
~ l ~ Chapter 5
, : · . ·Identification of inhibitors of Mycobacterium tuberculosis
Isocitrate Lyase

Cfiapter5 MtuiC£ In!tiDitors
5.1 Introduction
An important aspect in anti-TB drug discovery is the identification of new compounds
with the potential to reduce chemotherapy durations. Several lines of evidence suggest
that pathogenic mycobacteria primarily utilize fatty acids, rather than carbohydrates, as
carbon substrates during infection (Munoz-Elias & McKinney, 2005). Two pathways are
specifically required for utilization of fatty acids: the catabolic beta-oxidation cycle that
degrades fatty acids to acetyl-CoA (C2) units and the anaplerotic glyoxylate cycle. When
glycolytic substrates are absent or sparse, microorganisms rely on the glyoxylate cycle to
replenish TCA cycle intermediates during growth on fatty acid substrates. The glyoxylate
cycle in M tuberculosis apparently comprises a single gene encoding malate synthase
and two genes encoding isocitrate lyases (McKinney, et a/., 2000). Isocitrate lyases
catalyze an essential reaction of the glyoxylate shunt, an anaplerotic pathway that
bypasses the C02-generating steps of the tricarboxylic acid cycle and enables bacteria to
synthesize carbohydrates and replenish tricarboxylic acid cycle intermediates from fatty
acid-derived acetyl-coenzyme A (Sharma, eta/., 2000).
Isocitrate lyase can be broadly classified as oxo-acid-lyases, which specifically cleaves
carbon-carbon bond and catalyzes the reaction shown in Equation 1. M tuberculosis
codes for two isocitrate lyases ic/1 (Rv0467) and ic/2 (Rv1915), enzymes involved in
fatty acid metabolism (Munoz-Elias & McKinney, 2005). Recently, ic/1 has also been
shown to play role in propionate metabolism via methylcitrate cycle by functioning as 2-
methyisocitrate lyase (Gould. eta/., 2006).
ICL
Isocitrate succinate + glyoxylate (Eq. 1)
5.1.1 Domain Organization of M. tuberculosis ICL
The crystal structures of icll is available in apoenzyme form (Pdb code: 1F61) and with
bound substrate 3-Bromopyruvate (Pdb code: 1F8M). To analyze the domain
organization of both the ICLs, the homology model of ic/2 was prepared. The details of
113

Cliapter5 :MtuiC£ Infzi6itors
the methods used for ic/2 model development are described in experimental section. The
domain organization of both the ICL is found to be similar for the active site. The ICL
catalytic signature motif KKCGH (AA 193-203 in ic/1; AA 213-217 in icl2) present in
Domain I is conserved in icll as well as in ic/2 (Fig 5.1). Comparative analysis based on
icll crystal structure and icl2 homology model shows that domain II and IV are present in
icl2 while absent in icll. Domain I and III are present in both the homologs and both the
structures are similar with rmsd value of 1.4 A.
Fig 5.1 ICL Domain Organization. Superimposition of in si/ico-modeled ic/2 monomer
(in red) onto the X-ray crystal structure of icll monomer 1 F61 (in blue). All ICL contains
four domains. Domains I and III are present in all eukaryotic as well as prokaryotic ICLs.
Domain I contains the conserved catalytic motif KKCGH. Domain II is present in fungal
and plant ICLs and in mycobacterial ic/2, but absent in mycobacterial ic/1. Domain IV
(not shown) is unique to mycobacterial ic/2 and cannot be modeled as no homologous
structure is available in PDB (Protein Data Bank).
In M tuberculosis both the genes icll and ic/2 are essential for maintenance of
persistence in mouse models (Munoz-Elias & McKinney, 2005). Some other studies have
also implicated the role of glyoxylate cycle in the virulence of bacterial and fungal
pathogens (Idnurm & Howlett, 2002; Lorenz & Fink, 2001; Solomon, et al., 2004; Wang,
114

Cha.pter5 :Mtul CL I nlii6itors
eta/., 2003). Thus, the chemical inhibitors of this pathway could have broad therapeutic
utility. One of our lab's projects revolves around the identification of novel classes of
inhibitors against ic/1 from M tuberculosis (referred as MtuiCL) using virtual screening
techniques and structure elucidation of the inhibitor-enzyme complex. Against this
backdrop, the present chapter reports the identification of novel inhibitors against M
tuberculosis isocitrate lyase (ic/1) using molecular docking approaches. A virtual library
of compounds, belonging to three different classes' viz. galactosyl propanolamines,
cyclopropylphenyl methanes and nitrobutanoates, was evaluated by molecular docking
approaches using both the open and closed conformations of the enzyme. Promising
molecules were then evaluated for ICL inhibition using a high-throughput assay
developed by Dr. R. Shrivastava 's group, Microbiology Division, CDR!, Lucknow.
5.2 Experimental Section
The flowchart showing the systematic methodology used for identification of inhibitors
against MtuiCL is shown in Figure 5.2. The details of the compound synthesis and
enzyme assays are not discussed here as these are not part of the thesis. The compound
synthesis has been carried out by Dr. R. P. Tripathi 's group, MPC Division, CDR!,
Lucknow while the enzyme assays were carried out by Dr. R. Shrivastava 's Group,
Microbiology Division, CDR!, Lucknow.
5.2.1 Homology modeling of ic/2
The homology model of ic/2 was generated using X-ray structure of Aspergillus nidulans
Isocitrate lyase (PDB: 1DQY) and Mycobacterium tuberculosis ic/1 (PDB: 1F61) as
templates and Modeller8v2 (Marti-Renom, et a/., 2000) as modeling tool. Domain I and
III were modeled using M tuberculosis ic/1 while Domain II of ic/2 (AA 269-365) was
modeled after the A. nidulans ICL domain II. Domain N cannot be modeled as had no
homology to any of the known sequences in PDB (Berman, et a/., 2000). The structure
quality of modeled icl2 was verified using Procheck (Laskowski, et a/., 1996) and Whatif
server (Vriend, 1990). The stereochemical quality was satisfying all the parameters with
87.9% amino acid residues present in the core, 9.0% in allowed, 2.5% in generously
allowed and 0.6% in disallowed regions. The score expressing how well the backbone
115

Cliapter5 Mtul CL I nliiDitors
conformations of all residues (Ramachandran Z-score: -0.215), corresponding to the
known allowed areas in the Ramachandran plot was within expected ranges for well
refmed structures. Since the active site in icll is made of residues from two adjacent
subunits, the active site of icl2 was also modeled by generating its dimer using Homology
module of Insightll (M/s. Accelrys Inc.). Prior to virtual screening experiments, all the
polar hydrogen atoms were added to the model.
5.2.2 Virtual Screening Protocol
The docking simulations were performed by using Autodock v3 (Morris, et a/., 1998) and
GOLD v2.2 (Verdonk. eta/., 2003). A combination of Simulated Annealing, Lamarkian
Genetic Algorithm and Monte Carlo algorithms were used for ligand conformational
searching.
Target Preparation:
Target preparation for AutoDock: The available structures of isocitrate lyase from M.
tuberculosis were retrieved from Protein Data Bank (http://www.rcsb.org/pdb) (Pdb
code: 1F8M, 1F81, 1F61) where dimer 1F61 is in its open conformation while tetramer
1F8m and 1F81 are its closed conformation in complex with 3-bromopyruvate and 3-
nitropropionate respectively.
Structures with PDB codes 1 F61 and 1 F8M were selected as the docking template. Prior
to docking studies, crystallographic waters and heteroatoms were removed. Polar
hydrogens were added. Kollman charges were assigned to all atoms. A 60x60x60 3D and
80x80x80 3D affinity grid centered on the active site (CYS191) with 0.375 A spacing
were calculated for 1F61 and 1F8m respectively; for each of the following atom types C,
A( aromatic C ),N, 0, S, H, F, Cl, Brande by using Autogrid3.
Target preparation for Gold: All the heteroatoms were removed and all the hydrogens
were added to the docking templates using Insightll (M/s. Accelrys Inc.).
Autodock Docking Parameters:
Ligands were drawn and optimized using Builder module of lnsightll. The ligands
translation, rotation and internal torsions were assigned. For each run, following docking
parameters were used - Trials of 100 dockings, population size of 150, random starting
116

Cftapter5 !MtuiCL Inliibitors
position and conformation translation step ranges of 1.5 A, rotation step ranges 35,
elitism of 1, mutation rate of0.02, cross-over rate of0.8, local search rate of0.06 and 10
million energy evaluations. The jobs were distributed to the SGI ORIGIN 350 cluster.
Final docked conformations were clustered by tolerance of 1.5A RMSD. The top 10% of
compounds with best simulated binding energies within standard deviation of 2 Kcal/mol
were selected for synthesis and subsequent inhibition assays.
Gold Docking Parameters:
Gold v2.2 (Genetic Optimization of Ligand Docking) also samples the ligand
conformational space using a genetie algorithm (GA). However, in contrast to AutoDock,
it uses the atomic description of the protein (or a truncated binding site) and allows for
the hydroxyl (Ser, Thr, Tyr) and ammonium (Lys) hydrogen atoms to relax upon ligand
docking. All the heteroatoms were removed and all the hydrogens were added to the
receptor file. Ligand input files were prepared and optimized by Builder module of
lnsightll (M/s. Acclerys Inc.) and saved in mol2 format. Ligands were loaded in Gold
GUI using 'Adding all files in the directory' option.
Gold trials were performed using the' default parameters (1 X speed-up, 6/12 internal
potential, no flipping of amide bonds, etc.). All docking trials for GOLD v2.2 were
performed with active site radius of 10.0 A for both the molecules. Binding site was
defined using cavity. atoms file. This file contains the list of all solvent accessible atoms
making-up the protein active site. All the parameters used by GOLD (e.g. hydrogen bond
energies, atom radii and polarisabilities, torsion potentials, hydrogen bond directionalities
etc.) are specified in gold.params file. Configuration file gold.conf reads parameter
settings from a previously saved confi~uration file and loads the parameter values into
the front end of GOLD window. GoldScore were calculated and reranking was performed
for all the docked ligand conformations.
5.2.3 Enzyme Assays
Enzyme in vitro inhibition assays:
The ICL assay measured the synthesis of glyoxylate directly by its reaction with
phenylhydrazine and the formation of glyoxylate phenylhydrazone was monitored as
I17

Cftapter5 !Mtul C.L I nliibitors
increase in absorption at 324 run (Drxon & Kornberg, 1959). The reagents and steps
included 50 mM potassium phosphate buffer, pH 7.0, 6 mM MgCh, 4 mM phenyl
hydrazine, 12 mM L-cysteine, 3 mM threo-D(s)-isocitrate and purified enzyme 2J!l in 200
J.ll reaction volume. The reaction was monitored for 10 min as increase in absorption at
324 run. The known ICL inhibitor, 3-nitropropionate (Sigma) was used at 1 OOJ!M as the
positive control.
BACTEC assays:
Stock solutions of the test compounds pr.epared in DMSO at 1 mglml were sterilized by
passage through 0.22 J.lm filters and were added (50 J.ll) to 4 ml radiometric 7H12 broth;
(Becton Dickinson Instrument System US) to achieve final concentrations. Controls
received 50 J.ll DMSO. Isoniazid and rifampcin (Sigma Chemical Co., St. Louis, MO)
were included as positive drug controls. The vials were inoculated with 104 to 105
CFU/ml of M. tuberculosis H37Rv. An additional control was inoculated with 1:100
dilution of the inoculum to represent 1% of the bacterial population (1ff to 103 CFU/ml).
The vials were incubated at 3-t>C and GI (Growth index) readings were recorded daily
until GI in 1: 100 control had reached 30. The concentration of the drug producing fmal
GI reading lower than those in 1: 100 control was considered to have inhibited more than
90% of the bacteria and was defined as the MIC (Panda et al. 2004).
118

Cliapter5
Open Conformation
(1F61)
Virtual Screening Models
Open Conformation
(1F8M)
!Mtul C£ I nlii6itors
Virtual Screening with AutoDock & Gold
I Biological Testing
/Yovel ~ffolds
BACTEC
Optimization
Fig 5.2 Flowchart showing the systematic workflow used for identification of inhibitors against MtuiCLs.
119

Cliapter5 9vftul C£ I nhiDitors
5.3 Results
It has been established that inhibitors of tll:l.e enzymes isocitrate lyase (icll), responsible
for fatty acid metabolism in mycobacterium may lead to new class of anti TB drugs to
kill the persistent bacteria (Sharma, et al. 2000). Bromopyruvic acid, itaconate, and 3-
nitropropionates are known to inhibit this enzyme at one or the other concentration.
However, none of these molecules show 1my antitubercular activity, a plausible reason
being that these molecules do not meet the c::riterion for drug like molecules.
5.3.1 Virtual library generation and mole~cular docking with Gold and AutoDock
With the background knowledge that inhibitors of this enzyme are more likely to emerge
from the intermediate products formed in TCA cycle and glyoxalate shunt, parallely that
glycosyl amino acid and aromatic amines do possess significant antitubercular activity in
vitro (Tripathi, et al., 2002; Grover, et al., 2004; Dwivedi, et al., 2005; Srivastava, et al.
2006), it has been concluded from the studies that hydrophobicity plays very important
role in displaying antitubercular action. Some hydrophobic classes of compounds like
nitrobutanoates, galactosylpropanolamines and cyclopropyl derivatives were taken up for
our initial modeling and docking studies. The results indicated that the all the three
classes of compounds fitted well into the "binding site of MtuiCL. A virtual library of
about 200 compounds based on derivatives of all the three classes of compounds was
generated. All the parent scaffolds and some of the selected primary substitutions are
displayed in Figure 5.3. The parent scaffolds in virtual library design for cyclopropyl
derivatives were 4-Cyclopropyl-methyl phenol (Fig 5.3 A) and 4-(Cyclopropyl-hydroxy
methyl)-phenol (Fig 5.3 B); while the design principles were based on the aryloxyphenyl
cyclopropyl methanones series which were reported earlier as anti-mycobacterial agents
(Dwivedi, et al., 2005). The other parent s.caffolds were nitro butyric acid which was
designed based on the fact that the nitropropinates were known to inhibit icll (Sharma et
al. 2000) and Galactosyl propanolamines which were designed on the basis of
glycosylated beta amino acid series, another known anti- mycobacterial agents {Tripathi,
et al., 2002). The primary substitutions includes ethyl benzene, ethyl furan, Methyl
tetrahydro-furo-dioxole, propenyl benzene, 4-Chlorobenza.ldehyde and 3-
pyridylcarboxaldehyde, etc. while secondary substitutions were on the primary
12:0

ChapterS !Mtul CL I nliiiiitors
substitutions and includes methanol, methyl, hydroxyl, amines, dimethyl dioxolanes etc.
side chains.
RO RO
A OH
B
R=
0
CY' Cyclopropyl derivatives
H
Ar----_/N"''''"· OH
R=
Ar=4 Chlorobenzaldehyde = 3 pyridylcarboxaldehyde Nitrobutanoates
Galactosyl propanolamines
Fig 5.3 The scheme of virtual library design for the selected compounds reported as M tuberculosis ic/1 inhibitors.
121

ChapterS !Mtu!CL Inhibitors
These compounds were docked and scored in the first instance with Autodock using both
the open and closed conformation of the structures and the acceptable docking scores are
shown in Figure S.4. The in silico screening studies were used to prioritize compounds
and compounds with docking energy lower than -7 kcallmol were subsequently selected
for synthesis and enzyme assays. The compounds selected for synthesis from the virtual
library were augmented with a view to increase the structural diversity of the library. The
synthesized compounds were assayed initially for in vitro protein activity inhibition. The
in vitro inhibition results for the selected compounds are shown in Figure S.S and the
respective docking scores and structures are shown in Table SA & SB. Successful
compounds were then evaluated for inhibition of growth of M tuberculosis in BACTEC
assays (data not shown).
5.3.2 Synergistic Effect in ICL Inhibition
An examination of the docking energies of compounds against the two conformations of
the enzyme revealed that apparently some compounds exhibited a preference for a
particular conformation of MtbiCL. The docking results suggested that pairs of certain
compounds, each with better predicted affinity for 'closed' and 'open' ICL structural
conformations, should be more effective as inhibitors than individual compounds by
themselves. A closer analysis of the docking poses suggested that this might be due to the
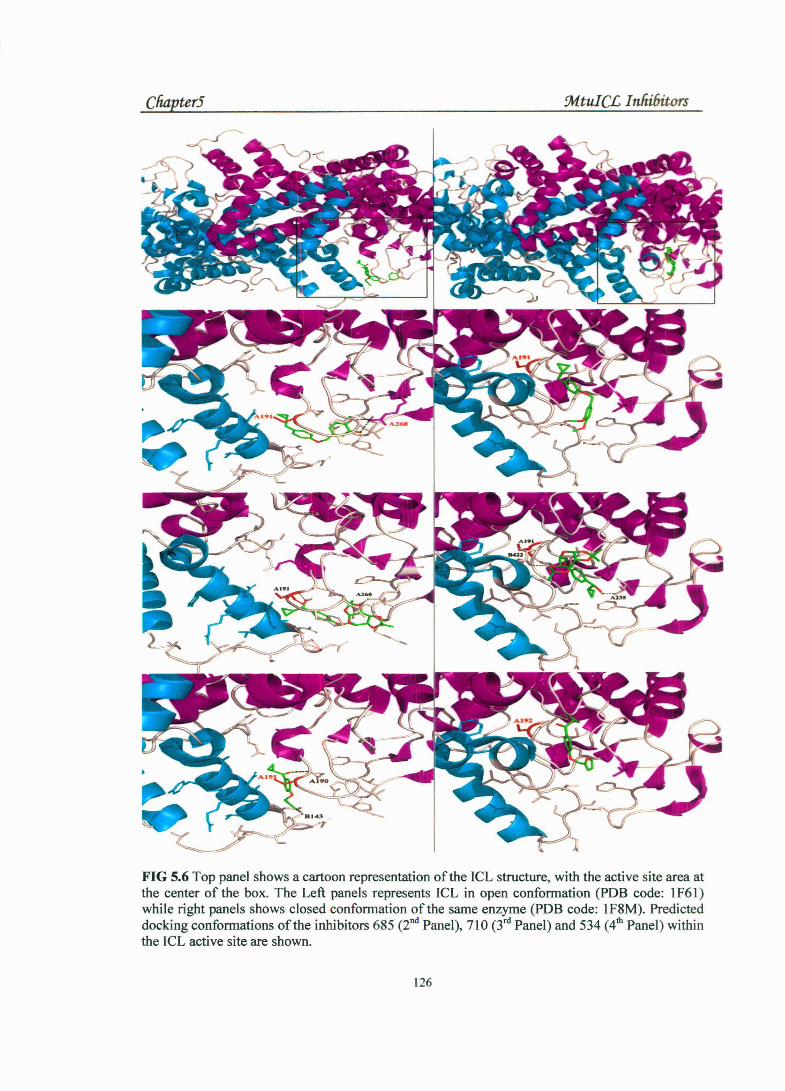
differences in the conformation of the active site loop which leads to better stabilization
of a docking pose with a particular conformation (Fig S.6). The ligands were selected for
further re-docking procedure with Gold instead of Autodock to further validate the
docking results. The Gold Scores were also biased towards one or the other
conformations of the enzyme {Table SA). We accordingly evaluated inhibition of
successful compounds alone and also in pairs to test for any synergistic effect. The in
vitro phenyhydrazone-based assay suggests that at least some compounds in pairs are
demonstrating a synergistic effect for enzyme inhibition and results are shown in Figure
S.S. This was further confirmed by the in vivo BACTEC assays based on M tuberculosis
growth under conditions of carbon deprivation. This novel approach offers to be effective
in the design of more potent inhibitors of theM tuberculosis isocitrate lyase (ic/1).
122

-N ',..)
0
-1
·2 - ·3 .J 0-4 ~ -5 ::J.o < -7 u~ ~ .--9
'-'·10 <I
r::
I U"'S\1 I 1~'61
. - -
-14~------------------------------------------------------------------------~ 1 3 S 7 9 11 13 15 17 19 21 23 25 2? 2931 D )5 37 39 41 43 45 47 .tg 51 Sl!S 57 59&1 63 6S 67 6911 73 ?5 n 79 8183 8S 87 89 91 93
___ ...,. COMPOUNl> ll>ENTIFIER
Figure 5.4 Docking energy graph. The plot between the binding energy calculated by AutoDock and compound identifiers. The two curves represent binding energy plots for the two different conformation of the enzyme and the compounds below the dashed lines were chosen for the inhibition assays. The compounds reported here are
indicated by an asterisk(*) viz. first* is 534, second* is 685, third* is 710 and fourth* is 689.

Cliapter5 9.1 tul CL I nliibitors
70
60 ~ •% INHIBITION H f-4 50 H IQ H
I 40 H
-.!. 30
20
10
0
!0<-, (c) ""(;;) "'\
~~ <-,
Fig 5.5 The plot showing in vitro inhibition (in percentage) of the compounds. The % inhibition on y-axis is plotted against the compound codes on x-axis. Some compounds are showing better in vitro enzyme inhibition in pairs. NP is nitropropionate.
Table SA Docking Energy table. Compounds with docking energy above nitropropionate (NPN) were selected for in-vitro assays. 1F61 is the open while 1F8M is the closed conformation of the ic/1. The docking score calculated by Autodock as well as Gold are shown in the table.
Compound Docking Scores S.No.
Autodock Docking Energy (kcallmol) GoldScore codes
I 1F61 1F8M ic/2 1F61 1F8M
1 NPN I -5.04 -8.02 -8.83 19.23 27.99
2 710 I -10.5 -11.07 -12.33 45.42 36.49
3 685 -10.8 -10.67 -13 .25 46.82 48.61 ~- - ·--
'
4 534 ' -8.87 -11.4 -11.28 40.62 -' -- ---- ~
5 689 -13.3 -12.12 -15.22 54.48 49.71
6 168 -8.42 -9.66 -11.78 35.32 26.95 ~ ~ - -- , ___ -
124

Cliapter5 9vf tul CL I nhi6itors
Table SB. Structures of the compounds shown in Table 5A. The first three compounds belongs to the cyclopropyl class, forth is combination of galctosylpropanolarnines and cyclopropyl while fifth molecule belongs to nitrobutanoates class of compounds.
S.No.
1
2
CompoundiD
710
4-( 4-Cyclopropylmethylphenoxymethyl )-1 ,2-dimethoxy
benzene
534
Cyclopropyl-[ 4-(furan-2-ylmethoxy)-phenyl]-methanone
689
Hexadecanoic acid cyclopropyl-3 {4-[5-(2,2-dimethyl-
4
5
[ 1,3 ]dioxolan-4-yl)-2,2-dimethyltetrahydro-furo[2,3-d][1 ,3 ]dioxol-
6-yloxy]-phenyl}-methyl ester
685 1-[3-( 4-Cyclopropylmethyl
phenoxy)-5,5-dimethylhexahydro-cyclopenta[b] furan-2-
yl ]-ethane-1 ,2-diol
168
3-Nitromethyl-5-phenyl-pent-4-enoic acid ethyl ester
Structures
___ ,,X ··'
# ........ ~
0
\. •+01111110
;:;;, ~
COOEt
125
Cliapter5 :M.tu!C£ Inlii6itors
FIG 5.6 Top panel shows a cartoon representation of the ICL structure, with the active site area at the center of the box. The Left panels represents ICL in open conformation (PDB code: 1F61) while right panels shows closed conformation ofthe same enzyme (PDB code: 1F8M). Predicted docking conformations of the inhibitors 685 (2nd Panel), 710 (3rd Panel) and 534 (4th Panel) within the ICL active site are shown.
126

Cfiapter5 9rftul CL I nfzi6itors
5.4 Discussion
The structure-based drug design strategy has been successfully used to identify three
novel classes of inhibitors viz. nitrobutanoates, cyclopropyls and galactosyl
propanaolamines active against MtuiCL. The docking results suggested that selected
compounds used in 'pairs' could be more effective for inhibition rather than individual
compounds. Interestingly, the in vitro inhibition assays support the apparent 'synergistic'
effect of pairs of molecules being better inhibitors than individual compounds. This was
further confirmed through the in vivo BACTEC assays. We are not aware of any other
group having identified such 'pairs of compounds' showing better inhibition compared to
single ICL inhibitors. The synergistic approach developed here offers a new paradigm in
the design of more potent inhibitors of the M tuberculosis isocitrate lyase. The results are
currently being refmed for the identification of more potent inhibitors of the enzyme.
Earlier, it was demonstrated that both the genes ic/1 and ic/2 are necessary for the
maintenance of persistence (Munoz-Elias and McKinney, 2005). Molecular modeling
and biochemical characterization suggested that both icl genes are similar in terms of
binding site architectures. It is therefore to be expected that inhibitors of the ic/1 gene
product should also be effective against its homolog ic/2. Indeed nitropropionate was
found to inhibit the activity of both the icl gene. products (Munoz-Elias and McKinney,
2005). The present evidence suggests that the present results should broadly hold true for
the ic/2 gene product too. Nevertheless, the group is purifying the MtuiCL2 protein and
the present results against MtuiCL1 will be verified against the homolog also.
5.5 References
Berman, H. M., eta/. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235-242.
Dwivedi, N., et a/. (2005) An efficient synthesis of aryloxyphenyl cyclopropyl methanones: a new class of anti-mycobacterial agents. Bioorg Med Chem Lett, 15, 4526-30.
Dixon, G. H. & Kornberg, H. L. (1959). Assay methods for key enzymes of the glyoxylate cycle. Biochem J, 72, 3.
Gould, T.A., et a/. (2006) Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol, 61, 940-947.
127

Cfiapter5 !MtuiCL Inhibitors
Grover, R.K.; et a/. (2004) Novel phenyl cyclopropyl methanones useful as Antitubercular agents Ind. Patent 0636DEL2004
Idnurm, A. & Howlett, B.J. (2002) Isocitrate lyase is essential for pathogenicity of the fungus Leptosphaeria maculans to canola (Brassica napus). Eukaryot Cell, 1, 719-724.
Laskowski, R.A., eta/. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR, 8, 477-486.
Lorenz, M.C. and Fink, G.R. (2001) The glyoxylate cycle is required for fungal virulence. Nature, 412, 83-86.
Marti-Renom, M.A., et al. (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct, 29, 291-325.
Morris, G.M., eta/. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem, 19, 1639-1662.
McKinney, J.D., eta/. (2000) Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature, 406, 735-738.
Munoz-Elias, E.J. & McKinney, J.D. (2005) Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med, 11, 638-644.
Panda, G., et a/. (2004) Diaryloxy methano phenanthrenes : a new class of antituberculosis agents. Bioorg Med Chem, 12, 5269-5276.
Sharma, V., et al. (2000) Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Bioi, 1, 663-668.
Solomon, P.S., et a/. (2004) Pathogenicity of Stagonospora nodorum requires malate synthase. Mol Microbiol, 53, 1065-1073.
Srivastava, R; et a/. (2006) Process for the preparation of novel 4-Nitrobtanoate Inhibitors of Isocitrate Lyase from Mycobacterium tuberculosis. Ind. Patent 067DEL2006.
Tripathi, R.P., eta/. (2002) Synthesis of glycosylated beta-amino acids as new class of antitubercular agents. Eur J Med Chem, 37, 773-781.
Verdonk, M.L., eta/. (2003) Improved protein-ligand docking using GOLD. Proteins, 52,609-623.
Vriend, G. (1990) WHAT IF: a molecular modeling and drug design program. J Mol Graph, 8, 52-56.
Wang, Z.Y., eta/. (2003) The glyoxylate cycle is required for temporal regulation of virulence by the plant pathogenic fungus Magnaporthe grisea. Mol Microbiol, 47, 1601-1612.
128
Recommended

















