Embed Size (px)
Citation preview

Journal of Inorganic Biochemistry 89 (2002) 142–148www.elsevier.com/ locate / jinorgbio
Vanadate-induced cell growth arrest is p53-dependent through activation ofp21 in C141 cells
a,b a a a,b ,*Zhuo Zhang , Chuanshu Huang , Jinxia Li , Xianglin ShiaHealth Effects Laboratory Division, National Institute for Occupational Safety and Health, Morgantown, WV 26505, USA
bDepartment of Basic Pharmaceutical Sciences, West Virginia University, Morgantown, WV 26506, USA
Received 27 September 2001; received in revised form 5 November 2001; accepted 6 November 2001
Abstract
Vanadium is widely used in industry. It is a potent toxic agent and carcinogen. The mechanisms involved in its toxicity andcarcinogenesis are still unclear. Improper cell growth is believed to be involved in cancer development. The present study investigated theregulation of p53 on vanadate-induced cell growth arrest using both p53 wild type C141 cells and p53 deficient embryo fibroblasts (p532 /2). On vanadate stimulation, C141 cells exhibited a dose- and time-dependent S phase arrest as determined by DNA content analysis.In contrast, vanadate was unable to increase the percentage of S phase in p53 2 /2 cells. Luciferase assay showed that vanadate inducedp53 activation in a dose- and time-dependent manner in p53 wild type C141 cells. Addition of pifithrin-a (PFT), a specific inhibitor ofp53, reduced the activation of p53 with a concomitant decrease in growth arrest at S phase. Western blotting analysis demonstrated thatvanadate caused a dose- and time-dependent increase of p21 level in C141 cells. Pretreatment of C141 cells with PFT decreased p21expression induced by vanadate while the p21 expression did not vary in vanadate stimulated p53 2 /2 cells. The results obtained fromthe present study suggest that vanadate is able to induce S phase arrest through p53- and p21-dependent pathway. 2001 ElsevierScience Inc. All rights reserved.
Keywords: Vanadate; Cell cycle; p53; p21
1. Introduction membrane receptors [3]. However, other mechanisms areplausible. For example, as a pro-oxidant, vanadate causes
Vanadium exists in water, rocks, and soils in low DNA damage, DNA strand breaks, and cell transformationconcentrations, and in coal and oil deposits in relatively [4,5].high concentrations. Vanadium is widely used in mining, The ability of cells to maintain genomic integrity is vitalsteel and steel-alloy manufacture, and in the chemical for cell survival and proliferation. Lack of fidelity in DNAindustry. Epidemiological studies have shown a correlation replication and maintenance can result in deleteriousbetween vanadate exposure and the incidence of lung mutations leading to cell death or, in multicellular organ-cancer [1,2]. Workers occupationally exposed to vanadium isms, cancer [6]. Minor cell damage is repaired by aare at risk as respirable particulates may penetrate deep temporary pause of cell cycle. If the damage is severe,into the tracheobronchial tree.Vanadium mimics the effects cells will undergo apoptosis and enter into a dormant G0
of insulin, and stimulates or inhibits several enzymes in state. Signal transduction pathways play a key role in thevivo and in vitro. Many studies have focused on the regulation of cell cycle progression and stabilization ofmitogenic effects of vanadate, which is mediated by DNA under genotoxic stress. It is known that signalinhibiting the activity of tyrosine phosphatase and trig- transduction pathways control the activation of transcrip-gering the autophosphorylation of tyrosine kinase on cell tion factors and the regulation of gene expression as well
as a temporary pause of cell progression to allow thedamaged DNA to be repaired. The mechanisms involved
*Corresponding author. Present address: Pathology and Physiology in the regulations of vanadate on cell cycle control remainResearch Branch, Health Effects Laboratory Division, National Institute
unknown.for Occupational Safety and Health, 1095 Willowdale Road, Morgan-The p53 tumor suppressor is a multifunctional proteintown, WV 26505, USA. Tel.: 11-304-285-6158; fax: 11-304-285-5938.
E-mail address: [email protected] (X. Shi). that exerts a variety of different effects and plays a central
0162-0134/01/$ – see front matter 2001 Elsevier Science Inc. All rights reserved.PI I : S0162-0134( 01 )00409-3

Z. Zhang et al. / Journal of Inorganic Biochemistry 89 (2002) 142 –148 143
role in the regulation of the normal cell cycle. A number of p53, the cells were pre-incubated with PFT for 0.5 h priordifferent stimuli are known to activate p53 [7]. Over- to the vanadate treatment.expression of the p53 protein was found to induce cellgrowth arrest associated with G /G checkpoint [8–10]0 1 2.4. Measurement of cell cycle /DNA contentand to induce apoptosis that occurs either through theG /G checkpoint or the S phase [11,12], or cell differen-0 1 DNA content in S phase was analyzed using flowtiation. Overexpression of the p53 protein has also been cytometry according to the methods described previouslysuggested to be associated with G /M checkpoint regula-2 [18,19]. Cells were first fixed and permeabilized with 70%tion [13,14]. Furthermore, p21 (WAF-1/CIP1) is known to ice-cold ethanol for more than 2 h, followed by incubationbe a transactivation target of p53, and is believed to with the freshly prepared staining buffer (0.1% Tritonmediate p53-induced growth arrest triggered by DNA X-100 in PBS, 200 mg/ml RNase A, and 20 mg/ml PI) fordamage [15,16]. Induction of p21 expression could serve 30 min at room temperature. Cell cycle analysis wasas an indicator for transactivation by p53, although p21 performed by flow cytometry with at least 10,000 cells forwas also shown to be transactivated by p53-independent each sample. The DNA content histogram was abstractedmechanisms [17]. and the percentage of cells in S phase was then calculated
The purpose of the present study was to identify the using ModFit LT software.possible mechanisms of vanadate-induced cell growtharrest. The specific questions to be addressed were: (a) 2.5. Luciferase assay for p53 activityDoes vanadate induce cell growth arrest? (b) If yes, doesp53 play an important role? (b) What is the mechanism 4After C141 p53 cells were 80–90% confluent, 1310involved in p53 regulated cell growth arrest? cells were added into 96-well plate with 100 ml medium in
each well. The cells were subjected to different treatmentsaccording to the experimental design. At various time
2. Materials and methods points, the cells were extracted with lysis buffer. The p53luciferase activity was measured using a luminometer
2.1. Reagents (moonlight 3010). The results were expressed as p53activity relative to the control.
Sodium metavanadate was from Aldrich (Milwaukee,WI). RNase A, DMEM and EMEM medium were from
2.6. Western blot analysisSigma (St. Louis, MO). Propidium iodide (PI) was fromMolecular Probes (Eugene, OR). Fetal bovine serum
Whole cell extracts were mixed with Tris-Glycine SDS(FBS) was purchased from Gibco BRL (Life Tech-sample buffer and then subjected to Tris-Glycine gelnologies, Gaithersburg, MD). Pifithrin-a (PFT) was pur-electrophoresis. The resolved proteins were transferred to achased from Alexis (San Diego, LA). Antibody to p21 wasPVDF membrane. A Western blot assay was performedfrom Santa Cruz Biotechnology (Santa Cruz, CA). Sec-using antibodies against p21 and secondary anti-rabbitondary AP linked anti-rabbit IgG was from Cell SignalingIgG. After reaction with ECF substrate, the signal was(Beverly, MA).detected using a Storm Scanner (Molecular Dynamics,Sunnyvale, CA).2.2. Cell culture
1 2.7. Statistical analysisThe JB6 P mouse epidermal cell line, C141 and itsstable p53 luciferase reporter plasmid transfectant, C141
All data were based on at least three independentp53 cells, were cultured in MEM medium containing 5%experiments. Cell growth arrest and relative p53 activityFBS, 2 mM L-glutamine and 25 mg/ml gentamicin in andata were presented as means6S.D. and analyzed usingincubator at 5% CO and 37 8C. p53-deficient embryo2one-way ANOVA with Scheffe’s test. A p-value less thanfibroblasts (p53 2 /2) derived from p53 gene knockout0.05 was considered statistically significant.mice were incubated in DMEM medium with 10% FBS, 2
mM L-glutamine and 25 mg/ml gentamicin at 5% CO and2
37 8C.3. Results
2.3. Treatments3.1. The effects of vanadate in cell cycle in C141 cells
For the time-course study, the cells were treated with 50 and p53 2 /2 cellsmM vanadate for 6, 12, 24, and 48 h. For the dose–response study, the cells were treated with 10, 25, 50, and To study vanadate-induced cell growth arrest, DNA100 mM vanadate for 24 h. For the inhibitory studies of content was used to measure the percentages of different
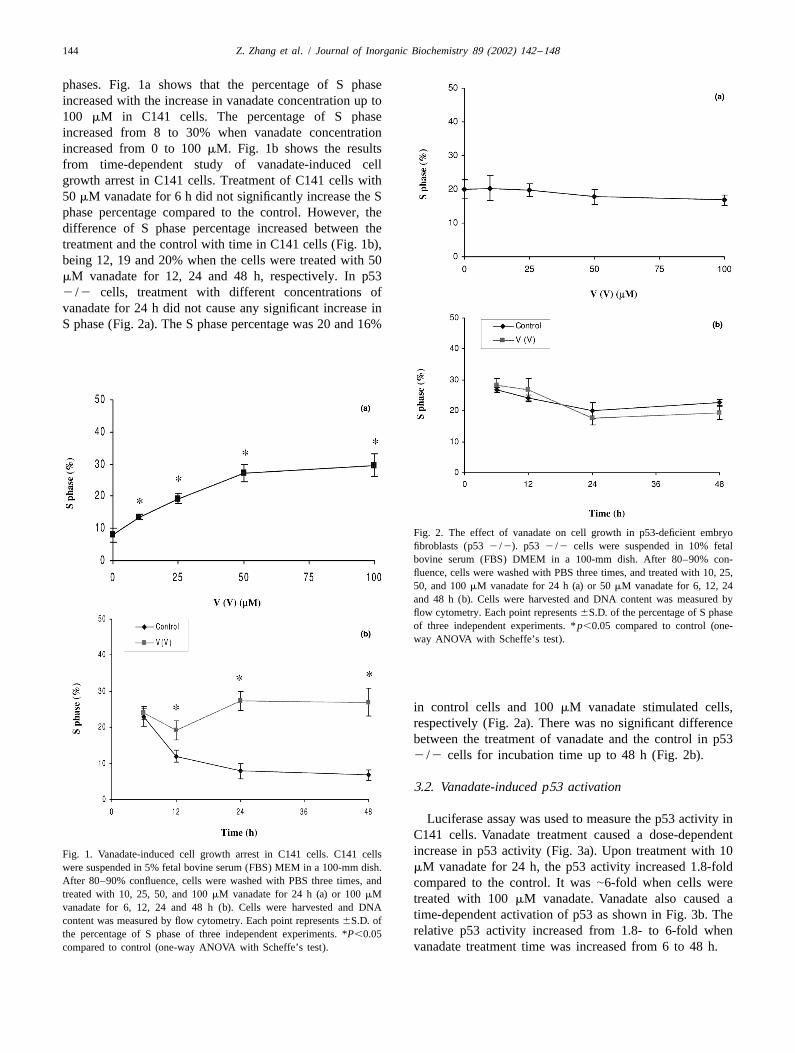
144 Z. Zhang et al. / Journal of Inorganic Biochemistry 89 (2002) 142 –148
phases. Fig. 1a shows that the percentage of S phaseincreased with the increase in vanadate concentration up to100 mM in C141 cells. The percentage of S phaseincreased from 8 to 30% when vanadate concentrationincreased from 0 to 100 mM. Fig. 1b shows the resultsfrom time-dependent study of vanadate-induced cellgrowth arrest in C141 cells. Treatment of C141 cells with50 mM vanadate for 6 h did not significantly increase the Sphase percentage compared to the control. However, thedifference of S phase percentage increased between thetreatment and the control with time in C141 cells (Fig. 1b),being 12, 19 and 20% when the cells were treated with 50mM vanadate for 12, 24 and 48 h, respectively. In p532 /2 cells, treatment with different concentrations ofvanadate for 24 h did not cause any significant increase inS phase (Fig. 2a). The S phase percentage was 20 and 16%
Fig. 2. The effect of vanadate on cell growth in p53-deficient embryofibroblasts (p53 2 /2). p53 2 /2 cells were suspended in 10% fetalbovine serum (FBS) DMEM in a 100-mm dish. After 80–90% con-fluence, cells were washed with PBS three times, and treated with 10, 25,50, and 100 mM vanadate for 24 h (a) or 50 mM vanadate for 6, 12, 24and 48 h (b). Cells were harvested and DNA content was measured byflow cytometry. Each point represents 6S.D. of the percentage of S phaseof three independent experiments. * p,0.05 compared to control (one-way ANOVA with Scheffe’s test).
in control cells and 100 mM vanadate stimulated cells,respectively (Fig. 2a). There was no significant differencebetween the treatment of vanadate and the control in p532 /2 cells for incubation time up to 48 h (Fig. 2b).
3.2. Vanadate-induced p53 activation
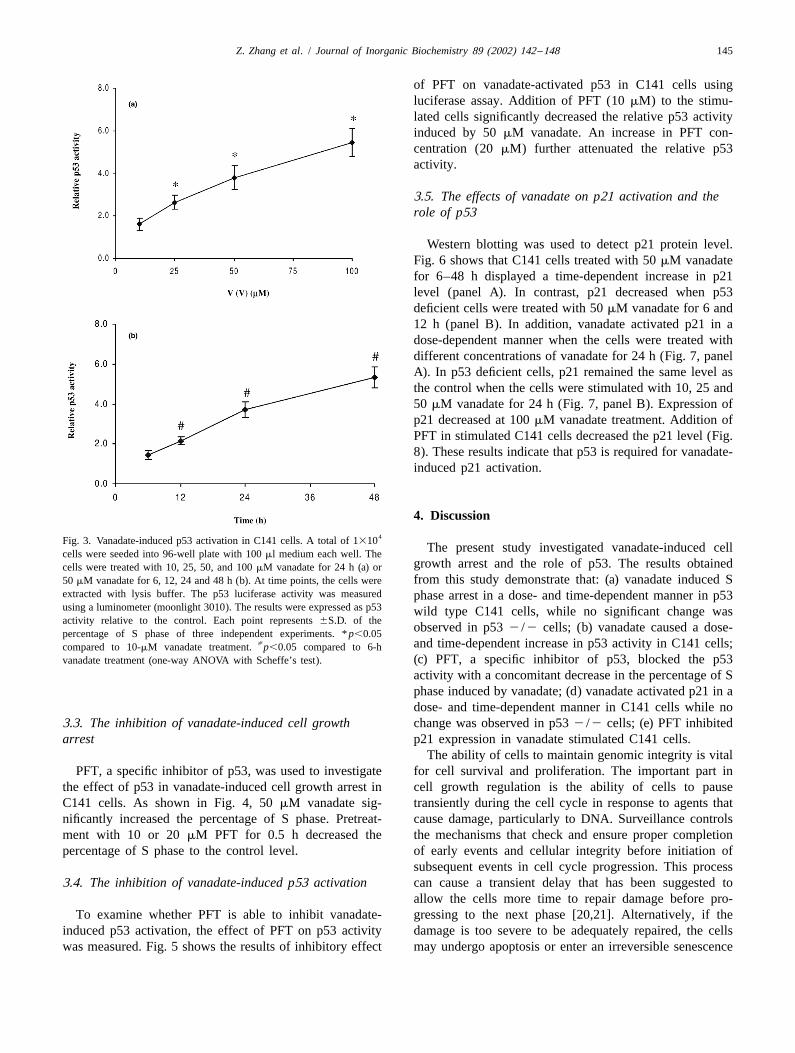
Luciferase assay was used to measure the p53 activity inC141 cells. Vanadate treatment caused a dose-dependentincrease in p53 activity (Fig. 3a). Upon treatment with 10Fig. 1. Vanadate-induced cell growth arrest in C141 cells. C141 cells
were suspended in 5% fetal bovine serum (FBS) MEM in a 100-mm dish. mM vanadate for 24 h, the p53 activity increased 1.8-foldAfter 80–90% confluence, cells were washed with PBS three times, and compared to the control. It was |6-fold when cells weretreated with 10, 25, 50, and 100 mM vanadate for 24 h (a) or 100 mM treated with 100 mM vanadate. Vanadate also caused avanadate for 6, 12, 24 and 48 h (b). Cells were harvested and DNA
time-dependent activation of p53 as shown in Fig. 3b. Thecontent was measured by flow cytometry. Each point represents 6S.D. ofrelative p53 activity increased from 1.8- to 6-fold whenthe percentage of S phase of three independent experiments. *P,0.05
compared to control (one-way ANOVA with Scheffe’s test). vanadate treatment time was increased from 6 to 48 h.
Z. Zhang et al. / Journal of Inorganic Biochemistry 89 (2002) 142 –148 145
of PFT on vanadate-activated p53 in C141 cells usingluciferase assay. Addition of PFT (10 mM) to the stimu-lated cells significantly decreased the relative p53 activityinduced by 50 mM vanadate. An increase in PFT con-centration (20 mM) further attenuated the relative p53activity.
3.5. The effects of vanadate on p21 activation and therole of p53
Western blotting was used to detect p21 protein level.Fig. 6 shows that C141 cells treated with 50 mM vanadatefor 6–48 h displayed a time-dependent increase in p21level (panel A). In contrast, p21 decreased when p53deficient cells were treated with 50 mM vanadate for 6 and12 h (panel B). In addition, vanadate activated p21 in adose-dependent manner when the cells were treated withdifferent concentrations of vanadate for 24 h (Fig. 7, panelA). In p53 deficient cells, p21 remained the same level asthe control when the cells were stimulated with 10, 25 and50 mM vanadate for 24 h (Fig. 7, panel B). Expression ofp21 decreased at 100 mM vanadate treatment. Addition ofPFT in stimulated C141 cells decreased the p21 level (Fig.8). These results indicate that p53 is required for vanadate-induced p21 activation.
4. Discussion4Fig. 3. Vanadate-induced p53 activation in C141 cells. A total of 1310
The present study investigated vanadate-induced cellcells were seeded into 96-well plate with 100 ml medium each well. Thegrowth arrest and the role of p53. The results obtainedcells were treated with 10, 25, 50, and 100 mM vanadate for 24 h (a) orfrom this study demonstrate that: (a) vanadate induced S50 mM vanadate for 6, 12, 24 and 48 h (b). At time points, the cells were
extracted with lysis buffer. The p53 luciferase activity was measured phase arrest in a dose- and time-dependent manner in p53using a luminometer (moonlight 3010). The results were expressed as p53 wild type C141 cells, while no significant change wasactivity relative to the control. Each point represents 6S.D. of the
observed in p53 2 /2 cells; (b) vanadate caused a dose-percentage of S phase of three independent experiments. * p,0.05[ and time-dependent increase in p53 activity in C141 cells;compared to 10-mM vanadate treatment. p,0.05 compared to 6-h
(c) PFT, a specific inhibitor of p53, blocked the p53vanadate treatment (one-way ANOVA with Scheffe’s test).
activity with a concomitant decrease in the percentage of Sphase induced by vanadate; (d) vanadate activated p21 in adose- and time-dependent manner in C141 cells while no
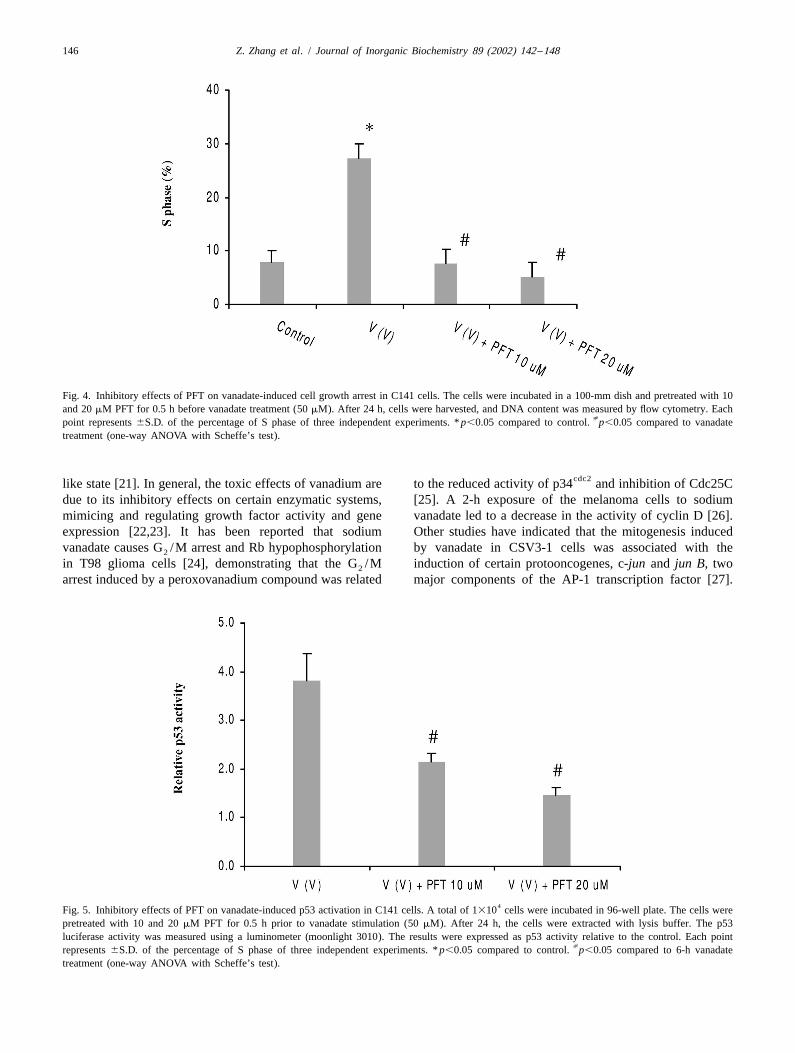
3.3. The inhibition of vanadate-induced cell growth change was observed in p53 2 /2 cells; (e) PFT inhibitedarrest p21 expression in vanadate stimulated C141 cells.
The ability of cells to maintain genomic integrity is vitalPFT, a specific inhibitor of p53, was used to investigate for cell survival and proliferation. The important part in
the effect of p53 in vanadate-induced cell growth arrest in cell growth regulation is the ability of cells to pauseC141 cells. As shown in Fig. 4, 50 mM vanadate sig- transiently during the cell cycle in response to agents thatnificantly increased the percentage of S phase. Pretreat- cause damage, particularly to DNA. Surveillance controlsment with 10 or 20 mM PFT for 0.5 h decreased the the mechanisms that check and ensure proper completionpercentage of S phase to the control level. of early events and cellular integrity before initiation of
subsequent events in cell cycle progression. This process3.4. The inhibition of vanadate-induced p53 activation can cause a transient delay that has been suggested to
allow the cells more time to repair damage before pro-To examine whether PFT is able to inhibit vanadate- gressing to the next phase [20,21]. Alternatively, if the
induced p53 activation, the effect of PFT on p53 activity damage is too severe to be adequately repaired, the cellswas measured. Fig. 5 shows the results of inhibitory effect may undergo apoptosis or enter an irreversible senescence
146 Z. Zhang et al. / Journal of Inorganic Biochemistry 89 (2002) 142 –148
Fig. 4. Inhibitory effects of PFT on vanadate-induced cell growth arrest in C141 cells. The cells were incubated in a 100-mm dish and pretreated with 10and 20 mM PFT for 0.5 h before vanadate treatment (50 mM). After 24 h, cells were harvested, and DNA content was measured by flow cytometry. Each
[point represents 6S.D. of the percentage of S phase of three independent experiments. * p,0.05 compared to control. p,0.05 compared to vanadatetreatment (one-way ANOVA with Scheffe’s test).
cdc2like state [21]. In general, the toxic effects of vanadium are to the reduced activity of p34 and inhibition of Cdc25Cdue to its inhibitory effects on certain enzymatic systems, [25]. A 2-h exposure of the melanoma cells to sodiummimicing and regulating growth factor activity and gene vanadate led to a decrease in the activity of cyclin D [26].expression [22,23]. It has been reported that sodium Other studies have indicated that the mitogenesis inducedvanadate causes G /M arrest and Rb hypophosphorylation by vanadate in CSV3-1 cells was associated with the2
in T98 glioma cells [24], demonstrating that the G /M induction of certain protooncogenes, c-jun and jun B, two2
arrest induced by a peroxovanadium compound was related major components of the AP-1 transcription factor [27].
4Fig. 5. Inhibitory effects of PFT on vanadate-induced p53 activation in C141 cells. A total of 1310 cells were incubated in 96-well plate. The cells werepretreated with 10 and 20 mM PFT for 0.5 h prior to vanadate stimulation (50 mM). After 24 h, the cells were extracted with lysis buffer. The p53luciferase activity was measured using a luminometer (moonlight 3010). The results were expressed as p53 activity relative to the control. Each point
[represents 6S.D. of the percentage of S phase of three independent experiments. * p,0.05 compared to control. p,0.05 compared to 6-h vanadatetreatment (one-way ANOVA with Scheffe’s test).

Z. Zhang et al. / Journal of Inorganic Biochemistry 89 (2002) 142 –148 147
G /G checkpoint, wild type p53 was associated with0 1
DNA repair activity, and this association prevented theentry into S phase [30,31]. At G checkpoint, p53 func-2
tions prior to mitosis and takes part in the spindlecheckpoint associated with mitosis [32]. p53 is importantfor securing the stability and integrity of the genome ofnormal cells and was suggested to be the ‘guardian of thegenome’ [33]. A variety of DNA insults were shown tostabilize the protein, which in turn can either cause growtharrest, permit the induction of the DNA repair process, orFig. 6. Time dependence of vanadate on p21 level in C141 cells and p53alternatively, direct cells to undergo apoptosis [34]. p532 /2 cells. Both types of cells were incubated in 6-well plate. The cells
were treated with 50 mM vanadate for different times. The whole cell was suggested to be a sensor of damaged DNA, and maylysates were collected for Western blotting using specific antibodies to be involved in the repair process [35]. It has been shownp21. Panel A represents C141 cells and panel B represents p53 2 /2 that cells containing wild type p53 alleles undergo G1cells. Lane 1, control; lane 2, 6 h; lane 3, 12 h; lane 4, 24 h; lane 5, 48 h.
arrest in response to g-radiation, whereas cells lackingThe results are representative of three separate experiments.functional p53 alleles enter S phase regardless of dose ofg-radiation [36]. It has been demonstrated that vanadate-mediated generation of reactive oxygen species (ROS)plays an important role in various adverse biologicaleffects induced by this metal [37,38]. Moreover, ROS arebelieved to be capable of damaging DNA [39]. Our earlierstudy showed that vanadate-induced apoptosis was p53dependent which was mediated by H O [40]. Our previ-2 2
ous study also suggested that ROS are mediators ofvanadate-induced cell growth arrest [28]. The results fromthe present study show that vanadate not only induces Sphase arrest in C141 cells, but also causes an increase in
Fig. 7. Dose–response study of vanadate on p21 level in C141 cells andp53 activity. Our study also showed that vanadate caused Sp53 2 /2 cells. The method used is the same as in Fig. 6. The cells werephase arrest in p53 1 /1 cells derived from mouse embryotreated with different concentrations of vanadate for 24 h. Panel A
represents C141 cells and panel B represents p53 2 /2 cells. Lane 1, fibroblasts similarly to that observed in C141 cells (un-control; lane 2, 10 mM vanadate; lane 3, 25 mM vanadate; lane 4 50 mM published observations). Due to lack of p53 in p53 2 /2vanadate; lane 5, 100 mM vanadate. The results are representative of three cells, vanadate failed to induce growth arrest at S phase.separate experiments.
Although the mechanism involved in the vanadate-inducedcell growth arrest remains to be investigated, it is knownOur earlier study demonstrated that vanadate induced G /2 that ROS generated by vanadate-mediated reactions causeM phase arrest in a dose- and time-dependent manner inDNA damage. This DNA damage may activate signalhuman epithelial cell line, A549 [28]. In the present study,transduction pathways, such as mitogen-activated proteinthe results showed that vanadate caused S phase arrest inkinase family, leading to an increase in p53 protein levelmouse epidermal C141 cells. The difference in phase arrestand its phosphorylation. PFT, an inhibitor of p53, mainlyinduced by vanadate may be cell line specific.inhibits p53 function [41]. The results obtained from theThe p53 tumor suppressor is a multifunctional proteinpresent study show that addition of 10 mM PFT decreasedthat exerts a variety of different effects and plays a centralthe p53 activity by 42% compared to the stimulated C141role in the regulation of the normal cell cycle [14,29].cells (treated with 50 mM vanadate for 24 h). TheOverexpression of this protein was found to induce growthinhibition was stronger when the concentration of PFT wasarrest associated with the G /G checkpoint [8–10]. At0 1 increased to 20 mM, being |60%. More importantly,pretreatment with PFT significantly decreased the per-centage of S phase induced by vanadate. The percentage ofS phase in stimulated cells was 27%. It was 8 or 5% afteraddition of PFT 10 or 20 mM, respectively. There was nosignificant difference in S phase between the PFT pre-treated, vanadate-stimulated cells and the control cells (S
Fig. 8. The inhibitory effects of PFT on p21 expression in C141 cells. phase, 8%). p21 (WAF-1/CIP1) is known to be a transacti-The cells were pretreated with 10 and 20 mM PFT for 0.5 h prior to vation target of p53, and has been suggested to mediatevanadate treatment (50 mM, 24 h). Western blotting was used to examine
p53-induced growth arrest triggered by DNA damage [42].the p21 level. Lane 1, control; lane 2, 50 mM vanadate for 24 h; lane 3,Induction of p21 expression could serve as an indicator for50 mM vanadate110 mM PFT for 24 h; lane 4, 50 mM vanadate120 mM
PFT for 24 h. The results are representative of three separate experiments. transactivation by p53, although p53-independent mecha-

148 Z. Zhang et al. / Journal of Inorganic Biochemistry 89 (2002) 142 –148
[12] M. Oren, Semin. Cancer Biol. 5 (1994) 221–227.nisms were also observed [14,29]. Induction of p21 may[13] R. Aloni-Grinstein, D. Schwartz, V. Rotter, EMBO J. 14 (1995)inhibit cell progression in two ways: (a) by inhibiting a
1392–1401.variety of cyclin /cdk complexes and (b) by inhibiting [14] V. Rotter, R. Aloni-Grinstein, D. Schwartz, N.B. Elkins, A. Simons,DNA synthesis through proliferation cell nuclear antigen R. Wolkowicz, M. Lavigne, P. Besserman, A. Kapon, N. Goldfinger,(PCNA) binding [43]. It is known that cyclin A accumu- Semin. Cancer Biol. 5 (1994) 229–236.
[15] W.S. EI-Deiry, T. Tokino, V.E. Velculescu, D.B. Levy, R. Parsons,lates at the G /S phase transition and persists through S1J.M. Tren, D. Lin, W.E. Mercer, K.W. Kinzler, B. Vogelstein, Cell 75phase. Cyclin A initially associates with cdk2 and then, in(1993) 817–825.
late S phase, associates with cdk1. Cyclin A-associated [16] J. Wade-Harper, G.R. Adami, N. Wei, K. Keyomarsi, S.J. Elledge,kinase activity is required for entry into S phase, comple- Cell 75 (1993) 805–816.tion of S phase, and entry into M phase [44–46]. The p21 [17] P. Michieli, M. Chedid, D. Lin, J.H. Pierce, W.E. Mercer, D. Givol,
Cancer Res. 54 (1994) 3391–3395.protein inhibits the activity of cyclin A/cdk2, which in[18] I. Nicoletti, G. Migliorati, M.C. Pagliacci, F. Grignani, C. Riccardi,turn phosphorylates the E2F heterodimerization DP1,
J. Immunol. Methods 139 (1991) 271–279.resulting in an inhibition of E2F DNA-binding activity [19] R. Sgonic, G. Wick, Int. Arch. Allergy Immunol. 105 (1994)[43]. The present study shows that vanadate indeed is able 327–332.to cause a dose- and time-dependent increase in p21 [20] L.H. Hartwell, T.A. Weinert, Science 246 (1989) 629–633.
[21] M.B. Kastan (Ed.), Checkpoint Controls and Cancer, Cold Springprotein level in C141 cells. In contrast, it was unable toHarbor Laboratory Press, Plainview, NY, 1997.activate p21 in p53 2 /2 cells regardless of dose or time
[22] T.F. Cruz, A. Morgan, W. Min, Mol. Cell. Biochem. 153 (1995)of vanadate treatment. Furthermore, addition of PFT 161–166.significantly decreased p21 protein level activated by [23] G. Swarup, S. Cohen, D.L. Garbers, Biochem. Biophys. Res.vanadate, indicating that vanadate-induced activation of Commun. 107 (1982) 1104–1109.
[24] L.S. Chin, S.F. Murray, D.H. Harter, P.F. Doherty, S.K. Singh, J.p21 is through p53-dependent pathway. Our preliminaryBiomed. Sci. 6 (1999) 213–218.data also showed that during p21 activation in p53 wild
[25] R. Faure, M. Vincent, M. Dufour, A. Shaver, B.I. Posner, J. Cell.type cells vanadate reduced both cyclin A and cdk2 Biochem. 58 (1995) 389–401.expression. This observation suggests that both activated [26] R.M. Strasberg, M. Rieber, Biochem. Biophys. Res. Commun. 216p21 binding to and inhibition of cdk2 are involved in (1995) 422–427.
[27] H. Wang, Z. Xie, R.E. Scott, Mol. Cell. Biochem. 168 (1997)vanadate-induced S phase arrest.21–30.In conclusion, the results obtained from the present
[28] Z. Zhang, C. Huang, J. Li, S.S. Leonard, R. Lanciotti, L. Butter-study demonstrate that vanadate is capable of inducing S worth, X. Shi, Arch. Biochem. Biophy. 392 (2001) 311–320.phase arrest in C141 cells that contain wild type p53 [29] N. Almog, V. Rotter, Biochim. Biophy. Acta 1333 (1997) F1–F27.alleles, and this interruption of cell cycle is p53-dependent. [30] X. Lu, D.P. Lane, Cell 75 (1993) 7491–7495.
[31] W.G. Nelson, M.B. Kastan, Mol. Cell. Biol. 14 (1994) 1815–1823.Activation of p53 leads to an increase in p21 expression,[32] S.M. Cross, C.A. Sanchez, C.A. Morgan, M.K. Schimke, S. Ramel,resulting in a pause in growth progression at S phase.
R.L. Idzerda, W.H. Raskind, B.J. Reid, Science 267 (1995) 1353–Thus, vanadate-induced S phase arrest is mediated by p53 1356.through activation of p21. [33] D.P. Lane, Nature 358 (1992) 15–16.
[34] L.R. Livingstone, A. White, J. Sprouse, E. Livanos, T. Jacks, T.D.Tlsy, Cell 70 (1992) 923–935.
[35] Y. Sanchez, J.S. Elledge, Bioassays 17 (1995) 545–548.References[36] L.A. Di, S.P. Linke, K. Clarkin, G.M. Wahl, Genes Dev. 8 (1994)
2540–2551.[1] P. Stock, Br. J. Cancer 14 (2000) 397–418. [37] J. Ye, M. Ding, S.S. Leonard, V.A. Robinson, L. Millecchia, X.[2] R.J. Hickey, E.P. Schoff, R.C. Clelland, Arch. Environ. Health 15 Zhang,V. Castranova,V.Vallyathan, X. Shi, Mol. Cell. Biochem. 202
(1967) 728–738. (1999) 9–17.[3] X. Yin, A.J. Davison, S.S. Tsang, Mol. Cell. Biochem. 115 (1992) [38] X. Shi, N.S. Dalal, Free Radic. Res. Commun. 17 (1992) 369–376.
85–96. [39] C.M. Payne, C. Bernsterin, H. Bersterin, Leuk. Lymphoma 19[4] X. Shi, H. Jiang, Y. Mao, J. Ye, U. Saffiotti, Toxicology 106 (1996) (1995) 43–93.
27–38. [40] C. Huang, Z. Zhang, M. Ding, J. Li, J. Ye, S.S. Leonard, H. Shen, Y.[5] A. Stern, X. Yin, S.S. Tsang, A.J. Davison, J. Moon, Biochem. Cell. Lu, V. Castranova, V. Vallyathan, X. Shi, J. Biol. Chem. 275 (2000)
Biol. 71 (1993) 104–112. 32516–32522.[6] R.E. Shackelford, W.K. Kaufmann, R.S. Paules, Environ. Health [41] E.A. Komarova, A.V. Gudkov, Biochemistry 65 (2000) 41–48.
Persp. 107 (1999) 5–24. [42] T. Hunter, Cell 75 (1993) 839–841.[7] J.L. Ko, C. Prives, Genes Dev. 10 (1996) 1054–1072. [43] D.G. Johnson, C.L. Walker, Annu. Rev. Pharmacol. Toxicol. 39[8] D. Michalovitz, O. Halevy, M. Oren, Cell 62 (1990) 671–680. (1999) 295–312.[9] L. Diller, J. Kassel, C.E. Nelson, N.A. Gryka, G. Litwak, M. [44] F. Girard, U. Strausfeld, A. Fernandez, N.J.C. Lamb, Cell 67 (1991)
Gebhardt, B. Bressac, M. Ozturk, S.J. Baker, B. Vogelstein, S.H. 1169–1179.Friend, Mol. Cell. Biol. 10 (1990) 5772–5781. [45] C.F. Lehner, P.H. O’Farrell, Cell 56 (1989) 957–968.
[10] G. Shaulsky, N. Goldfinger, A. Peled, V. Rotter, Proc. Natl. Acad. [46] D.H. Walker, J.L. Maller, Nature 354 (1991) 314–317.Sci. USA 88 (1991) 8982–8986.
[11] M. Yonish-Rouach, D. Grunwald, S. Wilder, A. Kimchi, E. May, J.J.Lawrence, P. May, M. Oren, Mol. Cell. Biol. 13 (1993) 1415–1423.





























