G.Tian, H.Lu, P. Liu, Y. Wang,and X. Gu. 2016. Qtl mapping of seedling traits in
cucumber using recombinant inbred lines. Plant Breeding 135(1): 124-129.
Wang, Z., Gerstein, M., Snyder, M. 2010. RNA-Seq: a revolutionary tool for transcriptomics. National Review on Genetics
10: 57-63.
Ye, J., L.Fang, H.Zheng, Y. Zhang, J.Chen, Z.Zhang, J.Wang, S.Li, R.Li, L.Bolund, and J.Wang 2006. WEGO: a web tool
for plotting GO annotations. Nucleic Acids Research 34(Web Server issue): W293.
Ye, Y., B.Liu, M.Zhao, K.Wu, W.Cheng, X. Chen, Q. Liu, Z.Liu, X. Fu, and Y. Wu. 2015. CEF1/OsMYB103L is involved
in GA-mediated regulation of secondary wall biosynthesis in rice. Plant Molecular Biology 89(5): 385-401.
Zhang, G. Y.; Long-Ting, S. I. and Dan-Dan, A. L. 2011. Qtl analysis for hypocotyl traits of cucumber seedlings under low
light stress. Acta Horticulturae Sinica 25(1): 81-93. (in Chinese)
Zhang, G., G.Guo, X.Hu, Y.Zhang, Q.Li, R.Li, R.Zhuang, Z.Lu, Z.He, X.Fang, L.Chen, W.Tian, Y.Tao, K. Kristiansen, X.
Zhang, S.Li, H.Yang, J.Wang, and J.Wang. 2010. Deep RNA sequencing at single base-pair resolution reveals high
complexity of the rice transcriptome. Genome Research 20: 646-654.
Zhang, Y., C.Yu, J. Lin, B. Liu, J.Wang, A. Huang, H.Li, and T. Zhao 2017. OsMPH1 regulates plant height and improves
grain yield in rice. Plos One 12(7): e0180825.
Zhao, W., X.Yang, H.Yu, W. Jiang, N.Sun, X. Liu, X.Zhang, Y.Wang and X. Gu. 2015. RNA-seq-based transcriptome
profiling of early nitrogen deficiency response in cucumber seedlings provides new insight into the putative nitrogen
regulatory network. Plant Cell Physiology 56(3): 455-467.
Zhong, R. and Z.H. Ye. 2012. MYB46 and MYB83 bind to the SMRE sites and directly activate a suite of transcription
factors and secondary wall biosynthetic genes. Plant Cell Physiology 53(2): 368-380.
Zhong, R., C. Lee, J. Zhou, R.L. McCarthy and Z. H. Ye. 2008. A battery of transcription factors involved in the regulation
of secondary cell wall biosynthesis in Arabidopsis. Plant Cell 20(10): 2763-2782.
Zhou, X. C., F. L. Jiang, C. H. Ming, H.U. Hong-Min, Q. Jiang, and W. U. Zhen. 2010. Effects of different temperature and
light treatments on cucumber seedling growth and content of photosynthetic pigments. Acta Agriculturae Jiangxi
22(12): 20-25. (in Chinese).
Zhou, Y., P. Yang, F. Cui, F. Zhang, X. Luo and Xie J. 2016. Transcriptome analysis of salt stress responsiveness in the
seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). Plos One 11(1): e0146242.
Comparative Transcriptome Sequencing of Cucumber Cultivars
51
Recommended