Embed Size (px)
Citation preview

J. CHEM. SOC. FARADAY TRANS., 1994, 90(12), 1581-1598 1581
What Woodward and Hoffmann Didn't Tell Us: The Failure of the Born-Oppenheimer Approximation in Competing Reaction Pathways
Gabriela C. G. Waschewsky, Phillip W. Kash, Tanya L. Myers, David C. Kitchen and Laurie J. Butler The James Franck Institute and Department of Chemistry, The University of Chicago, Chicago, lL 60637, USA
The experiments presented here identify a class of organic reactions, allowed by overall electronic symmetry but WoodwardHoffmann forbidden, in which the failure of the Born-Oppenheimer approximation results in a marked change in t h e expected branching between energetically allowed chemical bond fission channels.
W e first review crossed laser-molecular beam experiments on the competition between photodissociation pathways in bromoacetyl and bromopropionyl chloride at 248 nm and bromoacetone at 308 nm. In t h e com- petition between C-CI and C-Br fission in Br(CH,),COCI, the barrier to C-Br fission on t h e lowest 'A" potential-energy surface is formed from a weakly avoided electronic configuration crossing, so that non- adiabatic recrossing of t h e barrier dramatically reduces t h e branching to C- Br fission. The experimental results and supporting a b initio calculations investigate the strong intramolecular distance dependence of t h e electronic configuration interaction matrix elements which split the adiabats at the barrier to C-Br fission. The second set of experiments reviewed investigates the competition between C-C and C-Br bond fission in bromoacetone excited in the ' [n(O), .n*(C=O)] absorption, elucidating the role of molecular conformation in influencing t h e probability of adiabatically traversing t h e conical intersection along the C-C fission reaction coordinate.
The paper finishes by presenting new experiments on the photodissociation of chloroacetone at 308 nm which test the conclusions of the earlier work. Photofragment velocity and angular distribution measurements show that C-C fission competes with C-CI fission in th i s molecule, while only C-CI fission occurs in acetyl chloride upon '[n(O), n*(C=O)] excitation. W e investigate two contributing factors to understand t h e difference in branching. Ab initio calculations show that t h e splitting at t h e avoided crossing between the no.nE=o and t h e npc, a%=, configurations which forms the barrier to C-CI fission is smaller, on average, in trans-chloroacetone than in acetyl chloride, so the rate constant for C-CI fission is more suppressed by non-adiabatic recrossing of the reaction barrier. In addition, C-C fission can proceed more adiabatically from t h e gauche conformer of chloroacetone than from near-planar geometries in acetyl chloride owing to a conformation dependence of non-adiabatic recrossing near the conical intersection. A final measurement of t h e conformation population dependence of the branching investigates t h e second contributing factor.
1. Introduction Much of our predictive ability for the branching between chemical reaction pathways has relied on statistical transition-state theories'-3 or, in smaller systems, quantum scattering calculations4 on a single adiabatic potential-energy surface. The potential-energy surface gives the energetic bar- riers to each chemical reaction and allows prediction of the reaction rates. Yet the chemical reaction dynamics evolves on a single potential-energy surface only if the Born- Oppenheimer' separation of nuclear and electronic motion is valid. Thus, the adiabatic approximation, though challenged at the inception of transition-state theory,? is often now implicitly assumed to be valid for bimolecular and unimolecular reactions in the ground electronic ~ t a t e . ~ 9 ~ Its shortcomings are only widely recognized for ion-molecule, charge tran~fer,~ and other reactions involving obvious elec- tronic curve The experiments presented here demonstrate the importance of considering the possibility of non-adiabaticity at the transition state of any chemical reac- tion with a barrier along the reaction coordinate, including ground-state bimolecular reactions, and identify two classes of chemical reactions in which non-adiabatic effects are criti- cal.
t The adiabatic assumption was briefly discussed in ref. qa) and challenged by E. Rabinowitch in the discussion of the paper by Evans and Polanyi.6b The following paper6c also discussed the adia- batic assumption.
2. Reduction in Rate Constant from Non-adiabatic Recrossing of the Barrier
Recent has shown that the breakdown of the Born-Oppenheimer approximation at a reaction barrier formed from an avoided electronic configuration c r o ~ s i n g l ~ * ' ~ can dramatically reduce the rate constant for the chemical reaction and change the resulting branching between energetically allowed product channels. The qualit- ative mechanism for a reduction in the reaction rate due to non-adiabatic recrossing of the barrier is illustrated in Fig. 1 for an A-B bond fission reaction with a barrier along the forward and reverse reaction coordinates. Along the adia- batic reaction coordinate the dominant electronic configu- ration changes from one bonding in the A-B bond, on the reactant side of the barrier, to one repulsive in the A-B bond, after the barrier in the exit-channel region. The change in electronic wavefunction required to follow the adiabatic reaction coordinate near the barrier is considerable and can result in a failure of the Born-Oppenheimer approximation. If the splitting between the adiabatic electronic surfaces at the barrier along the reaction coordinate is small,' ' reflecting the weak configuration interaction between the bonding and repulsive electronic configurations, the molecule may not tra- verse the barrier adiabatically. Instead the electronic wave- function may retain the bonding configuration character, resulting in a non-adiabatic hop to the upper bond potential- energy surface at the avoided crossing. The molecule feels the bound wall of that potential instead of undergoing bond fission on the lower adiabat.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online / Journal Homepage / Table of Contents for this issue

1582 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
8-1 bonding configuration - repulsive configuration I mixed character
reaction coordinate Fig. 1 Schematic reaction coordinate for the A-BC --* A + BC reaction with a barrier to both the forward and reverse reaction. Along the lower adiabat, the dominant electronic configuration of the electronic wavefunction changes from bonding in character on the reactant side to repulsive (antibonding) in character on the product side. The figure also shows the upper bound adiabat formed from the avoided electronic crossing of the bonding and repulsive electronic configurations. If the Born-Oppenheimer approximation fails, the molecule retains its initial bonding electronic character at the barrier and makes a non-adiabatic 'hop' to the upper adiabat instead of proceeding to products on the lower adiabat. Figure repro- duced from ref. 12.
Several classes of reactions are susceptible to a reduction in the rate constant due to non-adiabatic recrossing of the reaction barrier. A completely analogous mechanism to that described above results in a non-adiabatic reduction in the rate constant for electron-transfer reaction^.^ In a statistical expression for the reaction rate constant k = K(k, T/h)exp( - E J k , T ) one may correct for this rec- rossing with a transmission factor K of less than 0116.~ '~~ (Traditionally," however, a reduced K has been used to correct for recrossing due to a large curvature of the potential-energy surface in the transition state region, not for recrossing due to non-adiabatic transitions.) S,2 reactions could also evidence a non-adiabatic reduction in the rate con- stant,'* because again the barrier is formed from an avoided electronic curve crossing,' but this possibility has not been closely investigated. The work reviewed in the next two sec- tions shows that the reduction in rate due to non-adiabatic recrossing of the barrier markedly changes the branching between competing bond fission pathways belonging to two classes of reactions in which the splitting between adiabats is anomalously small, Woodward-Hoffmann-forbidden reactions"." and reactions with a conical intersection" along the reaction coordinate.
3. The Importance of Non-adiabatic Recrossing in Woodward-Hoffmann-for bidden Reactions :
Br( CH,),COCl The experiments described in this section on the photoin- duced Woodward-Hoffmann-forbidden bond cleavage reac- tions of Br(CH,),COCl focus on how non-adiabatic recrossing of the low-energy barrier to C-Br fission barrier markedly decreases the branching to C-Br fission from that predicted with adiabatic transition-state theories. ''*" They further investigate the intramolecular distance dependence of the electronic configuration interaction matrix elements
which result in the splitting between the adiabats at the barrier to C-Br fission. We end by qualitatively outlining a simple molecular orbital picture for why this splitting is expected to be anomalously small for Woodward-Hoffmann- forbidden reactions, making these reactions so susceptible to a non-adiabatic reduction in reaction rate. Indeed, although the barrier is often cited as the reason why this class of reac- tions is unfavourable, the present results show that non- adiabatic recrossing may be the dominant reason why these reactions are 'forbidden'.
Non-adiabaticity in Product Branching in Br(CH,),COCI
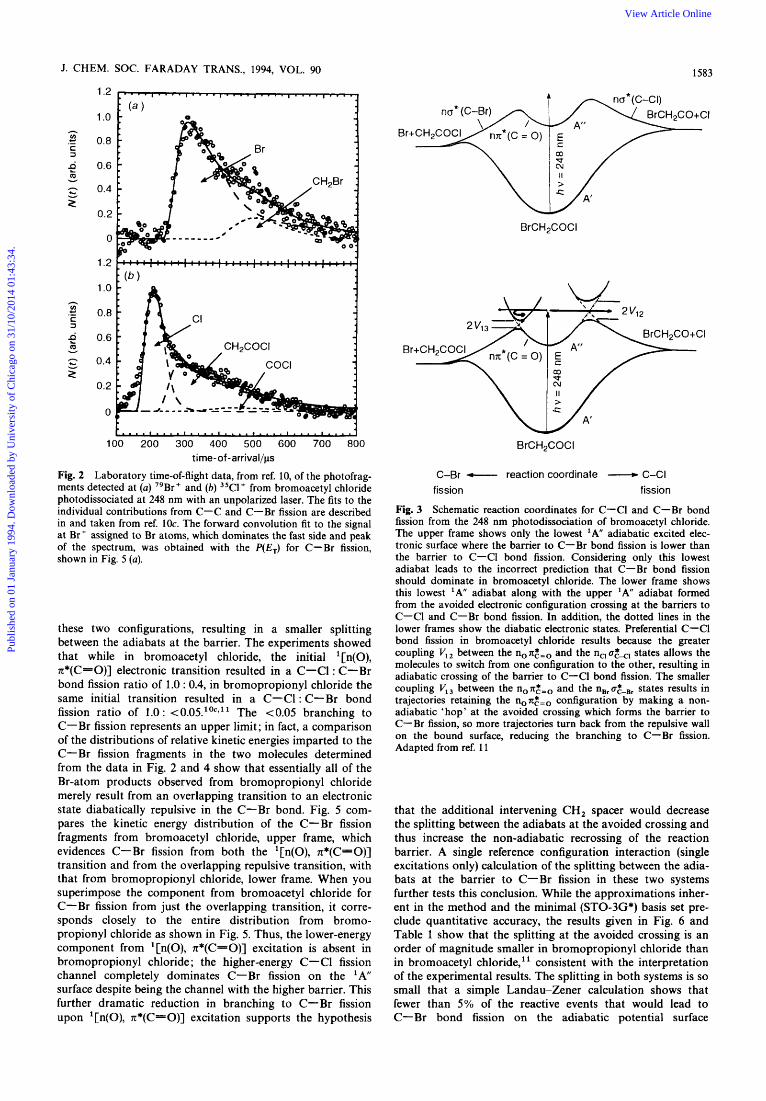
The first result of our experiments on the competition between photodissociation pathways in bromoacetyl and bromopropionyl chloride showed that the reaction channeI with the lowest energetic barrier need not be the major one; it can be so suppressed by non-adiabatic recrossing that a reaction channel with a much higher barrier can dominate. Excitation of BrCH,COCl to the lowest 'A" potential-energy surface via the '[n(O), n*(C=O)] transition at 248 nm results in a competition between C-Br and C-C1 fission, with a minor contribution from C-C fission." Although sta- tistical transition-state theories predict that, given compara- ble pre-exponential factors, the reaction pathway with the lowest energetic barrier, C-Br fission, should dominate, the experiments find the C-Cl bond cleaves preferentially by a ratio of C-C1 : C-Br = 1 : 0.4 in bromoacetyl chloride upon excitation of the '[n(O), n*(C=O)] transition.7 (The analysis requires separate identification of the C-Br fission resulting from the '[n(O), n*(C=O)] transition us. C-Br fission resulting from excitation of an overlapping electronic transition; this is described in the next paragraph.) Fig. 2 shows the time of flight from the photolysis region to the electron-bombardment ionization detector of the primary photofragments as measured with a molecular beam tech- nique described in Section 5. The model developed to explain the experimental results attributed the reduction in branching to C-Br fission on the ' A surface to non-adiabatic re- crossing of the barrier to C-Br fission. Fig. 3 shows that if the molecule retains the non:=o bonding configuration as it approaches the barrier to C-Br fission, it hops to the upper potential-energy surface at the avoided crossing and turns back towards the Franck-Condon region instead of pro- ceeding over the barrier to C-Br fission along the lower adiabatic reaction coordinate. The experiments on bromo- propionyl chloride described next test this model's predic- tions.
The experiments on bromopropionyl chloride were initiated to test whether we could further suppress the branching to C-Br fission by choosing a system where the splitting at the avoided crossing which forms the barrier to C-Br fission would be significantly reduced from that in bromoacetyl chlo- ride. This smaller splitting would enhance the probability of the barrier being non-adiabatically recrossed, further decreasing the rate constant for C-Br fission (see schematic reaction coordinate in Fig. 3). Because the barrier results from the avoided crossing of the no n&o and the npBr o&,, configurations, the additional intervening CH, group between the C=O and C-Br orbitals would necessarily reduce the electronic interaction matrix elements between
t Identification of the small fraction of C-C fission in bromo- acetyl chloride by using a comparison with bromoacetone allowed a better determination of the C-Br branching. See analysis in ref, 12.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90 1583
1 .o
0.8
0.6
0.4
0.2
0
1 .o
0.8
0.6
0.4
0.2
0
100 200 300 400 500 600 700 800 t ime-of -arrival/ps
Fig. 2 Laboratory time-of-flight data, from ref. 10, of the photofrag- ments detected at (a) 79Br+ and (b) 35Cl+ from bromoacetyl chloride photodissociated at 248 nm with an unpolarized laser. The fits to the individual contributions from C-C and C-Br fission are described in and taken from ref. 1Oc. The forward convolution fit to the signal at Br' assigned to Br atoms, which dominates the fast side and peak of the spectrum, was obtained with the P(E,) for C-Br fission, shown in Fig. 5 (a).
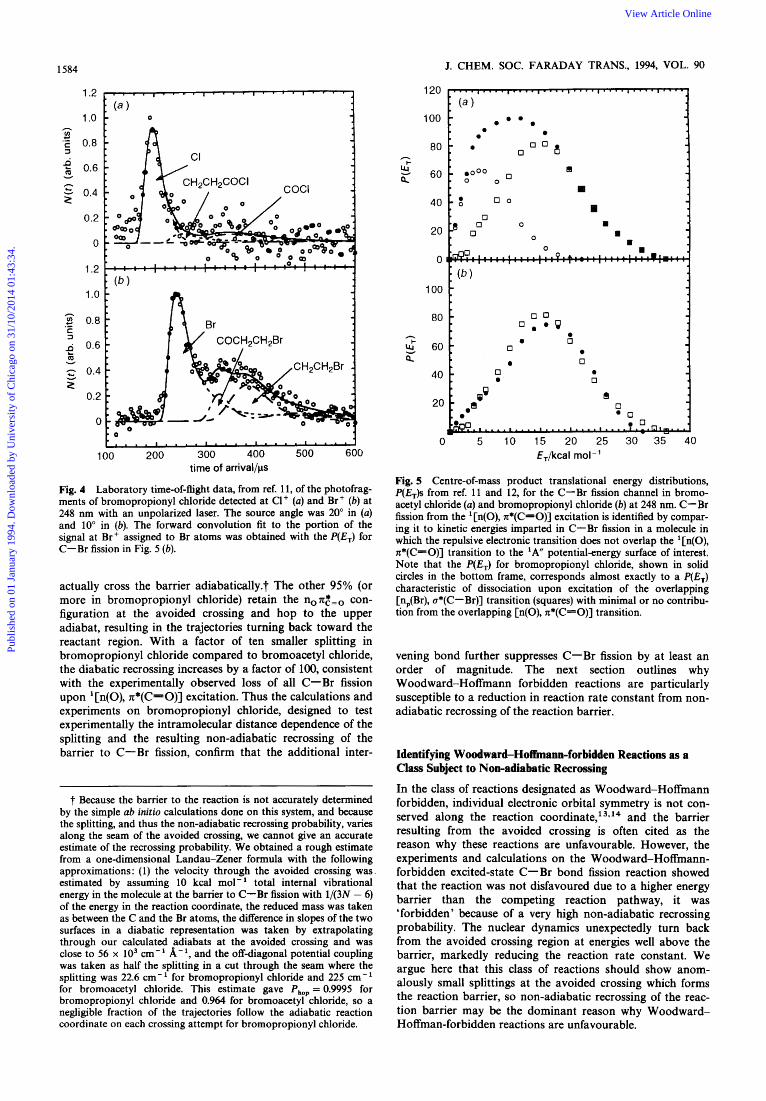
these two configurations, resulting in a smaller splitting between the adiabats at the barrier. The experiments showed that while in bromoacetyl chloride, the initial '[n(O), n*(C=O)] electronic transition resulted in a C-Cl : C-Br bond fission ratio of 1.0 : 0.4, in bromopropionyl chloride the same initial transition resulted in a C-Cl : C-Br bond fission ratio of 1.0 : <0.05.'0'*' ' The <0.05 branching to C-Br fission represents an upper limit; in fact, a comparison of the distributions of relative kinetic energies imparted to the C-Br fission fragments in the two molecules determined from the data in Fig. 2 and 4 show that essentially all of the Br-atom products observed from bromopropionyl chloride merely result from an overlapping transition to an electronic state diabatically repulsive in the C-Br bond. Fig. 5 com- pares the kinetic energy distribution of the C-Br fission fragments from bromoacetyl chloride, upper frame, which evidences C-Br fission from both the '[n(O), n*(C=O)] transition and from the overlapping repulsive transition, with that from bromopropionyl chloride, lower frame. When you superimpose the component from bromoacetyl chloride for C-Br fission from just the overlapping transition, it corre- sponds closely to the entire distribution from bromo- propionyl chloride as shown in Fig. 5. Thus, the lower-energy component from [n(O), n*(C=O)] excitation is absent in bromopropionyl chloride; the higher-energy C-Cl fission channel completely dominates C-Br fission on the 'A'' surface despite being the channel with the higher barrier. This further dramatic reduction in branching to C-Br fission upon ' [n(O), n*(C=O)] excitation supports the hypothesis
BrCH2COCI
BrCH,COCI
C-Br - reaction coordinate - C-CI fission fission
Fig. 3 Schematic reaction coordinates for C-Cl and C-Br bond fission from the 248 nm photodissociation of bromoacetyl chloride. The upper frame shows only the lowest ' A adiabatic excited elec- tronic surface where the barrier to C-Br bond fission is lower than the barrier to C-CI bond fission. Considering only this lowest adiabat leads to the incorrect prediction that C-Br bond fission should dominate in bromoacetyl chloride. The lower frame shows this lowest 'A" adiabat along with the upper 'A adiabat formed from the avoided electronic configuration crossing at the barriers to C-Cl and C-Br bond fission. In addition, the dotted lines in the lower frames show the diabatic electronic states. Preferential C-Cl bond fission in bromoacetyl chloride results because the greater coupling V,, between the n0n,*=, and the n,,a,*<, states allows the molecules to switch from one configuration to the other, resulting in adiabatic crossing of the bamer to C-CI bond fission. The smaller coupling V,, between the n0n,*=, and the nBra&Br states results in trajectories retaining the no x2.=o configuration by making a non- adiabatic 'hop' at the avoided crossing which forms the barrier to C-Br fission, so more trajectories turn back from the repulsive wall on the bound surface, reducing the branching to C- Br fission. Adapted from ref. 11
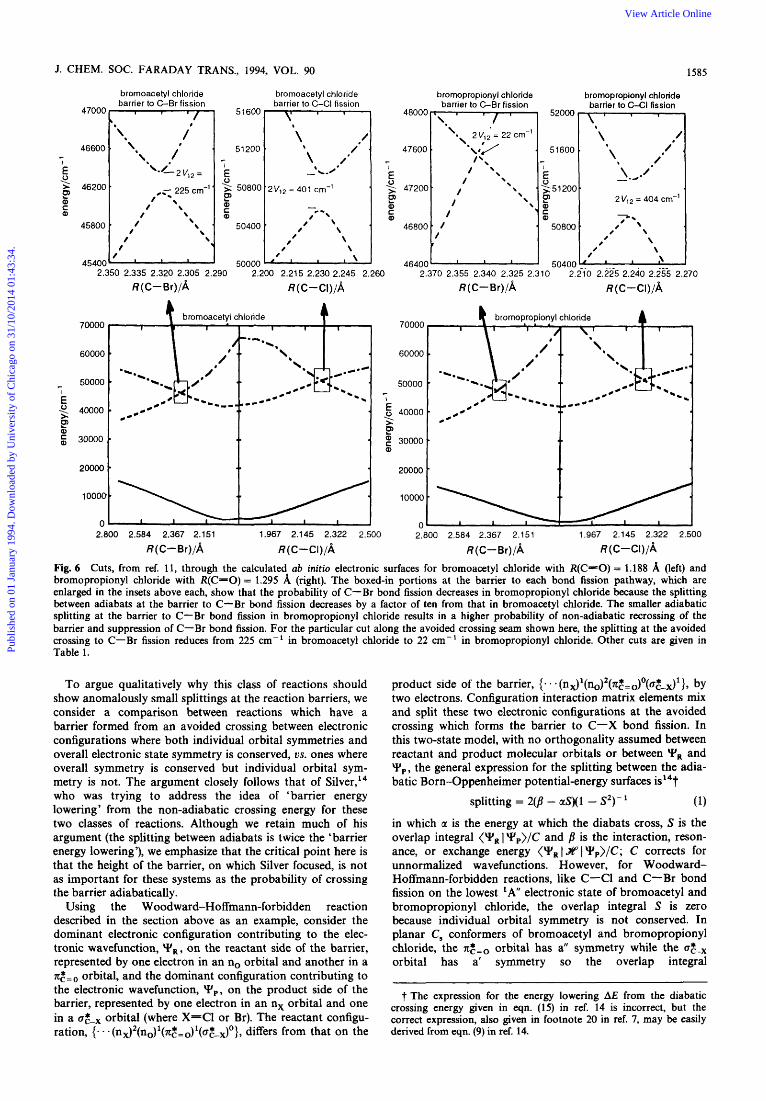
that the additional intervening CH, spacer would decrease the splitting between the adiabats at the avoided crossing and thus increase the non-adiabatic recrossing of the reaction barrier. A single reference configuration interaction (single excitations only) calculation of the splitting between the adia- bats at the barrier to C-Br fission in these two systems further tests this conclusion. While the approximations inher- ent in the method and the minimal (STO-3G*) basis set pre- clude quantitative accuracy, the results given in Fig. 6 and Table 1 show that the splitting at the avoided crossing is an order of magnitude smaller in bromopropionyl chloride than in bromoacetyl chloride," consistent with the interpretation of the experimental results. The splitting in both systems is so small that a simple Landau-Zener calculation shows that fewer than 5% of the reactive events that would lead to C-Br bond fission on the adiabatic potential surface
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
1584 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
h
c 'c 0.8 b
0.6 - 0.4 - 0.2 -
0 -
a CI
h
$ 0.8
0.6 - 0.4 - 0.2 -
0 -
.- C 3
4 U
v
2
100 200 300 400 500 600 time of arrival/ps
Fig. 4 Laboratory time-of-flight data, from ref. 11, of the photofrag- ments of bromopropionyl chloride detected at C1' (a) and Br+ (b) at 248 nm with an unpolarized laser. The source angle was 20" in (a) and 10" in (b). The forward convolution fit to the portion of the signal at Br+ assigned to Br atoms was obtained with the P(E,) for C-Br fission in Fig. 5 (b).
actually cross the barrier adiabatically.? The other 95% (or more in bromopropionyl chloride) retain the n,n.,*=, con- figuration at the avoided crossing and hop to the upper adiabat, resulting in the trajectories turning back toward the reactant region. With a factor of ten smaller splitting in bromopropionyl chloride compared to bromoacetyl chloride, the diabatic recrossing increases by a factor of 100, consistent with the experimentally observed loss of all C-Br fission upon '[n(O), n*(C=O)] excitation. Thus the calculations and experiments on bromopropionyl chloride, designed to test experimentally the intramolecular distance dependence of the splitting and the resulting non-adiabatic recrossing of the barrier to C-Br fission, confirm that the additional inter-
? Because the barrier to the reaction is not accurately determined by the simple ab initio calculations done on this system, and because the splitting, and thus the non-adiabatic recrossing probability, varies along the seam of the avoided crossing, we cannot give an accurate estimate of the recrossing probability. We obtained a rough estimate from a one-dimensional Landau-Zener formula with the following approximations: (1) the velocity through the avoided crossing was. estimated by assuming 10 kcal mol-' total internal vibrational energy in the molecule at the bamer to C-Br fission with 1/(3N - 6) of the energy in the reaction coordinate, the reduced mass was taken as between the C and the Br atoms, the difference in slopes of the two surfaces in a diabatic representation was taken by extrapolating through our calculated adiabats at the avoided crossing and was close to 56 x lo3 an-' A-', and the offdiagonal potential coupling was taken as half the splitting in a cut through the seam where the splitting was 22.6 cm-' for bromopropionyl chloride and 225 an-' for bromoacetyl chloride. This estimate gave Phop = 0.9995 for bromopropionyl chloride and 0.964 for bromoacetyl chloride, so a negligible fraction of the trajectories follow the adiabatic reaction coordinate on each crossing attempt for bromopropionyl chloride.
n I-
'u, Q
n I-
'u, k
401: O 0
a
100 1
0 5 10 15 20 25 30 35 40 E,/kcal rnol-'
Fig. 5 Centre-of-mass product translational energy distributions, P(E,)s from ref. 11 and 12, for the C-Br fission channel in bromo- acetyl chloride (a) and bromopropionyl chloride (b) at 248 nm. C-Br fission from the '[n(O), n*(C=O)] excitation is identified by compar- ing it to kinetic energies imparted in C-Br fission in a molecule in which the repulsive electronic transition does not overlap the '[n(O), n*(C=O)] transition to the 'Aff potentialenergy surface of interest. Note that the P(E,) for bromopropionyl chloride, shown in solid circles in the bottom frame, corresponds almost exactly to a P(E,) characteristic of dissociation upon excitation of the overlapping [n,(Br), o*(C-Br)] transition (squares) with minimal or no contribu- tion from the overlapping [n(O), n*(C=O)] transition.
vening bond further suppresses C-Br fission by at least an order of magnitude. The next section outlines why Woodward-Hoffmann forbidden reactions are particularly susceptible to a reduction in reaction rate constant from non- adiabatic recrossing of the reaction barrier.
Identifying Woodward-Hoftinann-forbidden Reactions as a Class Subject to Non-adiabatic Recrossing
In the class of reactions designated as Woodward-Hoffmann forbidden, individual electronic orbital symmetry is not con- served along the reaction ~oordinate , '~* '~ and the barrier resulting from the avoided crossing is often cited as the reason why these reactions are unfavourable. However, the experiments and calculations on the Woodward-Hoffmann- forbidden excited-state C-Br bond fission reaction showed that the reaction was not disfavoured due to a higher energy barrier than the competing reaction pathway, it was 'forbidden' because of a very high non-adiabatic recrossing probability. The nuclear dynamics unexpectedly turn back from the avoided crossing region at energies well above the barrier, markedly reducing the reaction rate constant. We argue here that this class of reactions should show anom- alously small splittings at the avoided crossing which forms the reaction barrier, so non-adiabatic recrossing of the reac- tion barrier may be the dominant reason why Woodward- Hoffman-forbidden reactions are unfavourable.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
52000
51600
I
E >512008
1585
-
*
bromoacetyl chloride brornoacetyl chloride barrier to C-Br fission barrier to C-CI fission
47000 I I 1 51600 K I . I
70000 I I I I 1 /
60000 *. - a /' I"'.
/ * , /*
-- 2- - - -1dId
/ *
P 5 30000 * - 30000- Q)
20000 ' 20000 .
46600 5
46200 *
45800
I 1
La1 ., '.
/--
/ 0 454OOL I I I 50000' ' 1 I \
brornopropionyl chloride barrier to C-Br fission
48000 b I 1 I I '.
46400
bromopropionyl chloride barrier to C-CI fission
7 \ /
2 V,, = 404 crn-'
50400 I 1 - _
2.350 2.335 2.320 2.305 2.290 2.200 2.215 2.230 2.245 2.260 2.370 2.355 2.340 2.325 2.310 2.510 2.225 2.240 2.255 2.270
R ( C- &)/A R(C-CI)/A R ( C- B r) /A R (C-Cl)/A
Fig. 6 Cuts, from ref. 11, through the calculated ab initio electronic surfaces for bromoacetyl chloride with R(C-0) = 1.188 A (left) and bromopropionyl chloride with R(C-0) = 1.295 8, (right). The boxed-in portions at the barrier to each bond fission pathway, which are enlarged in the insets above each, show that the probability of C-Br bond fission decreases in bromopropionyl chloride because the splitting between adiabats at the barrier to C-Br bond fission decreases by a factor of ten from that in bromoacetyl chloride. The smaller adiabatic splitting at the barrier to C-Br bond fission in bromopropionyl chloride results in a higher probability of non-adiabatic recrossing of the barrier and suppression of C-Br bond fission. For the particular cut along the avoided crossing seam shown here, the splitting at the avoided crossing to C-Br fission reduces from 225 cm-' in bromoacetyl chloride to 22 cm-' in bromopropionyl chloride. Other cuts are given in Table 1.
To argue qualitatively why this class of reactions should show anomalously small splittings at the reaction barriers, we consider a comparison between reactions which have a barrier formed from an avoided crossing between electronic configurations where both individual orbital symmetries and overall electronic state symmetry is conserved, us. ones where overall symmetry is conserved but individual orbital sym- metry is not. The argument closely follows that of Silver,I4 who was trying to address the idea of 'barrier energy lowering' from the non-adiabatic crossing energy for these two classes of reactions. Although we retain much of his argument (the splitting between adiabats is twice the 'barrier energy lowering'), we emphasize that the critical point here is that the height of the barrier, on which Silver focused, is not as important for these systems as the probability of crossing the barrier adiabatically.
Using the Woodward-Hoffmann-forbidden reaction described in the section above as an example, consider the dominant electronic configuration contributing to the elec- tronic wavefunction, VR, on the reactant side of the barrier, represented by one electron in an no orbital and another in a n& orbital, and the dominant configuration contributing to the electronic wavefunction, Yp, on the product side of the barrier, represented by one electron in an n, orbital and one in a o& orbital (where X-Cl or Br). The reactant configu- ration, {- - - (n.J2(no)1(7r~=o)1(a&Jo), differs from that on the
product side of the barrier, (. * (n.J'(n,)z(n~=o)o(o~x)l}, by two electrons. Configuration interaction matrix elements mix and split these two electronic configurations at the avoided crossing which forms the barrier to C-X bond fission. In this two-state model, with no orthogonality assumed between reactant and product molecular orbitals or between YR and Yp, the general expression for the splitting between the adia- batic Born-Oppenheimer potential-energy surfaces
(1) splitting = 2(8 - aSK1 - S2)- ' in which a is the energy at which the diabats cross, S is the overlap integral (YRIYp)/C and B is the interaction, reson- ance, or exchange energy ( Y R l ~ l ~ p ) / C ; C corrects for unnormalized wavefunctions. However, for Woodward- Hoffmann-forbidden reactions, like C-Cl and C-Br bond fission on the lowest 'A" electronic state of bromoacetyl and bromopropionyl chloride, the overlap integral S is zero because individual orbital symmetry is not conserved. In planar C, conformers of bromoacetyl and bromopropionyl chloride, the n:=o orbital has a" symmetry while the orbital has a' symmetry so the overlap integral
~~
7 The expression for the energy lowering AE from the diabatic crossing energy given in eqn. (15) in ref. 14 is incorrect, but the correct expression, also given in footnote 20 in ref. 7, may be easily derived from eqn. (9) in ref. 14.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online

1586
Table 1 coordinates, at a variety of C-0 bond lengths, in bromoacetyl and bromopropionyl chloride
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
Summary of ab initio calculations of the splitting between adiabats at the barriers along the C-Cl and C-Br bond fission reaction
Bromoacetyl chloride Barrier to C-Cl bond fission
energy/cm-' at barrier on 'A, referenced to minimum
R(C = OJA R(C-B,dA Ro,, at barrierldi in the ground state 2 V',/Cm - 1
1.935 2.232 50 548 401.7 1.188 1.313 1.935 2.382 44 192 88.7 1.388 1.588 1.935 2.553 53 264 1 160.0
Barrier to C-Br bond fission
1.935 2.464 44 763 57.3
energy/cm-' at barrier on ' A , referenced to minimum
R(C = oJA R(C-Br) at barrier/A R o I J A in the ground state 2 Vl Jcm -
1.188 2.3205 1.789 46 136 225.0 1.313 2.522 1.789 40051 137.9
1.588 2.777 1.789 50 513 67.8 1.388 2.627 1.789 40 821 121.0
Bromopropionyl chloride Barrier to C-Cl bond fission
~~
energy/cm-' at barrier on 'A", referenced to minimum
in the ground state Ro,, at barrier/A 2 Vl Jcm - R,c= o JA R ( ~ - ~ ~ JA 1.195 1.295 1.395 1.495
1.935 1.935 1.935 1.935
2.239 2.351 2.453 2.523
50 897 45 335 45 296 48 279
404.1 130.6 106.5 494.4
~
Barrier to C-Br bond fission
energy/cm-' at barrier on 'A", referenced to minimum
R,c = O J A R(C-Br) at barrier/A R(C4lJA in the ground state 2V1 &rn - ' 1.195 1.295 1.395 1.495
2.3439 2.503 2.626 2.7247
1.797 1.797 1.797 1.797
47 536 41 698 41 324 44 550
22.6 20.2 12.1 9.7
The first three columns show the C - 0 , C-Br and C-Cl bond lengths, in A, at the barrier. The fourth column shows the calculated energy of the lowest 'A" adiabatic electronic surface at the barrier, while the fifth column, labelled 2V12, shows the splitting between the two lowest 'A" states at the barrier. Table from ref. 11.
( x Z = ~ I C J Z - ~ ) = <a" I a') = 0. Similarly, the n, orbital has a'' symmetry while the no orbital has a' symmetry so the overlap integral (n, I no) = (a" 1 a') = 0. Because individual orbital symmetry is not conserved for Woodward-Hoffmann- forbidden reactions, all one-electron integrals that contribute to the resonance and exchange energy represented by /? above are also zero, leaving only two-electron integrals to mix and split the electronic states at the avoided cro~sing. '~
splitting in W-H-forbidden reactions = 2P2 (2)
Thus the splitting between the adiabats at the barrier is expected to be anomalously small for this class of reactions. Indeed, our simple electronic structure calculations show the splitting is on the order of hundreds of wavenumbers for both C-Br and C-CI fission in bromoacetyl chloride and on the order of only tens of wavenumbers for C-Br fission in bromopropionyl chloride. In addition, there is preliminary computational evidence that upon breaking the symmetry element that makes the reaction Woodward-Hoffmann- forbidden, as in the gauche conformer of these molecules or of bromoacetone' (see Table 2, later), the splitting increases considerably.
The marked intramolecular distance dependence of the splitting and resulting non-adiabatic recrossing probability for C-Br fission in Br(CH,),COCl can also be understood by analysing the configuration interaction matrix elements (equal to twice the off-diagonal potential coupling between two diabatic electronic configurations in the diabatic representation) using an orthogonal basis set. For singlets, they are:15b
The first matrix element, similar to that in a Forster model for intramolecular electronic energy transfer, decreases with the separation between chromophores due to the r12 in the denominator, while the second matrix element, similar to that in the Dexter energy-transfer mechanism, decreases roughly exponentially as the overlap densities no(l)nx(l) and n,*,0(2)acx(2) decrease. The reason for the parallel with energy-transfer theory is the implicit intramolecular elec- tronic energy transfer required along the adiabatic reaction coordinate for this system. To proceed over the barrier to
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online

J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90 1587
C-Br fission on the A" potential-energy surface, the elec- tronic wavefunction must change from no .nz=o in character to npBr t ~ , ? ~ ~ in character at the avoided crossing which forms the barrier to C-Br fission. If the off-diagonal potential coupling between these two non-adiabatic electronic configu- rations is small (due in part to the spatial separation between the C=O and C-Br orbitals), the electronic wavefunction can retain n7r,?j=o character as the molecule approaches the barrier to C-Br fission. Then, instead of going over the barrier on the adiabatic potential-energy surface it turns back from the attractive potential of the nnz=o diabat shown by the dotted line in Fig. 3, resulting in a smaller branching to C-Br fission than expected, as observed in the experiment. (This is equivalent to non-adiabatically hopping up to the upper adiabat, reversing direction on the attractive wall, and returning to the reactant upon non-adiabatic transition to the lower adiabat.) Indeed, electron-transfer reactions and intramolecular electronic energy-transfer processes are often described in a diabatic representation, where the rate is pro- portional to the square of the off-diagonal potential coupling matrix element ; the larger the off-diagonal potential coupling between diabats, the larger the splitting and thus the higher the probability of following the adiabatic electron-transfer reaction coordinate and the faster the electron-transfer rate.7 transfer rate.'
4. Conformation Dependence of Non-adiabatic Recrossing near a Conical Intersection: C - C fission in
CH,COCH,Br The importance of non-adiabatic effects at a conical intersec- tion has long been recogni~ed.'~?' 5,20 In condensed-phase photochemical processes where internal energy can be drained from the local system, they have been termed funnels, providing a region of an excited potential-energy surface where transition to the ground state can occur with high probability.'5 The experiments on the competition between C-Br and C-C photocleavage in anti- and gauche- bromoacetone described below ', elucidate two general fea- tures of reactions which must proceed past a conical intersection along either an excited-state or a ground-state adiabatic reaction coordinate (see schematic excited-state reaction coordinate for C-C fission in bromoacetone in Fig. 7), their dramatic conformation dependence and their essen- tial non-adiabatic character. We end with outlining why many reactions which are described as 'symmetry forbidden' are in fact symmetry forbidden only at a singularity on the multidimensional reactive potential-energy surface, so they are better described as forbidden due to the high non- adiabatic recrossing probabiity in the region of the conical intersection.
The study of bromoacetone'2 was initiated to investigate the conformation dependence of the non-adiabatic recrossing probability along the C-Br bond fission reaction coordinate, only to find that the conformation dependence of C-C fission through a conical intersection dominated the differ- ence in branching.? This paragraph outlines the three initial observations which led us to turn to considering the confor- mation dependence of C-C fission through a conical inter- section: (1) in a thermal population of bromoacetone
-f The bromoacetone and chloroacetone conformer which we refer to here as 'trans' or 'anti' should, in correct nomenclature, be called s-cis, as the dihedral angle between the C-CI(Br) and the C=O bond is zero and the C1 or Br and 0 atoms, having the highest atomic numbers, should determine the groups to which the geometry refers. However, we retain the non-standard name of trans for this conformer in order to remain consistent with several earlier studies.
% 5a
Fig. 7 Schematic excited-state reaction coordinate for C-C bond fission in bromoacetone (top). The cross-sections show the different regions of the conical intersection along the C-C fission reaction coordinate sampled by dissociating the anti us. the gauche conformer. The frames on the r.h.s. of the figure show three slices through the conical intersection: the lowest at planar geometry (atorsion = 0); the middle at close to planar geometries (small a) where the adiabats are weakly split, so non-adiabatic recrossing dominates the dynamics; and the uppermost at highly non-planar geometries, sampled by dis- sociative trajectories from the gauche conformer, where the adiabats are strongly split so C-C fission can proceed adiabatically. Adapted from ref. 12.
conformers, both C-C and C-Br fission occur; (2) the angular distribution of Br atoms shows C-Br fission occurs primarily from the minor anti conformer, not the dominant gauche one; and (3) low-level ab initio calculations show C-Br fission is more, not less, non-adiabatically suppressed in the anti conformer than in the gauche, so the conformation dependence of the C-C : C-Br branching ratio is not deter- mined by the C-Br fission reaction coordinate; it must reflect the conformation dependence of C-C fission. The photofragment time-of-flight spectra in Fig. 8 show the first result: photoexcitation of bromoacetone at 308 nm in the nnz,o absorption band, analogous to the absorption excited in bromoacetyl chloride above without the overlapping repulsive transition, results in both primary C-Br and primary C-C bond fission products. Linear momentum con- servation allows us to use the velocities of the Br atoms detected in the Br+ spectrum to identify the portion of the CH,CO+ spectrum in Fig. 8 (b) from the momentum- matched CH,COCH3 photoproduct cracking to CH,CO + in the mass spectrometer. The remaining signal in that spectrum is easily assigned to COCH, from primary C-C bond fission, as the COCH, product is also seen with the same distribution of arrival times, peaking near 340 ps, at the CH,CO+ parent ion and as this portion of the distribution is momentum-matched to a weak signal at CH,Br+ from CH,Br product. Armed with the knowledge that C-C and C-Br fission compete upon excitation at 308 nm, the angular distribution of the Br gave the second key result, our first probe of a conformation dependence to the branching. If there were no conformation dependence to the branching, the angular distribution of the Br atom fragments would be a
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
1588 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
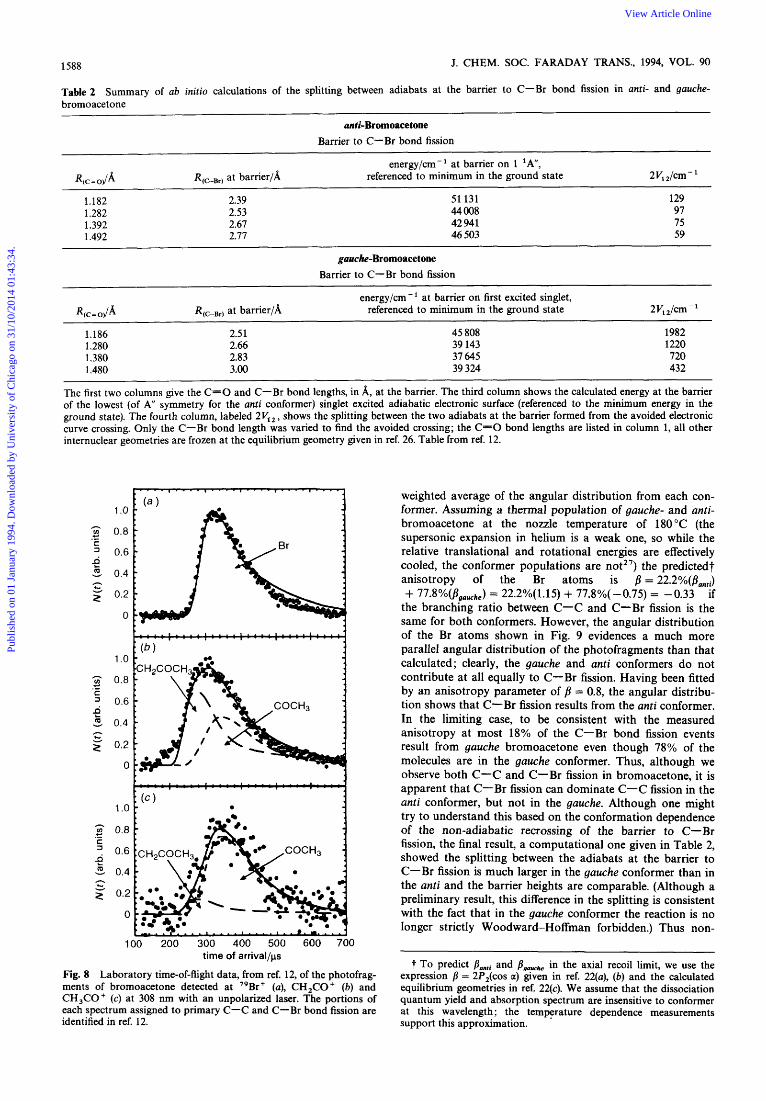
Table 2 bromoacetone
Summary of ab initio calculations of the splitting between adiabats at the barrier to C-Br bond fission in anti- and gauche-
anti-Bromoacetone Barrier to C-Br bond fission
~~~
energy/cm-' at barrier on 1 'A, referenced to minimum in the ground state R(C-Br) at barrier/A 2 Vl Jcm - R, = oJA
1.182 2.39 51 131 129 1.282 2.53 44 008 97 1.392 2.67 42 941 75 1.492 2.77 46 503 59
gauche-Bromoacetone Barrier to C- Br bond fission
energy/cm-' at barrier on first excited singlet, R(C-Br) at barrier/A referenced to minimum in the ground state 2 v, ,/cm - 1 R{c = o JA
1.186 2.51 45 808 1982
1.380 2.83 37 645 720 1.480 3.00 39 324 432
1.280 2.66 39 143 1220
The first two columns gve the C-0 and C-Br bond lengths, in A, at the barrier. The third column shows the calculated energy at the barrier of the lowest (of A" symmetry for the anti conformer) singlet excited adiabatic electronic surface (referenced to the minimum energy in the ground state). The fourth column, labeled 2V1, , shows the splitting between the two adiabats at the barrier formed from the avoided electronic curve crossing. Only the C-Br bond length was varied to find the avoided crossing; the C=O bond lengths are listed in column 1, all other internuclear geometries are frozen at the equilibrium geometry given in ref. 26. Table from ref. 12.
1 .o 0.8
0.6
0.4
0.2
0
100 200 300 400 500 600 700 time of arrival/ps
Fig. 8 Laboratory time-of-flight data, from ref. 12, of the photofrag- ments of bromoacetone detected at "Br+ (a), CH,CO+ (b) and CH,CO+ (c) at 308 nm with an unpolarized laser. The portions of each spectrum assigned to primary C-C and C-Br bond fission are identified in ref. 12.
weighted average of the angular distribution from each con- former. Assuming a thermal population of gauche- and anti- bromoacetone at the nozzle temperature of 180°C (the supersonic expansion in helium is a weak one, so while the relative translational and rotational energies are effectively cooled, the conformer populations are not2') the predicted? anisotropy of the Br atoms is = 22.2%(8,,,,) + 77.8%(8g,,h,) = 22.2%(1.15) + 77.8%( -0.75) = -0.33 if
the branching ratio between C-C and C-Br fission is the same for both conformers. However, the angular distribution of the Br atoms shown in Fig. 9 evidences a much more parallel angular distribution of the photofragments than that calculated; clearly, the gauche and anti conformers do not contribute at all equally to C-Br fission. Having been fitted by an anisotropy parameter of B = 0.8, the angular distribu- tion shows that C-Br fission results from the anti conformer. In the limiting case, to be consistent with the measured anisotropy at most 18% of the C-Br bond fission events result from gauche bromoacetone even though 78% of the molecules are in the gauche conformer. Thus, although we observe both C-C and C-Br fission in bromoacetone, it is apparent that C-Br fission can dominate C-C fission in the anti conformer, but not in the gauche. Although one might try to understand this based on the conformation dependence of the non-adiabatic recrossing of the barrier to C-Br fission, the final result, a computational one given in Table 2, showed the splitting between the adiabats at the barrier to C-Br fission is much larger in the gauche conformer than in the anti and the barrier heights are comparable. (Although a preliminary result, this difference in the splitting is consistent with the fact that in the gauche conformer the reaction is no longer strictly Woodward-Hoffman forbidden.) Thus non-
t To predict Bmri and #lglruche in the axial recoil limit, we use the expression #I = 2P,(cos a) given in ref. 22(a), (b) and the calculated equilibrium geometries in ref. 22(c). We assume that the dissociation quantum yield and absorption spectrum are insensitive to conformer at this wavelength ; the temperature dependence measurements support this approximation.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
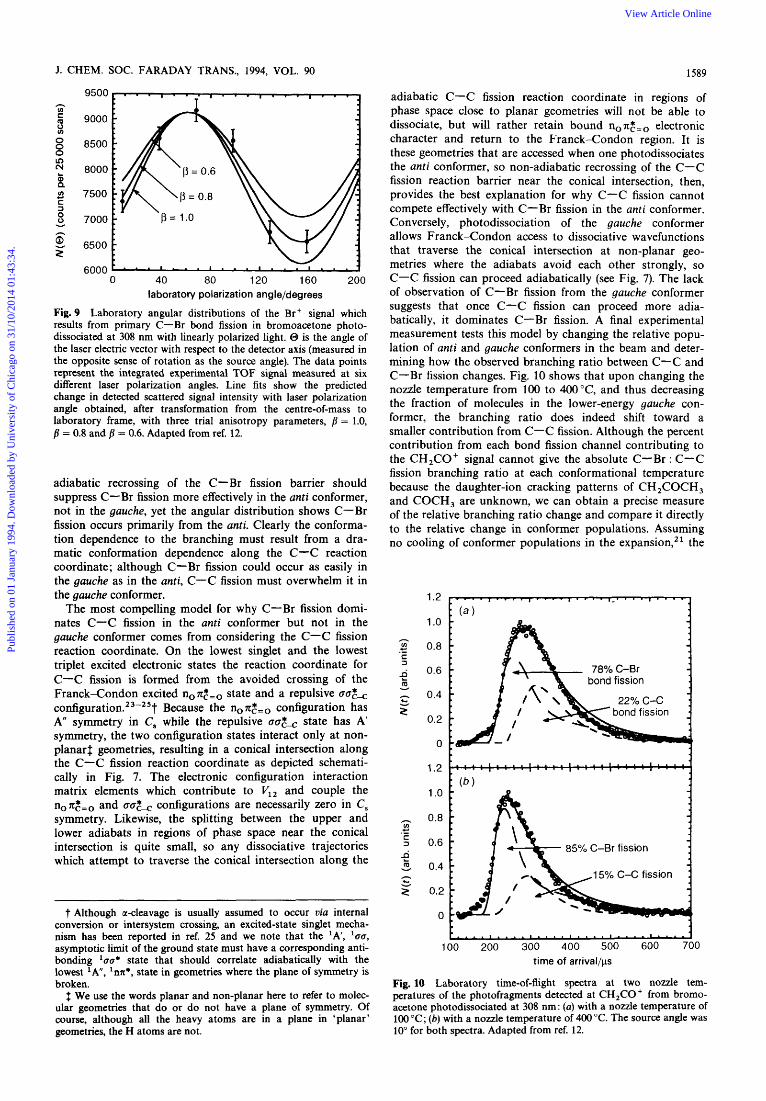
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90 1589
0 0 0 m N t P UJ
t C
s W
. . . , . . . I . . . I . . . I . . .
T
9000
8500
8000
7500
7000
6500
0 40 80 120 160 200 laboratory polarization angle/degrees
Fig. 9 Laboratory angular distributions of the Br+ signal which results from primary C-Br bond fission in bromoacetone photo- dissociated at 308 nm with linearly polarized light. @ is the angle of the laser electric vector with respect to the detector axis (measured in the opposite sense of rotation as the source angle). The data points represent the integrated experimental TUF signal measured at six different laser polarization angles. Line fits show the predicted change in detected scattered signal intensity with laser polarization angle obtained, after transformation from the centre-of-mass to laboratory frame, with three trial anisotropy parameters, j = 1.0, fi = 0.8 and /? = 0.6. Adapted from ref. 12.
adiabatic recrossing of the C-Br fission barrier should suppress C-Br fission more effectively in the anti conformer, not in the gauche, yet the angular distribution shows C-Br fission occurs primarily from the anti. Clearly the conforma- tion dependence to the branching must result from a dra- matic conformation dependence along the C-C reaction coordinate; although C-Br fission could occur as easily in the gauche as in the anti, C-C fission must overwhelm it in the gauche conformer.
The most compelling model for why C-Br fission domi- nates C-C fission in the anti conformer but not in the gauche conformer comes from considering the C-C fission reaction coordinate. On the lowest singlet and the lowest triplet excited electronic states the reaction coordinate for C-C fission is formed from the avoided crossing of the Franck-Condon excited n0n,*=, state and a repulsive a& c~nfigurat ion.~~-~ ' t Because the no n&o configuration has A" symmetry in C, while the repulsive a& state has A' symmetry, the two configuration states interact only at non- planart geometries, resulting in a conical intersection along the C-C fission reaction coordinate as depicted schemati- cally in Fig. 7. The electronic configuration interaction matrix elements which contribute to V,, and couple the n, n&o and aa& configurations are necessarily zero in C , symmetry. Likewise, the splitting between the upper and lower adiabats in regions of phase space near the conical intersection is quite small, so any dissociative trajectories which attempt to traverse the conical intersection along the
7 Although a-cleavage is usually assumed to occur via internal conversion or intersystem crossing, an excited-state singlet mecha- nism has been reported in ref. 25 and we note that the 'A, 'm, asymptotic limit of the ground state must have a corresponding anti- bonding 'OQ* state that should correlate adiabatically with the lowest 'A, h * , state in geometries where the plane of symmetry is broken.
1 We use the words planar and non-planar here to refer to molec- ular geometries that do or do not have a plane of symmetry. Of course, although all the heavy atoms are in a plane in 'planar' geometries, the H atoms are not.
adiabatic C-C fission reaction coordinate in regions of phase space close to planar geometries will not be able to dissociate, but will rather retain bound no . ~ l & electronic character and return to the Franck-Condon region. It is these geometries that are accessed when one photodissociates the anti conformer, so non-adiabatic recrossing of the C-C fission reaction barrier near the conical intersection, then, provides the best explanation for why C-C fission cannot compete effectively with C-Br fission in the anti conformer. Conversely, photodissociation of the gauche conformer allows Franck-Condon access to dissociative wavefunctions that traverse the conical intersection at non-planar geo- metries where the adiabats avoid each other strongly, so C-C fission can proceed adiabatically (see Fig. 7). The lack of observation of C-Br fission from the gauche conformer suggests that once C-C fission can proceed more adia- batically, it dominates C-Br fission. A final experimental measurement tests this model by changing the relative popu- lation of anti and gauche conformers in the beam and deter- mining how the observed branching ratio between C-C and C-Br fission changes. Fig. 10 shows that upon changing the nozzle temperature from 100 to 40O0C, and thus decreasing the fraction of molecules in the lower-energy gauche con- former, the branching ratio does indeed shift toward a smaller contribution from C-C fission. Although the percent contribution from each bond fission channel contributing to the CH,CO+ signal cannot give the absolute C-Br : C-C fission branching ratio at each conformational temperature because the daughter-ion cracking patterns of CH,COCH, and COCH, are unknown, we can obtain a precise measure of the relative branching ratio change and compare it directly to the relative change in conformer populations. Assuming no cooling of conformer populations in the expansion,21 the
h v)
C *.' .-
1.2
1 .o
0.8
0.6
0.4
0.2
0 ?
85% C-Br fission
15% C-C fission
100 200 300 400 500 600 700 time of arrival/ps
Fig. 10 Laboratory time-of-flight spectra at two n o d e tem- peratures of the photofragments detected at CH2CO+ from bromo- acetone photodissociated at 308 nm: (a) with a nozzle temperature of 100 "C; (b) with a nozzle temperature of 400 "C. The source angle was 10" for both spectra. Adapted from ref. 12.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online

1590 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
relative change in conformer population in heating the nozzle from 100 to 400°C is:
4 0 0 ” ~ : ( anti ) f C-Br)
t 100°C:
\4auchej,oo (0.34) (0.25)
predicted = 1.4
(4)
giving a predicted increase in the C-Br: C-C branching ratio of a factor of 1.4 if C-C fission dominates in the gauche conformer and C-Br fission dominates in the anti con- former. The experimentally observed change in branching ratio is quite similar :
observed (z) (E) = 1.6!
The result is in accord with the model presented in which dissociation from the anti conformer accesses regions of the C-C fission reaction coordinate near the conical intersec- tion, where it is non-adiabatically inhibited so cannot compete with C-Br fission, and dissociation from the gauche conformer proceeds through regions of the conical intersec- tion where the surfaces are further split from each other, so the C-C dissociation can proceed adiabatically and domi- nate C-Br fission.
We have avoided using the terminology ‘symmetry forbidden’ when describing the lack of C-C fission from the anti conformer because, although commonly used, this lan- guage obscures the physical reason why a reaction pathway that traverses a conical intersection might be unfavourable. Indeed, the reaction is ‘symmetry forbidden’ only through a singular point on the potential-energy surface;t even small zero-point bending or torsional motion of the anti conformer at the transition state puts amplitude at molecular geometries where the adiabatic correlation goes smoothly from reactants to products. The reason why C-C fission in the anti con- former is suppressed is that trajectories that attempt to undergo C-C fission near, but not at, the point of conical intersection sample a region of phase space where the reac- tant and product electronic configurations are not strongly coupled, so the configuration interaction splitting between the upper and lower adiabats is small. Instead of following the adiabatic reaction coordinate, along which the electronic wavefunction changes from no ~ r z = ~ to a& in character, the dissociative trajectory hops to the upper bound adiabat as it tries to traverse the C-C reaction barrier. Thus a reaction pathway through a conical intersection is not ‘symmetry forbidden ’ for most dissociative trajectories; rather, it is char- acterized by a high non-adiabatic recrossing probability.
5. Intramolecular Distance and Conformational Dependence of Non-adiabatic Recrossing: Competing
C - C and C-Cl fission in CH,COCH,Cl YS. - CH,COCl
The background work reviewed in the previous sections examined the competition between bond fission channels belonging to two classes of reactions highly susceptible to
t If one only considers two internuclear degrees of freedom, one obtains a single point of conical intersection as shown in Fig. 7. Of course, along any internuclear coordinate which does not break the plane of symmetry, this becomes a line of intersection.
non-adiabatic effects, Woodward-Hoffmann-forbidden reac- tions and reactions proceeding through a conical intersection. Two key results in that work motivated the new experiments on chloroacetone presented below. The results on C-Br fission in bromopropionyl chloride, when compared with bromoacetyl chloride, showed that the additional intervening CH, spacer reduces the splitting between the adiabatic potential-energy surfaces at the barrier to C- Br fission, so non-adiabatic recrossing further supresses C- Br fission by over an order of magnitude. The experiments on bromo- acetone investigated the conformation dependence of non- adiabaticity at a conical intersection, supporting a model in which non-adiabatic recrossing of the barrier to C-C bond fission depends strongly on molecular conformer. While the anti conformer accesses regions of the conical intersection where the splitting between adiabats is very small, the gauche conformer accesses regions where C-C fission can proceed adiabatically and dominate C-Br fission. Those two results led us to predict that in chloroacetone C-C fission may effectively compete with C-Cl fission upon nn* excitation. Although previous work on a closely related system, acetyl chloride,26 showed that only primary C-Cl fission occurs, the increased distance between the C=O and C-Cl orbitals in chloroacetone should reduce the splitting at the avoided crossing which forms the barrier to C-Cl on the A potential-energy surface; the increase in non-adiabatic re- crossing of the barrier to C-Cl fission may allow C-C fission to compete. In addition, while acetyl chloride disso- ciates from near-planar geometries where C-C fission would be suppressed by non-adiabatic recrossing of the conical intersection, chloroacetone offers a gauche conformer from which the C-C channel could proceed more adiabatically. Thus both effects lead us to the prediction that C-C fission should compete effectively with C-Cl fission in chloroace- tone. The following experiments and calculations test this prediction.
Experimental Method To measure the photofragment velocities and angular dis- tributions from the photodissociation of chloroacetone, ClCH,COCH, , we used a crossed laser-molecular beam apparat~s.~’*~* Upon photodissociation with a pulsed excimer laser, neutral dissociation products scatter from the crossing point of the laser and the molecular beam with velo- cities determined by the vector sum of the molecular beam velocity and the recoil velocity imparted in the dissociation. Those scattered into the acceptance angle of the differentially pumped detector travel 44.1 cm to an electron bombardment ionizer and are ionized by 200 eV electrons. After mass selec- tion with a quadrupole mass filter, the ions are counted with a Daly detector and multichannel scalar with respect to their time of flight (TOF) from the interaction region after the dis- sociating laser pulse. Upon subtraction of the calibrated ion flight time, forward convolution fitting of the TOF spectrum determines the distribution of energies released to relative product translation in the dissociation. The angular distribu- tion of the scattered photofragments is obtained with a lin- early polarized photolysis beam by measuring the variation in signal intensity with the direction of the electric vector of the laser in the molecular beam/detector scattering plane.
The molecular beam was formed by expanding gaseous chloroacetone, at its vapour pressure at 45 “C, seeded in He to give a total stagnation pressure of 300 Torr. The 0.076 mm diameter nozzle was heated to 200°C in the measurements to determine the angular distribution of the primary photofrag- ments. The peak beam velocity was 1.38 x lo5 cm s - l with a full-width-at-half-maximum of 18%. To determine the
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online

J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90 1591
changes in the relative branching between primary C-Cl and C-C bond fission as a function of the trans : gauche ratio in the parent molecular beam the data was retaken at two additional nozzle temperatures, 100°C and 400°C. To measure the velocity of the parent molecular beam in situ, the molecular beam source was rotated to point into the detector and a chopper wheel raised into the beam. To measure the velocities of the neutral photofragments, the molecular beam source is rotated to a different angle in the plane containing the beam and detector axis, a plane perpendicular to the laser beam propagation direction. Laser polarization angles and molecular beam source angles are given here with respect to the detector axis, the first defined as positive with clockwise rotation and the other as positive with counterclockwise rotation.
Time-of-flight and angular distribution measurements were made on chloroacetone photofragments at 308 nm. The source angle was maintained at 15" with respect to the detec- tor axis for the data with the 200°C nozzle, and 10" for the data with the 100°C and 400°C nozzle. The unpolarized laser power from a Questek 2640 excimer was typically 130 mJ per pulse at 308 nm, with the light focused to a 5 mm2 spot size at the crossing region of the laser and molecular beam. Polarized spectra typically were taken at 30 mJ per pulse for 308 nm. Quadrupole resolution was adjusted to 0.9 u FWHM for m/z+ = 35 (Cl') and to 1.1 u for m/z+ = 49 (ClCH,), m/z+ = 43 (CH,CO+) and m/z+ = 42 (CH,CO+). For the anisotropy measurements, we disperse the unpo- larized laser light into two linearly polarized components with a single-crystal quartz Pellin-Broca and use the hori- zontal component, rotating the polarization into the desired direction with a half-wave retarder. The polarization depen- dent signal, integrated in many repeated short scans and alternating between each laser polarization direction, required no additional normalization to laser power or detec- tor efficiency.
The strong signal observed at C1+ after 200000 shots evi- dences primary C-Cl fission. The momentum-matched CH,COCH, product was not detected at the parent ion after 200000 shots, but was observed in the strong signal for the CH,CO+ daughter ion after 400000 shots. The weaker signal at ClCHl after 700000 shots could be fitted entirely to ClCH2-COCH, fission. The energy distribution for this C-C fission also fit well to the slow shoulders in the signal at C1+ and CH,CO+. The weak signal at CH,CO+ after 400000 shots with a 100°C nozzle came largely from C-C fission, although the poor signal-to-noise allows for some contribution from the daughter ion of the CH,COCH, product from the C-Cl fission. At 200"C, after 500000 shots, the signal at CH,CO+ was even weaker, but showed a substantial contribution from C-C1 fission. No signal was seen after 300 OOO shots at 200 "C, at the mass corresponding to the ClCH,CO+ product of the other possible C-C fission channel, and the small signal after lo6 shots at the mass at 100°C is attributed to C-C1 and C-C fission from clusters formed in the expansion.
Computational Method
To help interpret the experimental results, we also present ab initio electronic structure calculations for acetyl chloride and chloroacetone using the GAUSSIAN 92 system of pro- g r a m ~ . ~ ~ We use second-order Msller-Plesset theory with full electron correlation and the 3-21G* basis set, with sym- metry constraints where appropriate, to obtain equilibrium geometries shown in Table 3 for chloroacetone (Fig. 11) and Table 4 for acetyl chloride (Fig. 12); all other calculations use
Table 3 Optimized ground-state geometries for trans- and gauche- chloroacetone
parameter trans gauche
r(c-Cl) r(C1 -C2)
r(C=O) r(C--H,) r(C--H,) r(C-H3) r(C--H,) r(C-H,) L (C- c- C1) L(C,-C2==0) L (C-c-C) L (C-C-H L(C-C-H,) L (C- C- H3) L (C - C - H4) L(C-C-H,) z(C1- c- c- 0) z(c-c-c-0)
r(c2-c3)
z(H -C-C=O) z(H,-C-C=O) .t( H 3-C- C-0) z(H,-C-C=O) z(H,-C-C=O)
1.796 1.540 1.531 1.245 1.093 1.093 1.092 1.096 1.096
111.8 123.4 112.8 109.6 109.6 108.5 110.3 110.3
0.0 180.0
- 120.1 120.1
0.0 120.1
- 120.1
1.817 1.540 1.523 1.251 1.092 1.089 1.092 1.095 1.095
11 1.1 120.1 116.0 110.9 108.5 108.5 1 10.2 110.1
- 123.4 181.5 117.5 - 4.7 - 2.2 118.2
- 122.6
In the MP2(FULL)/3-21G* optimization the trans conformer was constrained to have a plane of symmetry. Bond lengths are given in 8, and bond angles in degrees.
0 1 1
Fig. 11 Chloroacetone. The torsional dihedral angle z[CIC(2)C(l) = 03 of 0" corresponds to the trans conformer.z4
the STO-3G* basis set. Configuration interaction with single and double excitations (CISD) calculations provide electronic ground-state energies in the harmonic region of the C-Cl and C-0 stretching potentials. These energies are then fit to Morse potentials with estimated dissociation energies of 83.5
Table 4 Optimized ground-state geometry for acetyl chloride
r(c-CI) 4C-C) r(C=O) r(C-H,) r(C-H,) r(C--H,) L (C1- c = 0) L(C-c-0) L (C-C-H L (C- C- H 2 )
L (C- C- H 3)
1.822 1 1 S579 1.2447 1.1044 1.1041 1.1041
120.5 127.0 110.0 1 10.0 110.0
z( c - c = 0 - a) 180.0 7(H ,-C-C=O) 0.0 r(H,-C-C=O) 120.0 z(H,-C-C=O) - 120.0
In the MP2(FULL)/3-2AG* optimization the molecule was con- strained to have a plane of symmetry. Bond lengths are given in 8, and bond angles in degrees.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
1592
140
120
100
+ 80 h
luu 6 0 1
40
20
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
. . . . r - . . . i . . . . i . . . . i . . . . ~ . . . . .
0
; a -
0
0
0
a - 0
a . . . . l . . . . l . . . . i . . . . I . . . r l r . = . . . ' - . ~ . .
0
II
Fig. 12 Acetyl chloride
and 169 kcal mol-', respectively.? The ground state energy, as a function of the C-Cl and C=O stretching coordinates, is then assumed to be a sum of the two independent Morse potentials. Configuration interaction with single excitations (CIS) calculations provide excitation energies from the ground electronic state to the relevant excited electronic states. These CIS excitation energies are added to the ground-state Morse oscillator energies to construct the excited electronic state surfaces. Calculation of the excited electronic states provides the barrier height to C-C1 bond fission along the lowest adiabatic 'A" excited electronic state. In addition, the calculations also provide energetic splittings between the two lowest singlet adiabatic excited electronic states at the avoided crossing in the C-C1 bond fission channel for acetyl chloride and both the trans and gauche conformers of chloroacetone.
Because the 'no nz=o excited electronic state, accessed by a 308 nm photon, has a longer equilibrium C-0 bond length than in the ground electronic state, we present CIS calcu- lations of the avoided crossing region on the excited elec- tronic surfaces at a variety of C-0 bond lengths which represent the range of C=O stretching motion likely sampled by the dissociative wavefunction.
Identification of Primary Product Channels : Results and Analysis
Primary C-Cl and C-C Bond Fission in Chloroacetone When chloroacetone is excited at 308 nm in the '[n(O), n*(C=O)] absorption band, either the C-Cl bond, or the C-C bond will break; the data shows competition between the two bond fission channels. Fig. 13 shows the photofrag- ment TOF spectra taken at m/z+ = 49, ClCHl , with 700000 laser shots and a 200°C nozzle. All of the signal was attrib- uted to primary C-C fission:
ClCH,COCH, + hv(308 nm) -, ClCH, + COCH, (6)
Through forward convolution fitting of the ClCH; spectrum, a centre-of-mass (c.m.) translational energy distribution, P(E,) (Fig. 14) was derived for C-C bond fission which peaks near very low energies, but extends out near 5 kcal mol-' or approximately one-third of the 13 kcal mol-' 30
available energy. Fig. 15 shows the photofragment TOF spectrum taken at
m/z+ = 35, Cl', with 200000 laser shots and a 200°C nozzle. All of the signal except for a shoulder on the slow edge was attributed to primary C-Cl bond fission:
ClCH,COCH, + hv(308 nm) -+ C1+ CH,COCH, (7)
A forward convolution fit of the portion of the C1+ spectrum attributed to primary C-Cl bond fission gave a translational energy distribution (Fig. 16) peaking near 5 kcal mol-' indi- cating that a significant portion of the available energy goes
t For both molecules we use the typical C-0 (169 kcal mol-') and C-C (80 kcal mol-') bond energies reported in ref. 3qa). For the C-Cl bond energy in both chloroacetone and acetyl chloride, we use the 83.5 kcal mol- ' bond energy for acetyl chloride reported in ref. 3qb).
l m O E 1 3 0.8 1
100 200 300 400 500 600 700 time of arrival/ps
Fig. 13 Laboratory time-of-flight spectrum of the photofragments detected at 35C1CH: from chloroacetone photodissociated at 308 nm with an unpolarized laser. The source angle was 15". All of the signal results from primary C-C bond fission and is fit with a P(E,) shown in Fig. 14.
g W
h c W
P
0.8
0.6
0.4
0.2
0
100 200 300 400 500 600 700 time of arrival/ps
Fig. 15 Laboratory time-of-flight spectrum of the photofragments detected at 35Cl+ from chloroacetone photodissociated at 308 nm with an unpolarized laser. The source angle was 15". The contribu- tion from ClCH, fragments to the C1' spectrum was determined by fitting the signal with the P(E,) in Fig. 14 derived from forward con- volution fitting the ClCHi signal. The rest of the signal results from the C1 fragments produced in primary C-Cl bond fission and is fit with the P(E,) in Fig. 16.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
100
80
60
40
20
0
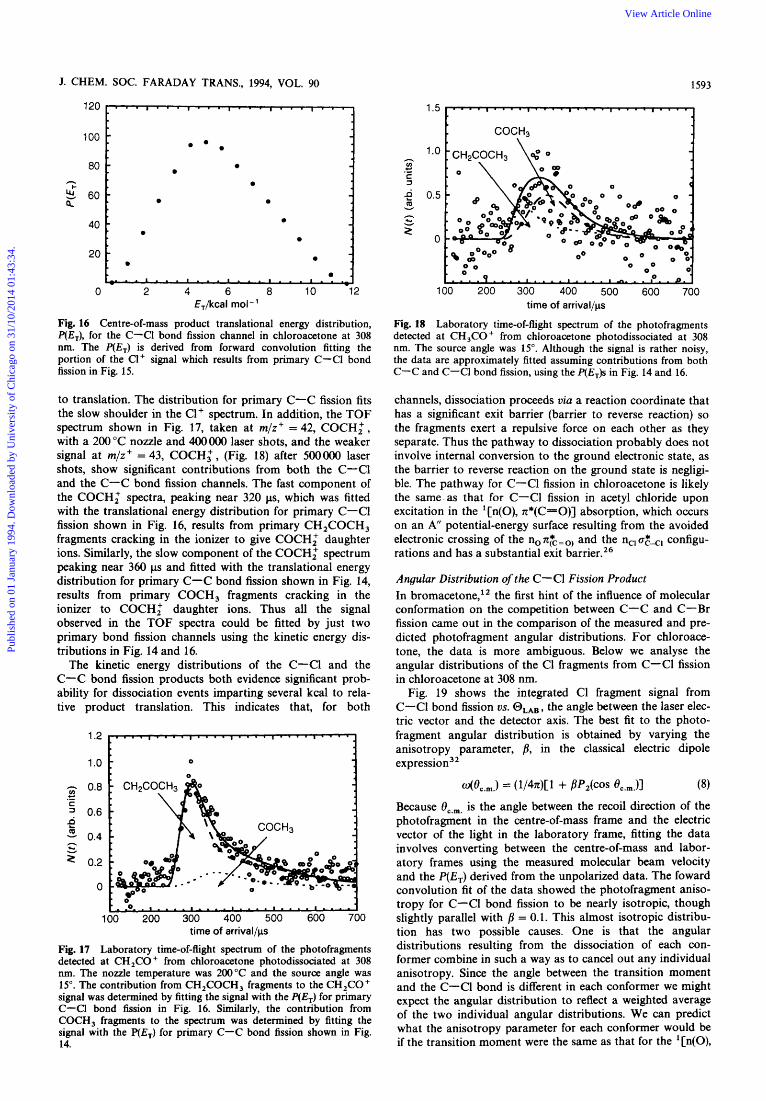
Fig. 16 Centre-of-mass product translational energy distribution, P(E,), for the C-Cl bond fission channel in chloroacetone at 308 nm. The P(E,) is derived from forward convolution fitting the portion of the C1+ signal which results from primary C-Cl bond fission in Fig. 15.
to translation. The distribution for primary C-C fission fits the slow shoulder in the C1+ spectrum. In addition, the TOF spectrum shown in Fig. 17, taken at m / z f = 42, COCH;, with a 200°C nozzle and 4OOOOO laser shots, and the weaker signal at m/z+ = 43, COCH;, (Fig. 18) after 500000 laser shots, show significant contributions from both the C-Cl and the C-C bond fission channels. The fast component of the COCH; spectra, peaking near 320 ps, which was fitted with the translational energy distribution for primary C-Cl fission shown in Fig. 16, results from primary CH,COCH, fragments cracking in the ionizer to give COCH; daughter ions. Similarly, the slow component of the COCH; spectrum peaking near 360 ps and fitted with the translational energy distribution for primary C-C bond fission shown in Fig. 14, results from primary COCH, fragments cracking in the ionizer to COCH; daughter ions. Thus all the signal observed in the TOF spectra could be fitted by just two primary bond fission channels using the kinetic energy dis- tributions in Fig. 14 and 16.
The kinetic energy distributions of the C-Cl and the C-C bond fission products both evidence significant prob- ability for dissociation events imparting several kcal to rela- tive product translation. This indicates that, for both
1.0 1
100 200 300 400 500 600 700 time of arrival/ps
Fig. 17 Laboratory time-of-flight spectrum of the photofragments detected at CH,CO + from chloroacetone photodissociated at 308 nm. The nozzle temperature was 200°C and the source angle was 15". The contribution from CH,COCH, fragments to the CH,CO+ signal was determined by fitting the signal with the P(E,) for primary C-CI bond fission in Fig. 16. Similarly, the contribution from COCH, fragments to the spectrum was determined by fitting the signal with the P(E,) for primary C-C bond fission shown in Fig. 14.
1.5
1 .o h v)
5 c .-
$ 0.5 a W
h
W c
z o
1593
100 200 300 400 500 600 700 time of arrival/ps
Fig. 18 Laboratory time-of-flight spectrum of the photofragments detected at CH3CO+ from chloroacetone photodissociated at 308 nm. The source angle was 15". Although the signal is rather noisy, the data are approximately fitted assuming contributions from both C-C and C-Cl bond fission, using the P(E,)s in Fig. 14 and 16.
channels, dissociation proceeds via a reaction coordinate that has a significant exit barrier (barrier to reverse reaction) so the fragments exert a repulsive force on each other as they separate. Thus the pathway to dissociation probably does not involve internal conversion to the ground electronic state, as the barrier to reverse reaction on the ground state is negligi- ble. The pathway for C-Cl fission in chloroacetone is likely the same.as that for C-Cl fission in acetyl chloride upon excitation in the [n(O), n*(C-O)] absorption, which occurs on an A potential-energy surface resulting from the avoided electronic crossing of the no n;=o, and the n,, o&-, configu- rations and has a substantial exit barrier.26
Angular Distribution of the C-C1 Fission Product In bromacetone,'* the first hint of the influence of molecular conformation on the competition between C-C and C-Br fission came out in the comparison of the measured and pre- dicted photofragment angular distributions. For chloroace- tone, the data is more ambiguous. Below we analyse the angular distributions of the C1 fragments from C-C1 fission in chloroacetone at 308 nm.
Fig. 19 shows the integrated C1 fragment signal from C-Cl bond fission us. OLAB, the angle between the laser elec- tric vector and the detector axis. The best fit to the photo- fragment angular distribution is obtained by varying the anisotropy parameter, B, in the classical electric dipole expressionJ2
Because Oc.m. is the angle between the recoil direction of the photofragment in the centre-of-mass frame and the electric vector of the light in the laboratory frame, fitting the data involves converting between the centre-of-mass and labor- atory frames using the measured molecular beam velocity and the P(E,) derived from the unpolarized data. The foward convolution fit of the data showed the photofragment aniso- tropy for C-Cl bond fission to be nearly isotropic, though slightly parallel with /3 = 0.1. This almost isotropic distribu- tion has two possible causes. One is that the angular distributions resulting from the dissociation of each con- former combine in such a way as to cancel out any individual anisotropy. Since the angle between the transition moment and the C-Cl bond is different in each conformer we might expect the angular distribution to reflect a weighted average of the two individual angular distributions. We can predict what the anisotropy parameter for each conformer would be if the transition moment were the same as that for the '[n(O),
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
1594 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
C
5: 0 0 0 In N
Q f cn
3
w
00 W
850
800
750
700
650
40 80 120 160 200 600; ' ' ' ' a ' ' ' ' ' ' '
laboratory polarization angle/degrees
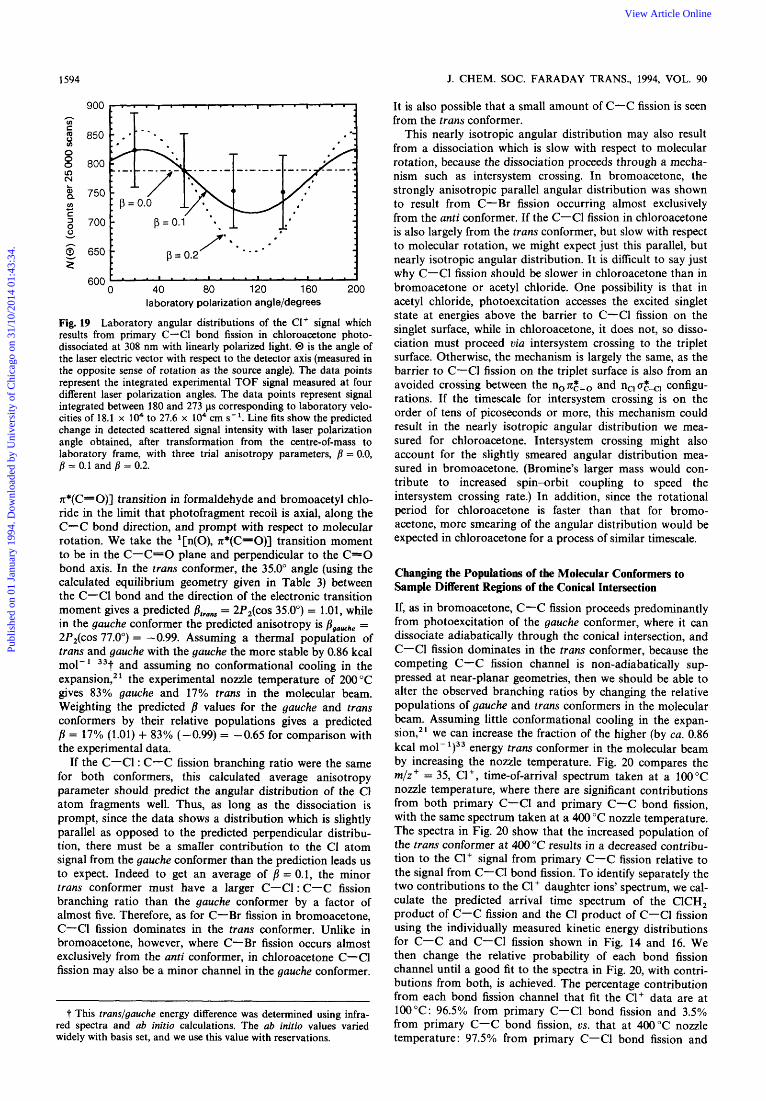
Fig. 19 Laboratory angular distributions of the C1+ signal which results from primary C-C1 bond fission in chloroacetone photo- dissociated at 308 nm with linearly polarized light. @ is the angle of the laser electric vector with respect to the detector axis (measured in the opposite sense of rotation as the source angle). The data points represent the integrated experimental TOF signal measured at four different laser polarization angles. The data points represent signal integrated between 180 and 273 ps corresponding to laboratory velo- cities of 18.1 x lo4 to 27.6 x lo4 cm s-'. Line fits show the predicted change in detected scattered signal intensity with laser polarization angle obtained, after transformation from the centre-of-mass to laboratory frame, with three trial anisotropy parameters, /I = 0.0, fi = 0.1 and fi = 0.2.
n*(C=O)] transition in formaldehyde and bromoacetyl chlo- ride in the limit that photofragment recoil is axial, along the C-C bond direction, and prompt with respect to molecular rotation. We take the '[n(O), n*(C50)J transition moment to be in the C-C=O plane and perpendicular to the C=O bond axis. In the trans conformer, the 35.0" angle (using the calculated equilibrium geometry given in Table 3) between the C-Cl bond and the direction of the electronic transition moment gives a predicted fit,,, = 2P2(c0s 35.0") = 1.01, while in the gauche conformer the predicted anisotropy is flgouehhe = 2P2(cos 77.0") = -0.99. Assuming a thermal population of trans and gauche with the gauche the more stable by 0.86 kcal mol-' 33f and assuming no conformational cooling in the expansion,2' the experimental nozzle temperature of 200 "C gives 83% gauche and 17% trans in the molecular beam. Weighting the predicted fi values for the gauche and trans conformers by their relative populations gives a predicted /? = 17% (1.01) + 83% (-0.99) = -0.65 for comparison with the experimental data.
If the C-Cl : C-C fission branching ratio were the same for both conformers, this calculated average anisotropy parameter should predict the angular distribution of the C1 atom fragments well. Thus, as long as the dissociation is prompt, since the data shows a distribution which is slightly parallel as opposed to the predicted perpendicular distribu- tion, there must be a smaller contribution to the Cl atom signal from the gauche conformer than the prediction leads us to expect. Indeed to get an average of f i = 0.1, the minor trans conformer must have a larger C-Cl : C-C fission branching ratio than the gauche conformer by a factor of almost five. Therefore, as for C-Br fission in bromoacetone, C-Cl fission dominates in the trans conformer. Unlike in bromoacetone, however, where C-Br fission occurs almost exclusively from the anti conformer, in chloroacetone C-Cl fission may also be a minor channel in the gauche conformer.
~- ~~
t This translgauche energy difference was determined using infra- red spectra and ab initio calculations. The ab initio values varied widely with basis set, and we use this value with reservations.
It is also possible that a small amount of C-C fission is seen from the trans conformer.
This nearly isotropic angular distribution may also result from a dissociation which is slow with respect to molecular rotation, because the dissociation proceeds through a mecha- nism such as intersystem crossing. In bromoacetone, the strongly anisotropic parallel angular distribution was shown to result from C-Br fission occurring almost exclusively from the anti conformer. If the C-CI fission in chloroacetone is also largely from the trans conformer, but slow with respect to molecular rotation, we might expect just this parallel, but nearly isotropic angular distribution. It is difficult to say just why C-Cl fission should be slower in chloroacetone than in bromoacetone or acetyl chloride. One possibility is that in acetyl chloride, photoexcitation accesses the excited singlet state at energies above the barrier to C-Cl fission on the singlet surface, while in chloroacetone, it does not, so disso- ciation must proceed via intersystem crossing to the triplet surface. Otherwise, the mechanism is largely the same, as the barrier to C-Cl fission on the triplet surface is also from an avoided crossing between the no n&o and n,, CJ&, configu- rations. If the timescale for intersystem crossing is on the order of tens of picoseconds or more, this mechanism could result in the nearly isotropic angular distribution we mea- sured for chloroacetone. Intersystem crossing might also account for the slightly smeared angular distribution mea- sured in bromoacetone. (Bromine's larger mass would con- tribute to increased spin-orbit coupling to speed the intersystem crossing rate.) In addition, since the rotational period for chloroacetone is faster than that for bromo- acetone, more smearing of the angular distribution would be expected in chloroacetone for a process of similar timescale.
Changing the Populations of the Molecular Conformers to Sample Different Regions of the Conical Intersection
If, as in bromoacetone, C-C fission proceeds predominantly from photoexcitation of the gauche conformer, where it can dissociate adiabatically through the conical intersection, and C-C1 fission dominates in the trans conformer, because the competing C-C fission channel is non-adiabatically sup- pressed at near-planar geometries, then we should be able to alter the observed branching ratios by changing the relative populations of gauche and trans conformers in the molecular beam. Assuming little conformational cooling in the expan- sioq2' we can increase the fraction of the higher (by ca. 0.86 kcal mol- energy trans conformer in the molecular beam by increasing the nozzle temperature. Fig. 20 compares the m/z+ = 35, C1+, time-of-arrival spectrum taken at a 100°C nozzle temperature, where there are significant contributions from both primary C-Cl and primary C-C bond fission, with the same spectrum taken at a 400°C nozzle temperature. The spectra in Fig. 20 show that the increased population of the trans conformer at 400°C results in a decreased contribu- tion to the Cl+ signal from primary C-C fission relative to the signal from C-Cl bond fission. To identify separately the two contributions to the C1+ daughter ions' spectrum, we cal- culate the predicted arrival time spectrum of the ClCH, product of C-C fission and the C1 product of C-Cl fission using the individually measured kinetic energy distributions for C-C and C-Cl fission shown in Fig. 14 and 16. We then change the relative probability of each bond fission channel until a good fit to the spectra in Fig. 20, with contri- butions from both, is achieved. The percentage contribution from each bond fission channel that fit the C1+ data are at 100°C: 96.5% from primary C-Cl bond fission and 3.5% from primary C-C bond fission, us. that at 400°C nozzle temperature: 97.5% from primary C-Cl bond fission and
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
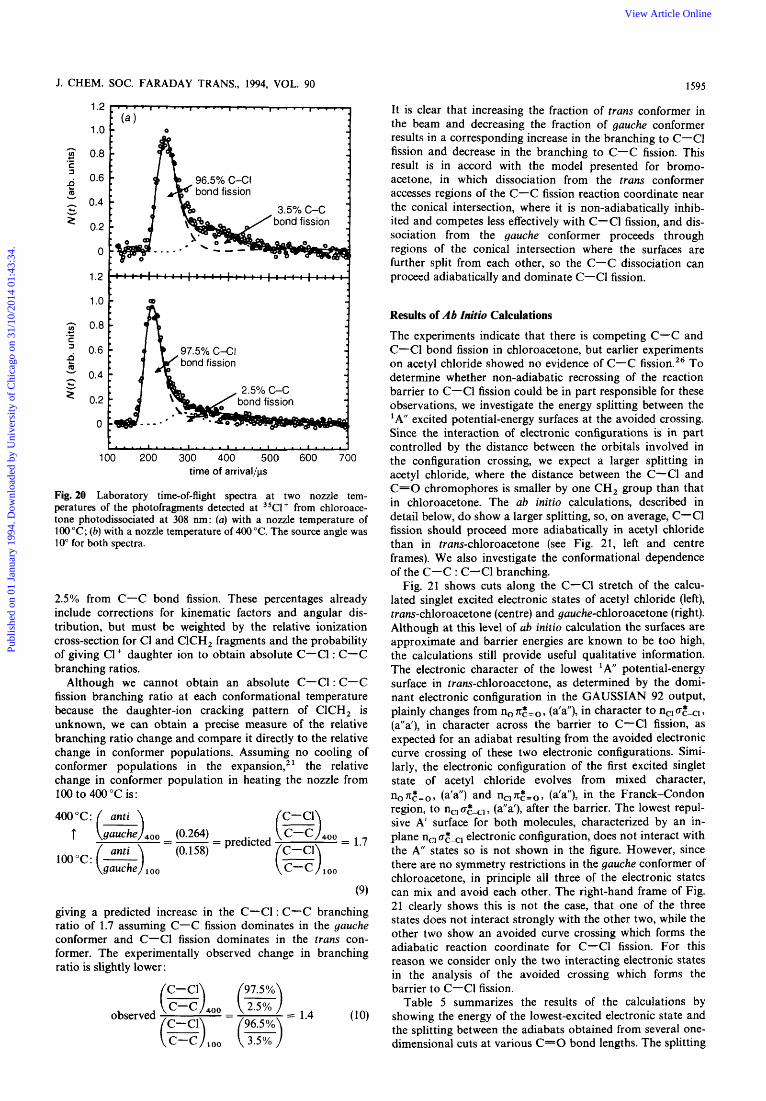
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90 1595
1.2 . . . . I . . . . ( . . . . ( . . . . , . . . . ( . . . .
1.0 - 0
: ( a ) b
5 0.8 - 0.6 - 0.4 -
.- C
4 Y
c W
0.2 1 0 -
h
CI v)
C .-
$ W
h c W
z
1.2
1 .o
0.8
0.6
0.4
0.2
0 c 4
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . i 100 200 300 400 500 600 700
time of arrival/ps
Fig. 20 Laboratory time-of-flight spectra at two nozzle tem- peratures of the photofragments detected at 35Cl+ from chloroace- tone photodissociated at 308 nm: (a) with a nozzle temperature of 100 “C; (b) with a nozzle temperature of 400 “C. The source angle was lo” for both spectra.
2.5% from C-C bond fission. These percentages already include corrections for kinematic factors and angular dis- tribution, but must be weighted by the relative ionization cross-section for Cl and ClCH, fragments and the probability of giving C1+ daughter ion to obtain absolute C-Cl : C-C branching ratios.
Although we cannot obtain an absolute C-Cl : C-C fission branching ratio at each conformational temperature because the daughter-ion cracking pattern of ClCH, is unknown, we can obtain a precise measure of the relative branching ratio change and compare it directly to the relative change in conformer populations. Assuming no cooling of conformer populations in the expansion,” the relative change in conformer population in heating the nozzle from 100 to 400°C is:
4 0 0 ” ~ : f anti \ fC-Cl\ t
100°C:
(0.264) (0.158)
predicted = 1.7
(9)
giving a predicted increase in the C-Cl : C-C branching ratio of 1.7 assuming C-C fission dominates in the gauche conformer and C-Cl fission dominates in the trans con- former. The experimentally observed change in branching ratio is slightly lower :
c-Cl 97.5%
(10) 400 = - observed - 1.4 c- c1 96.5% (10)
(C-CJ400 - (2.5% ) observed = la4
/c-Cl\ - - (c-c)loo (3.5%)
It is clear that increasing the fraction of trans conformer in the beam and decreasing the fraction of gauche conformer results in a corresponding increase in the branching to C-Cl fission and decrease in the branching to C-C fission. This result is in accord with the model presented for bromo- acetone, in which dissociation from the trans conformer accesses regions of the C-C fission reaction coordinate near the conical intersection, where it is non-adiabatically inhib- ited and competes less effectively with C-Cl fission, and dis- sociation from the gauche conformer proceeds through regions of the conical intersection where the surfaces are further split from each other, so the C-C dissociation can proceed adiabatically and dominate C-C1 fission.
Results of Ab Znitio Calculations
The experiments indicate that there is competing C-C and C-Cl bond fission in chloroacetone, but earlier experiments on acetyl chloride showed no evidence of C-C fission.26 To determine whether non-adiabatic recrossing of the reaction barrier to C-C1 fission could be in part responsible for these observations, we investigate the energy splitting between the ‘A” excited potential-energy surfaces at the avoided crossing. Since the interaction of electronic configurations is in part controlled by the distance between the orbitals involved in the configuration crossing, we expect a larger splitting in acetyl chloride, where the distance between the C-Cl and CEO chromophores is smaller by one CH, group than that in chloroacetone. The ab initio calculations, described in detail below, do show a larger splitting, so, on average, C-Cl fission should proceed more adiabatically in acetyl chloride than in trans-chloroacetone (see Fig. 21, left and centre frames). We also investigate the conformational dependence of the C-C : C-Cl branching.
Fig. 21 shows cuts along the C-C1 stretch of the calcu- lated singlet excited electronic states of acetyl chloride (left), trans-chloroacetone (centre) and gauche-chloroacetone (right). Although at this level of ab initio calculation the surfaces are approximate and barrier energies are known to be too high, the calculations still provide useful qualitative information. The electronic character of the lowest ‘A” potential-energy surface in trans-chloroacetone, as determined by the domi- nant electronic configuration in the GAUSSIAN 92 output, plainly changes from no ~ n , * = ~ , (a”’’), in character to n,, o&,, (a“a’), in character across the barrier to C-Cl fission, as expected for an adiabat resulting from the avoided electronic curve crossing of these two electronic configurations. Simi- larly, the electronic configuration of the first excited singlet state of acetyl chloride evolves from mixed character, no @ = o , (a’a”) and n,, X& o , (a”’’), in the Franck-Condon region, to ncla&-l, (a”a’), after the barrier. The lowest repul- sive A’ surface for both molecules, characterized by an in- plane na electronic configuration, does not interact with the A“ states so is not shown in the figure. However, since there are no symmetry restrictions in the gauche conformer of chloroacetone, in principle all three of the electronic states can mix and avoid each other. The right-hand frame of Fig. 21 clearly shows this is not the case, that one of the three states does not interact strongly with the other two, while the other two show an avoided curve crossing which forms the adiabatic reaction coordinate for C-Cl fission. For this reason we consider only the two interacting electronic states in the analysis of the avoided crossing which forms the barrier to C-Cl fission.
Table 5 summarizes the results of the calculations by showing the energy of the lowest-excited electronic state and the splitting between the adiabats obtained from several one- dimensional cuts at various C-0 bond lengths. The splitting
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
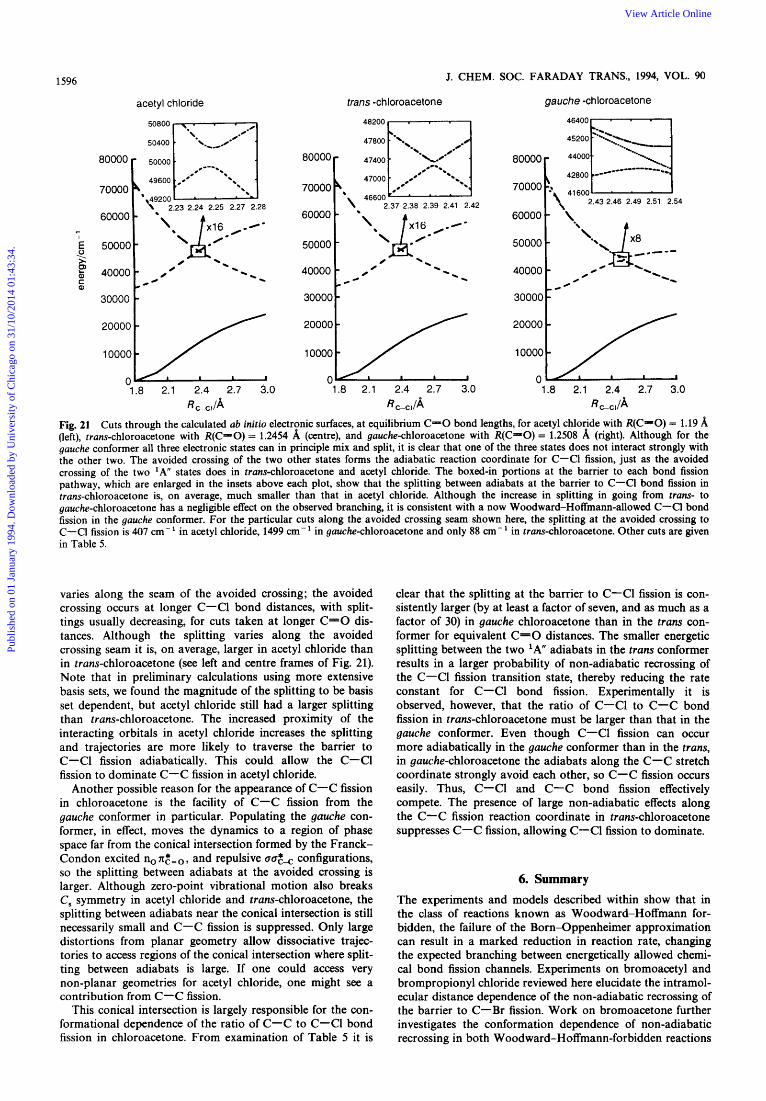
1596
80000
70000
60000
50000
40000
30000
c I
E Y > ; Q,
4
- -
- '* \ JP'- %
0 - \ 0
% - 0 - %
- *#I
acetyl chloride
50000
49600
2.23 2.24 2.25 2.27 2.28
80000
70000
60000
50000
40000
J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
trans -chloroacetone gauche -chloroacetone
48200 1-1
47400
46600 2.37 2.38 2.39 2.41 2.42
\
0
4 - % 0
80000
70000 2.43 2.46 2.49 2.51 2.54
30000 1 30000 1 20000y, ;ooool/,
10000
0 1.8 2.1 2.4 2.7 3.0 1.8 2.1 2.4 2.7 3.0
R,c,lA R,c,lA Fig. 21 Cuts through the calculated ab initio electronic surfaces, at equilibrium C-0 bond lengths, for acetyl chloride with R(C-0) = 1.19 A (left), trans-chloroacetone with R(C-0) = 1.2454 8, (centre), and gauche-chloroacetone with R(C-0) = 1.2508 A (right). Although for the gauche conformer all three electronic states can in principle mix and split, it is clear that one of the three states does not interact strongly with the other two. The avoided crossing of the two other states forms the adiabatic reaction coordinate for C-C1 fission, just as the avoided crossing of the two ' A states does in trans-chloroacetone and acetyl chloride. The boxed-in portions at the barrier to each bond fission pathway, which are enlarged in the insets above each plot, show that the splitting between adiabats at the barrier to C-Cl bond fission in trans-chloroacetone is, on average, much smaller than that in acetyl chloride. Although the increase in splitting in going from trans- to gauche-chloroacetone has a negligible effect on the observed branching, it is consistent with a now Woodward-Hoffmann-allowed C-C1 bond fission in the gauche conformer. For the particular cuts along the avoided crossing seam shown here, the splitting at the avoided crossing to C-Cl fission is 407 cm-' in acetyl chloride, 1499 m-' in gauche-chloroacetone and only 88 cm-' in trans-chloroacetone. Other cuts are given in Table 5.
varies along the seam of the avoided crossing; the avoided crossing occurs at longer C-C1 bond distances, with split- tings usually decreasing, for cuts taken at longer C-0 dis- tances. Although the splitting varies along the avoided crossing seam it is, on average, larger in acetyl chloride than in trans-chloroacetone (see left and centre frames of Fig. 21). Note that in preliminary calculations using more extensive basis sets, we found the magnitude of the splitting to be basis set dependent, but acetyl chloride still had a larger splitting than trans-chloroacetone. The increased proximity of the interacting orbitals in acetyl chloride increases the splitting and trajectories are more likely to traverse the barrier to C-Cl fission adiabatically. This could allow the C-Cl fission to dominate C-C fission in acetyl chloride.
Another possible reason for the appearance of C-C fission in chloroacetone is the facility of C-C fission from the gauche conformer in particular. Populating the gauche con- former, in effect, moves the dynamics to a region of phase space far from the conical intersection formed by the Franck- Condon excited no &, , and repulsive a o k configurations, so the splitting between adiabats at the avoided crossing is larger. Although zero-point vibrational motion also breaks C, symmetry in acetyl chloride and trans-chloroacetone, the splitting between adiabats near the conical intersection is still necessarily small and C-C fission is suppressed. Only large distortions from planar geometry allow dissociative trajec- tories to access regions of the conical intersection where split- ting between adiabats is large. If one could access very non-planar geometries for acetyl chloride, one might see a contribution from C-C fission.
This conical intersection is largely responsible for the con- formational dependence of the ratio of C-C to C-Cl bond fission in chloroacetone. From examination of Table 5 it is
clear that the splitting at the barrier to C-Cl fission is con- sistently larger (by at least a factor of seven, and as much as a factor of 30) in gauche chloroacetone than in the trans con- former for equivalent C-0 distances. The smaller energetic splitting between the two ' A adiabats in the trans conformer results in a larger probability of non-adiabatic recrossing of the C-C1 fission transition state, thereby reducing the rate constant for C-Cl bond fission. Experimentally it is observed, however, that the ratio of C-C1 to C-C bond fission in trans-chloroacetone must be larger than that in the gauche conformer. Even though C-C1 fission can occur more adiabatically in the gauche conformer than in the trans, in gauche-chloroacetone the adiabats along the C-C stretch coordinate strongly avoid each other, so C-C fission occurs easily. Thus, C-Cl and C-C bond fission effectively compete. The presence of large non-adiabatic effects along the C-C fission reaction coordinate in trans-chloroacetone suppresses C-C fission, allowing C-Cl fission to dominate.
6. Summary The experiments and models described within show that in the class of reactions known as Woodward-Hoffmann for- bidden, the failure of the Born-Oppenheimer approximation can result in a marked reduction in reaction rate, changing the expected branching between energetically allowed chemi- cal bond fission channels. Experiments on bromoacetyl and brompropionyl chloride reviewed here elucidate the intramol- ecular distance dependence of the non-adiabatic recrossing of the barrier to C-Br fission. Work on bromoacetone further investigates the conformation dependence of non-adiabatic recrossing in both Woodward-Hoffmann-forbidden reactions
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online
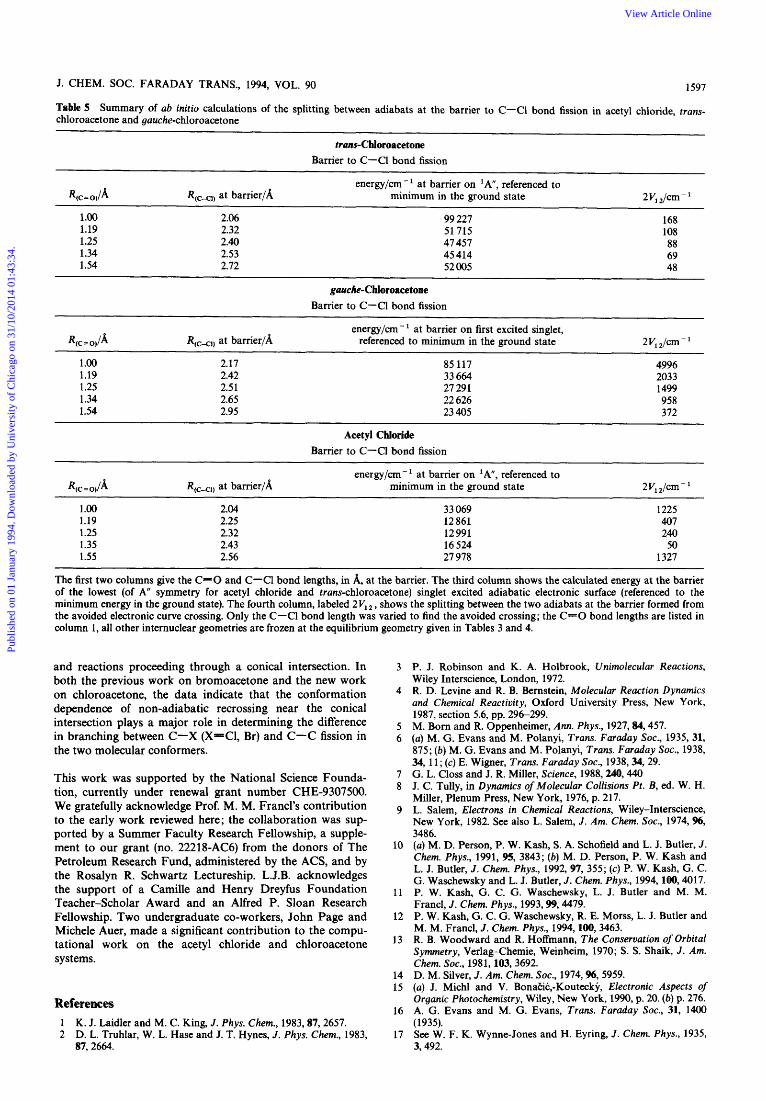
1597 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
Table 5 Summary of ab initio calculations of the splitting between adiabats at the barrier to C-C1 bond fission in acetyl chloride, trans- chloroacetone and gauche-chloroacetone
trans-Chloroacetone Barrier to C-C1 bond fission
energy/cm-' at barrier on 'A", referenced to Ro,, at barrier/A minimum in the ground state 2Vl2/cm-1
1 .oo 1.19 1.25 1.34 1.54
2.06 2.32 2.40 2.53 2.72
99 227 51 715 47 457 45 414 52 005
~~
168 108 88 69 48
gauche-Chloroacetone Barrier to C-C1 bond fission
energy/cm-' at barrier on first excited singlet, at barrier/A referenced to minimum in the ground state 2 V' - 1
1 .oo 1.19 1.25 1.34 1.54
2.17 2.42 2.51 2.65 2.95
85 117 33 664 27 291 22 626 23 405
4996 2033 1499 958 372
Acetyl Chloride Barrier to C-Cl bond fission
energy/cn-' at barrier on ' A , referenced to minimum in the ground state R ( c - l , at barrier/A
1.00 1.19 1.25 1.35 1.55
2.04 2.25 2.32 2.43 2.56
33 069 12 861 12991 16 524 27 978
1225 407 240 50
1327
The first two columns give the CEO and C-C1 bond lengths, in A, at the barrier. The third column shows the calculated energy at the barrier of the lowest (of A" symmetry for acetyl chloride and trans-chloroacetone) singlet excited adiabatic electronic surface (referenced to the minimum energy in the ground state). The fourth column, labeled 2VI2, shows the splitting between the two adiabats at the barrier formed from the avoided electronic curve crossing. Only the C-C1 bond length was varied to find the avoided crossing; the C-0 bond lengths are listed in column 1, all other internuclear geometries are frozen at the equilibrium geometry given in Tables 3 and 4.
and reactions proceeding through a conical intersection. In both the previous work on bromoacetone and the new work on chloroacetone, the data indicate that the conformation dependence of non-adiabatic recrossing near the conical intersection plays a major role in determining the difference in branching between C-X (X-C1, Br) and C-C fission in the two molecular conformers.
This work was supported by the National Science Founda- tion, currently under renewal grant number CHE-9307500. We gratefully acknowledge Prof. M. M. Francl's contribution to the early work reviewed here; the collaboration was sup- ported by a Summer Faculty Research Fellowship, a supple- ment to our grant (no. 22218-AC6) from the donors of The Petroleum Research Fund, administered by the ACS, and by the Rosalyn R. Schwartz Lectureship. L.J.B. acknowledges the support of a Camille and Henry Dreyfus Foundation Teacher-Scholar Award and an Alfred P. Sloan Research Fellowship. Two undergraduate co-workers, John Page and Michele Auer, made a significant contribution to the compu- tational work on the acetyl chloride and chloroacetone sys tems.
References 1 K. J. Laidler and M. C. King, J. Phys. Chem., 1983,87, 2657. 2 D. L. Truhlar, W. L. Hase and J. T. Hynes, J. Phys. Chem., 1983,
87,2664.
3
4
5 6
7 8
9
10
11
12
13
14 15
16
17
P. J. Robinson and K. A. Holbrook, Unimolecular Reactions, Wiley Interscience, London, 1972. R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics and Chemical Reactivity, Oxford University Press, New York, 1987, section 5.6, pp. 296-299. M. Born and R. Oppenheimer, Ann. Phys., 1927,84,457. (a) M. G. Evans and M. Polanyi, Trans. Faraday SOC., 1935, 31, 875; (b) M. G. Evans and M. Polanyi, Trans. Faraday SOC., 1938, 34 , l l ; (c) E. Wigner, Trans. Faraday SOC., 1938,34,29. G. L. Closs and J. R. Miller, Science, 1988,240,440 J. C. Tully, in Dynamics of Molecular Collisions Pt. B, ed. W. H. Miller, Plenum Press, New York, 1976, p. 217. L. Salem, Electrons in Chemical Reactions, Wiley-Interscience, New York, 1982. See also L. Salem, J. Am. Chem. SOC., 1974,%, 3486. (a) M. D. Person, P. W. Kash, S. A. Schofield and L. J. Butler, J . Chem. Phys., 1991, 95, 3843; (b) M. D. Person, P. W. Kash and L. J. Butler, J. Chem. Ph.ys., 1992, 97, 355; (c) P. W. Kash, G. C. G. Waschewsky and L. J. Butler, J. Chem. Phys., 1994,100,4017. P. W. Kash, G. C. G. Waschewsky, L. J. Butler and M. M. Francl, J . Chem. Phys., 1993,W, 4479. P. W. Kash, G. C. G. Waschewsky, R. E. Morss, L. J. Butler and M. M. Francl, J. Chem. Phys., 1994,100,3463. R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Verlag-Chemie, Weinheim, 1970; S. S. Shaik, J. Am. Chem. SOC., 1981,103,3692. D. M. Silver, J . Am. Chem. SOC., 1974,%, 5959. (a) J. Michl and V. Bona&iC,-Koutecky, Electronic Aspects of Organic Photochemistry, Wiley, New York, 1990, p. 20. (b) p. 276. A. G. Evans and M. G. Evans, Trans. Faraday SOC., 31, 1400 (1935). See W. F. K. Wynne-Jones and H. Eyring, J. Chem. Phys., 1935, 3,492.
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online

1598 J. CHEM. SOC. FARADAY TRANS., 1994, VOL. 90
18 19
20
21
22
23
24
25 26
27
J. K. Burdett, personal communication. See D. M. Cyr, G. A. Bishea, M. G. Scarton and M. A. Johnson, J. Chem. Phys., 1992, 97, 5911; S. S. Shaik, J. Am. Chem. SOC., 1982,104,2708. See, e q . S. L. Mielke, G. J. Tawa, D. G. Truhlar and D. W. Schwenke, J. Am. Chem. SOC., in the press. R. S. Ruoff, T. D. Klots, T. Emilsson and H. S. Gutowsky, J. Chem. Phys., 1990,93,3142. (a) G. E. Busch and K. R. Wilson, J. Chem. Phys., 1972, 56, 3638; (b) S. C. Yang and R. Bersohn, J. Chem. Phys., 1974, 61, 4400; (c) J. R. Durig, J. Lin and H. V. Phan, J . Ramun Spectrosc., 1992,23, 253. T . H. Lowry and K . S. Richardson, Mechanism and Theory in Organic Chemistry, Harper and Row, New York, 1987, p. 1041. See M. Reinsch and M. Klessinger, J. Phys. Org. Chem., 1990,3, 81, and references therein. N. C. Yang and E. D. Feit, J. Am. Chem. SOC., 1968,90,504. See M. D. Person, P. W. Kash and L. J. Butler, J. Phys. Chem., 1992,%, 2021 and references therein. Y. T. Lee, J. D. McDonald, P. R. LeBreton and D. R. Hersch- bach, Rev. Sci. Instrum., 1969,40, 1402.
28
29
30
31
32 33
M. D. Person, Ph.D. Thesis, Department of Chemistry, Uni- versity of Chicago, 1991. GAUSSIAN 92, Revision C, M. J. Frisch, G. W. Trucks, M. Head-Gordon, P. M. W. Gill, M. W. Wong, J. B. Foresman, B. G. Johnson, H. B. Schlegel, M. A. Robb, E. S. Replogle, R. Gomperts, J. L. Andres, K. Raghavachari, J. S. Binkley, C. Gon- zalez, R. L. Martin, D. J. Fox, D. J. Defrees, J. Baker, J. J. P. Stewart and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1992. (a) A. J. Gordon and R. A. Ford, The Chemist’s Companion, Wiley, New York, 1972; (b) J. A. Devore and H. E. ONeal, J. Phys. Chem., 1969,73,2644. K. Yates, S. L. Klemenko and I. G. Csizmadia, Spectrochim. Acta, Part A, 1969,25, 765. R. N. Zare, Mol. Photochem., 1972,4,1. J. R. Durig, J. Lin, C. L. Tolley and T. S. Little, Spectrochim. Acta, Part A, 1991,47, 105.
Paper 3/05223K; Received 27th August, 1993
Publ
ishe
d on
01
Janu
ary
1994
. Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
31/1
0/20
14 0
1:43
:34.
View Article Online



![Asymmetric [2,3]-Sigmatropic Rearrangement of Allylic ...11708/FULLTEXT01.pdfreaction, in accordance with the Woodward-Hoffmann rules.4,5 For instance, the ... rearrangement is believed](https://img.pdfslide.us/doc/110x75/5ab666257f8b9a86428d989a/asymmetric-23-sigmatropic-rearrangement-of-allylic-11708fulltext01pdfreaction.jpg)



![Lecture 8 Pericyclic ppt - staff.du.edu.egstaff.du.edu.eg/upfilestaff/447/courses/8447_1461417216__Lecture.8...(1) The Woodward-Hoffmann rules for [1,n] sigmatropic rearrangements](https://img.pdfslide.us/doc/110x75/5ab666257f8b9a86428d98ef/lecture-8-pericyclic-ppt-staffduedu-1-the-woodward-hoffmann-rules-for-1n.jpg)





















