Embed Size (px)
Citation preview

Yoshida Lab
M1 Yoshitaka Mino
Electronic structure of La2-
xSrxCuO4 calculated by self-interaction correction method

CONTENTS
Computational Materials Design First-principles calculation Local Density Approximation (LDA) Self-Interaction Correction (SIC) Previous study and my work Summary

COMPUTATIONAL MATERIALS DESIGN
Calculation & Simulation
Physical properties
New ideas
Experiments

FIRST-PRINCIPLES CALCULATION
Predict physical properties of materials ← Input parameters: Atomic Number and Atomic position !
Advantage Low costs Extreme conditions Ideal environment …
・・・

KOHN-SHAM THEORYWe map a many body problem on one electron problem with effective potential.
v(r)
ψi(r)
Kohn-Sham equation

LOCAL DENSITY APPROXIMATION (LDA)
We do not know the Exc and μxc and we need approximate expressions of them to perform electronic structure calculations.
For a realistic approximation, we refer homogeneous electron gas.
Exchange correlation energy
Exchange correlation energy of homogeneous electron gas
with the density at that position.
When the electron density changes in the space, we assume that the change is moderate and the electron density is locally homogeneous.
Local Density Approximation (LDA)

SUCCESS OF LDA
For almost of all materials, the LDA can describe electronic structures reasonably !
Calculated atomic volume (lattice constant) as a function of atomic number.
笠井英明,赤井久純,吉田博 編 ; 「計算機マテリアルデザイン」(大阪大学出版会)
O
Etotal
r(lattice constant)
a
Emin

SYSTEMATIC ERROR OF LDA
LDA has some errors in predicting material properties.
Underestimation of lattice constant.
Overestimation of cohesion energy.
Overestimation of bulk modulus.
Underestimation of band gap energy.
Predicting occupied localize states (d states) at too high energy.
...

PROBLEM IN LDA
J.P. Perdew, A. Zunger; Phys. Rev. B23 5048 (1981)
The exchange hole is distributed around the electron in the LDA. But in reality, the exchange hole is distributed around the nucleus.
The picture on the left shows the distribution of exchange hole at distance R from an electron which is located at r from the Ne nucleus.
r
-e
+e
electron
nuclear
exact
r
-e
+e
electron
nuclear
LDA
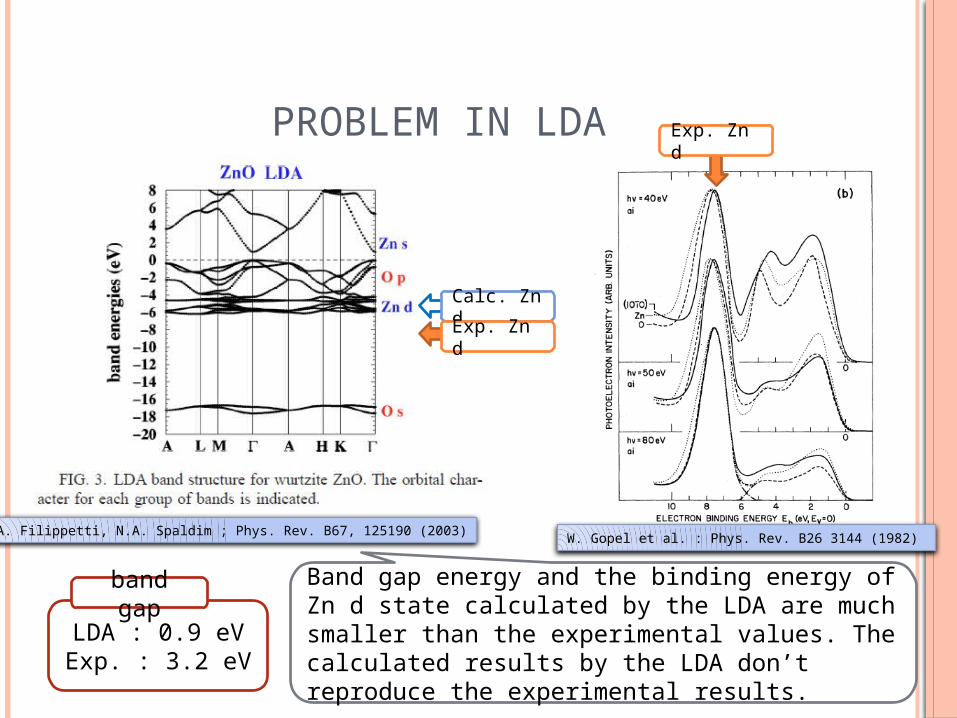
PROBLEM IN LDA
A. Filippetti, N.A. Spaldim ; Phys. Rev. B67, 125190 (2003) W. Gopel et al. : Phys. Rev. B26 3144 (1982)
LDA : 0.9 eVExp. : 3.2 eV
band gapBand gap energy and the binding energy of Zn d state calculated by the LDA are much smaller than the experimental values. The calculated results by the LDA don’t reproduce the experimental results.
Calc. Zn dExp. Zn d
Exp. Zn d

SELF-INTERACTION CORRECTION (SIC)
Coulomb interaction between electrons.
LDA
Self Coulomb interaction and self exchange correlation interaction don’t cancel each other perfectly.
exchange correlation energy
We need self-interaction correction (SIC) !

IMPROVEMENT BY SIC
J.P. Perdew, A. Zunger; Phys. Rev. B23 5048 (1981)
The SIC can reproduce exact solution reasonably.
The picture on the left shows the distribution of exchange hole located at R from an electron located at r from the nuclear about Ne.
r
-e
+e
electron
nuclear r
-e
+e
electron
nuclear
exact SIC

The result calculated by self-interaction correction (SIC) reproduces the experimental result of band gap and Zn d-state by photoemission spectroscopy much better than that of LDA.
LDA : 0.9 eVSIC : 3.5 eVExp. : 3.2 eV
band gap
Exp. Zn d
Calc. Zn d Exp. Zn
d
IMPROVEMENT BY SICA. Filippetti, N.A. Spaldim ; Phys. Rev. B67, 125190 (2003)A. Filippetti, N.A. Spaldim ; Phys. Rev. B67, 125190 (2003)
W. Gopel et al. : Phys. Rev. B26 3144 (1982)
Calc. Zn d
Exp. Zn d

PREVIOUS STUDY AND MY WORK
AFM : antiferromagnetism, PM : paramagnetism, SG : spin glass, I : insulator, M : metal, N : normal conductivity, SC : superconductivity, T : tetragonal, O : orthorhombic
Warren E. Pickett ; Rev. Mod. Phys. 61, 433 (1989)
La2CuO4
The pictures on the right is the phase diagram and crystal structure of La2-
xMxCuO4 which is produced experiments.

PREVIOUS STUDY AND MY WORK
The LDA predicts nonmagnetic and metallic ground state for La2CuO4.
I will reproduce the phase diagram of La2CuO4 from first-principles, particularly AFM and insulating region, by introducing SIC.
Experimentally, La2CuO4 is antiferromagnetic and insulating.The local magnetic moment is from 0.3 to 0.5 μB.
experimental results
calculated results
Warren E. Pickett ; Rev. Mod. Phys. 61, 433 (1989)
metal … Not corresponding
…

SUMMARY We can calculate electronic structures of many materials
from first-principles owing to the success of the local density approximation (LDA)
In general, the LDA is reasonable approximation for the exchange correlation potential.
The LDA results sometimes fails to reproduce experimental results, in particular when the system has moderately localized states due to the self-interaction.
So we introduce self-interaction correction (SIC) and the correction improve calculated results.
I will apply the SIC-LDA method to La2CuO4-based systems.

THE END . . .
THANK YOU FOR YOUR ATTENTION.



![MINO] - WordPress.com · JR Gifu Station → [JR Takayama Main Line・35 min] → Mino-Ota Station → [Nagaragawa Railway・37 min・¥1,130 in total] → Mino-shi Station With an](https://img.pdfslide.us/doc/110x75/60894f2505d04f710d20500e/mino-jr-gifu-station-a-jr-takayama-main-linef35-min-a-mino-ota-station.jpg)

























