
Hematology winter symposium in Kaohsiung Veterans General hospital
Dec. 17th, 2016 in Kaohsiung
Clinical Application of Anti-CML immunity in the era of TKI therapy
Hiroshi Fujiwara, MD,PhD
Department of Hematology, Clinical Immunology and Infectious Diseases,Graduate School of Medicine, Ehime University

COI Disclosure Information
Hiroshi Fujiwara, MD, Ph.D.
Honoraria ; BMSTravel accommodation ; BMS
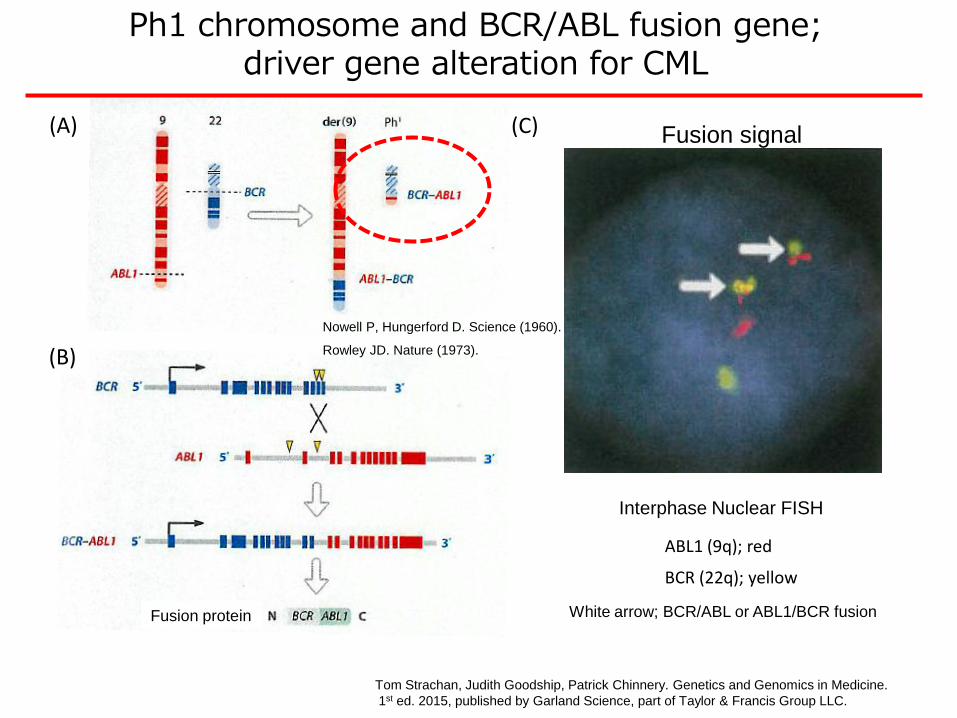
Interphase Nuclear FISH
ABL1 (9q); red
BCR (22q); yellow
White arrow; BCR/ABL or ABL1/BCR fusionFusion protein
Ph1 chromosome and BCR/ABL fusion gene; driver gene alteration for CML
Tom Strachan, Judith Goodship, Patrick Chinnery. Genetics and Genomics in Medicine.
1st ed. 2015, published by Garland Science, part of Taylor & Francis Group LLC.
(A)
(B)
(C) Fusion signal
Nowell P, Hungerford D. Science (1960).
Rowley JD. Nature (1973).

Douglas Hanahan , Robert A. Weinberg. Hallmarks of Cancer: The Next Generation. Cell, Volume 144, Issue 5, 2011, 646 - 674
Tactics of anticancer therapy based on characteristics of cancer cells
Selective TKI therapy
Allogeneic HSCT; the most powerful treatment option, however enforcing
pts to live with substantial Treatment-related Toxicity.
Target specific, i.e. less toxic systemically.
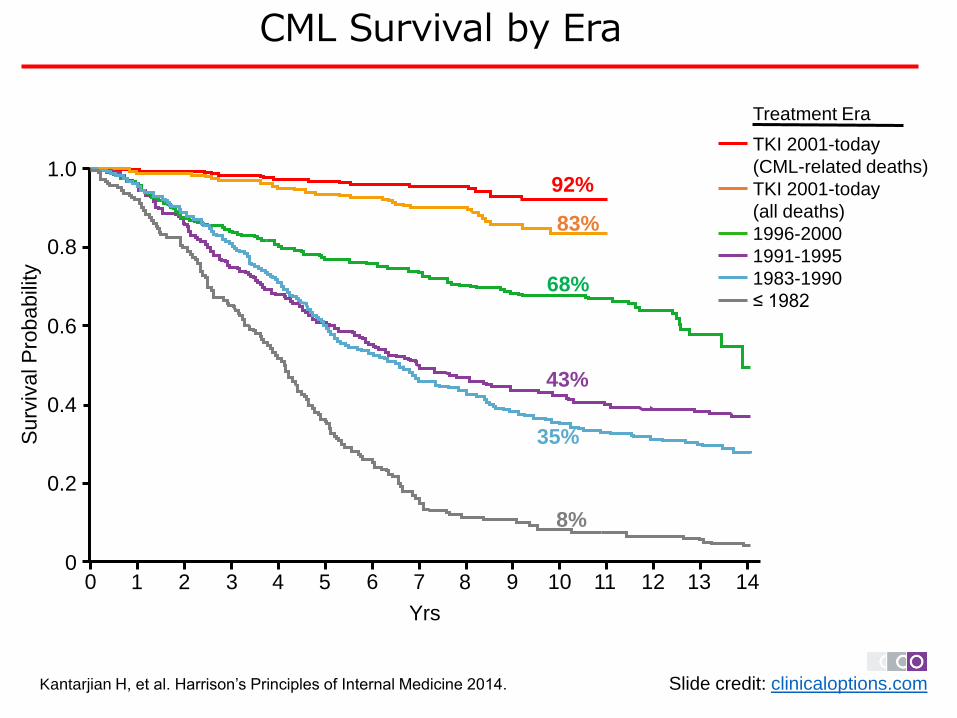
Kantarjian H, et al. Harrison’s Principles of Internal Medicine 2014. Slide credit: clinicaloptions.com
68%
92%
83%
43%
35%
8%
0
0.2
0.4
0.6
0.8
1.0
0 1 2 3 4 5
Yrs
Surv
ival P
robabili
ty
6 7 8 9 10 11 12 13 14
Treatment Era
TKI 2001-today
(CML-related deaths)
TKI 2001-today
(all deaths)
1996-2000
1991-1995
1983-1990
≤ 1982
CML Survival by Era
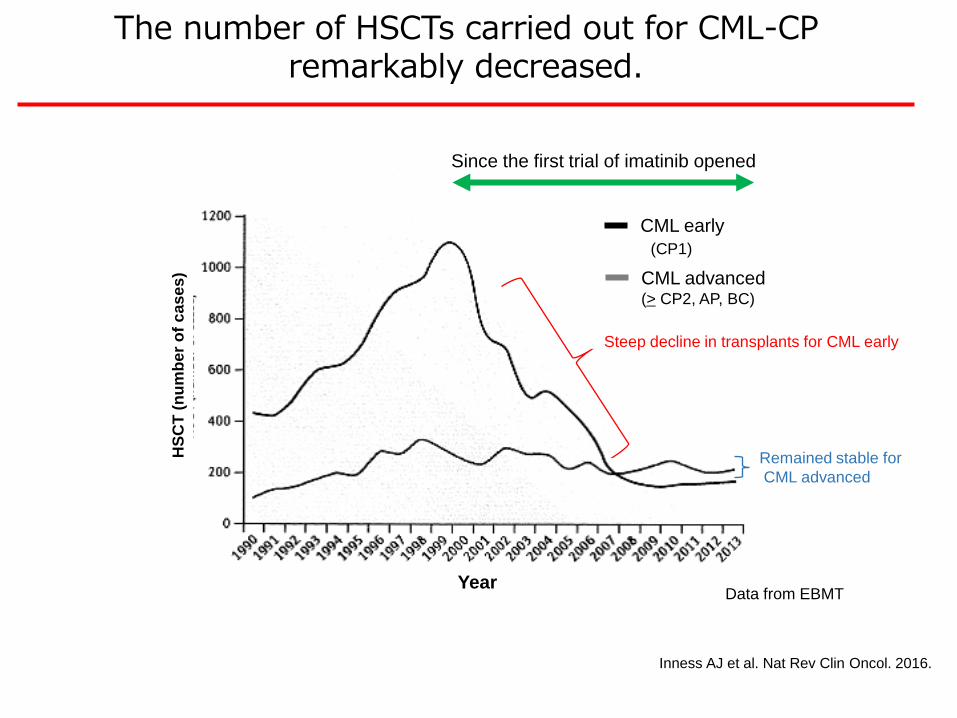
Inness AJ et al. Nat Rev Clin Oncol. 2016.
The number of HSCTs carried out for CML-CP remarkably decreased.
CML advanced(> CP2, AP, BC)
HS
CT
(n
um
be
r o
f c
as
es
)
Year
Since the first trial of imatinib opened
CML early
(CP1)
Steep decline in transplants for CML early
Remained stable for
CML advanced
Data from EBMT

Ponatinib vs SCT for CML With T315I Mutation
Ponatinib vs SCT
Median OS, Mos
Disease
Group Ponatinib SCT
P
Value
CP NR 103 .013
AP NR 56 .889
BP 7 11 .026
Ph+ ALL 7 32 .136
CP
AP
BP
Nicolini FE, et al. Blood. 2015;126. Abstract 480. Slide credit: clinicaloptions.com
0
20
40
60
80
100
OS
(%
)
0 12 24 36 48
Ponatinib
SCT
Mos
0
20
40
60
80
100
0 12 24 36 48
Mos
0
20
40
60
80
100
0 12 24 36 48
Mos
OS
(%
)O
S (
%)
Ponatinib
SCT
Ponatinib
SCT

Overall response rates to TKIs in pts with CML-CP
IM 1st line Tx.60% durable
response
40% eligible for
IM treatment
withdrawal
Up to 50%
achieve
Operational
cure
40% require
2nd TKI
50% durable
response
50% require
3rd TKI
50% durable
response
50% require
alternative
treatment
Around 10-15% of
initial group might
benefit from HSCT
2nd TKI
1st line Tx.
70-80% durable
response
20-30% require
3rd TKI
50% durable
response
50% require
alternative
treatment
Around 10-15% of
initial group might
benefit from HSCT
(A)
(B)
Inness AJ et al. Nat Rev Clin Oncol. 2016. modified.
Treatment
withdrawal
under evaluation

Clinical questions in the treatment of CML in the era of TKI
Withdrawal of TKI ?
MRD ; minimal or measurable? residual disease
How to manage after TKI withdrawal
/ Concept of “Treatment-Free Remission” “Operational Cure”
CML stem cell
Required alternative strategy ?
To pts with refractory to TKI or advanced disease w/o T315I mutation
Interfere the disease control after TKIs withdrawal
allo-HSCT Immunotherapy
Immunotherapy
> 3rd TKIs
NCCN(National Comprehensive Cancer Network)
Clinical Practice Guidelines in Oncology. 2012
European LeukemiaNet recommendations
for the management of chronic myeloid leukemia.
Baccarani M et al. Blood, 2013
“ Propose continuation of TKI treatment indefinitely
in all responders”
A challenge;
To establish a strategy to avoid the life-long dependency on TKIs.
….Secondary Malignancies, financial issues,
recognized and/or unrecognized AEs (for young pts),
impairment of QoL (pregnancy issue), …
e.g. For whom ?
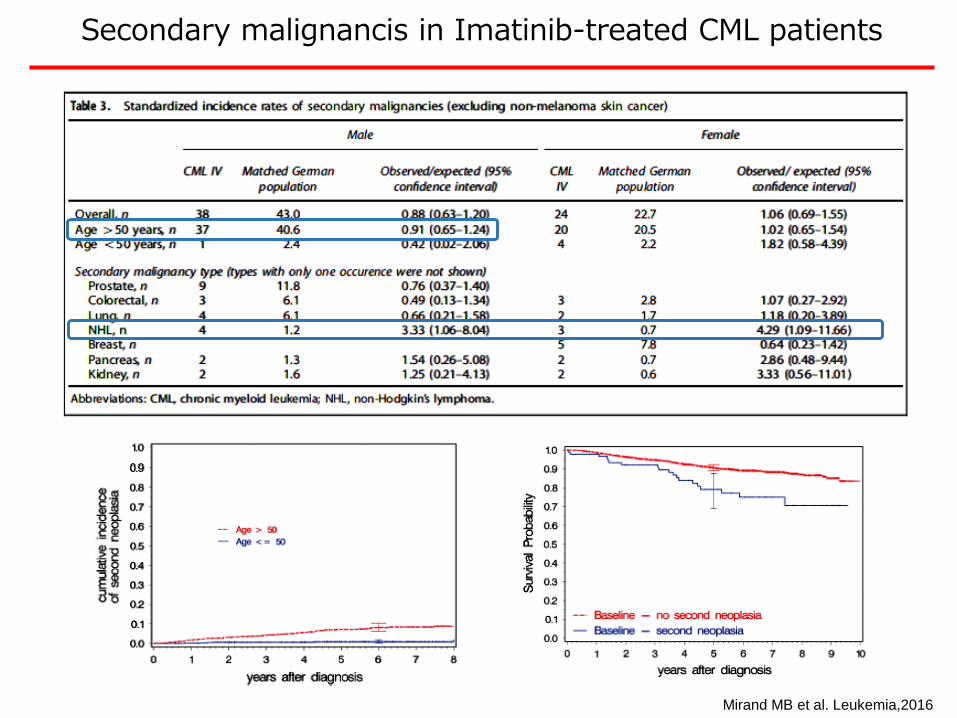
Secondary malignancis in Imatinib-treated CML patients
Mirand MB et al. Leukemia,2016

Tx.before Requisite TFR% discontinuation for discontinuation (m edian follow -up tim e)
IM discont. trialsSTIM 1 100 IFN then IM for>3y C M R for > 2y 39% (55 m o.)STIM 2 200 IM > 3y As for STIM prelim inary 46% @ 2yEU RO -SKI 809 IM , N IL, D AS M R4 for > 1y in progress
TKI for > 3y prelim inary 61% @ 0.5yIM /N IL/D AS discont. trialsSTO P 2G -TKI pilot 50 N IL or D AS C M R for m edian 29m o. in progress
prelim inary 61.1%EN ESTFreedom 175 N IL front-line M R4.5 for > 1y in progressEN ESTop 117 N IL 2nd-line >3y M R4.5 for > 1y in progress
EN ESTPath 1058 IM >2 and N IL M R4.5 for > 1y vs in progressM R4.5 for >2y RC T
Japanese trial* 88 D AS 1y as consoli. D M R, unclearly defined 49% @ 6m o.(63 discont.)
Study Pt. N o.
“Operational Cure”
; Prolonged survival in molecular remission without therapy
by Goldman JM et al. J Natl Compr Canc Netw. 2012
What does MRD in leukemia really mean?
Goldman JM, Gale RP. Leukemia 2014
Saußele S et al, Leukemia 2016, modified.

Levels of molecular response and corresponding log-reduction
and BCR-ABL transcript levels on the International Scale.
Michele Baccarani, and Simona Soverini Blood
2014;124:469-471
© 2014 by American Society of Hematology

NCCN Guidelines Version 1.2017
Chronic Myeloid Leukemia
Discontinuation of TKI therapy
Criteria for TKI Discontinuation
1) Age > 18 y. 2) CP with no prior history of AP or BC, 3) TKI Tx. > 3 y.4) Prior evidence of quantifiable BCR-ABL transcript, 5) Stable MR4; < 0.01% IS for > 2y.6) No history of resistance to any TKI, 7) Access to a reliable QPCR test with a sensitivity, of detection of > 4.5 logs that reports
results on the IS and provides results within 2w, monthly molecular monitoring for the first 6 mo. following discontinuation, bimonthly during months 7-24, and quarterly thereafter (indefinitely) for pts who remain in MMR (MR3; <0.1% IS),
8) Consultation with a CML Specialty Center to review the appropriateness for TKIdiscontinuation, including TKI withdrawal syndrome
9) Prompt resumption of TKI, with a monthly molecular monitoring for the first 6 mo. following resumption of TKI and every 3 mo. thereafter is recommended indefinitely for pts with a loss of MMR. For those who fail to achieve MMR after 6 mo. of TKI resumption, BCR-ABL1 kinase domain mutation testing should be performed, andmonthly molecular monitoring should be continued for another 6 mo..
Low risk (sokal score) CML at diagnosis
Timely available and highly sensitive (> 4.5 log sensitivity
below the standard baseline) MRD measurement
Optimal response achieved to prior TKI cessation.
Achievement of deep molecular remission > 2y
with prior TKI treatment >3y before the discontinuation
Discontinuation should be as a “clinical trial”
with Higher NK cell number
Bhalla S et al. Clin Lymphoma, Myeloma & Leukemia, 2016, modified
Although the depth of molecular response seems
associated with TFR (Treatment Free Remission) in
fact, the correlation between TFR and the “long-term
outcome” still remains to be elucidated.

A primitive subset of CD34+ CML cells survives complete Bcr-Abl inhibition in growth factor–free medium for 12 days.
Ashley Hamilton et al. Blood 2012;119:1501-1510
Chronic myeloid leukemia stem cells are not dependent on Bcr-Able kinase activity for their survival
NC
DAS 150nM
Untreated
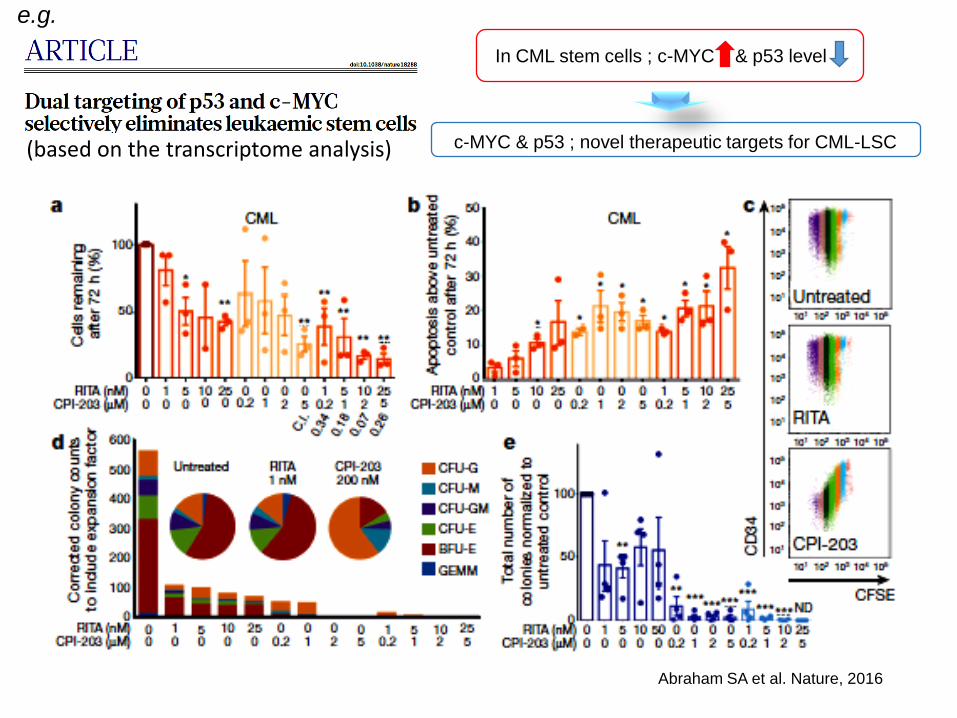
Abraham SA et al. Nature, 2016
In CML stem cells ; c-MYC & p53 level
c-MYC & p53 ; novel therapeutic targets for CML-LSC
e.g.
(based on the transcriptome analysis)
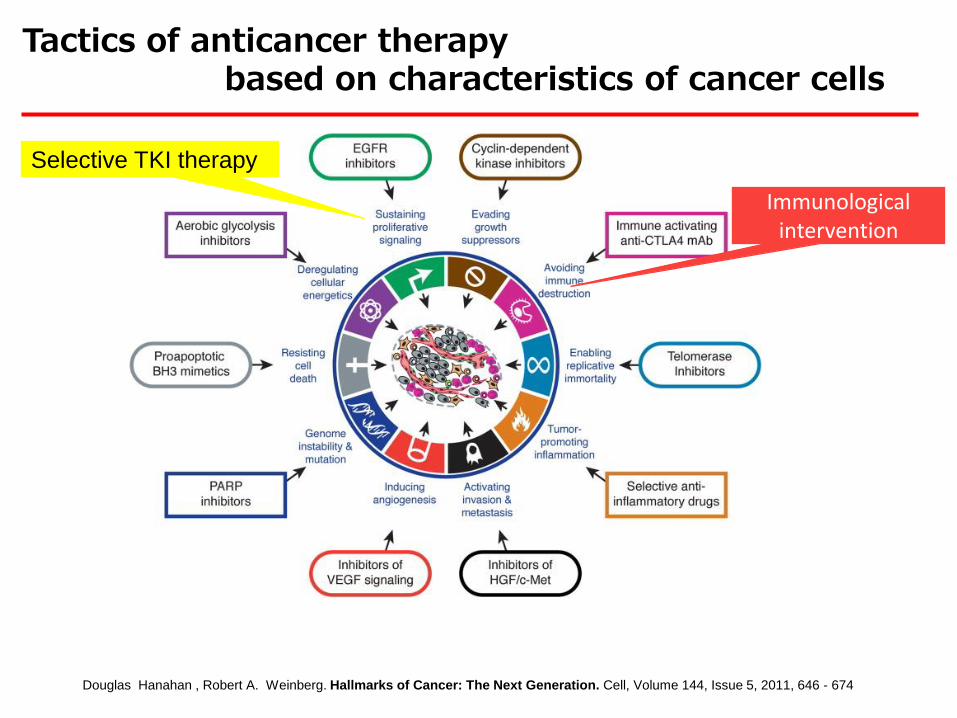
Douglas Hanahan , Robert A. Weinberg. Hallmarks of Cancer: The Next Generation. Cell, Volume 144, Issue 5, 2011, 646 - 674
Tactics of anticancer therapy based on characteristics of cancer cells
Selective TKI therapy
Immunological intervention

CTL: non-labeled moving cells, Leukemia cell : Green labeled
Dead cells: red labeled, Fibroblasts: by-stander cells
Biostation IM, NIKON Inc.Asai H. & Fujiwara H.
Leukemia-specific CD8+ T cells (CTLs) selectively killed leukemia cells.
CTL
CD8
CD3
α β
Garcia KC, et al. Science, 1996
TCR
HLA class I β2MG
Time-lapse study
Perforin/Granzyme
IFN-g/TNF-a/Fas-L
Being target-specific might reflect the less systemic toxicity
(on-target/off-tumor AE).
Immunotherapeutic strategy
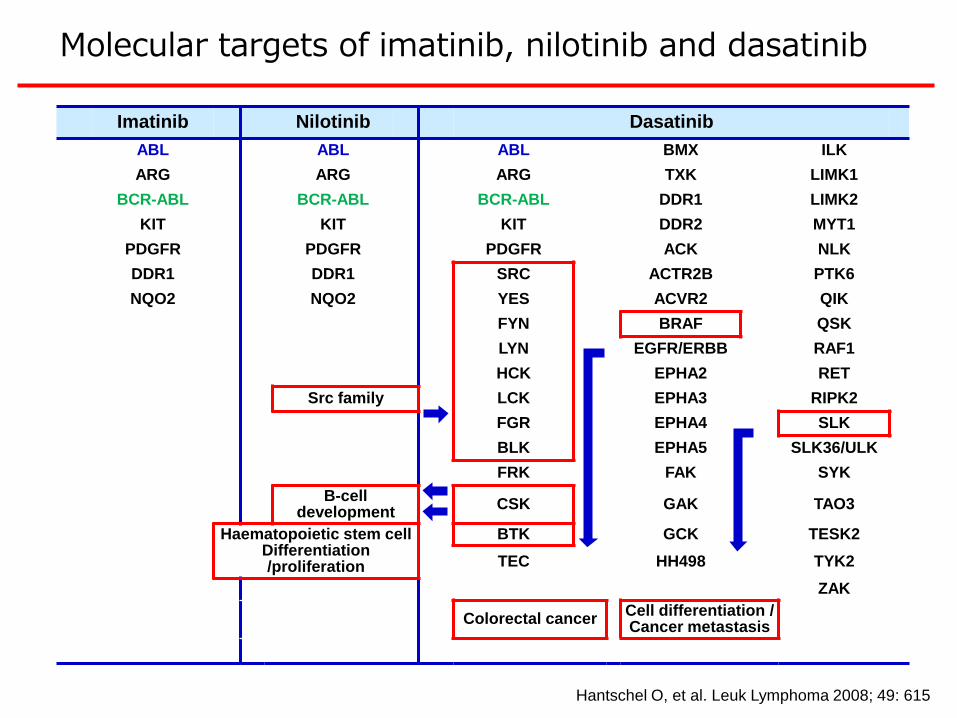
Hantschel O, et al. Leuk Lymphoma 2008; 49: 615
Imatinib Nilotinib Dasatinib
ABL ABL ABL BMX ILK
ARG ARG ARG TXK LIMK1
BCR-ABL BCR-ABL BCR-ABL DDR1 LIMK2
KIT KIT KIT DDR2 MYT1
PDGFR PDGFR PDGFR ACK NLK
DDR1 DDR1 SRC ACTR2B PTK6
NQO2 NQO2 YES ACVR2 QIK
FYN BRAF QSK
LYN EGFR/ERBB RAF1
HCK EPHA2 RET
Src family LCK EPHA3 RIPK2
FGR EPHA4 SLK
BLK EPHA5 SLK36/ULK
FRK FAK SYK
B-cell development
CSK GAK TAO3
Haematopoietic stem cell Differentiation/proliferation
BTK GCK TESK2
TEC HH498 TYK2
ZAK
Colorectal cancerCell differentiation / Cancer metastasis
Molecular targets of imatinib, nilotinib and dasatinib

Lo
g10
(BC
R-A
BL)
Time course (mo.)
Mathematical mode; Autologous immune responsemight interfere the variation of BCR-ABL transcripts
- 3.5 log reduction- 3.5 log reduction
- 3.5 log reduction
Clapp GD et al. Oncoimmunol. 2016. modified.
Based on data from IM first-line Tx.
Immune
window
drug
induced
Immune response-mediated disease suppression

Host immune responses are involved in the clinical course of CML pts treated with TKIs
Lymphocytosis
Schiffer CA et al. Cancer, 2016
NK cell increase
Christiansson L et al. Mol Cancer Ther., 2015
Christiansson L et al. Mol Cancer Ther., 2015
MDSCA
rg1
(ng/m
L )
IL-6
(lin
ear
NP
X )
IL-1
2 (
linear
NP
X )
IL-8
(pg/m
L )
Naka K and Ichinohe T. Stem Cell Investig 2016

Natural Killer cells in the clinical efficacy of TKIs
A phase I/II clinical trial of autologous cytokine-induced killer cells as adjuvant immunotherapy
for acute and chronic myeloid leukemia in clinical remission. Linn YC, et al. Cytother. 2012
Yeung DT, et al, Blood, 2015
Nievergall E, et al, Blood, 2014
NK cells seem involved in anti-CML efficacy mediated by TKIs
Adoptively transferred NK cells seemed less effective
ADCC mediated by NK cells might be able to kill CML stem cells
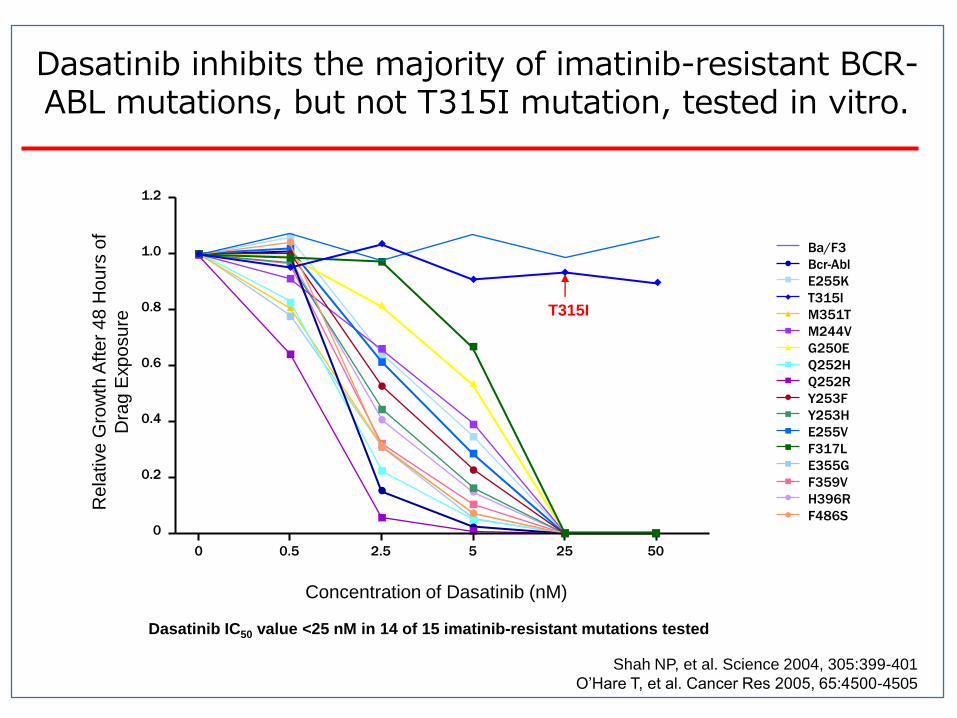
Dasatinib IC50 value <25 nM in 14 of 15 imatinib-resistant mutations tested
Ba/F3
Bcr-Abl
E255K
T315I
M351T
M244V
G250E
Q252H
Q252R
Y253F
Y253H
E255V
F317L
E355G
F359V
H396R
F486S
Concentration of Dasatinib (nM)
1.2
5025
0
0.2
0.4
0.6
0.8
1.0
52.50.50
T315I
Rela
tive G
row
th A
fter
48 H
ours
of
Dra
g E
xp
osu
re
Shah NP, et al. Science 2004, 305:399-401
O’Hare T, et al. Cancer Res 2005, 65:4500-4505
Dasatinib inhibits the majority of imatinib-resistant BCR-ABL mutations, but not T315I mutation, tested in vitro.
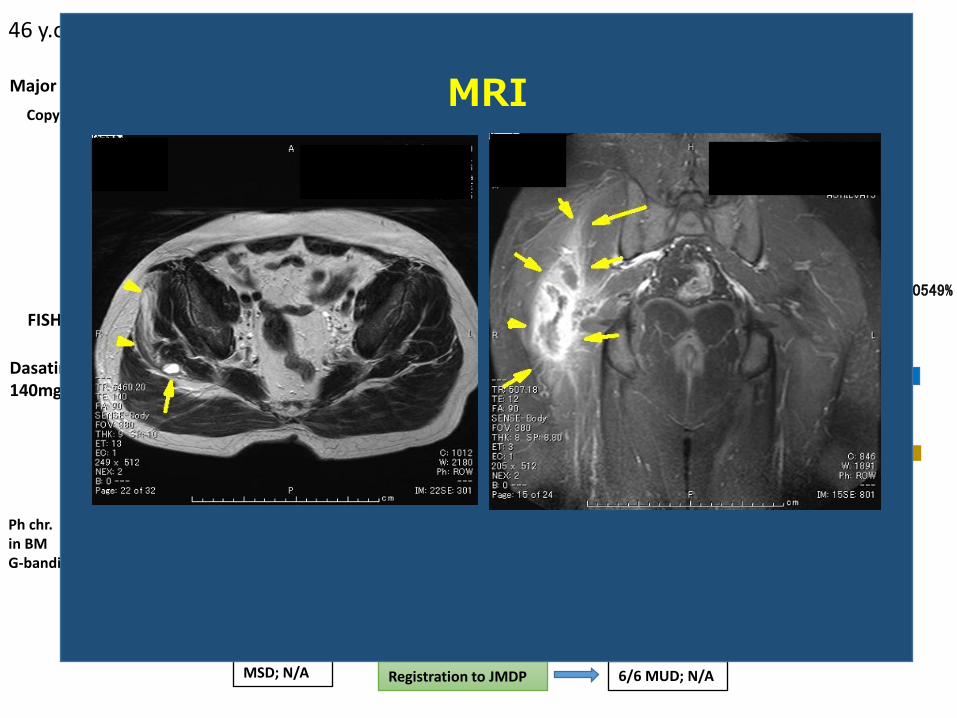
0
200
400
600
800
+3M +6M +9M +12M +15M +18M +21M +24M +27M +30M +33M +36M +39M
Nilotinib800mg/day
T315I(+)
Interferon α600 x10^4 U/day
CHR
Ph chr.in BMG-banding
20/20 0/20 0/20 1/20 0/20 2/20 4/20 1/20
CCyR Loss of CCyR MR3.0(MMR)
Dasatinib100mg/day
Dasatinib140mg/day
0% 0.8% 1.5% 0.1% 2.3% 5.0% 2.4% 2.1%
0.0854% 0.0664% 0.0783% 0.0549%
Major bcr-abl mRNA(IS)
Registration to JMDPMSD; N/A 6/6 MUD; N/A
Ineligible for the Ponatinib clinical trial
Clinical Course
Copy /assay
Major bcr-abl mRNA(TMA)
FISH (PB)
T315I(-)
46 y.o male ; CML-CP+ myeloid sarcoma (rt.hip)
MRI

Science 2013 Trends in Immunology 2016Nature 2014
Blood 2015;125:4017
CD19-CAR-T
Journal of clinical pharmacology, 2015,doi10.1002/jcph.591
Anti-PD1 mAb
Induction or
Adoptive transfer of
High quality effector cell
Removal of
immuno-regulatory elements
Tactics of immunotherapy for CML

Drawn by author
MDSC
Tactics of immunotherapy for CML-2
BCR-ABL1 derived antigens
Driver mutation = BCR-ABL1
Overexpressed
protein
LAA
CML cell
HLA1
A gene
B gene
PGPs;
PR3 (PR1)
NE (PR1)
Cath-G (CG-1)
WT1
AURKA
p210 BCR-ABL
CML specific AgC
ML
rela
ted
Ag
s
Treg.
HLA1
B gene
CML stem cell
anti-CML
-CAR-T
CD123
Primary Granular Proteins as therapeutic targets of cellular immunotherapy for CM-CP: lineage specific Ags
1 2 3 4 5 6
CG
PR3
GAPDH
HNE
Lane 1, 2; Normal G-CSF
mobilized PB
CD34+ cells
Lane 3, 4; CD34+ cells
from CML patient
Lane 6; HL60 as positive
control
Lane 6; dH2O as negetive
control
(PGP mRNAs detected by RT-PCR)
Fujiwara H et al, Clin Cancer Res, 2005

Molldrem J.J. et. al., Nat Med,2000
PR1 peptide-specific CD8 killed CML cells
Fujiwara H. et. al., Blood,2004
NE-specific CD8 killed CML progenitor cells
*
CD3+/ HNE
0
50
100
150
200
50:125:112.5:16.25:10:1
*
Untreated CD3+
CD3+/HNE with anti-class I
E:T ratio
Nu
mb
er
of
co
lon
ies
/10
e3
ce
lls
CTLs specific for PR1 peptide which derived from both PR3 and NE display tumoricidal activity against CML cells (CML-BM-CD34+ cells).
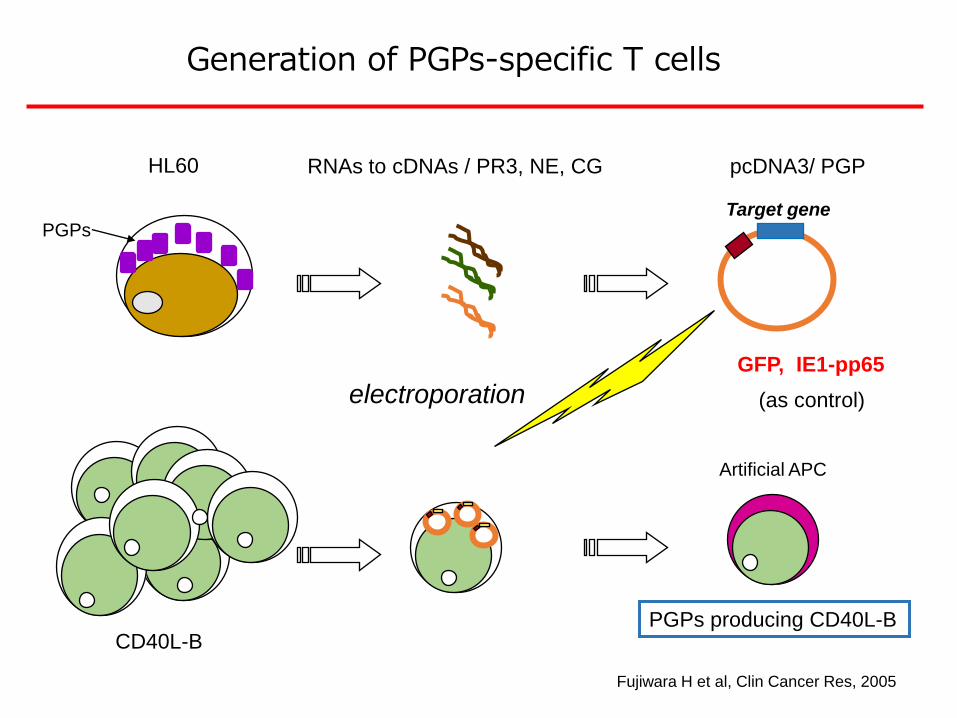
HL60 RNAs to cDNAs / PR3, NE, CG pcDNA3/ PGP
CD40L-B
electroporation
PGPs producing CD40L-B
GFP, IE1-pp65
(as control)
Target gene
Fujiwara H et al, Clin Cancer Res, 2005
Generation of PGPs-specific T cells
PGPs
Artificial APC

By PR3 expressing CD40B
By HNE expressing CD40B
By CG expressing CD40B
By mock transfected CD40B
Cyto
lyti
c a
cti
vit
y (
%)
E:T ratio
50:1 12.5:125:1
E:T ratio
CML cells Normal BM cells
0
10
20
30
40
50
60
70
50:1 12.5:125:1
- CML-CP (case# 5)-
Responder T-cells
Fujiwara H et al, Clin Cancer Res, 2005
Successful generation of PGPs-specific T cells derived from CML patient’s CD3+T cells at day 90 after allo-HSCT
using PCP gene-modified autologous CD40LBs

CML CML
Zhang M, et al, Clin Cancer Res, 2012, Alatrash G. Oncoimmunology, 2013,
Sergeeva A, et al, Leukemia, 2016, Qing M, et al, Cytother. 2016,
Ataie N, et al. J Mol Biol 2016, modified,
Pep.vaccine
+
adjuvant
Multiple pep.vaccine
+
adjuvant
SS
Va
Ca
Vb
Cb
Single target
Multiple targets
Adoptive transfer
VL VH
or
or
T
T
T
8F4-CAR
8F4-Bite
C
C
C
8F4 mAb
Up-dated strategy to target PGPs
ESK1-CAR
CML CML

Immunogenic determinant epitopederived from AURKA
1 51 132 383 402
H2N COOHKinase domain
KEN motif D-box activating motif D-box (PxxRxxL)
PNILRLYGYF HDATRVYLILEYAPLGTVYR ELQKLSKFDE191 230207 215
Ochi T, et al. Blood. 2009
ALLBMMCs(n=7)
AMLBMMCs(n=17)
NormalPBMCs(n=4)
PHA-blasts(n=3)
CMLBMMCs(n=10)
0
10
20
3040
120
Re
lative
exp
ressio
n o
f A
UR
KA
m
RN
A AURKAmRNA
%Specific lysis
Pt#1 (CML)
Pt#2 (CML)
Pt#4 (AMLM2)
Pt#5 (AMLM0)
Leukemia cells
Pt#6 (ALL L2)
0 20 40 60
7.0
19.1
9.4
9.5
27.3
HLA-A*0201
+
-
-
-
+
Pt#3 (CML) +
10.5
Chr. 20q
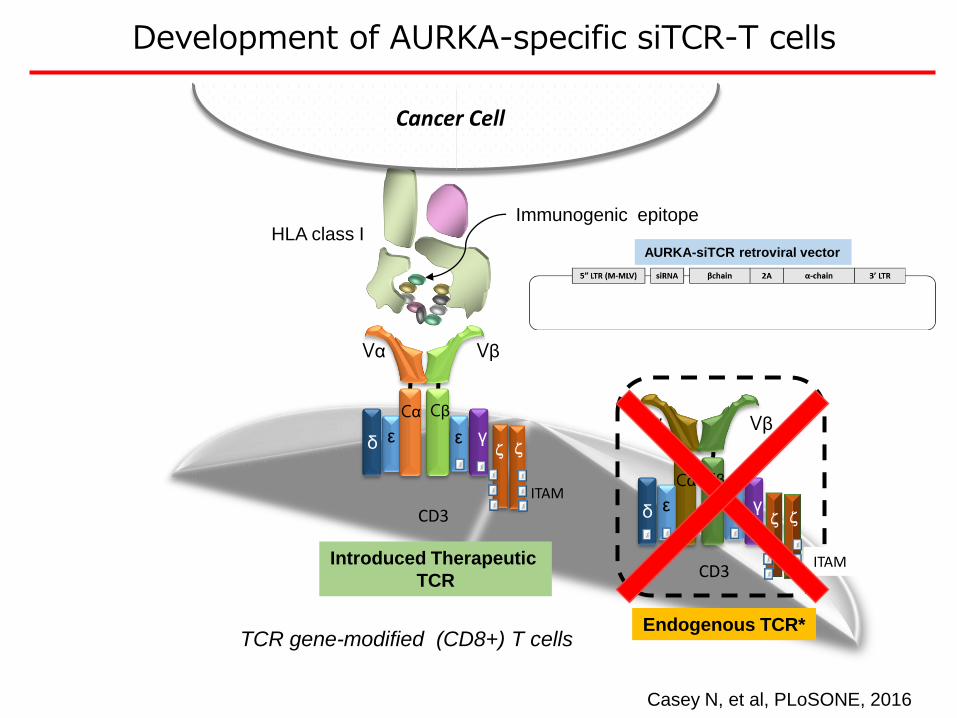
Vα Vβ
εδ
Cα Cβ
ε γz z
Vα Vβ
εδ
CD3
Cα Cβ
ε γz z
HLA class I
ITAM
TCR gene-modified (CD8+) T cells
Introduced Therapeutic
TCR
Endogenous TCR*
Immunogenic epitope
Cancer Cell
CD3ITAM
Development of AURKA-specific siTCR-T cells
Casey N, et al, PLoSONE, 2016
AURKA-siTCR retroviral vector
CML (BMMCs) Cord blood (CBMC)
34+38low
34+38low
CD34
CD
38
CML
CD34+
38low
Normal
PBMCs
(n=4)
0
5
10
15
CML
PBMCs
CML
whole
BMMCs
10.6
10.9
4.4
1.1 1
CBMC
CD34+
38low
Normal
PBMCs
(n=4)
Rela
tiv
e a
mo
un
t o
f
Au
rora
-Am
RN
A
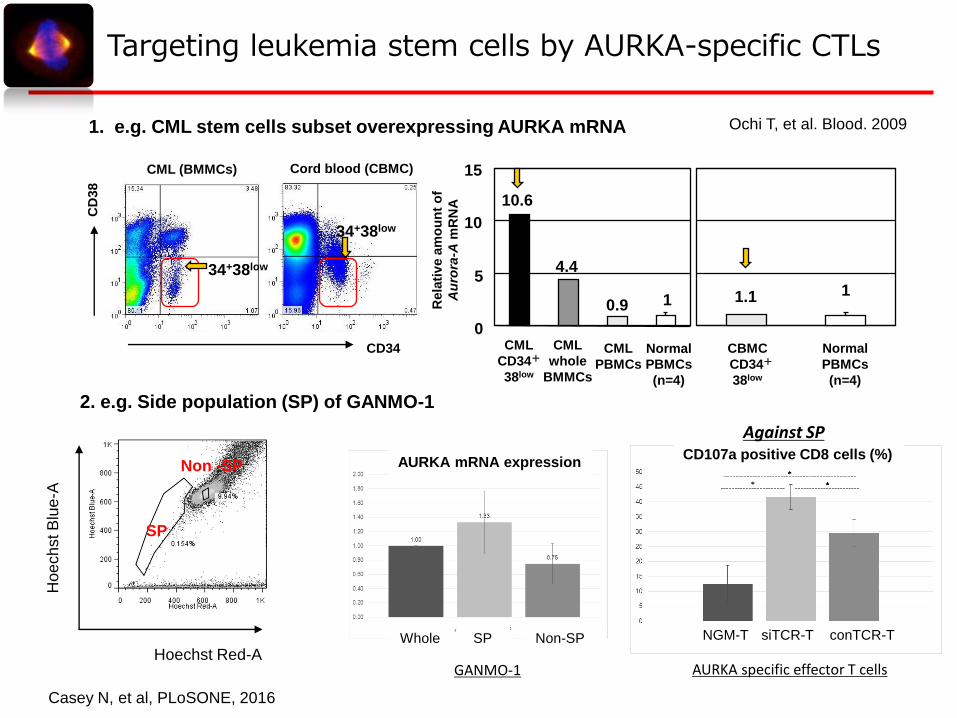
1. e.g. CML stem cells subset overexpressing AURKA mRNA Ochi T, et al. Blood. 2009
Hoechst Red-A
Hoe
ch
st B
lue
-A
2. e.g. Side population (SP) of GANMO-1
Targeting leukemia stem cells by AURKA-specific CTLs
AURKA mRNA expressionCD107a positive CD8 cells (%)
Whole SP Non-SP
SP
Non -SP
GANMO-1
NGM-T siTCR-T conTCR-T
AURKA specific effector T cells
Casey N, et al, PLoSONE, 2016
Against SP
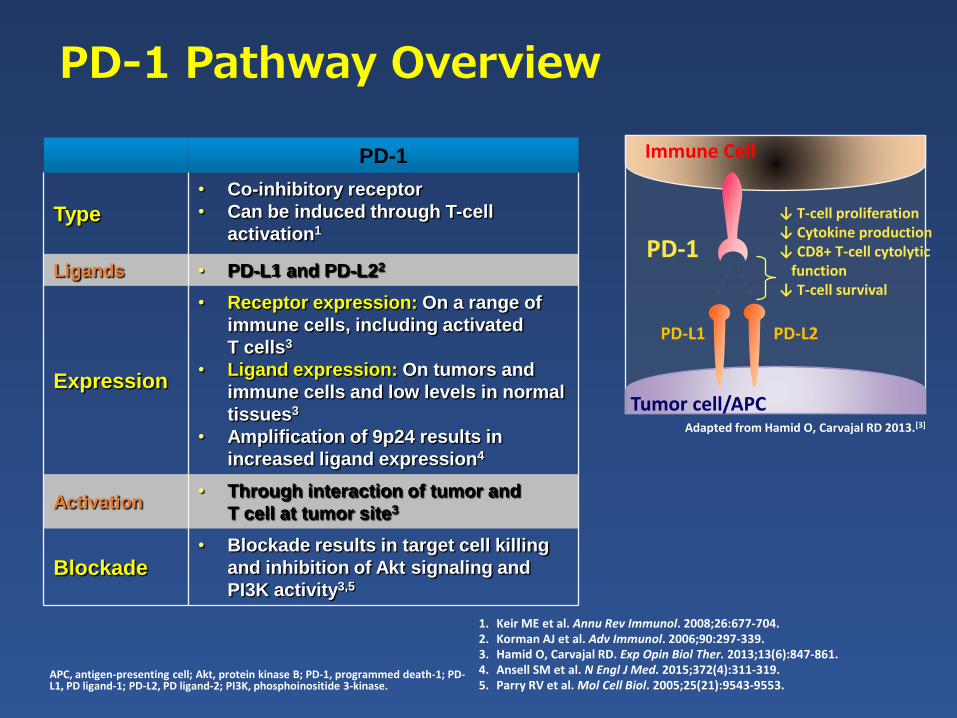
PD-1 Pathway Overview
PD-1
Type
• Co-inhibitory receptor
• Can be induced through T-cell
activation1
Ligands • PD-L1 and PD-L22
Expression
• Receptor expression: On a range of
immune cells, including activated
T cells3
• Ligand expression: On tumors and
immune cells and low levels in normal
tissues3
• Amplification of 9p24 results in
increased ligand expression4
Activation• Through interaction of tumor and
T cell at tumor site3
Blockade• Blockade results in target cell killing
and inhibition of Akt signaling and
PI3K activity3,5
1. Keir ME et al. Annu Rev Immunol. 2008;26:677-704. 2. Korman AJ et al. Adv Immunol. 2006;90:297-339. 3. Hamid O, Carvajal RD. Exp Opin Biol Ther. 2013;13(6):847-861.4. Ansell SM et al. N Engl J Med. 2015;372(4):311-319. 5. Parry RV et al. Mol Cell Biol. 2005;25(21):9543-9553.
APC, antigen-presenting cell; Akt, protein kinase B; PD-1, programmed death-1; PD-L1, PD ligand-1; PD-L2, PD ligand-2; PI3K, phosphoinositide 3-kinase.
PD-L1 PD-L2
PD-1
Immune Cell
Tumor cell/APC
↓ T-cell proliferation↓ Cytokine production↓ CD8+ T-cell cytolytic
function↓ T-cell survival
Adapted from Hamid O, Carvajal RD 2013.[3]
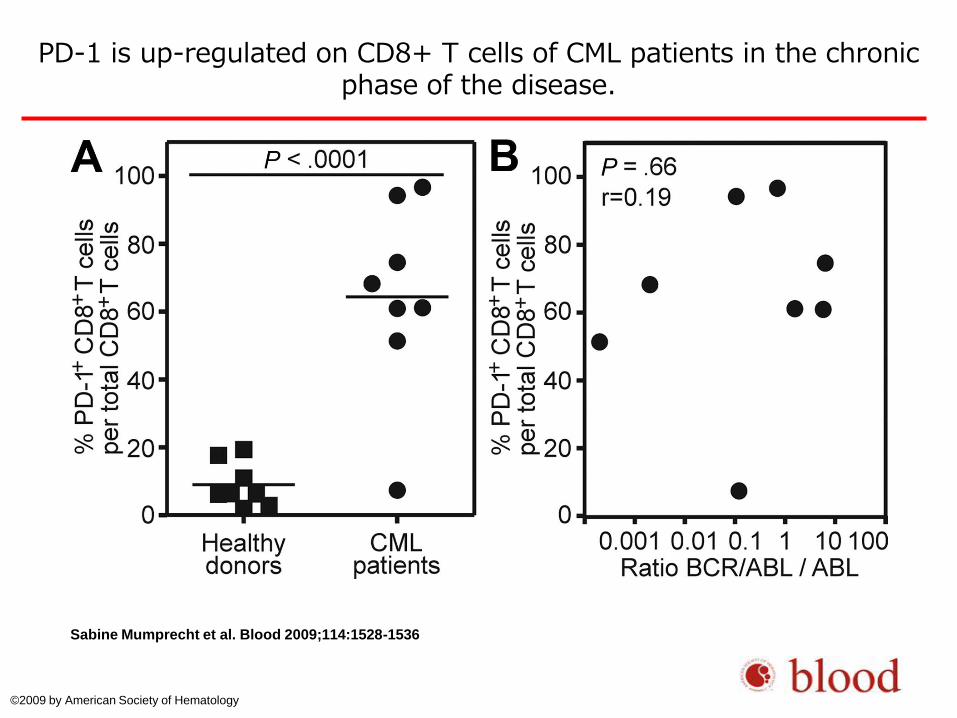
PD-1 is up-regulated on CD8+ T cells of CML patients in the chronic phase of the disease.
Sabine Mumprecht et al. Blood 2009;114:1528-1536
© 2009 by American Society of Hematology

DMSO
DMSO + IFN-g
CD274-PE(PD-L1)
CD274-PE(PD-L1)
MFI: 360 vs. 399
MFI: 393 vs. 1641
MOLM-13 (AML-M5a) LCL
DMSO
DMSO + IFN-g
MFI: 360 vs. 4689
MFI: 393 vs. 5250
Via IFN-g stimulation Constitutive expression
Maruta M, Fujiwara H, Unpublished data
Constitutiveexpression
PD-L1 expression in human leukemia cells
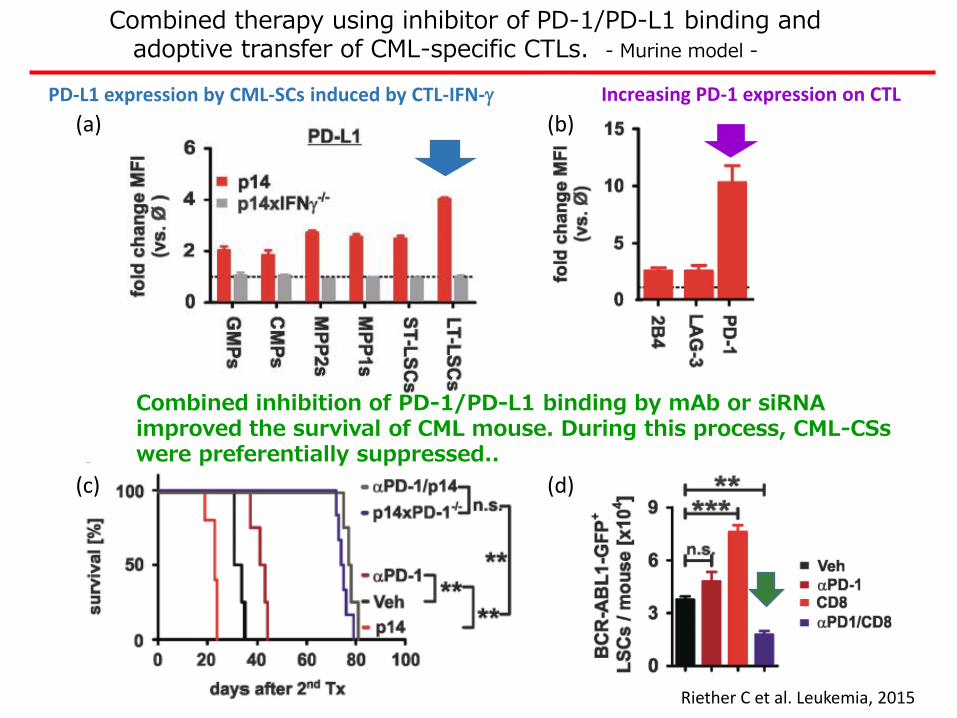
PD-L1 expression by CML-SCs induced by CTL-IFN-g Increasing PD-1 expression on CTL
Combined inhibition of PD-1/PD-L1 binding by mAb or siRNA improved the survival of CML mouse. During this process, CML-CSs were preferentially suppressed..
Combined therapy using inhibitor of PD-1/PD-L1 binding andadoptive transfer of CML-specific CTLs. - Murine model -
Riether C et al. Leukemia, 2015
(a) (b)
(c) (d)
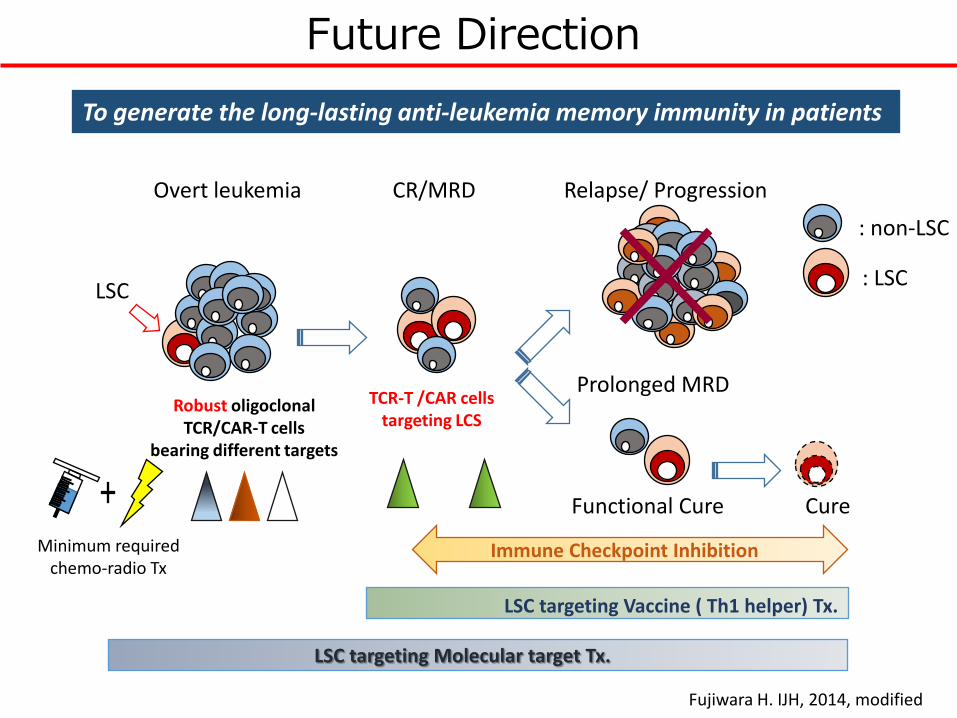
Future Direction
CR/MRDOvert leukemia Relapse/ Progression
Prolonged MRD
Cure
To generate the long-lasting anti-leukemia memory immunity in patients
LSC targeting Molecular target Tx.
LSC targeting Vaccine ( Th1 helper) Tx.
Immune Checkpoint Inhibition
TCR-T /CAR cells targeting LCS
Functional Cure
: LSC
: non-LSC
Robust oligoclonalTCR/CAR-T cells
bearing different targets
LSC
Minimum required chemo-radio Tx
Fujiwara H. IJH, 2014, modified

Take-home messages
In the era of TKIs, the prognosis of pts with CML-CP has beautifully improved.
Studies of clinical responses and underlying mechanisms again emphasized the
importance of host immune responses.
Immunological strategies would be beneficial not only for the enhancement
of efficacy of TKI therapy, e.g., maintenance after withdrawal of TKIs via
suppression of CML-SCs, but also for the breakthrough strategy of
TKI-resistant and aggressive CML, particularly for pts ineligible for allo-HSCT.
Furthermore, immunotherapy could be applied for disease relapse after allo-
HSCT.
Accumulated knowledge regarding anti-CML immunity during TKI treatment
will have to provide a scientific rationale not only for the treatment of leukemias,
but also for solid tumors.

Acknowledgement
Toshiki Ochi Kozo NagaiFumihiro OchiNicholas CaseyKazushi TanimotoTaichi AzumaMasaki Yasukawa
Kiyotaka Kuzushima
Sachiko OkamotoJunichi Mineno
Ehime University
Aichi Cancer Center
Takara Bio Inc.
Mie University
Hiroshi Shiku
Thank you very much for your attention !
A. John Barrett
NHBLI/NIH
Katayoun Rezvani
MDACC
Dogo Hotspring in Matsuyama, Ehime
Recommended





![Potential enhancement of host immunity and anti-tumor ......[22–24]. In addition, curcumin also enhances host anti-tumor immunity by mediating the restoration of T-cell populations,](https://img.pdfslide.us/doc/110x75/60f891b2f5b6860e284a41e2/potential-enhancement-of-host-immunity-and-anti-tumor-22a24-in-addition.jpg)











