Embed Size (px)
Citation preview

VOLUMETRIC ANALYSIS OF METALS IN GLACIAL ACETIC ACID
by
ALLAN TERENCE CASEY
A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF
MASTER OF SCIENCE i n the Department
of Chemistry
We accept t h i s thesis as conforming to the standard required from candidates for the degree of MASTER OF SCIENCE.
Members of the Department of Chemistry.
THE UNIVERSITY OF BRITISH COLUMBIA A p r i l , 1953

ABSTRACT
Recent work has demonstrated that certain metal s a l t s of
inorganic acids may e a s i l y be t i t r a t e d with perchloric acid
i n g l a c i a l acetic acid. To test the accuracy of the method,
a series of t i t r a t i o n s was run on the acetates of twenty-three
representative metals. The r e s u l t i n g curves show that accurate
reproducible end points may be obtained f o r the acetates of the
more ele c t r o p o s i t i v e metals, while those of aluminum, chromium
and iron y i e l d anomalous r e s u l t s . An explanation f o r t h i s i s
offered. The acetates of t i n , bismuth, antimony, uranium and
copper are too weak to be determined by t h i s method.
Metals, oxides, or other more d i f f i c u l t l y soluble com
pounds may be conveniently introduced into acetic acid solution
by f i r s t d i s s o l v i n g i n n i t r i c acid (or i n certain cases aqua
regi a ) , followed by the addition of s u f f i c i e n t acetic anhydride
to react with a l l the water and decompose a l l the n i t r a t e
present. Quantitative a n a l y t i c a l separations may be effected
using such common reagents as oxa l i c , n i t r i c , and t a r t a r i c
acid, hydrogen iodide and hydrogen sulphide. Hhey were used
i n analysing eleven metal mixtures, with accuracy comparable
to the more lengthy gravimetric process often required i n
aqueous solution.

ACKNOWLEDGEMENTS
I wish to express my sincere apprecia
t i o n to my research d i r e c t o r , Dr. K. Starke,
f o r his invaluable aid and encouragement
during the course of t h i s i n v e s t i g a t i o n .
I also wish to thank the International
Nickel Company fo r f i n a n c i a l support of t h i s
work, and Major Aluminum Products of Vancouver
f o r a sample of aluminum-magnesium a l l o y ;

TABLE OF CONTENTS
Page
I INTRODUCTION 1
II EXPERIMENTAL A. Materials and Apparatus
Solvents 12 Preparation of 'i'itrant 13 Indicator. 14 Potentiometer........ 14 Standardization of the Ti t r a n t 15
B. T i t r a t i o n of Metal Salts Sodium Acetate 16 Acetates of Potassium, Lithium, 16
Barium, Strontium, and Ammonium Magnesium Acetate 17 Acetates of Calcium, Manganese, and
Cadmium 17 Acetates of Cobalt and Nickel 17 zinc Acetate IS Acetates of Lead and S i l v e r IB Acetates of Iron, Chromium, and
Aluminum IB Acetates of Tin, Antimony, Copper,
Bismuth, and Uranium 1<3 T i t r a t i o n of Chloroacetates 19 T i t r a t i o n of Chlorides 19
C. Separation and Analysis of Metal Acetate Mixtures
S o l u b i l i t i e s of Metal Salts i n Acetic Acid 20
Separations Effected Zinc and Magnesium... 22 Zinc and Copper 23 Barium and Calcium 23 S i l v e r and Aluminum *. 23 Lead and Tin 24 Magnesium and Antimony; Magnesium and Lead, Magnesium and Copper 24 Nickel and Chromium 25 Cadmium and Magnesium 25 Tin and Magnesium. 25
D. Introduction of Metals Into Acetic Acid Solution 26

TABLE OF CONTENTS (continued) Page I I I RESULTS AND DISCUSSION
A. Determination of Potentiometric End Point 23
B. T i t r a t i o n of Metal Salts '30 C. S o l u b i l i t i e s i n Acetic Acid..... 33 D. Separations 36 E. Introduction of Metals into
Acetic Acid Solution. 3&
IV CONCLUSIONS 42
V BIBLIOGRAPHY 44

i v
TABLE OF vCONTENTS FOR TABLES
Page
Table I T i t r a t i o n of Metal Salts. 3 1
Table II P r e c i p i t a t i o n Reactions i n Acetic Acid... 3 4
Table I I I Results of S o l u b i l i t y Tests
With Sulphates 3 7
Table IV Results of T i t r a t i n g M e t a l l i c Mixtures... 3 9
Table V Reactivity of Amine Acetates, Acetic Acid, and the Chloroacetic Acids Toward Metals 4 0
Table VI Acids Tested Which Attack the Metals Considered With S u f f i c i e n t Rapidity to be of P r a c t i c a l Use 4 1

TABLE OF CONTENTS FOR FIGURES
Figure I
Figure I I A
Figure I I B
Figure I I I A
Figure I I I B
Fi g u r e IV A
Figur e IV B
Figur e V A
Figur e V B
Figure V I A
Figure V I B
Figure V I I A
Figure V I I B
Figure V I I I A
Figure V I I I B
Page R e l a t i v e Basic Strengths of C e r t a i n Acid Ions, Solvents and Organic Bases.. 10
P o t e n t i o m e t r i c T i t r a t i o n of Sodium Acetate w i t h P e r c h l o r i c A c i d 4# P r e c i s e Determination of the End Point f o r Figure I I A 49
P o t e n t i o m e t r i c T i t r a t i o n of Barium Acetate w i t h P e r c h l o r i c Acid 50
P r e c i s e Determination of the End Point f o r Figure I I I A..... 51
P o t e n t i o m e t r i c T i t r a t i o n of L i t h i u m and Strontium Acetates with P e r c h l o r i c Acid 52
P r e c i s e Determination of the End Poin t f o r Figure IV A 53
P o t e n t i o m e t r i c T i t r a t i o n of Magnesium Acetate w i t h P e r c h l o r i c t A c i d 54
P r e c i s e Determination of the End Point f o r Figure V A 55
P o t e n t i o m e t r i c T i t r a t i o n of Calcium Acetate w i t h P e r c h l o r i c Acid 56
P r e c i s e Determination of the End Point f o r Figure V I A 57
P o t e n t i o m e t r i c T i t r a t i o n of Cadmium and Manganese Acetates with P e r c h l o r i c Acid 53
P r e c i s e Determination of the End Poin t f o r Figure V I I A 59
P o t e n t i o m e t r i c T i t r a t i o n of Cobalt Acetate w i t h P e r c h l o r i c A c i d 60
P r e c i s e Determination of the End Point f o r Figure V I I I A 61

v i
TABLE OF CONTENTS FOR FIGURES (Continuted) Page
Figure IX A Potentiometric T i t r a t i o n of Lead Acetate with Perchloric Acid.. 62
Figure IX B Precise Determination of the End Point f o r Figure IX A 63
Figure X A Potentiometric T i t r a t i o n of Aluminum, • Chromium, and Iron (III) Acetates with Perchloric Acid 6 4
Figure X B Precise Determination of the End Points f o r Figure X A 65
Figure XI Curves Obtained from the Attempted Potentiometric T i t r a t i o n of Copper (II) Antimony ( I I I ) , and Tin (II) Acetates-with Perchloric Acid 6 6
Figure XII Curves Obtained from the Attempted Potentiometric T i t r a t i o n -of Bismuth (III) and Uranyl Acetates with Perchloric Acid 67
Figure XIII A Potentiometric T i t r a t i o n of Barium Chloroacetate with Perchloric Acid.... 68
Figure XIII B Precise Determination of the End Point f o r Figure XIII A 6 9

1
I . INTRODUCTION
U n t i l r e c e n t l y , w a t e r has been the s o l v e n t employed f o r
most a n a l y s e s , b o t h q u a l i t a t i v e and q u a n t i t a t i v e . However, i t
i s i m p o s s i b l e t o c a r r y out r e a c t i o n s , such a s n e u t r a l i z a t i o n ,
i n v o l v i n g a c i d s (or b a s e s ) a p p r e c i a b l y weaker t h a n w a t e r ,
s i n c e w a t e r , b e i n g a s t r o n g e r a c i d ( b a s e ) , w i l l r e a c t p r e f
e r e n t i a l l y w i t h t h e a d d e d r e a g e n t . F o r t h i s r e a s o n , i n t h e
l a s t t h r e e d e c a d e s , n o n - a q u e o u s s o l v e n t s have become i n
c r e a s i n g l y i m p o r t a n t i n a n a l y s i s , p a r t i c u l a r l y i n q u a n t i t a
t i v e d e t e r m i n a t i o n s , s i n c e a c i d - b a s e t i t r a t i o n s i n t h e s e
m e d i a p o s s e s s the a c c u r a c y , s p e e d , and s i m p l i c i t y r e q u i r e d
f o r good a n a l y t i c a l m e t h o d s .
I n o r d e r t o c h a r a c t e r i z e t h e p r o p e r t i e s o f i n t e r e s t i n
nonaqueous s o l u t i o n s , i t i s u s e f u l t o r e v i e w t h e p a r t i c u l a r
p r o p e r t i e s o f w a t e r w h i c h a r e r e s p o n s i b l e f o r t h e u n i q u e
p o s i t i o n i t o c c u p i e s among s o l v e n t s ( 3 4 ) . W a t e r , a v a i l a b l e
i n l a r g e q u a n t i t i e s , i s an e x c e l l e n t s o l v e n t f o r most c l a s s e s
o f s u b s t a n c e s , b o t h e l e c t r o l y t e s and n o n - e l e c t r o l y t e s . The
p u r e s o l v e n t i s a p o o r c o n d u c t o r o f e l e c t r i c i t y , but aqueous
s o l u t i o n s o f e l e c t r o l y t e s have h i g h c o n d u c t i v i t i e s . M o r e
o v e r , w a t e r has t h e a b i l i t y t o c o n v e r t p o t e n t i a l e l e c t r o l y t e s
i n t o t r u e e l e c t r o l y t e s . Many s u b s t a n c e s , such as s u l p h u r
d i o x i d e , • a m m o n i a , hydrogen c h l o r i d e , and t e r t i a r y a m i n e s ,
w h i c h by t h e m s e l v e s a r e n o n - e l e c t r o l y t e s , r e a c t w i t h w a t e r
t o g i v e s o l u t i o n s w h i c h a r e e x c e l l e n t c o n d u c t o r s . N e u t r a
l i z a t i o n r e a c t i o n s between s t r o n g a c i d s y i e l d i n g h y d r o n i u m

2
ions and bases y i e l d i n g hydroxyl ions occur e a s i l y i n water,
a process made possible by i t s l i m i t e d s e l f - i o n i z a t i o n . The
phenomenon of hydrolysis i s a c h a r a c t e r i s t i c reaction of water.
Not only do dissolved s a l t s undergo hydrolysis, but many sub
stances such as acyljchlorides, esters, and s a l t s of very
weak acids react chemically with the solvent i n such a way
that the o r i g i n a l solutes cannot be recovered d i r e c t l y .
The s u p e r i o r i t y of water as a general solvent i s re
f l e c t e d more p r e c i s e l y through i t s exceptional and anomalous
physical properties. The high values of i t s melting point,
b o i l i n g point, heat of vaporization, and d i e l e c t r i c constant,
and i t s low v i s c o s i t y , d i s t i n g u i s h i t as a solvent of the
highest general u t i l i t y . However, there are properties of
nonaqueous media which indicate that they are, i n c e r t a i n
spec i a l i z e d circumstances, superior t o water as a solvent.
One of the most s t r i k i n g of these properties i s the
high a c i d i t y displayed by solutions of HCIO^, H^SO^, and HCI, f o r example, i n aprotic l i q u i d s such as benzene or
chloroform, or i n acid solvents, such as g l a c i a l a c e t i c acid.
These compounds are strongly a c i d i c and completely ionized
i n aqueous solution, but are even more a c i d i c i n solvents of
l e s s e r i o n i z i n g power. Moreover, the acid strengths of
HCIO^, H^SO^, and HCI d i f f e r widely in these media, whereas
i n water they are indistinguishable f o r equal concentrations.
That such phenomena occur may at f i r s t seem surprising, but,
as the ensuing discussion shows, they arise as a l o g i c a l

consequence of the Bronsted-Lowry concept of acids and bases. The concept of electrolytic dissociation has dominated
the f i e l d of acid-base chemistry for over half a century. Until 1926 i t was considered to be the f u l l explanation of acid-base phenomena, although i t was realized that the theory could not be applied to solvents other than water. There were scattered reports of titrations in nonaqueous media (19,20,21^ but i t was not u n t i l Hantsch (30) published his series of investigations that a departure from the prevailing theory became evident. The appearance of the Bronsted-Lowry theory (2, 3, 4, 5, 42, 43) and the classic investigations by Conant, Hall, and Werner (7, 8, 26, 27, 28) on acidity in glacial acetic acid, put the following analysis on a sound experimental and theoretical basis.
It has been shown by Bronsted that a logically con- , sistent general measure of acidity i s the hydrogen-ion potential, or the closely related hydrogen-ion activity. It has .been found that the use of acti v i t i e s instead of potentials i s convenient, but i t should be noted that this does, not imply any knowledge of the concentration. Hydrogen-ion act i v i t y i s defined as an exponential function of the hydrogen-ion potential, which represents the work concerned in the reversible removal of hydrogen ions from a given system, or their addition to i t . High activity means loosely bound hydrogen ions; low activity implies strongly bound hydrogen • ions. A reaction proceeds by the transfer of hydrogen ions

4
from higher to lower a c t i v i t y , although hydrogen ions as such do not exist to a measurable extent i n the solution.'
Since only the r a t i o s of a c t i v i t i e s may be measured, one must always set up an a r b i t r a r y scale of numerical values. Usually d i f f e r e n t conventions are adopted f o r d i f f e r e n t solutions such that the a c t i v i t y c o e f f i c i e n t approaches unity at i n f i n i t e d i l u t i o n , but t h i s i s not sa t i s f a c t o r y when s o l utions of the same solute i n various solvents are considered. I f the same scale i s used f o r a set of solvents, then two solutions with the same hydrogen-ion potential ( i . e . the same true a c i d i t y ) w i l l have the same hydrogen-ion a c t i v i t y , no matter what the solvent, but i t also follows that the a c t i v i t y c o e f f i c i e n t w i l l not approach unity f o r i n f i n i t e l y d i l u t e solutions.
With t h i s understanding, following Hammett (29) we may consider a mathematical approach to the effect of solvent upon a c i d i t y . Let HX be the acid concerned; S the solvent; C the concentration; a the a c t i v i t y ; f the a c t i v i t y c o e f f i c i ent; and C a the stoichiometric concentration of HX. Regardl e s s of the solvent, we have
C x - J ~
but i f the solvent i s basic we also have
i - K5 C S H * (2)

If, as seems probable, Cy+. may be considered negligibly small
d= CHX +CKr = C„K -f CSH+ (3)
since the reaction between the acid and the solvent i s HX + S ;F=± X" •+ SH+. A combination of 1, 2 and 3 gives
2 TP ( V KHX I T S^HX /
Since au+ increases with increasing K y this equation re-n o cords the principle that decreased basicity of the solvent (greater Ky) means greater acidity, although i t means less ionization.
In order to see more clearly the effect of changes of the dielectric constant, we may consider two limiting cases of 4. When the second term under the radical sign i s large compared with one, we obtain
• & Sx~
This i s the case f o r a weak acid or.weakly basic solvent; the.reaction HX + S ' X" + SH+ i s incomplete and the acidity depends on both and Kj^, i.e. upon the properties of both acid and solvent. On the other hand, when the same term i s small the radical may be expanded and using only the linear term we have
*H+ c & Ks SSH* ( 6 )
Here ionization i s large and the acidity depends only on

6
the p r o p e r t i e s of th e . s o l v e n t . This represents the l e v e l l i n g e f f e c t of the s o l v e n t , mentioned by Hantsch and by Conant and H a l l .
I t i s now p o s s i b l e t o make a t l e a s t a q u a l i t a t i v e pred i c t i o n of the e f f e c t of the d i e l e c t r i c constant from t h e known e f f e c t of t h i s property upon the a c t i v i t y c o e f f i c i e n t s . The a c t i v i t y c o e f f i c i e n t of an i o n i s gre a t e r i n solvents of low than of high d i e l e c t r i c constants, as demonstrated by the extremely low s o l u b i l i t y of t r u e s a l t s i n such media. This i s p r e d i c t e d by the i n t e r i o n i c a t t r a c t i o n theory of e l e c t r o l y t e s . The changes i n a c t i v i t y c o e f f i c i e n t s of uncharged molecular species from so l v e n t to solvent are much smaller than the changes of the i o n i c a c t i v i t y coeff i c i e n t s . Therefore, i t f o l l o w s that r a t i o s such as ..'HX/..X~ o r . S/\ SH w i l l decrease with decreasing d i e l e c t r i c constant. Now, as an admittedly rough approximation, assume t h a t the change i n t h i s r a t i o between two d i f f e r e n t s o l v e n t s i s independent of the a c i d or base concerned. There i s some reason f o r b e l i e v i n g t h a t s p e c i f i c e f f e c t s upon the r a t i o w i l l be considerably smaller than on the i n d i v i d u a l a c t i v i t i e s , corresponding t o the impression that a c i d s and bases very s o l u b l e i n organic l i q u i d s w i l l have s a l t s which are s o l u b l e i n organic l i q u i d s .
^8H + ^HX I t then f o l l o w s t h a t the r a t i o - — - i n equation
5 w i l l be approximately u n i t y f o r a l l s o l v e n t s ; t h a t the hydrogen i o n a c t i v i t y of a weakly i o n i z e d a c i d i s independent

of the d i e l e c t r i c constant of the medium. It follows from
equation 6 that the hydrogen ion a c t i v i t y of a highly ion
ized acid w i l l be greater i n solvents of low than i n s o l
vents of high d i e l e c t r i c constant. For intermediate cases-,
a decrease i n the d i e l e c t r i c constant w i l l r e s u l t i n a more
or l e s s large increase i n the hydrogen-ion a c t i v i t y . •
From the work of Conant and H a l l , and from the above
discussion, i t appears that there are three p r i n c i p a l i n
fluences a f f e c t i n g the apparent strength of an acid i n a
given solvent. The f i r s t i s the " i n t r i n s i c strength? of
the acid, the general tendency of the molecule to l i b e r a t e
a proton. The constant expressing t h i s tendency may be re
garded as f i x e d and equal f o r a l l solvents at any given
temperature. Next, there i s the basic strength, or proto-
p h i l i c tendency, of the solvent, which determines to what
extent the true strength of the acid can manifest i t s e l f .
F i n a l l y there i s the d i e l e c t r i c constant of the solvent
which determines the extent to which the ions, products of
the reaction of the acid with the solvent, can e x i s t i n
dependently of each other'. Water i s a basic solvent of high
d i e l e c t r i c constant. Therefore, strong acids react com
p l e t e l y with i t , and the products exist as separate ions.
HC10 4 + H 20 x H^0+ + ClO^"*
The a c i d i t y of a d i l u t e solution i n water can never be very
high, however, because the acid molecules a l l react to pro-

duce hydronium ions, whose proton a c t i v i t y i s not very great.
In a non-basic solvent,, such as acetic acid or benzene,
the acid molecules w i l l combine with the solvent to a much
le s s extent, so that, the proton a c t i v i t y of t h e i r i n d i v i d u a l
molecules may give a high a c i d i t y to the solution, although,
because of the low d i e l e c t r i c constant there w i l l be few
(unassociated) ions. Although as yet no method exists to
compare the a c i d i t i e s of solutes i n d i f f e r e n t solvents i n a
completely rigorous manner, i t has become evident that these
predictions are f u l f i l l e d . Thus i t may well be possible to
prepare a series of solutions containing only moderate con
centrations of the strongest acids i n suitable solvents
whose true a c i d i t i e s range well into the m i l l i o n f o l d values
of those obtainable i n water solu t i o n at the same concen
t r a t i o n . As indicators can be found which are s e n s i t i v e i n
the range of high absolute a c i d i t y , i t should be possible to
develop a "pH technique" which w i l l extend to high negative
values. One must always remember, however, that the true
measure of a c i d i t y i s to be taken as the hydrogen ion
a c t i v i t y as measured with a suitable electrode, and not
other l e s s precise e f f e c t s .
These conclusions may be amplified by an i l l u s t r a t i v e
example. Many very weak anhydro bases, which do not form
s a l t s i n aqueous solutions, are neutralized by perchloric
or sulphuric a c i d i n g l a c i a l acetic acid solution. This i s
shown by the t i t r a t i o n curves obtained by Conant and H a l l

(7, 2 7 ) . It should be noted that i n the formation of s a l t s
from anhydro bases, no water i s eliminated, and so the
water a c t i v i t y i s not involved. Their r e s u l t s confirm ex
perimentally that, whether acid strength i s defined i n terms
of the r e l a t i v e hydrogen ion a c t i v i t i e s of t h e i r equimolal
solutions, or i n terms of salt formation with anhydro bases,
perchloric and sulphuric acids are stronger i n acetic a c i d
than i n water.
At present we have some information-about the behavior
of acids i n a wide variety, of solvents, of which ammonia, water,
and g l a c i a l a c e tic acid may be considered as representative
examples. In each medium, evidence points to the proton
being completely solvated. In comparing the three solvents,
one i s led to consider that the essential difference between
glacial„acetic acid and water i s the same as that between
water and ammonia. The a c e t i c acid molecule has l e s s a f f i n -+
i t y f o r a proton than the water molecule; the CH^COOH^
ion gives up i t s extra proton more r e a d i l y than H^0+.
The formation of a s a l t from an anhydro base i s the re
verse of the d i s s o c i a t i o n of the a c i d . The anhydro base (B),
the anion of the a c i d (X~) oand the solvent molecule (5) may +
a l l be regarded as competing f o r the proton H . Therefore
the following equations may be written:
X" + H + ,==1 HX (7)
B + H + BH + . (g)
.'S + H + SH + (9)

10. Probably i n most solvents the amount of unsolvated proton
is- small, but we may s t i l l formulate the reaction of s a l t
formation of B:
SH + + B ;==! BH + + S
i n terms of the competing reactions 8" and 9 on Page 9 . A
very weak base B (corresponding to a very strong acid BH )
w i l l only form stable s a l t s i n a solvent which has con
siderably l e s s a f f i n i t y for the proton than B i t s e l f . Also
i t i s obvious that s a l t formation can only occur with an
anion (X ) which w i l l release the proton to B. The i n t e r
r e l a t i o n of these three competing reactions i s i l l u s t r a t e d
i n F i g . 1 , due to Conant and H a l l ( 7 ) .
s <*
C 6H 50" NH3
£ RNH2
as
o RCOO" C 6H 5NH 2
i -X
u. O
CCI3COO" H 20
>- NH2C0NH2
z u .
CH3C0NH2
14.
«c
*
C7H7SO3' HSO - AcOH
C6H5NHCOCH3
« 11/ (C 6H 5) 3N AC z C I O 4
Acid Ion Solvent Base X~ S B
FIG. r Relative Basic Strengths of Certain Acid Ions
Solvents and Organic Bases

The position of some common acids, anhydro bases and
three solvents are indicated by the formulas. The scale i s
ar b i t r a r y ; lack of data s t i l l makes i t impossible to evaluate
exact r e l a t i o n s h i p .
Acids are strong only i n the solvents l y i n g above them
i n the diagram, e.g. perchloric i s strong i n a l l three s o l
vents; sulphuric and hydrochloric i n water and ammonia; and
RCOOH only i n ammonia. Anhydro bases are neutralized by one
equivalent of any acid which i s stronger than t h e i r nonium t t
ions, provided that the action occurs i n a solvent l y i n g
below the p o s i t i o n of thedons on the scale. - Thus RCONHg i s
neutralized only i n ac e t i c acid and only by perchloric ac i d .
I f any solvent i s found i n which triphenylamine may be t i t
rated, i t w i l l l i e beneath acetic acid on the diagram.
Since perchloric acid i s very strong i n a c e t i c a c i d ,
and since the ac i d i c nature of the medium prevents solvoly-
s i s of s a l t s of strong acids and weak bases, one may con
clude that acetic a c i d i s an excellent solvent f o r the
analysis of substances weakly basic i n nature. The work of
Conant and Ha l l provided the basis f o r a l l subsequent i n
vestigations, but i t has only been i n the past f i f t e e n years
that s u f f i c i e n t , interest has arisen i n the method to y i e l d
p r a c t i c a l dividends. In t h i s period, procedures have been
published f o r the determination of pyridine and various
pyramidines (4S); amines (1, 22, 23, 44, 61), amino acids
and alcohols (3S, 44, 47, 5#, 59, 60); oxazolines (44);

12
quinine and nicotine (31, 35); alkylene oxides (1); sulphon-
amides (61); basic nitrogen i n o i l s (71); and choline s a l t s
of carboxylic acids (45). More clo s e l y related to the sub
je c t of t h i s paper.was•the determination of the s o l u b i l i t i e s
of c e r t a i n inorganic s a l t s i n acetic acid by Davidson and h i s
co-workers (9-17, 2 4 , 2 5 ) and by Kolthoff (40), followed by
the successful analysis of metal s a l t s of carboxylic acids
and of s t r i c t l y inorganic s a l t s by P i f e r and Wollish ( 5 0 , 51
53), Higuchi and Concha (3;2, 33), and Markunas and Riddick
(44). Since Davidson has shown (9) that metathetical rea
ctions may proceed as e a s i l y i n a c e t i c acid as i n water,
these l a s t r esults indicate that rapid and accurate quanti-,
t a t i v e analyses could be c a r r i e d out on one or more metal -
compounds provided they could be e a s i l y introduced into
acetic acid solution and that methods of separation could be
found. It was the object of t h i s i n v e s t i g a t i o n to begin de
veloping such methods.
I I . EXPERIMENTAL
A. Materials and Apparatus'
For every t i t r a t i o n , the sample was dissolved i n an-
hydrous acetic acid. The ordinary C P . grade of commercial
acid was p u r i f i e d by the method of Kendall and Gross (37),
that i s , by adding the calculated amount of acetic anhydride
to react with the water present, then d i s t i l l i n g twice using
a t h i r t y - i n c h h e l i x type column. The f r a c t i o n b o i l i n g at
117-118°C. was collected; samples melted at l6.5°C. ( l i t .

13
16.6° ( 3 7 ) ) .
Two solvents were used for the ti t r a t i n g solution of perchloric acid: glacial acetic acid and 1, 4-dioxane. The , latt e r was reagent quality solvent, purified according to Vogel (63 ) , and d i s t i l l e d twice. The fraction boiling between 100.5°C. and.l01.5°C. was collected; samples had a melting point'of 11,8-11.9°'C. ( l i t . 12°C. ( 6 3 ) ) .
The titrant used in a l l cases was an approximately tenth normal solution of perchloric acid in either glacial acetic acid or dioxane. These were prepared as follows:
(a) Acetic acid solution. Approximately thirty-one ml. of a 60% aqueous solution
of perchloric acid were mixed with glacial acetic acid (500 ml.), chilled acetic anhydride (33 ml.) was added, and the solution diluted to two l i t e r s . It was necessary to allow,the solution to stand overnight (or preferably two to three days) to allow the anhydride to react with the water present. This solution could be used with both indicator and potentiometer.
(b) Dioxane solution. This solution was prepared in a,similar.manner, except
that no acetic anhydride was added. Occasionally the .sol^ ution had a brown color:, but this did not influence the acid strength. The solution contained a small amount of water and so could only be used .for potentiometric ti t r a t i o n s .

14
Indicator
A 0.5% solution of c r y s t a l v i o l e t i n g l a c i a l acetic acid
was found to be.satisfactory. One drop was s u f f i c i e n t f o r a .
f i f t y - f i v e ml. solution. The color change of c r y s t a l v i o l e t
i s not sharp, passing from bluer-violet through various shades
of blue and. green to yellow... The blue-green end point was
e s p e c i a l l y d i f f i c u l t t o detect when the t i t r a t i o n produced a
white p r e c i p i t a t e , as with potassium or ammonium s a l t s . The
best procedure was to t i t r a t e a sample to the end-point pot-
entiometrically, add the indicator and use t h i s solution as
a standard.
Potentiometer
Two instruments were used:
(a) A Beckman model G pH meter, s e r i a l number 1996,
equipped with a glass saturated aqueous calomel electrode
system. To obtain maximum s e n s i t i v i t y , the l a t t e r was of the
sleeve type. The s o l u t i o n inside the electrode was changed
frequently to prevent contamination by acetic acid d i f f u s i n g
i n .
The calomel electrode could not be used to t i t r a t e s o l
utions of lead or s i l v e r , since p r e c i p i t a t e s of the chloride
froze the ground glass surface of the electrode. This d i f
f i c u l t y was overcome by using a salt bridge consisting of a
U-tube, with ground glass stoppers at each end, f i l l e d with
a supersaturated solution of l i t h i u m acetate i n acetic acid,
with a l i t t l e added g e l a t i n to prevent c r y s t a l l i z a t i o n of

15
the s a l t . The U-tube connected the solution being analysed
to a saturated aqueous solution of potassium chloride, into
which the calomel electrode dipped. A l e s s cumbersome
arrangement involved the use of a s i l v e r - s i l v e r chloride
electrode as a reference electrode i n place of the calomel
electrode.
(b) A Fisher senior t i t r i m e t e r was used when a s e r i e s
of analyses to the same end point was to be run. The same
electrode systems as described above were employed.
Standardization of the Titrant
Sodium carbonate, or preferably potassium acid phthalate
( 5 7 ) , may be used as a primary standard. Approximately s i x
tenths of a gram were dissolved i n g l a c i a l acetic acid (55 ml.)
and t i t r a t e d either potentiometrically, or with c r y s t a l
v i o l e t indicator to a blue-green end-point.
Water acts as a weak base i n acetic a c i d . Its presence
i n the solution when t i t r a t i n g potentiometrically can be
tolerated up to 1.5% (44) but i t causes erroneous r e s u l t s
when using an indicator. To minimize absorption from the at
mosphere, i t was convenient to use a l i p l e s s beaker closed by
a t h i n rubber stopper through which a small hole was punched
to admit the burette t i p .
One must remember that organic l i q u i d s generally have a
high c o e f f i c i e n t of cubical expansion and hence temperature
changes w i l l have an appreciable effect on the normality of
the solution. Allowance was made f o r t h i s by assuming that

16
the N/lO solution i n acetic acid or dioxane had the same co
e f f i c i e n t as the pure solvent.
The metals and acetates were of the best obtainable
purity, and were used without further p u r i f i c a t i o n . Other
compounds were prepared as described.
B. T i t r a t i o n of Metal Salts
The metals studied were to be t i t r a t e d as acetates, so,
to determine the s e n s i t i v i t y and accuracy of the method, .-.
series of t i t r a t i o n s were c a r r i e d out on metal acetates as
described below. To. check the accuracy each s a l t was an
alysed by the method indicated, according t o the in s t r u c t i o n s
i n Vogel's "Quantitative Analysis" (64). The r e s u l t s of a l l
the determinations are l i s t e d i n Table 1.
Sodium Acetate
The anhydrous salt was dried f o r one hour at 110°C. A
convenient solution f o r t i t r a t i o n contained three m i l l i -
equivalents of the sa l t i n f o r t y m i l l i l i t e r s of acetic a c i d .
This concentration was used i n a l l subsequent analyses. It
was necessary to warm the mixture to effect rapid solutionj
the s o l u t i o n was then quickly c h i l l e d and t i t r a t e d immediately,
eithe r potentiometrically or t o the blue-green c r y s t a l v i o l e t
end-point. The gravimetric check was as sodium magnesium
uranyl acetate.
Acetates of Potassium, Lithium, Barium, Strontium, and
Ammonium These were analysed i n the same manner as sodium acetate.

Since ammonium acetate i s so hygroscopic, i t had to be re-
c r y s t a l l i z e d . This i s best done from methanol or acetic
acid, followed by washing with ether and drying i n a vacuum
desiccator at one centimeter pressure. A l l gave equally good
r e s u l t s with indicator or potentiometer methods. The s a l t s
were analysed gravimetrically as follows:
potassium as perchlorate lithium as sulphate barium as sulphate strontium as sulphate ammonium (volumetrically) by t r e a t i n g with
strong base, d i s t i l l i n g into excess standard acid and t i t r a t i n g the excess.
Magnesium Acetate
Visual determinations with the indicator gave poor re
s u l t s , since the end-point occurs at an i l l - d e f i n e d yellow-
green shade. The end point may be determined by comparing
the sample being analysed with a solution of in d i c a t o r +
magnesium perchlorate i n acetic acid i n a Coleman spectro
photometer. However, i t was easier and quicker to use the .
potentiometric method. The gravimetric check was as mag
nesium ammonium phosphate.
Acetates of Calcium, Manganese, and Cadmium
These may be t i t r a t e d potentiometrically but not with
the i n d i c a t o r . The gravimetric checks were as follows:
calcium as the oxide, manganese as manganese ammonium phosphate, cadmium as the pyridine thiocyanate complex.
Acetates of Cobalt and Nickel
For reasons unexplained, no s a t i s f a c t o r y end-point

18
could be obtained when t i t r a t i n g with the acetic acid solution
of perchloric acid. However, reproducible, 'but poorly-defined,
end-points were obtained with a solution of perchloric acid
i n dioxane. Both cobalt and n i c k e l were determined g r a v i -
m e t r i c a l l y as the pyridine thiocyanate complex.
Zinc Acetate
zinc acetate i s quantitatively insoluble i n acetic acid,
and so cannot be determined d i r e c t l y . It may be e a s i l y
analysed by adding a measured excess of HCIO^ solution, and
back t i t r a t i n g with standard sodium acetate solution. The
gravimetric check was as the pyridine thiocyanate complex.
Acetates of Lead and S i l v e r
The calomel electrode could not be used i n either det
ermination f o r reasons described before, so both analyses
were carried out using a s i l v e r - s i l v e r chloride electrode.
In addition, s i l v e r acetate i s only p a r t l y soluble i n acetic
acid, and had to be determined by the back t i t r a t i o n method
described f o r zinc acetate. As a check, lead was determined
as the molybdate and s i l v e r as the chloride.
Acetates of Iron, Chromium and Aluminum
Although reproducible end-points were obtained, they
yielded anomalous r e s u l t s , so that the method could not be
applied d i r e c t l y . ' The r e s u l t s are discussed further i n the
next section.
Acetates of Tin, Antimony, Copper, Bismuth and Uranium
These acetates behave as i f they were e s s e n t i a l l y

19
undissociated i n acetic acid, and could not be t i t r a t e d by
t h i s method. Since t i n (II) usually occurs as the chloride,'
which in t e r f e r e s with the t i t r a t i o n , i t was removed by
adding the required amount of a standard solution of lead
acetate.
T i t r a t i o n of Chloroacetates
Several chloroacetates were prepared according to the
method of Kastle and Keiser (57) . They were a l l soluble i n
acetic acid, and, except for the ammonium s a l t , were analy
sed by the same method described previously f o r acetates.
Each was also estimated gravimetrically by the same method as
f o r the corresponding acetates, except f o r ammonium chloro-
acetate. Its solution i n acetic acid and i n water changed
strength continuously, possibly as a result of conversion
into glycine, and could not be t i t r a t e d .
T i t r a t i o n of Chlorides
The presence of the chloride ion interfered with the
t i t r a t i o n , a d i f f i c u l t y which was overcome by amethod due
to P i f e r and Wollish (50, 5 2 ) . A s l i g h t excess of a solution
of mercuric acetate i n acetic acid (6 gm. per 100 ml.) was
added t o the solution to be t i t r a t e d . The halide was re
moved as HgX2, and the acetate ion lib e r a t e d could then be
determined. Mercuric acetate i s e s s e n t i a l l y undissociated
i n acetic a c i d (46) and the excess therefore did not i n t e r
f e r e . However, on standing, or on the addition of acetic
anhydride, a complex mercuric compound i s formed (46)

preventing the desired reaction. Hence the so l u t i o n should
be f r e s h l y prepared, and.appreciable amounts of a c e t i c
anhydride must not be present i n the solution to be t i t r a t e d .
(The amount of acetic anhydride present may e a s i l y be de
termined (6, 41, 49).)
C. Separation and Analysis of Metal Acetate Mixtures
S o l u b i l i t i e s of Metal Salts i n Acetic Acid
Zinc acetate was found to be quantitatively insoluble
i n a c e t i c acid, and could be separated from other s a l t s
which were completely soluble by di s s o l v i n g the mixture i n
water, adding the r e q u i s i t e amount of acetic anhydride, and
f i l t e r i n g .
Various common reagents were tested as p r e c i p i t a t i n g
agents with twenty-one representative metals' as outlined
below. Whenever the metal acetate was only p a r t l y soluble
i n acetic a c i d , the pr e c i p i t a t e was f i l t e r e d o f f and the
resi d u a l solution tested. In a l l cases, a solution of one
mil l i e q u i v a l e n t of metal acetate i n twenty m i l l i l i t e r s was
used.
Whenever a pr e c i p i t a t e was produced upon the addition
of one of the test reagents, i t was removed by centrifuging
and the l i q u i d tested f o r completeness of p r e c i p i t a t i o n as
follows: The solution was evaporated almost to dryness
twice with excess concentrated n i t r i c acid to remove a l l
organic matter, and the residue taken up i n d i l u t e n i t r i c
acid. The s o l u t i o n was then tested for the presence of the

21 appropriate ion according to the methods outlined i n
Treadweil-Hall (62).
Hydrochloric, Hydriodic, and Hydrosulphuric Acids
In each case the dried gas was passed through the s o l
ution u n t i l the l a t t e r was saturated.
Sulphuric Acid
S u f f i c i e n t c h i l l e d acetic anhydride was added t o c h i l l e d
reagent grade acid to react with a l l the water present. The
mixture was allowed to stand out of contact with a i r f o r
twenty four hours to allow the reaction to go to completion,
r e s u l t i n g i n a yellow-tinged very viscous l i q u i d . Enough
of t h i s was added t o each te s t solution to give a f i v e - f o l d
excess. Those which did not react were allowed t o stand f o r
two days, a f t e r which a portion of each was d i l u t e d with
5% water.
N i t r i c Acid
Reagent grade f i f t e e n molar acid was added to each
tes t solution to give a f i v e - f o l d excess.
Phosphoric Acid
Reagent grade 85% acid was added to each solution to
give about a f i v e - f o l d excess.
Benzoic, Oxalic, S a l i c y l i c . T a r t a r i c Acids
These are not very soluble i n acetic a c i d at room
temperature, so a saturated s o l u t i o n at 100°C. was used,
the test solutions also being maintained at t h i s temperature.
About a f i v e - f o l d excess was added i n each case.

The r e s u l t s of these two hundred ten t e s t s are summarized
i n Table 2, Since the r e s u l t s with the sulphuric a c i d were
at variance with those of Davidson (9) i n that only three
metals gave pr e c i p i t a t e s , the following additional t e s t s
were carried out.
The sulphates of Mg, Mn(II), C r ( I I I ) , Fe(III), Ca, Co,
Nr and Sn(II) were refluxed with g l a c i a l acetic acid f o r four
hours, a f t e r which the l i q u i d was tested f o r the presence of
the appropriate ion i n the manner described before. Then
the sulphates were dissolved i n water, and s u f f i c i e n t a c etic
anhydride added to react with a l l of i t , producing the same
anhydrous medium as i n the f i r s t set of t e s t s . The r e s u l t s
of these experiments are given i n Table I I I .
Separations Effected
On the basis of the foregoing t e s t s , the following
a n a l y t i c a l separations were carr i e d out:
Zinc and Magnesium
Three m i l l i e q u i v a l e n t s each of the two metals were
mixed and dissolved i n d i l u t e n i t r i c a c i d (5 ml.). The
solution was d i l u t e d with a c e t i c acid (15 ml.) and heated
to b o i l i n g . Acetic anhydride (33 ml.) was added cautiously,
the mixture being boiled u n t i l the yellow color disappeared.
The beaker was covered with aluminum f o i l , the suspension d i
gested on a steam bath f o r ten minutes, c h i l l e d i n i c e -
water, and centrifuged. The magnesium solution was t i t r a t e d
immediately. The zinc may be determined as described above,
or by difference.

23
zinc and Copper
The metals were separated by the same procedure as above.
The zinc was determined as described before, and the copper
obtained by difference.
Barium and Calcium
Three m i l l i e q u i v a l e n t s of each metal were dissolved i n
n i t r i c acid (5 ml.), the solu t i o n d i l u t e d with acetic acid
(15 ml.), heated to b o i l i n g , and acetic anhydride (33 ml.)
cautiously added. The solution was boiled u n t i l 'the yellow
color disappeared, then c h i l l e d i n i c e water. Concentrated
n i t r i c acid (2 ml.) was added, and the solution vigorously
s t i r r e d . The p r e c i p i t a t e of barium n i t r a t e was removed by
centrifuging, and the calcium solution immediately t i t r a t e d .
The barium may be determined by difference, or by decomposing
the n i t r a t e with a c e t i c anhydride, and t i t r a t i n g i n the
usual way.
S i l v e r and Aluminum
Three m i l l i e q u i v a l e n t s each of the two metals were
brought into s o l u t i o n as above, except that concentrated
n i t r i c acid was used. The solution was c h i l l e d and saturated
with dried H2S gas. The s i l v e r sulphide was removed by c e n t r i
fuging, dissolved i n d i l u t e n i t r i c acid, and the solution
boiled to expel the l i b e r a t e d HgS. S u f f i c i e n t a c e t i c an
hydride was added t o react with the water and decompose the
n i t r a t e . T h i r t y - f i v e m i l l i l i t e r s of standard perchloric
acid solution were added, and the excess back-titrated with

standard sodium acetate solution, using s i l v e r - s i l v e r chlor
ide and glass electrodes.
Lead-Tin
The procedure was exactly the same as i n the preceding
separation; great care had to be exercised not to allow
any water to enter the system u n t i l the separation was com
plet e , since stannous sulphide would p r e c i p i t a t e i f water
was present. The lead sulphide was removed by centrifuging,
treated and analysed as the s i l v e r sulphide above.
Magnesium and Antimony; Magnesium and Lead; Magnesium and
Copper
The lead, antimony, and copper were removed with hydrogen
sulphide; the procedure, being exactly the same as i n the
preceding case, except that aqua regia was required to d i s
solve the antimony. S u f f i c i e n t mercuric acetate reagent
(6 gm. per 100 ml.) was added to the magnesium solution,
a f t e r removing the sulphide,, to react with a l l the chloride
present. The magnesium may then be determined i n the usual
manner.
If accuracy greater than four percent i s not required,
the magnesium may be t i t r a t e d d i r e c t l y i n the presence of the
antimony. These r e s u l t s were generally of the order of
k% too high.
S i m i l a r l y , magnesium may be t i t r a t e d d i r e c t l y i n the
presence of copper; the r e s u l t s were of the order of &%
too high.

Nickel-Chromium
About three m i l l i e q u i v a l e n t s of each metal were mixed '
and dissolved i n d i l u t e hydrochloric (5 ml.) acid (since
n i t r i c acid renders chromium passive). The solution was
heated to b o i l i n g and a c e t i c anhydride {'j>0 ml.) cautiously
added. A f i v e - f o l d excess of a hot solution of oxa l i c acid
i n acetic acid was added, the beaker covered and c h i l l e d : i n
i c e water. The pr e c i p i t a t e was removed by centrifuging,
evaporated twice to dryness with excess concentrated n i t r i c
acid, and the residue taken up i n d i l u t e n i t r i c a c i d . The
solution was treated with acetic anhydride i n the usual
manner, and the n i c k e l t i t r a t e d with perchloric acid i n
dioxane..
Cadmium and Magnesium
A solution of three m i l l i e q u i v a l e n t s of each metal was
made up i n the usual way, c h i l l e d , and saturated with dry
hydrogen iodide gas. The pr e c i p i t a t e was removed by c e n t r i
fuging, washed, and the r e s u l t i n g solution of magnesium
t i t r a t e d .
Tin and Magnesium
A solution of three m i l l i e q u i v a l e n t s of each metal i n
acetic acid, was prepared i n the usual way, but kept at 100°C.
A . f i v e - f o l d excess of oxalic a c i d solution i n acetic acid
was added, and the mixture c h i l l e d . The p r e c i p i t a t e of mag
nesium oxalate was.removed' by centrifuging, evaporated to .
dryness twice with concentrated n i t r i c acid, and the residue

taken up i n d i l u t e n i t r i c acid. Acetic anhydride was added
to react with the water and decompose the n i t r i c acid, a f t e r
which the magnesium was t i t r a t e d i n the usual way. •
The r e s u l t s of a l l these determinations are summarized
i n Table IV i n the next section.
D. Introduction of Metals Into Acetic Acid Solution
Acetic acid i t s e l f i s e f f e c t i v e i n dissolving only the
most elec t r o p o s i t i v e of the metals. I f , however, the acid
strength could be increased, more metals could be introduced
d i r e c t l y into the organic phase. A possible method of
accomplishing t h i s suggests i t s e l f from the following con
siderations:
I f AB i s a very weak organic base and HX a weak acid,
then a mixture of AB and HX w i l l remain a mixture (or s o l
ution) of one i n the other with no chemical reaction occur
r i n g . On the other hand, i f AB i s a strong organic base
and HX a strong a c i d , a s a l t w i l l be produced on mixing:
AB + HX -> ABH+ + X"
With a f a i r l y weak aci d (say CH^COOH) and a f a i r l y strong
base (piperidine f o r example) one would obtain a compound of
the sort AB...H+...X~ with a hydrogen bond, and a greater or
l e s s e r " a c t i v i t y " depending on the strength of the base and
the temperature. I f the a c t i v i t y i s s u f f i c i e n t l y great, the
p o s i t i v e ion may be displaced by metals of high enough
p o t e n t i a l .
To test t h i s theory, twenty f i v e representative metals

27
were treated with ammonium acetate, piperidinium acetate,
and dicyclohexylammonium acetate, the l a t t e r t o give a higher
b o i l i n g s a l t . Test tube experiments only were carried out,
to cover the ground as rapidl y as possible. No attempt was
made to determine the extent of,the reaction i f the metal
did not dissolve completely. The res u l t s of the tests are
summarized i n Table 17 and Table V.
Preparation of Piperidinium Acetate
Piperidine ( 4 9 . 4 ml.) and ace t i c acid ( 2 8 . 6 ml.) were
dissolved i n separate portions of acetone. Mixing immedi
ately produced a w e l l - c r y s t a l l i z e d white p r e c i p i t a t e . The
compound was not stable toward heat; drying at 5 5 ° C caused
i t to turn yellow. Therefore, a f t e r f i l t e r i n g and washing
with ether, the s a l t was stored i n vacuo u n t i l dry. The
y i e l d was 7 1 . 5 gm. (98%) of a s o l i d melting at 1 0 1 - 1 0 4 ° C .
with decomposition.
Preparation of Decyclohexylammonium Acetate
Dicyclohexylamine ( 1 0 0 . 7 ml.) and acetic acid ( 2 8 . 6 ml.)
were dissolved i n separate portions of acetone. Mixing pro
duced a powdery white p r e c i p i t a t e i n 98% y i e l d , melting at
1 1 6 - 1 1 8 ° C .
A second method of increasing the a c t i v i t y of an acid
i s t o introduce electro-negative atoms into the molecule, as
i n the chloroacetic acids. To determine the extent of t h i s
increase i n a c t i v i t y a survey was made of the r e a c t i v i t y of
the twenty-five metals mentioned toward pure monochloroacetic,

28
d i c h l o r o a c e t i c , and t r i c h l o r o a c e t i c acids and equimolar s o l
utions of each i n acetic acid. Each test was c a r r i e d out
from room temperature to the b o i l i n g point of the a c i d con
sidered. These r e s u l t s are also summarized i n Table IV and
Table V. . ' .
However, f o r reasons discussed i n the next secion, these
acids were unsatisfactory f o r general use, and the f o l l o w i n g
method was adopted. To introduce the metals into acetic acid
solution, s u f f i c i e n t n i t r i c acid was used to bring the metal(s)
into aqueous solution, followed by s u f f i c i e n t acetic anhyd
ride to react with a l l the water added and to decompose a l l
the n i t r a t e present. In c e r t a i n cases, aqua regia was re
quired, but i n general t h i s method was fast and e f f i c i e n t .
I I I . RESULTS AND DISCUSSION
A. Determination of the Potentiometric End-Point
The t i t r a t i o n curves obtained are i l l u s t r a t e d i n
plates jr~XJ|i8. The f i r s t i n each set i s a plot of volume
of t i t r a n t v. e.m.f. reading. The end point i s , of course,
the i n f l e c t i o n point on the curve, but i n t i t r a t i o n s the
p o t e n t i a l break was not sharp enough to determine the end
point accurately from the graph. A more precise method i s
to plot -^-| , the change i n potential per unit volume, v.
V, the volume added near the end point, as was done i n the
second graph i n each set. Since the point of i n f l e c t i o n

i n the t i t r a t i o n curve has the condition "7 7 = o, the dV^ '
corresponding point on the d i f f e r e n t i a l curve must be a
maximum, i . e . (~) = o. I f the l a t t e r curve i s not
symmetrical, the end-point may be found by extrapolation
(39), assuming that i n a small region about the end-point dE
varies l i n e a r l y with V. For a l l but the most basic of av the metal acetates, the s e n s i t i v i t y of the t i t r a t i o n did
not j u s t i f y extrapolating the volume to f r a c t i o n s of a drop.
The end points are indicated on a l l the graphs.
An attempt was made to calculate the error involved i n
the t i t r a t i o n s on a th e o r e t i c a l basis, by adapting the
method of Roll e r (54-56) . This was found to be impossible,
however, because of a lack of data on d i s s o c i a t i o n constants
i n acetic acid.
The s e n s i t i v i t y of the method may be increased by d r i l
l i n g a hole i n the side of the beaker and i n s e r t i n g one
electrode h o r i z o n t a l l y so that the t i p i s very close to that
of the other electrode, which i s i n the usual v e r t i c a l pos
i t i o n . The increase i n s e n s i t i v i t y i s caused by a decrease
i n e l e c t r i c a l resistance.
Since fluorpsulphonic acid i s too active to exist i n
water, i t was thought i t might be stronger i n acetic acid
than perchloric acid, and hence give better defined end-
points with the weaker bases. However, t h i s was not the
case; the a c i d i t y , as measured by the po t e n t i a l , was l e s s
than that of perchloric acid. Moreover the .end-point -

30
obtained when the fluorosulphonic acid was used as t i t r a n t
was very poorly defined'.
• B. T i t r a t i o n of Metal Salts
The r e s u l t s obtained from the t i t r a t i o n s are summarized'
i n Table I; the t i t r a t i o n curves are i l l u s t r a t e d i n Plates
I T - XJU B . The t i t r a t i o n curves show the same' changes
as one proceeds from strong to weak bases as i s found i n
water. Acetates of the f i r s t and second group metals show '
a pronounced pote n t i a l "break* at the end-point, while those
of metals usually considered "weak" i n water show a les s
pronounced break. An exception was ammonium acetate, which
i n acetic acid i s as strong as potassium acetate. In the
case of ni c k e l cobalt and cadmium, the accuracy of the t i t
r a t i o n was not always as great as the res u l t s i n Table I
might indicate, since the end-point was poorly defined.
Contrary to the res u l t s published by P i f e r and Wollish
(51) no end-point could be found f o r bismuth, antimony and
t i n , and a very low value was obtained f o r the percentages
of i r o n . Inspection of Plates iSJ r t N I >SI shows that the
curves obtained f o r bismuth, antimony, copper, t i n , and
uranium are of the same form as that f o r the solvent alone
i . e . t h e i r acetates are bases too weak to be t i t r a t e d i n
acetic acid with perchloric a c i d .
The curves f o r chromium, iron and aluminum yielded
end-points showing approximately 11-12% purity, while the
gravimetric analysis gave r e s u l t s close to 100%. This i n
spite of the fac t that the poten t i a l breaks are more

TABLE I TITRATION OF.METAL SALTS
Salt % Purity Found Gravimetric Determination
NaOAc 99.59 99.55 99.50 99.58 99.52
KOAc 99.56 99.41 99.49 99.48 99.47
LiOAc 99.73 99.70 99.77 99.75 99.60
Ba(0Ac) 2 99.40 99.47 99.49 99.41 99.46
Sr(0Ac) 2 99.65 99.60 99.49 99.41 99.46
NH^OAc 97.58 98.08 97.58 97.73 97.95
Mg(0Ac) 2 99.82 99.69 99.71 99.85 99.74
Mn(0Ac) 2 99.69 99.31 99.88 99.62 99.73
Ca(0Ac) 2 98.95 99.06 99.00 98.96 99.05
Cd(0Ac) 2 98.72 98.25 98.55 9 8 . 6 0 - 98.72
Co(0Ac)2 91.35 91.0*5 91.17 91.06 91.00
Ni(0Ac) 2 93.61 93.30 93.21 93.29 93.42
Zn(0Ac) 2 97.73 97.50 97.26 97.40 97.30
Pb(0Ac) 2 98.36 98.49 98.57 98.30 98.33
Ag(0Ac) 2 „ 99.72 100.13 99.62 99.83 99.76
Fe(OAc) 3l i 11.36 11.39 11.59 98.96 98.90
C r ( O A c ) 3i 11.71 11.50 11.44 99.65 99.43
Al(OAc ) 3 S 11.06 11.23 11.15 99.03 99.15
ClCH 2 C00Na 97.96 98.13 98.07 98.10 98.00
C1CH2C00K 97.83 99.00 99.01 98.96 98.89
(GlCH 2 C00) 2Ba 97.36 97.30 . 97.46 97.50 97.35
(ClCH 2 C00) 2Mg 97.35 97.46 97.33 97.49 97.46
A F i r s t column i n M % purity found" had concentration of three m i l l i e q u i v a l e n t s of sample per 4 5 ml. of solvent, the second s i x m i l l i e q u i v a l e n t s and the t h i r d nine m i l i i e q u i v a l e n t s per 4 5 ml. Each value i n "% purity found' represents the mean of two determinations. "

3 2
c l e a r l y defined than those f o r cetain metals whose curves
y i e l d reasonable r e s u l t s . A possible explanation f o r these
low r e s u l t s may be i n the work of Weinland et. a l ( 6 5 - 7 0 ) ,
who found that solutions of. chromic, and f e r r i c s a l t s i n
acetic acid give r i s e to numerous complexes many of which
were quite stable. , One of these has the form . [cr^OH.)^
(AcO)^J AcO. T i t r a t i n g t h i s with perchloric acid solution
would result i n the compound ^Cr-^OH^tAcO^.J CIO^,. that
i s , only one molecule of HCIO^ has been consumed f o r three
atoms of chromium instead of the expected three molecules
of HCIO^ f o r one atom of chromium. , This would give r i s e to
a r e s u l t 1 1 . 1 1 % of the theoretical,_not f a r from the values
obtained. Moreover, the values obtained remained e s s e n t i a l l y
constant over a three-fold increase i n concentration,, which
would indicate that a d e f i n i t e compound or set of compounds
i s involved, under the conditions at hand, and not an
equilibrium mixture. Further work along t h i s , l i n e would
promise to y i e l d very i n t e r e s t i n g r e s u l t s .
When the end-point i s i n d i s t i n c t or poorly defined'in
acetic acid alone, i t may be improved, as shown recently by
P i f e r et a l . , ( 5 3 ) by the addition of a miscible organic
l i q u i d of low d i e l e c t r i c constant. ..This was i l l u s t r a t e d by
the improvement of the end-point f o r cobalt, n i c k e l , and
cadmium acetates when a solution of.perchloric acid i n
dioxane was used as a t i t r a n t . The reason f o r the increase
i n s e n s i t i v i t y i s that perchloric acid i s l e s s ionized i n

the solvent of lower d i e l e c t r i c constant, and hence, as
shown i n the introduction, the solution has a higher a c i d i t y .
C. S o l u b i l i t i e s i n Acetic Acid
Zinc acetate i s the only one quantitatively insoluble
i n a c e t i c acid; those of s i l v e r and thorium, aluminium and
bismuth are only p a r t l y soluble. When an aqueous solution
of arsenious a c i d i s treated with'racetic anhydride, a white
p r e c i p i t a t e forms, but i t i s doubtful i f t h i s i s arsenic
acetate. The re s u l t s of the tests with possible p r e c i p i t
ating reagents, as summarized i n Table I I , have several
i n t e r e s t i n g features. In many cases the s o l u b i l i t i e s p a r a l -
l e l l t h o s e i n the aqueous system,, but there are some out
standing exceptions. For instance, strontium and barium
n i t r a t e s are precipitated immediately and quantit a t i v e l y
upon the addition of concentrated n i t r i c acid or a solution
of sodium n i t r a t e i n acetic acid. Lead i s also p r e c i p i
tated, but not quantitatively, and magnesium p r e c i p i t a t e s
a f t e r standing overnight.
Although barium chloride was insoluble i n a c e t i c acid,
strontium chloride was very soluble, i n comparison with
other chlorides. Cadmium chloride also p r e c i p i t a t e d r e a d i l y ,
but tended to dissolve i n excess HCI. P r e c i p i t a t i o n was not
quantitative, but was when hydrogen iodide i s used.
Hydrogen sulphide gave p r e c i p i t a t e s which were l e s s
c o l l o i d a l than those obtained i n water i n certa i n cases,
notably cadmium lead and s i l v e r . (In general, p r e c i p i t a t e s

TABLE II
RESULTS OF ADDING THE REAGENTS LISTED TO A OF 1 MILLIEQUIVALENT OF THE METAL ACETATE IN
SOLUTION ACETIC ACID
Reagent Ca Mg Th A l U Mn Cr Mo Fe Cd Co Na Sn Pb Sb Bi As Se Te Ag Sr Ba
Anhydrous I^SO^ - - - x - - - - X X
- H2 S 0 4 + 5% H 20 X x - - - 0 - - - - X X X
15N HNO3 a - - - - - - -Dry HCl gas - - _ 0 x - - - - 0 fa
Dry HI gas - - x x - - - - X
Dry H 2S gas O O X X X X X X X X 0 X
85% H 3P0 4 x fa fa 0 fa fa x 0
S a l i c y l i c acid 0
Benzoic Acid
T a r t a r i c Acid X X X O X X X X X X X
Oxalic acid X X X X X X X X
LEGEND '•" x Quantitative p r e c i p i t a t i o n . 0 Non-quantitative p r e c i p i t a t i o n & Quantitative p r e c i p i t a t i o n on standing several hours. fa Non-quantitative p r e c i p i t a t i o n on standing several hours.
Not tested.
•F-

formed i n a c e t i c a c i d are not well c r y s t a l l i z e d ) . A l l the
metals found i n group II i n the usual a n a l y t i c a l scheme pre
c i p i t a t e d r e a d i l y except t i n , which remained i n solution as
long as no water was present. (Addition of 1% water caused
formation of a brown precipitate.) Nickel and cobalt s u l
phides, which do not p r e c i p i t a t e i n a c i d i c aqueous solutions,
p r e c i p i t a t e d more slowly than the metals of group II , but
p r e c i p i t a t i o n was quantitative a f t e r f i f t e e n minutes. There
was also a reaction with chromium to produce a small amount
of grey-green c o l l o i d a l p r e c i p i t a t e . This was somewhat sur
p r i s i n g , as there was no such reaction with i r o n or aluminum,
and prevented the separation of other metals from chromium
using hydrogen sulphide.
Although no quantitative p r e c i p i t a t i o n s resulted from
addition of s a l i c y l i c acid, color changes i n several cases
showed complexes had been formed. Iron changed from red to
dark brown, cobalt from red to rust brown, n i c k e l from green
to yellow, and the solutions of aluminum and antimony from
c o l o r l e s s to yellow. The molybdenum sample became deep red
v i o l e t , with the formation of a very f i n e brown p r e c i p i t a t e .
Other substituents on the benzene nucleus might r e s u l t i n the
formation of insoluble complexes.
Benzoic acid produced no p r e c i p i t a t e with any metals,
but t a r t a r i c and oxalic acids were very e f f e c t i v e , almost
a l l the p r e c i p i t a t i o n s being quantitative. Although
p r e c i p i t a t i o n i s best done i n a hot solution with these

reagents and s a l i c y l i c acid because of s o l u b i l i t y considerations, precipitates were better crystallized when brought down in the cold in acetic acid. However, almost a l l were colloidal in nature, and so a centrifuge was much preferred to a f i l t e r for separation.
Perhaps the most unusual phenomena occurred with sulphuric acid and the sulphates. The results of the experiments carried out are summarized in Table III.. According to Davidson (14) sulphates in general are insoluble in acetic acid, and metals precipitate on the. addition of anhydrous sulphuric acid. Table I I I shows that four hours refluxing failed to dissolve an appreciable amount of any of the sulphates tested, but on the other hand, Table II shows that most metals failed to be precipitated upon the addition of sulphuric acid. Moreover, when the sulphates were dissolved in water and the solution treated with acetic anhydride, two, chromium and iron,, failed to precipitate at a l l , and the precipitation of several others was far from complete. These results show that one cannot rely on the behavior of the sulphates, and that sulphuric acid i s not a good precipe itating agent f o r this reason.
D. Separations As mentioned before, precipitates formed in hot acetic
acid solution are very finely divided, and often d i f f i c u l t to separate from the f i l t r a t e , even in a high-speed centrifuge. Precipitates formed in the cold settle more readily,

37
TABLE I I I
RESULTS OF SOLUBILITY TESTS WITH SULPHATES
Sulphate Result of Refluxing with G l a c i a l Acetic Acid
Result of d i s s o l v i n g i n water and adding acetic anhydride.
. Mg Insoluble Quantitative p r e c i p i t a t i o n
Mn(II) Insoluble P r e c i p i t a t i o n almost quantitative
Cr Insoluble No p r e c i p i t a t i o n
Fe(III) Insoluble P a r t i a l p r e c i p i t a t i o n on addition of l a s t 5%"of acetic anhydride, but pre c i p i t a t e dissolves on b o i l i n g
Ca Insoluble Quantitative p r e c i p i t a t i o n
Co Insoluble P a r t i a l p r e c i p i t a t i o n <
Ni Insoluble P a r t i a l p r e c i p i t a t i o n
Sn Insoluble P r e c i p i t a t i o n almost quantitative.

but are not well c r y s t a l l i z e d and clog f i l t e r paper. Thus a
centrifuge should be used whenever possible.
The r e s u l t s obtained are l i s t e d i n Table IV. They
show that good precision and accuracy are obtainable with t h i s
method, often with appreciable saving of time. The complete
analysis of an a l l o y of cadmium and magnesium, f o r example,
requires approximately f o r t y - f i v e minutes, i f a standard per
c h l o r i c acid solution i s a v a i l a b l e . I f a rough estimation
of a binary a l l o y of copper or antimony with a t i t r a t a b l e
metal i s required, i t may be completed i n f i f t e e n minutes by
t i t r a t i n g the metal i n the presence of the copper or antimony.
E. Introduction of Metals Into Acetic Acid Solution
The r e s u l t s of the tests carried out with the amine
acetates, acetic acid, and the chloroacetic acids are sum
marized i n Tables V and VI. Ammonium acetate and p i p e r i d -
inium acetate were completely i n e f f e c t i v e i n attacking metals
other than calcium and magnesium, and these reactions were
very slow. For t h i s reason they are not included i n t h e % t a b l e
The r e s u l t s with chloroacetic acids show that even
elements below hydrogen i n the electromotive series (such as
copper and mercury) are attacked vigorously by these acids.
According to Doughty (18) the reaction between copper and
t r i c h l o r o a c e t i c acid i s not a simple one. A variety of com
pounds, depending on the temperature and concentration, are
produced, such as copper chloroacetate, copper dichloroacetate
chloroform and other products.

39
TABLE IV RESULTS OF TITRATING METALLIC MIXTURES
Mixture T h e o r e t i c a l % of Metal Determined
% Found
Zn-Mg Mg determined
27.11 2 7 . 0 0 26.95 26.98
Zn-Cu Zn determined
5 0 . 7 0 50.59 5 0 . 5 0 50.65
Ba-Ca Ca determined
22.58 22.56 22.75 22.69
Ag-Al Ag determined
7 9 . 9 9 79.86 79.91 79.81
Pb-Sn Pb determined
6 3 . 5 8 63.42 63.49 63.36
Mg-Sb Mg determined
I 6 . 6 5 1 6 . 4 8 16.53 16.57
Mg-Sb Mg determined i n presence of Sb
16.65 17.53 17.67 17.73
Mg-Cu Mg determined
27.67 27.38 27.46 2 7 . 5 5
Mg-Cu Mg determined i n presence of Cu
27.67 29.58 29.65 29.71
Mg-Pb Mg determined
10.50 10.57 1 0 . 4 0 10.48
Ni-Cr N i determined
5 3 . 03 52.85 52.89 52.93
Cd-Mg Mg determined
17.79 17.60 17.69 17 ".59
Sn-Mg Mg determined
1 7 .00 1 6 . 8 7 16.91 17.10

TABLE V. REACTIVITY OF AMINE ACETATES, ACETIC ACID CHLOROACETIC ACIDS TOWARDS METALS
AND THE
Metal Dicyclo- Pure Equimolar Equimolar Equimolar Pure Pure Pure hexyl- acetic soln. of soln. of soln. of chloro dichloro t r i c h l o r o ammonium acid chloroacetic dichloroacetic t r i c h l o r o a c e t i c acetic acetic acetic acetate i n acetic i n acetic i n acetic acid . acid acid
Ca ++ +++ +++ +++ +++ +++ +++ +++ Mg ++ +++ +++ +++ +++ +++ +++ +++ Th — + _ _ — — _
A l _ _ +++ _ + + + ++ U _ + +++ + ++ + ++ Mn + +++ +++ +++ +++ +++ +++ +++ Zn + ++ +++ +++ +++ +++ +++ +++ Cr _ ++ +++ +++ +++ ++ +++ Mo _ + + + ++ + + +++ Fe ++ * +++ • +++- ++.+ ++ +++ Cd mm +++ + + ++ —
Co + + + ++ ++ + + ++ Ni _ _ + — + —
Sn _ +++ +++ ++ +++ Pb +++ +++ + ++ +++ Sb + + + ++ +++ Bi - - — - — •. - + ++ As — _ - - - -Cu S o
+ ± - - ++ +++ u c Te _ _ — — - - -Ag _ - - - mm
Hg — . . - - - -W • _ — — — - - -S i - - - - - — —
LEGEND: +++ vigorous reaction + slow reaction - no apparent reaction ++ moderate reaction ± slight' reaction
O

41
TABLE VI
ACIDS TESTED WHICH ATTACK THE METALS CONSIDERED WITH SUFFICIENT RAPIDITY. TO BE OF PRACTICAL USE
i
Reagent Metals which dissolve r e a d i l y
Dicyclohexyl-ammonium acetate
None
Piperidinium acetate :None :
Ammonium acetate Ca
Acetic Acid Ca, Mg, Mn, Fe
Equimolar chloroacetic i n acetic Ca, Mg, A l , Mn, Zn //' Cr, Fe
Equimolar dichloroacetic i n .acetic Ca, Mg, U, Mn, Zn., .Cr,, Fe,
Cd, Sn, Pb // Co
Equimolar t r i c h l o r o a c e t i c i n acetic
Ca, Mg, U, Mn, Zn, Cr, Fe, Sn, Pb // Mo, Co, Hg
Pure chloroacetic Ca, Mg, Zn, Mn, Cr, Fe // U
Pure dichloroacetic Ca, Mg, Mn, Zn // Cr, Fe, Cd, 'Sh/ Pb, Sb * 1 : '
Pure t r i c h l o r o a c e t i c Ca, Mg, Mn, Zn, Cr, Mo, Fe, Sn, Pb, Sb, Cu // A l , U, Co, Bi
Those elements ; a f t e r the double stroke iri the table react much les s rapidly than those before i t .

1+2
Beside possible undesirable side reactions, another
disadvantage of these acids from the point of view of t i t
rations was the large quantity of acid often required to
dissolve the metal completely. Because of the l i m i t e d s o l
u b i l i t y of chloroacetic and t r i c h l o r o a c e t i c acids i n acetic
acid, the r e s u l t i n g mixture was often not completely s o l
uble i n acetic acid. Moreover, the table shows certain
common a l l o y s would not react r e a d i l y with a s i n g l e a c i d .
For these reasons the method of introducing metals into
acetic acid solution using n i t r i c acid and acetic anhydride
was preferred. Using t h i s method, a l l o y s corresponding to
the metal acetate mixtures discussed previously may r e a d i l y
be analysed i n a minimum of time and with good accuracy.
IV. CONCLUSIONS
Alloys, and mixtures of the l e s s soluble metal com
pounds, may be conveniently introduced into a c e t i c acid
solution using n i t r i c acid (or aqua regia) and acetic an
hydride. Quantitative separations may often be effected
i n t h i s medium using common reagents such as hydrogen s u l
phide and hydrogen iodide, n i t r i c , oxalic and t a r t a r i c acids.
The constituents may then be t i t r a t e d r a p i d l y and accurately
with a standard solution of perchloric a c i d i n acetic a c i d .
In spite of the interest shown i n a n a l y t i c a l methods
such as the one described, comparatively l i t t l e i s known
of the chemistry of acetic acid solutions. Much work needs

to be done on the determination of d i s s o c i a t i o n constants,
on s o l u b i l i t y r e l a t i o n s h i p s , and on the founding of a more
rigorous t h e o r e t i c a l approach to the question of a c i d i t y i n
non-aqueous solutions. With a clear insight into these
questions, t h i s type of analysis may prove to be of much
wider a p p l i c a b i l i t y than i s now imagined.
i i t

44
BIBLIOGRAPHY
1 . Blumrich, K. and Bandei, G. Agnew. Chem. 54 *• 3 7 4 . 1 9 4 1 .
2 . Bronsted, J. N. Rec. trav. chim. 42 : 7 1 8 . 1 9 2 3 .
3 . Bronsted, J. N. J. Phys. Chem. 30 : 7 7 7 . 1926.
4 . Bronsted, J. N. Chem. Rev. 5 : 2 8 4 . 1 9 2 8 .
5 . Bronsted, J. N. Ber. 6 l : 2 0 4 9 . 1 9 2 8 .
6 . Calcott, W. S., English, F. L., and Wilbur, 0 . G. Ind. Eng. Chem. 17 : 9 4 2 . 1 9 2 5 .
7 . Conant, J. B. and H a l l , N. F. J. Am. Chem. Soc. 49 3 0 6 2 . 1927.
8 . Conant, J. B. and Werner, T. H. J. Am. Chem. Soc. 52 :
4 4 3 6 . 1 9 3 0 .
9 . Davidson, A. W. J. Am. Chem. Soc. 50 : 1 8 9 0 . 1 9 2 8 .
1 0 . Davidson, A. W. Trans. Kans. Acad. S c i . 31 : 6 0 . 1 9 2 8 .
1 1 . Davidson, A. W. and Chappell, W. J . Am. Chem. Soc 55 -3 5 3 1 . 1 9 3 3 .
1 2 . Davidson, A. W. and Chappell, W. J. Am. Chem. Soc. 60 : 2 0 4 3 . 1938
1 3 . Davidson, A. W. and Geer, H. J. Am. Chem. Soc. 55 :
6 4 2 . 1 9 3 3 .
1 4 . Davidson, A. W. and Geer, H. J. Am. Chem. Soc. 60 : 1211. 1938.
1 5 . Davidson, A. W. and Griswold, E. J. Am. Chem. Soc. 53 : 1 3 4 1 . 1 9 3 1 .
16. Davidson, A. W. and Griswold, E. J. Am. Chem. Soc. 57 : 4 2 3 . 1 9 3 5 .
17. Davidson, A. W. and M c A l l i s t e r , W. H. J. Am. Chem. Soc. 52 : 5 0 7 . 1 9 3 0 .
IS. Doughty, H. W. and Freeman, B. J. Am. Chem. Soc. 44 • 636. 1922.
1 9 . F o l i n , 0 . and Flanders, F. F. J. Am. Chem. Soc. 34 7 7 4 . 1 9 1 2 .

45
2 0 .
2 1 .
2 2 .
2 3 .
24.
25 .
2 6 .
2 7 .
2 8 .
2 9 .
3 0 .
3 1 .
3 2 .
33;
34 .
3 5 .
3 6 .
3 7 .
F o l i n , 0 . and Flanders, F. F. 257. 1912.
F o l i n , 0 . and Wentworth, A. H. 421. 1909-10.
J . B i o l . Chem. 11 :
J. B i o l . Chem. 7
F r i t z , J. 3. ,Anal. Chem. 22 : 578. 1950.
F r i t z , J. S. Anal. Chem. 22 : 102$. 1950.
J. Am. Chem. Soc.
J. Am. Chem. Soc.
Ha l l , N. F. J. Am. Chem. Soc. 52 : 5115. 1930.
Griswold, E. and Olson, F. V. 59 : 1894. 1937.
Griswold, E. and van Horne, W. 67 : 763. ' 1945.
H a l l , N. F. and Conant, J. B. J. Am. Chem. Soc. 49 : 3 0 4 7 . 1927 .
H a l l , N.F. and Werner, T. H. J. Am. Chem. Soc. 50 : 2367 . 1928 .
Hammett, L. P. J. Am. Chem. Soc. 50 : 2666 . 1928 .
A. (a) Ber. 58 : 612. 1925. (b] Ber. 58 : 941." 1925. (c; Ber. 59 : 793. 1926. (d) Ber. 59 1096. 1926. (e! Ber. 60 : 1933. 1927. (f: Z. Electrochem. 29 : 221. 1923. (g) Z. Electrochem. 30 : 194. 1924. (hi Z. Electrochem. 31 : 167. 1925.
Z. phys. Chem. 125 : 251. 1927.
L. J . Am. Pharm. Assoc . 31 : 9 . 1942.
Higuchi, T. and Concha, J. J. Am. Pharm. Assoc. Sc. Ed, 40 : 173. 1951. '
Higuchi, T. and Concha, J. Science. 113 210. 1951.
Jander, G. Die Chemie i n Wasser&hnlichen LSsungmitteln. J. Springer. B e r l i n . 1949.
Kahane, E. B u l l . Soc. Chim. France. 18 : 9 2 . 1951.
Kastle, J. H. and Keiser, B. C. Am. Chem. J. 15 : 471. . 1893.
Kendall, J. C. and Gross, G. H. J. Am. Chem. Soc. 43 ': 1426. 1921.

46
3 8 . Kolb, J. J. and'Toennies, G. J. Biol.-Chem. 144 : 1 9 3 . 1 9 4 2 .
3 9 . Kolthoff, I. M. and Laitenen, H. A. pH and Elect r o T i t r a t i o n s . John Wiley and Sons. New York. 1 9 3 1 .
4 0 . Kolthoff, I. M. and Willman, A. J. Am. Chem. Soc. $6 : 1 0 1 4 . 1 9 3 4 .
4 1 . Levine, V. E. Science. 52 : 2 0 7 . 1 9 2 0 .
42. Lowry, T. M. Chem. and Ind. 42 : 4 3 . 1 9 2 3 .
4 3 . Lowry, T. M. Trans. Faraday S o c , 20 : 1 8 . 1 9 2 4 .
4 4 . Markunas, P. C. and Riddick, J. A. Anal. Chem. 23 : 3 3 7 . 1 9 5 1 .
4 5 . Markunas, P. C. and Riddick, J. A. Anal. Chem. 24 : 3 1 2 . 1 9 5 2 .
4 6 . Marsh, J. F. and Struthers, R. de J. F. J. Chem. Soc. 1927 : 2 6 5 8 .
4 7 . Nadeau, G. F..and Branchen, L. E. J.:Am. Chem. Soc. - 57 : 1 3 6 3 . 1 9 3 5 .
4 8 . Oppenheim, J. C. Proc. Soc. Chem. Ind. V i c t o r i a 45 : 6 4 7 . N 1 9 4 5 .
4 9 . Orton, K. J. and Bradfield, A. E. J. Chem. Soc. 1927 : 9 8 3 .
5 0 . P i f e r , C. W. and Wollish, E. G. Anal. Chem. 24 : 3 0 0 . 1 9 5 2 .
5 1 . P i f e r , C. W. and Wollish,.E. G. Anal. Chem. 24 : 5 1 9 . 1 9 5 2 .
5 2 . P i f e r , C. W..and Wollish, E. G. J. Am. Pharm. Assoc. 40 : 6 0 9 . 1 9 5 2 .
5 3 . P i f e r , C. W. and Wollish, E. G., and Schmall, M. Anal..Chem. 25 : 3 1 0 . 1 9 5 3 .
54. Roller, P. S. J. Am. Chem. Soc. 50
5 5 . Roller, P. i i . J. Am. Chem. Soc. 54
56. Roller, P. S. J. Am. Chem. Soc. 57
1 . 1 9 2 8 .
3 4 8 5 . 1 9 3 2 .
9 8 . 1 9 3 5 .
5 7 . Seaman, W. and Allen, E. Anal. Chem. 23 : 592'. 1 9 5 1 .

47
58. Sakami, W.-and Toennies, G. J. B i o l . Chem. 144 • " 203. 1942.
59. Toennies, G. and Callan, R. J . ' B i o l . Chem. 125 :
259. 1938. 60. Toennies, G. and Kolb, J. J. J. Biol."Chem. * 144
219. 1942.' 6 1 . Tomicek, 0. C o l l e c t i o n . Czechoslov. Chem. Communs.
13 : 116. 1948. 62. Treadwell, F. P. and H a l l , W. T. A n a l y t i c a l Chemistry,
Vol. 1.' John Wiley and Sons, New York. 1937.
63. Vogel, A. I; P r a c t i c a l Organic Chemistry. Longmans, Green & Company, New York. 1948. P. 175.
6 4 . Vogel, A. I. Quantitative Inorganic Analysis. Longmans, Green & Company, New York. 1948.
65. Weinland, R. Ber. 41 3236. 1908. • • • •
66. Weinland, R. and Dinkelacher, P. Ber. 42 : 2997. 1909.
67. Weinland, R. and Gussman, E. Ber. 42 : 3881. 1909.
68. Weinland, R.-and Gussman, E. Z. anorg. Chem. 66 :
157; 1910. 69. Weinlandj R;, Kessler, K., and Bayerl,. A. Z. anorg.
Chem.-132 : 209. 1924. 70; Weinland, R; and 0. Loebich. Z. anorg. allgem. Chem.
151 : 271. 1926. 71; Wittman,G.- Agnew. Chem. A60 : 330. "1948;-
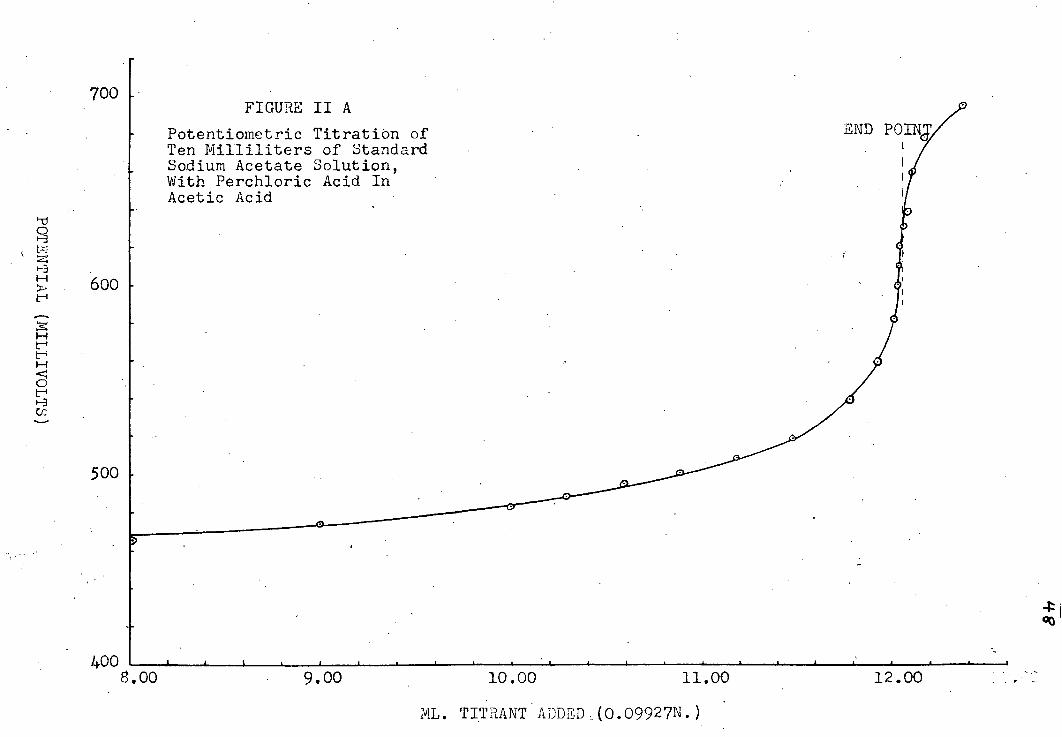
700 FIGURE I I A
Po t e n t i o m e t r i c T i t r a t i o n of Ten M i l l i l i t e r s of Standard Sodium Acetate S o l u t i o n , With P e r c h l o r i c Acid In Ac e t i c Acid
600
500
400 _i i i i i i _
8.00 9.00 10.00 11.00
ML. TITRANT'ADDED,(0.09927N.)
12.00
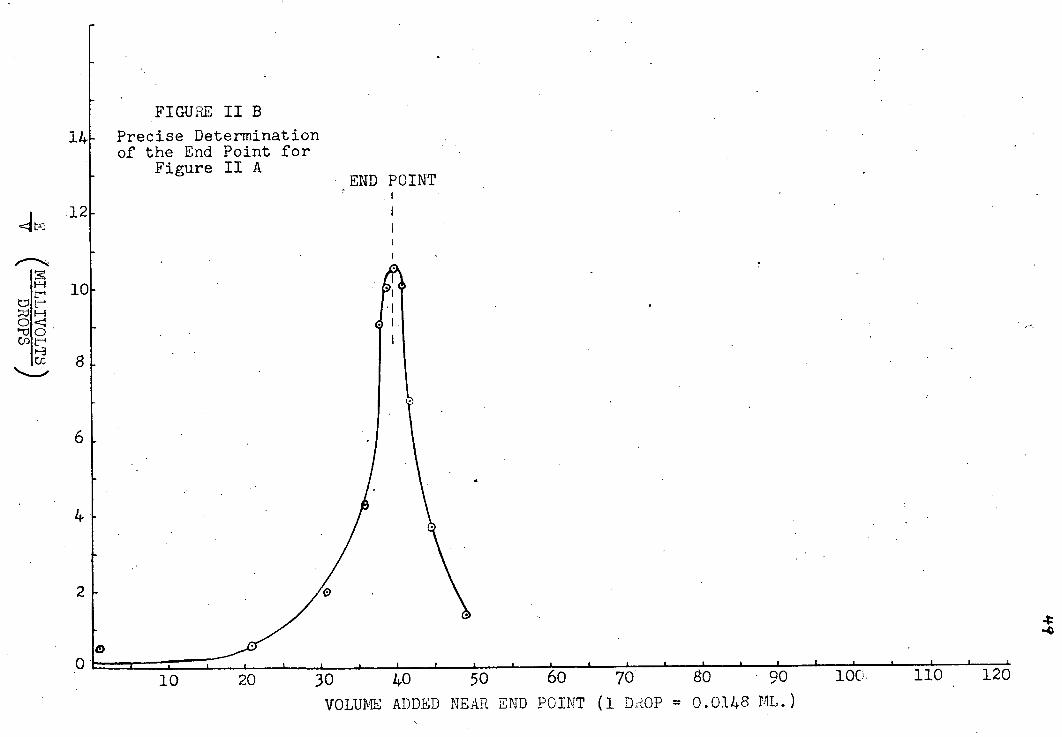
FIGURE II B
S HH L
L a
LL
o < O CO
•-3
1 4
12
10
8
0
Precise Determination of the End Point f o r
Figure II A END POINT
. i
• i i 1 1 1 1 1 30 40 50 60 70 VOLUME ADDED NEAR END POINT (1 DROP
10 20 80 90 0.0148 ML.)
100 110 120

FIGURE I I I A T i t r a t i o n (potentiometric) of Approximately Three M i l l i e q u i v a l e n t s of Barium Acetate With Perchloric Acid i n Acetic Acid
END POINT
600-
-i • * • • • 23.00 24.00 25.00 26 .00
ML. TITRANT ADDED (0.09927N.)
27.00

i i t- 1
C3 t-< ?G M O < •n C CO
i-5 cc
1 4
1 2
10
8
0
FIGURE I I I B Precise Determination of the End Point f o r
Figure I I I A
Gr
1 0
END POINT
20 3 0 40 50 6 0 7 0 80 9 0 100 1 1 0
VOLUME ADDED NEAR END POINT ( l DROP = 0.0148 ML.)

FIGURE IV A
700
600
500
P o t e n t i o m e t r i c T i t r a t i o n o f Approximately Three M i l l i -e q u i v a l e n t s of L i t h i u m and Strontium A c e t a t e s i n A c e t i c A c i d With P e r c h l o r i c A c i d .
Ammonium and Potassium Ac e t a t e s Each Y i e l d A S i m i l a r Curve. END POINT
END POINT
8
460 _ i i i i i i
23.00 24.00 25.00 26.00 27.00
ML. TITRANT A D D E D (0.09927N,)

FIGUREIV B 14 f- Precise Determination of
the End Point f o r Figure U A
12
2ND POINT
10
8
0 1
0 10 20. 30 40 50 60 70 80 90 100 • 110
VOLUME ADDED NEAR END POINT (1 DROP = 0.014^ ML.)
120
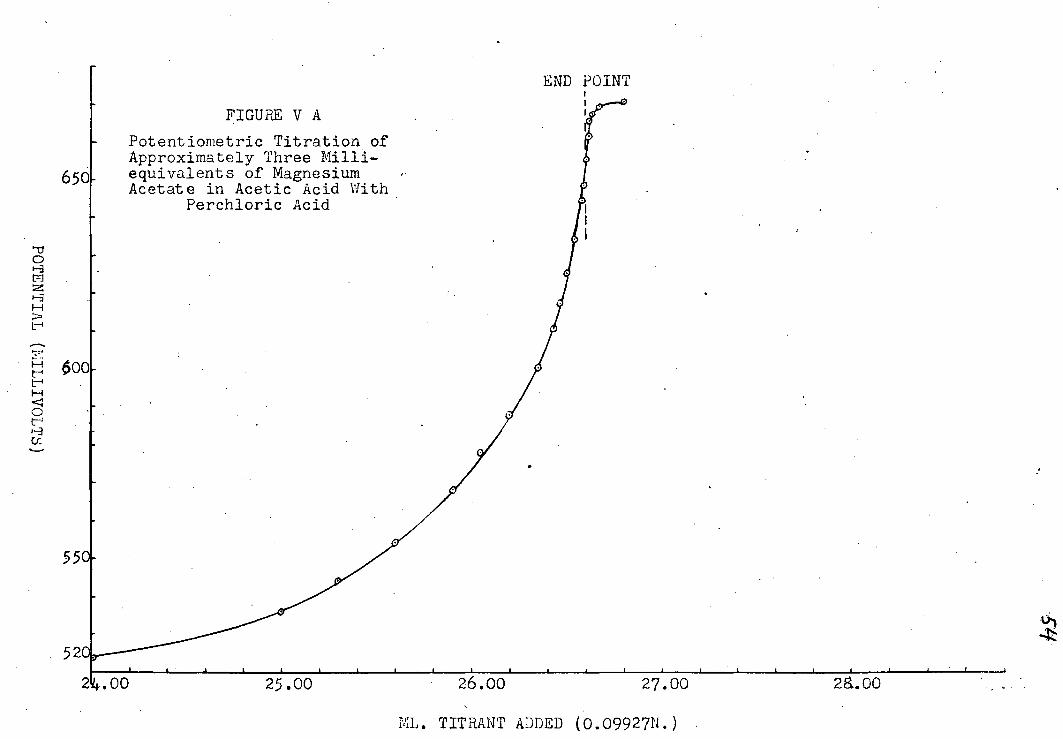
END POINT
650
FIGURE V A Potentiometric T i t r a t i o n of Approximately Three M i l l i equivalents of Magnesium Acetate i n Acetic Acid With
Perchloric Acid
$00
550-
520
2^.00 25.00 26.00 27.00 2&.00
ML. TITRANT ADDED ( 0 . 0 9 9 2 7 N . )

I6d FIGURE V B
Precise Determination of the End Point f o r Figure V A
4* 11A
§ 3
M
l-i < o
cc
DJ3
ao
40
20
0
E N D POINT
• • - i 1 i
0 10 20 30 40 50 60 70 80 90 100
V O L U M E A D D E D N E A R E N D P O I N T ( l D R O P = 0 . 0 1 4 8 M L . )
110 120

FIGURE VI A Potentiometric T i t r a t i o n of
• Approximately Three M i l l i - END POINT, equivalents of Calcium Acetate ^ 1
i n Acetic Acid With Perchloric ! Acid
I
1 1 ' ' 1 ' i i 1 1 1 1 i i i i i i • i •
2 3 . 0 0 . . 2 4 . 0 0 2 5 . 0 0
ML. TITRANT ADDED (0.09927N.)

I
8
21
11
FIGURE VI B P r e c i s e Determination of the End Point f o r
Figure VI A END POINT'
0 i i i i i i i i i i , i l i s 1
lio o 10 20 30 40 50 60 70 80 90 100
VOLUME ADDED NEAR END POINT (1 DROP = 0.0148 ML.)

• FIGURE VII A
ML. TITRANT ADDED (0 .100SN.)

FIGURE VII B 3.5
3 . 0
<|M 2 . 5
g P 2 . 0 '
ait-1 o 00
< o •-3 o: 1 . 5
1.0L
0 . 5
0 0
Precise Determination of the En<i Points f o r
Figure VII A
END POINT
i i
10 2 0 3 0 — J 40 ' 50 6 0 7 0 SO 90
VOLUME ADDED NE2|R END POINT ( 1 DROP = 0 . 0 1 0 9 M L . )
1 0 0
-0
_i i i — . — i u_ 1 1 1 0 1 2 0

ML. TITRANT ADDED (0.1031 N.)

END POINT i FIGURE VIII B
'5:'
0
Precise Determination of the.End Point f o r
Figure VIII A
-flu
0 10 20 30 40 50 60 70 90 100 110 120
VOLUME ADDED NEAR END POINT ( 1 DROP = 0 . 0 1 3 8 ML.)

END POINT FIGURE IX A i
i
3 0 . 0 0
ML. TITRANT ADDED ( 0 . 1 1 1 6 N)

F I G U R E IX B

700
FIGURE X A Potentiometric Determination of
2.00 > 3.00 4.00
ML. TITRANT ADDED (0.09927 N.)

FIGURE X B
M tr*
a t r 1
o O
Cr. tr* >-3 GO
0
Precise.Determination of the End Points f o r
Figure X A 1
END POINT I
END POINT i
0 1 0 20 3 0 40 50 60 7 0 8 0 9 0 1 0 0
VOLUME ADDED NEAR END POINT (1 DROP = 0.0148 ML.)

FIGURE XI Curves Obtained from the Attempted Potentiometric Determination of Copper ( I I ) , Antimony ( I I I ) , and Tin (II) Acetates i n Acetic Acid With Perchloric Acid. The Curve Obtained Upon Adding Perchloric Acid to Acetic Acid Alone i s Included f o r
Reference.
1.00 00 3.00 4.00
ML. TITRANT ADDED (0.09927 N.)

7 0 0 -
6 0 0
5 0 0 -
460L 0
F I G U R E
Curves Obtained From the Attempted Potentiometric Determination of Bismuth (III) and Uranyl Acetates i n Acetic Acid With Perchloric Acid. The Curve Obtained Upon Adding Perchloric Acid to Acetic Acid Alone i s Included f o r Reference..
1 . 0 0 2 . 0 0 3 . 0 0
l i L . TITRANT ADDED ( 0 . 0 9 9 2 7 N )
4 . 0 0

750 FIGURE XIII A
Potentiometric Determination of Approximately Three M i l l i e q u i v a l e n t s of Barium Chloroacetate i n Acetic Acid With Perchloric Acid, The Curves Obtained f o r the Other Chloroacetates Tested Were Similar to the Curves Obtained f o r the Corresponding Acetates
:ND POINT I
o
S 7 0 0
H
t-1
f M < O
IS.
650
6U0 26.00 27.00 2&\00
ML. rp T rp X J . RANT ADDED (0.09927 N)

<4K
M tr 1
a M
o <! •x; O CO t~<
>-3 CO
14
12
10
2
0 0
FIGURE X I I I B P r e c i s e D e t e r m i n a t i o n of
the End Poi n t for Fi g u r e 'XIII A
10
END POINT
I I L .
20 30 -V&0 50 60 70 80 90 100 VOLUME ADDED NEAR END POINT (0.0148 ML. = 1 DROP)
110




























