Embed Size (px)
Citation preview

© Ste
fan
Hoh
man
n 20
00-
2004
YEAST GENETICS AND MOLECULAR BIOLOGY
The yeast Saccharomyces cerevisiae: habitate and use Other yeasts Yeast is a eukaryote: the yeast cell Yeast has a sexual cycle and an exciting sex life Yeast genetics: basics Yeast genetics: crossing yeast strains Yeast genetics: making mutants Cloning yeast genes: vectors Cloning yeast genes by complementation Deleting genes in yeast Smart gene deletions and transposon mutagenesis Getting further: more genes/proteins Model systems studied in yeast Yeast biotechnology

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast information resources WWW
There is unfortunately no real text book on yeast genetics and molecular biology Genetic Techniques for Biological Research by Corinne Michels gives a brief
overview on yeast genetics and summarises genetic approaches Yeast Gene Analysis by Brown and Tuite is a book about methods There are excellent resources on the WWW and many individual group pages with
interesting information and even movies! Check out the course link page For instance, there is kind of a text book on the Internet: http://
www.phys.ksu.edu/gene/chapters.html This site: http://genome-www.stanford.edu/Saccharomyces/VL-yeast.html links to
various types of basic information on yeast genetics This site links to more than 700 hundred yeast labs all over the world
http://genome-www.stanford.edu/Saccharomyces/yeastlabs.html The Stanford Saccharomyces Genome database under http://genome-
www.stanford.edu/Saccharomyces has information on all yeast genes including links and information to other yeast genome projects and global analysis projects

© Ste
fan
Hoh
man
n 20
00-
2004
The yeast Saccharomyces cerevisiae:habitate and use
Yeast lives on fruits, flowers and other sugar containing substrates Yeast copes with a wide range of environmental conditions:
Temperatures from freezing to about 55°C are tolerated Yeasts proliferate from 12°C to 40°C Growth is possible from pH 2.8-8.0 Almost complete drying is tolerated (dry yeast) Yeast can still grow and ferment at sugar concentrations of 3M (high osmoti pressure) Yeast can tolerate up to 20% alcohol
Saccharomyces cerevisiae is the main organism in wine productionbesides other yeasts; reason is the enormous fermentation capacity, low pH and high ethanol tolerance
Saccharomyces cerevisiae (carlsbergensis) is the beer yeastbecause it ferments sugar to alcohol even in the presence of oxygen, lager yeast ferments at 8°C
Saccharomyces cerevisiae is the yeast used in bakingbecause it produces carbon dioxide from sugar very rapidly
Saccharomyces cerevisiae is used to produce commercially important proteinsbecause it can be genetically engineered, it is regarded as safe and fermentation technology is highly advanced
Saccharomyces cerevisiae is used for drug screening and functional analysisbecause it is a eukaryote but can be handled as easily as bacteria
Saccharomyces cerevisiae is the most important eukaryotic cellular model systembecause it can be studied by powerful genetics and molecular and cellular biology; many important features of the eukaryotic cell have first been discovered in yeastHence S. cerevisiae is used in research that aims to find out features and mechanisms of the function of the living cell AND in to improve existing or to generate new biotechnological processes

© Ste
fan
Hoh
man
n 20
00-
2004
Other important yeasts
Schizosaccharomyces pombe, the fission yeast; important model organisms in molecular and cellular biology; used for certain fermentations
Kluyveromyces lactis, the milk yeast; model organismsome biotech importance due to lactose fermentation
Candida albicans, not a good model since it lacks a sexual cycle; but studied intensively because it is human pathogen
Saccharomyces carlsbergensis and Saccharomyces bayanus are species closely related to S. cerevisiae; brewing and wine making
Pichia stipidis, Hansenula polymorpha, Yarrovia lipolytica have smaller importance for genetic studies (specilaised features such as peroxisome biogenesis are studied), protein production hosts
Filamentous fungi, a large group of genetic model organisms in genera like Cryptococcus, Aspergillus, Neurospora...., biotechnological importance, includes human pathogens. Also S. cerevisiae can grow in a filamentous form.

© Ste
fan
Hoh
man
n 20
00-
2004
Saccharomyces cerevisiae is a eukaryote
Belongs to fungi, ascomycetes Unicellular organism with ability to produce
pseudohyphae S. cerevisiae divides by budding (hence:
budding yeast) while Schizosaccharomyces pombe divides by fission (hence: fission yeast)
Budding results in two cells of unequal size, a mother (old cell) and a daughter (new cell)
Yeast life is not indefinite; yeast cells age and mothers die after about 30-40 dividions
Cell has a eukaryotic structure with different organelles:
Cell wall consisting of glucans, mannans and proteins
Periplasmic space with hydrolytic enzymes Plasma membrane consisting of a phospholipid
bilayer and many different proteins Nucleus with nucleolus Vacuole as storage and hydrolytic organelle Secretory pathway with endoplasmic reticulum,
Golgi apparatus and secretory vesicles Peroxisomes for oxidative degradation Mitochondria for respiration
A yeast cells is about 4-7m largeThe ”eyes” at the bottom are bud scars
© Ste
fan
Hoh
man
n 20
00-
2004
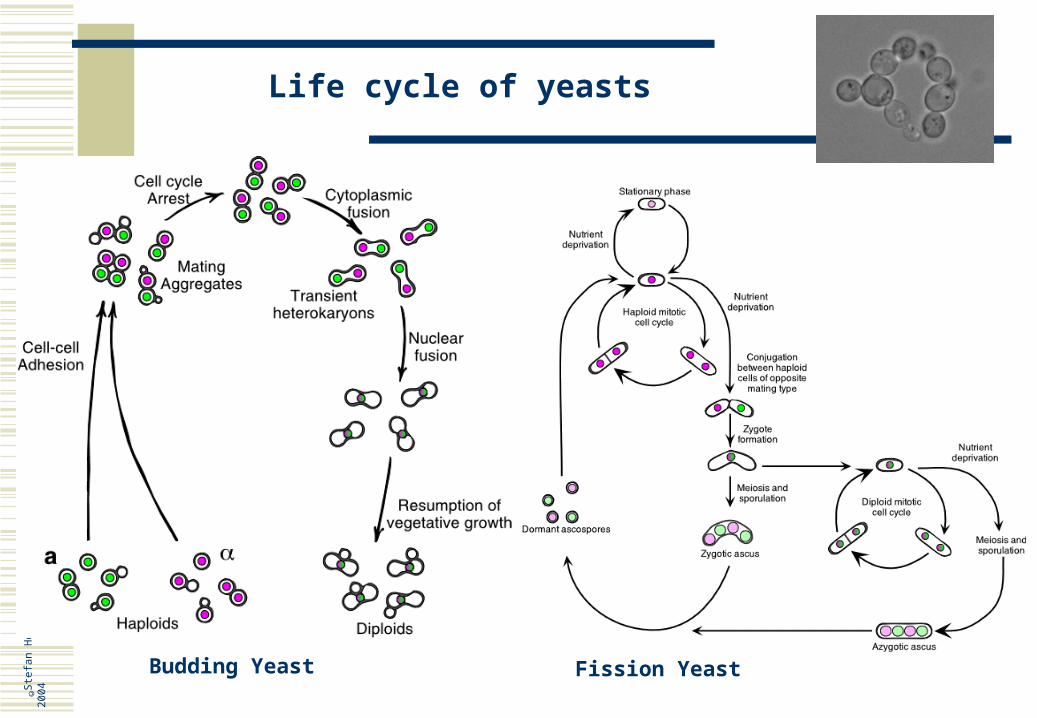
Life cycle of yeasts
Budding Yeast Fission Yeast
© Ste
fan
Hoh
man
n 20
00-
2004
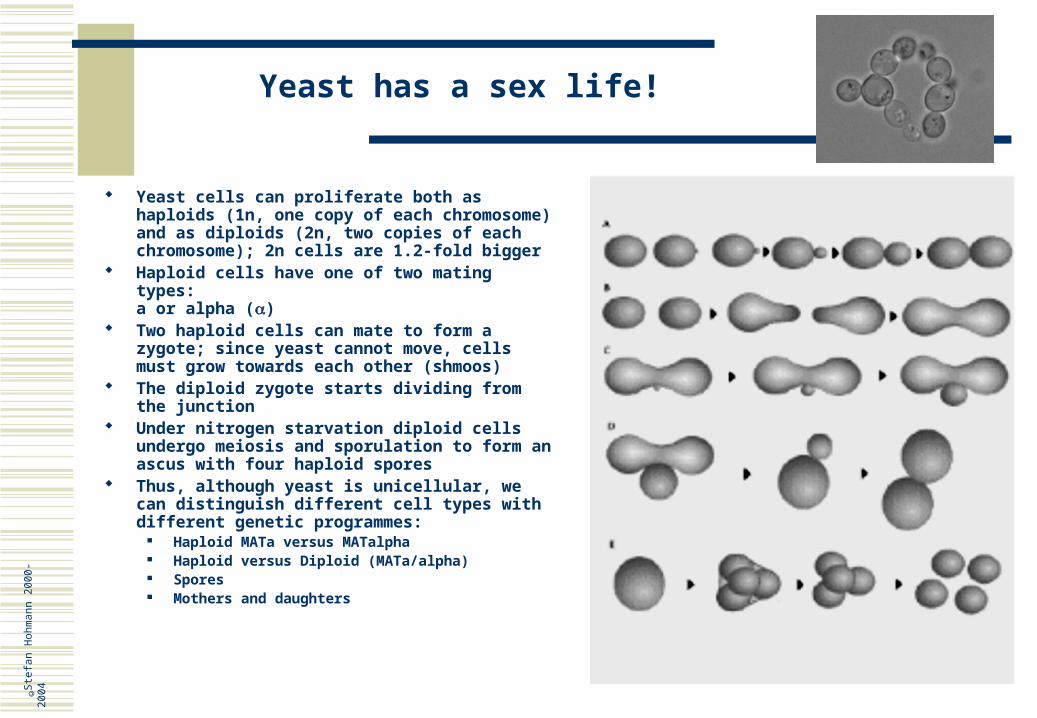
Yeast has a sex life!
Yeast cells can proliferate both as haploids (1n, one copy of each chromosome) and as diploids (2n, two copies of each chromosome); 2n cells are 1.2-fold bigger
Haploid cells have one of two mating types:a or alpha ()
Two haploid cells can mate to form a zygote; since yeast cannot move, cells must grow towards each other (shmoos)
The diploid zygote starts dividing from the junction
Under nitrogen starvation diploid cells undergo meiosis and sporulation to form an ascus with four haploid spores
Thus, although yeast is unicellular, we can distinguish different cell types with different genetic programmes:
Haploid MATa versus MATalpha Haploid versus Diploid (MATa/alpha) Spores Mothers and daughters
© Ste
fan
Hoh
man
n 20
00-
2004
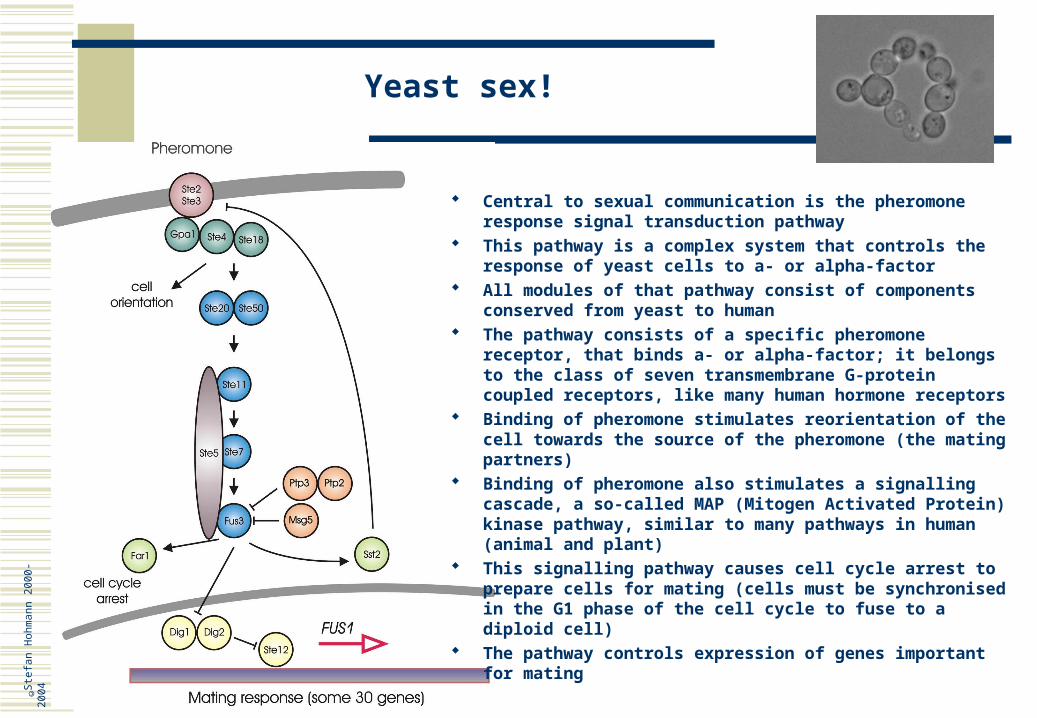
Yeast sex!
Central to sexual communication is the pheromone response signal transduction pathway
This pathway is a complex system that controls the response of yeast cells to a- or alpha-factor
All modules of that pathway consist of components conserved from yeast to human
The pathway consists of a specific pheromone receptor, that binds a- or alpha-factor; it belongs to the class of seven transmembrane G-protein coupled receptors, like many human hormone receptors
Binding of pheromone stimulates reorientation of the cell towards the source of the pheromone (the mating partners)
Binding of pheromone also stimulates a signalling cascade, a so-called MAP (Mitogen Activated Protein) kinase pathway, similar to many pathways in human (animal and plant)
This signalling pathway causes cell cycle arrest to prepare cells for mating (cells must be synchronised in the G1 phase of the cell cycle to fuse to a diploid cell)
The pathway controls expression of genes important for mating
© Ste
fan
Hoh
man
n 20
00-
2004
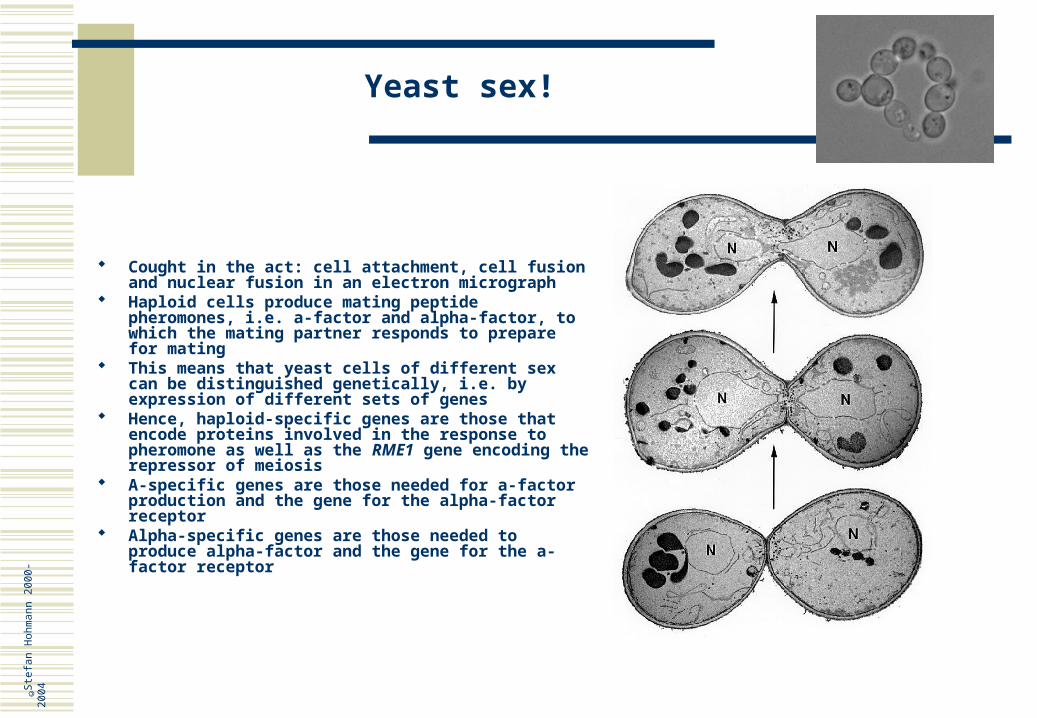
Yeast sex!
Cought in the act: cell attachment, cell fusion and nuclear fusion in an electron micrograph
Haploid cells produce mating peptide pheromones, i.e. a-factor and alpha-factor, to which the mating partner responds to prepare for mating
This means that yeast cells of different sex can be distinguished genetically, i.e. by expression of different sets of genes
Hence, haploid-specific genes are those that encode proteins involved in the response to pheromone as well as the RME1 gene encoding the repressor of meiosis
A-specific genes are those needed for a-factor production and the gene for the alpha-factor receptor
Alpha-specific genes are those needed to produce alpha-factor and the gene for the a-factor receptor
© Ste
fan
Hoh
man
n 20
00-
2004
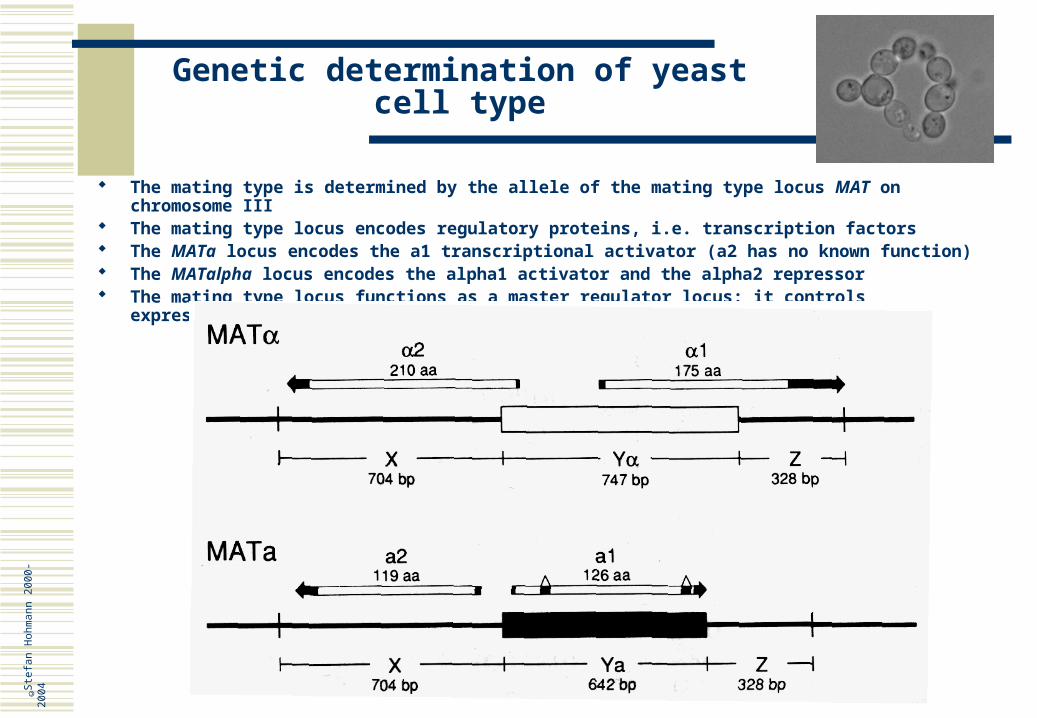
Genetic determination of yeast cell type
The mating type is determined by the allele of the mating type locus MAT on chromosome III The mating type locus encodes regulatory proteins, i.e. transcription factors The MATa locus encodes the a1 transcriptional activator (a2 has no known function) The MATalpha locus encodes the alpha1 activator and the alpha2 repressor The mating type locus functions as a master regulator locus: it controls expression of many
genes

© Ste
fan
Hoh
man
n 20
00-
2004
Gene expression that determines the mating type
In alpha cells the alpha1 activator stimulates alpha-specific genes and the alpha2 repressor represses a-specific genes
In a cells alpha-specific genes are not activated and a-specific genes are not repressed (they use a different transcriptional activitor to become expressed)
In diploid cells the a1/alpha2 heteromeric repressor represses expression of alpha1 and hence alpha-specific genes are not activated. A-specific genes and haploid-specific genes are repressed too.
One such haploid-specific gene is RME, encoding the repressor of meiosis. Although it is not expressed in diploids the meiosis and sporulation programme will only start once nutrients become limiting
Taken together, cell type is determined with very few primary transcription factors that act individually or in combination.
This is a fundamental principle and is conserved in multicellular organisms for the determination of different cell types: homeotic genes (in fact, a1 is a homeobox factor)

© Ste
fan
Hoh
man
n 20
00-
2004
In nature, yeast cells always grow as diploids, probably because this increases their chance to survive mutation of an essential gene (because there is another copy)
Under nitrogen starvation, diploid cells sporulate and then haploid spores germinate, provided that they have received functional copies of all essential genes
This often means that only a single spore (if any) of a tetrad survives How to make sure that this single spore can find a mating partner to form a diploid again? The
answer is mating type switch! After the first division the mother cell switches mating type and mates with its daughter to form
a diploid, which then of course is homozygous for all genes and starts a new clone of cells If mating type can be switched and diploid is the prefered form, why then sporulate and have
mating types? There are probably several reasons: (1) Spores are hardy and survive very harsh conditions (2)
Sporulation is a way to ”clean” the genome from accumulated mutations (3) Meiosis is a way to generate new combinations of alleles, which may turn out to be advantageous, i.e. better than the previous one (4) Sometimes cells may find a mating partner from a different tetrad and form a new clone, with possibly advantageous allele combination
In order to do yeast genetics and to grow haploid cells in the laboratory, mating type switch must be prevented: all laboratory strains are HO mutants and can not switch
So how does this mysterious switch of sex work?
Haploids and dipoids in nature and laboratory
© Ste
fan
Hoh
man
n 20
00-
2004
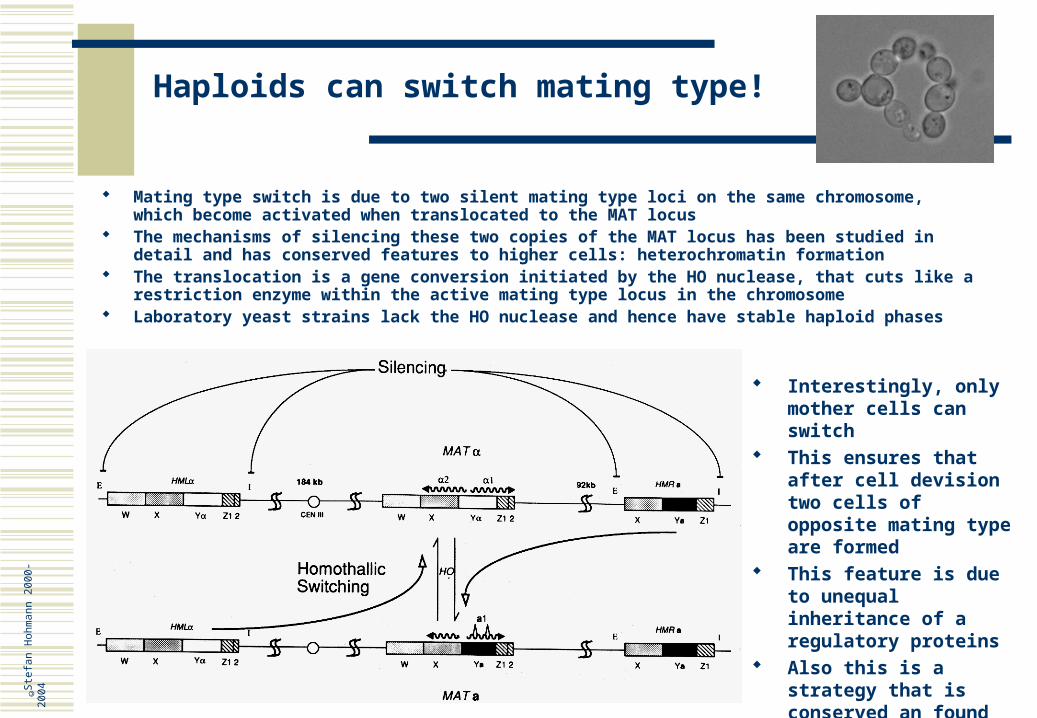
Haploids can switch mating type!
Mating type switch is due to two silent mating type loci on the same chromosome, which become activated when translocated to the MAT locus
The mechanisms of silencing these two copies of the MAT locus has been studied in detail and has conserved features to higher cells: heterochromatin formation
The translocation is a gene conversion initiated by the HO nuclease, that cuts like a restriction enzyme within the active mating type locus in the chromosome
Laboratory yeast strains lack the HO nuclease and hence have stable haploid phases
Interestingly, only mother cells can switch
This ensures that after cell devision two cells of opposite mating type are formed
This feature is due to unequal inheritance of a regulatory proteins
Also this is a strategy that is conserved an found in differentiation of cell types in multicellular organisms
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: the genetic material
The S. cerevisiae nuclear genome has 16 chromosomes In addition, there is a mitochondrial genome and a plasmid,
the 2micron circle The yeast chromosomes contain centromeres and
telomeres, which are simpler than those of higher eukaryotes
The haploid yeast genome consists of about 12,500 kb and was completely sequenced as early 1996 (first complete genome sequence of a eukaryote)

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: the genetic material
The yeast genome is predicted to contain about 6,200 genes, annotation is, however, still ongoing
There is substantial ”gene redundancy”, which originates from an ancient genome duplication
This means that there are many genes for which closely related homologue exist, which often are differentially regulated
The most extreme example are sugar transporter genes; there are more than twenty
Roughly 1/3 of the genes has been characterised by genetic analysis, 1/3 shows homology hinting at their biochemical function and 1/3 is not homologous to other genes or only to other uncharacterised genes
Only a small percentage of yeast genes has introns, very few have more than one; mapping of introns is not complete
The intergenic space between genes is only between 200 and 1,000bp
The largest known regulatory sequences are spread over about 2,800bp (MUC1/FLO11)
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genome analysis
A joint goal of the yeast research community: determination of the function of each and every gene
For this, there are several large projects and numerous approaches Micro array analysis: simultaneous determination of the expression
of all genes Micro array analysis to determine the binding sites in the genome
for all transcription factors Yeast deletion analysis: a complete set of more than 6,000 deletion
mutants is available for research Various approaches to analyse the properties of these mutants All yeast genes have been tagged to green fluorescent protein
(GFP) to allow protein detection and microscopic localisation Different global protein interaction projects are ongoing

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: nomenclature
Yeast genes have names consisting of three letters and up to three numbers:GPD1, HSP12, PDC6...Usually they are meaningful (or meaningless) abbreviations
Wild type genes are written with capital letters in italics: TPS1, RHO1, CDC28... Recessive mutant genes are written with small letters in italics: tps1, rho1, cdc28 Mutant alleles are designated with a dash and a number: tps1-1, rho1-23, cdc28-2 If the mutation has been constructed, i.e. by gene deletion, this is indicated and the genetic
marker used for deletion too: tps1::HIS3 The gene product, a protein, is written with a capital letter at the beginning and not in italics;
often a ”p” is added at the end: Tps1p, Rho1p, Cdc28p Many genes have of course only be found by systematic sequencing and as long as their
function is not determined they get a landmark name: YDR518C, YML016W..., where Y stands for ”yeast” The second letter represents the chromosome (D=IV, M=XIII....) L or R stand for left or right chromosome arm The three-digit number stands for the ORF counted from the centromere on that chromosome arm C or W stand for ”Crick” or ”Watson”, i.e. indicate the strand or direction of the ORF
Some genes do not follow this nomenclature: you heard already about: HO, MATa, MAT

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: markers and strains
Genetic markers are used to follow chromosomes in genetic crosses, to select diploids in genetic crosses, to select transformants in transformation with plasmids or integration of genes into the genome
Commonly genetic markers cause auxotrophies: HIS3, URA3, TRP1, LEU2, LYS2, ADE2
The ade2 mutation has a specific useful feature: cells turn red The first markers in yeast genetics were fermentation markers, i.e. genes
that confer the ability to catabolise certain substrates: SUC, MAL, GAL SUC genes (SUC1-7) encode invertase (periplasmic enzyme) and can be
located on different chromosomes in different yeast strains (telomere location)
MAL loci (MAL1-6) encode each three genes: maltase, maltose transporter and a transcriptional activator; also telomer location
GAL genes encode the enzymes needed to take up galactose and convert it to glucose-6-phosphate
Like in E. coli also certain antibiotic resistance markers can be used in transformation: kanamycin resistance, kanR
There are many yeast strains in use in the laboratories: W303-1A, S288C, 1278b, SK1, BY4741....
Their specific properties can be quite different and are different to wild or industrial strains
The full genotype of our favourite strain W303-1A reads like this:
MATa leu2-3/112 ura3-1 trp1-1 his3-11/15 ade2-1 can1-100 GAL SUC2 mal0

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: crossing strains
Yeast genetics is based on the possibility to cross two haploid strains with different mutations and of opposite mating type to a diploid strain
The diploid can then be investigated, for instance if one wants to find out if the two haploid strains had mutations in the same or different genes
The diploid can be sporulated to form tetrads, tetrads can be dissected using a micromanipulator and spores form individual colonies, and hence can be investigated
In the past, such genetic crosses were done a lot in order to map genes on chromosomes: the frequency with which two mutations recombined (i.e. resulted in spores carrying both mutations or spores without any of the two mutations) is a measure for the genetic distance
The last genetic map (before the genome was sequenced) encompassed more than 1,000 genes and turned out to be very accurate (also thanks to the enormous capacity of yeast for genetic recombination)
Today genetic crosses are used to generate yeast strains with new combination of mutations, for instance double, triple....mutations – for this it is useful to know some principles of genetic crosses and gene segregation
And even today with the genome fully sequenced we often perform genetic screens for new mutations, for instance to find genes/proteins that function in the same pathway/molecular system than an already known gene/protein – then genetic analysis of the mutants one obtained is the first and essential step in characterisation
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: crossing strains
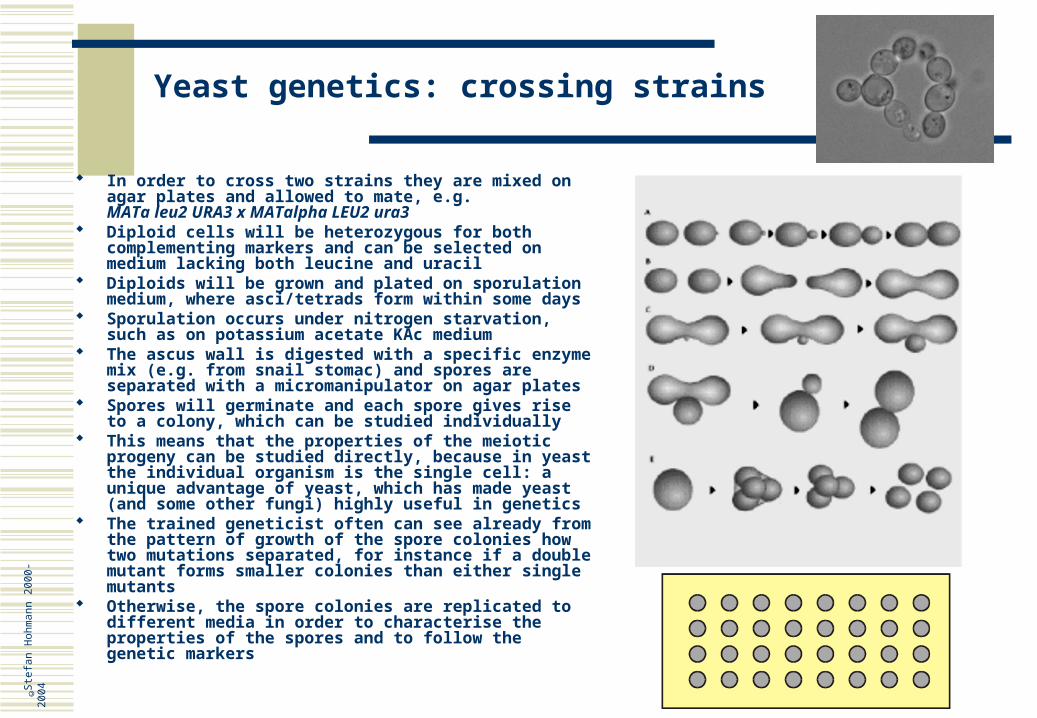
In order to cross two strains they are mixed on agar plates and allowed to mate, e.g.MATa leu2 URA3 x MATalpha LEU2 ura3
Diploid cells will be heterozygous for both complementing markers and can be selected on medium lacking both leucine and uracil
Diploids will be grown and plated on sporulation medium, where asci/tetrads form within some days
Sporulation occurs under nitrogen starvation, such as on potassium acetate KAc medium
The ascus wall is digested with a specific enzyme mix (e.g. from snail stomac) and spores are separated with a micromanipulator on agar plates
Spores will germinate and each spore gives rise to a colony, which can be studied individually
This means that the properties of the meiotic progeny can be studied directly, because in yeast the individual organism is the single cell: a unique advantage of yeast, which has made yeast (and some other fungi) highly useful in genetics
The trained geneticist often can see already from the pattern of growth of the spore colonies how two mutations separated, for instance if a double mutant forms smaller colonies than either single mutants
Otherwise, the spore colonies are replicated to different media in order to characterise the properties of the spores and to follow the genetic markers
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: crossing strains
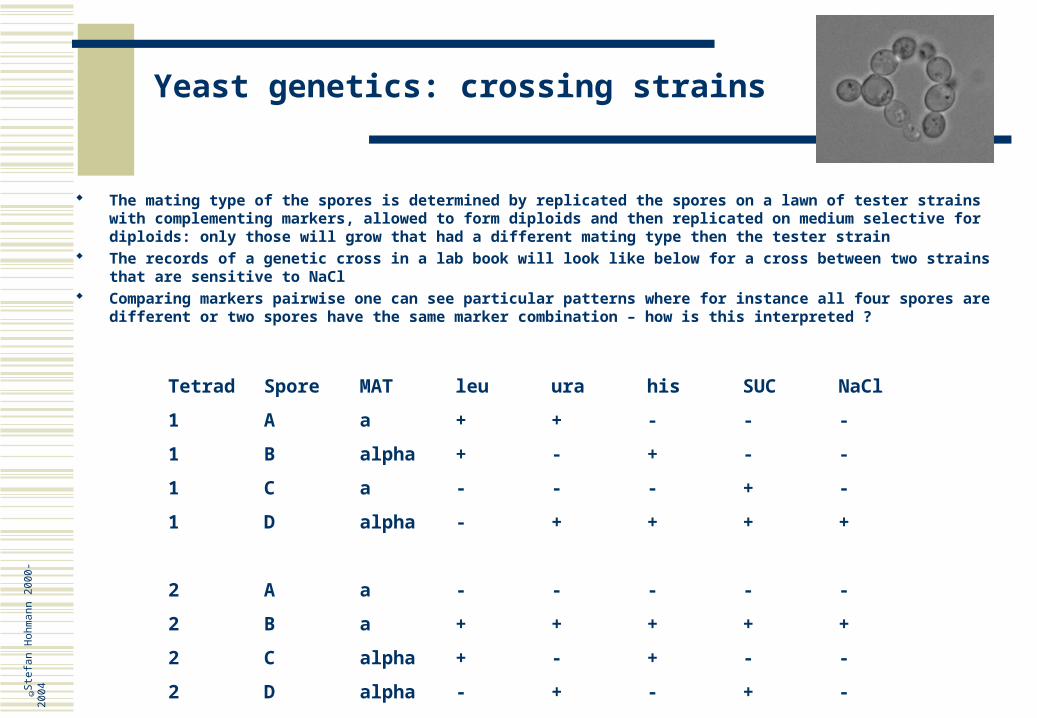
The mating type of the spores is determined by replicated the spores on a lawn of tester strains with complementing markers, allowed to form diploids and then replicated on medium selective for diploids: only those will grow that had a different mating type then the tester strain
The records of a genetic cross in a lab book will look like below for a cross between two strains that are sensitive to NaCl
Comparing markers pairwise one can see particular patterns where for instance all four spores are different or two spores have the same marker combination – how is this interpreted ?
Tetrad Spore MAT leu ura his SUC NaCl
1 A a + + - - -
1 B alpha + - + - -
1 C a - - - + -
1 D alpha - + + + +
2 A a - - - - -
2 B a + + + + +
2 C alpha + - + - -
2 D alpha - + - + -
© Ste
fan
Hoh
man
n 20
00-
2004
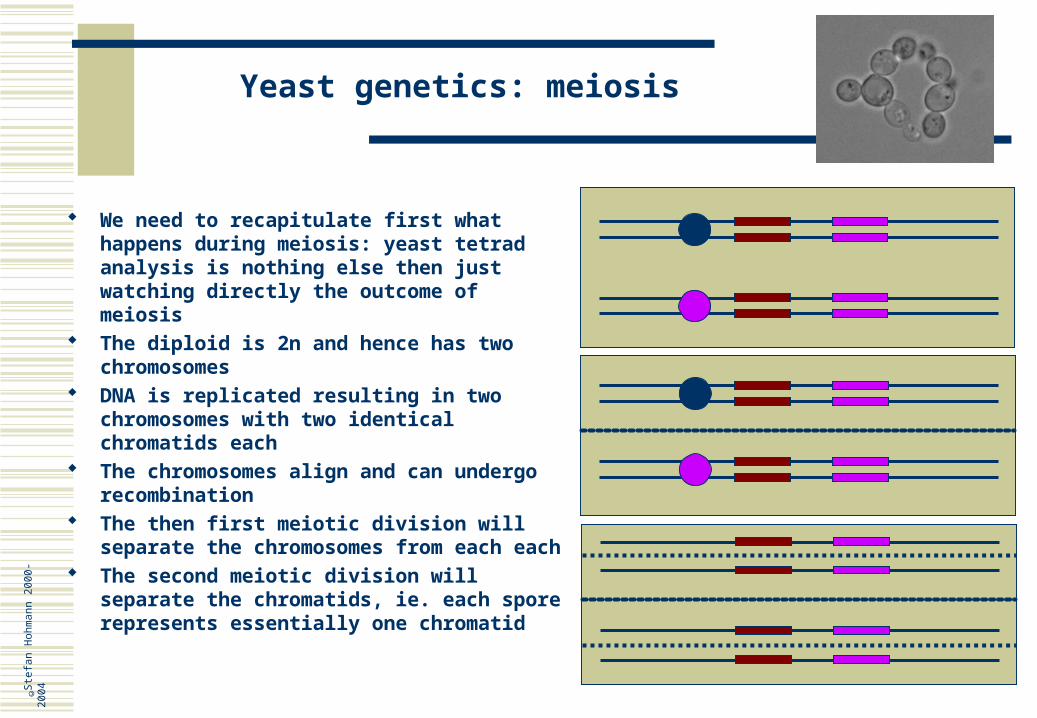
We need to recapitulate first what happens during meiosis: yeast tetrad analysis is nothing else then just watching directly the outcome of meiosis
The diploid is 2n and hence has two chromosomes
DNA is replicated resulting in two chromosomes with two identical chromatids each
The chromosomes align and can undergo recombination
The then first meiotic division will separate the chromosomes from each each
The second meiotic division will separate the chromatids, ie. each spore represents essentially one chromatid
Yeast genetics: meiosis

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: the outcome of a cross
Let us now imagine that LEU2 and URA3 are close together on the same chromosome
LEU2
LEU2
ura3
ura3
leu2
leu2
URA3
URA3
LEU2 ura3
LEU2 ura3
leu2 URA3
leu2 URA3
In the likely case that no cross-over occurs between the two markers all haploid spores will just look like the parental haploid strains
There are only two different types of spores, i.e. (leu-plus ura-minus) and (leu-minus ura-plus) spores
Hence such a tetrad is called a parental ditype PD
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: cross over
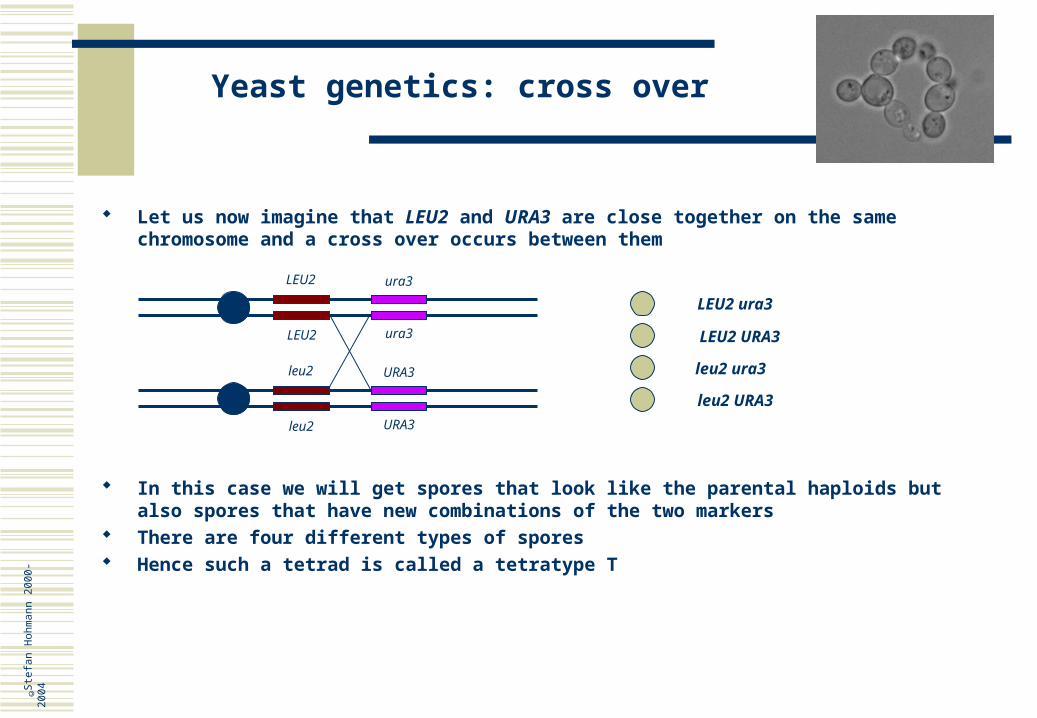
Let us now imagine that LEU2 and URA3 are close together on the same chromosome and a cross over occurs between them
LEU2
LEU2
ura3
ura3
leu2
leu2
URA3
URA3
LEU2 ura3
LEU2 URA3
leu2 ura3
leu2 URA3
In this case we will get spores that look like the parental haploids but also spores that have new combinations of the two markers
There are four different types of spores Hence such a tetrad is called a tetratype T

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: double cross over
Let us now imagine that LEU2 and URA3 are close together on the same chromosome and two cross over occur between them such that four DNA strands are involved
LEU2
LEU2
ura3
ura3
leu2
leu2
URA3
URA3
LEU2 URA3
LEU2 URA3
leu2 ura3
leu2 ura3
In this case we will get only spores that look different from the parental haploids There are two different types of spores Hence such a tetrad is called non parental ditype NPD
Since with close linkage it is most likely that no cross over occurs and least likely that two cross over occur the proportion of tetrads would be PD > T > NPD and the relative numbers can be used to map genetic distances. For mapping one investigated hundreds of tetrads from the same cross. This has been done extensively in the past and the last genetic map from 1995 comprised about 1,000 locations
To generate new combination of mutations (such as leu2 ura3) one will have to dissect the more tetrads the closer the two genes are, and this can be estimated based on the physical distance (in kb), which relates well to the genetic distance (in cM, centi Morgan). For two close genes (1cM, i.e. 1% recombinant spores) one would have to dissect at least 25 tetrads, statistically

© Ste
fan
Hoh
man
n 20
00-
2004
Crossing with markers on different chromosomes
Let us now imagine that LEU2 and URA3 are on different chromosomes
LEU2
LEU2
leu2
leu2
LEU2 ura3
LEU2 ura3
leu2 URA3
leu2 URA3
Different chromosomes assort randomly in the first meiotic division For this reason two types of tetrads become equally frequent, the parental and the non-parental
ditype, PD and NPD Hence, linked and unlinked genes can easily be distinguished in tetrad analysis because with
unlinked genes PD = NPD while with linked genes PD>>NPD.
ura3
ura3
URA3
URA3
LEU2 URA3
LEU2 URA3
leu2 ura3
leu2 ura3

© Ste
fan
Hoh
man
n 20
00-
2004
Crossing with markers on different chromosomes
Let us now imagine that LEU2 and URA3 are on different chromosomes and a crossing over occurs between a centromere and a marker
LEU2
LEU2
leu2
leu2
LEU2 ura3
LEU2 URA3
leu2 ura3
leu2 URA3
Now the different alleles of URA3 will only be separated in the second meiotic division The result is a tetratype tetrad T The above situation means also that if markers are distant from the centromere many Ts will
occur while if both markers are close to the centromere few Ts will occur. What is the outcome of double cross-overs with four or with three strands? Due to the possibility of double cross-overs the proportion between different tetrad types for
unlinked genes that are not centromere-linked becomes 1:1:4 for PD:NPD:T This also means that one out of four spores will be recombinant, i.e. in order to obain the new
combination of genes (leu2 ura3) one only needs to dissect one tetrad, statistically
ura3
ura3
URA3
URA3

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: making mutants
Mutations that enhance or abolish the function of a certain protein are extremely useful to study cellular systems
The phenotype of mutations (i.e. the properties of the mutant) can tell a lot about the function of a gene, protein or pathway
This approach is valid even with the genome sequenced and even with the complete deletion set available: point mutations can have different properties than deletion mutants
Random versus targetted mutations In random mutagenesis one tries to link genes to a certain
function/role; this identifies new genes or new functions to known genes
Hence in random mutagenesis usually the entire genome is targetted
Random mutagenesis is also possible for a specific protein (whose genes is then mutated in vitro); in this case one wishes to identify functional domains
In targetted mutagenesis one knocks out or alters a specific gene by a combination of in vitro and in vivo manipulation
Induced versus spontaneous mutations Mutations can be induced by treating cells with a mutagen; this
can of course give multiple hits per cell Spontaneous mutations ”just occur” at a low frequency and it is
likely that there is only one hit per cell

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast genetics: finding mutants
Screening versus selection When screening for mutants one tests clone by clone to find
interesting mutants For that, one usually plates many cells and tries to find mutants
because they are unable to grow on a certain medium after replica-plating or because they develop a colour
For screening, mutations are usually induced to increase their frequency
Still: screening requires hundreds of perti dishes and commonly more than 10,000 clones to be scored
To develop a new selection system is the art of genetic analysis
When selecting for mutants one has established a condition under which the mutant phenotype confers a growth advantage
In other words, the intellectual challenge is to design conditions and /or strains such that the mutant grows, but the wild type does not
A smart screening system allows one to go for spontaneous mutations, because up to 108 cells can easily be spread on one plate
Selection systems are often based on resistance to inhibitors We try to train our students to watch out for any such
opportunity to find conditions that allow to select for new mutants with interesting properties to advance the understanding of the system under study
Wild type
hog1
sko1
aca1 aca2
hog1 aca1 aca2
hog1 sko1
hog1 sko1 aca1 aca2
YPD YPD + 0.4M NaCl
Wild type
aca2
hog1
hog1 aca2
YPD YPD + 0.4M NaCl

© Ste
fan
Hoh
man
n 20
00-
2004
Once mutants have been identified they need to be characterised and the genes affected have to be identified; this requires the following steps
A detailed phenotypic analysis, i.e. testing also for other phenotypes than the one used in screening/selection
Establishing if a mutant is dominant or recessive Placing the mutants into complementation groups. Usually one complemetation
group is equivalent to one gene Cloning the gene by complementation.
Yeast genetics: characterising mutants
© Ste
fan
Hoh
man
n 20
00-
2004
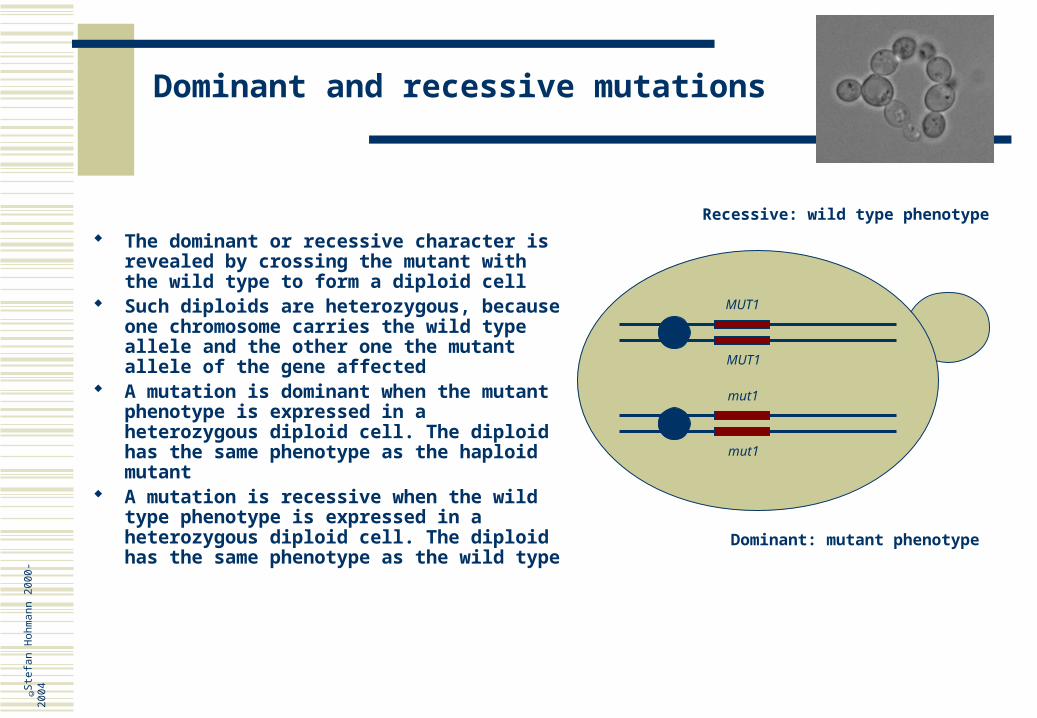
Dominant and recessive mutations
MUT1
MUT1
mut1
mut1
Dominant: mutant phenotype
Recessive: wild type phenotype
The dominant or recessive character is revealed by crossing the mutant with the wild type to form a diploid cell
Such diploids are heterozygous, because one chromosome carries the wild type allele and the other one the mutant allele of the gene affected
A mutation is dominant when the mutant phenotype is expressed in a heterozygous diploid cell. The diploid has the same phenotype as the haploid mutant
A mutation is recessive when the wild type phenotype is expressed in a heterozygous diploid cell. The diploid has the same phenotype as the wild type
© Ste
fan
Hoh
man
n 20
00-
2004
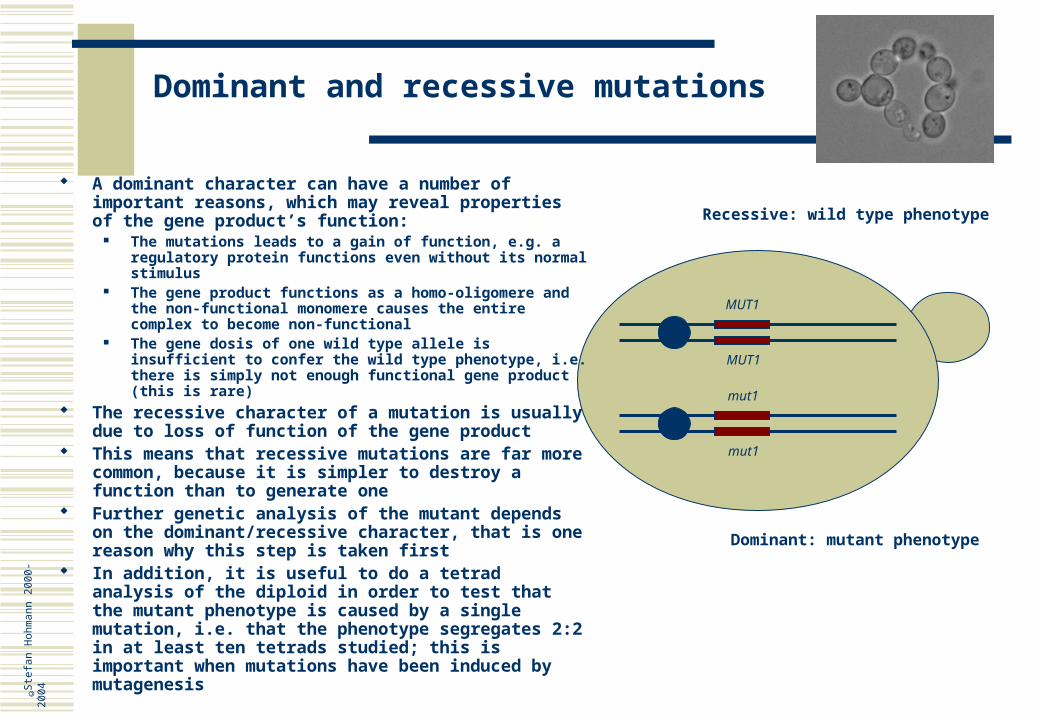
Dominant and recessive mutations
MUT1
MUT1
mut1
mut1
Dominant: mutant phenotype
Recessive: wild type phenotype
A dominant character can have a number of important reasons, which may reveal properties of the gene product’s function:
The mutations leads to a gain of function, e.g. a regulatory protein functions even without its normal stimulus
The gene product functions as a homo-oligomere and the non-functional monomere causes the entire complex to become non-functional
The gene dosis of one wild type allele is insufficient to confer the wild type phenotype, i.e. there is simply not enough functional gene product (this is rare)
The recessive character of a mutation is usually due to loss of function of the gene product
This means that recessive mutations are far more common, because it is simpler to destroy a function than to generate one
Further genetic analysis of the mutant depends on the dominant/recessive character, that is one reason why this step is taken first
In addition, it is useful to do a tetrad analysis of the diploid in order to test that the mutant phenotype is caused by a single mutation, i.e. that the phenotype segregates 2:2 in at least ten tetrads studied; this is important when mutations have been induced by mutagenesis
© Ste
fan
Hoh
man
n 20
00-
2004
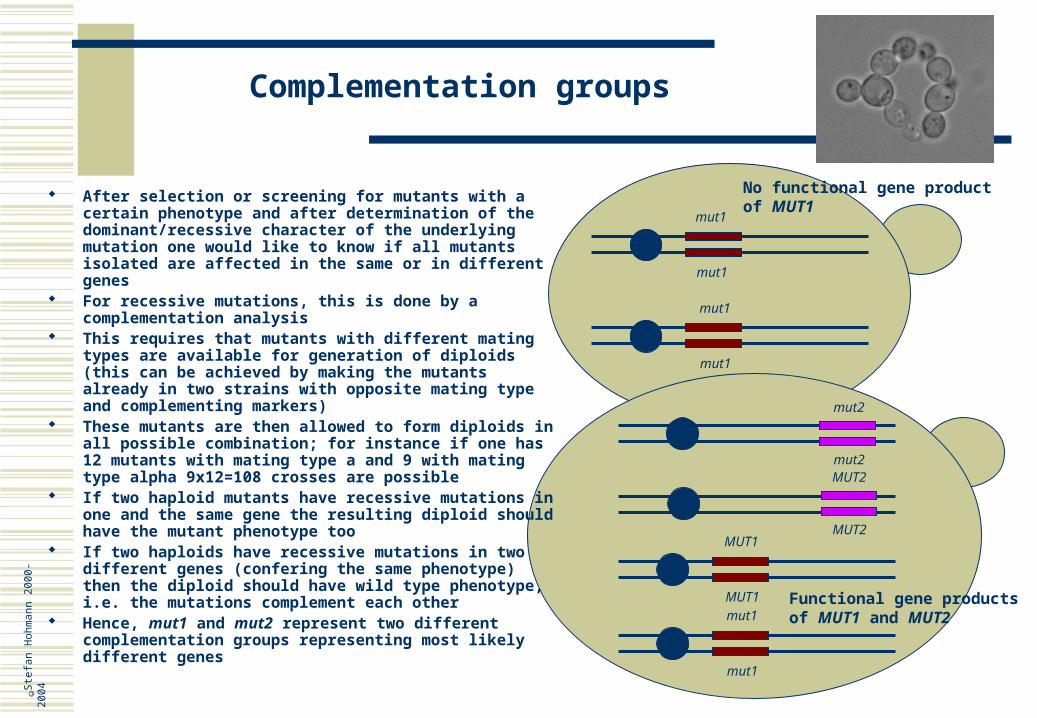
Complementation groups
mut1
mut1
mut1
mut1
MUT1
MUT1
mut1
mut1
mut2
mut2MUT2
MUT2
After selection or screening for mutants with a certain phenotype and after determination of the dominant/recessive character of the underlying mutation one would like to know if all mutants isolated are affected in the same or in different genes
For recessive mutations, this is done by a complementation analysis
This requires that mutants with different mating types are available for generation of diploids (this can be achieved by making the mutants already in two strains with opposite mating type and complementing markers)
These mutants are then allowed to form diploids in all possible combination; for instance if one has 12 mutants with mating type a and 9 with mating type alpha 9x12=108 crosses are possible
If two haploid mutants have recessive mutations in one and the same gene the resulting diploid should have the mutant phenotype too
If two haploids have recessive mutations in two different genes (confering the same phenotype) then the diploid should have wild type phenotype, i.e. the mutations complement each other
Hence, mut1 and mut2 represent two different complementation groups representing most likely different genes
No functional gene productof MUT1
Functional gene productsof MUT1 and MUT2
© Ste
fan
Hoh
man
n 20
00-
2004
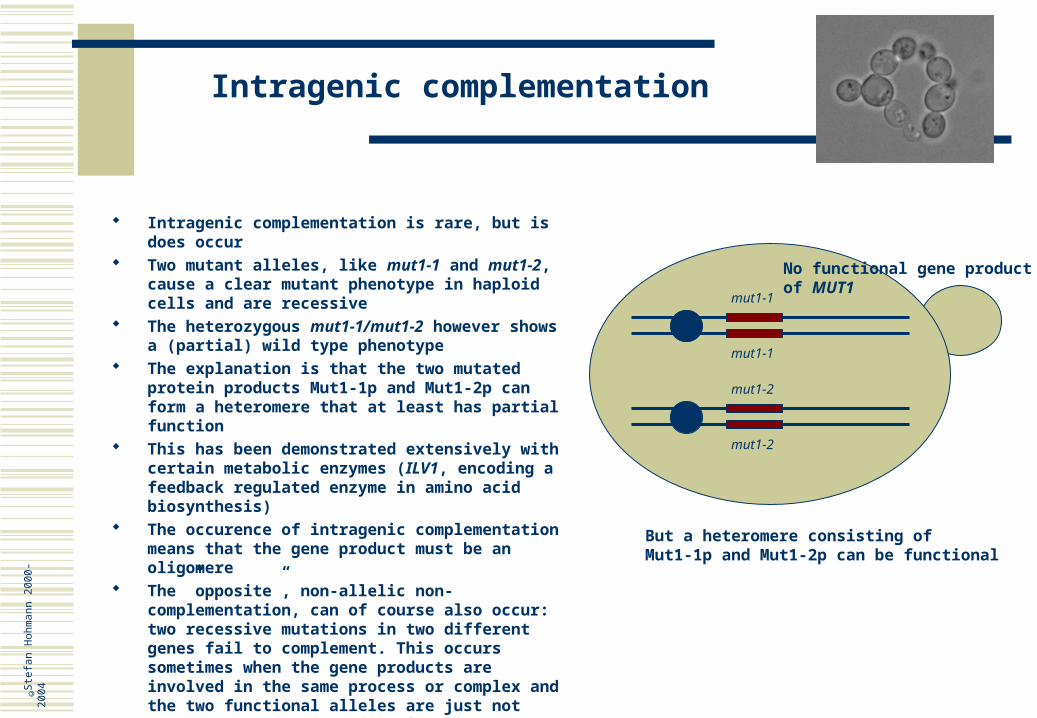
Intragenic complementation
mut1-1
mut1-1
mut1-2
mut1-2
Intragenic complementation is rare, but is does occur Two mutant alleles, like mut1-1 and mut1-2, cause a
clear mutant phenotype in haploid cells and are recessive
The heterozygous mut1-1/mut1-2 however shows a (partial) wild type phenotype
The explanation is that the two mutated protein products Mut1-1p and Mut1-2p can form a heteromere that at least has partial function
This has been demonstrated extensively with certain metabolic enzymes (ILV1, encoding a feedback regulated enzyme in amino acid biosynthesis)
The occurence of intragenic complementation means that the gene product must be an oligomere
The ”opposite”, non-allelic non-complementation, can of course also occur: two recessive mutations in two different genes fail to complement. This occurs sometimes when the gene products are involved in the same process or complex and the two functional alleles are just not enough to confer full functionality
No functional gene productof MUT1
But a heteromere consisting ofMut1-1p and Mut1-2p can be functional

© Ste
fan
Hoh
man
n 20
00-
2004
Cloning in yeast
The era of yeast molecular genetics started as early as 1978, when S. cerevisiae was first transformed successfully with foreign DNA
There are numerous transformation protocols but all are at least three orders of magnitude less efficient as transformation in E. coli
Yeast can maintain replicating plasmids but the copy number is much smaller than in E. coli, usually between one and 50 per cell
Yeast can maintain more than one type of plasmid at the same time. This can complicate gene cloning from a library. It can also be very useful to transform yeast with two different plasmids simultaneously, for instance for a method called plasmid shuffling
Cloning and plasmid preparation from yeast is very ineffective Therefore, cloning in yeast uses E. coli as a plasmid production
system: Plasmids are constructed in vitro Plasmids are transformed into E. coli and the constructions are
confirmed, just in the same way as when working with bacteria Plasmids are produced in bacteria.... ....and then transformed into yeast
Hence we work with so-called yeast-E. coli shuttle vectors On the other hand, yeast has a very efficient and reliable system
for homologous recombination, which can be used for cloning
pUC182686 bp
AP r
ALPHAP(BLA)
P(LAC)
ORI
Ava I (435)
Bam H I (430)
Eco R I (451)
Hin d III (400)
Pst I (416)
Sma I (437)
Xma I (435)
Apa LI (178)
Apa LI (1121)
Apa LI (2367)
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast-E. coli shuttle vectors
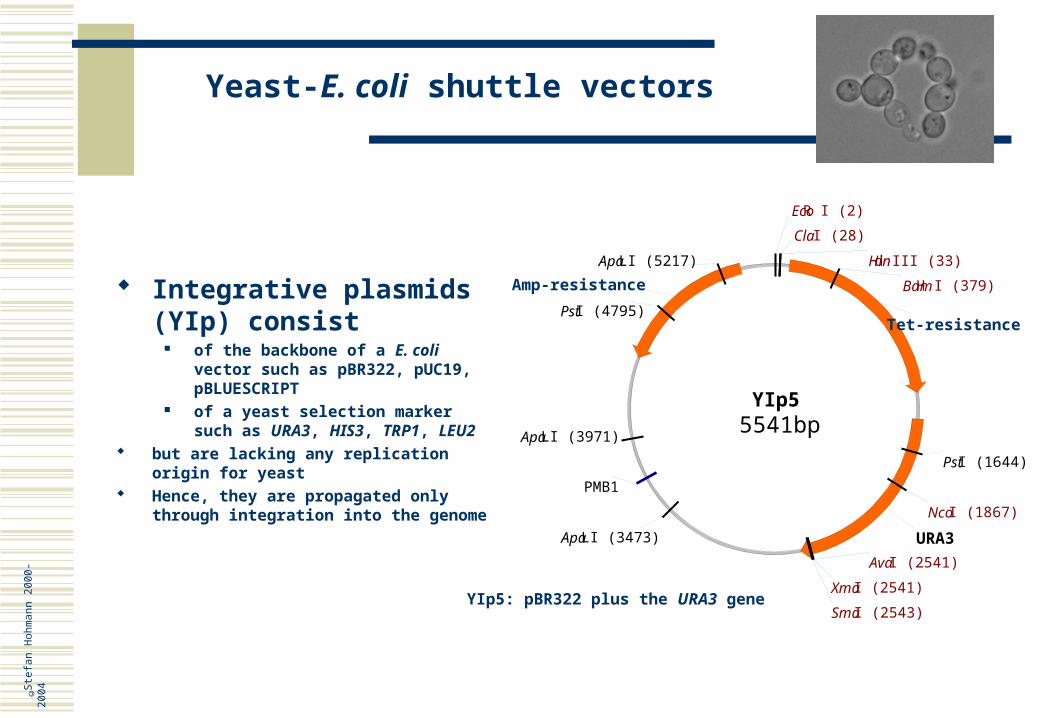
Integrative plasmids (YIp) consist
of the backbone of a E. coli vector such as pBR322, pUC19, pBLUESCRIPT
of a yeast selection marker such as URA3, HIS3, TRP1, LEU2
but are lacking any replication origin for yeast
Hence, they are propagated only through integration into the genome
YIp55541bp
URA3
PMB1
Ava I (2541)
BamH I (379)
Cla I (28)
EcoR I (2)
Hind III (33)
Nco I (1867)
Sma I (2543)
Xma I (2541)
Pst I (1644)
Pst I (4795)
Apa LI (3473)
Apa LI (3971)
Apa LI (5217)
Amp-resistance
Tet-resistance
YIp5: pBR322 plus the URA3 gene
© Ste
fan
Hoh
man
n 20
00-
2004
Integration of plasmids into the yeast genome
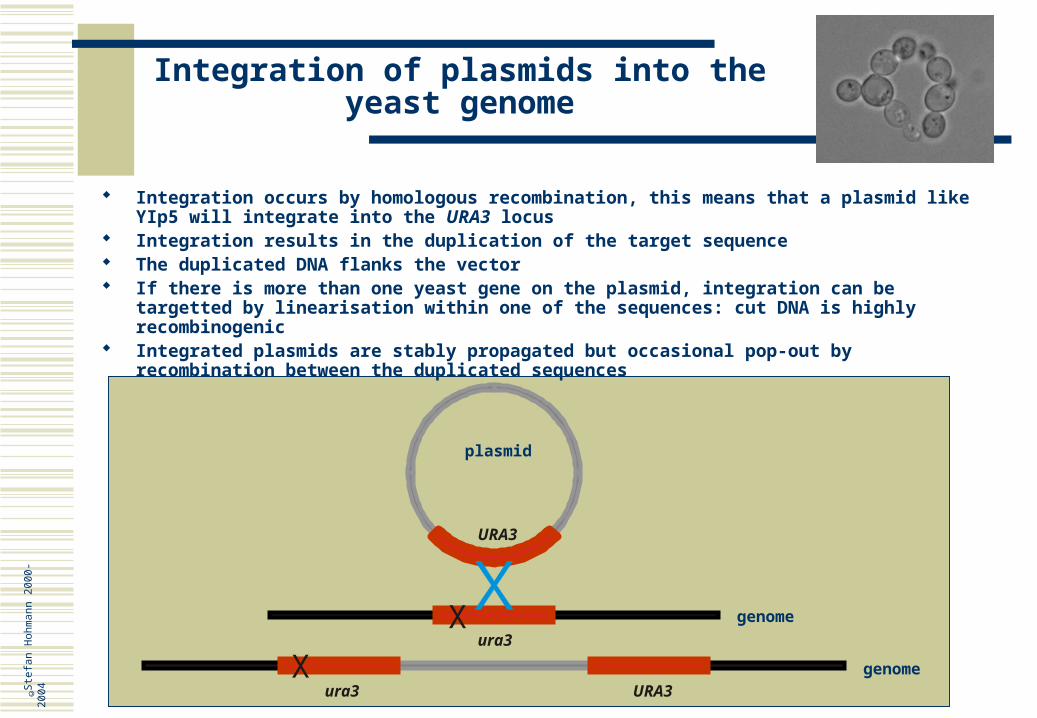
Integration occurs by homologous recombination, this means that a plasmid like YIp5 will integrate into the URA3 locus
Integration results in the duplication of the target sequence The duplicated DNA flanks the vector If there is more than one yeast gene on the plasmid, integration can be targetted by
linearisation within one of the sequences: cut DNA is highly recombinogenic Integrated plasmids are stably propagated but occasional pop-out by recombination between
the duplicated sequences
URA3ura3X
URA3
ura3X
plasmid
genome
genome
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast-E. coli shuttle vectors
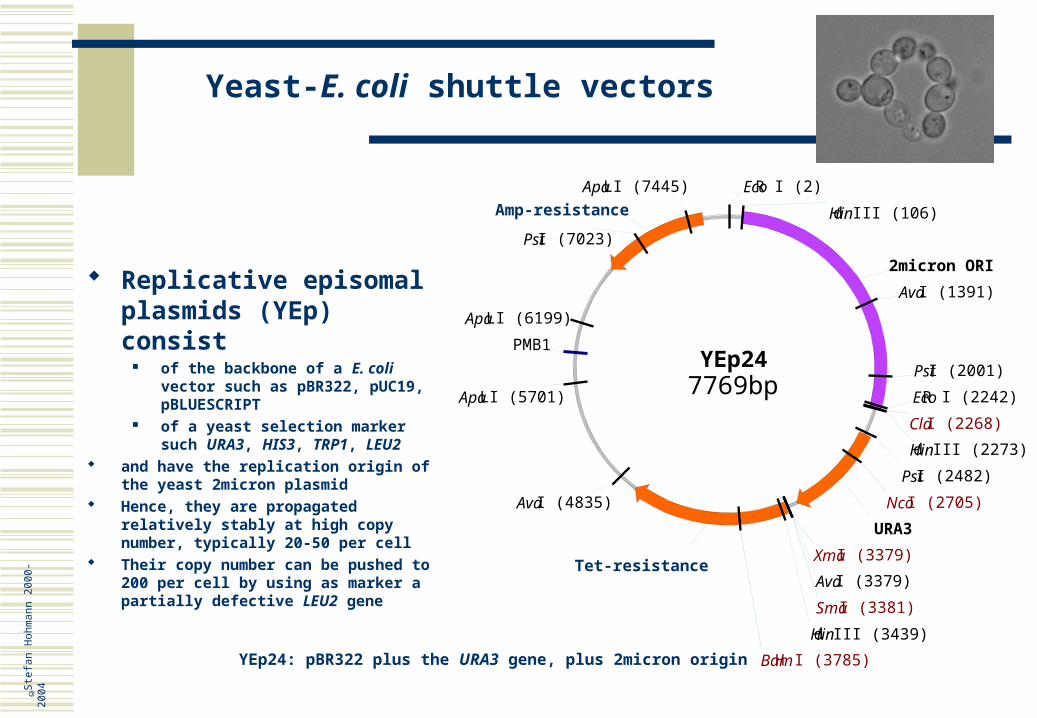
Replicative episomal plasmids (YEp) consist
of the backbone of a E. coli vector such as pBR322, pUC19, pBLUESCRIPT
of a yeast selection marker such URA3, HIS3, TRP1, LEU2
and have the replication origin of the yeast 2micron plasmid
Hence, they are propagated relatively stably at high copy number, typically 20-50 per cell
Their copy number can be pushed to 200 per cell by using as marker a partially defective LEU2 gene
YEp24: pBR322 plus the URA3 gene, plus 2micron origin
YEp247769bp
URA3
PMB1
2micron ORI
BamH I (3785)
Cla I (2268)
Nco I (2705)
Sma I (3381)
Xma I (3379)
EcoR I (2)
EcoR I (2242)Apa LI (5701)
Apa LI (6199)
Apa LI (7445)
Ava I (1391)
Ava I (3379)
Ava I (4835)
Hind III (106)
Hind III (2273)
Hind III (3439)
Pst I (2001)
Pst I (2482)
Pst I (7023)
Amp-resistance
Tet-resistance
© Ste
fan
Hoh
man
n 20
00-
2004
Yeast-E. coli shuttle vectors
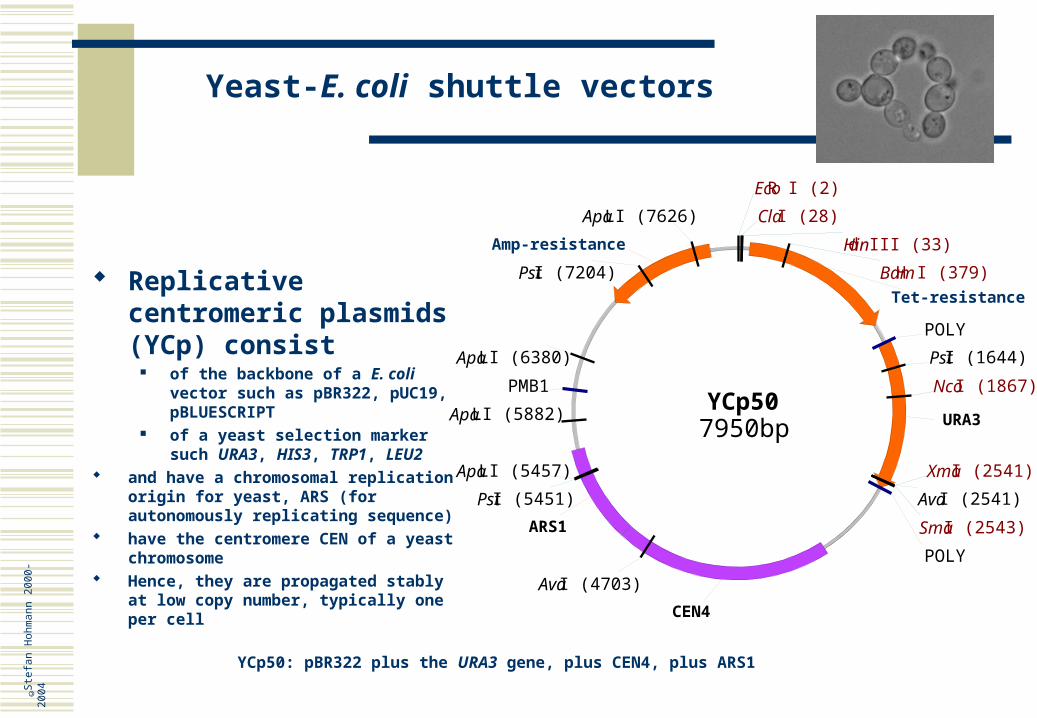
Replicative centromeric plasmids (YCp) consist
of the backbone of a E. coli vector such as pBR322, pUC19, pBLUESCRIPT
of a yeast selection marker such URA3, HIS3, TRP1, LEU2
and have a chromosomal replication origin for yeast, ARS (for autonomously replicating sequence)
have the centromere CEN of a yeast chromosome
Hence, they are propagated stably at low copy number, typically one per cell
Amp-resistance
Tet-resistance
YCp507950bp URA3
POLY
CEN4
ARS1
POLY
PMB1
BamH I (379)
Cla I (28)
EcoR I (2)
Hind III (33)
Nco I (1867)
Sma I (2543)
Xma I (2541)
Ava I (2541)
Ava I (4703)
Pst I (1644)
Pst I (5451)
Pst I (7204)
Apa LI (5457)
Apa LI (5882)
Apa LI (6380)
Apa LI (7626)
YCp50: pBR322 plus the URA3 gene, plus CEN4, plus ARS1

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast-E. coli shuttle vectors
Plasmid series are based on an E. coli cloning vector such as
pUC19 or pBLUESCRIPT have one out of three or four different yeast
markers come as YIp, YCp and YEp for convenience
pRS4235797 bp
HIS3
AP r
LACZ'
MCS
P(LAC)
T7 P
T3 P
2 MICRON
F1 ORI
PMB1
Bam H I (2143)
Cla I (2108)
Eco R I (2125)
Sma I (2139)
Xma I (2137)
Pst I (1188)
Pst I (2135)
Apa LI (178)
Apa LI (2891)
Apa LI (4137) Ava I (2092)
Ava I (2137)
Ava I (4680)
Hin d III (809)
Hin d III (996)
Hin d III (2113)
pRS3134967 bp
AP r
HIS3
LACZ'
CEN6
MCS
ARSH4
T3 P
T7 P
PMB1
F1 ORI
Bam H I (2110)
Cla I (2147)
Eco R I (2128)
Sma I (2118)
Xma I (2116)
Ava I (2116)
Ava I (2161)
Pst I (1187)
Pst I (2126)
Apa LI (178)
Apa LI (2888)
Apa LI (4134) Hin d III (808)
Hin d III (995)
Hin d III (2140)
YIps are used for integration only YCps are used for low copy
expression YEps are used for overexpression

© Ste
fan
Hoh
man
n 20
00-
2004
Cloning by complementation
Frequently when one has isolated a number of mutants and classified them into complementation groups the nature of the gene is not known (and this is still often the case even though the genome sequence is known!)
To identify the gene it is cloned from a gene library by complementation of the mutation A gene library is a large population of plasmids containing different fragments of genomic
yeast DNA, cumulatively representing the entire yeast genome Such libraries are constructed by digesting the entire yeast DNA partially with a nuclease such
as Sau3A (cutting site GATC), which cuts frequently; this strategy generates many overlapping fragments and it ensures that all genes are functionally represented; Sau3A fragments can be cloned into BamHI (GGATCC) cut plasmids; all available yeast libraries are done that way
If the fragments cloned are 5-9kb on average, 2,000 plasmids represent the genome once and 10,000 plasmids give a more than 90% probability that all genes are functionally represented
The library is transformed into the yeast mutant of interest Transformants are screened or selected for restoration of the wild type phenotype Plasmids are prepared from positive clones, transformed into E. coli and further analysed;
some sequence information reveals the identity of the clone Retransformation into the yeast mutant verifies that the plasmid contains a truly
complementing gene; this is necessary because yeast cells can take up more than one kind of plasmid

© Ste
fan
Hoh
man
n 20
00-
2004
Cloning by complementation
Cloning by complementation sounds like a straightforward approach but there are quite a few caveats to it
First of all, it can only be done with recessive mutants For cloning of genes with dominant mutants, a gene library has to be prepared from each
mutant and transformed into the wild type strain; transformants showing the mutant phenotype are then screened or selected
In addition, complementation of a mutation does not mean that the cloned gene is indeed the one that is defective in the mutant – it could be a multi-copy suppressor
This can even happen with centromeric vectors, because selective pressure can drive up the copy number of even these plasmids
A multi-copy suppressor is a gene that overcomes the primary defect in the mutant when expressed at high levels; this is a common phenomenon
It is in fact so common that it is a useful approach to clone new genes starting from a certain mutant – we return to that
To demonstrate that the cloned gene is the one that is mutated in the mutant, a deletion mutant has to be constructed by homologous recombination using the cloned gene as template
If the original and the deletion mutant have the same phenotype, this is good evidence that the two genes are the same
Final proof is obtained by crossing the two mutants; if the diploid has the mutant phenotype too and all spores isolated form the diploid as well, this is proof that the two genes are the same
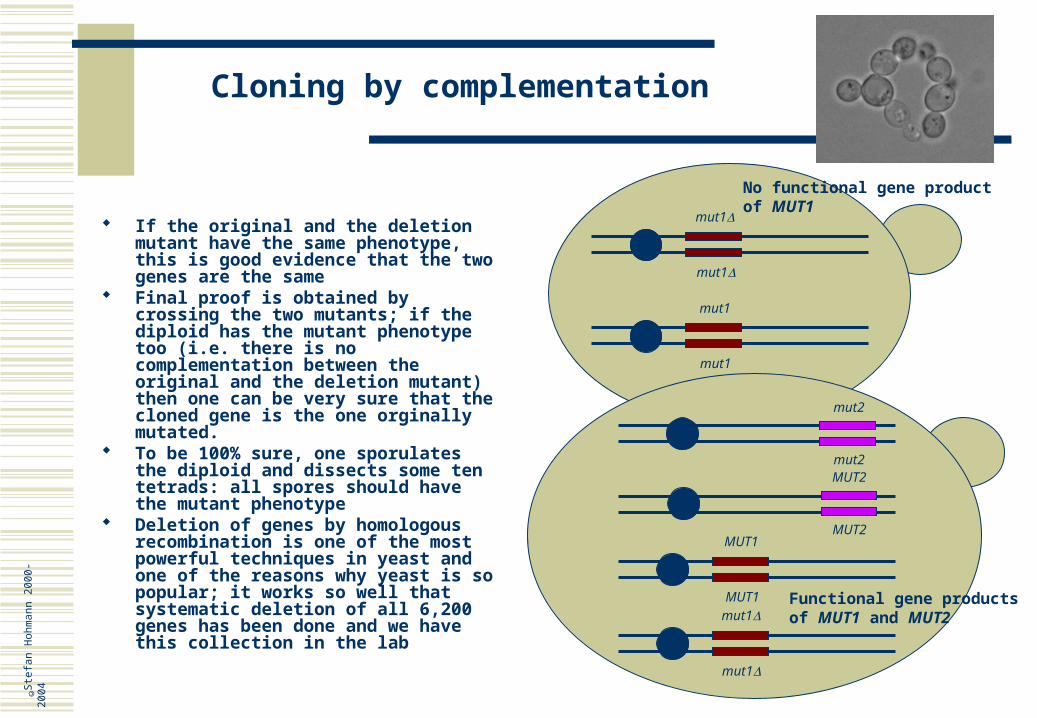
Deletion of genes by homologous recombination is one of the most powerful techniques in yeast and one of the reasons why yeast is so popular; it works so well that systematic deletion of all 6,200 genes has been done and we have this collection in the lab
© Ste
fan
Hoh
man
n 20
00-
2004
Cloning by complementation
If the original and the deletion mutant have the same phenotype, this is good evidence that the two genes are the same
Final proof is obtained by crossing the two mutants; if the diploid has the mutant phenotype too (i.e. there is no complementation between the original and the deletion mutant) then one can be very sure that the cloned gene is the one orginally mutated.
To be 100% sure, one sporulates the diploid and dissects some ten tetrads: all spores should have the mutant phenotype
Deletion of genes by homologous recombination is one of the most powerful techniques in yeast and one of the reasons why yeast is so popular; it works so well that systematic deletion of all 6,200 genes has been done and we have this collection in the lab
mut1
mut1
mut1
mut1
MUT1
MUT1
mut1
mut1
mut2
mut2MUT2
MUT2
No functional gene productof MUT1
Functional gene productsof MUT1 and MUT2
© Ste
fan
Hoh
man
n 20
00-
2004
Deleting a yeast gene
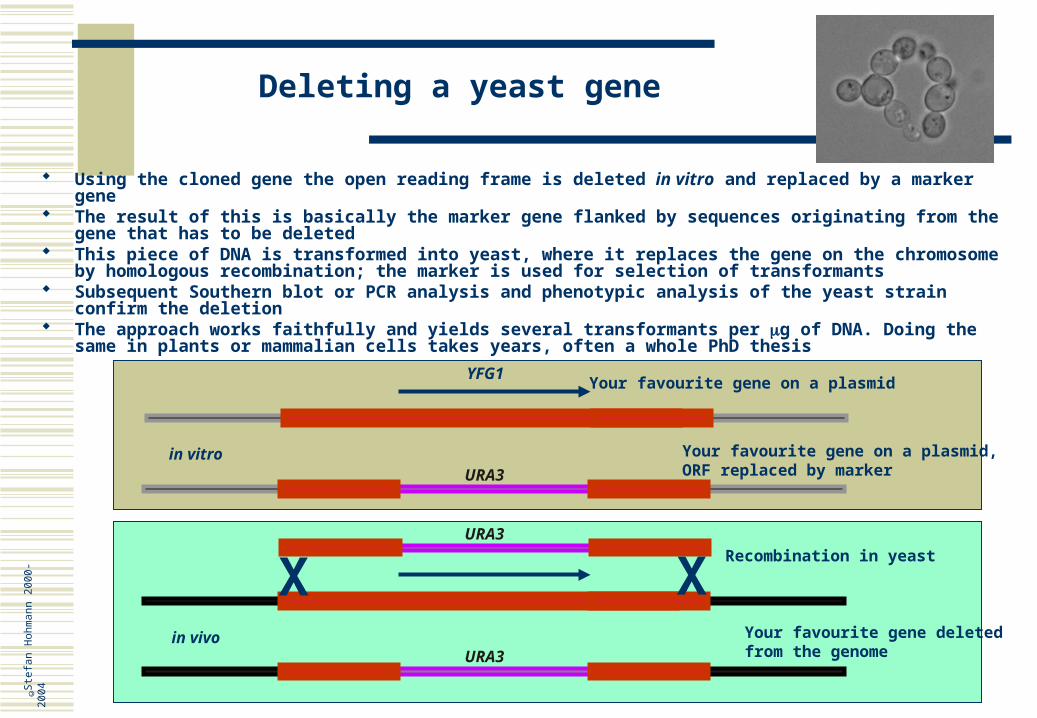
Using the cloned gene the open reading frame is deleted in vitro and replaced by a marker gene The result of this is basically the marker gene flanked by sequences originating from the gene that has
to be deleted This piece of DNA is transformed into yeast, where it replaces the gene on the chromosome by
homologous recombination; the marker is used for selection of transformants Subsequent Southern blot or PCR analysis and phenotypic analysis of the yeast strain confirm the
deletion The approach works faithfully and yields several transformants per g of DNA. Doing the same in plants
or mammalian cells takes years, often a whole PhD thesis
YFG1Your favourite gene on a plasmid
URA3
Your favourite gene on a plasmid,ORF replaced by marker
Your favourite gene deletedfrom the genome
X XRecombination in yeast
URA3
URA3
in vitro
in vivo

© Ste
fan
Hoh
man
n 20
00-
2004
Deleting a yeast gene
There are a number of different ways to generate the piece of DNA for yeast transformation, i.e. the marker flanked by fragments with DNA from YFG1
It can be done using restriction enzymes and DNA ligation It can be done by PCR/restriction/ligation; the entire plasmid is amplified by PCR with the exception of the
ORF; restriction sites in the PCR primers generate a site where the marker can be cloned in It can be done by PCR without any cloning step; in two separate PCR reactions the flanking regions of
YFG1 are amplified and used in a second round as primers to amplify the marker gene; this requires the primers to be designed accordingly (see below)
It can also be done with long PCR primers, in which only the marker is amplified and recombination is mediated by the primer sequences; as little as 30bp can be enough to mediate recombination; in such cases the use of a heterologous marker is recommended to make integration in the right place more reliable
The latter two approaches do not even require the gene to be cloned!! A gene deletion project hence may take only a couple of days
YFG1 First PCR to amplify the flanking parts of your favourite gene
URA3 Final PCR product ready for transformation
URA3
Second PCR to amplify the marker
© Ste
fan
Hoh
man
n 20
00-
2004
Smart gene deletion
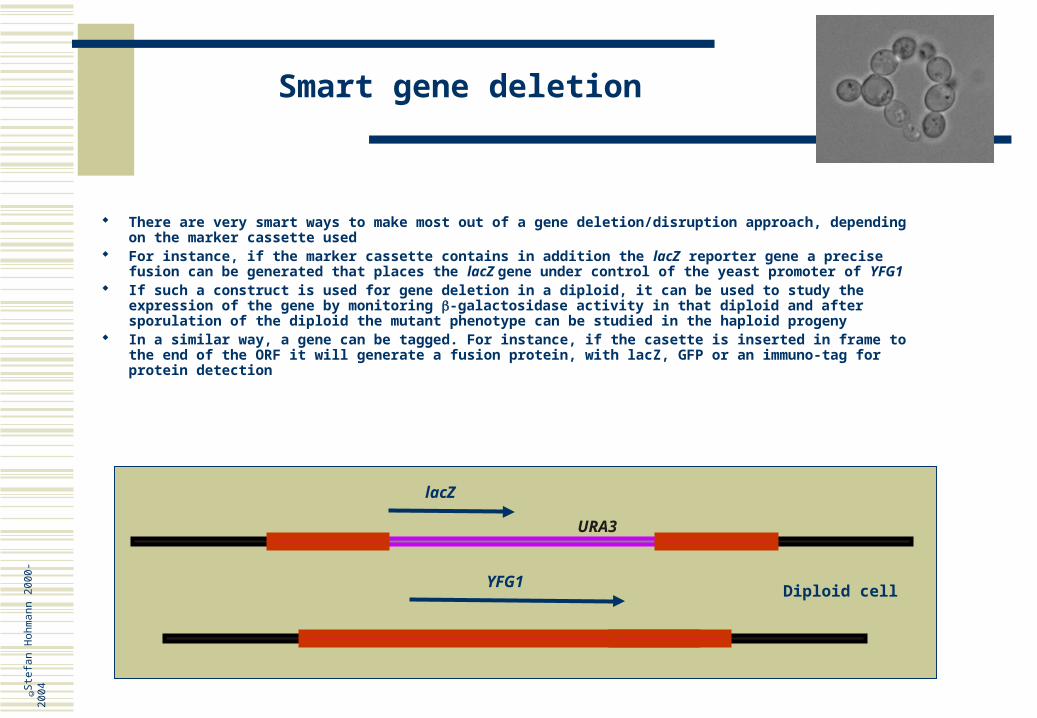
There are very smart ways to make most out of a gene deletion/disruption approach, depending on the marker cassette used
For instance, if the marker cassette contains in addition the lacZ reporter gene a precise fusion can be generated that places the lacZ gene under control of the yeast promoter of YFG1
If such a construct is used for gene deletion in a diploid, it can be used to study the expression of the gene by monitoring -galactosidase activity in that diploid and after sporulation of the diploid the mutant phenotype can be studied in the haploid progeny
In a similar way, a gene can be tagged. For instance, if the casette is inserted in frame to the end of the ORF it will generate a fusion protein, with lacZ, GFP or an immuno-tag for protein detection
URA3
lacZ
YFG1Diploid cell
© Ste
fan
Hoh
man
n 20
00-
2004
Smart gene deletion
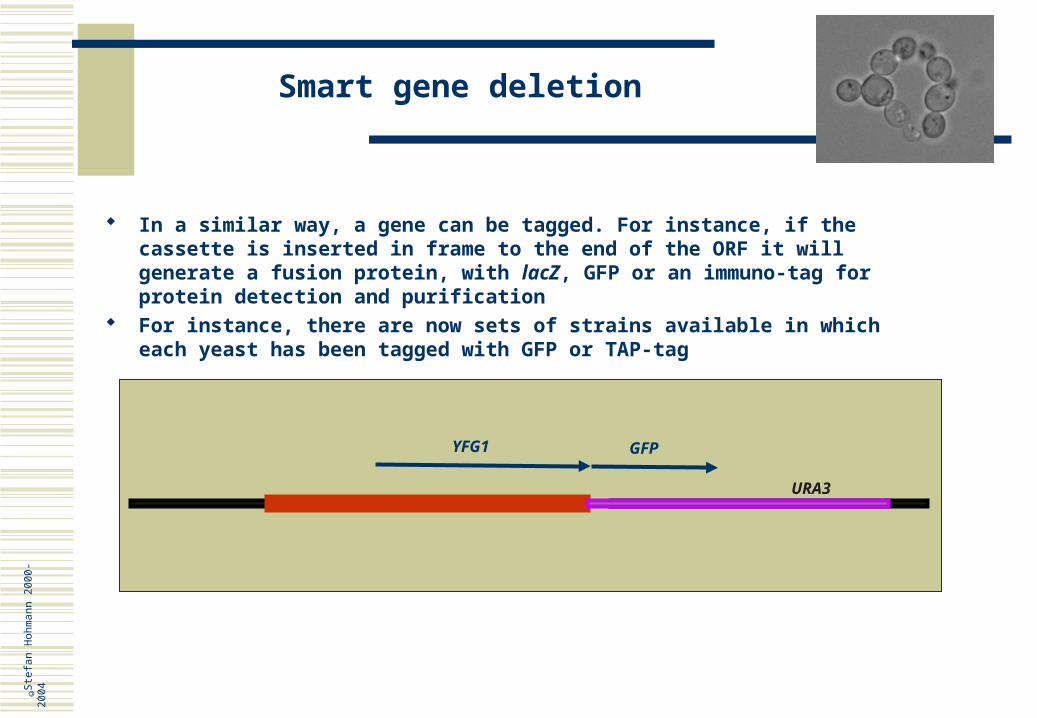
In a similar way, a gene can be tagged. For instance, if the cassette is inserted in frame to the end of the ORF it will generate a fusion protein, with lacZ, GFP or an immuno-tag for protein detection and purification
For instance, there are now sets of strains available in which each yeast has been tagged with GFP or TAP-tag
YFG1
URA3
GFP
© Ste
fan
Hoh
man
n 20
00-
2004
There are some ways to delete a yeast gene without leaving any trace behind, i.e. no marker gene
This is very important if one wants to re-use the marker in order to make many deletions in one and the same strain (there are strains with more than 20 deletions!)
It is also important for industrial yeast strains; when one wants to engineer those at the end no foreign DNA should be left behind (but for hardliners on genetic engineering the intermediate presence of foreign DNA ina yeast is already ”dangerous”)
All these methods use homologous recombination a second time, i.e. to pop-out the integrated DNA again
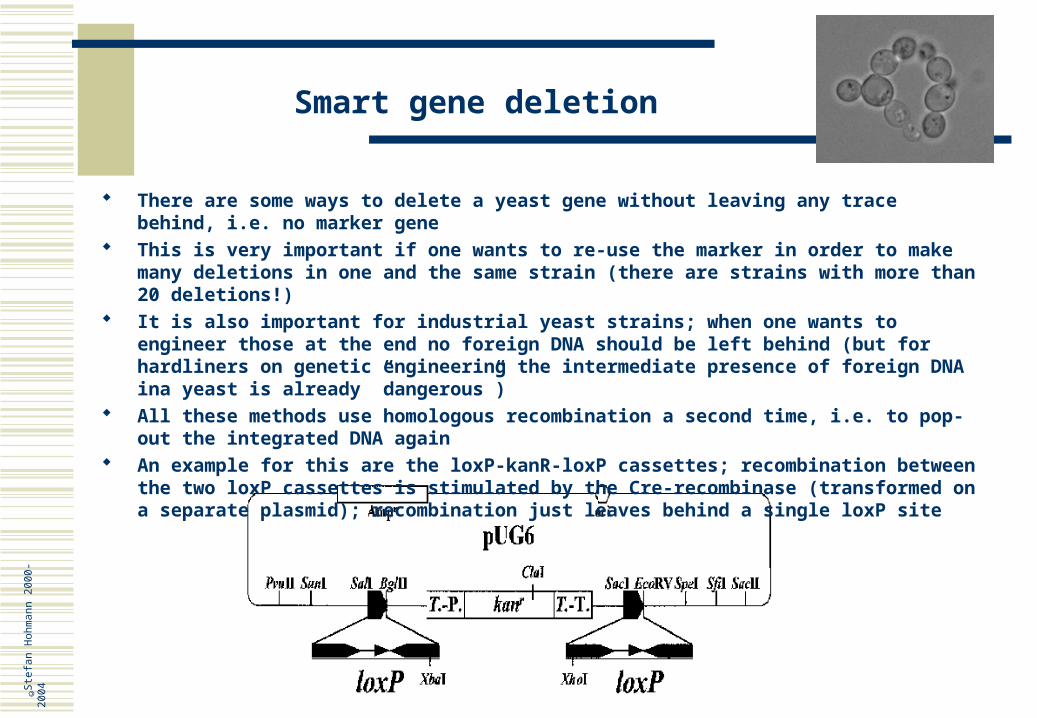
An example for this are the loxP-kanR-loxP cassettes; recombination between the two loxP cassettes is stimulated by the Cre-recombinase (transformed on a separate plasmid); recombination just leaves behind a single loxP site
Smart gene deletion
© Ste
fan
Hoh
man
n 20
00-
2004
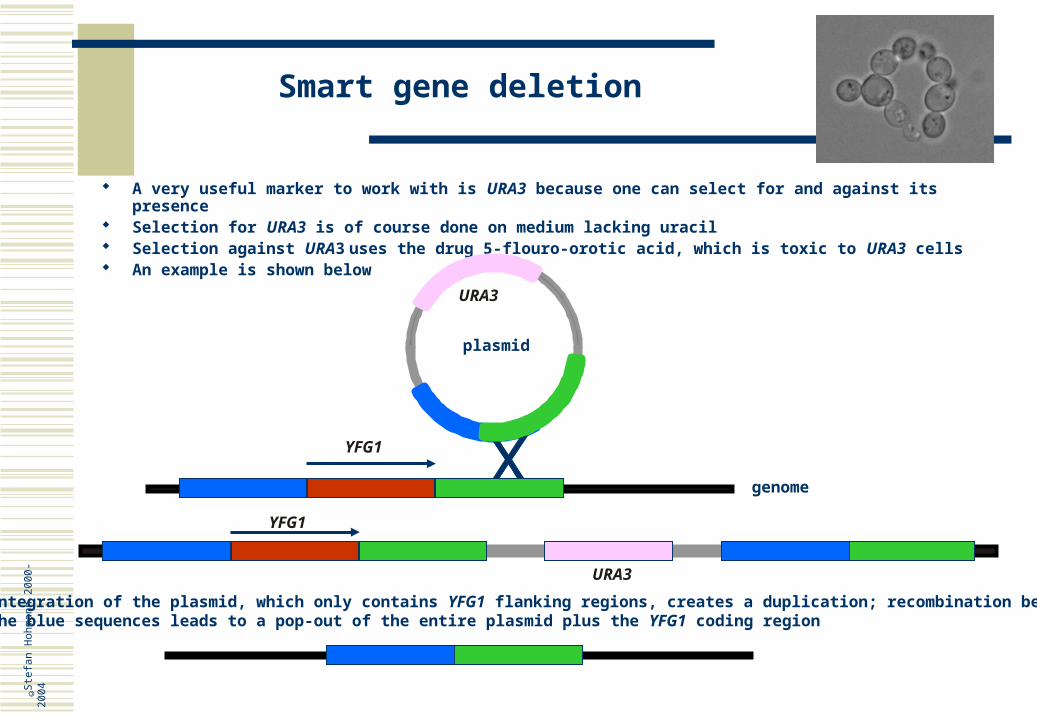
Smart gene deletion
A very useful marker to work with is URA3 because one can select for and against its presence Selection for URA3 is of course done on medium lacking uracil Selection against URA3 uses the drug 5-flouro-orotic acid, which is toxic to URA3 cells An example is shown below
URA3
URA3
plasmid
genome
YFG1
YFG1
Integration of the plasmid, which only contains YFG1 flanking regions, creates a duplication; recombination betweenthe blue sequences leads to a pop-out of the entire plasmid plus the YFG1 coding region

© Ste
fan
Hoh
man
n 20
00-
2004
How to deal with essential genes
We have discussed now random chemical and targetted mutagenesis; an obvious question is: how can we identify and work with mutations in genes whose products are essential for the cell (and that is about 1/3)? A mutation that knocks out the function of that protein kills the cell and it is difficult to work with dead cells....
For chemical mutagenesis the most common approach is to work with conditional mutations; usually these are mutations where the gene product functions at a lower temperature, like 25°C, but not at higher temperature, like 37°C; the mutant is temperature-sensitive; many essential cellular functions have been identified through ts mutants
To determine in gene deletion experiments if a gene is essential, the deletion is done in a diploid; if after sporulation only two spores survive and if all living spores do not have the marker used for the deletion, the gene is regarded as essential
One can work with mutants in essential genes. Principally, the mutant is transformed with plasmid that expresses the relevant gene conditionally.
For instance a plasmid contains the essential gene under the control of the promoter of the GAL1 gene; this promoter is ”on” on galactose medium but ”off” on glucose medium; when shifting cells to glucose one can study at least for some time the properties of the cells...and watch them dying (yfg1 pGAL1-YFG1)
To analyse the function of in vitro generated point mutants, one can use plasmid shuffling. For this, the mutant is first transformed with the wild type gene and then with a mutant gene. The plasmid with the wild type gene carries URA3 as selectable marker, which can be forced to be lost on medium with 5-FOA. If the mutant grows on 5-FOA medium, the mutant allele is functional (yfg1 pURA3::YFG1 pLEU2::yfg1-1).

© Ste
fan
Hoh
man
n 20
00-
2004
From gene disruption to transposon mutagenesis
The gene deletion/disruption technique has been taken a step further to be used in random mutagenesis
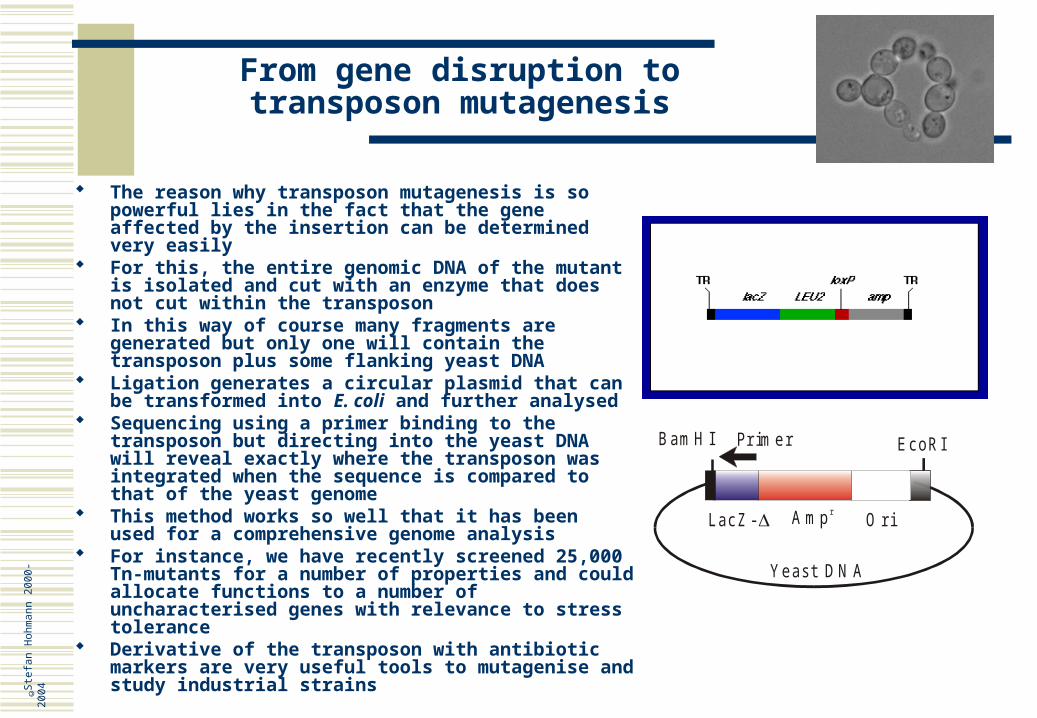
For this a gene library is first constructed as discussed before such that the inserted yeast DNA can be cut out with NotI, an enzyme that only cuts a very few times in the yeast genome
Then this library is mutagenised with a transposon in E. coli, where the Tn randomly integrates into the yeast DNA
Subsequently, the entire mix of NotI fragments is transformed into yeast where it is expected to replace genes; with about 30,000 yeast clones a more then 90% coverage of the genome is achieved
The Tn used is a quite sophisticated example of such a transposon, that can be partially cut out again through the lox-sites. This creates a tag, which allows immunolocalisation of the gene product
TR: Tn3 terminal inverted repeats Xa: Factor Xa cleavage recognition site loxR: lox site, target for Cre recombinase lacZ: 5'-truncated lacZ gene encoding -
galactosidase URA3 gene from S. cerevisiae tet: tetracycline resistance gene res: Tn3 site for resolution of transposition
intermediate loxP: lox site, target for Cre recombinase 3xHA: Hemagglutinin (HA) triple epitope tag
© Ste
fan
Hoh
man
n 20
00-
2004
From gene disruption to transposon mutagenesis
The reason why transposon mutagenesis is so powerful lies in the fact that the gene affected by the insertion can be determined very easily
For this, the entire genomic DNA of the mutant is isolated and cut with an enzyme that does not cut within the transposon
In this way of course many fragments are generated but only one will contain the transposon plus some flanking yeast DNA
Ligation generates a circular plasmid that can be transformed into E. coli and further analysed
Sequencing using a primer binding to the transposon but directing into the yeast DNA will reveal exactly where the transposon was integrated when the sequence is compared to that of the yeast genome
This method works so well that it has been used for a comprehensive genome analysis
For instance, we have recently screened 25,000 Tn-mutants for a number of properties and could allocate functions to a number of uncharacterised genes with relevance to stress tolerance
Derivative of the transposon with antibiotic markers are very useful tools to mutagenise and study industrial strains
Y east D N A
L acZ - A mp r
E coR IBamH I
O r i
Pr imer
© Ste
fan
Hoh
man
n 20
00-
2004
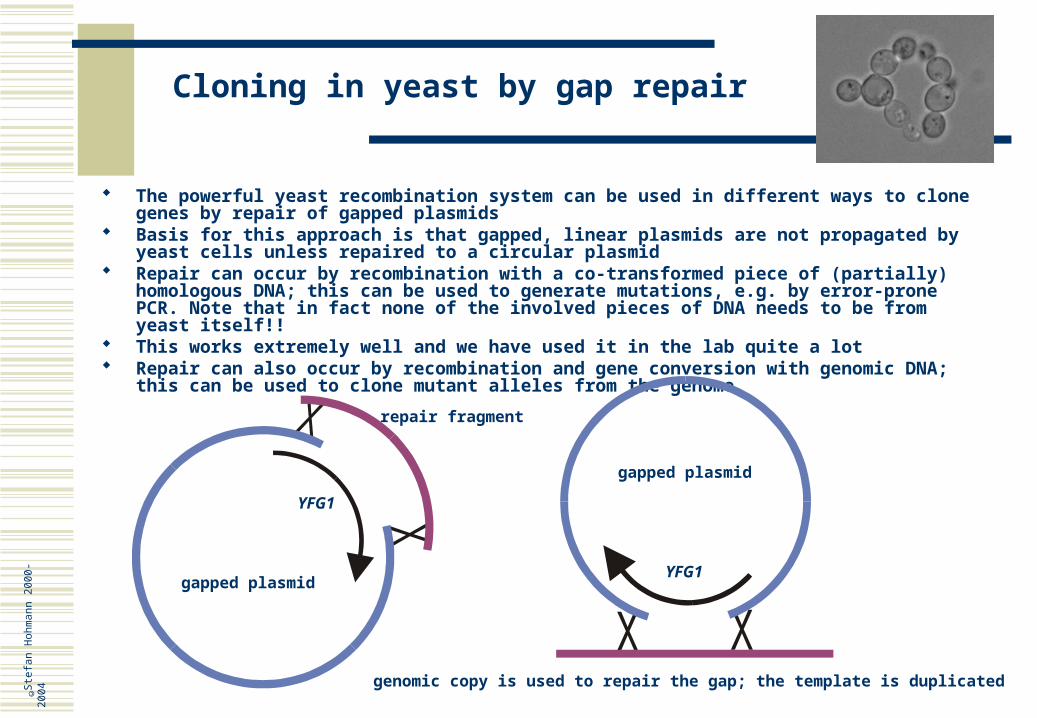
Cloning in yeast by gap repair
The powerful yeast recombination system can be used in different ways to clone genes by repair of gapped plasmids
Basis for this approach is that gapped, linear plasmids are not propagated by yeast cells unless repaired to a circular plasmid
Repair can occur by recombination with a co-transformed piece of (partially) homologous DNA; this can be used to generate mutations, e.g. by error-prone PCR. Note that in fact none of the involved pieces of DNA needs to be from yeast itself!!
This works extremely well and we have used it in the lab quite a lot Repair can also occur by recombination and gene conversion with genomic DNA; this can be
used to clone mutant alleles from the genome
X
Xgapped plasmid
repair fragment
X X
gapped plasmid
genomic copy is used to repair the gap; the template is duplicated
YFG1
YFG1
© Ste
fan
Hoh
man
n 20
00-
2004
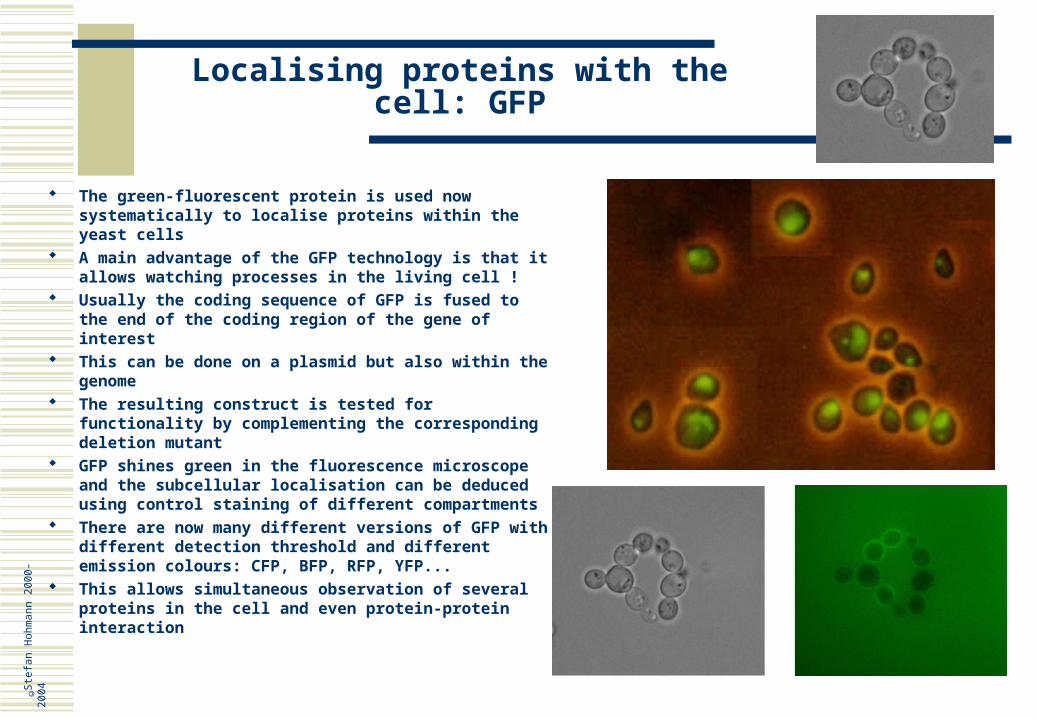
Localising proteins with the cell: GFP
The green-fluorescent protein is used now systematically to localise proteins within the yeast cells
A main advantage of the GFP technology is that it allows watching processes in the living cell !
Usually the coding sequence of GFP is fused to the end of the coding region of the gene of interest
This can be done on a plasmid but also within the genome
The resulting construct is tested for functionality by complementing the corresponding deletion mutant
GFP shines green in the fluorescence microscope and the subcellular localisation can be deduced using control staining of different compartments
There are now many different versions of GFP with different detection threshold and different emission colours: CFP, BFP, RFP, YFP...
This allows simultaneous observation of several proteins in the cell and even protein-protein interaction

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: isolating more genes
So far we have discussed different ways to generate mutations in yeast: chemical random mutagenesis random targetted mutagenesis with transposon-tagged DNA targetted deletion/disruption of yeast genes
and we have discussed some methods to study and engineer genes in yeast by fusion with a reporter gene to monitor gene expression by fusion with an epitope or with GFP to study the protein level or protein localisation
The power of genetic analysis lies in the possibility to use one gene/mutant to isolate further genes, which encode proteins involved in the same or in parallel or related cellular processes
The same genetic approaches can be used to allocate different genes/proteins to the same (or to different) cellular functions and to sort them in an order, for instance within a signalling pathway
Such approaches to get further include Multi-copy suppression Suppressor mutation Synthetic lethality The yeast two-hybrid system
All these systems are used in multiple variations; the intellectual challenge is to find the conditions that allow the approach to be used

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: suppressors
Definition: a reversion of a mutation means that the primary lesion is repaired and hence the orginal, wild-type situation is restored; obviously, a deletion mutant never can revert
Definition: a suppressor is a gene or mutation that (partially) overcomes the effect caused by a given mutation; hence a suppressor is a second-site genetic alteration that somehow restores (partially) the wild type situation
Suppressors can be intragenic, i.e. a second mutation in the same gene/protein can restore (partial) functionality of the gene product; again, this is only possible with point mutations and not with deletion mutants
More common are extragenic suppressor and we will discuss multi-copy suppressors and suppressor mutations
How a suppressor functions differs of course a lot from system to system but usually the analysis of the suppressor function provides a lot of important information
Principally, a suppressor either activates (or represses) the system affected by the primary mutation in another way or activates (or represses) an alternative, partially redundant system
Suppressors are useful as we discuss them here but at the same time can be annoying: yeast mutants that poorly grow can easily generate suppressors, something one has to be aware of when working with such mutants

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: multi-copy suppression
Multi-copy suppression is based on overexpression of a gene, usually on a multi-copy plasmid or via ectopic expression from a strong promoter
A multi-copy (or gene dosage) suppressor is a gene, which, when expressed at high levels, overcomes (some of) the effects of a certain mutation
Multi-copy suppression as a tool in gene discovery is exciting in a way: you hardly ever know what you will get.....
Generally, however, one expects genes whose products function downstream in the same pathway or in a parallel pathway
A nice thing about multi copy suppression: you get to the gene right away! Turning the argumentation around, if one knows from other genetic experiments that two genes are
functionally related, multi-copy suppression is a way to sort two proteins within a pathway within an epsitasis analysis: only a gene whose product functions downstream of the mutation can suppress in multi-copy
XPbs2p and Hog1p are in the same pathway and Hog1p is activated by Pbs2p. Overexpressed Hog1p may confer sufficient activity to mediate the required function even in the absence of Hog1p.
X
common target
Two parallel pathways share one or several common targets. Overexpression and hence higher activity of the parallel pathway may be sufficient to activate the target.

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: suppressor mutations
An extragenic suppressor mutation alters a different gene product such that the, or one of the, effects of a certain mutation are overcome
Like with multi-copy suppression there are many ways in which this can happen and the outcome of such an approach is often quite surprising but very informative
Typical suppressor mutations are those that activate a gene product downstream of the primary lesion in the same pathway; since such mutations cause a gain of function they are usually dominant
Other typical suppressor mutations knock out a repressor downstream in the same or in a parallel pathway; since such mutations cause a loss of function they are recessive
A suppressor mutation may also activate or inactivate pathways/systems that affect in some way the same physiological system than the primary lesion
If a given protein is part of a multimeric complex and the primary mutation is a point mutation, extragenic suppressor mutations might occur such that protein interactions are restored; hence this is a method to identify interacting proteins
XPbs2p and Hog1p are in the same pathway and Hog1p is activated by Pbs2p. A mutation that renders Hog1p active even without activation would suppress the pbs2 mutation and is probably dominant
The pathway ultimately inactivates a negative regulator, e.g. the repressor Sko1p; knock out of the repressor could overcome inactivation of the pathway; the mutation is most likely recessive
X
XSko1
© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: synthetic lethality
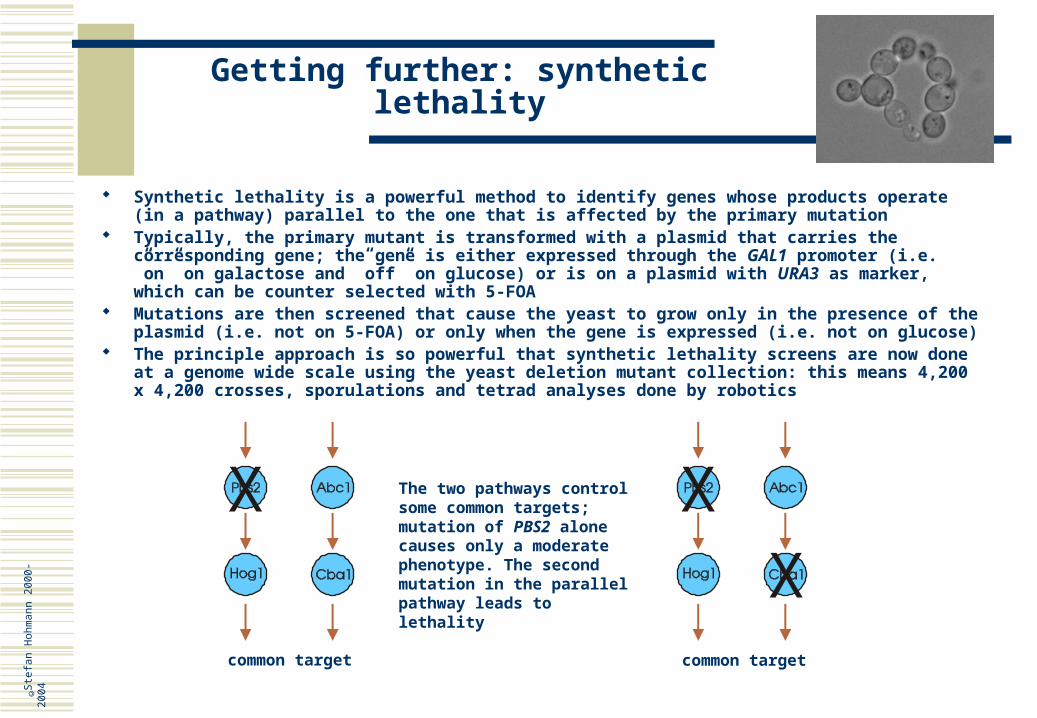
Synthetic lethality is a powerful method to identify genes whose products operate (in a pathway) parallel to the one that is affected by the primary mutation
Typically, the primary mutant is transformed with a plasmid that carries the corresponding gene; the gene is either expressed through the GAL1 promoter (i.e. ”on” on galactose and ”off” on glucose) or is on a plasmid with URA3 as marker, which can be counter selected with 5-FOA
Mutations are then screened that cause the yeast to grow only in the presence of the plasmid (i.e. not on 5-FOA) or only when the gene is expressed (i.e. not on glucose)
The principle approach is so powerful that synthetic lethality screens are now done at a genome wide scale using the yeast deletion mutant collection: this means 4,200 x 4,200 crosses, sporulations and tetrad analyses done by robotics
XX
common target
X
common target
The two pathways control some common targets; mutation of PBS2 alone causes only a moderate phenotype. The second mutation in the parallel pathway leads to lethality

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: epistasis I
The concepts of suppressor analysis and synthetic lethality are also the basis for a powerful tool of genetics, epistasis analysis
In a way it is similar to complementation analysis (How many different genes in the mutant collection cause the same phenotype?) as epistasis analysis asks the question: how many genes/proteins are involved in the same genetic system/pathway and in which order do they function?
The basic idea is to combine two mutations in the same cell, i.e. to generate a double mutant; the phenotype of the double mutant may reveal if the two gene products work in the same or in parallel pathways and they may reveal the order within a pathway.
XX
common target
Let us first assume mutation in all these four proteins cause similar phenotypes, such as moderate sensitivity to salt
When we combine the hog1 and the pbs2 in a hog1 pbs2 double mutant then we would expect that the double mutant has the same level of sensitivity as each single mutant; we would conclude that they function in the same pathway
When we combine the pbs2 and the cba1 mutation in a pbs2 cba1 double mutant we would expect a strongly enhanced sensitivity of the double mutant as compared to the single mutants; we would conclude that Hog1p and Cba1p work in different, though parallel pathways

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: epistasis II
Let us now assume that deletion of PBS2 (and of HOG1) causes sensitivity to high salt concentrations while deletion of SKO1 causes higher tolerance to salt in the medium
If those proteins act in the same pathway there are different possibilities for the phenotype of the pbs2 sko1 or hog1 sko1 double mutant
If Sko1p were downstream of Pbs2p and Hog1p we would expect that the double mutant is tolerant, i.e. has the same phenotype as the sko1 single mutant: sko1 would be epistatic (”dominant over”) to pbs2 and hog1 (and this is really the case)
If Sko1p were upstream of Hog1p and Pbs2p we would expect that the double mutant pbs2 sko1 and hog1 sko1 is sensitive to salt
X
XSko1

© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: epistasis III
Also multi copy suppression or activating mutations are useful tools in epistasis analysis
Suppression by overexpression can only work for a gene/protein functioning downstream of the primary lesion, as indicated here for Hog1p; overexpression of PBS2 would not suppress a hog1 mutation
In a similar way, an activating mutation of HOG1 can suppress the salt sensitivity of a pbs2 mutant, but not vice versa, and this is indeed exactly how it works
The epistasis concept has been used in very many examples to analyse the order of events in signalling pathways and other cellular systems: if the phenotype of the double mutant resembles that of one of the single mutants the latter gene product functions further downstream in the system, i.e. closer to the physiological effect
X
X
© Ste
fan
Hoh
man
n 20
00-
2004
Getting further: two-hybrid system
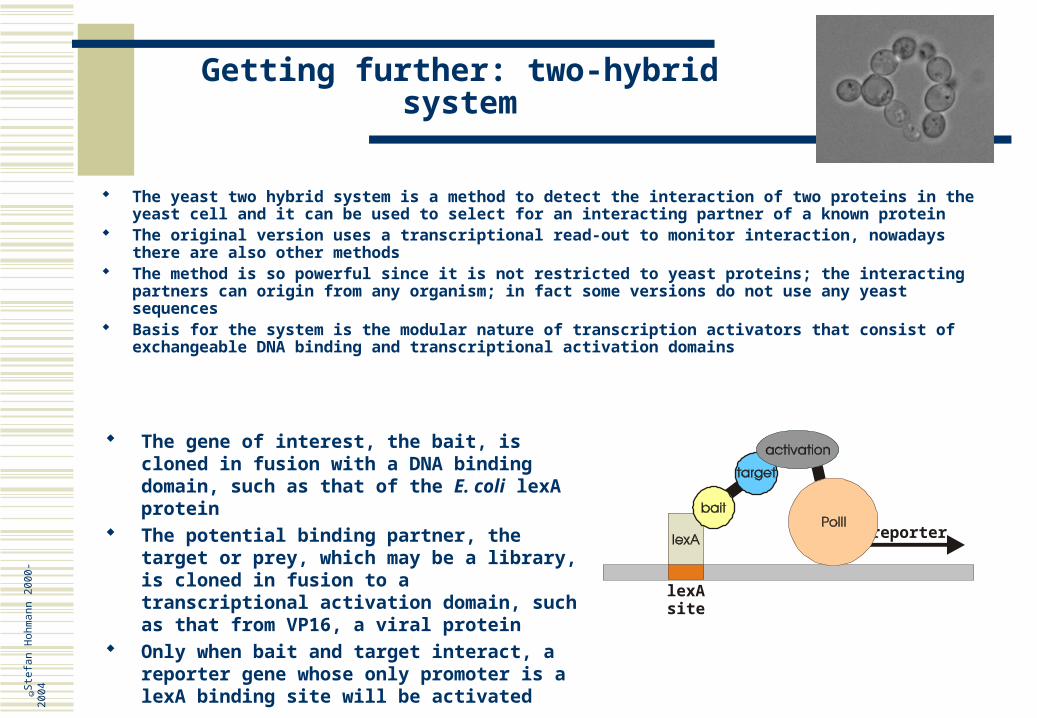
The yeast two hybrid system is a method to detect the interaction of two proteins in the yeast cell and it can be used to select for an interacting partner of a known protein
The original version uses a transcriptional read-out to monitor interaction, nowadays there are also other methods
The method is so powerful since it is not restricted to yeast proteins; the interacting partners can origin from any organism; in fact some versions do not use any yeast sequences
Basis for the system is the modular nature of transcription activators that consist of exchangeable DNA binding and transcriptional activation domains
lexAsite
reporter
The gene of interest, the bait, is cloned in fusion with a DNA binding domain, such as that of the E. coli lexA protein
The potential binding partner, the target or prey, which may be a library, is cloned in fusion to a transcriptional activation domain, such as that from VP16, a viral protein
Only when bait and target interact, a reporter gene whose only promoter is a lexA binding site will be activated

© Ste
fan
Hoh
man
n 20
00-
2004
Application of the yeast two-hybrid system
The possible applications to the two-hybrid system are absolutely tremendous The system can be used to detect interaction between two proteins The system can be used to characterise the domains and residues in the two proteins that
mediate interaction; this can be done by mutagenesis and the use of a counterselectable reporter, such as URA3
The system can of course be used to find interaction partners The system can be used to find proteins that regulate the interaction between two proteins The system can be used to screen for drugs that inhibit the interaction between two proteins The system is actually used to construct an genome-wide map of protein interactions in yeast;
using laboratory robots 6000 bait strains are crossed to 6000 prey strains to study all possible protein intercations
etc................
© Ste
fan
Hoh
man
n 20
00-
2004
Genetic analysis in action: the HOG pathway
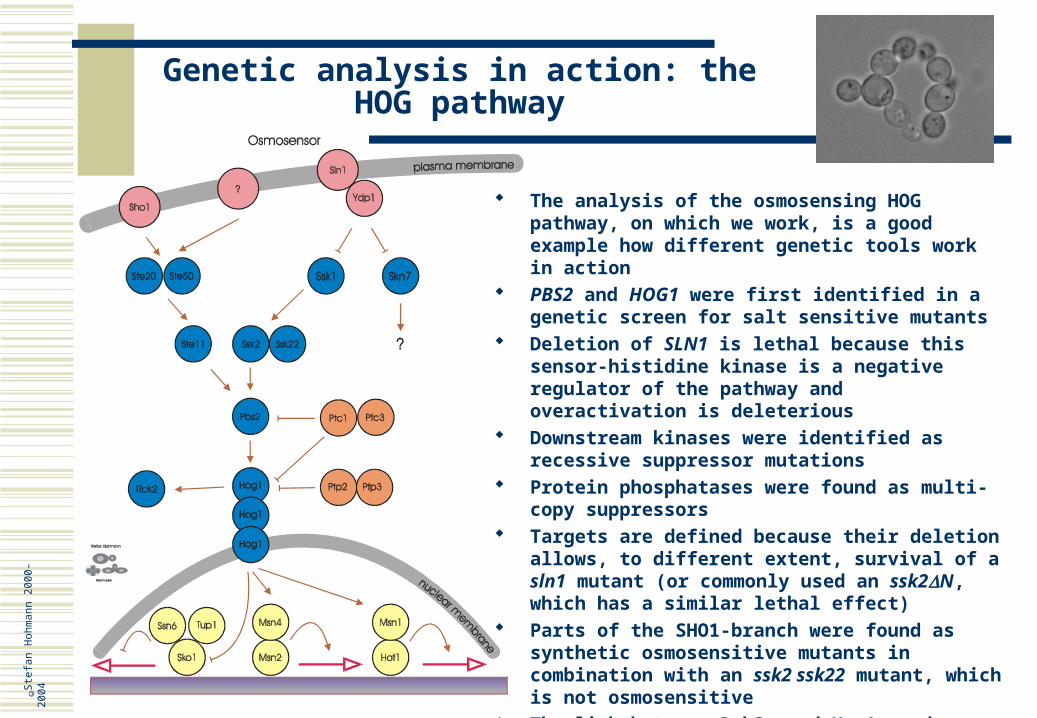
The analysis of the osmosensing HOG pathway, on which we work, is a good example how different genetic tools work in action
PBS2 and HOG1 were first identified in a genetic screen for salt sensitive mutants
Deletion of SLN1 is lethal because this sensor-histidine kinase is a negative regulator of the pathway and overactivation is deleterious
Downstream kinases were identified as recessive suppressor mutations
Protein phosphatases were found as multi-copy suppressors
Targets are defined because their deletion allows, to different extent, survival of a sln1 mutant (or commonly used an ssk2N, which has a similar lethal effect)
Parts of the SHO1-branch were found as synthetic osmosensitive mutants in combination with an ssk2 ssk22 mutant, which is not osmosensitive
The link between Rck2p and Hog1p and between Hog1p and Hot1p was found in two-hybrid screens
© Ste
fan
Hoh
man
n 20
00-
2004
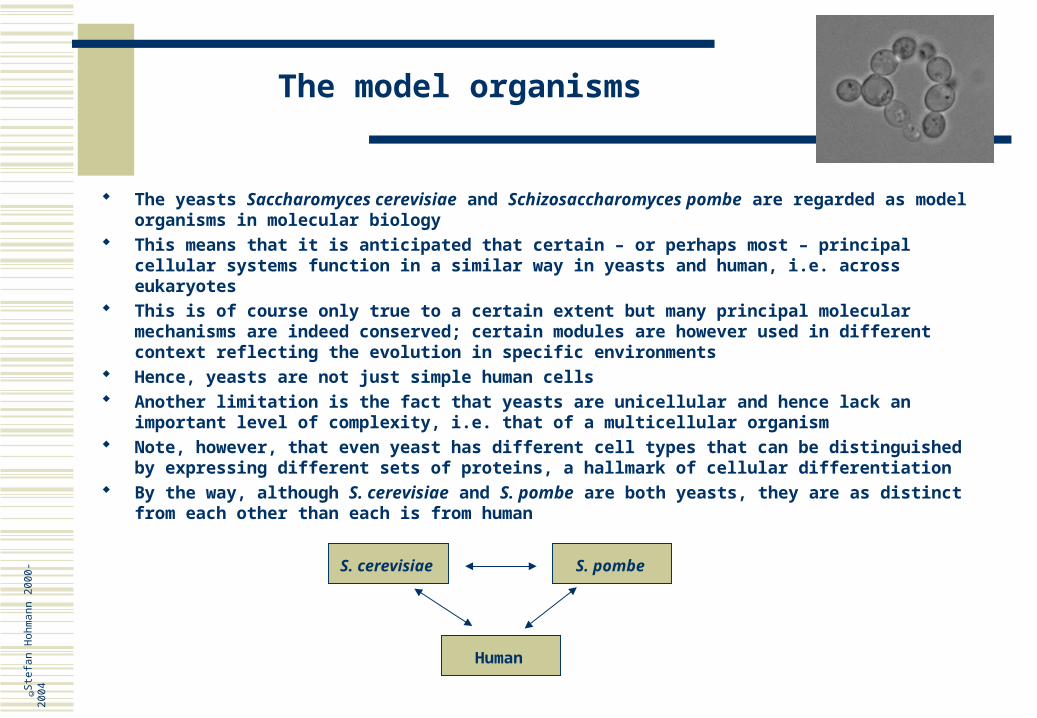
The model organisms
The yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe are regarded as model organisms in molecular biology
This means that it is anticipated that certain – or perhaps most – principal cellular systems function in a similar way in yeasts and human, i.e. across eukaryotes
This is of course only true to a certain extent but many principal molecular mechanisms are indeed conserved; certain modules are however used in different context reflecting the evolution in specific environments
Hence, yeasts are not just simple human cells Another limitation is the fact that yeasts are unicellular and hence lack an important level of
complexity, i.e. that of a multicellular organism Note, however, that even yeast has different cell types that can be distinguished by expressing
different sets of proteins, a hallmark of cellular differentiation By the way, although S. cerevisiae and S. pombe are both yeasts, they are as distinct from
each other than each is from human
S. cerevisiae S. pombe
Human
© Ste
fan
Hoh
man
n 20
00-
2004
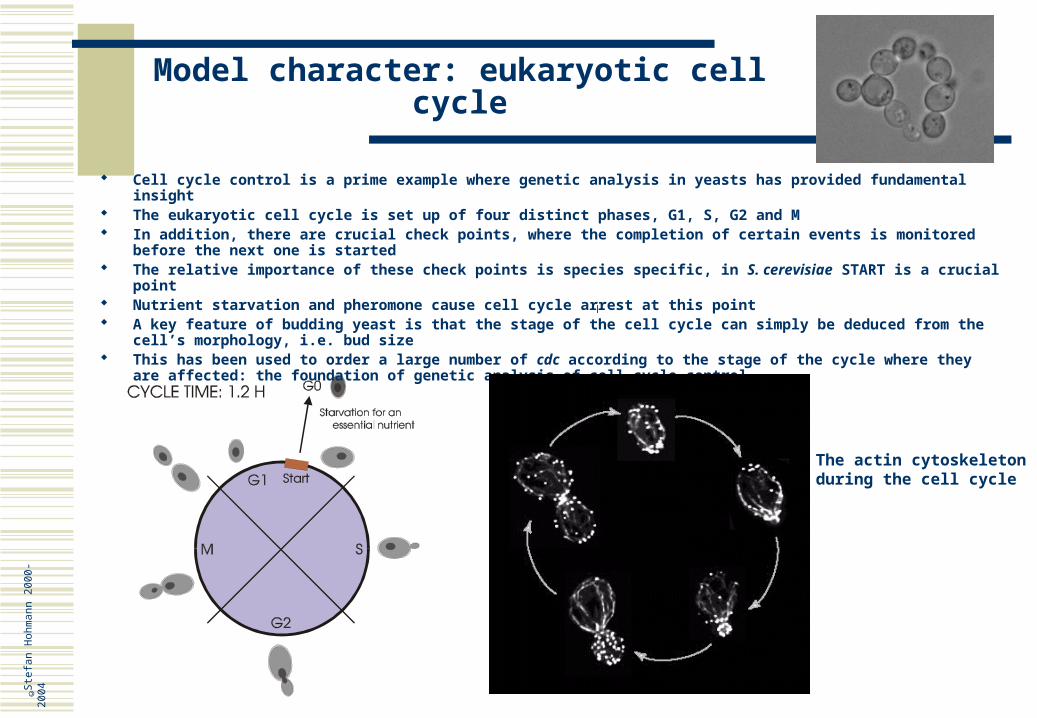
Model character: eukaryotic cell cycle
Cell cycle control is a prime example where genetic analysis in yeasts has provided fundamental insight The eukaryotic cell cycle is set up of four distinct phases, G1, S, G2 and M In addition, there are crucial check points, where the completion of certain events is monitored before the next
one is started The relative importance of these check points is species specific, in S. cerevisiae START is a crucial point Nutrient starvation and pheromone cause cell cycle arrest at this point A key feature of budding yeast is that the stage of the cell cycle can simply be deduced from the cell’s
morphology, i.e. bud size This has been used to order a large number of cdc according to the stage of the cycle where they are affected: the
foundation of genetic analysis of cell cycle control
The actin cytoskeleton during the cell cycle

© Ste
fan
Hoh
man
n 20
00-
2004
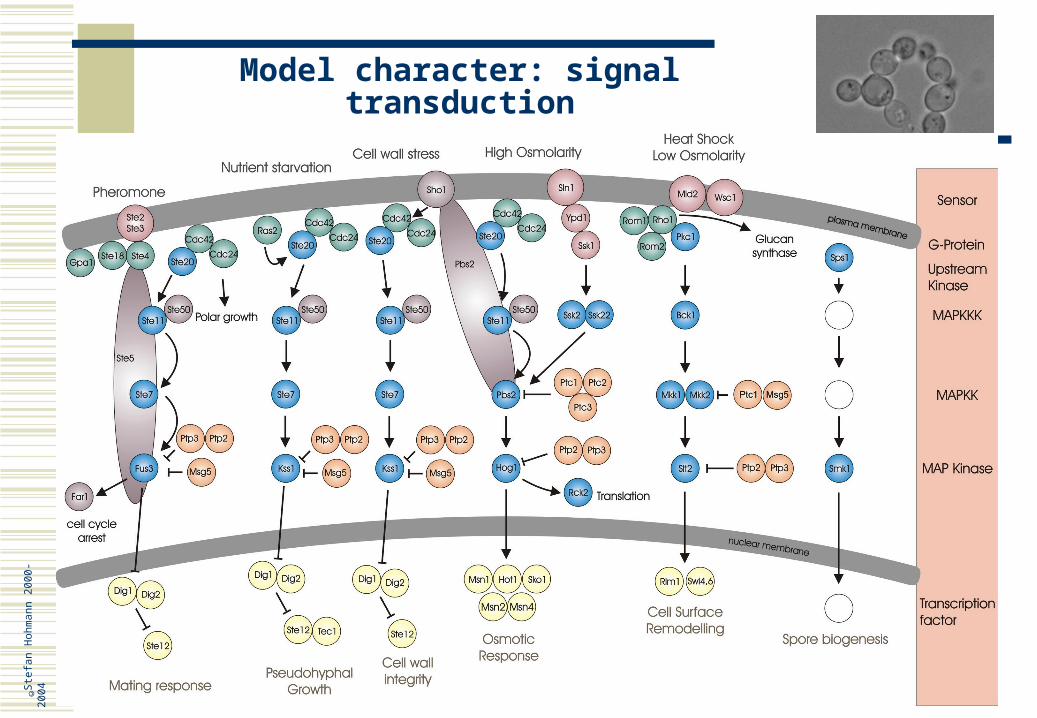
Model character: signal transduction
The principles of signal transduction are well conserved among eukaryotic cells For instance, animals and fungi use cAMP as a second messenger and it seems
that cAMP mediates nutritional signals For instance, all eukaryotic cells have common classes of signalling proteins, such
as G-protein couples receptors, a type of hormone receptors; the yeast pheromone receptors belong to this class
A prototypical eukaryotic signalling system are MAP (mitogen activated protein) kinase cascades; these are modules of three protein kinases that typically control gene expression; the module is used in many signalling pathways responsive to different stimuli and hence controlled by different sensing mechanisms
S. cerevisiae has at least six such pathways, which together control cellular morphology and responses to pheromone and environmental stress
Genetic analysis in yeast has and is contributing greatly to the understanding of how these pathways function
There are of course also limitations to the model character; for instance S. cerevisiae is lacking receptor tyrosine kinases or nuclear receptors, important classes of mammalian hormone receptors
© Ste
fan
Hoh
man
n 20
00-
2004
Model character: signal transduction
© Ste
fan
Hoh
man
n 20
00-
2004
Model character: morphology switch
We have already pointed out that yeast cells can switch their morphology This switch requires a MAP kinase pathway and nutritional signals; also cAMP plays a role The yeast pseudohyphal switch (or invasive growth in haploids) is a model system for
morphogenesis Most importantly, a morphological switch is associated with pathogenesis for instance of
Candida albicans and hence much research is focussed on the basic mechanisms S. cerevisiae may use the switch and co-expression of polysaccharide degrading enzymes to
penetrate plant tissues
© Ste
fan
Hoh
man
n 20
00-
2004
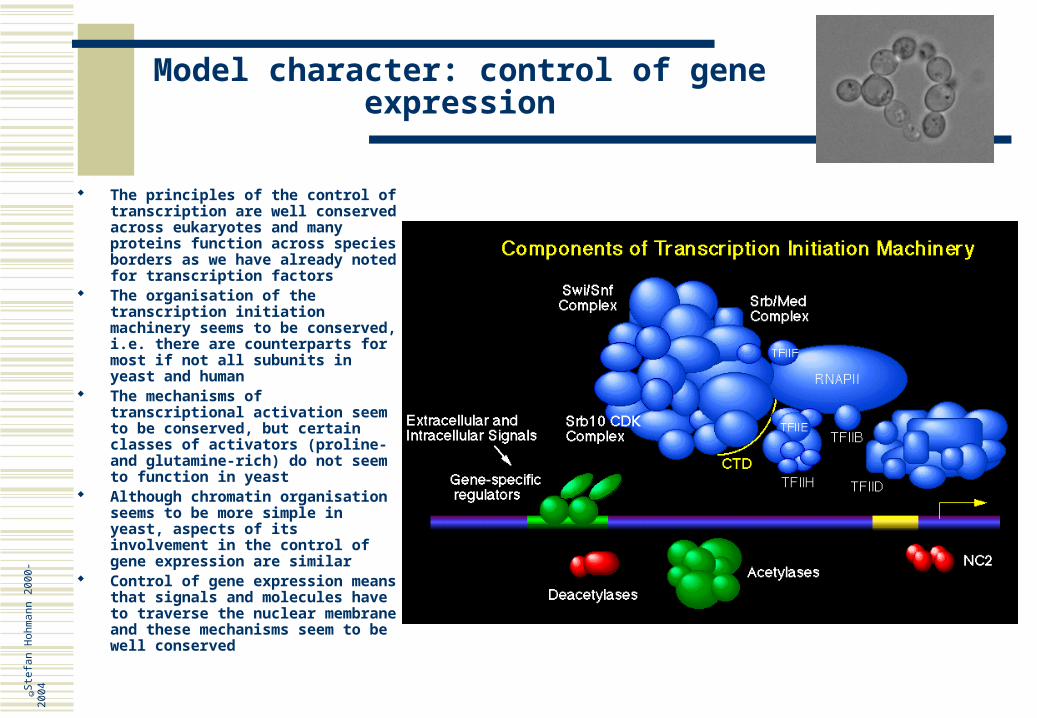
Model character: control of gene expression
The principles of the control of transcription are well conserved across eukaryotes and many proteins function across species borders as we have already noted for transcription factors
The organisation of the transcription initiation machinery seems to be conserved, i.e. there are counterparts for most if not all subunits in yeast and human
The mechanisms of transcriptional activation seem to be conserved, but certain classes of activators (proline- and glutamine-rich) do not seem to function in yeast
Although chromatin organisation seems to be more simple in yeast, aspects of its involvement in the control of gene expression are similar
Control of gene expression means that signals and molecules have to traverse the nuclear membrane and these mechanisms seem to be well conserved

© Ste
fan
Hoh
man
n 20
00-
2004
Model character: vesicular transport
Vesicular transport, i.e. the mechanisms that control the trafficking of proteins and membranes is another feature that is highly conserved across eukaryotes
Temperature sensitive sec mutants have been sorted according to the stage where transport stops (using electron micoscopy) and this has been the foundation for genetic analysis
In addition, transport to the vacuole and endocytosis are studied by genetic analysis combined with biochemistry and cell biology

© Ste
fan
Hoh
man
n 20
00-
2004
Model character: proteasome
The proteasome is a multi protein complex conserved in eukaryotes
It is located in the cytoplasm and the nucleus and controls degradtion of proteins that have been ubiquitinated
The 26S proteasome consist of a 20S catalytic and a 19/22S regulatory subunit
The 20S proteasome is composed of 14 different proteins and all genes are known in yeast
The yeast 20S complex has been purified and the X-ray structure has been determined

© Ste
fan
Hoh
man
n 20
00-
2004
Model character: the unexpected
Prions Have of course been in the focus of interest through mad cow disease Yeast also has two systems that seem to have all features of prions! This means they are genetic
elements, alleles of known genes, that behave as non-Mendelian genetic elements: PSI+ (Sup35p), a protein involved in translation termination and URE3 (Ure2p), a regulator of nitrogen metabolism
Ageing Is a process very much assocated with multicellular organisms Yeast cells have a pre-determined life span, i.e. mother cells die after a certain number of divisions The ageing process in yeast seems to have some features in common with that of human, for instance
the accumulation of rDNA circles There is also a ”common” gene, WRN (Werner’s syndrom) in human and SGS1 in yeast; the genes are
homologous and mutations causes premature ageing in human and yeast, respectively
Cell type determination As discussed earlier, yeast develops different cell types determined by different gene expression pattern

© Ste
fan
Hoh
man
n 20
00-
2004
Functional genomics
The term functional genomics is not very well defined; since it is a nice term to attract funding these days many people call functional genomics what they have done for ages
Strictly, it should probably mean ”the determination of the function of previously uncharacterised genes identified by genome sequencing”
This aspect is indeed addressed in a systematic way in yeast by at least two different projects; their goal is the construction of deletion strains for all 6,200 genes and an initial phenotypic characterisation; the set is complete
Functional information can also come through other approaches; for instance, the yeast two-hybrid system is used to construct a complete protein interaction map
Transposon mutagenesis is used to tag a large number of yeast proteins to determine their localisation
Functional information also comes from expression analysis Expression of proteins is studied by 2D gel electrophoresis, which can resolve some 1,000
different yeast proteins Analysis of the expression of all 6,200 yeast genes has now become reality allowing a
comprehensive picture of transcriptional changes depending on conditions or in certain mutants
© Ste
fan
Hoh
man
n 20
00-
2004
Functional genomics: transcriptional profiling
Transcriptional profiling in yeast is reality now and a number of articles using the technology have appeared
A large data collection is generated in Stanford covering a number of growth conditions
Another large collection generated by Rick Young’s lab concerns effects of mutations in certain components of the transcription initiation machinery
We have used transcriptional profiling to study signal transdution in stress responses

© Ste
fan
Hoh
man
n 20
00-
2004
From functional genomics to systems biology
Systems biology goes a step further then functional analysis: the goal of systems biology is to describe the operation of the entire cell with all its proteins
In a more narrow definition, systems biology combines mathematical and experimental approaches to achieve a better understanding of biological networks and systems
Systems biology is a multidisciplinary approach involving biologists, engineers and mathematicians
There are two principle goals within systems biology: (1) to describe the wiring network of all proteins in the cell and (2) to decsribe the dynamic operation in the cell
Reconstruction of the wiring network uses all available data such as genetic, gene expression, protein interaction data to connect proteins with each other
Dynamic modelling and experimentation aims at decribing the overriding rules how e.g. metabolism and signalling dynamically operate
We use such approaches to understand how signalling pathways operate

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast biotechnology: fermentation industry
The yeast fermentation industry, comprising baking, brewing, wine making and industrial alcohol production, is still the biggest BioTech business world-wide
Industrial yeast strains are usually difficult to work with because they are diploid, polyploid or even aneuploid; many appear to be cross-species hybrids
There are many possible improvements to the fermentation processes, where the biology of yeast is the limiting factor; hence there are many attempts to improve yeasts
Wine yeasts: ability to perform the malolactic fermentation, which is normally performed by lactic acid bacteria (faster and more reliable production); ability to degrade polysaccharides that disturb filtration; ability to hydrolyse saccharides, which contain flavour compounds in glycosidic bonds (improved flavour); ability to kill competing bacteria and yeasts (cleaner fermentation and wine taste); osmotic and alcohol tolerance; better productivity and less byproducts during starvation
Beer yeast: ability to degrade polysaccharides (better filtration and low calory beer); reduced production of acetoin and butanediol (reduced maturation time); increased osmotolerance (high gravity brewing leading to less tank volume)
Distiller’s yeast: increased alcohol yield (less glycerol) and tolerance Baker’s yeast: ability to degrade different sugars at once through diminished catabolite repression (better
leavening); freeze-tolerance after fermentation initiation (frozen doughs); high osmotolerance (high-sugar doughs)
In the food industry attempt are done in parallel using classical genetics (where possible) and genetic engineering; public perception has so far not allowed to use genetically engineered yeasts in the food industry

© Ste
fan
Hoh
man
n 20
00-
2004
Yeast biotechnology: heterologous expression
The production of proteins is of interest for several purposes: For research, such as for purification and structural analysis For industry, such as for the production of enzymes for the food and paper industry or for research and
diagnostics For the pharmaceutical industry for the production of vaccines
There are a number of different expression hosts, such as bacteria and yeasts Yeast have the advantage that they may (or may not) perform the same or at least similar post-
translation modifications, such as glycosylation Yeast usually reaches only a lower level of expression: up to more than 50% of the cellular
protein have been obtained in E. coli systems but no more than 10-20% even in the yery best yeast system
The apparently most productive known yeast is the species Pichia pastoris; it catabolises methanol and the promoter for methanol oxidase is extremely strong and can be induced by methanol
In S. cerevisiae one usually uses the promoters of genes encoding glycolytic enzymes such as PGK1 and TPI1 or a regulated promoter such as that of GAL1
The advantage of S. cerevisiae is that so much is known about its molecular biology and one can device genetic screens to improve protein production and secretion
Recently we have developed a yeast strain that does not make ethanol but rather more biomass; we try to market that strain through a start-up company
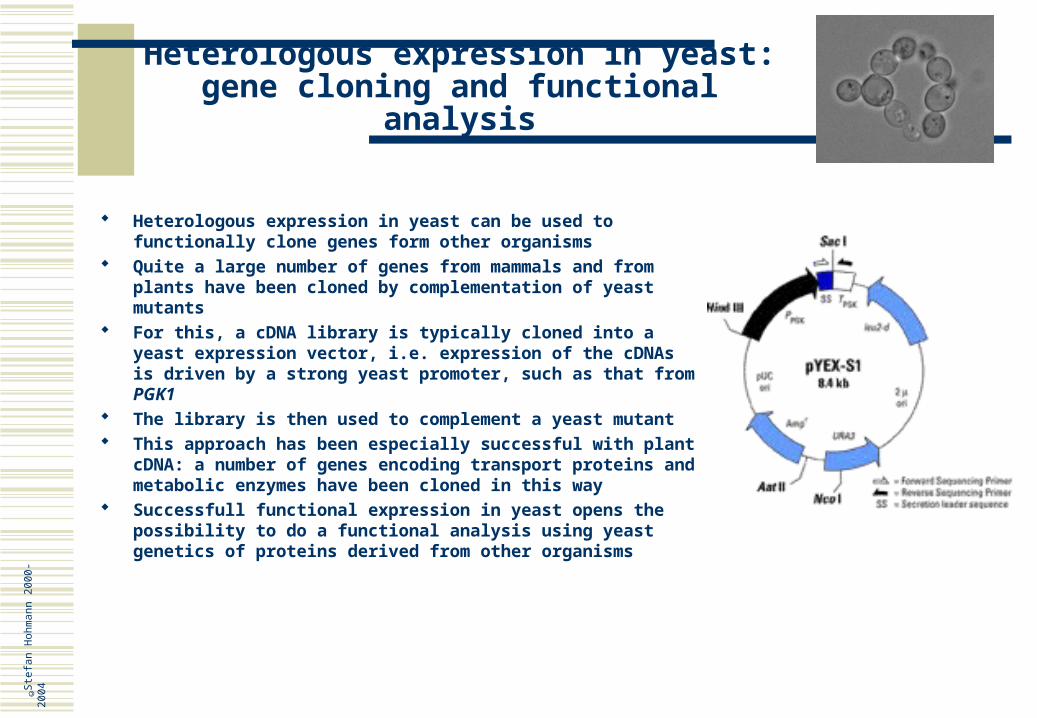
© Ste
fan
Hoh
man
n 20
00-
2004
Heterologous expression in yeast: gene cloning and functional analysis
Heterologous expression in yeast can be used to functionally clone genes form other organisms
Quite a large number of genes from mammals and from plants have been cloned by complementation of yeast mutants
For this, a cDNA library is typically cloned into a yeast expression vector, i.e. expression of the cDNAs is driven by a strong yeast promoter, such as that from PGK1
The library is then used to complement a yeast mutant This approach has been especially successful with plant
cDNA: a number of genes encoding transport proteins and metabolic enzymes have been cloned in this way
Successfull functional expression in yeast opens the possibility to do a functional analysis using yeast genetics of proteins derived from other organisms
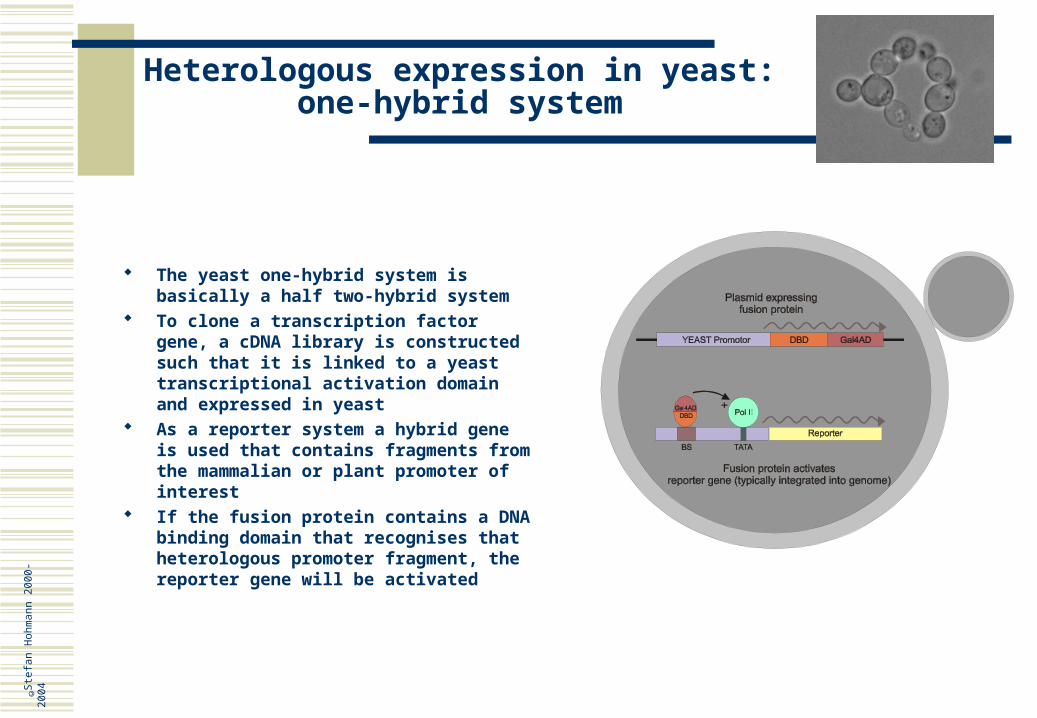
© Ste
fan
Hoh
man
n 20
00-
2004
Heterologous expression in yeast: one-hybrid system
The yeast one-hybrid system is basically a half two-hybrid system
To clone a transcription factor gene, a cDNA library is constructed such that it is linked to a yeast transcriptional activation domain and expressed in yeast
As a reporter system a hybrid gene is used that contains fragments from the mammalian or plant promoter of interest
If the fusion protein contains a DNA binding domain that recognises that heterologous promoter fragment, the reporter gene will be activated
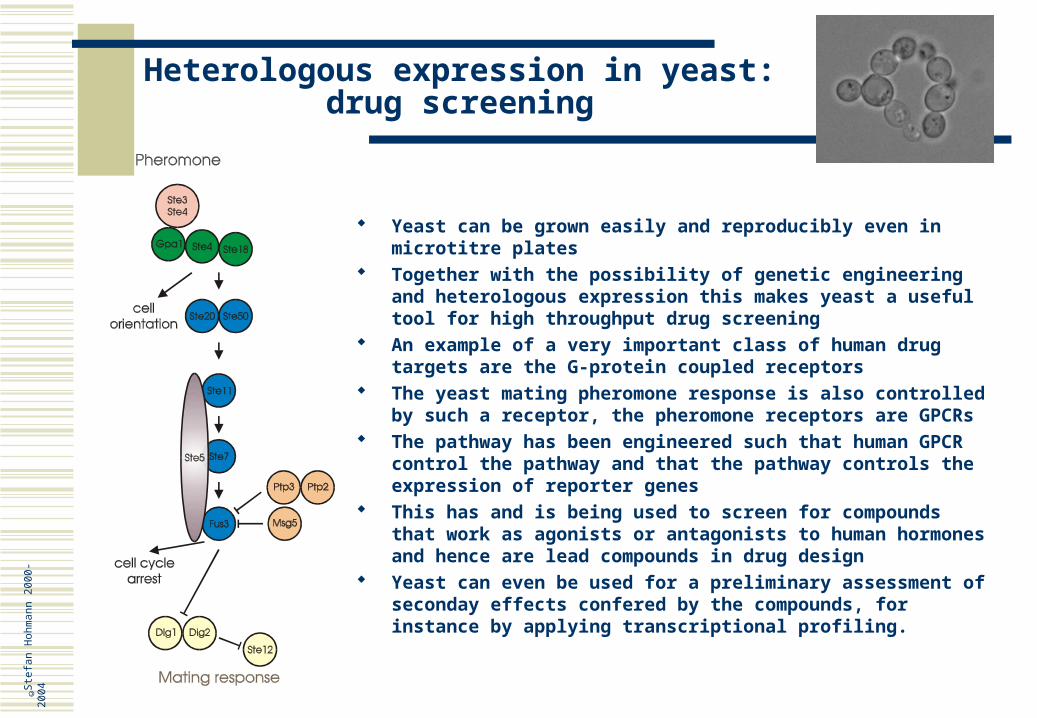
© Ste
fan
Hoh
man
n 20
00-
2004
Heterologous expression in yeast: drug screening
Yeast can be grown easily and reproducibly even in microtitre plates
Together with the possibility of genetic engineering and heterologous expression this makes yeast a useful tool for high throughput drug screening
An example of a very important class of human drug targets are the G-protein coupled receptors
The yeast mating pheromone response is also controlled by such a receptor, the pheromone receptors are GPCRs
The pathway has been engineered such that human GPCR control the pathway and that the pathway controls the expression of reporter genes
This has and is being used to screen for compounds that work as agonists or antagonists to human hormones and hence are lead compounds in drug design
Yeast can even be used for a preliminary assessment of seconday effects confered by the compounds, for instance by applying transcriptional profiling.


















