
J Clin Pathol 1983;36:1298-1301
A case of longstanding primary sclerosing cholangitisBM GOUDIE, GG BIRNIE, G WATKINSON, RNM MACSWEEN
From the Gartnavel General Hospital, and the University Department of Pathology, Western Infirmary,Glasgow
SUMMARY A 45-year-old man is described in whom there is currently ERCP and histologicalevidence of primary sclerosing cholangitis (PSC). A liver biopsy obtained 29 years ago showssimilar histological features confirming that he had PSC at that time. This case indicates that PSCmay follow a relatively benign course.
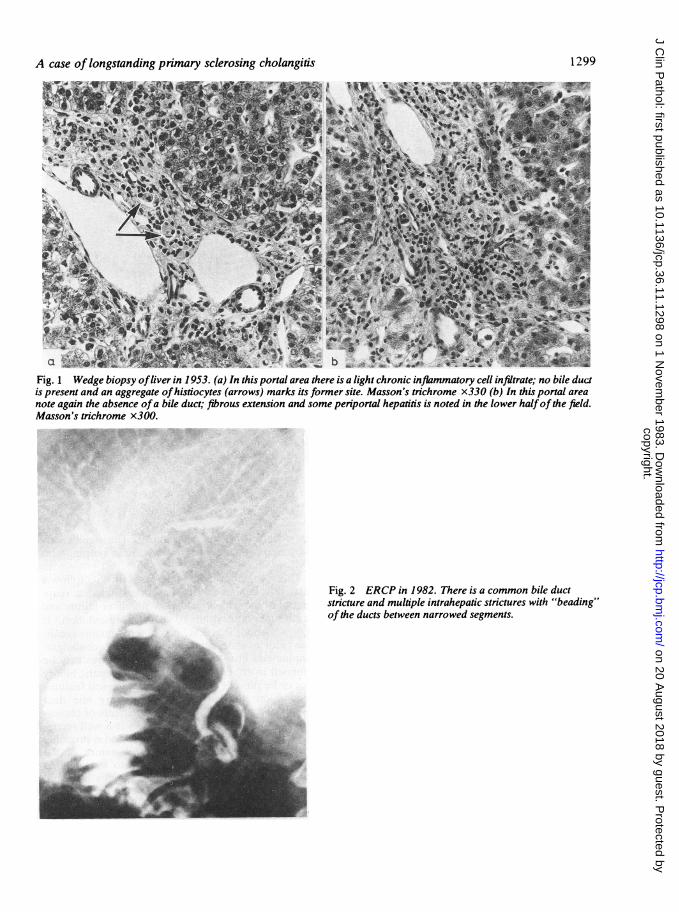
Case reportIn 1952 this patient presented at the age of 16 yrwith a two-year history of diarrhoea and crampinglower abdominal pain. He was found to be clinicallyanaemic, physically and sexually underdevelopedand to have liver enlargement. Investigationconfirmed an iron deficiency anaemia due to occultgastrointestinal blood loss but barium enema andsigmoidoscopy were normal. Associated liver dis-ease was suggested by an increase of the alkalinephosphate to 28 King Armstrong Units (KAU)-normal range s 25 KAU in this age group. A yearlater his general condition had not improved, theliver was still enlarged and the alkaline phosphatasewas still raised (30 KAU). Diagnostic laparotomywas undertaken and revealed an enlarged scarredliver. The extra-hepatic biliary tree was found to becompletely normal with no evidence of gallstones.No operative procedure was attempted on the com-mon bile duct and cholangiography was not per-formed. A wedge biopsy of liver was taken. Thisshowed some portal fibrosis and a few attenuatedportal-portal fibrous septa. There was a light chronicinflammatory cell infiltrate in the portal tracts(Fig. 1); the significant observation was a definitepaucity of small bile ducts, small histiocytic aggre-gates being present at their previous site (Fig. la);some mild focal periportal inflammation was notedin some areas (Fig. lb) and there were smallperiportal accumulations of copper-associated pro-tein demonstrable on the orcein stain.Three years later he developed severe diarrhoea
and rectal bleeding. Sigmoidoscopy and bariumenema showed typical features of ulcerative colitis.Despite an initial improvement with steroid therapyhis condition deteriorated, and necessitated electivecolectomy with ileorectal anastomosis in April 1957.
Accepted for publication 21 June 1983
The alkaline phosphatase was 56 KAU at this time.Some months later he developed paralyticpoliomyelitis which produced a residual paraplegia.For the next eight years he remained generallywell-adapting to his neurological deficit and matur-ing normally. He trained as a civil servant and worksto the present day. In 1965 he had a recurrence ofdiarrhoea and underwent anterior resection of therectum. Histological examination confirmed con-tinuing active ulcerative colitis.He remained well until 1981 when he developed
painless jaundice with the passage of pale stools anddark urine. He had no fever. On clinical examina-tion there was hepatosplenomegaly but no stigmataof chronic liver disease. The liver function tests wereabnormal: bilirubin 50 ,molV1, alkaline phosphatase169 IU/I (normal range 35-130 IU/l). This episodeof jaundice resolved spontaneously but he continuedto have raised alkaline phosphatase (360 IU/l) andtransaminase activities (serum AST 76 U/l, serumALT 149 U/l). The serum albumin remainednormal. Serological testing for antimitochondrialantibody was negative, the IgM level was normaland there was no eosinophilia.An ERCP (Fig. 2) demonstrated a stricture in the
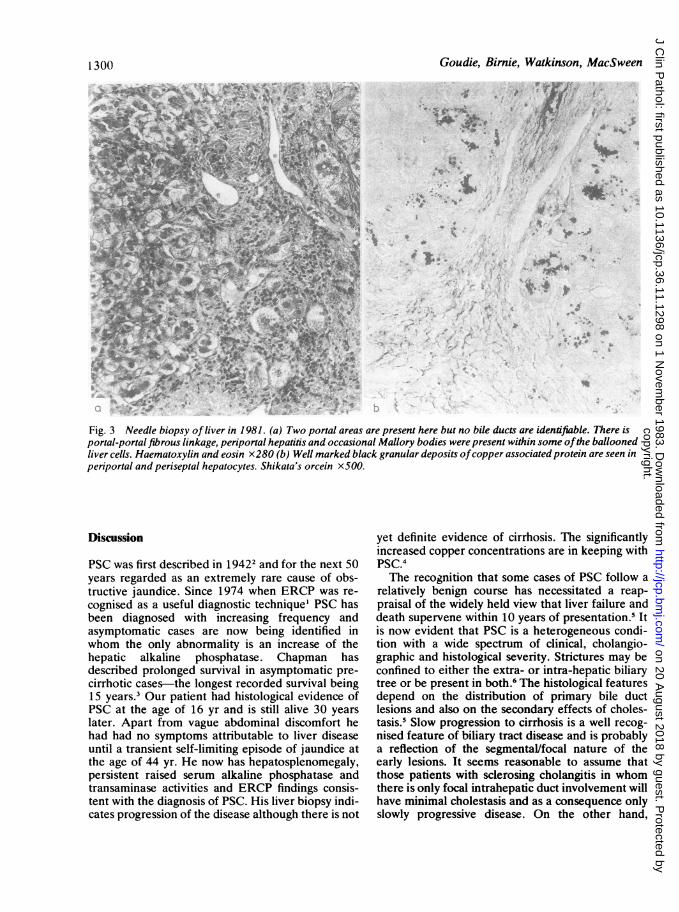
common bile duct with widespread stricturing andbeading of the intra-hepatic biliary tree-featureswhich are characteristic of sclerosing cholangitis.' Apercutaneous liver biopsy showed more marked por-tal tract fibrosis with fibrous septum formation but adiagnosis of cirrhosis could not be made with cer-tainty. In the majority of portal tracts no bile ductscould be identified; there was well marked peripor-tal hepatitis with ballooning of liver cells, occasionalMallory bodies and heavy deposits of copper associ-ated protein were demonstrated on the orcein stain(Fig. 3). The liver copper content was estimated byneutron activation analysis and found to be 18 timesnormal at 440 ppm (dryweight).
1298
copyright. on 20 A
ugust 2018 by guest. Protected by
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.36.11.1298 on 1 N
ovember 1983. D
ownloaded from
A case of longstanding primary sclerosing cholangitis
Fig. 1 Wedge biopsy ofliver in 1953. (a) In this portal area there is a light chronic inflammatory cell infiltrate; no bile ductis present and an aggregate ofhistiocytes (arrows) marks its former site. Masson's trichrome x330 (b) In this portal areanote again the absence ofa bile duct; fibrous extension and some periportal hepatitis is noted in the lower halfof the field.Masson's trichrome x300.
Fig. 2 ERCP in 1982. There is a common bile ductstricture and multiple intrahepatic strictures with "beading"ofthe ducts between narrowed segments.
1299
copyright. on 20 A
ugust 2018 by guest. Protected by
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.36.11.1298 on 1 N
ovember 1983. D
ownloaded from
Goudie, Birnie, Watkinson, MacSween
b4' F$~'
V.S
Jo ,
1, i
As4e *
We' 8f tr
.* ;.*..-.*-.,-
Fig. 3 Needle biopsy ofliver in 1981. (a) Two portal areas are present here but no bile ducts are identifiable. There isportal-portal fibrous linkage, periportal hepatitis and occasional Mallory bodies were present within some ofthe balloonedliver cells. Haematoxylin and eosin x280 (b) Well marked black granular deposits ofcopper associated protein are seen inperiportal and periseptal hepatocytes. Shikata's orcein x500.
Discussion
PSC was first described in 19422 and for the next 50years regarded as an extremely rare cause of obs-tructive jaundice. Since 1974 when ERCP was re-
cognised as a useful diagnostic technique' PSC hasbeen diagnosed with increasing frequency andasymptomatic cases are now being identified inwhom the only abnormality is an increase of thehepatic alkaline phosphatase. Chapman hasdescribed prolonged survival in asymptomatic pre-cirrhotic cases-the longest recorded survival being15 years.3 Our patient had histological evidence ofPSC at the age of 16 yr and is still alive 30 yearslater. Apart from vague abdominal discomfort hehad had no symptoms attributable to liver diseaseuntil a transient self-limiting episode of jaundice atthe age of 44 yr. He now has hepatosplenomegaly,persistent raised serum alkaline phosphatase andtransaminase activities and ERCP findings consis-tent with the diagnosis of PSC. His liver biopsy indi-cates progression of the disease although there is not
yet definite evidence of cirrhosis. The significantlyincreased copper concentrations are in keeping withPSC.4The recognition that some cases of PSC follow a
relatively benign course has necessitated a reap-praisal of the widely held view that liver failure anddeath supervene within 10 years of presentation.5 Itis now evident that PSC is a heterogeneous condi-tion with a wide spectrum of clinical, cholangio-graphic and histological severity. Strictures may beconfined to either the extra- or intra-hepatic biliarytree or be present in both.6 The histological featuresdepend on the distribution of primary bile ductlesions and also on the secondary effects of choles-tasis.5 Slow progression to cirrhosis is a well recog-nised feature of biliary tract disease and is probablya reflection of the segmental/focal nature of theearly lesions. It seems reasonable to assume thatthose patients with sclerosing cholangitis in whomthere is only focal intrahepatic duct involvement willhave minimal cholestasis and as a consequence onlyslowly progressive disease. On the other hand,
1300
i"Is
.i 4 N 'r, .
I
`
6
. X
*o- .
4 v
44, .. 4. . ." I
.41
copyright. on 20 A
ugust 2018 by guest. Protected by
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.36.11.1298 on 1 N
ovember 1983. D
ownloaded from

A case of longstanding primary sclerosing cholangitis 1301
patients in whom there is marked cholestasis, as theresult of widespread intrahepatic or major duct stric-tures, would be expected to sustain more severehepatocellular damage and to have a poorer prog-nosis. The activity of an associated colitis does notappear to be related to the progression of PSC andcolectomy does not confer protection against itsdevelopment.4 At the present time there isinsufficient information about the aetiology andnatural history of the disease to permit prediction ofprognosis in individual patients.The inability to anticipate the course of disease in
asymptomatic cases has important implicationswhere the use of potentially toxic drugs is being con-sidered. Clearly any therapeutic trial must be con-ducted on a carefully controlled basis.
We wish to thank Mr Gordon Gillespie for perform-ing the ERCP.
References
Elias E, Summerfield JA, Dick K, Sherlock S. Endoscopicretrograde cholangio-pancreatography in the diagnosis ofjaundice associated with ulcerative colitis. Gastroenteirology1974;67:907-11.
2 Delbet P. Retrecissement du choledoque: Cholecysto-duodenostomie. Bull Mem Soc Nation Chirurg1924;50:1144-6.
3Chapman RWG, Burroughs AK, Bass NM, Sherlock S. Long-standing asymptomatic primary sclerosing cholangitis: Reportof three cases. Dig Dis Sci 1981;26:778-82.
4 Chapman RWG, Arborgh BAM, Rhodes JM, et al. Primarysclerosing cholangitis: A review of its features, cholangiogra-phy and hepatic histology. Gut 1980;21:870-7.
5 Lefkowitch JH. Primary sclerosing cholangitis. Arch Intern Med1982;142:1157-60.
6 Wiesner RH, La Russo NF. Clinicopathologic features of thesyndrome of primary sclerosing cholangitis. Gastroenterology1980;79:200-6.
Requests for reprints to: Professor RNM MacSween, Uni-versity Department of Pathology, Western Infirmary,Glasgow GI1 6NT, Scotland.
copyright. on 20 A
ugust 2018 by guest. Protected by
http://jcp.bmj.com
/J C
lin Pathol: first published as 10.1136/jcp.36.11.1298 on 1 N
ovember 1983. D
ownloaded from
Recommended

















