
Allostery in GPCRs: ‘MWC’ revisitedMeritxell Canals, Patrick M. Sexton and Arthur Christopoulos
Drug Discovery Biology, Monash Institute of Pharmaceutical Sciences and Department of Pharmacology, Monash University,
Australia
Review
G protein-coupled receptors (GPCRs) constitute the larg-est family of receptors in the genome and are the targetsfor at least 30% of current medicines. In recent years,there has been a dramatic increase in the discovery ofallosteric modulators of GPCR activity and a growingappreciation of the diverse modes by which GPCRs canbe regulated by both orthosteric and allosteric ligands.Interestingly, some of the contemporary views of GPCRfunction reflect characteristics that are shared by proto-typical allosteric proteins, as encompassed in the classicMonod–Wyman–Changeux (MWC) model initially pro-posed for enzymes and subsequently extended to otherprotein families. In this review, we revisit the MWCmodel in the context of emerging structural, functionaland operational data on GPCR allostery.
GPCRs as therapeutic targetsG protein-coupled receptors (GPCRs) constitute the largestgroup of cell surface signal-transducing proteins (�2% ofthe human genome). Endogenous GPCR ligands are ex-traordinarily diverse, ranging from small ions to amines,lipids and large peptides [1]. Phylogenetically, GPCRs canbe classified into five main families, but structurally theyare commonly subdivided into three: Family A (rhodopsin-like) GPCRs represent the largest group and, thus far, theonly one for which high-resolution crystal structures havebeen reported [2–7]. Family B GPCRs are utilized by largepeptides such as calcitonin, secretin and glucagon. FamilyC GPCRs recognize small molecules such as glutamate,calcium, g-aminobutyric acid (GABA) and some taste mole-cules.
GPCRs have long represented model systems throughwhich key principles of drug–receptor interactions havebeen developed. Nonetheless, until recently, GPCR-baseddrug discovery largely treated these receptors as blackboxes, with the main focus being on discovering moleculesthat mimic or inhibit the actions of endogenous hormonesor neurotransmitters; by analogy to the substrate bindingsite in enzymes, the agonist binding site on GPCRs (andother receptors) is termed the orthosteric site [8]. Thisstrategy has, of course, been successful for numerousGPCRs [1]. However, the attrition rate of modern drugdiscovery is higher than ever, and the development of moreselective compounds as probes for understanding GPCRfunction and possible drug leads remains a challenge. Oneof the key issues in this regard is the fact that many GPCRsshare high sequence homology within the orthosteric siteacross receptor subtypes; targeting this site alone is un-likely to yield highly subtype-selective compounds. An
Corresponding author: Christopoulos, A. ([email protected]).
0968-0004/$ – see front matter � 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.tibs.2011.
explosion in technologies that have facilitated highthroughput and deeper analyses of GPCR cellular functionhave also resulted in the realization that the same ligandacting at the same receptor can display substantial diver-sity in functional outcomes depending on the cellularcontext and the assay type, highlighting the disconnectthat can exist between cellular and clinical efficacy if the‘incorrect’ functional assay is used to study and validatedrug action.
The issues described above are consistent with thebehavior of allosteric proteins. Indeed, during the lastdecade, the idea of targeting allosteric sites as a novelapproach to GPCR drug discovery has become a major topicin receptor pharmacology and within the pharmaceuticalindustry [9]. However, the mechanistic understanding ofallostery at GPCRs still lags behind studies of other pro-tein families. This is due to a variety of reasons, includingthe difficulty in obtaining high-resolution structural infor-mation on GPCRs and their interacting partners (ligandsand proteins) and the complex and promiscuous nature ofGPCR functionality. Nonetheless, many of these chal-lenges are now being addressed. For instance, the recentsolution of a number of Family A GPCR crystal structureshas provided some unexpected insights into the nature ofligand binding pockets on these receptors [2–7,10]. It isthus timely to re-consider allostery within the context ofcontemporary views of GPCR structure and function. Spe-cifically, the concept of allosteric proteins, first proposed byMonod, Wyman, Jacob and Changeux and then formalizedas the classic Monod–Wyman–Changeux (MWC) model[11,12], identified a number of key characteristics associ-ated with allosteric proteins. These MWC characteristics ofallosteric proteins can be summarized as: (i) they areoligomeric; (ii) they display an axis of symmetry; (iii) theyexist in multiple equilibria (even in the absence of ligand)and the transition from one state to another involves aconservation of the symmetry of the individual subunitscomprising the complex; and (iv) they possess multiplebinding sites, and ligand binding to any of these sitesstabilizes a subset of conformational states at the expenseof others. This article considers to what extent thesebehaviors can be generalized to GPCRs.
Allosteric proteins are oligomericThe first experimental observations of allosterism werefrom studies of enzymes and ion channels that displayedcooperative behavior due to an oligomeric architecture[13]. The existence of dimers or higher order oligomershas since been confirmed at all other receptor classes [14].By contrast, GPCRs were traditionally considered to be
08.005 Trends in Biochemical Sciences, December 2011, Vol. 36, No. 12 663
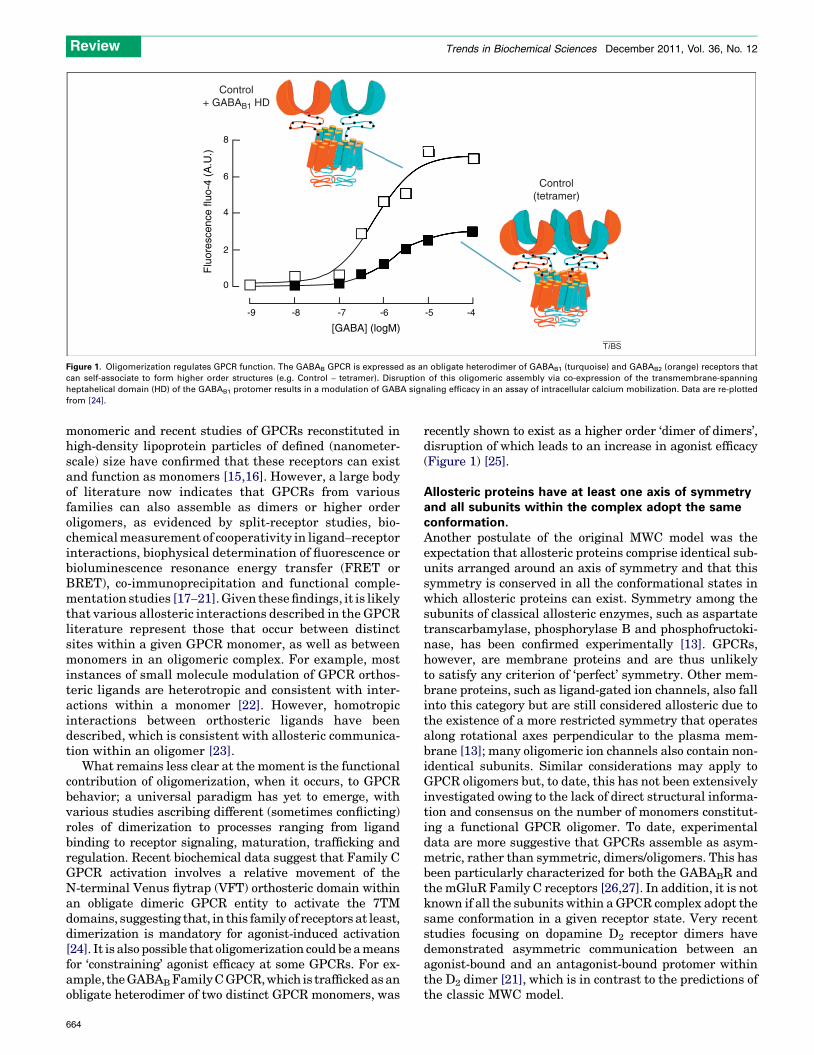
Control(tetramer)
Control+ GABAB1 HD
[GABA] (logM)
-9
0
2
4
6
8
-8 -7 -6 -5 -4
Flu
ores
cenc
e flu
o-4
(A.U
.)
TiBS
Figure 1. Oligomerization regulates GPCR function. The GABAB GPCR is expressed as an obligate heterodimer of GABAB1 (turquoise) and GABAB2 (orange) receptors that
can self-associate to form higher order structures (e.g. Control – tetramer). Disruption of this oligomeric assembly via co-expression of the transmembrane-spanning
heptahelical domain (HD) of the GABAB1 protomer results in a modulation of GABA signaling efficacy in an assay of intracellular calcium mobilization. Data are re-plotted
from [24].
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
monomeric and recent studies of GPCRs reconstituted inhigh-density lipoprotein particles of defined (nanometer-scale) size have confirmed that these receptors can existand function as monomers [15,16]. However, a large bodyof literature now indicates that GPCRs from variousfamilies can also assemble as dimers or higher orderoligomers, as evidenced by split-receptor studies, bio-chemical measurement of cooperativity in ligand–receptorinteractions, biophysical determination of fluorescence orbioluminescence resonance energy transfer (FRET orBRET), co-immunoprecipitation and functional comple-mentation studies [17–21]. Given these findings, it is likelythat various allosteric interactions described in the GPCRliterature represent those that occur between distinctsites within a given GPCR monomer, as well as betweenmonomers in an oligomeric complex. For example, mostinstances of small molecule modulation of GPCR orthos-teric ligands are heterotropic and consistent with inter-actions within a monomer [22]. However, homotropicinteractions between orthosteric ligands have beendescribed, which is consistent with allosteric communica-tion within an oligomer [23].
What remains less clear at the moment is the functionalcontribution of oligomerization, when it occurs, to GPCRbehavior; a universal paradigm has yet to emerge, withvarious studies ascribing different (sometimes conflicting)roles of dimerization to processes ranging from ligandbinding to receptor signaling, maturation, trafficking andregulation. Recent biochemical data suggest that Family CGPCR activation involves a relative movement of theN-terminal Venus flytrap (VFT) orthosteric domain withinan obligate dimeric GPCR entity to activate the 7TMdomains, suggesting that, in this family of receptors at least,dimerization is mandatory for agonist-induced activation[24]. It is also possible that oligomerization could be a meansfor ‘constraining’ agonist efficacy at some GPCRs. For ex-ample, the GABABFamily C GPCR, which is trafficked as anobligate heterodimer of two distinct GPCR monomers, was
664
recently shown to exist as a higher order ‘dimer of dimers’,disruption of which leads to an increase in agonist efficacy(Figure 1) [25].
Allosteric proteins have at least one axis of symmetryand all subunits within the complex adopt the sameconformation.Another postulate of the original MWC model was theexpectation that allosteric proteins comprise identical sub-units arranged around an axis of symmetry and that thissymmetry is conserved in all the conformational states inwhich allosteric proteins can exist. Symmetry among thesubunits of classical allosteric enzymes, such as aspartatetranscarbamylase, phosphorylase B and phosphofructoki-nase, has been confirmed experimentally [13]. GPCRs,however, are membrane proteins and are thus unlikelyto satisfy any criterion of ‘perfect’ symmetry. Other mem-brane proteins, such as ligand-gated ion channels, also fallinto this category but are still considered allosteric due tothe existence of a more restricted symmetry that operatesalong rotational axes perpendicular to the plasma mem-brane [13]; many oligomeric ion channels also contain non-identical subunits. Similar considerations may apply toGPCR oligomers but, to date, this has not been extensivelyinvestigated owing to the lack of direct structural informa-tion and consensus on the number of monomers constitut-ing a functional GPCR oligomer. To date, experimentaldata are more suggestive that GPCRs assemble as asym-metric, rather than symmetric, dimers/oligomers. This hasbeen particularly characterized for both the GABABR andthe mGluR Family C receptors [26,27]. In addition, it is notknown if all the subunits within a GPCR complex adopt thesame conformation in a given receptor state. Very recentstudies focusing on dopamine D2 receptor dimers havedemonstrated asymmetric communication between anagonist-bound and an antagonist-bound protomer withinthe D2 dimer [21], which is in contrast to the predictions ofthe classic MWC model.

Box 1. Intracellular allosteric sites
Although most small molecule GPCR allosteric sites have been
proposed (or assumed) to exist on the extracellular surface, some
sites have been identified on the intracellular side of the receptors.
For example, small molecule inhibitors of the chemokine receptors
have been identified that act at sites on the cytoplasmic tail of CCR4
and CXCR2. Studies of domain swapping between CCR4 and CCR5
receptors demonstrated that inhibition of signal transduction by a
series of pyrazinyl sulfonamides is conferred through the CCR4
C-terminal domain [69]. Similarly, studies of domain swapping with
CXCR1 and CXCR2 receptors have demonstrated that sensitivity to
two nonpeptide CXCR2 antagonists is conferred by the C-terminal
domain [70]. These studies suggest that such an intracellular site
might exist across the chemokine receptor family and could
potentially be exploited for drug discovery. Other studies have
recently reported the discovery of pepducins, which are created by
attaching a lipidated group such as an acyl chain to a peptide
corresponding to a portion of one of the intracellular loops of the
GPCR [71]. The activity of pepducins appears to require an intact
receptor, and studies on a protease-activated receptor 1 (PAR1)-
directed pepducin have suggested that the C-terminal tail of the
receptor is likely to be an interaction site [71]. Thus, pepducins can
be regarded as novel allosteric modulators of GPCR function. Both
agonistic and antagonistic pepducins have been developed against
a variety of GPCRs, including protease-activated receptors (PAR1,
-2 and -4), the chemokine receptors (CXCR1, -2 and -4) and the
sphingosine-1-phosphate receptor (S1P3) [71]. To date, pepducins
have shown efficacy in the control of platelet-dependent hemostasis
and thrombosis, tumor growth, invasion, angiogenesis and sepsis in
animal models [72].
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
Allosteric proteins have multiple ligand binding sitesIn agreement with the MWC model for allosteric proteins,GPCRs possess topographically distinct binding domainsfor recognition of the receptor’s natural ligand and forinteraction with intracellular effector molecules responsi-ble for propagating the stimulus imparted by receptoragonists; the average distance between the extracellularand intracellular domains of GPCRs is 40–60 A. The pro-totypical example of an allosteric interaction between twodistinct domains on a GPCR is that involving the G proteinitself, which is characterized by positive cooperativitybetween the binding of an agonist and the (nucleotide-free)Ga subunit of the G protein [28]. However, the list of GPCRinteracting proteins is constantly expanding [29], withnew roles identified for proteins such as GPCR kinases(GRKs), arrestins and receptor activity modifying proteins(RAMPs) as signal transducers or modulators, often inde-pendently of the actions of G proteins [30,31]. These find-ings underscore the vectorial nature of informationtransfer across GPCRs [32].
One point of divergence between ligand binding atGPCRs compared to many other allosteric proteins is thatthe binding domains are not typically situated at subunitinterfaces. Despite a common structural architecture, thesubstantial diversity of endogenous ligands recognized byGPCRs suggests that these receptors are sufficiently ‘plas-tic’ such that a canonical orthosteric domain within onetype of (monomeric) GPCR can represent a potential allo-steric domain at another type of GPCR. Numerous muta-genesis and chimeric receptor studies have providedevidence to support this notion [22]. Recently solved crystalstructures have also confirmed directly that rhodopsin andbiogenic amine GPCRs bind endogenous ligands within theTM helical bundle of the receptor [7,33–35], whereas thechemokine CXCR4 receptor binds the peptide CVX15through more extracellular contacts (relative to the TMpocket) that utilize the loop regions of the receptor [6].Conversely, prototypical allosteric modulators of biogenicamine GPCRs bind within the extracellular loops and thetop of the TM domains [36,37], whereas small moleculemodulators of the chemokine receptors are thought to bindfurther in the TM regions relative to orthosteric peptidechemokine ligands [38]. Perhaps the most striking separa-tion between extracellular orthosteric and allosteric siteshas been observed in Family C GPCRs. Endogenous ago-nists for this family of receptors bind to the large VFTdomain that comprises their extracellular N-terminus andis linked to the distant 7TM domain by a large cysteine-richspacer region. Most synthetic allosteric modulators for thisclass of receptors have been shown to utilize the TMdomain to modulate ligand function in the orthostericVFT domain [39].
The preceding discussion does not rule out the potentialfor lateral information transfer between two orthosteric(or allosteric) sites across a GPCR dimer. By and large,however, these binding events do not occur at subunitinterfaces. One interesting exception is the calcitoningene-related peptide (CGRP) receptor, which is an obligateheterodimer comprising the GPCR, calcitonin-receptor-like receptor and the accessory protein RAMP1. Recently,a small molecule inhibitor of this receptor, BIBN4096BS,
was described that gains its selectivity by targeting theGPCR–RAMP interface of this receptor [40]. Given theincreasing focus on intracellular interacting partners ofGPCRs, it is likely that novel allosteric small moleculeswill be discovered that specifically target GPCR–proteininterfaces to mediate their effects. Indeed, reports are nowemerging describing small molecules and peptidesthat interact with intracellular allosteric sites of GPCRs(Box 1).
Allosteric proteins exist in multiple conformationalequilibria in the absence of ligandsA key feature of the classic MWC model is the presence ofmore than one conformational state of the allosteric pro-tein in the absence of ligands [11]. These states exist in adynamic equilibrium and the isomerization of the proteinfrom one state to the other is often referred to as theallosteric transition. The simplest case of this modeldescribes receptor activity in terms of two global states,one active and one inactive; it is thus common to see thismodel referred to as a two state model. An expectation ofreceptors that behave according to such a mechanism is theability to display functional activity in the absence ofligand, a phenomenon termed constitutive receptor activi-ty. Although this remained a contentious issue for manyyears, experimental evidence for activation of GPCRs inthe absence of ligand was presented in the late 1980s forthe d opioid receptor [41] and is now acknowledged as anaturally occurring phenomenon for nearly all GPCRs [42].This has led to a re-evaluation of the phenomenologicaldescriptors applied to GPCR-targeting compounds;agonists promote a GPCR active state over and abovethose that occur spontaneously, whereas ligands with a
665
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
preferential affinity for an inactive state are defined asinverse agonists [43].
The recognition that GPCRs can adopt constitutivelyactive states has led to a number of key findings with directbearing on drug discovery and development. For example,although the extent of constitutive activity displayed bymany receptors is limited under most physiological condi-tions, numerous diseases are known to arise as a conse-quence of mutations that lead to constitutive GPCRactivity, such as retinitis pigmentosa, precocious puberty,nonautoimmune hyperthyroidism and autosomal domi-nant hypocalcemia [44]. In such instances, an inverseagonist would be the preferred therapeutic agent and drugscreens are routinely modified to increase the likelihood ofdetecting inverse agonism, either through over-expression
-10 -9 -8 -7 -6
0
20
40
60
80
Log [ACh] (M)
pE
RK
1/2
(% F
BS
(10
%))
M4 mAChR
-13 -12 -11 -10 -9 -8
0
20
40
60
80
100
120 (i)
(ii)
2
4
6
8
10
Free receptors NMS-occrecepto
[3H]NMSexperiments
[3H]Dimethyexperime
logα
Gallamine pK-values
pK
Log [ACh] (M)
AC
h-s
tim
ula
ted
Ca2+
flu
x(%
AC
h m
ax)
-1
(a)
(b)
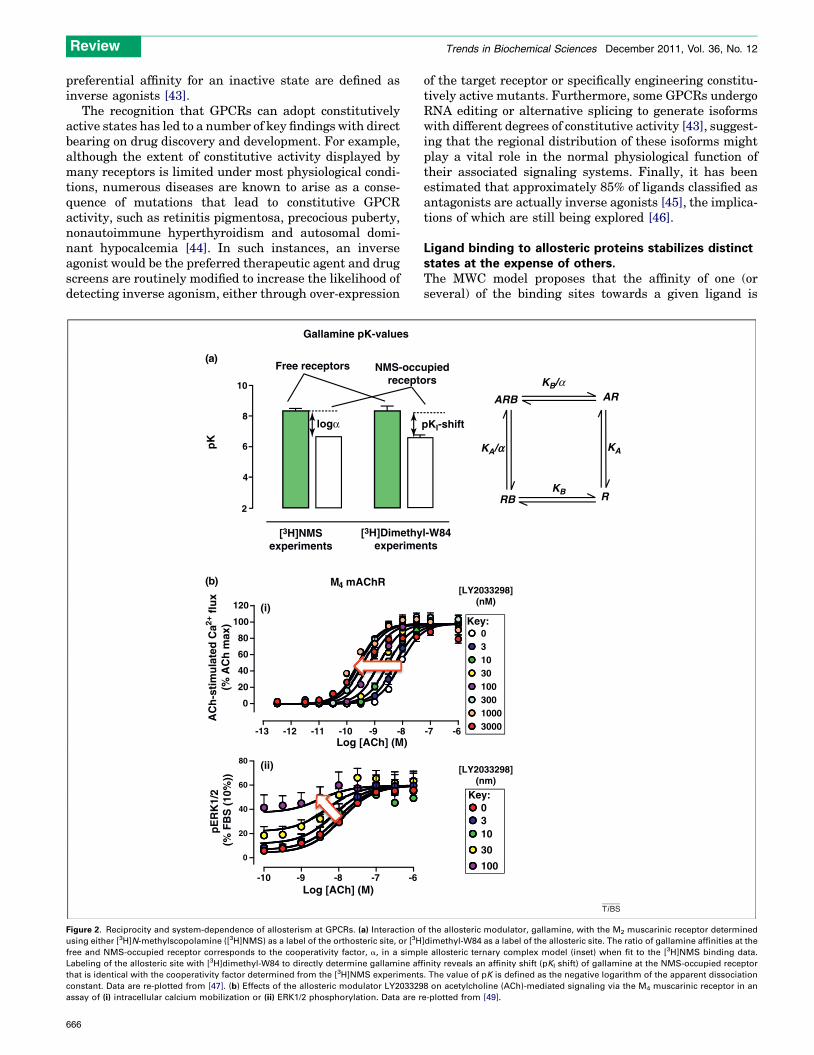
Figure 2. Reciprocity and system-dependence of allosterism at GPCRs. (a) Interaction o
using either [3H]N-methylscopolamine ([3H]NMS) as a label of the orthosteric site, or [3H
free and NMS-occupied receptor corresponds to the cooperativity factor, a, in a simp
Labeling of the allosteric site with [3H]dimethyl-W84 to directly determine gallamine af
that is identical with the cooperativity factor determined from the [3H]NMS experiments
constant. Data are re-plotted from [47]. (b) Effects of the allosteric modulator LY203329
assay of (i) intracellular calcium mobilization or (ii) ERK1/2 phosphorylation. Data are r
666
of the target receptor or specifically engineering constitu-tively active mutants. Furthermore, some GPCRs undergoRNA editing or alternative splicing to generate isoformswith different degrees of constitutive activity [43], suggest-ing that the regional distribution of these isoforms mightplay a vital role in the normal physiological function oftheir associated signaling systems. Finally, it has beenestimated that approximately 85% of ligands classified asantagonists are actually inverse agonists [45], the implica-tions of which are still being explored [46].
Ligand binding to allosteric proteins stabilizes distinctstates at the expense of others.The MWC model proposes that the affinity of one (orseveral) of the binding sites towards a given ligand is
0Key:
Key:
310
30
100
[LY2033298](nm)
RB
KA/α
ARB AR
R
KA
KB
KB/α
-7 -6
upiedrs
l-W84nts
pKI-shift
03103010030010003000
[LY2033298](nM)
TiBS
f the allosteric modulator, gallamine, with the M2 muscarinic receptor determined
]dimethyl-W84 as a label of the allosteric site. The ratio of gallamine affinities at the
le allosteric ternary complex model (inset) when fit to the [3H]NMS binding data.
finity reveals an affinity shift (pKI shift) of gallamine at the NMS-occupied receptor
. The value of pK is defined as the negative logarithm of the apparent dissociation
8 on acetylcholine (ACh)-mediated signaling via the M4 muscarinic receptor in an
e-plotted from [49].

Box 2. Stimulus bias at GPCRs with multiple endogenous
ligands
The existence of probe dependence and stimulus bias in the
interactions between GPCRs, orthosteric and allosteric ligands has
substantial implications with regards to physiology and drug
discovery. The chemokine receptor family provides one of the best
known examples of receptors with multiple endogenous ligands.
Chemokines and their receptors are central to the inflammatory
process and are attractive therapeutic targets. To date, 50 chemo-
kine ligands and 19 functional receptors have been described. Few
chemokine GPCRs bind a single ligand, and often a given ligand can
bind to several different receptors. However, such assessments are
merely descriptions of possible physical interactions, not explana-
tions of biological interactions in vivo. Indeed, an example
demonstrating that different chemokines binding to the same
receptor elicit different responses is provided by the CCR7 receptor
and its endogenous ligands CCL19 and CCL21 [73]. These two
ligands differ in terms of the GRK isoform they activate; CCL19
activates both GRK3 and GRK6, whereas CCL21 activates only GRK6.
Functionally, such differential GRK activation leads to differential
receptor phophorylation and desensitization patterns. The Family B
GLP-1 receptor (GLP-1R) is another example of a GPCR that binds
multiple endogenous ligands. This GPCR is a key regulator of insulin
secretion and a major therapeutic target for the treatment of
diabetes. GLP-1R responds to at least four distinct endogenous
GLP-1 variants (the full-length peptide GLP-1 (1-37), the truncated
form GLP-1 (7-37) and the corresponding amidated counterparts) as
well as to the related peptide oxyntomodulin. Recently, stimulus
bias has been identified in the signaling profile of these orthosteric
peptides [74]. Importantly, the same study found distinct (probe-
dependent) pharmacological profiles when investigating the allos-
teric modulation of the GLP-1R by a synthetic small molecule
modulator and quercetin, a naturally occurring flavonol [74]. These
recent observations illustrate the importance of orthosteric probe
selection when screening for allosteric modulators in drug dis-
covery programs.
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
altered when a transition occurs from one state to another;ligands then have a selective bias towards one state rela-tive to another. Conformational selection by one type ofligand (e.g. orthosteric) in the presence of a second type(e.g. allosteric) will manifest as a change in the macroscopicbinding or functional properties of each ligand. At equilib-rium, such interactions are reciprocal and should thusbe evident irrespective of which site on the receptor(orthosteric or allosteric) is being probed experimentally(Figure 2a) [47].
Another important consequence of the MWC model thatis often unappreciated in the GPCR field is the expectationthat all ligands, whether they bind to orthosteric or allo-steric sites, should display some degree of either agonismor inverse agonism depending on whether they selectactive or inactive receptor states [22,48]. Yet, this is notalways observed. One explanation is that the observedpharmacological behavior of a ligand is as much a propertyof the experimental assay system as it is the molecularproperty of the compound. Orthosteric sites have likelyevolved to more efficiently facilitate ligand-mediatedtransition between receptor states, whereas the energybarriers can be higher for allosteric ligands; if the effectis small, it may simply not be detected due to assaythreshold limitations. However, in an over-expressedreceptor system, or an assay monitoring a sensitive oramplified response, low level allosteric agonism can bereadily unmasked. For example, the allosteric modulatorLY2033298 (Figure 2b) displays only positive modulationof acetylcholine function at the M4muscarinic receptor in acalcium mobilization assay, whereas it is both a robustallosteric agonist and a positive modulator of acetylcholinein an assay monitoring M4 receptor-mediated ERK1/2phosphorylation [49].
The presence of additional receptor states can alsoaccount for deviations from the expected behaviordescribed above. Indeed, the notion that GPCRs adoptmultiple biologically relevant active states has becomewidespread in recent years owing to the accumulation ofdata indicating that a given ligand at the same receptorcan mediate markedly different functional outcomesdepending on the signal pathway under investigation[50,51]. This phenomenon has been dubbed stimulus-bias,biased agonism or functional selectivity [52]. Mechanisti-cally, stimulus bias reflects the ability of ligands to selectcertain sets of GPCR active states at the expense of others.In accordance with the MWC principles, this bias is recip-rocal, in that it can also be imposed on the ligands by theintracellular effectors with which the GPCR can interact.For example, a recent study using purified b2 adrenergicreceptors reconstituted in high-density lipoprotein parti-cles found that, although the agonist isoproterenol promotedformation of receptor–Gs protein complexes and the inverseagonist ICI-118,551 prevented their formation, the inverseagonist had minimal effect on preformed receptor–Gs
complexes [53]. This finding indicates that the inverse ago-nist cannot bind to the preformed receptor–G protein com-plex because the G protein biases the receptor conformationaway from the inactive state towards the active state. Theconcept of stimulus bias has also been extended to ligand-selective activation of G protein-independent pathways
[51,54] and such observations are having a profound impacton GPCR-based drug discovery because they suggest thatultimate clinical efficacy might be determined by a certainmix of biased efficacies and that drug screens focusing on asingle functional endpoint will lead to optimization of anincorrect compound structure-activity series. Moreover, thefact that many GPCRs recognize multiple endogenousligands (Box 2) suggests that stimulus bias is a pharmaco-logical phenomenon and could play a vital role in physiologyas well.
Importantly, studies are beginning to more directlyvisualize the existence of multiple GPCR states. Examplesinclude spectroscopy-based measurements of environmen-tally sensitive fluorophores at the intracellular ends of aGPCR [55,56], BRET studies of receptor–G protein rear-rangements, [57], NMR studies of dynamic changes inGPCR extracellular regions in the presence of differentclasses of ligand [58] and the solution of high-resolutioncrystal structures of both inactive and active state receptor–ligand complexes [4,5,10,33–35]. The requirements for thestabilization of an active GPCR state are evident whencomparing the cytoplasmic ends of the b2 adrenergic recep-tor from the crystal structures solved of the receptor incomplex with an inverse agonist, an irreversible agonistor an agonist together with a nanobody that mimics theeffect of the G protein in promoting an active state (Figure 3).Of note, both the inverse agonist and irreversible (butpositive) agonist-bound states are virtually superimposable
667

β2-AR-NB80 (“Active”) β2-AR-FAUC50 (“Irreversible agonist”)
β2-AR-Cz (“Inactive”)
β2-ARβ2-ARβ2-AR
NB
TM6TM7
TM2
TM1
TM4
TM3
TM5
TiBS
11.4 Å
Figure 3. Stabilization of an active GPCR state requires both agonist and intracellular effector binding. Superimposed molecular models of the cytoplasmic surface of the b2
adrenergic receptor from high-resolution crystal structures of an inactive state bound to the inverse agonist carazolol (blue; PDB 2RH1), a state containing the high-efficacy,
irreversibly bound agonist FAUC50 (purple; PDB 3PDS) and a state bound to both a high-affinity, high-efficacy agonist (BI 167107) and a camelid nanobody (NB80) that
promotes an active state by mimicking the effects of the G protein (orange; PDB 3P0G). Note the large movement in TM6 associated with receptor activation is seen only
when both an agonist and a nanobody are bound to the receptor.
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
in these regions, indicating that stabilization of the activereceptor requires conformational selection by both theextracellular activating ligand and an intracellular effector.
Operational characteristics of allosteric interactions inGPCRsAlthough GPCRs fulfill some criteria for allosteric proteinsas described by the MWC model, the field is still largely at astage where the phenomenology exceeds detailed mecha-nistic and structural understanding. This is changing but,in the meantime, approaches by which the molecularproperties underlying allosteric GPCR modulation canbe operationally linked to observed behaviors in biologicalsystems remain necessary. These operational approachescannot be used to prove that an allosteric modulator fol-lows the MWC model at the molecular level, but provide auseful foundation for subsequent studies. In this regard,there are at least four key operational characteristics ofGPCR allostery:
Saturability of effect.
The most obvious consequence of an allosteric mechanismis a limit to the pharmacological effect mediated by anallosteric modulator depending on the cooperativitybetween orthosteric and allosteric sites. If the cooperativ-ity is limited, then the effect of the modulator is to essen-tially ‘fine-tune’ a physiological response in either apositive or negative direction to a new set-point whilemaintaining the spatial and temporal characteristics ofnatural endogenous signaling. This also represents a dis-tinct advantage in terms of reducing the risks associatedwith overdose when compared to orthosteric ligands [48].
668
Probe dependence
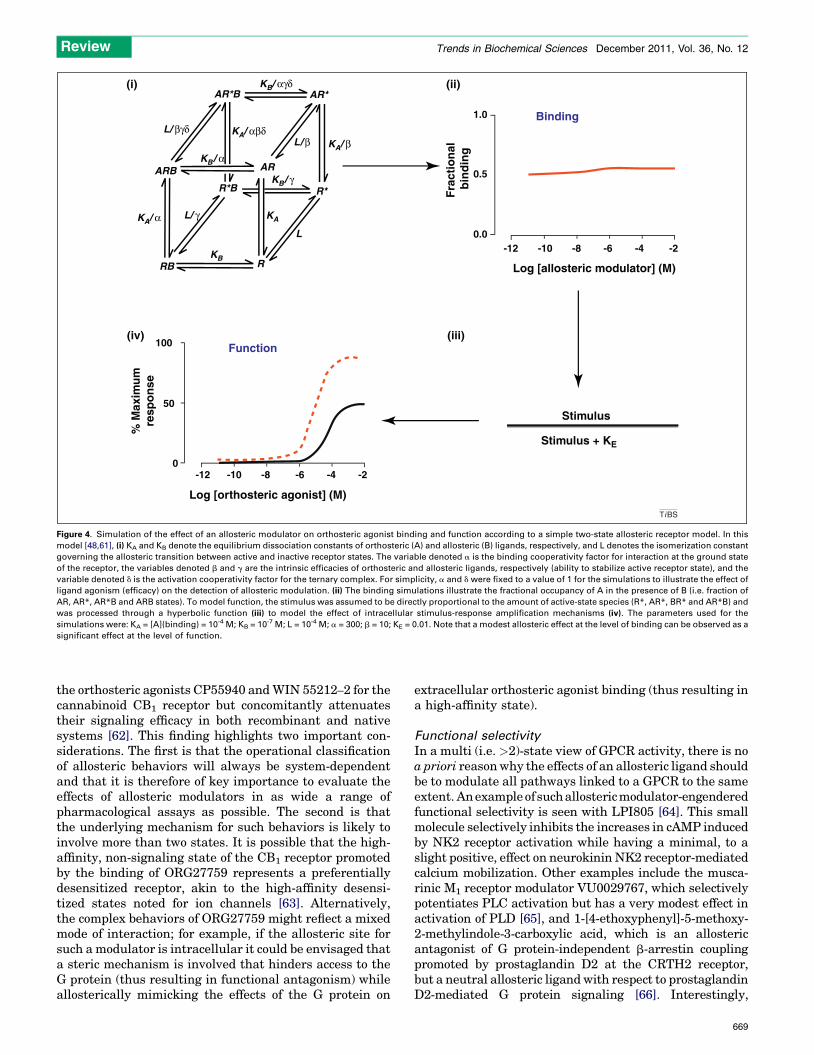
Probe dependence is a phenomenon whereby the allostericeffect can change depending on the nature of the orthostericligand used to ‘probe’ receptor function [32]. For example,the allosteric modulator of the chemokine CCR5 receptoraplaviroc can robustly inhibit the binding of the agonist[125I]MIP-1a (CCL3) but not that of the agonist[125I]RANTES (CCL5) [59], yet is an effective antagonistof both compounds in cell-based signaling assays. Anotherexample is that of the allosteric modulator of the M4musca-rinic receptor LY2033298, which shows robust potentiationof some muscarinic agonists, such as acetylcholine andoxotremorine, but not others, such as xanomeline [60].The simplest explanation for probe dependence in termsof the MWC model is that the degree of allosteric modulationwill correlate with the degree of agonism (positive orinverse) [11,48,61]. This can be illustrated by a theoreticalsimulation of the interaction between a weak agonist and anallosteric modulator within a simple allosteric two-statemodel (Figure 4); at the level of ligand binding, the interac-tion with the agonist appears almost neutral At the level ofligand function, however, positive allosteric modulation isevident owing to the additional influence of cellular ampli-fication signaling mechanisms, modeled in this instanceby assuming a hyperbolic stimulus-response relationshiplinking occupancy to observed response.
Differential modulation of ligand affinity and efficacy
The fact that GPCRs can adopt more than one biologicallyactive state highlights the potential for more complexmanifestations of allosteric behaviors. For example, theallosteric modulator ORG27759 enhances the affinity of
Binding
Function
1.0
KA/ β
KA
L
R*BKB/ γ
KB/ α
KB
KB/ αγδ
KA/ α
KA/ αβδ
L/ γ
L/ βγδ
ARB
AR*B
R*
RRB
AR
L/ β
AR*
0.5
0.0
100
50
0-12 -10 -8 -6 -4 -2
-12 -10 -8 -6 -4 -2
Log [allosteric modulator] (M)
Stimulus
Stimulus + KE
(i)
(iv) (iii)
(ii)
Log [orthosteric agonist] (M)
% M
axim
um
resp
on
se
Fra
ctio
nal
b
ind
ing
TiBS
Figure 4. Simulation of the effect of an allosteric modulator on orthosteric agonist binding and function according to a simple two-state allosteric receptor model. In this
model [48,61], (i) KA and KB denote the equilibrium dissociation constants of orthosteric (A) and allosteric (B) ligands, respectively, and L denotes the isomerization constant
governing the allosteric transition between active and inactive receptor states. The variable denoted a is the binding cooperativity factor for interaction at the ground state
of the receptor, the variables denoted b and g are the intrinsic efficacies of orthosteric and allosteric ligands, respectively (ability to stabilize active receptor state), and the
variable denoted d is the activation cooperativity factor for the ternary complex. For simplicity, a and d were fixed to a value of 1 for the simulations to illustrate the effect of
ligand agonism (efficacy) on the detection of allosteric modulation. (ii) The binding simulations illustrate the fractional occupancy of A in the presence of B (i.e. fraction of
AR, AR*, AR*B and ARB states). To model function, the stimulus was assumed to be directly proportional to the amount of active-state species (R*, AR*, BR* and AR*B) and
was processed through a hyperbolic function (iii) to model the effect of intracellular stimulus-response amplification mechanisms (iv). The parameters used for the
simulations were: KA = [A](binding) = 10-4 M; KB = 10-7 M; L = 10-4 M; a = 300; b = 10; KE = 0.01. Note that a modest allosteric effect at the level of binding can be observed as a
significant effect at the level of function.
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
the orthosteric agonists CP55940 and WIN 55212–2 for thecannabinoid CB1 receptor but concomitantly attenuatestheir signaling efficacy in both recombinant and nativesystems [62]. This finding highlights two important con-siderations. The first is that the operational classificationof allosteric behaviors will always be system-dependentand that it is therefore of key importance to evaluate theeffects of allosteric modulators in as wide a range ofpharmacological assays as possible. The second is thatthe underlying mechanism for such behaviors is likely toinvolve more than two states. It is possible that the high-affinity, non-signaling state of the CB1 receptor promotedby the binding of ORG27759 represents a preferentiallydesensitized receptor, akin to the high-affinity desensi-tized states noted for ion channels [63]. Alternatively,the complex behaviors of ORG27759 might reflect a mixedmode of interaction; for example, if the allosteric site forsuch a modulator is intracellular it could be envisaged thata steric mechanism is involved that hinders access to theG protein (thus resulting in functional antagonism) whileallosterically mimicking the effects of the G protein on
extracellular orthosteric agonist binding (thus resulting ina high-affinity state).
Functional selectivity
In a multi (i.e. >2)-state view of GPCR activity, there is noa priori reason why the effects of an allosteric ligand shouldbe to modulate all pathways linked to a GPCR to the sameextent. An example of such allosteric modulator-engenderedfunctional selectivity is seen with LPI805 [64]. This smallmolecule selectively inhibits the increases in cAMP inducedby NK2 receptor activation while having a minimal, to aslight positive, effect on neurokinin NK2 receptor-mediatedcalcium mobilization. Other examples include the musca-rinic M1 receptor modulator VU0029767, which selectivelypotentiates PLC activation but has a very modest effect inactivation of PLD [65], and 1-[4-ethoxyphenyl]-5-methoxy-2-methylindole-3-carboxylic acid, which is an allostericantagonist of G protein-independent b-arrestin couplingpromoted by prostaglandin D2 at the CRTH2 receptor,but a neutral allosteric ligand with respect to prostaglandinD2-mediated G protein signaling [66]. Interestingly,
669

Constitutive activity
Oligomerization
System-dependentallosteric modulation/agonism
Stimulus bias
AgonismQuiescent
system
Sensitivesystem
Inverse agonism
+ [Drug]
Log [drug]Log [drug]
R**
R
R*
A
AB
B
Log [orthosteric ligand]
+ Modulator
Basal
Res
po
nse
Res
po
nse
R**
Res
po
nse
R*
Res
po
nse
TiBS
Figure 5. Summary of GPCR paradigms that reflect characteristics shared with the MWC model for allosteric proteins. GPCRs can self-associate, possess multiple
topographically distinct binding sites and isomerize between multiple active (R* and R**) and inactive (R) states in the absence of ligand. These molecular properties can
give rise to distinct pharmacological paradigms, as given in purple.
Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
pathway-biased allosteric modulation might be involved insome pathophysiological conditions. For example, a case ofacquired hypocalciuric hypercalcemia has been reportedthat is mediated by an autoantibody to the extracellularcalcium-sensing GPCR that allosterically enhances Gq/11-mediated phosphoinositide accumulation but allostericallyinhibits Gi/o-mediated ERK1/2 phosphorylation [67]. Mostrecently, the meningogoccus Neisseria meningitides hasbeen reported to allosterically activate the b2 adrenergicreceptor through a b-arrestin-biased pathway in endothelialcells and, in doing so, increase cellular permeability to allowpassage of the infecting organism [68].
Concluding remarksAlthough the initial MWC model for allosteric proteins wasderived from observations of enzyme function, it is evidentthat GPCRs also fulfill some of the criteria for allostericproteins; surprisingly few studies have attempted to quan-titatively assess the pharmacological actions of GPCRallosteric ligands in terms of the key properties and pre-dictions of this model. Nonetheless, the main operationalcharacteristics that have been ascribed to GPCR allostericmodulators can be shown to have mechanistic correlateswithin the MWC scheme (Figure 5). However, the contem-porary view of GPCRs as dynamic entities that adoptmultiple biologically relevant conformations highlightsthe fact that the MWC model itself might need to beextended to accommodate the multi-state behavior thatis observed in this large protein family. It is envisaged that
670
a deeper understanding of the inter-relationships betweenquantitative allosteric receptor models and observed sys-tems behavior of allosteric ligands can facilitate structure-activity studies and novel drug discovery opportunities.
AcknowledgementsArthur Christopoulos is a Senior Research Fellow and Patrick M. Sextonis a Principal Research Fellow of the National Health and MedicalResearch Council (NHMRC) of Australia. Meritxell Canals is a MonashResearch Fellow. Work in the authors’ laboratory is funded, in part, byNHMRC Program grant no. 519461 (to A.C. and P.M.S.), NHMRC Projectgrant nos 545974 (to A.C.), APP1002180 (to P.M.S.) and APP1011796 (toM.C.), and Australian Research Council Discovery grant nosDP110100687 (toA.C.) and DP0985210 (to P.M.S.).
References1 Overington, J. et al. (2006) How many drug targets are there? Nat. Rev.
Drug Discov. 5, 993–9962 Chien, E. et al. (2010) Structure of the human dopamine D3 receptor in
complex with a D2/D3 selective antagonist. Science 330, 1091–10953 Shimamura, T. et al. (2011) Structure of the human histamine H1
receptor complex with doxepin. Nature 475, 65–704 Cherezov, V. et al. (2007) High-resolution crystal structure of an
engineered human beta2-adrenergic G protein-coupled receptor.Science 318, 1258–1265
5 Jaakola, V. et al. (2008) The 2.6 A crystal structure of a human A2Aadenosine receptor bound to an antagonist. Science 322, 1211–1217
6 Wu, B. et al. (2010) Structures of the CXCR4 chemokine GPCR withsmall-molecule and cyclic peptide antagonists. Science 330, 1066–1071
7 Rosenbaum, D.M. et al. (2011) Structure and function of an irreversibleagonist-b2 adrenoceptor complex. Nature 469, 236–240
8 Neubig, R. et al. (2003) International Union of PharmacologyCommittee on Receptor Nomenclature and Drug Classification.

Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
XXXVIII. Update on terms and symbols in quantitative pharmacology.Pharmacol. Rev. 55, 597–606
9 Conn, P. et al. (2009) Allosteric modulators of GPCRs: a novelapproach for the treatment of CNS disorders. Nat. Rev. Drug Discov.8, 41–54
10 Rasmussen, S. et al. (2011) Crystal structure of the b2 adrenergicreceptor–Gs protein complex. Nature DOI: 10.1038/nature10361
11 Monod, J. et al. (1965) On the nature of allosteric transitions: aplausible model. J. Mol. Biol. 12, 88–118
12 Monod, J. et al. (1963) Allosteric proteins and cellular control systems.J. Mol. Biol. 6, 306–329
13 Changeux, J.P. and Edelstein, S.J. (2005) Allosteric mechanisms ofsignal transduction. Science 308, 1424–1428
14 Changeux, J.P. and Edelstein, S.J. (1998) Allosteric receptors after 30years. Neuron 21, 959–980
15 Whorton, M. et al. (2007) A monomeric G protein-coupled receptorisolated in a high-density lipoprotein particle efficiently activates its Gprotein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687
16 Kuszak, A.J. et al. (2009) Purification and functional reconstitution ofmonomeric mu-opioid receptors: allosteric modulation of agonistbinding by Gi2. J. Biol. Chem. 284, 26732–26741
17 Kobilka, B.K. et al. (1988) Chimeric alpha 2-,beta 2-adrenergicreceptors: delineation of domains involved in effector coupling andligand binding specificity. Science 240, 1310–1316
18 Fung, J. et al. (2009) Ligand-regulated oligomerization of [beta]2-adrenoceptors in a model lipid bilayer. EMBO J. 28, 3315–
332819 Milligan, G. and Bouvier, M. (2005) Methods to monitor the
quaternary structure of G protein-coupled receptors. FEBS J. 272,2914–2925
20 Springael, J.Y. et al. (2007) Allosteric properties of G protein-coupledreceptor oligomers. Pharmacol. Ther. 115, 410–418
21 Han, Y. et al. (2009) Allosteric communication between protomers ofdopamine class A GPCR dimers modulates activation. Nat. Chem. Biol.5, 688–695
22 May, L. et al. (2007) Allosteric modulation of G protein-coupledreceptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51
23 Smith, N.J. and Milligan, G. (2010) Allostery at G protein-coupledreceptor homo- and heteromers: uncharted pharmacologicallandscapes. Pharmacol. Rev. 62, 701–725
24 Rondard, P. et al. (2008) Functioning of the dimeric GABA(B) receptorextracellular domain revealed by glycan wedge scanning. EMBO J. 27,1321–1332
25 Maurel, D. et al. (2008) Cell-surface protein-protein interactionanalysis with time-resolved FRET and snap-tag technologies:application to GPCR oligomerization. Nat. Methods 5, 561–567
26 Galvez, T. et al. (2001) Allosteric interactions between GB1 and GB2subunits are required for optimal GABA(B) receptor function. EMBO J.20, 2152–2159
27 Goudet, C. et al. (2005) Asymmetric functioning of dimericmetabotropic glutamate receptors disclosed by positive allostericmodulators. J. Biol. Chem. 280, 24380–24385
28 Ehlert, F.J. (1985) The relationship between muscarinic receptoroccupancy and adenylate cyclase inhibition in the rabbit myocardium.Mol. Pharmacol. 28, 410–421
29 Bockaert, J. et al. (2004) GPCR interacting proteins (GIP). Pharmacol.Ther. 103, 203–221
30 DeWire, S. et al. (2007) b-Arrestins and cell signaling. Annu. Rev.Physiol. 69, 483–510
31 Ritter, S. and Hall, R. (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830
32 Kenakin, T. (2009) 7TM receptor allostery: putting numbers toshapeshifting proteins. Trends Pharmacol. Sci. 30, 460–469
33 Rasmussen, S.G.F. et al. (2011) Structure of a nanobody-stabilizedactive state of the b2 adrenoceptor. Nature 469, 175–180
34 Xu, F. et al. (2011) Structure of an agonist-bound human A2Aadenosine receptor. Science 332, 322–327
35 Scheerer, P. et al. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502
36 Gregory, K. et al. (2010) Identification of orthosteric and allostericsite mutations in M2 muscarinic acetylcholine receptors thatcontribute to ligand-selective signaling bias. J. Biol. Chem. 285,7459–7474
37 Avlani, V.A. et al. (2010) Orthosteric and allosteric modes of interactionof novel selective agonists of the M1 muscarinic acetylcholine receptor.Mol. Pharmacol. 78, 94–104
38 Jensen, P.C. et al. (2008) Positive versus negative modulation of differentendogenous chemokines for CC-chemokine receptor 1 by small moleculeagonists through allosteric versus orthosteric binding. J. Biol. Chem.283, 23121–23128
39 Knoflach, F. et al. (2001) Positive allosteric modulators of metabotropicglutamate 1 receptor: characterization, mechanism of action, andbinding site. Proc. Natl. Acad. Sci. U.S.A. 98, 13402–13407
40 Hay, D.L. et al. (2006) Determinants of 1-piperidinecarboxamide, N-[2-[[5-amino-l-[[4-(4-pyridinyl)-l-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-dibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazolinyl) (BIBN4096BS) affinity for calcitonin gene-related peptide and amylin receptors–the role of receptor activitymodifying protein 1. Mol. Pharmacol. 70, 1984–1991
41 Costa, T. and Herz, A. (1989) Antagonists with negative intrinsicactivity at delta opioid receptors coupled to GTP-binding proteins.Proc. Natl. Acad. Sci. U.S.A. 86, 7321–7325
42 Costa, T. and Cotecchia, S. (2005) Historical review: negative efficacyand the constitutive activity of G-protein-coupled receptors. TrendsPharmacol. Sci. 26, 618–624
43 Stewart, G. et al. (2011) Inverse agonists. In Encyclopedia ofPsychopharmacology (Stolerman, I.P., ed.), pp. 661–669, Springer
44 Smit, M.J. et al. (2007) Pharmacogenomic and structural analysis ofconstitutive g protein-coupled receptor activity. Annu. Rev. Pharmacol.Toxicol. 47, 53–87
45 Kenakin, T. (2004) Allosteric modulators: the new generation ofreceptor antagonist. Mol. Interv. 4, 222–229
46 Parra, S. and Bond, R.A. (2007) Inverse agonism: from curiosity toaccepted dogma, but is it clinically relevant? Curr. Opin. Pharmacol. 7,146–150
47 Trankle, C. et al. (1999) Using a radioalloster to test predictions of thecooperativity model for gallamine binding to the allosteric site ofmuscarinic acetylcholine M(2) receptors. Mol. Pharmacol. 56, 962–
96548 Keov, P. et al. (2011) Allosteric modulation of G protein-coupled
receptors: a pharmacological perspective. Neuropharmacology 60,24–35
49 Leach, K. et al. (2009) Molecular mechanisms of action and invivo validation of an M4 muscarinic acetylcholine receptorallosteric modulator with potential antipsychotic properties.Neuropsychopharmacology 35, 855–869
50 Azzi, M. et al. (2003) b-Arrestin-mediated activation of MAPKby inverse agonists reveals distinct active conformations for Gprotein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 100,11406–11411
51 Wisler, J.W. et al. (2007) A unique mechanism of b-blocker action:carvedilol stimulates b-arrestin signaling. Proc. Natl. Acad. Sci. U.S.A.104, 16657–16662
52 Stallaert, W. et al. (2011) Ligand functional selectivity andquantitative pharmacology at G-protein-coupled receptors. Exp.Opin. Drug Discov. 6, 811–825
53 Yao, X.J. et al. (2009) The effect of ligand efficacy on the formation andstability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. U.S.A.106, 9501–9506
54 Pierce, K. and Lefkowitz, R. (2001) Classical and new roles ofb-arrestins in the regulation of G-protein-coupled receptors. Nat.Rev. Neurosci. 2, 727–733
55 Peleg, G. et al. (2001) Single-molecule spectroscopy of the b(2)adrenergic receptor: observation of conformational substates in amembrane protein. Proc. Natl. Acad. Sci. U.S.A. 98, 8469–8474
56 Ghanouni, P. et al. (2001) Agonist-induced conformational changes inthe G-protein-coupling domain of the b2 adrenergic receptor. Proc.Natl. Acad. Sci. U.S.A. 98, 5997–6002
57 Gales, C. et al. (2006) Probing the activation-promoted structuralrearrangements in preassembled receptor-G protein complexes. Nat.Struct. Mol. Biol. 13, 778–786
58 Bokoch, M. et al. (2010) Ligand-specific regulation of the extracellularsurface of a G-protein-coupled receptor. Nature 463, 108–112
59 Watson, C. et al. (2005) The CCR5 receptor-based mechanism of actionof 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol.Pharmacol. 67, 1268–1282
671

Review Trends in Biochemical Sciences December 2011, Vol. 36, No. 12
60 Suratman, S. et al. (2011) Impact of species variability and ‘‘probe-dependence’’ on the detection and in vivo validation of allostericmodulation at the M4 muscarinic acetylcholine receptor. Br. J.Pharmacol. 162, 1659–1670
61 Hall, D.A. (2000) Modeling the functional effects of allostericmodulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol. Pharmacol. 58, 1412–1423
62 Price, M.R. (2005) Allosteric modulation of the cannabinoid CB1receptor. Mol. Pharmacol. 68, 1484–1495
63 Taly, A. et al. (2009) Nicotinic receptors: allosteric transitions andtherapeutic targets in the nervous system. Nat. Rev. Drug Discov. 8,733–750
64 Maillet, E.L. et al. (2007) A novel, conformation-specific allostericinhibitor of the tachykinin NK2 receptor (NK2R) with functionallyselective properties. FASEB J. 21, 2124–2134
65 Marlo, J. et al. (2009) Discovery and characterization of novel allostericpotentiators of M1 muscarinic receptors reveals multiple modes ofactivity. Mol. Pharmacol. 75, 577–588
66 Mathiesen, J. et al. (2005) Identification of indole derivatives exclusivelyinterfering with a G protein-independent signaling pathway of theprostaglandin D2 receptor CRTH2. Mol. Pharmacol. 68, 393–402
67 Makita, N. et al. (2007) An acquired hypocalciuric hypercalcemiaautoantibody induces allosteric transition among active human
672
Ca-sensing receptor conformations. Proc. Natl. Acad. Sci. U.S.A.104, 5443–5448
68 Coureuil, M. et al. (2010) Meningococcus hijacks a b2-adrenoceptor/b-arrestin pathway to cross brain microvasculature endothelium. Cell143, 1149–1160
69 Andrews, G. et al. (2008) An intracellular allosteric site for a specificclass of antagonists of the CC chemokine G protein-coupled receptorsCCR4 and CCR5. Mol. Pharmacol. 73, 855–867
70 Salchow, K. et al. (2010) A common intracellular allosteric binding sitefor antagonists of the CXCR2 receptor. Br. J. Pharmacol. 159,1429–1439
71 Tressel, S.L. et al. (2011) Pharmacology, biodistribution, and efficacy ofGPCR-based pepducins in disease models. Methods Mol. Biol. 683,259–275
72 Sevigny, L.M. et al. (2011) Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc. Natl. Acad.Sci. U.S.A. 108, 8491–8496
73 Whalen, E.J. et al. (2011) Therapeutic potential of b-arrestin- andG protein-biased agonists. Trends Mol. Med. 17, 126–139
74 Koole, C. et al. (2010) Allosteric ligands of the glucagon-like peptide 1receptor (GLP-1R) differentially modulate endogenous and exogenouspeptide responses in a pathway-selective manner: implications fordrug screening. Mol. Pharmacol. 78, 456–465
Recommended

















