Embed Size (px)
Citation preview

www.454.com
GS Junior System – First Results

www.454.com
IMPORTANT NOTICEIntended Use
Unless explicitly stated otherwise, all Roche Applied Science and 454 Life Sciences products and services
referenced in this presentation / document are intended for the following use:
For Life Science Research Only.
Not for Use in Diagnostic Procedures.

www.454.com
Hemorrhagic Fever Virus Discovery in Native Host
www.454.com
http://www.ncbi.nlm.nih.gov/pubmed/21544192

www.454.com
Hemorrhagic Fever Virus Discovery in Native Host
• Darted Red Colobus monkey in the wild in Kibale National Park, Uganda
• Collected blood sample, isolated viral RNA/DNA
• Sequenced on GS Junior System
• Assembled using CLC genomics assembler, screened out host contigs
• Identified two novel SHFV (simian hemorrhagic fever virus) strains
• Generated near full-length viral sequences by filling in short gaps with PCR/Sanger sequencing and 3’RACE
• Significant findings:– Not one, but TWO divergent SHFV viruses were present in one individual– Red Colobus monkey is a native reservoir for these pathogenic viruses– DNA was isolated from a healthy animal, demonstrating that these viruses
can hide in apparently healthy individuals – Consequences for human contact, spreading viruses through research
colonies
www.454.com

www.454.com
Plant Pathogen Sequencing
www.454.com
http://www.ncbi.nlm.nih.gov/pubmed/21131493

www.454.com
Plant Pathogen Sequencing
• Erwina amylovora, fire blight pathogen, isolated from blackberry in Illinois
• Commercial apple and pear blight, reported in 1790s
• 3.81 Mb genome, 53% GC, three circular plasmids
• Sequenced using 3/8 of GS FLX run and one GS Junior run (equal to four GS Junior runs)
• 31x coverage, 375 bp avg. read length
• Assembled by 454 GS De Novo Assembler into 29 contigs, gaps closed in silico using LaserGene
• Used GenDB to assign gene function for 3869 coding sequences
• Comparative genomics with related strains
www.454.com

www.454.com
Rare Variant Detection for HIV-1Saliou et al. Antimicrob. Agents Chemother April 2011
www.454.com

www.454.com
Why Detect HIV Variants?
• HIV variants or “quasispecies” can use CCR5 and/or CXCR4 cell-surface receptors to enter cells
• Drugs that block CCR5 receptors work only if CXCR4-binding variants are absent
• As a result, there are tests to be sure that there are no CXCR4 binding viral variants before administering this class of HIV drugs to an individual

www.454.com
Why use 454 Sequencing System?Potential to deliver speed, ease of use, cost savings• Current high sensitivity assays can detect viral variants at 0.3%, but
are slow, expensive and difficult
• Current Sanger sequencing assays are rapid, cheap but cannot detect quasi-species below 10-20%
• Sensitivity at 0.3% can best predict treatment outcomes
• 454 Sequencing Systems can deliver sequencing specificity for ~25 samples in one GS Junior run

www.454.com
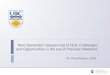
Experimental Design
• 415 base cDNA amplicon covering V3 env. region of HIV-1
• Nested RT-PCR to generate amplicons with MIDs
• 23 individual samples obtained ~3,500 reads/sample, sequenced in one GS Junior run
• GS AVA software used to align to reference
• Processed the reads using third party prediction software
• Detected quasispecies to 0.6% reliably
• Calculated mean error rate of .000853 for pyrosequencing from control plasmids!
www.454.com
Results
www.454.com
Summary
- 84,000 reads
- 23 samples
- 0.6% detection limit
Critical Factors
- 415 bp amplicon
- 1600 or more reads per sample
Detection limited by software that predicts phenotype

www.454.com
First Publication using GS Junior System Data

www.454.com
Summary of Results
• Sequencing of MHC class I transcripts in macaques to discover all expressed transcripts from common class I haplotypes
• Sequenced 3 amplicons from ~440 to 620 bases
• Combination experiment– 7 individuals on GS FLX System, 3 using GS Junior System– Identified all sequences found previously – Discovered 2x more haplotypes than with previous Sanger-based
approach
• 440-600 base amplicons allow resolution of haplotypes that are impossible with 190 base amplicons

www.454.com
GS Junior SystemPrimary applications
• de novo sequencing– sequencing of whole microbial, viral and other small genomes
• Targeted sequencing– Using sequence capture, PCR, amplicons, transcriptome cDNA
sequencing– Genotyping, rare variant detection, somatic mutation detection,
disease associated genes, genomic regions
• Metagenomics– characterization of complex environmental samples (16s rRNA
and shotgun)
www.454.com
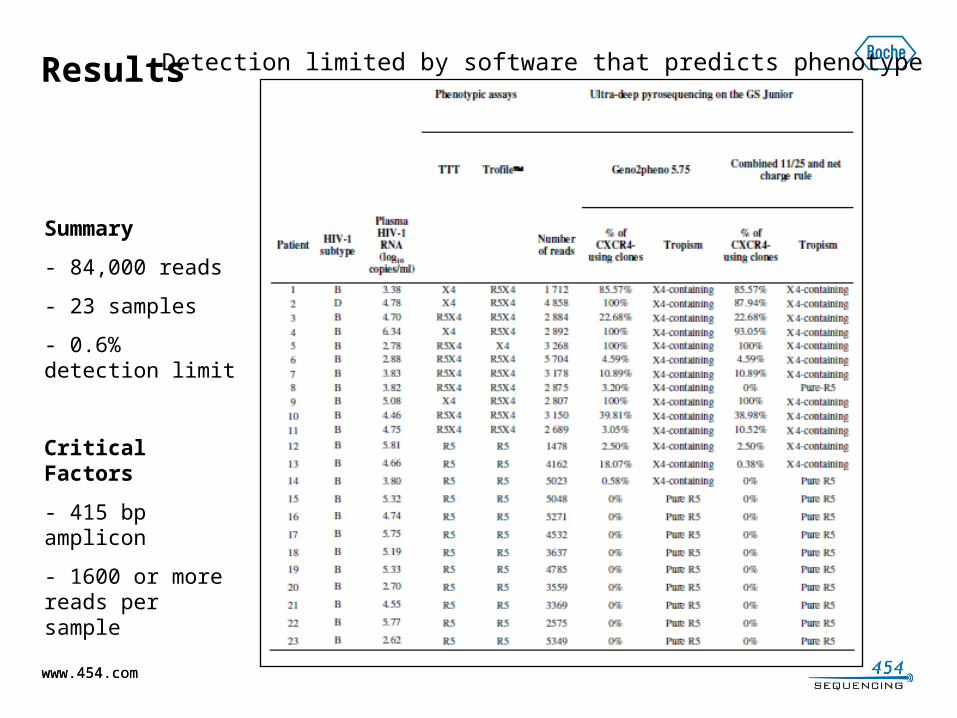
Whole Genome Shotgun SequencingSequencing of three representative bacterial genomes
System GS FLX
GS Junio
r GS FLXGS Junior GS FLX
GS Junior
Organism E. coli K-12 T. thermophilus C. jejuni
Genome Size(in Kb) 4563 2120 1600Avg. ContigSize (in Kb) 39 58 44 53 49 46N50
ContigSize(in Kb) 84 112 112 121 115 95Largest
ContigSize (in Kb) 209 352 474 578 304 173Number OfContigs 115 78 48 40 33 35
de novo Assemblies at 25x coverage using GS Junior and GS FLX Titanium reads
www.454.com
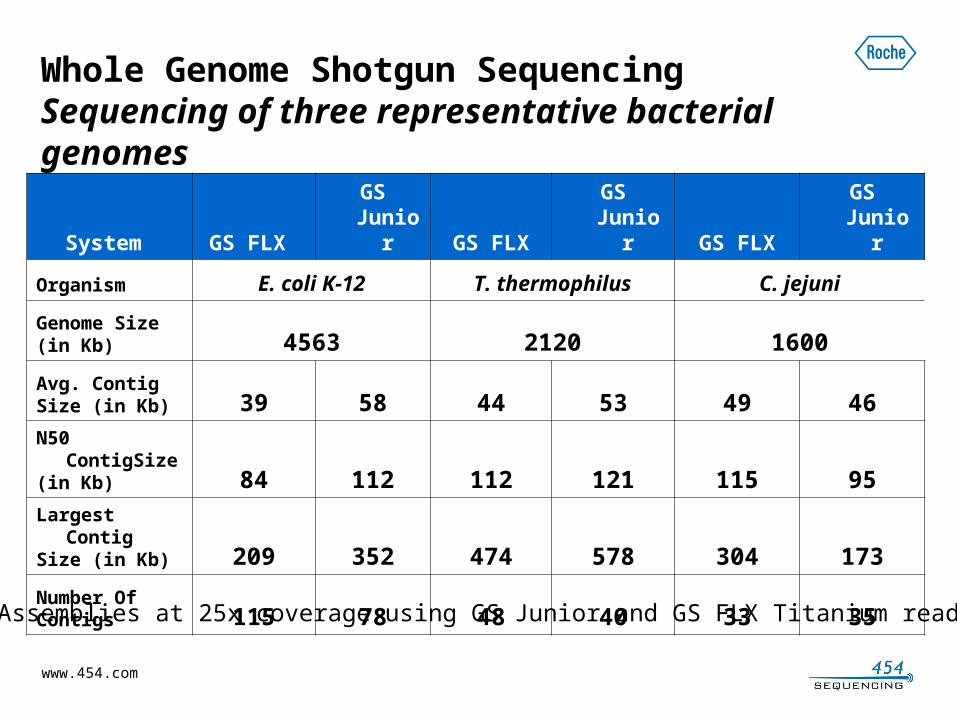
Data from GS Junior System Shotgun RunsVariety of different microbes, early access site dataRun
Passed Filter Reads
Avg Length Total Bases
1 117,636 445.1 52,350,2542 83,045 323.6 26,867,0863 90,415 386.7 34,954,1014 128,225 350.6 44,939,6535 43,321 353.2 15,297,8286 66,100 367.2 24,265,4077 100,335 433.4 43,475,7248 79,145 394.8 31,242,8759 109,894 422.6 46,430,503
10 108,779 437.8 47,613,70811 94,605 457.4 43,271,23312 61,975 398.7 24,706,55713 99,273 384.2 38,134,16514 115,776 429.5 49,716,84915 115,972 419.3 48,622,87416 115,031 414.4 47,661,170
Average 95,595 401 38,721,874
3kb paired end- 1M base genome, 1 run, one scaffold
www.454.com
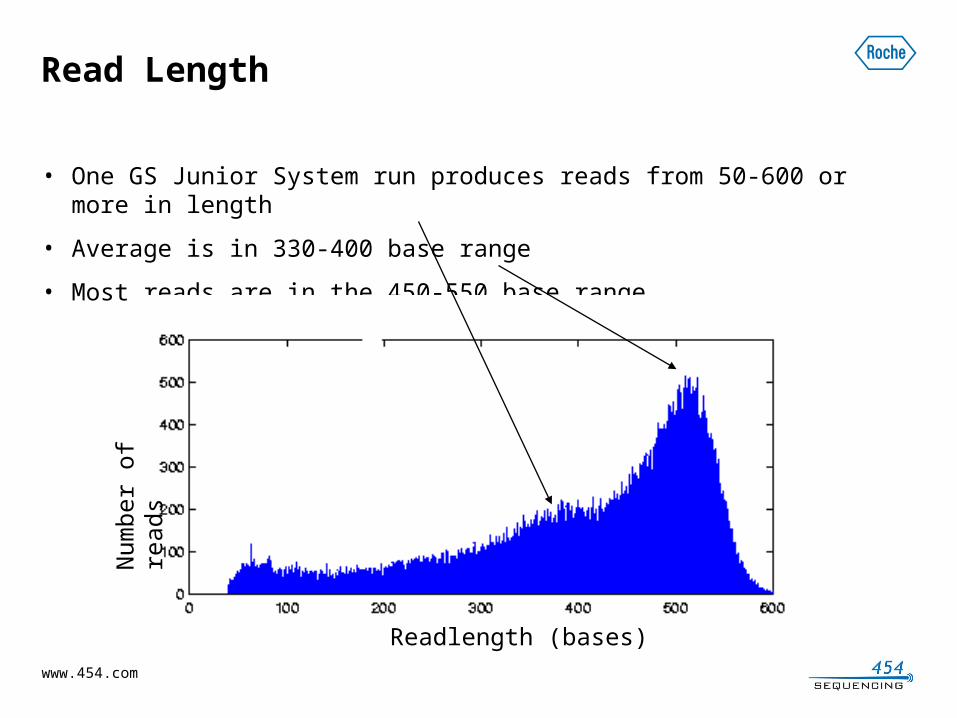
Read Length
• One GS Junior System run produces reads from 50-600 or more in length
• Average is in 330-400 base range
• Most reads are in the 450-550 base range
Num
ber
of
read
s
Readlength (bases)

www.454.com
CFTR Exon Resequencing on GS Junior System
Experimental design:• 11 Coriell samples with known
mutations in CF gene• Each sample was MID-labeled (11
MIDs)• Amplified all 27 coding exons with
34 amplicons • Mixed 11x34 = 374 amplicons• Sequenced in 1 GS Junior System
run• Average coverage 182x• 96% of the reads mapped back to
the CF gene region
Numbers of reads per amplicon(across 11 samples)
0
100
200
300
400
500
600
0 50 100 150 200 250 300 350 400
374 Individual Amplicons#
of R
ead
s
Coverage graph: range 27-551x
Since multiplex PCR reactions could not be normalized, PCR efficiency dictated the coverage levels for each amplicon
www.454.com
Sizes of actual amplicons
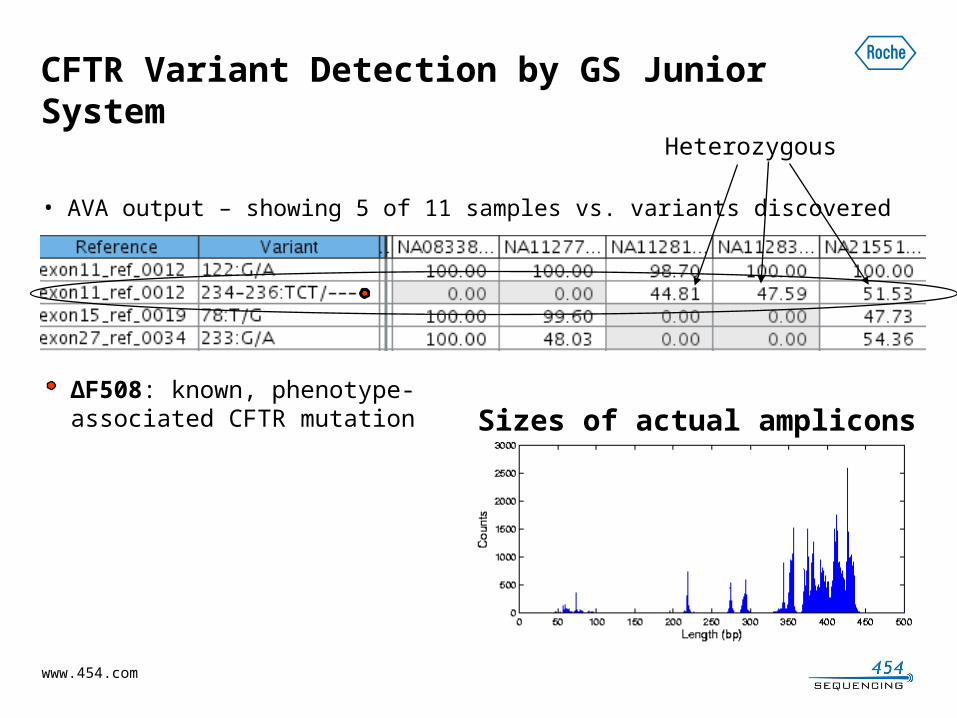
CFTR Variant Detection by GS Junior System
• AVA output – showing 5 of 11 samples vs. variants discovered
ΔF508: known, phenotype-associated CFTR mutation
Heterozygous

www.454.com
GS Junior and GS FLX reads are equivalent CFTR Variant Detection ΔF508
R668C
known, phenotype-associated CFTR mutation
Synonymous
same mutation detected in two separate, overlapping, amplicons
www.454.com
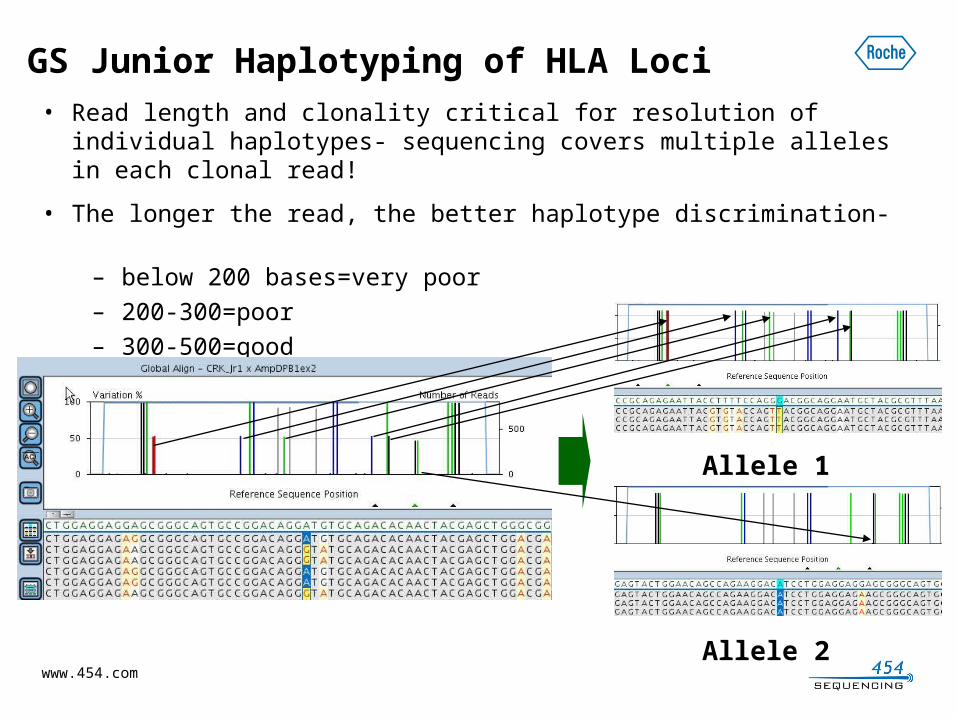
GS Junior Haplotyping of HLA Loci• Read length and clonality critical for resolution of individual
haplotypes- sequencing covers multiple alleles in each clonal read!
• The longer the read, the better haplotype discrimination- – below 200 bases=very poor– 200-300=poor– 300-500=good– 500-800=excellent
Allele 1
Allele 2

www.454.com
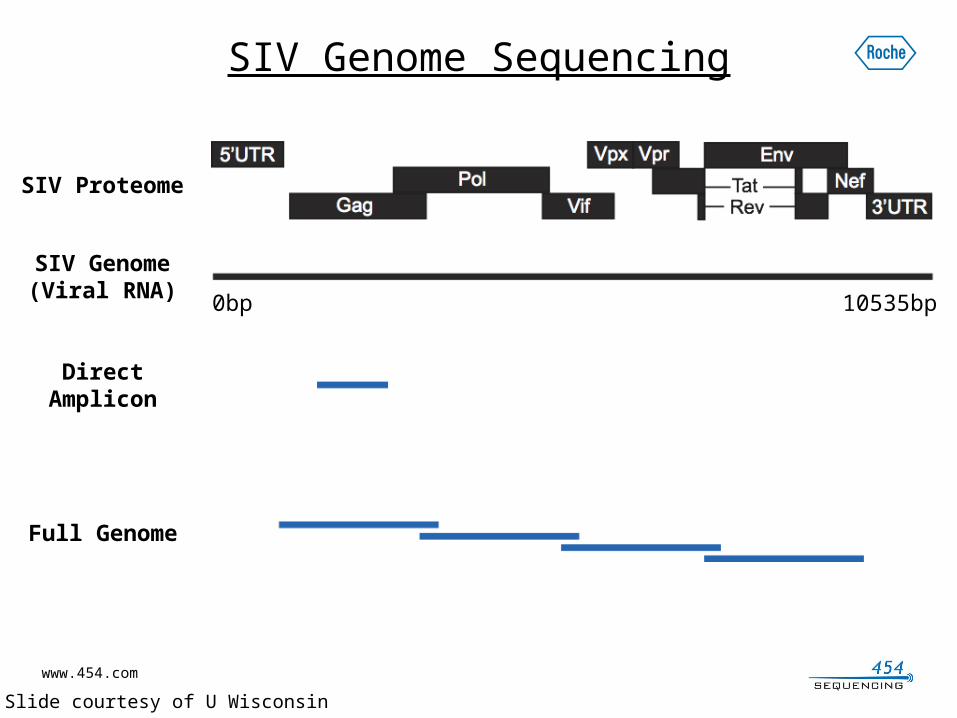
Studying SIV using GS Junior System
• Ben Burwitz in Dave O’Connor’s lab, Univ. of Wisconsin
• Follow changes in GAG gene as virus evolves to evade immune response
• Find genome-wide mutations in viral pool
Simian Immunodeficiency Virus
Rhesusmacaque

www.454.com
Amplicon Sequencing- Basic Amplicon454 amplicon design using tailed primers
454 Titanium B-primer (21 bp)
MID
MID
key
key
A
B
454 Titanium A-primer (21 bp)
Sequence of interest
Locus-specific PCR amplification
200-600 bp
emPCR Amplification and sequencing
• Long reads required to sequence through the locus specific primer, enable haplotyping over longer distances
• 100s to 1000s of amplicon clones sequenced simultaneously

www.454.com
Amplicon Sequencing- Long Range AmpliconsUsing long range amplicons for whole viral or other genomic region sequencing
Sequence of interest
Locus-specific long range PCR amplification
1,500-15,000 or more bp
emPCR Amplification and sequencing
MIDkey
A BMID
key
454 Titanium A-primer (21 bp)
454 Titanium B-primer (21 bp)
Ligate sheared amplicon into 454 primers using gDNA protocol
Shear to 400-600 bases using gDNA protocol
BA
BA
BA
www.454.com
SIV Genome Sequencing
SIV Genome(Viral RNA)
0bp 10535bp
Direct Amplicon
SIV Proteome
Full Genome
* Slide courtesy of U Wisconsin
www.454.com
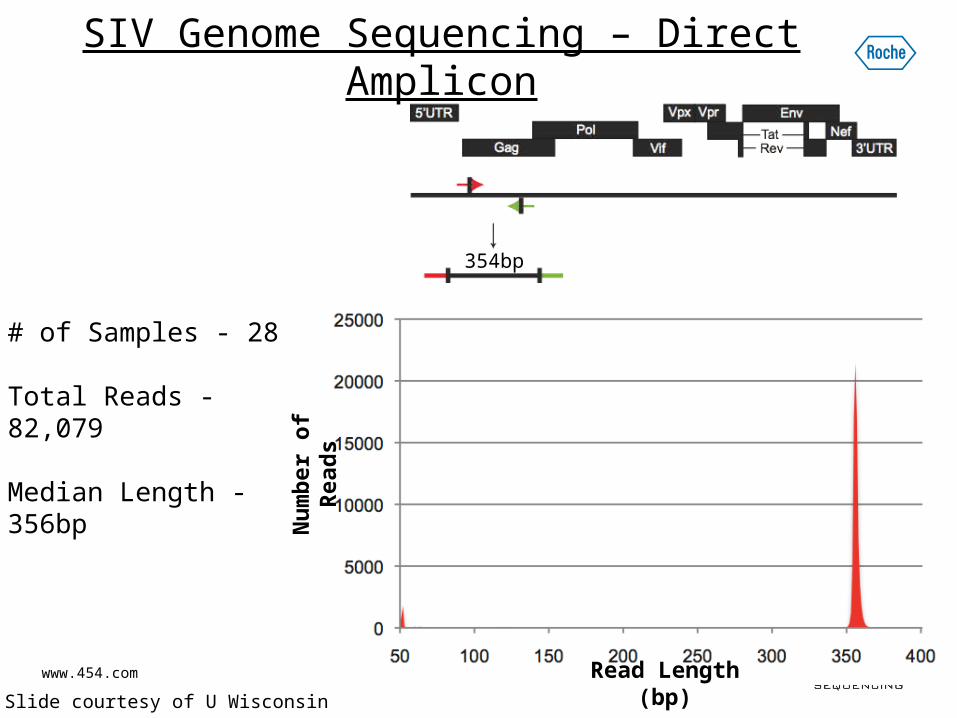
SIV Genome Sequencing – Direct Amplicon
Read Length (bp)
Nu
mb
er o
f R
ead
s
354bp
# of Samples - 28
Total Reads - 82,079
Median Length - 356bp
* Slide courtesy of U Wisconsin
www.454.com
Viral Mutations in the Structural SIV Protein Gag evolve to escape immune response
Mutations in the SIV protein Gag affect viral fitness- Gag protein is the ‘particle making machine’
* Slide courtesy of U Wisconsin
www.454.com
Viral Mutations in the Structural SIV Protein Gag evolve to escape immune response
Mutations in the SIV protein Gag affect viral fitness- Gag protein is the ‘particle making machine’
* Slide courtesy of U Wisconsin

www.454.com
SIV Genome Sequencing
SIV Genome(Viral RNA)
0bp 10535bp
Direct Amplicon
SIV Proteome
Full Genome
* Slide courtesy of U Wisconsin
www.454.com
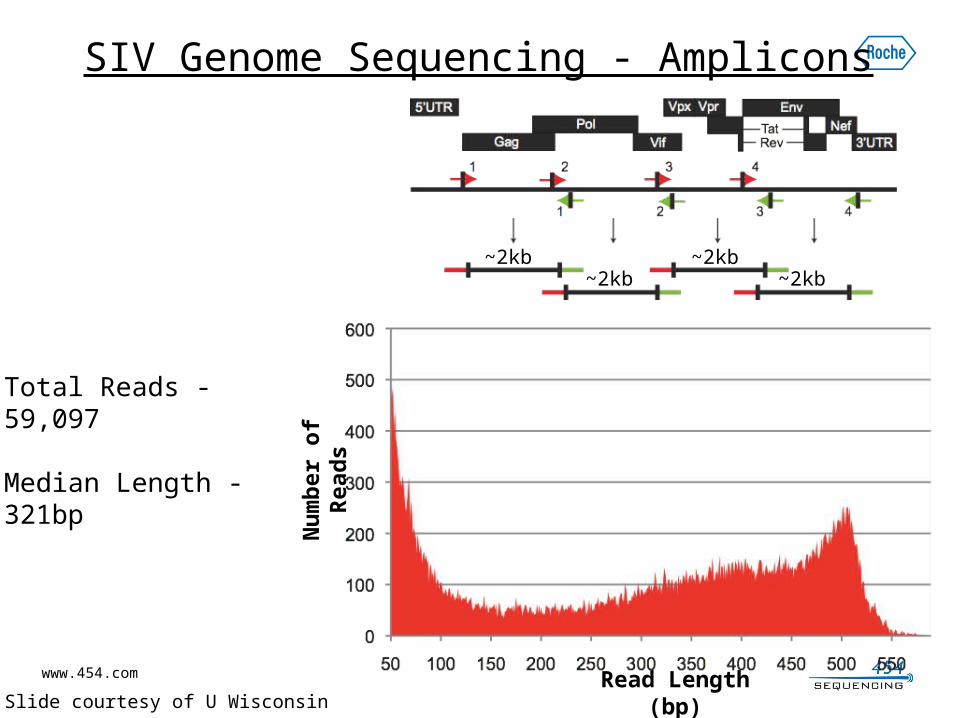
SIV Genome Sequencing - Amplicons
Read Length (bp)
Nu
mb
er o
f R
ead
s
Total Reads - 59,097
Median Length - 321bp
~2kb~2kb
~2kb~2kb
* Slide courtesy of U Wisconsin
www.454.com
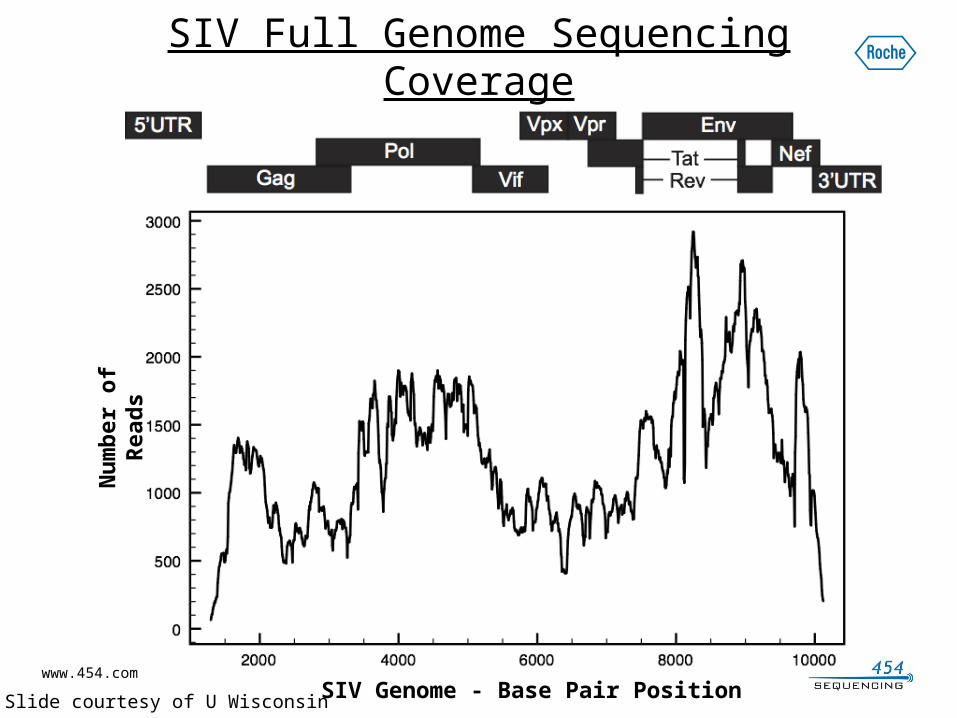
SIV Full Genome Sequencing Coverage
SIV Genome - Base Pair Position
Nu
mb
er o
f R
ead
s
* Slide courtesy of U Wisconsin

www.454.com
454 Sequencing System vs. Sanger
Animal 1 Animal 2 Animal 3
* Slide courtesy of U Wisconsin

www.454.com
Ben’s Conclusions
•GS Junior System detects low frequency genetic variants that are missed by traditional Sanger sequencing
•A bench-top GS Junior System improves turn around time and can be readily adapted to small academic lab settings
Acknowledgements
Ben BurwitzRoger WisemanShelby O’Connor
Dawn DudleyJulie Karl
Simon LankCharlie BurnsEricka BeckerBen Bimber
Dave O’Connor
O’Connor Lab
Watkins Lab
Jonah SachaMatt ReynoldsNick ManessNancy WilsonDavid Watkins
www.454.com
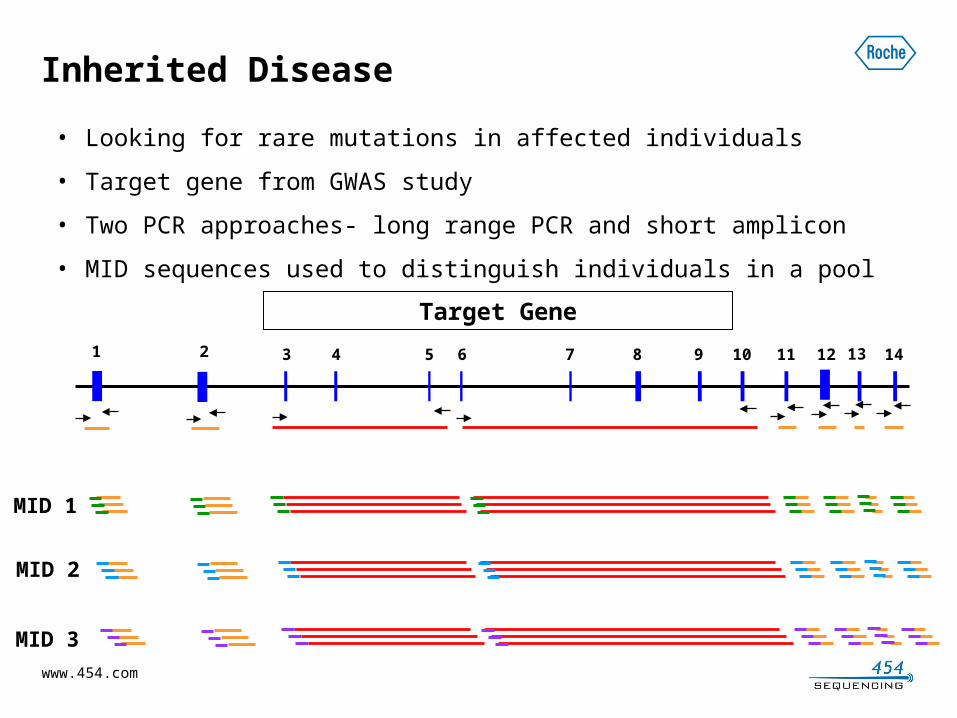
Inherited Disease
• Looking for rare mutations in affected individuals
• Target gene from GWAS study
• Two PCR approaches- long range PCR and short amplicon
• MID sequences used to distinguish individuals in a pool
Target Gene
1 2 3 4 5 6 7 8 9 10 11 12 13 14
MID 1
MID 2
MID 3
www.454.com
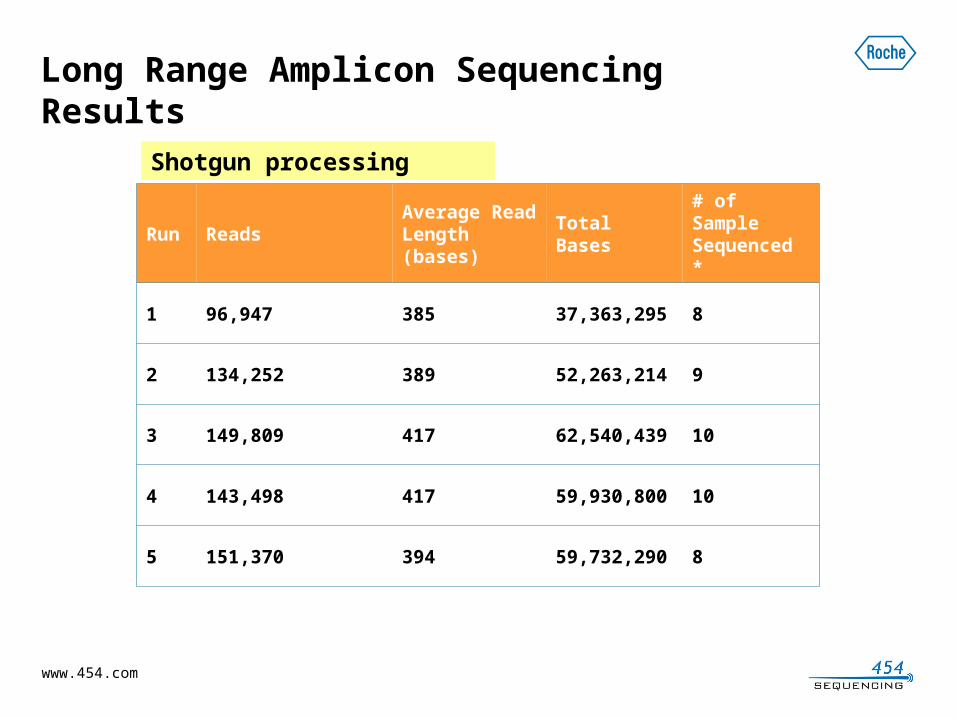
Long Range Amplicon Sequencing Results
Run
ReadsAverage Read Length (bases)
Total Bases
# of Sample Sequenced *
1 96,947 385 37,363,295 8
2 134,252 389 52,263,214 9
3 149,809 417 62,540,439 10
4 143,498 417 59,930,800 10
5 151,370 394 59,732,290 8
Shotgun processing
www.454.com
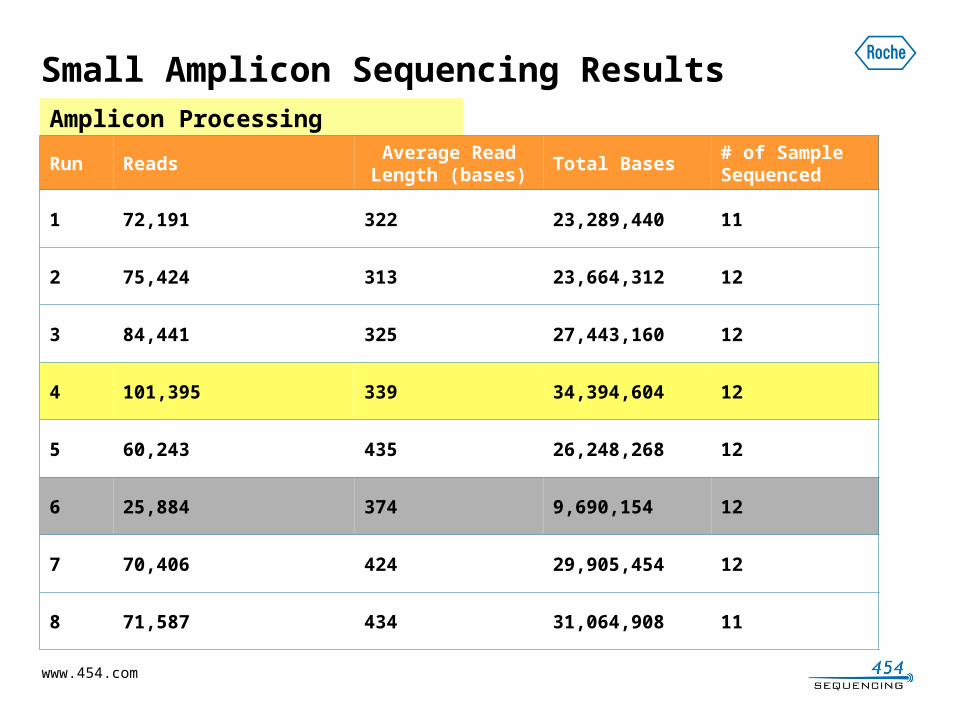
Small Amplicon Sequencing ResultsAmplicon Processing
Run ReadsAverage Read Length (bases)
Total Bases# of Sample Sequenced
1 72,191 322 23,289,440 11
2 75,424 313 23,664,312 12
3 84,441 325 27,443,160 12
4 101,395 339 34,394,604 12
5 60,243 435 26,248,268 12
6 25,884 374 9,690,154 12
7 70,406 424 29,905,454 12
8 71,587 434 31,064,908 11

www.454.com
Amplicon Coverage- Accurate Pooling Required!
Ind
ivid
ual
Sam
ple
sAmplicons
Poor performing Sample
Poor Performing AmpliconSampling Variability
Poorly Pooled Amplicon
www.454.com
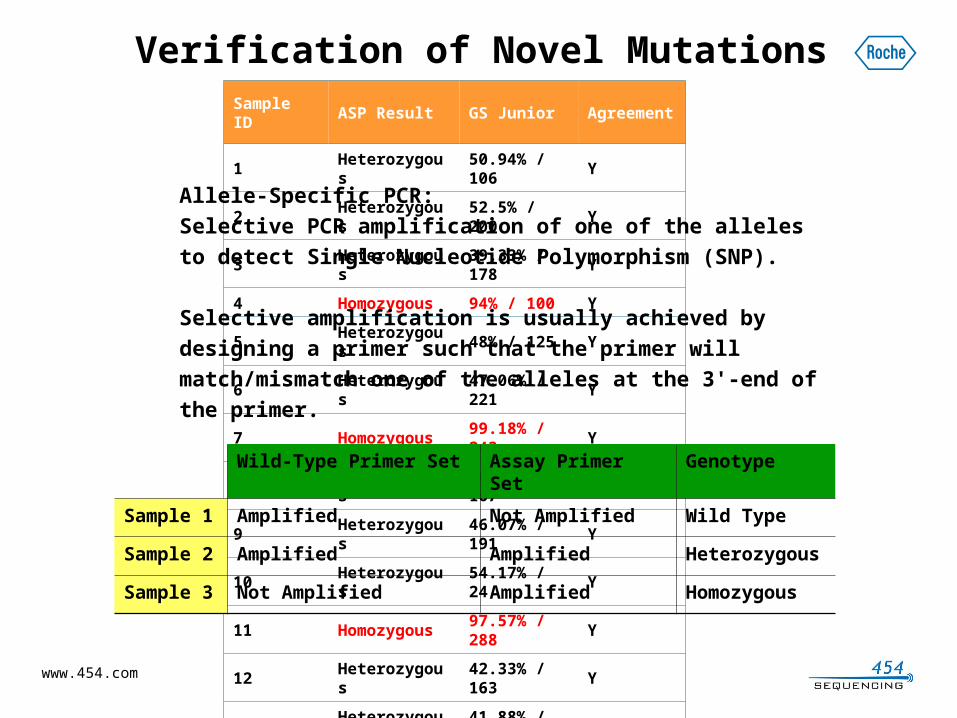
Sample ID
ASP Result GS JuniorAgreement
1Heterozygous
50.94% / 106
Y
2Heterozygous
52.5% / 200
Y
3Heterozygous
39.33% / 178
Y
4Homozygous
94% / 100 Y
5Heterozygous
48% / 125 Y
6Heterozygous
47.06% / 221
Y
7Homozygous
99.18% / 243
Y
8Heterozygous
46.71% / 167
Y
9Heterozygous
46.07% / 191
Y
10Heterozygous
54.17% / 24
Y
11Homozygous
97.57% / 288
Y
12Heterozygous
42.33% / 163
Y
13Heterozygous
41.88% / 191
Y
14Heterozygous
47.02% / 151
Y
15Heterozygous
48.07% / 441
Y
16Heterozygous
17.86% / 252
N
17Heterozygous
50.32% / 157
Y
18Heterozygous
16.18% / 272
Y
19Heterozygous
14.85% 330
Y
Allele-Specific PCR:
Selective PCR amplification of one of the alleles to detect
Single Nucleotide Polymorphism (SNP).
Selective amplification is usually achieved by designing a
primer such that the primer will match/mismatch one of the
alleles at the 3'-end of the primer.
Wild-Type Primer Set Assay Primer Set Genotype
Sample 1 Amplified Not Amplified Wild Type
Sample 2 Amplified Amplified Heterozygous
Sample 3 Not Amplified Amplified Homozygous
Verification of Novel Mutations

www.454.com
Pathogen Discovery on the GS Junior System•Case from Sandton, South Africa
•Infected paramedic during transfer, nurse at hospital, cleaning staff, and nurse of paramedic- 4/5 did not survive
Serum and tissue samples from victims were subjected to unbiased pyrosequencing, yielding within 72 hours of sample receipt, multiple discrete sequence fragments that represented approximately 50% of a prototypic arenavirus genome.
•Recapitulated GS FLX System study in single GS Junior System run
•250 Hits to LuJo Virus covering 57% of the L-segment and 79% of the S-segment

www.454.com
Coming Soon
• GS Junior System Publications in – Metagenomic characterization of human environments– Whole Genome Sequencing of bacterial pathogens– Rare variant discovery in human disease- GWAS follow up
experiments– Viral pathogen sequencing– Many more!

www.454.com
GS Junior System First ResultsDisclaimer & Trademarks
Disclaimer:
For life science research only. Not for use in diagnostic procedures.
Trademarks:
454, 454 LIFE SCIENCES, 454 SEQUENCING, EMPCR, GS FLX, GS FLX TITANIUM, GS JUNIOR and SEQCAP are trademarks of Roche.
Other brands or product names are trademarks of their respective holders.

www.454.com