8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 1/11
O R I G I N A L A R T I C L E
Role of cagA-Positive Helicobacter pylori on Cell Proliferation,Apoptosis, and Inflammation in Biliary Cells
Wongwarut Boonyanugomol • Chariya Chomvarin • Seung-Chul Baik •
Jea-Young Song • Chariya Hahnvajanawong • Kyung-Mi Kim • Myung-Je Cho •
Woo-Kon Lee • Hyung-Lyun Kang • Kwang-Ho Rhee • Banchob Sripa
Received: 28 September 2010 / Accepted: 19 November 2010 / Published online: 22 December 2010
Ó Springer Science+Business Media, LLC 2010
Abstract
Background and Aims The pathogenesis of Helicobacter
pylori in the human hepatobiliary system has not been
clearly elucidated. We compared the effects of H. pylori
cagA? and cagA- mutant strains on cell proliferation,
apoptosis, and inflammation in a cholangiocarcinoma
(CCA) cell line (KKU-100).
Methods MTT and BrdU were used to determine cell
viability and DNA synthesis, respectively. The results were
further investigated by RT-PCR and Western-blot analysis.
The production of interleukin-8 (IL-8) was measured by
ELISA assay.
Results At low H. pylori inocula (cell-bacteria ratio of
1:1), the H. pylori cagA? strain showed a significant
stimulation in KKU-100 cell growth (109 ± 1.79%) and
DNA synthesis (131 ± 3.39%) than did the H. pylori
cagA- strain (95 ± 3.06% and 120 ± 2.32%, respec-
tively), through activation of the anti-apoptotic bcl-2 gene,
MAP kinase and NF- jB cascade. By contrast, at high
H. pylori inocula (cell-bacteria ratio of 1:200), the
H. pylori cagA? strain showed a significant reduction in
KKU-100 cell survival (49 ± 2.47%) and DNA synthesis
(49 ± 1.14%) than did the H. pylori cagA- strain (60 ±
1.30% and 75 ± 4.00%, respectively), by increased iNOS ,
p53 and bax, while decreased bcl-2. Additionally, caspase-
8 and -3 protein were activated. The H. pylori cagA? strain
had significantly stronger effect on IL-8 production than
did the cagA- strain.
Conclusions These results suggest that the H. pylori
cagA? strain may play an important role in the development
K.-M. Kim
e-mail: [email protected]
M.-J. Cho
e-mail: [email protected]
W.-K. Lee
e-mail: [email protected]
H.-L. Kange-mail: [email protected]
K.-H. Rhee
e-mail: [email protected]
B. Sripa
Department of Pathology, and Liver Fluke and
Cholangiocarcinoma Research Center, Faculty of Medicine,
Khon Kaen University, Khon Kaen 40002, Thailand
e-mail: [email protected]
Electronic supplementary material The online version of thisarticle (doi:10.1007/s10620-010-1512-y ) contains supplementarymaterial, which is available to authorized users.
W. Boonyanugomol Á C. Chomvarin (&) Á C. Hahnvajanawong
Department of Microbiology, and Liver Fluke and
Cholangiocarcinoma Research Center, Faculty of Medicine,
Khon Kaen University, Khon Kaen 40002, Thailand
e-mail: [email protected]
W. Boonyanugomol
e-mail: [email protected]
C. Hahnvajanawonge-mail: [email protected]
S.-C. Baik Á J.-Y. Song Á K.-M. Kim Á M.-J. Cho Á W.-K. Lee Á
H.-L. Kang Á K.-H. Rhee
Department of Microbiology, and Research Institute of Life
Science, Gyeongsang National University College of Medicine,
Chiram-dong 90, Jinju, Gyeongsangnam-do 660-751,
Republic of Korea
e-mail: [email protected]
J.-Y. Song
e-mail: [email protected]
123
Dig Dis Sci (2011) 56:1682–1692
DOI 10.1007/s10620-010-1512-y

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 2/11
of biliary cancer by disturbing cell proliferation, apoptosis,
and promoting cell inflammation in the CCA cell line.
Keywords Helicobacter pylori Á Cell proliferation Á
Apoptosis Á Inflammation Á Hepatobiliary diseases Á
Cholangiocarcinoma
Introduction
Cholangiocarcinoma (CCA) is a fatal bile duct epithelial
cancer. The highest incidence of this primary liver cancer
in the world has been reported in northeast Thailand [1].
The infection with the liver fluke Opisthorchis viverrini is
intimately related to pathogenesis of CCA in this region
[2]. However, pathogenesis of CCA or other biliary cancers
in non-liver fluke endemic areas is still poorly understood.
Recently, a new synergistic role of bacterial infection
including Helicobacter spp. in the hepatobiliary tract has
been proposed to be involved in severe liver diseasesincluding cancer [3]. Therefore, it is useful to explore roles
of Helicobacter in biliary cancer development.
The entero-hepatic Helicobacter spp. was detected in
the hepatobiliary tract of humans [4]. Among this genus,
H. pylori was the most common species detected in
patients with hepatobiliary diseases [5–7]. H. pylori was
detected more often in both CCA and hepatocellular car-
cinoma (HCC) patients than in patients with non-malignant
tumors and the control group [6, 8], suggesting a correla-
tion between H. pylori and liver cancer development.
Partial DNA sequences of H. pylori from liver samples
(HCC, CCA) clustered separately from gastric H. pylori
strains [9]. The animal model with C57BL/6 mice dem-
onstrated that prolonged infection with H. pylori resulted in
the development of HCC [10]. Additionally, the cytotoxin-
associated gene A (cagA) was reported to be associated
with liver tract diseases [11]. Therefore, we hypothesize
that the H. pylori cagA? strain could be a co-factor in the
development of liver diseases, especially CCA.
The cagA is an important virulence gene of H. pylori
associated with the pathogenesis of H. pylori infection
[12]. CagA, a product of cagA, has an effect on biological
activities such as cell motility, cytoskeleton rearrangement,
cell proliferation, and apoptosis [13, 14]. Moreover, cagA
can induce interleukin-8 (IL-8) gene expression via mito-
gen-activating protein kinases (MAPK) and NF- jB in both
CagA tyrosine phosphorylation-dependent and -indepen-
dent manners [15]. IL-8, a proinflammatory cytokine, can
induce neutrophil and monocyte infiltration leading to
inflammation [16]. Additionally, IL-8 has been shown to
activate multiple intracellular signaling pathways, leading
to an increase in proliferation and survival of cancer
cells [16].
H. pylori has been reported to induce apoptosis as well
as to increase cellular proliferation in gastric epithelial
cells [17–19]. Recently, an in vitro study showed that a
putatively more virulent strain of H. pylori induced apop-
tosis in a hepatocyte cell line by inhibiting DNA synthesis
and activating caspase-3 at high doses of bacteria, while
low doses of H. pylori increase DNA synthesis [20].
However, the mechanisms by which H. pylori (especiallycagA? strain) in biliary cells including cell proliferation,
induction of apoptosis and inflammation have not been
extensively studied. In this study, therefore, we investi-
gated the molecular mechanisms of H. pylori cagA? and
cagA- strains in cell proliferation, induction of apoptosis,
and pro-inflammatory cytokine production (IL-8) in biliary
cells.
Materials and Methods
Bacterial Strains
H. pylori (Korean strain 51) cagA wild-type (cagA?) and
cagA isogenic mutant (cagA-) strains were obtained from
the H. pylori Korean-type culture collection (Gyeongsang
National University, School of Medicine). The two strains
were grown on Brucella agar supplemented with 10%
bovine serum for 24 h at 37°C under microaerophilic
conditions [21].
Cell Culture
The human CCA cell line (KKU-100) was cultured in Ham
F12 medium containing 10% heat-inactivated FBS,
100 units/ml of penicillin, and 100 lg/ml of streptomycin
at 37°C in a humidified incubator containing 5% CO2 [22].
Cytotoxicity by MTT Assay
The effect of H. pylori on cell viability was determined by
the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetra-
zoliumbromide] assay. KKU-100 cells (8 9 104 cells/well)
were seeded in a 96-well microtiter plate and incubated at
37°C for 24 h. After co-culturing with H. pylori at low
inocula (cell-bacteria ratio of 1:1) to a high inocula (1:200)
for 24 h, MTT assay was performed according to the
procedure of Kim et al. (2007). After removing the
supernatant, formazan crystal was dissolved in dimethyl-
sulfoxide (DMSO), and the optical density of each well
was measured by a microplate reader at a wavelength of
540 nm. Culture medium without H. pylori was used as
the control. The absorbance value was calculated as a
percentage of cell viability compared to the control.
Dig Dis Sci (2011) 56:1682–1692 1683
123

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 3/11
Measurement of DNA Synthesis by BrdU Assay
After co-culturing KKU-100 cells with H. pylori at low
inocula (cell-bacteria ratio of 1:1) to a high inocula (1:200)
in 96-well microtiter plates for 24 h, BrdU (5-bromo-
20-deoxyuridine) incorporation was performed as previ-
ously described [20]. Chromogenic substrate tetra-meth-
ylbenzidine (TMB) was then added and optical density wasmeasured using a microplate reader at a wavelength of
520 nm. Culture medium without H. pylori was used as the
control. The absorbance value was calculated as a per-
centage of DNA synthesis compared to the control.
Determination of Apoptotic Cells
Nuclear staining using 4,6-diamidino-2-phenylindole
(DAPI) was performed to determine chromatin condensa-
tion and fragmentation. KKU-100 cells (1 9 106 cells/
well) were grown in a cell culture dish at 37°C for 24 h andco-cultured with medium alone (control) or H. pylori
(1:200) for 12 and 24 h. The treated cells were washed with
PBS, fixed with methanol, stained with 1 lg/ml of DAPI at
37°C for 15 min and observed under a fluorescence
microscope (Olympus, Japan). Apoptotic cell death was
enumerated by counting a total of 500 cells, and the result
was expressed as a percentage of apoptotic cells.
DNA fragmentation was assessed using agarose gel
electrophoresis. Cells treated with H. pylori (1:200) were
washed with PBS, and DNA extracted by QIAampÒ DNA
Mini Kit (Qiagen, USA) according to the manufacturer’s
instructions. DNA was electrophoresed in a 1.5% agarose
gel and the DNA band was visualized using a UV-illumi-
nator (Bio-Rad, USA).
RT-PCR for iNOS and Apoptosis-Related Gene
Expression
KKU-100 cells (1 9 106 cells) were cultured in a cell
culture dish for 24 h. After co-culturing with medium alone
(control) or H. pylori (1:1 or 1:200), total RNA was
extracted from the control and treated cells using TRIzolÒ
reagent (Invitrogen, Grand Island, NY, USA) according tothe manufacturer’s instructions. The quantity of RNA and
its purity were measured with a spectrophotometer.
RT-PCR of iNOS , p53, bax, bcl-2, caspase-3, and
GAPDH were performed as previously described [23–25].
The PCR primers are presented in Table 1. The PCR
product was electrophoresed in 1.5% agarose, stained with
ethidium bromide, and visualized under UV light. The
intensity of the PCR product bands was quantified using
Quantities One software version 4.6.2 (Bio-Rad, USA).
The intensity of PCR product band was normalized to
GAPDH intensity and data were expressed as fold changed
compared to control (intensity of PCR product band of H. pylori-treated cells divided by PCR product band of
control).
Western-Blot Analysis for Protein-Involved Apoptosis
and Cell Proliferation
After co-culturing KKU-100 cells (1 9 106) with medium
alone (control) or H. pylori (1:1 or 1:200) for 0, 6, 12, and
24 h, the protein extraction and Western blotting were
performed as previously described [21]. After blocking, the
membranes were incubated with the following antibodies:
Bax, Bcl-2, Bcl-xL, Fas, activated caspase-8, cytochrome
c, b-actin (Santa Cruz Biotechnology, CA, USA), activated
caspase-9, activated and -3, cleaved PARP, pMAPK,
Table 1 Primers used
for RT-PCRGenes Primer sequences Product
size (bp)
References
iNOS 50-CGGTGCTGTATTTCCTTACGAGGCGACGAAGG-3 0
50-GGTGCTGCT TGT TAGGAGGTCAAGTAAAGGGC-30
259 [24]
p53 50-TTCTTGCATTCTGGGACAGCC-3 0
50-GCCTCATTCAGCTCTCGGAAC-3 0
650 [24]
bax 50-TGGCAGCTGACATGTTTTCTGAC-3 0
50-CGTCCCAACCACCCTGGTCT-3 0
195 [24]
bcl-2 50-CTGTACGGCCCCAGCATGCG-3 0
50-GCTTTGTTTCATGGTACATC-3 0
231 [23]
caspase-3 50-TTCAGAGGGGATCGTTGTAGAAGTC-3 0
50-CAAGCTTGTCGGCATACTGTTTCAG-3 0
264 [25]
GAPDH 50-CGGAGT CAACGGATT TGGTCGTAT-30
50-AGCCTTCTCCATGGTGGTGAAGAC-3 0
306 [24]
1684 Dig Dis Sci (2011) 56:1682–1692
123
8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 4/11
NF- jB (Cell Signaling Technology, MA, USA) overnight
at 4°C. HRP-conjugated secondary antibody was added,
and protein was visualized using the enhanced chemilu-
minescence (ECL) system (Thermo Scientific, IL, USA).
The intensity of the protein bands was quantified using
Quantities One software version 4.6.2 (Bio-Rad, USA).
The intensity of protein band was normalized to b-actin
intensity and data expressed as fold changed compared tocontrol (intensity of protein band of H. pylori-treated cells
divided by protein band of control).
Measurement of IL-8 Production for Cell Inflammation
KKU-100 cells were co-cultured with H. pylori at a cell-
bacteria ratio of 1:1 or 1:200 then the supernatant was
collected and centrifuged at 7,000 rpm for 10 min to
eliminate unattached bacteria. The concentration of IL-8
was measured using IL-8 enzyme-link immunosorbent
assay (ELISA) (BioSource International) according to
the manufacturer’s instructions. Culture medium without
H. pylori was used as the control. The absorbance value
was calculated as a concentration of IL-8 compared to
standard IL-8.
Statistical Analysis
All of the data were reported as means ± SE. Differences
between the untreated control, and the H. pylori cagA? and
cagA- strains were analyzed using Student’s t test.
p\ 0.05 and p\ 0.01 were considered significant.
Results
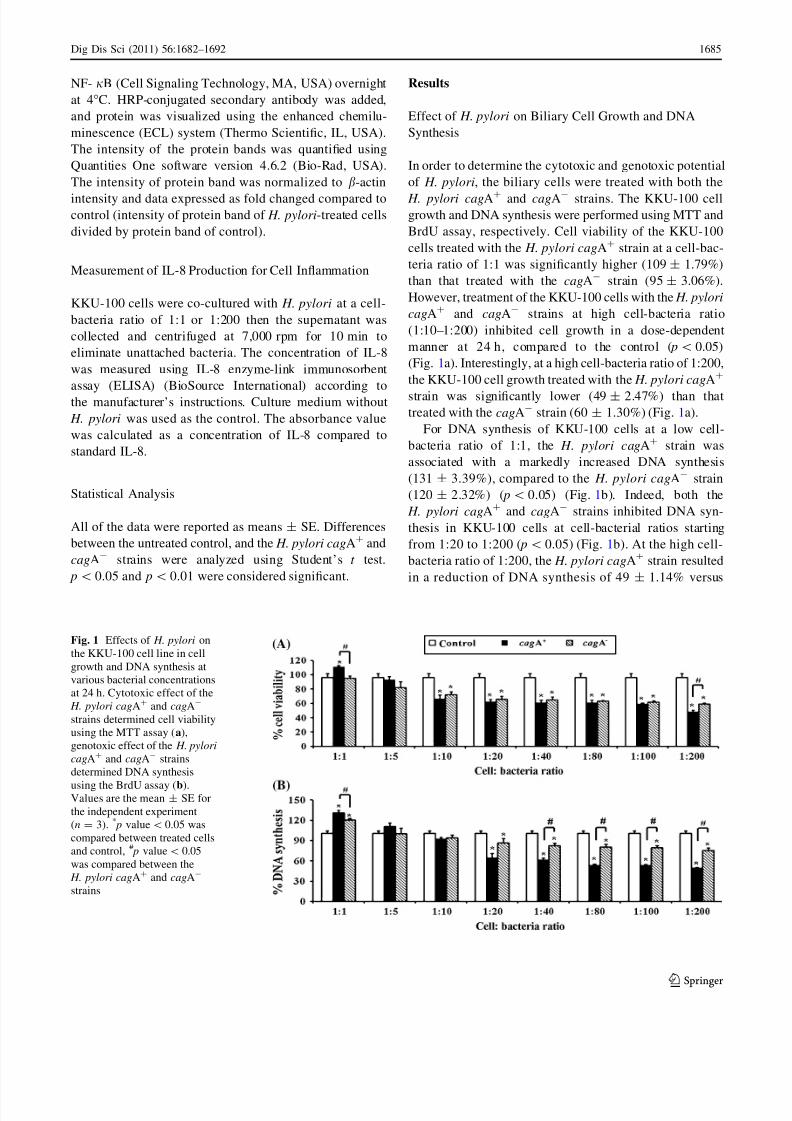
Effect of H. pylori on Biliary Cell Growth and DNA
Synthesis
In order to determine the cytotoxic and genotoxic potential
of H. pylori, the biliary cells were treated with both the
H. pylori cagA? and cagA- strains. The KKU-100 cellgrowth and DNA synthesis were performed using MTT and
BrdU assay, respectively. Cell viability of the KKU-100
cells treated with the H. pylori cagA? strain at a cell-bac-
teria ratio of 1:1 was significantly higher (109 ± 1.79%)
than that treated with the cagA- strain (95 ± 3.06%).
However, treatment of the KKU-100 cells with the H. pylori
cagA? and cagA- strains at high cell-bacteria ratio
(1:10–1:200) inhibited cell growth in a dose-dependent
manner at 24 h, compared to the control ( p\ 0.05)
(Fig. 1a). Interestingly, at a high cell-bacteria ratio of 1:200,
the KKU-100 cell growth treated with the H. pylori cagA?
strain was significantly lower (49 ± 2.47%) than thattreated with the cagA- strain (60 ± 1.30%) (Fig. 1a).
For DNA synthesis of KKU-100 cells at a low cell-
bacteria ratio of 1:1, the H. pylori cagA? strain was
associated with a markedly increased DNA synthesis
(131 ± 3.39%), compared to the H. pylori cagA- strain
(120 ± 2.32%) ( p\ 0.05) (Fig. 1b). Indeed, both the
H. pylori cagA? and cagA- strains inhibited DNA syn-
thesis in KKU-100 cells at cell-bacterial ratios starting
from 1:20 to 1:200 ( p\ 0.05) (Fig. 1b). At the high cell-
bacteria ratio of 1:200, the H. pylori cagA? strain resulted
in a reduction of DNA synthesis of 49 ± 1.14% versus
Fig. 1 Effects of H. pylori on
the KKU-100 cell line in cell
growth and DNA synthesis at
various bacterial concentrations
at 24 h. Cytotoxic effect of the
H. pylori cagA? and cagA-
strains determined cell viability
using the MTT assay (a),
genotoxic effect of the H. pylori
cagA? and cagA- strains
determined DNA synthesis
using the BrdU assay (b).Values are the mean ± SE for
the independent experiment
(n = 3). * p value\0.05 was
compared between treated cells
and control, # p value\ 0.05
was compared between the
H. pylori cagA? and cagA-
strains
Dig Dis Sci (2011) 56:1682–1692 1685
123

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 5/11
75 ± 4.00% in the H. pylori cagA- strain ( p\ 0.05)
(Fig. 1b).
H. pylori Induced Biliary Cell Proliferation
Determination of H. pylori-induced biliary cell prolifera-
tion was performed using RT-PCR and Western blotting to
determine bcl-2, bax, p53 gene expression, and phosphor-ylated MAPK and NF-jB protein expression. At low
inocula of the H. pylori cagA? and cagA- strains (cell-
bacteria ratio of 1:1), the expression of pro-apoptotic
genes, bax and p53, were not significantly changed com-
pared to control (Fig. 2a). Interestingly, at 6, 12, and 24 h
of treatment, the increase in the anti-apoptotic gene bcl-2 in
the KKU-100 cells exposed to the H. pylori cagA? strain
was significantly higher than in the cells treated with the
H. pylori cagA- strain (Fig. 2a).
Figure 2b illustrates the time dependent of phosphor-
ylated MAP kinase and NF- jB activation following
exposure of the KKU-100 cells to the H. pylori cagA?
and cagA- strains at a low inoculation (1:1). Phosphor-
ylation of the MAP kinase was markedly activated in the
KKU-100 cells treated with the H. pylori cagA? strain,
while in the H. pylori cagA--treated cells showed
slightly activated. The activation of the NF- jB was
clearly evident within 15 min of the KKU-100 cells
treated with the H. pylori cagA? strain, but cells treated
with the H. pylori cagA- strain exhibited marked acti-
vation at 30 min.
H. pylori Induces Biliary Cell Apoptosis
Determination of Apoptotic Morphology
Inhibition of cell growth and DNA synthesis was found in
the KKU-100 cells exposed to high doses of bacteria;
a cell-bacteria ratio of 1:200 was used to determine biliary
cell apoptosis. Apoptotic cell death was determined usingDAPI staining and DNA fragmentation assay. Using DAPI
staining, cells with chromatin condensation and nuclear
fragmentation, the typical characteristics of apoptosis, were
observed in KKU-100 cells treated both with the H. pylori
cagA? and cagA- (Fig. 3a, middle and right panels).
However, the control cells showed nuclei with homoge-
neous chromatin distribution (Fig. 3a, left panel). At 24 h,
the number of apoptotic cells resulting from treatment with
the H. pylori cagA? was significantly higher than in cells
treated with the H. pylori cagA- strain (Fig. 3b). Typical
DNA fragmentation, using agarose gel electrophoresis, was
observed in the cells exposed to both the H. pylori cagA?
and cagA- strains (Fig. 3c).
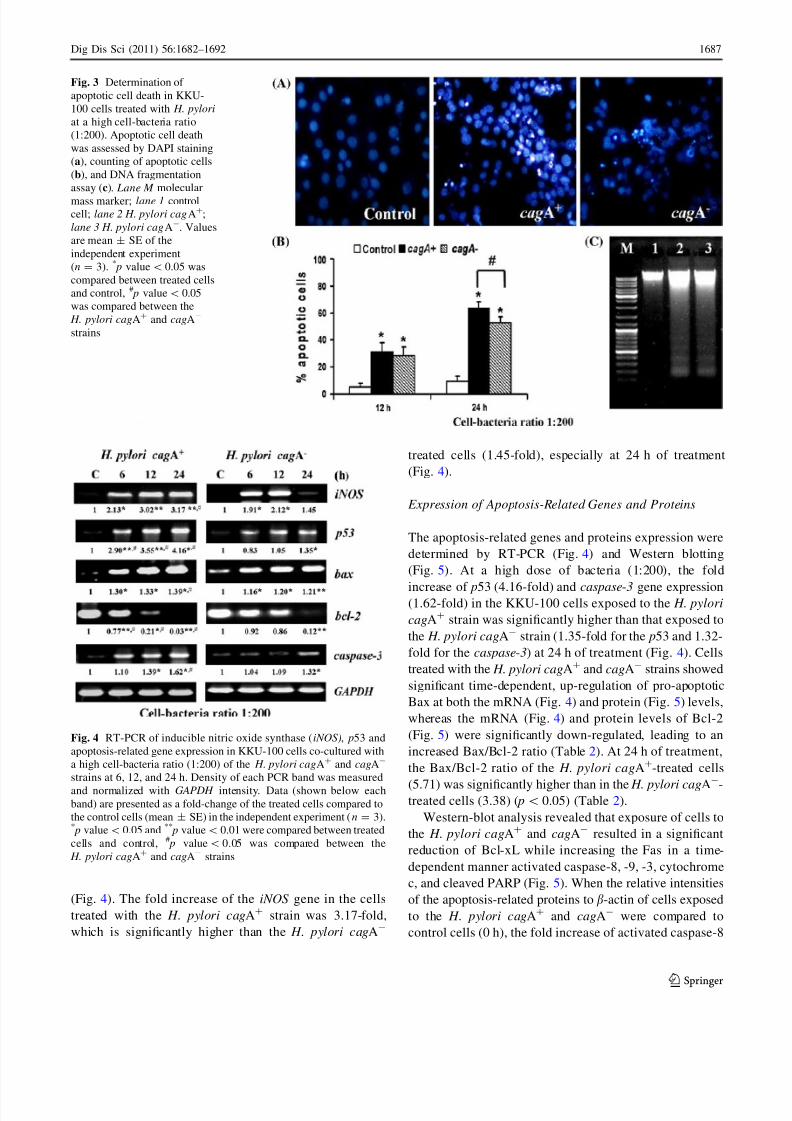
Expression of iNOS Gene
To investigate the role of the iNOS gene involved in pro-
grammed cell death, the mRNA level was evaluated. Using
RT-PCR analysis, a significance of the fold increase of
iNOS mRNA in cells treated with the H. pylori cagA? and
cagA- was observed at a cell-bacteria ratio of 1:200
Fig. 2 Role of H. pylori on cellproliferation in KKU-100 cells,
co-cultured with the H. pylori
cagA? and cagA- strains at a
low inoculum (1:1); apoptosis-
related gene expression (a), and
activation of MAP kinase and
NF- jB protein expression (b).
Density of each PCR and
protein band was measured and
normalized with GAPDH and
b-actin intensity, respectively.
Data (shown below each band)
are presented as a fold-change
of the treated cells compared to
the control cells (mean ± SE)in the independent experiment
(n = 3). * p value\ 0.05 was
compared between treated cells
and control, # p value\ 0.05
was compared between the
H. pylori cagA? and cagA-
strains
1686 Dig Dis Sci (2011) 56:1682–1692
123
8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 6/11
(Fig. 4). The fold increase of the iNOS gene in the cells
treated with the H. pylori cagA? strain was 3.17-fold,
which is significantly higher than the H. pylori cagA-
treated cells (1.45-fold), especially at 24 h of treatment
(Fig. 4).
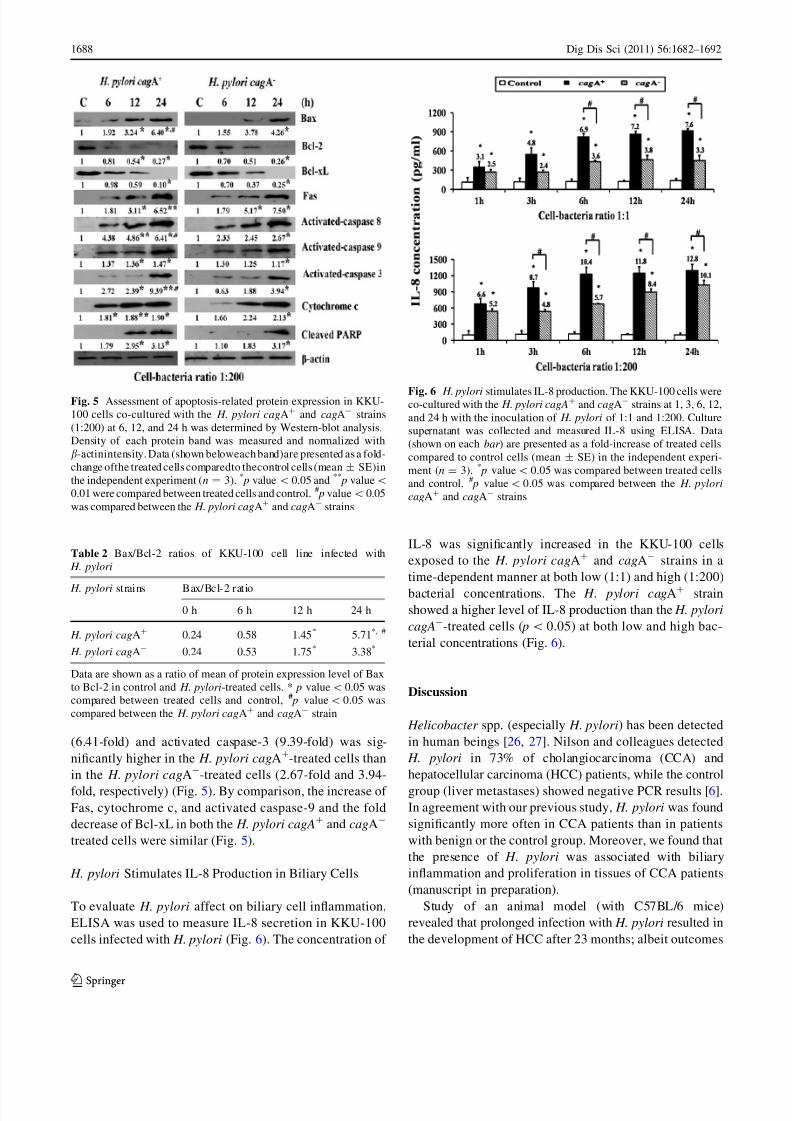
Expression of Apoptosis-Related Genes and Proteins
The apoptosis-related genes and proteins expression were
determined by RT-PCR (Fig. 4) and Western blotting
(Fig. 5). At a high dose of bacteria (1:200), the fold
increase of p53 (4.16-fold) and caspase-3 gene expression(1.62-fold) in the KKU-100 cells exposed to the H. pylori
cagA? strain was significantly higher than that exposed to
the H. pylori cagA- strain (1.35-fold for the p53 and 1.32-
fold for the caspase-3) at 24 h of treatment (Fig. 4). Cells
treated with the H. pylori cagA? and cagA- strains showed
significant time-dependent, up-regulation of pro-apoptotic
Bax at both the mRNA (Fig. 4) and protein (Fig. 5) levels,
whereas the mRNA (Fig. 4) and protein levels of Bcl-2
(Fig. 5) were significantly down-regulated, leading to an
increased Bax/Bcl-2 ratio (Table 2). At 24 h of treatment,
the Bax/Bcl-2 ratio of the H. pylori cagA?-treated cells
(5.71) was significantly higher than in the H. pylori cagA-
-treated cells (3.38) ( p\ 0.05) (Table 2).
Western-blot analysis revealed that exposure of cells to
the H. pylori cagA? and cagA- resulted in a significant
reduction of Bcl-xL while increasing the Fas in a time-
dependent manner activated caspase-8, -9, -3, cytochrome
c, and cleaved PARP (Fig. 5). When the relative intensities
of the apoptosis-related proteins to b-actin of cells exposed
to the H. pylori cagA? and cagA- were compared to
control cells (0 h), the fold increase of activated caspase-8
Fig. 3 Determination of
apoptotic cell death in KKU-
100 cells treated with H. pylori
at a high cell-bacteria ratio
(1:200). Apoptotic cell death
was assessed by DAPI staining
(a), counting of apoptotic cells
(b), and DNA fragmentation
assay (c). Lane M molecular
mass marker; lane 1 control
cell; lane 2 H. pylori cagA?;
lane 3 H. pylori cagA-. Values
are mean ± SE of the
independent experiment
(n = 3). * p value\0.05 was
compared between treated cells
and control, # p value\ 0.05
was compared between the
H. pylori cagA? and cagA-
strains
Fig. 4 RT-PCR of inducible nitric oxide synthase (iNOS), p53 and
apoptosis-related gene expression in KKU-100 cells co-cultured with
a high cell-bacteria ratio (1:200) of the H. pylori cagA? and cagA-
strains at 6, 12, and 24 h. Density of each PCR band was measured
and normalized with GAPDH intensity. Data (shown below each
band) are presented as a fold-change of the treated cells compared to
the control cells (mean ± SE) in the independent experiment (n = 3).* p value\0.05 and **
p value\ 0.01 were compared between treated
cells and control, # p value\ 0.05 was compared between the
H. pylori cagA?
and cagA-
strains
Dig Dis Sci (2011) 56:1682–1692 1687
123
8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 7/11
(6.41-fold) and activated caspase-3 (9.39-fold) was sig-
nificantly higher in the H. pylori cagA?-treated cells than
in the H. pylori cagA--treated cells (2.67-fold and 3.94-
fold, respectively) (Fig. 5). By comparison, the increase of
Fas, cytochrome c, and activated caspase-9 and the fold
decrease of Bcl-xL in both the H. pylori cagA? and cagA-
treated cells were similar (Fig. 5).
H. pylori Stimulates IL-8 Production in Biliary Cells
To evaluate H. pylori affect on biliary cell inflammation,
ELISA was used to measure IL-8 secretion in KKU-100
cells infected with H. pylori (Fig. 6). The concentration of
IL-8 was significantly increased in the KKU-100 cells
exposed to the H. pylori cagA? and cagA- strains in a
time-dependent manner at both low (1:1) and high (1:200)
bacterial concentrations. The H. pylori cagA? strain
showed a higher level of IL-8 production than the H. pylori
cagA--treated cells ( p\0.05) at both low and high bac-
terial concentrations (Fig. 6).
Discussion
Helicobacter spp. (especially H. pylori) has been detected
in human beings [26, 27]. Nilson and colleagues detected
H. pylori in 73% of cholangiocarcinoma (CCA) and
hepatocellular carcinoma (HCC) patients, while the control
group (liver metastases) showed negative PCR results [6].
In agreement with our previous study, H. pylori was found
significantly more often in CCA patients than in patients
with benign or the control group. Moreover, we found that
the presence of H. pylori was associated with biliary
inflammation and proliferation in tissues of CCA patients
(manuscript in preparation).
Study of an animal model (with C57BL/6 mice)
revealed that prolonged infection with H. pylori resulted in
the development of HCC after 23 months; albeit outcomes
Fig. 5 Assessment of apoptosis-related protein expression in KKU-
100 cells co-cultured with the H. pylori cagA? and cagA- strains
(1:200) at 6, 12, and 24 h was determined by Western-blot analysis.
Density of each protein band was measured and normalized with
b-actinintensity. Data (shown beloweach band)are presented as a fold-
change ofthe treated cells comparedto thecontrol cells (mean ± SE)in
the independent experiment (n = 3). * p value\ 0.05 and **
p value\
0.01 were compared between treated cells and control. # p value\ 0.05
was compared between the H. pylori cagA?
and cagA-
strains
Table 2 Bax/Bcl-2 ratios of KKU-100 cell line infected with
H. pylori
H. pylori strains Bax/Bcl-2 ratio
0 h 6 h 12 h 24 h
H. pylori cagA? 0.24 0.58 1.45* 5.71*, #
H. pylori cagA- 0.24 0.53 1.75* 3.38*
Data are shown as a ratio of mean of protein expression level of Bax
to Bcl-2 in control and H. pylori-treated cells. * p value\0.05 was
compared between treated cells and control, # p value\ 0.05 was
compared between the H. pylori cagA? and cagA- strain
Fig. 6 H. pylori stimulates IL-8 production. The KKU-100 cells wereco-cultured with the H. pylori cagA
? and cagA- strains at 1, 3, 6, 12,
and 24 h with the inoculation of H. pylori of 1:1 and 1:200. Culture
supernatant was collected and measured IL-8 using ELISA. Data
(shown on each bar ) are presented as a fold-increase of treated cells
compared to control cells (mean ± SE) in the independent experi-
ment (n = 3). * p value\ 0.05 was compared between treated cells
and control. # p value\ 0.05 was compared between the H. pylori
cagA? and cagA- strains
1688 Dig Dis Sci (2011) 56:1682–1692
123

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 8/11
depended on the diversity of H. pylori strains [10]. Addi-
tionally, H. pylori inoculated orally in the C57BL/6 mice
resulted in the development of severe gastric mucosal
inflammation, mild to moderate hepatitis, and gallbladder
mucosa thickening with mild submucosal lymphocytic
infiltration [28]. Goo and colleagues showed that H. pylori
promotes hepatic fibrosis and increases hepatocytes pro-
liferation by stimulation of a-smooth muscle actin(a-SMA) and transforming growth factor-b1 (TGF-b1),
suggesting that H. pylori induces the development of liver
cirrhosis [29].
Studies of other Helicobacter spp. infections such as
H. hepaticus—the causative agent of hepatocellular carci-
noma in mice [30]—revealed that inoculated A/JCr mice
developed chronic hepatitis and hepatocellular neoplasms,
which led to hepatocellular carcinoma [30, 31]. While
H. hepaticus produces toxins that cause cytotoxic activity in
a mouse liver cell line [32] and a cytolethal distending toxin
(Cdt), which has DNase activity [33], H. pylori produces
many toxins such as CagA and VacA. We therefore proposethat the enteric Helicobacter spp., including H. pylori, may
be the infectious factor involved in the development of
hepatobiliary diseases, including liver carcinoma.
A trigger in the development of cancer is chronic
infection and the resultant long-term inflammation. The
inflammation can be caused by stimulation of many cyto-
kine productions [34]. H. pylori infection is well known as
a cause of gastric inflammation and to stimulate pro-
inflammatory cytokines production (i.e., TNF-alpha, IFN-
gamma, IL-1, IL-6, IL-8) [35]. In inflammatory conditions,
iNOS can be induced, in almost any cell type, through
stimulation by inflammatory cytokines and/or bacterial
products (i.e., lipopolysaccharide) leading to nitric oxide
(NO) production [36]. The transcription factors, including
nuclear factor kappa-B (NF-jB), tumor suppressor protein
p53, and proinflammatory CXC chemokine (IL-8) have
been shown to be regulated by the NO [37–39].
IL-8 was shown to activate multiple intracellular sig-
naling pathways leading to an increase in proliferation and
survival of cancer cells [16]. In this study, using the KKU-
100 cell line, we found that at the low cell-bacteria ratio
(1:1), the H. pylori cagA? strain significantly enhanced
IL-8 production, DNA synthesis and cell proliferation
compared to the H. pylori cagA- strain. These results
imply that the H. pylori cagA? can stimulate IL-8 pro-
duction, which in turn increased DNA synthesis and cell
proliferation of the KKU-100 cells. Our results are similar
to a report of a virulent H. pylori strain (cagA? / vacA? /
babA? / oipA?) that at low concentrations induced higher
DNA synthesis on a hepatocyte cell line (Huh7) than a less
virulent strain [20]. However, no significant difference
between gastric epithelial cells treated with the type I
cytotoxic strains (cagA? / vacA?) and non-cytotoxic strains
(cagA- / vacA-) in the induction of apoptosis and cell
proliferation was reported [18]. The discrepancies of
results may depend on the specific H. pylori strains used in
each study.
Many studies failed to culture H. pylori from hepatob-
iliary samples [8, 11, 28]. It could be that the number of
bacteria was too small (since the bacteria were not in their
usual niche) [8, 9], indicating a low concentration of H. pylori infection. However, the detection of H. pylori
using immunohistochemistry confirms the true colonization
of H. pylori in the liver [8, 40]. We propose that there is a
low density of H. pylori infection in the biliary cells and
that persistent H. pylori infection could lead to subtle
modulations in cell replication as a previous report in
hepatocytes [20]. An increasing rate of DNA synthesis at
low dose of H. pylori may be a mechanism of the cell to
compensate the high rate of cell death [20]. Taken together,
our results suggest that at a low concentration of H. pylori,
CagA may play an important role in DNA synthesis and
proliferation of biliary cells.To understand the molecular mechanism in KKU-100
cell proliferation, several gene and protein expressions
were examined. Our results indicate that at the low cell-
bacteria ratio of 1:1, the KKU-100 cells treated with the
H. pylori cagA? strain had a marked, time-dependent,
increase in intracellular signaling molecules; viz., anti-
apoptotic bcl-2 gene expression, phosphorylated MAP
kinase and NF- jB protein expression, while the gene
expression levels of bax and p53 were not changed. These
effects may be due to an interaction of CagA with several
intracellular signaling molecules, MAPK [13] and NF- jB,
leading to the activation of many genes (i.e., growth-pro-
moting genes and anti-apoptotic genes) which might con-
tribute to the malignant transformation [14].
In a previous report, high concentration of NO, pro-
ceeding mainly by the iNOS pathway cause DNA damage
[41] and induce apoptosis [36, 42]. The tumor suppressor
protein (p53) has a dual role in NO-mediated apoptosis
because p53 protects the cell from apoptosis at low NO
concentrations and stimulates apoptosis at high NO con-
centrations [41]. H. pylori was reported as a cause of
gastric epithelial AGS cell line damage by inducing iNOS
and p53-dependent apoptosis [24]. In our study, at a high
cell-bacteria ratio (1:200), the H. pylori cagA? strain
showed significantly stronger growth inhibition, decreased
DNA synthesis, induced apoptosis, increased iNOS , and
p53 gene expression in the KKU-100 cell line than the
H. pylori cagA- strain. These results agree with previous
reports on various cell lines [20, 24, 43].
Apoptosis induced through the extrinsic pathway
involves death receptor stimulation and activation of cas-
pase-8 and -3 [44]. Our results showed a higher up-regu-
lation of activated caspase-8 and -3 protein expression in
Dig Dis Sci (2011) 56:1682–1692 1689
123

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 9/11
the KKU-100 cell line co-cultured with the H. pylori
cagA? strain than the H. pylori cagA- strain. Our findings
agree with the previous studies in epithelial intestinal cells
and in gastric cell lines co-cultured with the toxigenic
H. pylori strains (vacA? / cagA?) than the non-toxigenic
H. pylori strains (vacA- / cagA-) [45, 46].
In the intrinsic signaling pathway, the Bcl-2 family of
proteins plays major roles in apoptotic regulation throughthe mitochondrial pathway [47]. Our study found a sig-
nificant increase in the pro-apoptotic protein, Bax, and a
significant decrease in the anti-apoptotic proteins Bcl-2 and
Bcl-xL, in KKU-100 cells treated with the H. pylori cagA?
strain leading to an increased Bax/Bcl-2 ratio induced
apoptosis as a previous report [48]. The Bax protein
interacts with the Bcl-2 [47] thereby the release of cyto-
chrome c via pore-forming activity at the mitochondrial
outer membrane [49]. Cytochrome c forms a complex with
Apaf-1 and pro-caspase-9, leading to activation of caspase-
9 and -3 and DNA fragmentation [50, 51]. Our results
showed a higher increase in the level of cytochrome c,activated caspase-9 and -3 and cleaved PARP in KKU-100
cells treated with both H. pylori strains compared to the
control. These results suggest that H. pylori (especially the
H. pylori cagA? strain) induces apoptosis in the KKU-100
cell line through both a receptor/ligand interaction and
mitochondria-mediated apoptosis.
At the high cell-bacteria ratio of 1:200, the H. pylori
cagA? strain enhanced iNOS gene expression and IL-8
production in KKU-100 cells at a significantly higher level
than the H. pylori cagA- strain, and this agrees with a
previous study [52]. Since iNOS can cause NO production,
leading to DNA damage and up-regulation of IL-8, it may
increase proliferation and survival of cancer cells [16]. It is,
therefore, possible that in an environment with high NO
and IL-8 level, cells with damaged DNA are stimulated to
proliferate, which leads to the proliferation of unrepaired
DNA-containing cells that may be involved in tumor
development.
Our data indicate that CagA play an important role in
apoptosis-induced by H. pylori in KKU-100 cells. These
findings agree with other studies of gastroduodenal dis-
eases that show that infection with H. pylori cagA? strains
may have a role in atrophy development [17] and induction
of apoptosis [46]. Recently, the apoptosis-inducing factors
of H. pylori have been reported to play an important role in
the pathogenesis [21, 53, 54]. We propose that other vir-
ulence factors of H. pylori are also involved in apoptosis on
KKU-100 cells. In order to clarify whether CagA influ-
ences apoptosis in biliary cells, a purified CagA protein
should be used to examine its exact role in apoptosis.
Disturbance of the cell death process by apoptosis-
enhanced cell turnover will result in increased prolifera-
tion, which will increase the probability of accumulating
genetic alterations and may ultimately lead to tumor
development [55]. In this study, we proposed the role of
H. pylori (especially cagA? strain) on biliary cells in a
different density of bacteria. Our results implicate that both
low and high doses of H. pylori infection are involved in
cancer development of hepatobiliary cells by stimulation of
cell proliferation and induction of apoptosis pathway.
However, based on evidence that the density of Helico-bacter in hepatobiliary tract is too low, we thus speculate
that development of biliary cancer may relate in a low
number of H. pylori infections. This result agrees with our
previous study in the presence of H. pylori associated with
biliary cell inflammation and proliferation in tissues of
CCA patients.
In conclusion, our study shows that H. pylori has mul-
tiple effects on biliary cells, including: (1) stimulation of
DNA synthesis and increased cell proliferation via activa-
tion of MAP kinase and NF- KB at low doses of bacteria;
(2) induction of apoptosis through both the extrinsic and
mitochondria-dependent apoptosis pathways at high dosesof bacteria; and, (3) stimulation of IL-8 production. We
conclude that CagA play an important role in the carci-
nogenesis of the biliary cancer through the disturbance of
cellular homeostasis by stimulation of cell proliferation and
induction of apoptosis (which depends on bacterial con-
centration) and promotes inflammation by stimulating pro-
inflammatory cytokine IL-8 production. Further studies
regarding the other mechanisms of H. pylori in the CCA
cell line are warranted. Additionally, the in vivo roles of
H. pylori on the hepatobiliary tract need to be elucidated.
Acknowledgments I would like to thank the Commission onHigher Education, Thailand, for supporting with grant funds under the
program Strategic Scholarships for Frontier Research Network for the
Join Ph.D. Program Thai Doctoral degree for this research and also
thank Khon Kaen University for supporting some parts of this work. I
am grateful to the Helicobacter pylori Research Center, Department
of Microbiology, Gyeongsang National University School of Medi-
cine, for providing H. pylori strains and materials used in my
experiments. I also thank the Liver Fluke and Cholangiocarcinoma
Research Center, Khon Kaen University for providing the cell line. I
thank Mr. Bryan Roderick Hamman for assistance with the English-
language presentation of the manuscript.
References
1. Sripa B, Pairojkul C. Cholangiocarcinoma: lessons from Thai-
land. Curr Opin Gastroenterol. 2008;24:349–356.
2. Sripa B, Kaewkes S, Sithithaworn P et al. Liver fluke induces
cholangiocarcinoma. PLoS Med . 2007;4:e201.
3. Pellicano R, Menard A, Rizzetto M, Megraud F. Helicobacter
species and liver diseases: association or causation? Lancet Infect
Dis. 2008;8:254–260.
4. Fox JG, Dewhirst FE, Shen Z, et al. Hepatic Helicobacter species
identified in bile and gallbladder tissue from Chileans with
chronic cholecystitis. Gastroenterology . 1998;114:755–763.
1690 Dig Dis Sci (2011) 56:1682–1692
123

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 10/11
5. Avenaud P, Marais A, Monteiro L, et al. Detection of Helico-
bacter species in the liver of patients with and without primary
liver carcinoma. Cancer . 2000;89:1431–1439.
6. Nilsson HO, Mulchandani R, Tranberg KG, Stenram U, Wad-
strom T. Helicobacter species identified in liver from patients
with cholangiocarcinoma and hepatocellular carcinoma. Gastro-
enterology. 2001;120:323–324.
7. Pellicano R, Mazzaferro V, Grigioni WF, et al. Helicobacter
species sequences in liver samples from patients with and without
hepatocellular carcinoma. World J Gastroenterol. 2004;10:
598–601.
8. Huang Y, Fan XG, Wang ZM, et al. Identification of Helico-
bacter species in human liver samples from patients with primary
hepatocellular carcinoma. J Clin Pathol. 2004;57:1273–1277.
9. Abu Al-Soud W, Stenram U, Ljungh A, et al. DNA of Helico-
bacter spp. and common gut bacteria in primary liver carcinoma.
Dig Liver Dis. 2008;40:126–131.
10. Wang X, Willen R, Svensson M, Ljungh A, Wadstrom T. Two-
year follow-up of Helicobacter pylori infection in C57Bl/6 and
Balb/ca mice. Apmis. 2003;111:514–522.
11. Tiwari SK, Khan AA, Ibrahim M, Habeeb MA, Habibullah CM.
Helicobacter pylori and other Helicobacter species DNA in
human bile samples from patients with various hepato-biliary
diseases. World J Gastroenterol. 2006;12:2181–2186.
12. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helico-
bacter pylori infection. Clin Microbiol Rev. 2006;19:449–490.
13. Hatakeyama M. The role of Helicobacter pylori CagA in gastric
carcinogenesis. Int J Hematol. 2006;84:301–308.
14. Hatakeyama M. SagA of CagA in Helicobacter pylori patho-
genesis. Curr Opin Microbiol. 2008;11:30–37.
15. Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB
activation and potentiation of proinflammatory responses by the
Helicobacter pylori CagA protein. Proc Natl Acad Sci USA.
2005;102:9300–9305.
16. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin
Cancer Res. 2008;14:6735–6741.
17. Cabral MM, Mendes CM, Castro LP, et al. Apoptosis in Heli-
cobacter pylori gastritis is related to cagA status. Helicobacter .
2006;11:469–476.
18. Zhang ZW, Patchett SE, Farthing MJ. Role of Helicobacter
pylori and p53 in regulation of gastric epithelial cell cycle phase
progression. Dig Dis Sci. 2002;47:987–995.
19. Moss SF, Sordillo EM, Abdalla AM, et al. Increased gastric
epithelial cell apoptosis associated with colonization with cagA?
Helicobacter pylori strains. Cancer Res. 2001;61:1406–1411.
20. Ito K, Yamaoka Y, Yoffe B, Graham DY. Disturbance of apop-
tosis and DNA synthesis by Helicobacter pylori infection of
hepatocytes. Dig Dis Sci. 2008;53:2532–2540.
21. Kim KM, Lee SG, Park MG, et al. Gamma-glutamyl-
transpeptidase of Helicobacter pylori induces mitochondria-
mediated apoptosis in AGS cells. Biochem Biophys Res Commun.
2007;355:562–567.
22. Sripa B, Leungwattanawanit S, Nitta T, et al. Establishment and
characterization of an opisthorchiasis-associated cholangiocarci-noma cell line (KKU-100). World J Gastroenterol. 2005;11:
3392–3397.
23. Greenlund LJ, Korsmeyer SJ, Johnson EM Jr. Role of bcl-2 in the
survival and function of developing and mature sympathetic
neurons. Neuron. 1995;15:649–661.
24. Perfetto B, Buommino E, Canozo N, et al. Interferon-gamma
cooperates with Helicobacter pylori to induce iNOS-related
apoptosis in AGS gastric adenocarcinoma cells. Res Microbiol.
2004;155:259–266.
25. Winter RN, Kramer A, Borkowski A, Kyprianou N. Loss of
caspase-1 and caspase-3 protein expression in human prostate
cancer. Cancer Res. 2001;61:1227–1232.
26. Lin TT, Yeh CT, Wu CS, Liaw YF. Detection and partial
sequence analysis of Helicobacter pylori DNA in the bile sam-
ples. Dig Dis Sci. 1995;40:2214–2219.
27. Myung SJ, Kim MH, Shim KN, et al. Detection of Helicobacter
pylori DNA in human biliary tree and its association with
hepatolithiasis. Dig Dis Sci. 2000;45:1405–1412.
28. Huang Y, Tian XF, Fan XG, Fu CY, Zhu C. The pathological
effect of Helicobacter pylori infection on liver tissues in mice.
Clin Microbiol Infect . 2009;15:843–849.
29. Goo MJ, Ki MR, Lee HR, et al. Helicobacter pylori promotes
hepatic fibrosis in the animal model. Lab Invest . 2009;89:
1291–1303.
30. Ward JM, Fox JG, Anver MR, et al. Chronic active hepatitis and
associated liver tumors in mice caused by a persistent bacterial
infection with a novel Helicobacter species. J Natl Cancer Inst .
1994;86:1222–1227.
31. Boutin SR, Rogers AB, Shen Z, et al. Hepatic temporal gene
expression profiling in Helicobacter hepaticus-infected A/JCr
mice. Toxicol Pathol. 2004;32:678–693.
32. Taylor NS, Fox JG, Yan L. In vitro hepatotoxic factor in Heli-
cobacter hepaticus, H. pylori and other Helicobacter species.
J Med Microbiol. 1995;42:48–52.
33. Avenaud P, Castroviejo M, Claret S, et al. Expression and
activity of the cytolethal distending toxin of Helicobacter hepa-
ticus. Biochem Biophys Res Commun. 2004;318:739–745.
34. Balkwill F, Mantovani A. Inflammation and cancer: back to
virchow? Lancet . 2001;357:539–545.
35. Bodger K, Crabtree JE. Helicobacter pylori and gastric inflam-
mation. Br Med Bull. 1998;54:139–150.
36. Pfeilschifter J, Eberhardt W, Beck KF. Regulation of gene
expression by nitric oxide. Pflugers Arch. 2001;442:479–486.
37. Beck KF, Eberhardt W, Frank S, et al. Inducible NO synthase:
role in cellular signalling. J Exp Biol. 1999;202:645–653.
38. Brown Z, Robson RL, Westwick J. L-arginine/nitric oxide path-
way: a possible signal transduction mechanism for the regulation
of the chemokine IL-8 in human mesangial cells. Adv Exp Med
Biol. 1993;351:65–75.
39. Allen RG, Tresini M. Oxidative stress and gene regulation. Free
Radic Biol Med . 2000;28:463–499.
40. Ito K, Nakamura M, Toda G, et al. Potential role of Helicobacter
pylori in hepatocarcinogenesis. Int J Mol Med . 2004;13:221–227.
41. Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis
and tumour progression. Lancet Oncol. 2001;2:149–156.
42. Brune B, von Knethen A, Sandau KB. Nitric oxide and its role in
apoptosis. Eur J Pharmacol. 1998;351:261–272.
43. Obst B, Wagner S, Sewing KF, Beil W. Helicobacter pylori
causes DNA damage in gastric epithelial cells. Carcinogenesis.
2000;21:1111–1115.
44. Gupta S. Molecular signaling in death receptor and mitochondrial
pathways of apoptosis (review). Int J Oncol. 2003;22:15–20.
45. Rudi J, Kuck D, Strand S, et al. Involvement of the CD95 (APO-
1/Fas) receptor and ligand system in Helicobacter pylori-induced
gastric epithelial apoptosis. J Clin Invest . 1998;102:1506–1514.
46. Le’Negrate G, Ricci V, Hofman V, et al. Epithelial intestinal cellapoptosis induced by Helicobacter pylori depends on expression
of the cag pathogenicity island phenotype. Infect Immun.
2001;69:5001–5009.
47. Gogvadze V, Orrenius S. Mitochondrial regulation of apoptotic
cell death. Chem Biol Interact . 2006;163:4–14.
48. Perlman H, Zhang X, Chen MW, Walsh K, Buttyan R. An ele-
vated bax/bcl-2 ratio corresponds with the onset of prostate epi-
thelial cell apoptosis. Cell Death Differ . 1999;6:48–54.
49. Narita M, Shimizu S, Ito T, et al. Bax interacts with the perme-
ability transition pore to induce permeability transition and
cytochrome c release in isolated mitochondria. Proc Natl Acad
Sci USA. 1998;95:14681–14686.
Dig Dis Sci (2011) 56:1682–1692 1691
123

8/2/2019 Caspase Aline 2
http://slidepdf.com/reader/full/caspase-aline-2 11/11
50. Elmore S. Apoptosis: a review of programmed cell death. Toxicol
Pathol. 2007;35:495–516.
51. Bras M, Queenan B, Susin SA. Programmed cell death via
mitochondria: different modes of dying. Biochemistry (Mosc).
2005;70:231–239.
52. Crabtree JE, Farmery SM, Lindley IJ, et al. CagA/cytotoxic
strains of Helicobacter pylori and interleukin-8 in gastric epi-
thelial cell lines. J Clin Pathol. 1994;47:945–950.
53. Kawahara T, Teshima S, Kuwano Y, et al. Helicobacter pylori
lipopolysaccharide induces apoptosis of cultured guinea pig
gastric mucosal cells. Am J Physiol Gastrointest Liver Physiol.
2001;281:G726–G734.
54. Kuck D, Kolmerer B, Iking-Konert C, et al. Vacuolating cyto-
toxin of Helicobacter pylori induces apoptosis in the human
gastric epithelial cell line AGS. Infect Immun. 2001;69:
5080–5087.
55. Schulte-Hermann R, Bursch W, Grasl-Kraupp B, et al. Role
of active cell death (apoptosis) in multi-stage carcinogenesis.
Toxicol Lett . 1995;82–83:143–148.
1692 Dig Dis Sci (2011) 56:1682–1692
123
Recommended