
Association Between Invisible Basal Ganglia and ZNF335 Mutations: A Case ReportRieko Sato, MD, a, b Jun-ichi Takanashi, MD, PhD, c Yu Tsuyusaki, MD, d Mitsuhiro Kato, MD, PhD, e, f Hirotomo Saitsu, MD, PhD, g Naomichi Matsumoto, MD, PhD, g Takao Takahashi, MD, PhDb
aDepartment of Pediatrics, National Hospital Organization
Tokyo Medical Center, Tokyo, Japan; bDepartment of
Pediatrics, Keio University School of Medicine, Tokyo,
Japan; cDepartment of Pediatrics, Yachiyo Medical Center,
Tokyo Women's Medical University, Yachiyo, Japan; dDepartment of Child Neurology, Kanagawa Children’s
Medical Center, Yokohama, Japan; eDepartment of
Pediatrics, Yamagata University Faculty of Medicine,
Yamagata, Japan; fDepartment of Pediatrics, Showa
University School of Medicine, Tokyo, Japan; and gDepartment of Human Genetics, Yokohama City University
Graduate School of Medicine, Yokohama, Japan
Dr Sato was an attending pediatrician, designed
the study, collected clinical information, and wrote
the paper; Dr Takanashi designed the study and
examined MRI; Dr Tsuyusaki was an attending
pediatrician and collected clinical information;
Dr Kato performed initial genetic testing, including
Sanger sequencing; Dr Saitsu performed exome
sequencing and in silico analysis; Dr Matsumoto
designed the study; Dr Takahashi designed the
study and reviewed the manuscript; and all authors
approved the fi nal manuscript for submission and
agree to be accountable for all aspects of the study.
DOI: 10.1542/peds.2016-0897
Accepted for publication May 24, 2016
Address correspondence to Takao Takahashi, MD,
PhD, Department of Pediatrics, Keio University
School of Medicine, 35 Shinanomachi, Shinjuku-ku,
Tokyo 160-8582, Japan. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online,
1098-4275).
Copyright © 2016 by the American Academy of
Pediatrics
FINANCIAL DISCLOSURE: The authors have
indicated they have no fi nancial relationships
relevant to this article to disclose.
ZNF335, a nuclear zinc finger protein,
is essential for methylation and
expression of brain-specific genes. 1
ZNF335 was reported in 2012 as a
causative gene for microcephaly. 1
Yang et al 1 identified homozygous
ZNF335 mutations in 7 microcephaly
patients of 1 consanguineous Arab–
Israeli pedigree, suggesting that it
demonstrates autosomal recessive
inheritance. Because only this single
pedigree has been reported to date,
the key clinical features associated
with ZNF335 mutations remain
unknown. In this study, we report a
second family with ZNF335 mutations
and describe the associated clinical
features.
CASE PRESENTATION
The proband of this study was a
33-month-old girl. She was the
first child of nonconsanguineous
Japanese parents. No family members
had microcephaly or presented
developmental delay. Prenatal history
was unremarkable. She was delivered
vaginally at full term without
asphyxia. Her birth weight was 3030 g
(+0.9 SD), length was 49.5 cm (+0.5 SD),
and head circumference was 32.0 cm
(−0.6 SD). She had a systolic murmur
and was diagnosed with a ventricular
septal defect (3 mm).
At 3 months of age, she was admitted
to our hospital because of an afebrile
abstractZNF335 was first reported in 2012 as a causative gene for microcephaly.
Because only 1 consanguineous pedigree has ever been reported, the key
clinical features associated with ZNF335 mutations remain unknown. In
this article, we describe another family harboring ZNF335 mutations. The
female proband was the first child of nonconsanguineous Japanese parents.
At birth, microcephaly was absent; her head circumference was 32.0 cm
(−0.6 SD). At 3 months, microcephaly was noted, (head circumference,
34.0 cm [−4.6 SD]). Brain MRI showed invisible basal ganglia, cerebral
atrophy, brainstem hypoplasia, and cerebellar atrophy. At 33 months, (head
circumference, 41.0 cm [−5.1 SD]), she had severe psychomotor retardation.
After obtaining informed consent from her parents, we performed exome
sequencing in the proband and identified 1 novel and 1 known mutation
in ZNF335, namely, c.1399T>C (p.C467R) and c.1505A>G (p.Y502C),
respectively. The mutations were individually transmitted by her parents,
indicating that the proband was compound heterozygous for the mutations.
Her brain imaging findings, including invisible basal ganglia, were similar to
those observed in the previous case with ZNF335 mutations. We speculate
that invisible basal ganglia may be the key feature of ZNF335 mutations.
For infants presenting with both microcephaly and invisible basal ganglia,
ZNF335 mutations should be considered as a differential diagnosis.
CASE REPORTPEDIATRICS Volume 138 , number 3 , September 2016 :e 20160897
To cite: Sato R, Takanashi J, Tsuyusaki Y, et al.
Association Between Invisible Basal Ganglia and
ZNF335 Mutations: A Case Report. Pediatrics. 2016;
138(3):e20160897
by guest on August 27, 2018www.aappublications.org/newsDownloaded from
SATO et al
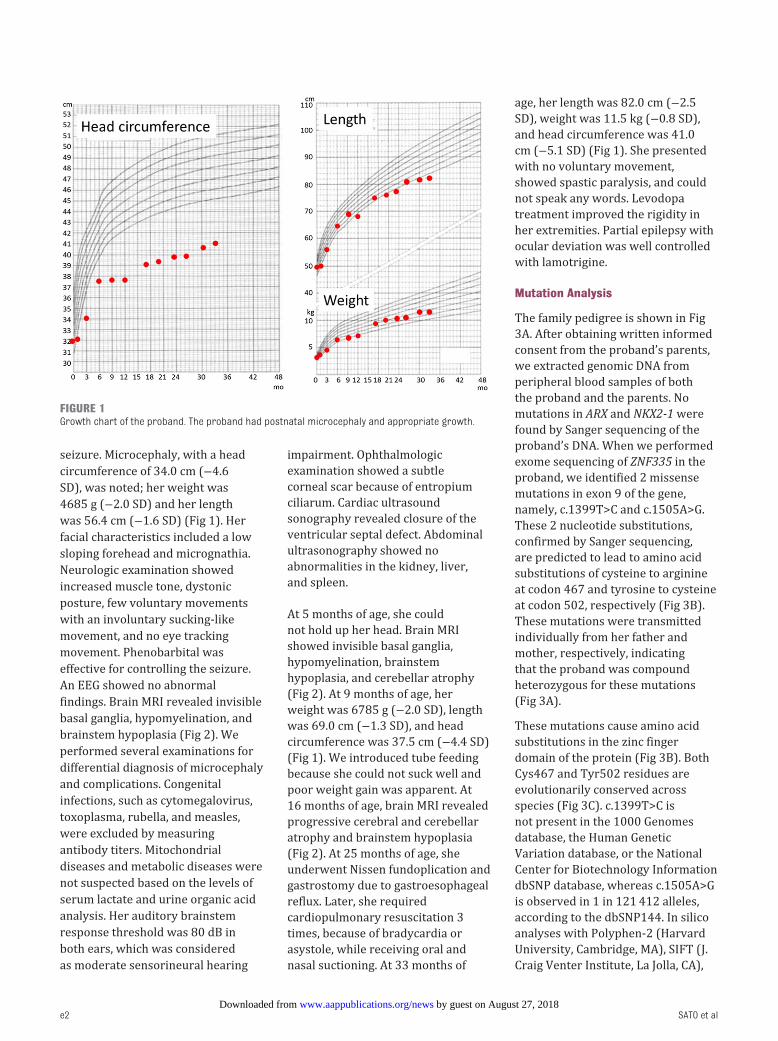
seizure. Microcephaly, with a head
circumference of 34.0 cm (−4.6
SD), was noted; her weight was
4685 g (−2.0 SD) and her length
was 56.4 cm (−1.6 SD) ( Fig 1). Her
facial characteristics included a low
sloping forehead and micrognathia.
Neurologic examination showed
increased muscle tone, dystonic
posture, few voluntary movements
with an involuntary sucking-like
movement, and no eye tracking
movement. Phenobarbital was
effective for controlling the seizure.
An EEG showed no abnormal
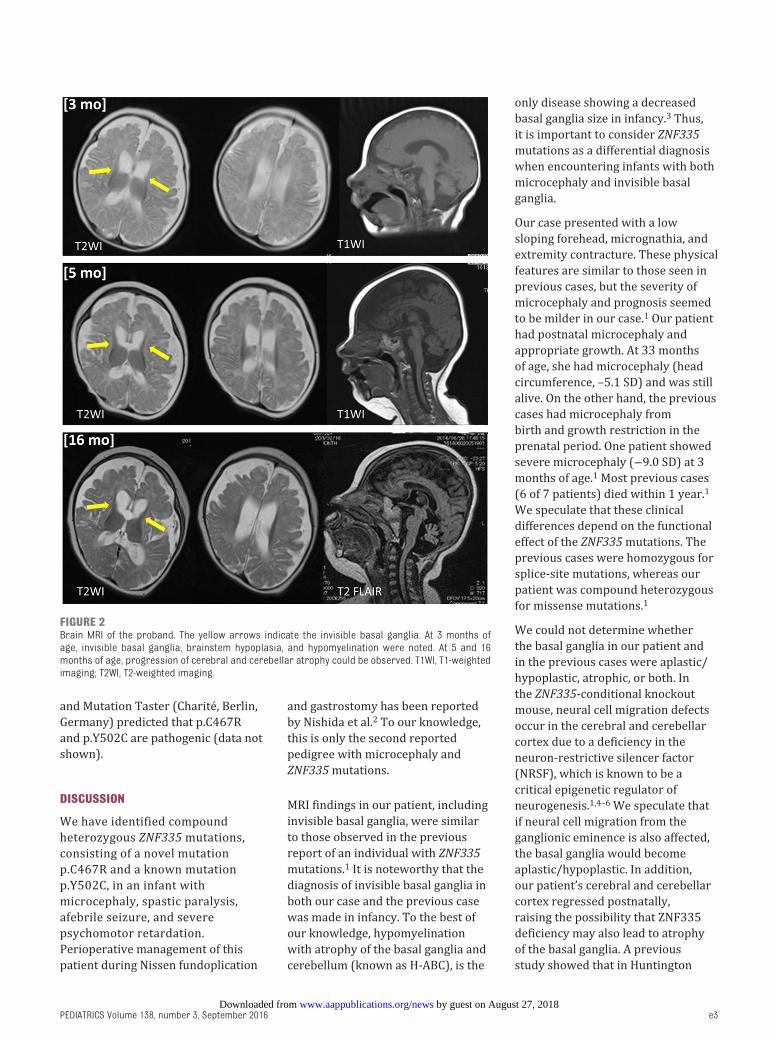
findings. Brain MRI revealed invisible
basal ganglia, hypomyelination, and
brainstem hypoplasia ( Fig 2). We
performed several examinations for
differential diagnosis of microcephaly
and complications. Congenital
infections, such as cytomegalovirus,
toxoplasma, rubella, and measles,
were excluded by measuring
antibody titers. Mitochondrial
diseases and metabolic diseases were
not suspected based on the levels of
serum lactate and urine organic acid
analysis. Her auditory brainstem
response threshold was 80 dB in
both ears, which was considered
as moderate sensorineural hearing
impairment. Ophthalmologic
examination showed a subtle
corneal scar because of entropium
ciliarum. Cardiac ultrasound
sonography revealed closure of the
ventricular septal defect. Abdominal
ultrasonography showed no
abnormalities in the kidney, liver,
and spleen.
At 5 months of age, she could
not hold up her head. Brain MRI
showed invisible basal ganglia,
hypomyelination, brainstem
hypoplasia, and cerebellar atrophy
( Fig 2). At 9 months of age, her
weight was 6785 g (−2.0 SD), length
was 69.0 cm (−1.3 SD), and head
circumference was 37.5 cm (−4.4 SD)
( Fig 1). We introduced tube feeding
because she could not suck well and
poor weight gain was apparent. At
16 months of age, brain MRI revealed
progressive cerebral and cerebellar
atrophy and brainstem hypoplasia
( Fig 2). At 25 months of age, she
underwent Nissen fundoplication and
gastrostomy due to gastroesophageal
reflux. Later, she required
cardiopulmonary resuscitation 3
times, because of bradycardia or
asystole, while receiving oral and
nasal suctioning. At 33 months of
age, her length was 82.0 cm (−2.5
SD), weight was 11.5 kg (−0.8 SD),
and head circumference was 41.0
cm (−5.1 SD) ( Fig 1). She presented
with no voluntary movement,
showed spastic paralysis, and could
not speak any words. Levodopa
treatment improved the rigidity in
her extremities. Partial epilepsy with
ocular deviation was well controlled
with lamotrigine.
Mutation Analysis
The family pedigree is shown in Fig
3A. After obtaining written informed
consent from the proband’s parents,
we extracted genomic DNA from
peripheral blood samples of both
the proband and the parents. No
mutations in ARX and NKX2-1 were
found by Sanger sequencing of the
proband’s DNA. When we performed
exome sequencing of ZNF335 in the
proband, we identified 2 missense
mutations in exon 9 of the gene,
namely, c.1399T>C and c.1505A>G.
These 2 nucleotide substitutions,
confirmed by Sanger sequencing,
are predicted to lead to amino acid
substitutions of cysteine to arginine
at codon 467 and tyrosine to cysteine
at codon 502, respectively ( Fig 3B).
These mutations were transmitted
individually from her father and
mother, respectively, indicating
that the proband was compound
heterozygous for these mutations
( Fig 3A).
These mutations cause amino acid
substitutions in the zinc finger
domain of the protein ( Fig 3B). Both
Cys467 and Tyr502 residues are
evolutionarily conserved across
species ( Fig 3C). c.1399T>C is
not present in the 1000 Genomes
database, the Human Genetic
Variation database, or the National
Center for Biotechnology Information
dbSNP database, whereas c.1505A>G
is observed in 1 in 121 412 alleles,
according to the dbSNP144. In silico
analyses with Polyphen-2 (Harvard
University, Cambridge, MA), SIFT (J.
Craig Venter Institute, La Jolla, CA),
e2
FIGURE 1Growth chart of the proband. The proband had postnatal microcephaly and appropriate growth.
by guest on August 27, 2018www.aappublications.org/newsDownloaded from
PEDIATRICS Volume 138 , number 3 , September 2016
and Mutation Taster (Charité, Berlin,
Germany) predicted that p.C467R
and p.Y502C are pathogenic (data not
shown).
DISCUSSION
We have identified compound
heterozygous ZNF335 mutations,
consisting of a novel mutation
p.C467R and a known mutation
p.Y502C, in an infant with
microcephaly, spastic paralysis,
afebrile seizure, and severe
psychomotor retardation.
Perioperative management of this
patient during Nissen fundoplication
and gastrostomy has been reported
by Nishida et al. 2 To our knowledge,
this is only the second reported
pedigree with microcephaly and
ZNF335 mutations.
MRI findings in our patient, including
invisible basal ganglia, were similar
to those observed in the previous
report of an individual with ZNF335
mutations. 1 It is noteworthy that the
diagnosis of invisible basal ganglia in
both our case and the previous case
was made in infancy. To the best of
our knowledge, hypomyelination
with atrophy of the basal ganglia and
cerebellum (known as H-ABC), is the
only disease showing a decreased
basal ganglia size in infancy. 3 Thus,
it is important to consider ZNF335
mutations as a differential diagnosis
when encountering infants with both
microcephaly and invisible basal
ganglia.
Our case presented with a low
sloping forehead, micrognathia, and
extremity contracture. These physical
features are similar to those seen in
previous cases, but the severity of
microcephaly and prognosis seemed
to be milder in our case. 1 Our patient
had postnatal microcephaly and
appropriate growth. At 33 months
of age, she had microcephaly (head
circumference, –5.1 SD) and was still
alive. On the other hand, the previous
cases had microcephaly from
birth and growth restriction in the
prenatal period. One patient showed
severe microcephaly (−9.0 SD) at 3
months of age. 1 Most previous cases
(6 of 7 patients) died within 1 year. 1
We speculate that these clinical
differences depend on the functional
effect of the ZNF335 mutations. The
previous cases were homozygous for
splice-site mutations, whereas our
patient was compound heterozygous
for missense mutations. 1
We could not determine whether
the basal ganglia in our patient and
in the previous cases were aplastic/
hypoplastic, atrophic, or both. In
the ZNF335-conditional knockout
mouse, neural cell migration defects
occur in the cerebral and cerebellar
cortex due to a deficiency in the
neuron-restrictive silencer factor
(NRSF), which is known to be a
critical epigenetic regulator of
neurogenesis. 1, 4 – 6 We speculate that
if neural cell migration from the
ganglionic eminence is also affected,
the basal ganglia would become
aplastic/hypoplastic. In addition,
our patient’s cerebral and cerebellar
cortex regressed postnatally,
raising the possibility that ZNF335
deficiency may also lead to atrophy
of the basal ganglia. A previous
study showed that in Huntington
e3
FIGURE 2Brain MRI of the proband. The yellow arrows indicate the invisible basal ganglia. At 3 months of age, invisible basal ganglia, brainstem hypoplasia, and hypomyelination were noted. At 5 and 16 months of age, progression of cerebral and cerebellar atrophy could be observed. T1WI, T1-weighted imaging; T2WI, T2-weighted imaging.
by guest on August 27, 2018www.aappublications.org/newsDownloaded from

disease characterized by striatal
and cerebral atrophy, aberrant
accumulation of NRSF in the nucleus
would lead to loss of expression of
neuronal genes regulated through the
neuron-restrictive silencer element.7
This implies that fine regulation of
NRSF is essential for maintenance
of the striatum and cerebral cortex.
Therefore, we speculate that invisible
basal ganglia in patients with ZNF335
mutations may result from aplasia/
hypoplasia and atrophy of the basal
ganglia via a deficiency in NRSF.
In summary, we report an infant with
microcephaly and ZNF335 mutations.
Invisible basal ganglia may be the key
clinical feature of ZNF335 mutations.
ACKNOWLEDGMENTS
We thank the family for
participating in this study. We
also thank Drs A. James Barkovich,
Masayuki Sasaki, Osamu Komiyama,
Tomohide Goto, Kazuki Yamazawa,
and Takeshi Sato for fruitful
discussions.
SATO et al e4
ABBREVIATION
NRSF: neuron-restrictive silencer
factor
FUNDING: This work was supported in part by a grant for Research on Measures for Intractable Diseases from the Japan Agency for Medical Research and
Development.
POTENTIAL CONFLICT OF INTEREST: The authors have indicated they have no potential confl icts of interest to disclose.
REFERENCES
1. Yang YJ, Baltus AE, Mathew RS, et al.
Microcephaly gene links trithorax and
REST/NRSF to control neural stem cell
proliferation and differentiation. Cell.
2012;151(5):1097–1112
2. Nishida T, Mihara T, Horiki T, Ka K.
Anaesthesia and orphan disease:
primary autosomal recessive
microcephaly-10 caused by a mutation
in the ZNF335 gene: A case report. Eur
J Anaesthesiol. 2015; 33(7):543–545
3. Hamilton EM, Polder E, Vanderver
A, et al; H-ABC Research Group.
Hypomyelination with atrophy of the
basal ganglia and cerebellum: further
delineation of the phenotype and
genotype-phenotype correlation. Brain.
2014;137(Pt 7):1921–1930
4. Chong JA, Tapia-Ramírez J, Kim S,
et al. REST: a mammalian silencer
protein that restricts sodium channel
FIGURE 3A, Pedigree of the family. c.1399T>C and c.1505A>G in ZNF335 were transmitted individually from the parents, indicating that the proband was compound heterozygous for these mutations. B, Protein structure of ZNF335. c.1399T>C and c.1505A>G are predicted to lead to amino acid substitutions of cysteine to arginine at codon 467, and of tyrosine to cysteine at codon 502, respectively. C467R and Y502C are located in the zinc fi nger domain of the protein. C, Comparison of conservation of Cys467 and Try502 residues across species. These 2 residues are evolutionarily conserved across species.
by guest on August 27, 2018www.aappublications.org/newsDownloaded from

PEDIATRICS Volume volume , number issue , pub-date e5
gene expression to neurons. Cell.
1995;80(6):949–957
5. Schoenherr CJ, Anderson DJ. Silencing
is golden: negative regulation in the
control of neuronal gene transcription.
Curr Opin Neurobiol. 1995;5(5):566–571
6. Sun YM, Greenway DJ, Johnson R,
et al. Distinct profi les of REST
interactions with its target genes
at different stages of neuronal
development. Mol Biol Cell.
2005;16(12):5630–5638
7. Zuccato C, Tartari M, Crotti A,
et al. Huntingtin interacts with
REST/NRSF to modulate the
transcription of NRSE-controlled
neuronal genes. Nat Genet.
2003;35(1):76–83
by guest on August 27, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2016-0897 originally published online August 18, 2016; 2016;138;Pediatrics
Naomichi Matsumoto and Takao TakahashiRieko Sato, Jun-ichi Takanashi, Yu Tsuyusaki, Mitsuhiro Kato, Hirotomo Saitsu,
Report Mutations: A CaseZNF335Association Between Invisible Basal Ganglia and
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/138/3/e20160897including high resolution figures, can be found at:
Referenceshttp://pediatrics.aappublications.org/content/138/3/e20160897#BIBLThis article cites 7 articles, 1 of which you can access for free at:
Subspecialty Collections
subhttp://www.aappublications.org/cgi/collection/neurologic_disorders_Neurologic Disordershttp://www.aappublications.org/cgi/collection/neurology_subNeurologyhttp://www.aappublications.org/cgi/collection/genetics_subGeneticsfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on August 27, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2016-0897 originally published online August 18, 2016; 2016;138;Pediatrics
Naomichi Matsumoto and Takao TakahashiRieko Sato, Jun-ichi Takanashi, Yu Tsuyusaki, Mitsuhiro Kato, Hirotomo Saitsu,
Report Mutations: A CaseZNF335Association Between Invisible Basal Ganglia and
http://pediatrics.aappublications.org/content/138/3/e20160897located on the World Wide Web at:
The online version of this article, along with updated information and services, is
1073-0397. ISSN:60007. Copyright © 2016 by the American Academy of Pediatrics. All rights reserved. Print
the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on August 27, 2018www.aappublications.org/newsDownloaded from
Recommended

















