Embed Size (px)
Citation preview

UvA-DARE is a service provided by the library of the University of Amsterdam (http://dare.uva.nl)
UvA-DARE (Digital Academic Repository)
Developing novel strategies for treating esophageal cancer: Dendritic cells immunotherapy, Tcell responses and an exploration of the tumor microenvironmentMilano, F.
Link to publication
Citation for published version (APA):Milano, F. (2007). Developing novel strategies for treating esophageal cancer: Dendritic cells immunotherapy, Tcell responses and an exploration of the tumor microenvironment.
General rightsIt is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s),other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons).
Disclaimer/Complaints regulationsIf you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, statingyour reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Askthe Library: http://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam,The Netherlands. You will be contacted as soon as possible.
Download date: 07 May 2019

Developing novel strategies for treating
esophageal cancer: Dendritic Cells immunotherapy,
T cells responses and an exploration
of the tumor microenvironment.
Francesca Milano
Develop
ing n
ovel strategies for treating esop
hageal can
cer Fran
cesca Milan
o

Developing novel strategies for treating
esophageal cancer: Dendritic Cells immunotherapy,
T cell responses and an exploration
of the tumor microenvironment.
Milano_opm_v5.indd 1 16-08-2007 15:42:10

Developing novel strategies for treating esophageal cancer: Dendritic Cells immunotherapy, T cells responses and an exploration of the tumor microenvironment. By Francesca Milano, Thesis university of Amsterdam with references with summary in Dutch.
© 2007 Francesca Milano, Amsterdam, The NetherlandsNo part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without written permission of the author.
Layout by M. Stokking and N. Buitendijk, Ponsen & Looijen b.v., Wageningen
Printed by Ponsen & Looijen b.v., Wageningen.
ISBN: 9789064641596
The study described in this thesis was supported by Astra Zeneca, J.E. Juriaanse Stichting, Novartis, Abbott, Boston Scientific, Stichting Nationaal Fonds Tegen Kanker voor onderzoek naar reguliere en alternatieve therapieën, BD Biosciences, University of Amsterdam en de Nederlandse Vereniging voor Gastroenterologie (NVGE, sectie experimentele Gastroenterologie).
Milano_opm_v5.indd 2 16-08-2007 15:42:10

Developing novel strategies for treating
esophageal cancer: Dendritic Cells immunotherapy,
T cell responses and an exploration
of the tumor microenvironment.
ACADEMISCH PROEFSCHRIFT
ter verkrijging van de graad van doctor aan de Universiteit van Amsterdam op gezag van de Rector Magnificus
prof.dr. D.C. van den Boomten overstaan van een door het college voor promoties ingestelde
commissie, in het openbaar te verdedigen in de Aula der Universiteit op dinsdag 9 oktober 2007, te 10:00 uur
door
Francesca Milano
geboren te Perugia, Italië
Milano_opm_v5.indd 3 16-08-2007 15:42:10

Promotiecommissie:
Promotor(es): Prof. Dr. M.P.Peppelenbosch Prof. Dr. G.E.E. Boeckxstaens
Copromotor(es): Dr. K.K.Krishnadath
Overige leden: Prof. drs. J.F.W.M. Bartelsman Prof. dr. J.P. Medema Prof. dr. D.J. Richel Dr. L.J.M. de Vries Prof. dr. J.J.B. van Lanschot Prof. dr. P. Fockens
Faculteit der Geneeskunde
Milano_opm_v5.indd 4 16-08-2007 15:42:10

Contents
F. Milano: “Developing novel strategies for treating esophageal cancer: Dendritic Cell therapy, T cell responses and an exploration of the tumor microenvironment”.
Chapter 1 General Introduction ............................................................................ 7
Chapter 2 An Improved Protocol for Generation of ImmunoPotent Dendritic Cells through Direct Electroporation of CD14+ Monocytes
Journal of Immunological Methods 2007 ........................................ 41
Chapter 3 An ex-vivo read out for evaluation of Dendritic Cell induced autologous CTL responses against esophageal cancer
Cancer Immunology and Immunotherapy 2007 ............................ 61
Chapter 4 Characterization of the esophageal cancer microenvironment: a balance towards tumor or towards anticancer immuneresponse Submitted ............................................................................................. 81
Chapter 5 Trastuzumab synergistically acts with HER2 specific CTLs to induce tumor cell lysis in Esophageal Adenocarcinoma cell lines
Manuscript in preparation ............................................................... 101
Chapter 6 Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells.”
Gastroenterology 2007 ...................................................................... 121
Chapter 7 Summary, general discussion and future perspective ................. 141
Appendices Samenvatting ..................................................................................... 149 Dankwoorden ................................................................................... 153 List of publications ............................................................................ 157 Curriculum Vitae ............................................................................... 159
Milano_opm_v5.indd 5 16-08-2007 15:42:10

Milano_opm_v5.indd 6 16-08-2007 15:42:11

Chapter 1
General introduction
&
outline of the thesis
Francesca Milano, Kausilia K. Krishnadath, Maikel P.Peppelenbosch
Milano_opm_v5.indd 7 16-08-2007 15:42:11

Milano_opm_v5.indd 8 16-08-2007 15:42:11

Chapter 1
9
Developing novel strategies for treating esophageal cancer: Dendritic Cell immunotherapy, induction of T cell responses and an exploration of the tumor microenvironment.
INTRODUCTION
Esophageal carcinoma already was described at the beginning of the 19th century, and the first successful surgical resection was performed in 1913 by Frank Torek. Worldwide, esophageal cancer is the seventh leading cause of cancer death, squamous cell carcinoma being responsible for 95% of the cases. Esophageal cancer is relatively rare in Western countries such as Western Europe and in the United States. In the US esophageal cancer, ranks 19th among cancers in incidence, with 13,900 new cases per year, however, it is a particularly deadly malignancy, ranking 10th in cancer deaths, with 13,000 deaths per year11. In the Netherlands, the incidence of Esophageal cancer increased from 6.4/100000 in 1994 to 8.9/100000 in 2003. Esopahgeal adenocarcinoma more commonly affects men than women, with lifetime risks of 0.75 and 0.26%, respectively24. Until the 1970s, over 90% of esophageal cancers were squamous cell carcinoma, with adenocarcinoma of the esophagus being relatively rare1. From then on the incidence of esophageal adenocarcinoma has been steadily increasing and, by the early 1990s, adenocarcinoma has become the most common cell type of esophageal cancer among whites5,6. On the other hand, squamous cell cancers still predominates in blacks, and in most Eastern countries its incidence seems to be increasing710. Currently in the Netherlands, esophageal adenocarcinoma accounts for more than 50% of all new cases of esophageal cancer. Its pathogenesis is linked to gastroesophageal reflux disease (GERD), and development of a metaplastic precursor lesion known as Barrett’s esophagus1114.
Risk factors for esophageal adenocarcinoma While risks factors for squamous cell carcinoma of the esophagus have been identified, such as tobacco, alcohol, diet, poor socioeconomic status, the risk factors associated with esophageal adenocarcinoma seems more complex7,15. Barrett’s esophagus is regarded to be the precursor lesion and is associated with an increased risk for developing adenocarcinoma1618. Norman Barrett is credited with the discovery in 1950 of columnar epithelial cells lining the lower esophagus19. Barrett originally believed that the columnar lining was the result of a short esophagus that drew the stomach tissue up into the thoracic cavity20. Barrett’s esophagus is now recognized as a metaplastic condition in which squamous epithelial cells are replaced by intestinaltype of columnar cells as a consequence of gastroesophageal reflux disease (GERD)21,22. Esophagitis, secondary to gastroesophageal reflux disease, is the most common medical condition in Western countries with 30% of adults complaining of heartburn at least once per month,
Milano_opm_v5.indd 9 16-08-2007 15:42:11

Chapter 1
10
a third of whom will have endoscopic evidence of esophagitis, of which 40% will improve spontaneously, while 50% have persistent esophagitis, and 10% develops metaplasia23. It is supposed that reflux induces damage to squamous epithelial cells and causes (stem) cell proliferation, resulting in the replacement of squamous cells with columnar cells. This cell conversion may initially progress through the appearance of simple columnar epithelium before specialized intestinal cells are evident14,24. Although different types of metaplasia may subsequently develop, at present Barrett’s esophagus is defined as an incompletely differentiated specialized columnar epithelium and is characterized by the presence of goblet cells. Barrett’s esophagus represents the first step leading to the development of EAC21,2527. In 1952, Morson and Belcher first described a patient with adenocarcinoma of the esophagus arising in columnar epithelium with goblet cells. In 1975, Naef et al. emphasized the malignant potential of Barrett’s esophagus28. Metaplastic Barrett segments may vary in length, and are divided into short segments (less than 3 cm) or long segments (more than 3 cm). A long metaplastic segment represents the most important risk factor for progression to adenocarcinoma29. The annual incidence of esophageal adenocarcinoma in patients with Barrett’s esophagus is estimated at 0.5% to 1%3032. Histopathologic classification of dysplasia grade in Barrett’s esophagus [negative, indefinite, lowgrade dysplasia (LGD), and highgrade dysplasia (HGD)] is the standard method of risk stratification3335. HGD has the highest morphologic
association with progression to esophageal adenocarcinoma, and may be further subdivided into focal or diffuse types36,37. A large number of genetic and epigenetic events contribute to the metaplasiadysplasiaadenocarcinoma sequence, in the multistep process of malignant transformation38. Most of the genes involved in the development of adenocarcinoma are normally responsible for cell cycle control. Alterations of the strictly controlled cell cycle may lead to dysregulated proliferation, and, subsequently, to cancer3944.
Clinical presentation and diagnosis of esophageal adenocarcinoma In a recent study, the most common presenting symptoms of EAC are dysphagia (74%), weight loss (57%) gastrointestinal reflux (20%) odynophagia (16%) and dyspnea (12%)45,46. Unfortunately, symptoms generally indicate relatively advanced disease. Early cancers are relatively asymptomatic and may be discovered as incidental findings at endoscopy. Dysphagia does not occur until the esophageal lumen is significantly compromised, and this therefore often points to the presence of a relatively advanced stage cancer. Generally dysphagia directs patients to endoscopy, which offers the opportunity for diagnosis, tissue sampling, and, when possible, therapy. Once an esophageal cancer is documented, staging is performed based on the TNM classification of the American Committee on cancer and the International Union Against Cancer. At first, it is advised to perform a computed tomography scan (CT) of
Milano_opm_v5.indd 10 16-08-2007 15:42:12

Chapter 1
11
the abdomen, chest and pelvis to detect possible metastatic formations. If none is found, endoscopic ultrasonography (EUS) should be performed, to establish the status of the tumor (T and N status). It has been previously demonstrated that this procedure has the highest accuracy for T and N status classification47. The addition of fine needle aspiration of suspicious nodes increases the accuracy of N status to 93%. Positron Emission tomography (PET) may be useful for further staging of the EAC. In general, at the time of presentation up to 50% of patients have stage IV disease and are therefore not considered for surgical treatment.
Treatment optionsThe survival rate of patients with EAC is poor. Asymptomatic small tumors confined to the esophageal mucosa or submucosas are detected only by chance. Until recently, surgery was the only treatment of choice for these small tumors48, 49. Novel non invasive treatment options for removing mucosal cancers with a high success rate are endoscopic mucosal resection with or without ablative therapies5052. Once symptoms such as dysphagia and weight loss are present, esophageal cancers have usually invaded the muscularis propria or beyond and may have metastasized to lymph nodes or other organs. Treatment strategies for EAC have been largely focused on surgery with curative intent48 53, 54. Patients with localized disease are chosen to undergo surgery. Nevertheless, patients undergoing resection have a median survival of only about 15 to 18 months, corresponding to a five years survival of less than 20%55, 56, therefore surgery with curative intent can be viewed as palliative in the majority of cases. To improve the outcomes of patients undergoing resection, the use of chemotherapy, radiation therapy, or both as adjuvant or neoadjuvant treatments in conjunction with surgery have been explored. Survival highly depends on stage of disease, for instance, there is an overall 5 years survival rate of 85% for stage I, and of 65% for stage IIa disease57, 58, while stage IIb and III have 5 years survival rates of 51 and 14%, respectively59. Outcomes for more advanced stages are considerably worst and palliative strategies are the only consideration. Endoscopic treatment modalities such as stents or laser therapy play an important role in palliation of dysphagia, prevention of aspiration, and improvement in quality of life for many of these patients60,61. After endoscopic palliation of adenocarcinoma at the distal esophagus or gastric cardia, mean survival has been around 140 days62. A recent study pointed out that palliative radiotherapy had similar or even slightly better results63. Improvement of surgical results over time has been attributed variably to change in epidemiology, patient selection, staging methods, surgical technique, and the use of additional treatments55.
Milano_opm_v5.indd 11 16-08-2007 15:42:12

Chapter 1
12
Combinations of surgery, radio- and chemotherapyBased on existing trials, there was no clear evidence that preoperative radiotherapy improves the survival of patients with potentially resectable esophageal cancer64,65. It has been previously reported that definitive radiation therapy in combination with chemotherapy compared to radiation therapy alone resulted in an improvement in 5year survival for the combined modality group (27% vs. 0%)66, while in a phase III trial comparing different dose of radiations, a higher radiation dose did not increase survival or local/regional control67. It has been previously demonstrated that chemotherapy using 5FU or cisplatin in combination with radiotherapy, followed by surgery compared to resection alone resulted in a modest patient survival improvement6870. Many attempts have been done to explore the effect of combined therapy compared to surgery alone. The effects of preoperative chemotherapy have been evaluated in a randomized trial comparing this regiment to surgery alone. It was demonstrated that preoperative chemotherapy with a combination of cisplatin and fluorouracil did not improve overall survival among patients with epidermoid cancer or adenocarcinoma of the esophagus.71
Despite several attempts to improve the outcome of EAC patients, no significant progress have been made and it is still a challenge to find novel and effective treatment to advance in the cure of EAC.69,72
Immunotherapy: the beginning of a new eraIn the past decades, a novel approach has been considered to be applicable to cancer therapy: immunotherapy using vaccines. The concept of immunotherapy is based on the body’s natural defence system, which protects us against a variety of diseases.
For many years, it was commonly believed that the immune system was effective only in combating infectious diseases caused by invading agents such as bacteria and viruses. More recently, evidence was found in favour of the hypothesis that the immune system may play a central role in protecting the body against cancer and in combating cancer that has already developed. The exact role is not well understood, but there is evidence that in many cancer patients the immune system slows down the growth and spread of tumors (Finn, O.J., Preventive role of existent immunity and developmental strategies for preventive vaccines, AACR 2007, Los Angeles, CA).
This concept came out for the first time in the 19th century, when Dr. William Coley from the New York Memorial Hospital, showed that he could control the growth of some cancers. He cured several advanced cancers with injections of a mixed vaccine of streptococcal and staphylococcal bacteria known as Coley’s toxin73. The disillusion with conventional medicine, led him to wonder whether nature held a cure for cancer. He started to follow a case of a patient who presented recurrent sarcoma to the cheek. The extensive wound after surgery could not be closed and skin grafts were unsuccessful. Ironically, this failure to close the wound
Milano_opm_v5.indd 12 16-08-2007 15:42:12

Chapter 1
13
would play a key part in the patient’s eventual cure. The wound became severely infected with erysipelas (Streptococcus pyogenes) and the patient developed a high fever. Little could be done to stop the infection, yet surprisingly, after each attack of fever the ulcer improved, the tumor shrank, and finally disappeared completely. Coley continued his studies based on the theory that nature can protect against cancer. Despite this concept, the tide began to turn against “Nature’s remedy” for cancer during the 20th century. Firstly, cancer surgery, like any other operation, became a sterile procedure after acceptance of Lister’s aseptic techniques in the late 1800s; secondly, by the time of Coley’s death in 1936, radiotherapy had become an established cancer treatment, and chemotherapy was rapidly gaining acceptance.
These treatments could be more easily standardised than Coley’s approach74.Several decades later, the new era of tumor immunology began, when data were
provided supporting the historical and recent evidence concerning the antagonisms between acute bacterial infections or their toxins and cancer and allied diseases. New data were provided for renewed incentives to undertake clinical programmes with mixed bacterial vaccines in many countries73. A mile stone in the concept of immunotherapy was introduced with the theory of Thomas and Burnett, in the midpoint of the twentieth century. They pioneered the famous concept of “immunesurveillance of cancer”, to describe a mechanism that protects immunecompetent hosts against tumors. Thomas and Burnett defined this as an “evolutionary necessity” and the existence of a mechanism to eliminate or inactivate dangerous mutated cells. (Burnet, F.M. Immunological factors in the process of carcinogenesis. Br. Med. Bull. 20, 154−158 (1964)75. With the availability of inbred strains of mice, the idea that tumors were immunologically distinguishable from normal cells could be critically tested. The demonstration that mice could be immunized against syngeneic transplants of tumor induced by chemical carcinogens, viruses or other means established the existence of “tumor specific antigens”76.
Several research groups started to perform experiments, aimed by the wish to demonstrate the consistency and reliability of the concept, but this idea was largely abandoned when no differences in primary tumor development were found between athymic nude mice and syngeneic wildtype mice77. Experimental tests failed to provide strong evidence for the mechanism of cancer immunosurveillance, and because of this discordance, and most of the time disappointing results, the conclusion was drawn that tumors are not sufficiently distinct from normal tissue to activate the immune system75. However, subsequent observations that nude mice do not completely lack functional T cells78 and that two components of the immune system, IFNγ and perforin, help to prevent tumor formation in mice have led to renewed interest in a tumorsuppressor role for the immune response.77 In addition, new experimental approaches, such as gene targeting and transgenic mouse technology, and the possibility to produce highly specific monoclonal antibodies able to block immune components were introduced. Herewith, interest in the tumor
Milano_opm_v5.indd 13 16-08-2007 15:42:13
Chapter 1
14
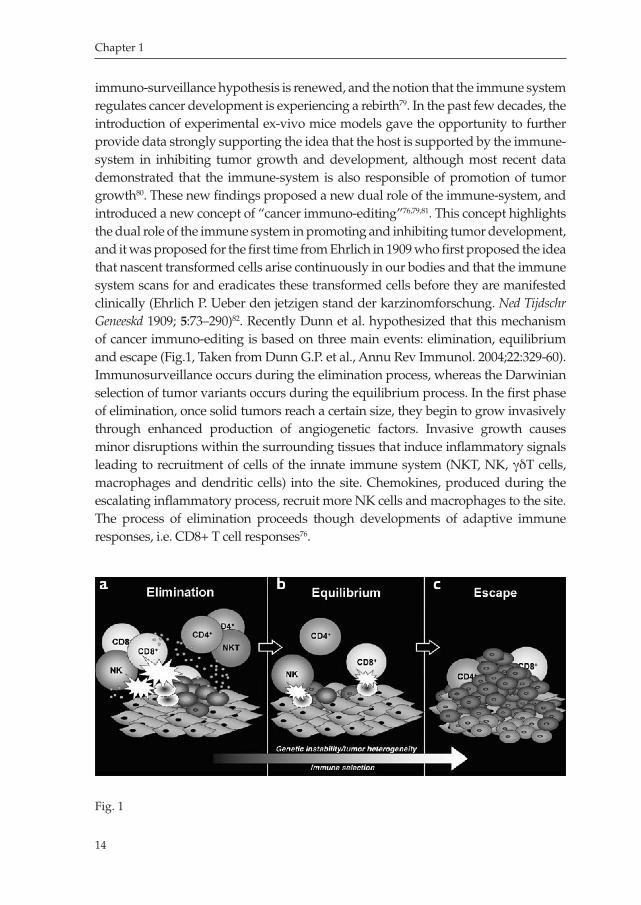
immunosurveillance hypothesis is renewed, and the notion that the immune system regulates cancer development is experiencing a rebirth79. In the past few decades, the introduction of experimental exvivo mice models gave the opportunity to further provide data strongly supporting the idea that the host is supported by the immunesystem in inhibiting tumor growth and development, although most recent data demonstrated that the immunesystem is also responsible of promotion of tumor growth80. These new findings proposed a new dual role of the immunesystem, and introduced a new concept of “cancer immunoediting”76,79,81. This concept highlights the dual role of the immune system in promoting and inhibiting tumor development, and it was proposed for the first time from Ehrlich in 1909 who first proposed the idea that nascent transformed cells arise continuously in our bodies and that the immune system scans for and eradicates these transformed cells before they are manifested clinically (Ehrlich P. Ueber den jetzigen stand der karzinomforschung. Ned Tijdschr Geneeskd 1909; 5:73–290)82. Recently Dunn et al. hypothesized that this mechanism of cancer immunoediting is based on three main events: elimination, equilibrium and escape (Fig.1, Taken from Dunn G.P. et al., Annu Rev Immunol. 2004;22:32960). Immunosurveillance occurs during the elimination process, whereas the Darwinian selection of tumor variants occurs during the equilibrium process. In the first phase of elimination, once solid tumors reach a certain size, they begin to grow invasively through enhanced production of angiogenetic factors. Invasive growth causes minor disruptions within the surrounding tissues that induce inflammatory signals leading to recruitment of cells of the innate immune system (NKT, NK, γδT cells, macrophages and dendritic cells) into the site. Chemokines, produced during the escalating inflammatory process, recruit more NK cells and macrophages to the site. The process of elimination proceeds though developments of adaptive immune responses, i.e. CD8+ T cell responses76.
Fig. 1
Milano_opm_v5.indd 14 16-08-2007 15:42:14

Chapter 1
15
The next step in cancer immunoediting proceeds to the equilibrium phase in which a continuous sculpting of tumor cells produces cells resistant to immune effector cells. This process leads to the immune selection of tumor cells with reduced immunogenicity. These cells are more capable of surviving in an immunocompetent host, which explains the apparent paradox of tumor formation in immunologically intact individuals76,82,83. On the other hand, some mice models of cancer have shown that the inflammatory actions of the immune system can promote tumor development and/or growth, suggesting a strict correlation between inflammation and tumor progression84,85. Furthermore, tumors evolve mechanisms to escape immune control by a process called immune editing, which provides a selective pressure in the tumor microenvironment that can lead to malignant progression79,86,87.This results in clinically observable malignant disease that, if left unchecked, results in the death of the host79.
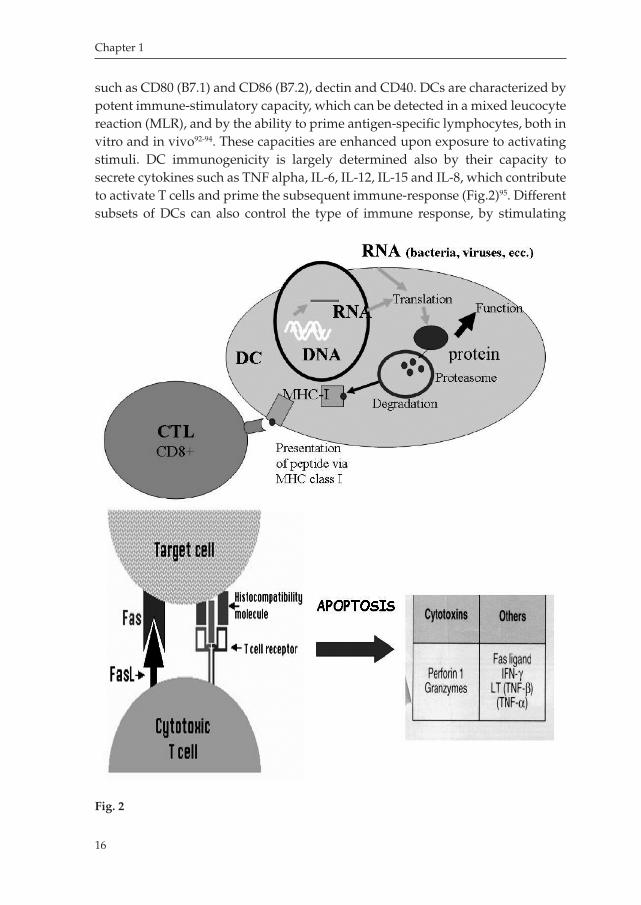
Dendritic Cells: a potent tool for cancer immunotherapyThe Dendritic Cell (DC) system of antigenpresenting cells (APCs), is the initiator and modulator of the immune response. First visualized as Langerhans cells (LCs) in the skin in 1868, the characterization of DCs began only 25 years ago. It was known that ‘accessory’ cells were necessary to generate a primary antibody response in culture, but it was only once DCs were identified and purified from contaminating lymphocytes and macrophages that their distinct function as APCs became apparent88. DCs are unique APCs, because they are the only ones that are able to induce primary immune responses, thus permitting establishment of immunological memory. DC progenitors in the bone marrow give rise to circulating precursors that home to tissues, where they reside as immature cells with high phagocytic capacity. Following tissue damage, immature DCs capture antigens (Ags) and subsequently migrate to the lymphoid organs, where they select rare Agspecific T cells, thereby initiating immune responses. DCs present Ags to CD4+ Thelper cells, which in turn regulate the immune effectors, including Agspecific CD8+ cytotoxic T cells and B cells, as well as non– Agspecific macrophages, eosinophils, and Natural Killer cells (Fig.2). Moreover, DCs educate effector cells to home to the site of tissue injury89.
DCs are typically defined on a combination of parameters that include morphology, phenotype, cytokine secretion pattern, immunostimulatory capacity, chemokine and chemokine receptor pattern, and migration in response to chemotactic stimuli88,90,91. The DC phenotype varies, depending on stage of maturation and differentiation. For example, CD1a is preferentially expressed on human myeloid DCs, whereas CD83 is typically upregulated in response to activation stimuli such as tumor necrosis factor alpha, Toll like receptor ligands, Cytidylyl2p,5pphosphoryl guanosine (CpG), double stranded RNA, prostaglandin E2, or T cell derived signals including CD40 ligand and IFNγ. DCs express as well costimulatory molecules
Milano_opm_v5.indd 15 16-08-2007 15:42:14
Chapter 1
16
such as CD80 (B7.1) and CD86 (B7.2), dectin and CD40. DCs are characterized by potent immunestimulatory capacity, which can be detected in a mixed leucocyte reaction (MLR), and by the ability to prime antigenspecific lymphocytes, both in vitro and in vivo9294. These capacities are enhanced upon exposure to activating stimuli. DC immunogenicity is largely determined also by their capacity to secrete cytokines such as TNF alpha, IL6, IL12, IL15 and IL8, which contribute to activate T cells and prime the subsequent immuneresponse (Fig.2)95. Different subsets of DCs can also control the type of immune response, by stimulating
Fig. 2
Milano_opm_v5.indd 16 16-08-2007 15:42:15

Chapter 1
17
helper or cytotoxic Tcells that in turn will produce different types of cytokines, for instance: IFNγ production by Thelper 1 cells, IL4 by Thelper 2, or IL10 by regulatory Tcells. This means that DCs have a complex constitution that under different micro environmental conditions can induce divergent immune responses, on one hand they can induce immunity, and on the other hand they may mediate tolerance96. At least three distinct types of DCs exist: myeloid DCs, lymphoid DCs and Langerhans cells (LCs)97. Evidence for the myeloid origin of DCs comes mainly from in vitro studies in which myeloidcommitted precursors give rise to both granulocytes/monocytes and myeloid DCs under the influence of granulocyte/ macrophage colonystimulating factor (GMCSF)98,99. Transfer of a population of purified lymphoid precursors into irradiated hosts resulted in the development of T cells, B cells, NK cells, and DCs that express CD8α, but not cells of the myeloid lineage100. Three subsets of DC precursors circulate in the blood: CD14+ monocytes, lineage negative CD11c+ precursor DCs, and CD11c− precursor DCs101. Monocytes can differentiate into cells displaying features of immature DCs or macrophages in response to GMCSF and IL4 or MCSF, respectively102. The CD11c+ subsets contain precursors of interstitial DCs, LCs and macrophages103. Distinct factors regulate the survival and differentiation of CD11c−IL3Rα+ DC precursors, originally described as plasmacytoid T cells or plasmacytoid monocytes104106. These cells die rapidly after isolation and are critically dependent on IL3 for survival and CD40L for maturation.
The role of T helper cellsDCs are highly capable of inducing Tcell responses, and the interaction of these two cell types, is determinant for the type of immune response that will be raised. The capacity of the DCs to stimulate Tcells depends on many factors, such as surface expression of costimulatory molecules, and the cytokine microenvironment. It is curious to note that T cells may play an important role in activating DCs, thus further enhancing the T cell stimulatory capacity of the DCs107. It has been demonstrated indeed that CD40 ligand, expressed by Tcells, increase DC viability and induce DC maturation108. DCs can produce IL12, a key cytokine for the generation of Thl responses. On the other hand, DC function can be suppressed by the production of IL10, which induces apoptosis, reduced capacity to stimulate Tcells and decreased production of IL12109,110. While development of IFNγ and IL2 producing Th1 cells leads to protective antitumor immune responses, Th2 cells producing IL4 and IL10 may be associated with nonprotective responses. Furthermore, as Th1 and Th2 cells have been considered as representing the two extremes of Tcell polarization111, a third CD4+ Tcell population has been recently singled out. These Tcells, named the ‘T regulatory cells (Tr1) has been demonstrated to produce high levels of the cytokine interleukin17 (IL17) , which has a major role in various models of
Milano_opm_v5.indd 17 16-08-2007 15:42:15

Chapter 1
18
immunemediated tissue injury. With this observation, a new perspective was introduced in the world of immunology112. Analysis of Tcell clones isolated from the mouse CD4+Tcell populations that were repeatedly stimulated with OVA peptide and splenic APCs in the presence of IL10 showed that half of the CD4+Tcell clones displayed this cytokine profile. They produced high levels of IL10 and undetectable levels of IL2 and IL4. The observation that Tr1 cells secrete high levels of the immunosuppressive cytokine IL10, and low levels of the Tcell growthpromoting cytokines IL2 and IL4, suggest that antigenspecific activation of Tr1 cells may result in inhibition of antigenspecific proliferation of other T cells. Indeed, coculture experiments using a transwell system confirmed this notion for both human and mouse T cells113. The recent identification of these Tr1 cells actually develops numerous observations of ‘suppressor’ CD4+ Tcells that may also play an important role in tumor ‘anergy’. The inhibitory factor appears to be IL10, a cytokine capable of converting DC/APC function to the induction of antigenspecific anergy, thus leading to the state of tolerance against tumor tissue114.
The choice of antigen sourceMost, if not all, tumors are antigenic and a majority of identified tumor associated antigens (TAA) are nonmutated, overexpressed or inappropriately expressed, tissue differentiation antigens111. Another category of TAAs represented in several tumors are reexpressed embryonic antigens and growth factors, such as MAGE, gp100, and HER2, which present an aberrant expression, or overexpression, mostly via gene amplification. The most notable examples are in melanoma in which major proteins implicated in the tumorspecific immune response are nonmutated reexpressed embryonic antigens, e.g., MAGE and gp100. Tumor antigens can be as well classified according to the type of immune response they elicit: humoral, cellular, CD4+ (T helper), or CD8+ cytotoxic T lymphocyte (CTL) responses. As will be discussed below, the fact that a tumor antigen elicits a tumorspecific immune response does not necessarily mean that the immune response will cause the rejection of the tumor in vivo. Thus, from a vaccination perspective, the question is which tumor antigen can or is better at inducing a clinically beneficial response. Such antigens are referred as “tumor rejection antigens”. Tumor antigens can be poor, intermediate, or strong tumor rejection antigens, describing quantitatively the effect of the immune response on tumor growth115. Several different strategies have been extensively explored to deliver antigens into exvivo manipulated of DCs to enhance their potency to stimulate T cells94,116118. DCs can be loaded with synthetic peptide epitopes derived from known TAAs such as MUC1, HER2/neu, tyrosinase, telomerase, CEA, p53, MAGE, or MelanA/MART5,119122. Major drawbacks of using peptides are the restriction to some HLA class I alleles, the need to
Milano_opm_v5.indd 18 16-08-2007 15:42:16

Chapter 1
19
determine the expression of the target antigen by a particular tumor, the limited number of defined TAA and the possibility that targeting single or few tumor epitopes may impede the detection of tumor cells that may have downregulated expression of these antigens95,123. Moreover, additional biologically important epitopes may be missed, as binding affinity for a particular HLA allele does not translate directly into immunogenicity. Using MHC class I–restricted peptides also ignores the role of MHC class II–restricted T helper cells in initiating and sustaining an immune response124. Another strategy which can be applied to deliver tumor antigens to DCs is based on gene transfer methods that result in antigen processing in the MHC class I pathway of DC and presentation to CD8+ T cells. DCs can be transduced with DNA, with or without liposomal encapsulation, which has been tried with varying success125. RNA derived from tumors or encoding tumor antigens can also be transfected into DCs leading to the generation of primarily class I MHC restricted responses126,127. When these approaches are used, the vaccine may contain multiple antigens, increasing the probability of inducing immunity to more than one tumorassociated antigen127
129. Although the target proteins are initially undefined, with this approach, the immunogen can be identified afterwards. Theoretically, with such an approach there is increased potential for the induction of a destructive autoimmune response to antigens expressed on normal tissues. In practice, there is a ‘self defence’ mechanism against the auto immune responses through selective destruction of Tcell that recognize autoantigens in the thymus during fetal development. Furthermore induction of autoimmune response by such antigens presumably would be limited to tumor cells bearing epitopes of the self antigens and TAAs, limiting the risk of collateral damage to normal tissues130. If such proteins arise in the tumor after thymic development, antigenreactive T cells could exist in the repertoire as they may have avoided thymic deletion. The mechanisms involved in these observations are poorly understood and future studies will enlighten us on this matter. One important limitation of most studies is that not all exvivo observations of immune responses can be extrapolated to the human situation. While animal models have been used frequently to examine the immunogenicity of tumorassociated antigens, there are as well significant caveats in extrapolating animal data to humans. For instance, many of the animal tumor antigen models use human proteins that are foreign to the animal (e.g. carcinoembryonic antigen, or HER2/ neu in mice), which by definition will induce a strong immune response in the animal. Transgenic tumor models could be used to address this issue although differences in T cell receptor repertoire remain an important confounding factor.
Milano_opm_v5.indd 19 16-08-2007 15:42:16

Chapter 1
20
DC dose, frequency and route of injections.In human trials published so far, several different dosages at different intervals were investigated without giving outstanding differences in results. It has previously been demonstrated that in vitrogenerated mature, in contrast to immature DCs, efficiently migrate into the Tcell areas of lymph nodes of melanoma patients and this difference was confirmed by in vitro studies. These findings demonstrate that the ability of DCs to induce an efficient immune response correlates with their ability to migrate both in vitro and in vivo131, and this phenomenon is correlated with molecular changes, for instance upregulation of antigenpresenting MHC molecules and costimulatory molecules132, a switch in chemokine receptor expression with downregulation of receptors for inflammatory chemokines, and upregulation of receptors for chemokines produced in secondary lymphoid organs133. Another important parameter to consider when preparing a vaccination strategy is the ability of the DCs to migrate from the site of injection to the lymph nodes to be able to encounter resting T cells and prime immune responses. In 2003 de Vries et al. demonstrated that regardless whether DCs are injected intranodal or intradermal, mature DCs are migratory both in vitro and in vivo131. Despite clear evidence of the capacity of the injected DCs to migrate to lymph nodes, the optimal route for administering DC vaccines is still a subject of debate 134. Historically, melanomas were the first cancers treated by DC therapy and traditionally intradermal vaccination is the most frequently applied route of vaccination. Several animal and human studies however show effective anticancer response in case of intratumoral injections135,136. In a randomized phase I trial patients with metastatic melanoma received peptide pulsed DCs either intravenously, intranodally or intradermally. The intranodal route induced significantly higher rates for the novo development of CD8+ T cells as determined by MHC Tetramer staining compared with the other routes137. DC therapy for treating malignancies within the gastrointestinal tract including esophageal cancers has a relatively short history. These cancers have the unique feature that they arise from the mucosa. The esophagus is furthermore provided with an extensive network of lymphatics. This is reflected in aggressiveness of these cancers characterized by early lymph node metastasis of even small mucosal cancers138. It is very likely that intramucosal vaccination for treating esophageal cancer may be as effective or perhaps superior to intranodal injections. Despite the frequently raised criticism that intranodal injection might greatly disturb the node architecture, migration to subsequent nodes has been observed and follows the physiological path through lymph vessels139,140. For esophageal cancer the best route for administering the vaccines is yet not known, therefore, intranodal versus intramucosal administration of peptideloaded DC vaccines remains to be explored.
Milano_opm_v5.indd 20 16-08-2007 15:42:16

Chapter 1
21
Results from clinical trials employing Dendritic Cell immunotherapyThe first attempt to use DCs vaccines was done by Hsu et al to treat Bcell lymphoma141. After this pioneer study, many others attempted to develop strategies to apply and improve DCs immunotherapy for cancer patients, particularly for melanoma and prostate cancer142147. Although the general experience suggest now that DCs can be administered safely, still clinical responses, either complete remission, partial remission, or stable disease, can be observed in only few patients148151. O’Rourke et al. observed complete remission in three patients after vaccination in melanoma patients using DCs loaded with irradiated autologous tumor cells149. Holtl and colleagues showed that loading mature DCs with tumor lysates, 2 out of 35 renal cell carcinoma patients could respond completely, one partially, and 7 could reach stable disease148. SchulerThurner and colleagues included 16 melanoma patient in a study employing mature DCs loaded with melanoma peptides, and could observe one complete remission, and 8 partially recovered patients142. Although these studies are hardly comparable because of the differences in criteria of inclusions, type of antigen source, route of vaccine delivery, the results are indicating that DCs are safe, immunogenic, and induces Tcell responses and remission in few patients. Many more studies have to be performed to improve such an appealing strategy for curing cancer, especially to understand what determines such unsatisfactory results in terms of clinical responses.
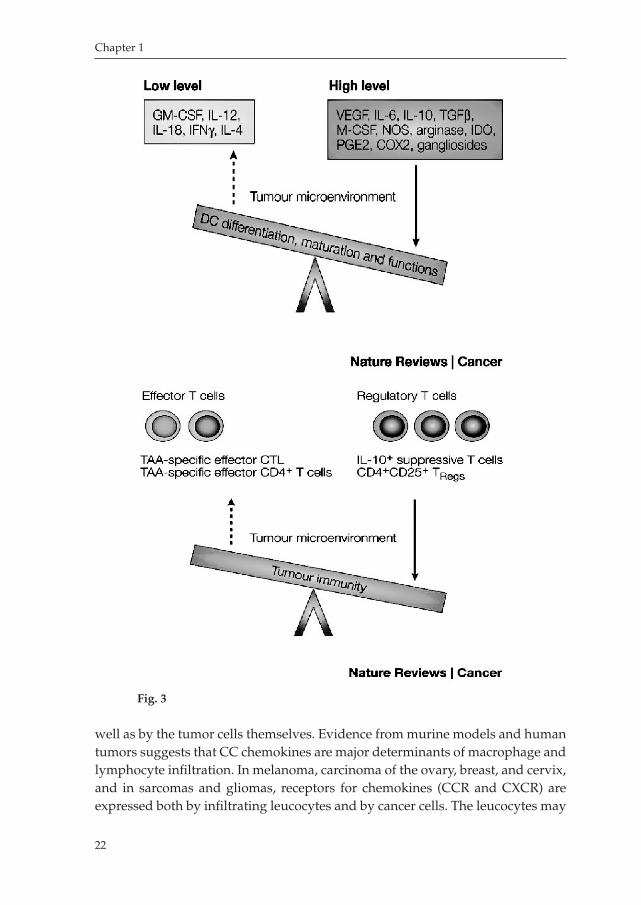
The Tumor MicroenvironmentDespite major advances in the understanding the components of the immune system, it has been broadly demonstrated that the poor clinical outcomes and low efficacy of immunotherapy against cancer and the failure in curing cancer is due to an immunosuppressive network, created as a consequence of pathological interactions between cancer cells and host immune cells80,152. Tumor resistance is a result of an alteration in the production of specific factors, for instance costimulatory and coinhibitory molecules, an imbalanced activation of effector Tcells and regulatory Tcells (Fig.3, taken from W.Zou, Nat Rev Cancer. 2005 Apr;5(4):26374). This balance is especially altered in patient with advanced stage cancer, which presents high levels of inflammatory molecules, cytokine, chemokines, and tumor infiltrating Tcells80. There is rising evidence that the tumor microenvironment certainly inhibits the development and function of DCs, by overexpressing immunosuppressive molecules such as COX2, TGFbeta, IL6, MCSF, IL10, PGE2, and VEGF153,154,155; on the other hand, there is a lack of immunostimulatory factors, such as GMCSF, IFNγ and IL4. This environment enables cancer cells to escape from effector Tcells responsible of recognition and attack of the tumor mass. Furthermore, chemokines are secreted into the tumor microenvironment by tumorinfiltrating inflammatory cells as
Milano_opm_v5.indd 21 16-08-2007 15:42:17
Chapter 1
22
well as by the tumor cells themselves. Evidence from murine models and human tumors suggests that CC chemokines are major determinants of macrophage and lymphocyte infiltration. In melanoma, carcinoma of the ovary, breast, and cervix, and in sarcomas and gliomas, receptors for chemokines (CCR and CXCR) are expressed both by infiltrating leucocytes and by cancer cells. The leucocytes may
Fig. 3
Milano_opm_v5.indd 22 16-08-2007 15:42:17

Chapter 1
23
lose receptor expression once they are exposed to inflammatory cytokines in the tumor microenvironment, as shown for CCR2 on tumor associated macrophages in ovarian cancer84. Recently, it has been demonstrated that regulatory Tcell mediated immunosuppression is one of the crucial tumor immuneevasion mechanisms and the main obstacle of successful tumor immunotherapy156158. It has been demonstrated that depleting Tregulatory cells (Tregs) in human cancer, including ovarian cancer, in combination with supplied therapies, improves immunity and may be therapeutic159,160. CD25 is constitutively expressed in effector Tcells and Tregs. Early studies demonstrated that in vivo administration of a CD25specific antibody suppressed growth of several progressive tumors. Elimination of CD25expressing T cells, which constitute 510% of peripheral CD4+ T cells in normal naive mice, elicited potent immune responses to syngeneic tumors in vivo160. Tumors employ several strategies to evade immuneresponse, including tumorinduced impairment of antigen presentation, the activation of negative costimulatory signals, and the elaboration of immunosuppressive factors. Moreover, recent years have witnessed increasing interest in immunosuppressive cells of myeloid origin, especially about their role in cancer immuneescape. Immunosuppressive myeloid cells accumulate in large numbers in tumorbearing mice, in several experimental models, as well as in patients with breast, lung, prostate, kidney, head and neck, and other types of cancer, they are produced in response to a variety of tumorderived cytokines and are a heterogeneous mixture of myeloid cells at different stages of differentiation161. For this reason, it is of basic importance to find new strategies to move the balance of all these factors in favor of the tumor immunity, either through boosting the activation of specific potent cytotoxic T cells, exploring novel strategies for enhancing DC therapy, or providing effector cytokines such as IL2, IFNalpha, IL12, or by blocking the suppressive environment via inhibition of immunosuppressive cells, such as T regs, myeloid derived suppressor cells, plasmacytoid DCs, immature DCs, and inhibition of immunosuppressive molecules, such as COX2, IL6, PGE2, TGFbeta, EGF, Cyclin D1and many more.
Combining DC therapy with anticancer drugs targeting several known substratesIt is anticipated that the efficacy of DC therapy will greatly benefit through combination with anticancer drugs that either modulate the tumor environment or inhibit growth factors. Therefore, the efficacy of several strategies combining DC vaccines and several applicable and ready available anticancer drugs against several pharmacological substrates known to be highly expressed in esophageal adenocarcinomas, such as Her2/neu, COX2, VEGF and TGFbeta, are currently being tested. Several anticancer drugs can potentially be interesting to be tested in combination with DC immunotherapy induced CTL responses
Milano_opm_v5.indd 23 16-08-2007 15:42:18

Chapter 1
24
It has been previously shown that Celecoxib, a specific inhibitor of COX2, reduces cell viability and induces apoptosis in vitro and, furthermore, evaluation in vivo has shown that Celecoxib administered as neoadjuvant treatment in patients with esophageal adenocarcinoma induced a decrease in COX2 expression, without particular adverse effects162. Another important mechanism of tumor progression is angiogenesis, mediated by several factors, in particular by vascular endothelial growth factor (VEGF), which is most essential proangiogenic growth factors expressed by most cancercell types and certain tumor stromal cells. It has been demonstrated that blocking this mechanism via specific antibodies against VEGF, such as Bevacizumab, significantly improve clinical benefit for several solid tumor patients163. The first report of a large, prospective randomized trial of antiVEGF therapy in patients with metastatic breast cancer (MBC), which demonstrated the benefit of adding the monoclonal antibody Bevacizumab to the chemotherapeutic agent paclitaxel was performed in 2005. The success of this trial provided proof of principle that inhibition of angiogenesis has the potential to enhance the effectiveness of treatment of breast cancers, and perhaps also for other solid tumors bearing VEGF overexpression. Therefore, it is of interest to evaluated the use of bevacizumab in esophageal cancer, which has high expression of this factor164.
Immunotherapy in combination with blocking of the HER-2 oncoprotein. It is now generally accepted that although many advances have been made in the field of cancer vaccination using DCs, the present clinical outcomes of patient trials anticipate that DC therapy alone for treating advanced stage disease will be below expectation. Therefore, the possibility to combine the efficacy of DC therapy with other treatment strategies to improve clinical responses is attractive. Several approaches have been explored, such as the combination of chemotherapy and immunotherapy165168, or strategies to increase immunity for instance through combining immunotherapy with depletion of CD25+CD4+FOXP3+ T cells160,
169. A potent immunomodulatory drug that might be eligible to be combined with DC immunotherapy is Herceptin (Trastuzumab). Trastuzumab is a fully humanized antibody designed to block the function of the HER2 oncoprotein specifically targeting the HER2 extracellular domain. Trastuzumab, as such, induces growth inhibition against HER2–overexpressing tumors. CerbB2, the (onco) gene that codes for HER2, can be overactivated by gene amplification
with an increased expression of the normal gene product. As a protein which is normally found in cells and is overexpressed in tumor, the HER2/neu protein represents a typical example of a rather new concept in tumor immunology, which suggests that selfproteins can serve as tumor antigens170. Thus, a current issue for the development of cancer vaccines is how best to induce Tcell immunity to “self” tumor antigens171. It has been reported in several studies that HER2/neu is overexpressed in 30 to 80% of esophageal adenocarcinomas172175, and in
Milano_opm_v5.indd 24 16-08-2007 15:42:18

Chapter 1
25
many other gastrointestinal cancers176,177. Lately, there has been much attention to HER2/neu expression in human breast cancer. It is known that HER2/neu overexpression can be found in 3040% of this common malignancy178,179. In several studies it has been demonstrated that selected HER2 peptides are immunogenic as they are processed as TAAs and can induce specific CTL responses against cancer cells in vitro and in vivo171,178,180184. These reports support the hypothesis that HER2 immuno targeting might form an attractive strategy to treat HER2 overexpressing tumors. The humanized monoclonal antibody Trastuzumab exhibits potent growth inhibitory activity against HER2/neu overexpressing tumors185. It has been demonstrated that Trastuzumab has survival effects on HER2/neu overexpressing breast cancer186,187. The use of Trastuzumab in combination with for instance chemotherapeutic agents has been reported to significantly improve treatment efficacy188. Although many studies demonstrated the efficacy of Trastuzumab as treatment for improving clinical outcomes, the mechanisms through which this antibody works is still unclear189. Amongst the mechanisms that have been proposed are: the reduction of PI3K pathway activation, thus promoting arrest of proliferation and apoptosis190; the inhibition of angiogenesis191; inhibiton of HER2 cleavage192 and antibodydependent cellular cytotoxicity (ADCC)185,193,194. Several questions have to be answered regarding the mechanism of this antibody, however, there is sufficient evidence that support the importance of developing combinatorial stategies including HER2/neu blockade for improving cancer patient outcomes.
Aim and structure of this thesisThe aim of this thesis is to investigate novel and more advantageous strategies to treat esophageal cancer, particularly focusing on Dendritic Cell (DC) immunotherapy. DC immunotherapy is an attractive strategy which could represent an alternative treatment approach for such an aggressive cancer. DC immunotherapy is still under development and in numerous aspects needs to be further optimized in preclinical studies. This thesis aimed at resolving a number of important difficulties in this field, in order to prepare for a phase I/II clinical trial. In order to achieve this purpose, we explored different strategies.
Chapter 2 aims on improving the efficiency manufacturing DCs from peripheral blood monocytes. This chapter describes a methodology to electroporate freshly isolated monocytes to obtain mature DCs, which in turns will be capable of presenting antigens to T lymphocytes and induce an immune response to those antigens. We demonstrated that this method is effective in obtaining immunepotent antigen presenting DCs with a proper immunophenotype, and with a proper immunostimulatory capacity and migration ability.
Chapter 3 aims on developing a better human model to monitor for DC mediated CTL responses. This chapter reports the establishment of a unique ex
Milano_opm_v5.indd 25 16-08-2007 15:42:18

Chapter 1
26
vivo autologous readout method, in which primary cell cultures from esophageal cancer and normal tissues of patients are established. Subsequently using the strategy described in chapter 2, autologous DCs and CTLs are obtained and immuneresponse, and potential adverse effects, are monitored in the autologous cultures of the esophageal cancer patients.
Chapter 4 reports a characterization of the immunological and molecular esophageal tumor microenvironment, which is of basic importance to monitor when applying immunotherapy to cancer patients. In addition, these factors represent also an explanation of why cancer vaccines at this point do not have convincing and successful results.
Chapter 5 aims on evaluating DC immunotherapy and Trastuzumab as a combinatorial strategy for treating esophageal adenocarcinoma patients. This chapter reports our findings about the action of Trastuzumab, the humanized monoclonal antibody against HER2, in EAC cell lines which overexpress HER2, and the prospective to use this antibody as a combinatorial treatment in EAC patients.
Chapter 6 describes the potential role of BMP4, a factor previously found by our group to be upregulated in Barrett biopsies, in the mechanism of development of intestinal nonspecialized columnar type of cells, which in turn can develop to a specified columnar type of epithelium typical of Barrett’s Esophagus.
Milano_opm_v5.indd 26 16-08-2007 15:42:19

Chapter 1
27
REFERENCES
1. Elton, E. Esophageal cancer. Dis Mon, 51: 664684, 2005.
2. Weir, H. K., Thun, M. J., Hankey, B. F., Ries, L. A., Howe, H. L., Wingo, P. A., Jemal, A., Ward,
E., Anderson, R. N., and Edwards, B. K. Annual report to the nation on the status of cancer,
19752000, featuring the uses of surveillance data for cancer prevention and control. J Natl
Cancer Inst, 95: 12761299, 2003.
3. Pera, M. Trends in incidence and prevalence of specialized intestinal metaplasia, barrett’s
esophagus, and adenocarcinoma of the gastroesophageal junction. World J Surg, 27: 9991008;
discussion 10061008, 2003.
4. Gamliel, Z. Incidence, epidemiology, and etiology of esophageal cancer. Chest Surg Clin N
Am, 10: 441450, 2000.
5. Bohnenkamp, H. R., Coleman, J., Burchell, J. M., TaylorPapadimitriou, J., and Noll, T. Breast
carcinoma cell lysatepulsed dendritic cells crossprime MUC1specific CD8+ T cells identi
fied by peptideMHCclassI tetramers. Cell Immunol, 231: 112125, 2004.
6. Bollschweiler, E., Wolfgarten, E., Gutschow, C., and Holscher, A. H. Demographic variations
in the rising incidence of esophageal adenocarcinoma in white males. Cancer, 92: 549555,
2001.
7. Blot, W. J. and McLaughlin, J. K. The changing epidemiology of esophageal cancer. Semin
Oncol, 26: 28, 1999.
8. Wang, J. M., Xu, B., Rao, J. Y., Shen, H. B., Xue, H. C., and Jiang, Q. W. Diet habits, alcohol
drinking, tobacco smoking, green tea drinking, and the risk of esophageal squamous cell
carcinoma in the Chinese population. Eur J Gastroenterol Hepatol, 19: 171176, 2007.
9. Wu, M., Zhao, J. K., Hu, X. S., Wang, P. H., Qin, Y., Lu, Y. C., Yang, J., Liu, A. M., Wu, D. L.,
Zhang, Z. F., Frans, K. J., and van ‘t Veer, P. Association of smoking, alcohol drinking and
dietary factors with esophageal cancer in high and lowrisk areas of Jiangsu Province, China.
World J Gastroenterol, 12: 16861693, 2006.
10. Hashibe, M., Boffetta, P., Janout, V., Zaridze, D., Shangina, O., Mates, D., SzeszeniaDabrows
ka, N., Bencko, V., and Brennan, P. Esophageal cancer in Central and Eastern Europe: tobacco
and alcohol. Int J Cancer, 120: 15181522, 2007.
11. Todd, J. A., de Caestecker, J., and Jankowski, J. Gastroesophageal reflux disease and bile
acids. J Pediatr Gastroenterol Nutr, 36: 172174, 2003.
12. Atherfold, P. A. and Jankowski, J. A. Molecular biology of Barrett’s cancer. Best Pract Res Clin
Gastroenterol, 20: 813827, 2006.
13. van Blankenstein, M., Bohmer, C. J., and Hop, W. C. The incidence of adenocarcinoma in Bar
rett’s esophagus in an institutionalized population. Eur J Gastroenterol Hepatol, 16: 903909,
2004.
14. Krishnadath, K.K. Novel findings in the pathogenesis of esophageal columnar metaplasia or
Barrett’s esophagus. Curr Opin Gastroenterol, 23: 440445, 2007.
15. Lee, C. H., Lee, J. M., Wu, D. C., Hsu, H. K., Kao, E. L., Huang, H. L., Wang, T. N., Huang,
M. C., and Wu, M. T. Independent and combined effects of alcohol intake, tobacco smoking
Milano_opm_v5.indd 27 16-08-2007 15:42:19

Chapter 1
28
and betel quid chewing on the risk of esophageal cancer in Taiwan. Int J Cancer, 113: 475482,
2005.
16. Demeester, S. R., Peters, J. H., and Demeester, T. R. Barrett’s esophagus. Curr Probl Surg, 38:
558640, 2001.
17. Reid, B. J., Levine, D. S., Longton, G., Blount, P. L., and Rabinovitch, P. S. Predictors of
progression to cancer in Barrett’s esophagus: baseline histology and flow cytometry identify
low and highrisk patient subsets. Am J Gastroenterol, 95: 16691676, 2000.
18. van Blankenstein, M., Looman, C. W., Hop, W. C., and Bytzer, P. The incidence of adenocarci
noma and squamous cell carcinoma of the esophagus: Barrett’s esophagus makes a difference.
Am J Gastroenterol, 100: 766774, 2005.
19. Barrett, N. R. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br J Surg, 38: 175
182, 1950.
20. Watson, A. Barrett’s oesophagus50 years on. Br J Surg, 87: 529531, 2000.
21. Cossentino, M. J. and Wong, R. K. Barrett’s esophagus and risk of esophageal adenocarci
noma. Semin Gastrointest Dis, 14: 128135, 2003.
22. Spechler, S. J., Zeroogian, J. M., Antonioli, D. A., Wang, H. H., and Goyal, R. K. Prevalence of
metaplasia at the gastrooesophageal junction. Lancet, 344: 15331536, 1994.
23. Mann, N. S., Tsai, M. F., and Nair, P. K. Barrett’s esophagus in patients with symptomatic
reflux esophagitis. Am J Gastroenterol, 84: 14941496, 1989.
24. Jankowski, J. A., Wright, N. A., Meltzer, S. J., Triadafilopoulos, G., Geboes, K., Casson, A. G.,
Kerr, D., and Young, L. S. Molecular evolution of the metaplasiadysplasiaadenocarcinoma
sequence in the esophagus. Am J Pathol, 154: 965973, 1999.
25. Jenkins, G. J., Doak, S. H., Parry, J. M., D’Souza, F. R., Griffiths, A. P., and Baxter, J. N. Genetic
pathways involved in the progression of Barrett’s metaplasia to adenocarcinoma. Br J Surg,
89: 824837, 2002.
26. Fitzgerald, R. C. Molecular basis of Barrett’s oesophagus and oesophageal adenocarcinoma.
Gut, 55: 18101820, 2006.
27. Glickman, J. N., Blount, P. L., Sanchez, C. A., Cowan, D. S., Wongsurawat, V. J., Reid, B. J., and
Odze, R. D. Mucin core polypeptide expression in the progression of neoplasia in Barrett’s
esophagus. Hum Pathol, 37: 13041315, 2006.
28. Patti, M. G., Gorodner, M. V., Galvani, C., Tedesco, P., Fisichella, P. M., Ostroff, J. W., Bagate
los, K. C., and Way, L. W. Spectrum of esophageal motility disorders: implications for diagno
sis and treatment. Arch Surg, 140: 442448; discussion 448449, 2005.
29. Reid, B. J. and Weinstein, W. M. Barrett’s esophagus and adenocarcinoma. Annu Rev Med, 38:
477492, 1987.
30. Drewitz, D. J., Sampliner, R. E., and Garewal, H. S. The incidence of adenocarcinoma in Bar
rett’s esophagus: a prospective study of 170 patients followed 4.8 years. Am J Gastroenterol,
92: 212215, 1997.
31. Hage, M., Siersema, P. D., van Dekken, H., Steyerberg, E. W., Dees, J., and Kuipers, E. J. Oeso
phageal cancer incidence and mortality in patients with longsegment Barrett’s oesophagus
after a mean followup of 12.7 years. Scand J Gastroenterol, 39: 11751179, 2004.
Milano_opm_v5.indd 28 16-08-2007 15:42:20

Chapter 1
29
32. Provenzale, D., Schmitt, C., and Wong, J. B. Barrett’s esophagus: a new look at surveillance
based on emerging estimates of cancer risk. Am J Gastroenterol, 94: 20432053, 1999.
33. Flejou, J. F. Barrett’s oesophagus: from metaplasia to dysplasia and cancer. Gut, 54 Suppl 1:
i612, 2005.
34. Haggitt, R. C. Pathology of Barrett’s esophagus. J Gastrointest Surg, 4: 117118, 2000.
35. Hameeteman, W., Tytgat, G. N., Houthoff, H. J., and van den Tweel, J. G. Barrett’s esophagus:
development of dysplasia and adenocarcinoma. Gastroenterology, 96: 12491256, 1989.
36. Srivastava, A., Hornick, J. L., Li, X., Blount, P. L., Sanchez, C. A., Cowan, D. S., Ayub, K.,
Maley, C. C., Reid, B. J., and Odze, R. D. Extent of lowgrade dysplasia is a risk factor for the
development of esophageal adenocarcinoma in Barrett’s esophagus. Am J Gastroenterol, 102:
483493; quiz 694, 2007.
37. Buttar, N. S., Wang, K. K., Sebo, T. J., Riehle, D. M., Krishnadath, K. K., Lutzke, L. S., An
derson, M. A., Petterson, T. M., and Burgart, L. J. Extent of highgrade dysplasia in Barrett’s
esophagus correlates with risk of adenocarcinoma. Gastroenterology, 120: 16301639, 2001.
38. Barrett, M. T., Sanchez, C. A., Prevo, L. J., Wong, D. J., Galipeau, P. C., Paulson, T. G., Rabi
novitch, P. S., and Reid, B. J. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat
Genet, 22: 106109, 1999.
39. Kimura, T., Maesawa, C., Ikeda, K., Wakabayashi, G., and Masuda, T. Mutations of the epi
dermal growth factor receptor gene in gastrointestinal tract tumor cell lines. Oncol Rep, 15:
12051210, 2006.
40. Koppert, L. B., Wijnhoven, B. P., van Dekken, H., Tilanus, H. W., and Dinjens, W. N. The mole
cular biology of esophageal adenocarcinoma. J Surg Oncol, 92: 169190, 2005.
41. van Baal, J. W., Milano, F., Rygiel, A. M., Bergman, J. J., Rosmolen, W. D., van Deventer, S. J.,
Wang, K. K., Peppelenbosch, M. P., and Krishnadath, K. K. A comparative analysis by SAGE
of gene expression profiles of Barrett’s esophagus, normal squamous esophagus, and gastric
cardia. Gastroenterology, 129: 12741281, 2005.
42. Hong, M. K., Laskin, W. B., Herman, B. E., Johnston, M. H., Vargo, J. J., Steinberg, S. M., Al
legra, C. J., and Johnston, P. G. Expansion of the Ki67 proliferative compartment correlates
with degree of dysplasia in Barrett’s esophagus. Cancer, 75: 423429, 1995.
43. Mueller, J., Werner, M., and Siewert, J. R. Malignant progression in Barrett’s esophagus: pa
thology and molecular biology. Recent Results Cancer Res, 155: 2941, 2000.
44. Miller, C. T., Moy, J. R., Lin, L., Schipper, M., Normolle, D., Brenner, D. E., Iannettoni, M. D.,
Orringer, M. B., and Beer, D. G. Gene amplification in esophageal adenocarcinomas and Bar
rett’s with highgrade dysplasia. Clin Cancer Res, 9: 48194825, 2003.
45. Daly, J. M., Fry, W. A., Little, A. G., Winchester, D. P., McKee, R. F., Stewart, A. K., and
Fremgen, A. M. Esophageal cancer: results of an American College of Surgeons Patient Care
Evaluation Study. J Am Coll Surg, 190: 562572; discussion 572563, 2000.
46. Peracchia, A., Bonavina, L., Via, A., and Incarbone, R. Current trends in the surgical treatment
of esophageal and cardia adenocarcinoma. J Exp Clin Cancer Res, 18: 289294, 1999.
47. Rosch, T. Endosonographic staging of esophageal cancer: a review of literature results. Gas
trointest Endosc Clin N Am, 5: 537547, 1995.
Milano_opm_v5.indd 29 16-08-2007 15:42:20

Chapter 1
30
48. Gee, D. W. and Rattner, D. W. Management of gastroesophageal tumors. Oncologist, 12: 175
185, 2007.
49. Rudiger Siewert, J., Feith, M., Werner, M., and Stein, H. J. Adenocarcinoma of the esophago
gastric junction: results of surgical therapy based on anatomical/topographic classification in
1,002 consecutive patients. Ann Surg, 232: 353361, 2000.
50. Bergman, J. J. Endoscopic resection for treatment of mucosal Barrett’s cancer: time to swing
the pendulum. Gastrointest Endosc, 65: 1113, 2007.
51. Sharma, V. K., Wang, K. K., Overholt, B. F., Lightdale, C. J., Fennerty, M. B., Dean, P. J., Ples
kow, D. K., Chuttani, R., Reymunde, A., Santiago, N., Chang, K. J., Kimmey, M. B., and Flei
scher, D. E. Balloonbased, circumferential, endoscopic radiofrequency ablation of Barrett’s
esophagus: 1year followup of 100 patients. Gastrointest Endosc, 65: 185195, 2007.
52. Prasad, G. A., Wang, K. K., Joyce, A. M., Kochman, M. L., Lutzke, L. S., and Borkenhagen,
L. S. Endoscopic therapy in patients with Barrett’s esophagus and portal hypertension. Gas
trointest Endosc, 65: 527531, 2007.
53. Stein, H. J. and Siewert, J. R. Improved prognosis of resected esophageal cancer. World J Surg,
28: 520525, 2004.
54. Wu, P. C. and Posner, M. C. The role of surgery in the management of oesophageal cancer.
Lancet Oncol, 4: 481488, 2003.
55. Law, S., Kwong, D. L., Kwok, K. F., Wong, K. H., Chu, K. M., Sham, J. S., and Wong, J. Im
provement in treatment results and longterm survival of patients with esophageal cancer:
impact of chemoradiation and change in treatment strategy. Ann Surg, 238: 339347; discus
sion 347338, 2003.
56. Lepage, C., Bouvier, A. M., Manfredi, S., Coatmeur, O., Cheynel, N., and Faivre, J. Trends in
incidence and management of esophageal adenocarcinoma in a welldefined population.
Gastroenterol Clin Biol, 29: 12581263, 2005.
57. Tseng, E. E., Wu, T. T., Yeo, C. J., and Heitmiller, R. F. Barrett’s esophagus with high grade
dysplasia: surgical results and longterm outcomean update. J Gastrointest Surg, 7: 164170;
discussion 170161, 2003.
58. Korst, R. J., Rusch, V. W., Venkatraman, E., Bains, M. S., Burt, M. E., Downey, R. J., and
Ginsberg, R. J. Proposed revision of the staging classification for esophageal cancer. J Thorac
Cardiovasc Surg, 115: 660669; discussion 669670, 1998.
59. Portale, G., Hagen, J. A., Peters, J. H., Chan, L. S., DeMeester, S. R., Gandamihardja, T. A.,
and DeMeester, T. R. Modern 5year survival of resectable esophageal adenocarcinoma:
single institution experience with 263 patients. J Am Coll Surg, 202: 588596; discussion 596
588, 2006.
60. Xinopoulos, D., Dimitroulopoulos, D., Tsamakidis, K., Korkolis, D., Fotopoulou, A., Bazinis,
A., Kontis, M., Vasilopoulos, P., and Paraskevas, E. Palliative treatment of advanced esopha
geal cancer with metalcovered expandable stents. A costeffectiveness and quality of life
study. J Buon, 10: 523528, 2005.
61. Homs, M. Y., Steyerberg, E. W., Kuipers, E. J., van der Gaast, A., Haringsma, J., van Blanken
stein, M., and Siersema, P. D. Causes and treatment of recurrent dysphagia after selfexpan
Milano_opm_v5.indd 30 16-08-2007 15:42:20

Chapter 1
31
ding metal stent placement for palliation of esophageal carcinoma. Endoscopy, 36: 880886,
2004.
62. Sihvo, E. I., Luostarinen, M. E., and Salo, J. A. Fate of patients with adenocarcinoma of the
esophagus and the esophagogastric junction: a populationbased analysis. Am J Gastroente
rol, 99: 419424, 2004.
63. Dipetrillo, T., Milas, L., Evans, D., Akerman, P., Ng, T., Miner, T., Cruff, D., Chauhan, B., Iannitti,
D., Harrington, D., and Safran, H. Paclitaxel poliglumex (PPXXyotax) and concurrent radiation
for esophageal and gastric cancer: a phase I study. Am J Clin Oncol, 29: 376379, 2006.
64. Arnott, S. J., Duncan, W., Gignoux, M., Girling, D. J., Hansen, H. S., Launois, B., Nygaard, K.,
Parmar, M. K., Roussel, A., Spiliopoulos, G., Stewart, L. A., Tierney, J. F., Mei, W., and Rugang,
Z. Preoperative radiotherapy in esophageal carcinoma: a metaanalysis using individual patient
data (Oesophageal Cancer Collaborative Group). Int J Radiat Oncol Biol Phys, 41: 579583, 1998.
65. Blackstock, A. W., Aklilu, M., Lovato, J., Farmer, M. R., Mishra, G., Melin, S. A., Oaks, T.,
Geisinger, K., and Levine, E. A. Pathologic complete response may not represent the optimal
surrogate for survival after preoperative therapy for esophageal cancer. Int J Gastrointest
Cancer, 37: 714, 2006.
66. Cooper, J. S., Guo, M. D., Herskovic, A., Macdonald, J. S., Martenson, J. A., Jr., AlSarraf, M.,
Byhardt, R., Russell, A. H., Beitler, J. J., Spencer, S., Asbell, S. O., Graham, M. V., and Leich
man, L. L. Chemoradiotherapy of locally advanced esophageal cancer: longterm followup of
a prospective randomized trial (RTOG 8501). Radiation Therapy Oncology Group. Jama, 281:
16231627, 1999.
67. Minsky, B. D., Pajak, T. F., Ginsberg, R. J., Pisansky, T. M., Martenson, J., Komaki, R., Oka
wara, G., Rosenthal, S. A., and Kelsen, D. P. INT 0123 (Radiation Therapy Oncology Group
9405) phase III trial of combinedmodality therapy for esophageal cancer: highdose versus
standarddose radiation therapy. J Clin Oncol, 20: 11671174, 2002.
68. Walsh, T. N., Noonan, N., Hollywood, D., Kelly, A., Keeling, N., and Hennessy, T. P. A compa
rison of multimodal therapy and surgery for esophageal adenocarcinoma. N Engl J Med, 335:
462467, 1996.
69. Kim, D. W., Blanke, C. D., Wu, H., Shyr, Y., Berlin, J., Beauchamp, R. D., and Chakravarthy,
B. Phase II study of preoperative paclitaxel/cisplatin with radiotherapy in locally advanced
esophageal cancer. Int J Radiat Oncol Biol Phys, 67: 397404, 2007.
70. Wright, C. D., Wain, J. C., Lynch, T. J., Choi, N. C., Grossbard, M. L., Carey, R. W., Moncure, A.
C., Grillo, H. C., and Mathisen, D. J. Induction therapy for esophageal cancer with paclitaxel
and hyperfractionated radiotherapy: a phase I and II study. J Thorac Cardiovasc Surg, 114:
811815; discussion 816, 1997.
71. Kelsen, D. P., Ginsberg, R., Pajak, T. F., Sheahan, D. G., Gunderson, L., Mortimer, J., Estes, N.,
Haller, D. G., Ajani, J., Kocha, W., Minsky, B. D., and Roth, J. A. Chemotherapy followed by
surgery compared with surgery alone for localized esophageal cancer. N Engl J Med, 339:
19791984, 1998.
72. Reynolds, J. V., Ravi, N., Hollywood, D., Kennedy, M. J., Rowley, S., Ryan, A., Hughes, N.,
Carey, M., and Byrne, P. Neoadjuvant chemoradiation may increase the risk of respiratory
Milano_opm_v5.indd 31 16-08-2007 15:42:21

Chapter 1
32
complications and sepsis after transthoracic esophagectomy. J Thorac Cardiovasc Surg, 132:
549555, 2006.
73. Nauts, H. C. Bacteria and cancerantagonisms and benefits. Cancer Surv, 8: 713723, 1989.
74. Hoption Cann, S. A., van Netten, J. P., and van Netten, C. Dr William Coley and tumour
regression: a place in history or in the future. Postgrad Med J, 79: 672680, 2003.
75. Gilboa, E. The promise of cancer vaccines. Nat Rev Cancer, 4: 401411, 2004.
76. Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. Cancer immunoediting:
from immunosurveillance to tumor escape. Nat Immunol, 3: 991998, 2002.
77. Shankaran, V., Ikeda, H., Bruce, A. T., White, J. M., Swanson, P. E., Old, L. J., and Schreiber,
R. D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour
immunogenicity. Nature, 410: 11071111, 2001.
78. Maleckar, J. R. and Sherman, L. A. The composition of the T cell receptor repertoire in nude
mice. J Immunol, 138: 38733876, 1987.
79. Dunn, G. P., Old, L. J., and Schreiber, R. D. The three Es of cancer immunoediting. Annu Rev
Immunol, 22: 329360, 2004.
80. Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic
relevance. Nat Rev Cancer, 5: 263274, 2005.
81. Smyth, M. J., Dunn, G. P., and Schreiber, R. D. Cancer immunosurveillance and immunoedi
ting: the roles of immunity in suppressing tumor development and shaping tumor immuno
genicity. Adv Immunol, 90: 150, 2006.
82. Kim, R., Emi, M., and Tanabe, K. Cancer immunoediting from immune surveillance to im
mune escape. Immunology, 121: 114, 2007.
83. Bui, J. D. and Schreiber, R. D. Cancer immunosurveillance, immunoediting and inflammation:
independent or interdependent processes? Curr Opin Immunol, 19: 203208, 2007.
84. Balkwill, F. and Mantovani, A. Inflammation and cancer: back to Virchow? Lancet, 357: 539
545, 2001.
85. Coussens, L. M. and Werb, Z. Inflammation and cancer. Nature, 420: 860867, 2002.
86. Kim, R., Emi, M., Tanabe, K., and Arihiro, K. Tumordriven evolution of immunosuppressive
networks during malignant progression. Cancer Res, 66: 55275536, 2006.
87. Smyth, M. J., Godfrey, D. I., and Trapani, J. A. A fresh look at tumor immunosurveillance and
immunotherapy. Nat Immunol, 2: 293299, 2001.
88. Banchereau, J. and Steinman, R. M. Dendritic cells and the control of immunity. Nature, 392:
245252, 1998.
89. Banchereau, J., Briere, F., Caux, C., Davoust, J., Lebecque, S., Liu, Y. J., Pulendran, B., and
Palucka, K. Immunobiology of dendritic cells. Annu Rev Immunol, 18: 767811, 2000.
90. Berger, T. G., Feuerstein, B., Strasser, E., Hirsch, U., Schreiner, D., Schuler, G., and Schuler
Thurner, B. Largescale generation of mature monocytederived dendritic cells for clinical
application in cell factories. J Immunol Methods, 268: 131140, 2002.
91. Ardavin, C., Amigorena, S., and Reis e Sousa, C. Dendritic cells: immunobiology and cancer
immunotherapy. Immunity, 20: 1723, 2004.
92. Sallusto, F. and Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human
Milano_opm_v5.indd 32 16-08-2007 15:42:21

Chapter 1
33
dendritic cells is maintained by granulocyte/macrophage colonystimulating factor plus inter
leukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med, 179: 11091118, 1994.
93. Brossart, P., Grunebach, F., Stuhler, G., Reichardt, V. L., Mohle, R., Kanz, L., and Brugger, W.
Generation of functional human dendritic cells from adherent peripheral blood monocytes by
CD40 ligation in the absence of granulocytemacrophage colonystimulating factor. Blood, 92:
42384247, 1998.
94. Dhodapkar, M. V., Krasovsky, J., Steinman, R. M., and Bhardwaj, N. Mature dendritic cells
boost functionally superior CD8(+) Tcell in humans without foreign helper epitopes. J Clin
Invest, 105: R9R14, 2000.
95. Nencioni, A. and Brossart, P. Cellular immunotherapy with dendritic cells in cancer: current
status. Stem Cells, 22: 501513, 2004.
96. Banchereau, J., SchulerThurner, B., Palucka, A. K., and Schuler, G. Dendritic cells as vectors
for therapy. Cell, 106: 271274, 2001.
97. Shortman, K., Vremec, D., Corcoran, L. M., Georgopoulos, K., Lucas, K., and Wu, L. The link
age between Tcell and dendritic cell development in the mouse thymus. Immunol Rev, 165:
3946, 1998.
98. Inaba, K., Inaba, M., Romani, N., Aya, H., Deguchi, M., Ikehara, S., Muramatsu, S., and Stein
man, R. M. Generation of large numbers of dendritic cells from mouse bone marrow cultures
supplemented with granulocyte/macrophage colonystimulating factor. J Exp Med, 176:
16931702, 1992.
99. Saunders, D., Lucas, K., Ismaili, J., Wu, L., Maraskovsky, E., Dunn, A., and Shortman, K.
Dendritic cell development in culture from thymic precursor cells in the absence of granulo
cyte/macrophage colonystimulating factor. J Exp Med, 184: 21852196, 1996.
100. Wu, L., Li, C. L., and Shortman, K. Thymic dendritic cell precursors: relationship to the T
lymphocyte lineage and phenotype of the dendritic cell progeny. J Exp Med, 184: 903911,
1996.
101. Bell, D., Young, J. W., and Banchereau, J. Dendritic cells. Adv Immunol, 72: 255324, 1999.
102. Caux, C., Vanbervliet, B., Massacrier, C., DezutterDambuyant, C., de SaintVis, B., Jacquet,
C., Yoneda, K., Imamura, S., Schmitt, D., and Banchereau, J. CD34+ hematopoietic progeni
tors from human cord blood differentiate along two independent dendritic cell pathways in
response to GMCSF+TNF alpha. J Exp Med, 184: 695706, 1996.
103. Ito, T., Inaba, M., Inaba, K., Toki, J., Sogo, S., Iguchi, T., Adachi, Y., Yamaguchi, K., Amakawa,
R., Valladeau, J., Saeland, S., Fukuhara, S., and Ikehara, S. A CD1a+/CD11c+ subset of human
blood dendritic cells is a direct precursor of Langerhans cells. J Immunol, 163: 14091419, 1999.
104. Grouard, G., Rissoan, M. C., Filgueira, L., Durand, I., Banchereau, J., and Liu, Y. J. The enig
matic plasmacytoid T cells develop into dendritic cells with interleukin (IL)3 and CD40li
gand. J Exp Med, 185: 11011111, 1997.
105. Siegal, F. P., Kadowaki, N., Shodell, M., FitzgeraldBocarsly, P. A., Shah, K., Ho, S., Antonenko,
S., and Liu, Y. J. The nature of the principal type 1 interferonproducing cells in human blood.
Science, 284: 18351837, 1999.
106. Cella, M., Jarrossay, D., Facchetti, F., Alebardi, O., Nakajima, H., Lanzavecchia, A., and
Milano_opm_v5.indd 33 16-08-2007 15:42:22

Chapter 1
34
Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large
amounts of type I interferon. Nat Med, 5: 919923, 1999.
107. Cella, M., Sallusto, F., and Lanzavecchia, A. Origin, maturation and antigen presenting func
tion of dendritic cells. Curr Opin Immunol, 9: 1016, 1997.
108. Caux, C., Massacrier, C., Vanbervliet, B., Dubois, B., Van Kooten, C., Durand, I., and Ban
chereau, J. Activation of human dendritic cells through CD40 crosslinking. J Exp Med, 180:
12631272, 1994.
109. Koch, F., Stanzl, U., Jennewein, P., Janke, K., Heufler, C., Kampgen, E., Romani, N., and Schu
ler, G. High level IL12 production by murine dendritic cells: upregulation via MHC class II
and CD40 molecules and downregulation by IL4 and IL10. J Exp Med, 184: 741746, 1996.
110. Ludewig, B., Graf, D., Gelderblom, H. R., Becker, Y., Kroczek, R. A., and Pauli, G. Spontane
ous apoptosis of dendritic cells is efficiently inhibited by TRAP (CD40ligand) and TNFal
pha, but strongly enhanced by interleukin10. Eur J Immunol, 25: 19431950, 1995.
111. NouriShirazi, M., Banchereau, J., Fay, J., and Palucka, K. Dendritic cell based tumor vaccines.
Immunol Lett, 74: 510, 2000.
112. Steinman, L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis
of T cellmediated tissue damage. Nat Med, 13: 139145, 2007.
113. Groux, H., O’Garra, A., Bigler, M., Rouleau, M., Antonenko, S., de Vries, J. E., and Roncarolo,
M. G. A CD4+ Tcell subset inhibits antigenspecific Tcell responses and prevents colitis.
Nature, 389: 737742, 1997.
114. Enk, A. H., Jonuleit, H., Saloga, J., and Knop, J. Dendritic cells as mediators of tumorinduced
tolerance in metastatic melanoma. Int J Cancer, 73: 309316, 1997.
115. Gilboa, E. The makings of a tumor rejection antigen. Immunity, 11: 263270, 1999.
116. Maier, T., TunKyi, A., Tassis, A., Jungius, K. P., Burg, G., Dummer, R., and Nestle, F. O. Vac
cination of patients with cutaneous Tcell lymphoma using intranodal injection of autologous
tumorlysatepulsed dendritic cells. Blood, 102: 23382344, 2003.
117. Muller, M. R., Grunebach, F., Kayser, K., Vogel, W., Nencioni, A., Brugger, W., Kanz, L., and
Brossart, P. Expression of her2/neu on acute lymphoblastic leukemias: implications for the
development of immunotherapeutic approaches. Clin Cancer Res, 9: 34483453, 2003.
118. Nair, S. K., Snyder, D., Rouse, B. T., and Gilboa, E. Regression of tumors in mice vaccinated with
professional antigenpresenting cells pulsed with tumor extracts. Int J Cancer, 70: 706715, 1997.
119. Brossart, P., Wirths, S., Brugger, W., and Kanz, L. Dendritic cells in cancer vaccines. Exp He
matol, 29: 12471255, 2001.
120. Schuler, G., SchulerThurner, B., and Steinman, R. M. The use of dendritic cells in cancer im
munotherapy. Curr Opin Immunol, 15: 138147, 2003.
121. Brossart, P., Stuhler, G., Flad, T., Stevanovic, S., Rammensee, H. G., Kanz, L., and Brugger, W.
Her2/neuderived peptides are tumorassociated antigens expressed by human renal cell
and colon carcinoma lines and are recognized by in vitro induced specific cytotoxic T lymp
hocytes. Cancer Res, 58: 732736, 1998.
122. Babatz, J., Rollig, C., Lobel, B., Folprecht, G., Haack, M., Gunther, H., Kohne, C. H., Ehninger,
G., Schmitz, M., and Bornhauser, M. Induction of cellular immune responses against carcin
Milano_opm_v5.indd 34 16-08-2007 15:42:22

Chapter 1
35
oembryonic antigen in patients with metastatic tumors after vaccination with altered peptide
ligandloaded dendritic cells. Cancer Immunol Immunother, 55: 268276, 2006.
123. Tuyaerts, S., Aerts, J. L., Corthals, J., Neyns, B., Heirman, C., Breckpot, K., Thielemans, K.,
and Bonehill, A. Current approaches in dendritic cell generation and future implications for
cancer immunotherapy. Cancer Immunol Immunother, 2007.
124. Fong, L. and Engleman, E. G. Dendritic cells in cancer immunotherapy. Annu Rev Immunol,
18: 245273, 2000.
125. Tuting, T., Wilson, C. C., Martin, D. M., Kasamon, Y. L., Rowles, J., Ma, D. I., Slingluff, C. L.,
Jr., Wagner, S. N., van der Bruggen, P., Baar, J., Lotze, M. T., and Storkus, W. J. Autologous
human monocytederived dendritic cells genetically modified to express melanoma antigens
elicit primary cytotoxic T cell responses in vitro: enhancement by cotransfection of genes
encoding the Th1biasing cytokines IL12 and IFNalpha. J Immunol, 160: 11391147, 1998.
126. Strobel, I., Berchtold, S., Gotze, A., Schulze, U., Schuler, G., and Steinkasserer, A. Human den
dritic cells transfected with either RNA or DNA encoding influenza matrix protein M1 differ
in their ability to stimulate cytotoxic T lymphocytes. Gene Ther, 7: 20282035, 2000.
127. Van Tendeloo, V. F., Ponsaerts, P., Lardon, F., Nijs, G., Lenjou, M., Van Broeckhoven, C., Van
Bockstaele, D. R., and Berneman, Z. N. Highly efficient gene delivery by mRNA electroporation in
human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to elec
troporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood, 98: 4956, 2001.
128. Heiser, A., Maurice, M. A., Yancey, D. R., Coleman, D. M., Dahm, P., and Vieweg, J. Human
dendritic cells transfected with renal tumor RNA stimulate polyclonal Tcell responses against
antigens expressed by primary and metastatic tumors. Cancer Res, 61: 33883393, 2001.
129. Cisco, R. M., AbdelWahab, Z., Dannull, J., Nair, S., Tyler, D. S., Gilboa, E., Vieweg, J., Daaka,
Y., and Pruitt, S. K. Induction of human dendritic cell maturation using transfection with
RNA encoding a dominant positive tolllike receptor 4. J Immunol, 172: 71627168, 2004.
130. Mayordomo, J. I., Loftus, D. J., Sakamoto, H., De Cesare, C. M., Appasamy, P. M., Lotze, M.
T., Storkus, W. J., Appella, E., and DeLeo, A. B. Therapy of murine tumors with p53 wildtype
and mutant sequence peptidebased vaccines. J Exp Med, 183: 13571365, 1996.
131. De Vries, I. J., Krooshoop, D. J., Scharenborg, N. M., Lesterhuis, W. J., Diepstra, J. H., Van
Muijen, G. N., Strijk, S. P., Ruers, T. J., Boerman, O. C., Oyen, W. J., Adema, G. J., Punt, C. J.,
and Figdor, C. G. Effective migration of antigenpulsed dendritic cells to lymph nodes in
melanoma patients is determined by their maturation state. Cancer Res, 63: 1217, 2003.
132. Mellman, I. and Steinman, R. M. Dendritic cells: specialized and regulated antigen processing
machines. Cell, 106: 255258, 2001.
133. Sallusto, F., Lanzavecchia, A., and Mackay, C. R. Chemokines and chemokine receptors in
Tcell priming and Th1/Th2mediated responses. Immunol Today, 19: 568574, 1998.
134. BarrattBoyes, S. M. and Figdor, C. G. Current issues in delivering DCs for immunotherapy.
Cytotherapy, 6: 105110, 2004.
135. Kuwashima, N., Nishimura, F., Eguchi, J., Sato, H., Hatano, M., Tsugawa, T., Sakaida, T.,
Dusak, J. E., FellowsMayle, W. K., Papworth, G. D., Watkins, S. C., Gambotto, A., Pollack, I.
F., Storkus, W. J., and Okada, H. Delivery of dendritic cells engineered to secrete IFNalpha
Milano_opm_v5.indd 35 16-08-2007 15:42:23

Chapter 1
36
into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines:
dependence on apoptotic pathways. J Immunol, 175: 27302740, 2005.
136. Tanaka, K., Ito, A., Kobayashi, T., Kawamura, T., Shimada, S., Matsumoto, K., Saida, T., and
Honda, H. Intratumoral injection of immature dendritic cells enhances antitumor effect of
hyperthermia using magnetic nanoparticles. Int J Cancer, 116: 624633, 2005.
137. Bedrosian, I., Mick, R., Xu, S., Nisenbaum, H., Faries, M., Zhang, P., Cohen, P. A., Koski, G., and
Czerniecki, B. J. Intranodal administration of peptidepulsed mature dendritic cell vaccines
results in superior CD8+ Tcell function in melanoma patients. J Clin Oncol, 21: 38263835, 2003.
138. Kitamura, K., Ikebe, M., Morita, M., Matsuda, H., Kuwano, H., and Sugimachi, K. The evalu
ation of submucosal carcinoma of the esophagus as a more advanced carcinoma. Hepatogas
troenterology, 40: 236239, 1993.
139. Jonuleit, H., GieseckeTuettenberg, A., Tuting, T., ThurnerSchuler, B., Stuge, T. B., Paragnik,
L., Kandemir, A., Lee, P. P., Schuler, G., Knop, J., and Enk, A. H. A comparison of two types of
dendritic cell as adjuvants for the induction of melanomaspecific Tcell responses in humans
following intranodal injection. Int J Cancer, 93: 243251, 2001.
140. Adema, G. J., de Vries, I. J., Punt, C. J., and Figdor, C. G. Migration of dendritic cell based
cancer vaccines: in vivo veritas? Curr Opin Immunol, 17: 170174, 2005.
141. Hsu, F. J., Benike, C., Fagnoni, F., Liles, T. M., Czerwinski, D., Taidi, B., Engleman, E. G., and
Levy, R. Vaccination of patients with Bcell lymphoma using autologous antigenpulsed den
dritic cells. Nat Med, 2: 5258, 1996.
142. SchulerThurner, B., Schultz, E. S., Berger, T. G., Weinlich, G., Ebner, S., Woerl, P., Bender, A.,
Feuerstein, B., Fritsch, P. O., Romani, N., and Schuler, G. Rapid induction of tumorspecific
type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopre
served, peptideloaded monocytederived dendritic cells. J Exp Med, 195: 12791288, 2002.
143. Banchereau, J., Palucka, A. K., Dhodapkar, M., Burkeholder, S., Taquet, N., Rolland, A.,
Taquet, S., Coquery, S., Wittkowski, K. M., Bhardwaj, N., Pineiro, L., Steinman, R., and Fay, J.
Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor
derived dendritic cell vaccine. Cancer Res, 61: 64516458, 2001.
144. Smithers, M., O’Connell, K., MacFadyen, S., Chambers, M., Greenwood, K., Boyce, A., Ab
dulJabbar, I., Barker, K., Grimmett, K., Walpole, E., and Thomas, R. Clinical response after
intradermal immature dendritic cell vaccination in metastatic melanoma is associated with
immune response to particulate antigen. Cancer Immunol Immunother, 52: 4152, 2003.
145. Butterfield, L. H., Ribas, A., Dissette, V. B., Amarnani, S. N., Vu, H. T., Oseguera, D., Wang, H.
J., Elashoff, R. M., McBride, W. H., Mukherji, B., Cochran, A. J., Glaspy, J. A., and Economou,
J. S. Determinant spreading associated with clinical response in dendritic cellbased immuno
therapy for malignant melanoma. Clin Cancer Res, 9: 9981008, 2003.
146. Fong, L., Brockstedt, D., Benike, C., Breen, J. K., Strang, G., Ruegg, C. L., and Engleman, E. G.
Dendritic cellbased xenoantigen vaccination for prostate cancer immunotherapy. J Immunol,
167: 71507156, 2001.
147. Heiser, A., Coleman, D., Dannull, J., Yancey, D., Maurice, M. A., Lallas, C. D., Dahm, P.,
Niedzwiecki, D., Gilboa, E., and Vieweg, J. Autologous dendritic cells transfected with pros
Milano_opm_v5.indd 36 16-08-2007 15:42:23

Chapter 1
37
tatespecific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin
Invest, 109: 409417, 2002.
148. Holtl, L., ZelleRieser, C., Gander, H., Papesh, C., Ramoner, R., Bartsch, G., Rogatsch, H., Barsoum,
A. L., Coggin, J. H., Jr., and Thurnher, M. Immunotherapy of metastatic renal cell carcinoma with
tumor lysatepulsed autologous dendritic cells. Clin Cancer Res, 8: 33693376, 2002.
149. O’Rourke, M. G., Johnson, M., Lanagan, C., See, J., Yang, J., Bell, J. R., Slater, G. J., Kerr, B. M.,
Crowe, B., Purdie, D. M., Elliott, S. L., Ellem, K. A., and Schmidt, C. W. Durable complete cli
nical responses in a phase I/II trial using an autologous melanoma cell/dendritic cell vaccine.
Cancer Immunol Immunother, 52: 387395, 2003.
150. Timmerman, J. M., Czerwinski, D. K., Davis, T. A., Hsu, F. J., Benike, C., Hao, Z. M., Taidi, B.,
Rajapaksa, R., Caspar, C. B., Okada, C. Y., van Beckhoven, A., Liles, T. M., Engleman, E. G.,
and Levy, R. Idiotypepulsed dendritic cell vaccination for Bcell lymphoma: clinical and im
mune responses in 35 patients. Blood, 99: 15171526, 2002.
151. Fong, L., Hou, Y., Rivas, A., Benike, C., Yuen, A., Fisher, G. A., Davis, M. M., and Engleman,
E. G. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor
immunotherapy. Proc Natl Acad Sci U S A, 98: 88098814, 2001.
152. Rabinovich, G. A., Gabrilovich, D., and Sotomayor, E. M. Immunosuppressive Strategies that
are Mediated by Tumor Cells. Annu Rev Immunol, 25: 267296, 2007.
153. Kryczek, I., Lange, A., Mottram, P., Alvarez, X., Cheng, P., Hogan, M., Moons, L., Wei, S., Zou,
L., Machelon, V., Emilie, D., Terrassa, M., Lackner, A., Curiel, T. J., Carmeliet, P., and Zou, W.
CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in
human ovarian cancers. Cancer Res, 65: 465472, 2005.
154. Sombroek, C. C., Stam, A. G., Masterson, A. J., Lougheed, S. M., Schakel, M. J., Meijer, C. J.,
Pinedo, H. M., van den Eertwegh, A. J., Scheper, R. J., and de Gruijl, T. D. Prostanoids play a
major role in the primary tumorinduced inhibition of dendritic cell differentiation. J Immu
nol, 168: 43334343, 2002.
155. Gabrilovich, D. I., Chen, H. L., Girgis, K. R., Cunningham, H. T., Meny, G. M., Nadaf, S., Ka
vanaugh, D., and Carbone, D. P. Production of vascular endothelial growth factor by human
tumors inhibits the functional maturation of dendritic cells. Nat Med, 2: 10961103, 1996.
156. Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol, 6: 295
307, 2006.
157. Zhou, G. and Levitsky, H. I. Natural regulatory T cells and de novoinduced regulatory T
cells contribute independently to tumorspecific tolerance. J Immunol, 178: 21552162, 2007.
158. Wang, H. Y. and Wang, R. F. Regulatory T cells and cancer. Curr Opin Immunol, 19: 217223,
2007.
159. Barnett, B., Kryczek, I., Cheng, P., Zou, W., and Curiel, T. J. Regulatory T cells in ovarian
cancer: biology and therapeutic potential. Am J Reprod Immunol, 54: 369377, 2005.
160. Shimizu, J., Yamazaki, S., and Sakaguchi, S. Induction of tumor immunity by removing
CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immu
nol, 163: 52115218, 1999.
161. Gabrilovich, D. I., Bronte, V., Chen, S. H., Colombo, M. P., Ochoa, A., OstrandRosenberg, S.,
Milano_opm_v5.indd 37 16-08-2007 15:42:24

Chapter 1
38
and Schreiber, H. The terminology issue for myeloidderived suppressor cells. Cancer Res, 67:
425; author reply 426, 2007.
162. Tuynman, J. B., Buskens, C. J., Kemper, K., ten Kate, F. J., Offerhaus, G. J., Richel, D. J., and
van Lanschot, J. J. Neoadjuvant selective COX2 inhibition downregulates important oncoge
nic pathways in patients with esophageal adenocarcinoma. Ann Surg, 242: 840849, discus
sion 849850, 2005.
163. Traina, T. A., Rugo, H. S., and Dickler, M. Bevacizumab for advanced breast cancer. Hematol
Oncol Clin North Am, 21: 303319, 2007.
164. von Rahden, B. H., Stein, H. J., PuhringerOppermann, F., and Sarbia, M. cmyc amplification
is frequent in esophageal adenocarcinoma and correlated with the upregulation of VEGFA
expression. Neoplasia, 8: 702707, 2006.
165. Albanell, J. and Gascon, P. Small molecules with EGFRTK inhibitor activity. Curr Drug Tar
gets, 6: 259274, 2005.
166. Baselga, J., Albanell, J., Ruiz, A., Lluch, A., Gascon, P., Guillem, V., Gonzalez, S., Sauleda, S.,
Marimon, I., Tabernero, J. M., Koehler, M. T., and Rojo, F. Phase II and tumor pharmacody
namic study of gefitinib in patients with advanced breast cancer. J Clin Oncol, 23: 53235333,
2005.
167. PiccartGebhart, M. J., Procter, M., LeylandJones, B., Goldhirsch, A., Untch, M., Smith, I., Gi
anni, L., Baselga, J., Bell, R., Jackisch, C., Cameron, D., Dowsett, M., Barrios, C. H., Steger, G.,
Huang, C. S., Andersson, M., Inbar, M., Lichinitser, M., Lang, I., Nitz, U., Iwata, H., Thoms
sen, C., Lohrisch, C., Suter, T. M., Ruschoff, J., Suto, T., Greatorex, V., Ward, C., Straehle, C.,
McFadden, E., Dolci, M. S., and Gelber, R. D. Trastuzumab after adjuvant chemotherapy in
HER2positive breast cancer. N Engl J Med, 353: 16591672, 2005.
168. Redjal, N., Chan, J. A., Segal, R. A., and Kung, A. L. CXCR4 inhibition synergizes with cyto
toxic chemotherapy in gliomas. Clin Cancer Res, 12: 67656771, 2006.
169. Sutmuller, R. P., van Duivenvoorde, L. M., van Elsas, A., Schumacher, T. N., Wildenberg, M.
E., Allison, J. P., Toes, R. E., Offringa, R., and Melief, C. J. Synergism of cytotoxic T lympho
cyteassociated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor
therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte
responses. J Exp Med, 194: 823832, 2001.
170. Nanda, N. K. and Sercarz, E. E. Induction of antiselfimmunity to cure cancer. Cell, 82: 1317,
1995.
171. Disis, M. L., Grabstein, K. H., Sleath, P. R., and Cheever, M. A. Generation of immunity to the
HER2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide
based vaccine. Clin Cancer Res, 5: 12891297, 1999.
172. Jankowski, J., Coghill, G., Hopwood, D., and Wormsley, K. G. Oncogenes and oncosuppres
sor gene in adenocarcinoma of the oesophagus. Gut, 33: 10331038, 1992.
173. Geddert, H., Zeriouh, M., Wolter, M., Heise, J. W., Gabbert, H. E., and Sarbia, M. Gene ampli
fication and protein overexpression of cerbb2 in Barrett carcinoma and its precursor lesions.
Am J Clin Pathol, 118: 6066, 2002.
174. Dahlberg, P. S., Jacobson, B. A., Dahal, G., Fink, J. M., Kratzke, R. A., Maddaus, M. A., and
Milano_opm_v5.indd 38 16-08-2007 15:42:24

Chapter 1
39
Ferrin, L. J. ERBB2 amplifications in esophageal adenocarcinoma. Ann Thorac Surg, 78: 1790
1800, 2004.
175. Reichelt, U., Duesedau, P., Tsourlakis, M., Quaas, A., Link, B. C., Schurr, P. G., Kaifi, J. T., Gros,
S. J., Yekebas, E. F., Marx, A., Simon, R., Izbicki, J. R., and Sauter, G. Frequent homogeneous
HER2 amplification in primary and metastatic adenocarcinoma of the esophagus. Mod
Pathol, 20: 120129, 2007.
176. Ooi, A., Takehana, T., Li, X., Suzuki, S., Kunitomo, K., Iino, H., Fujii, H., Takeda, Y., and Dobashi,
Y. Protein overexpression and gene amplification of HER2 and EGFR in colorectal cancers: an
immunohistochemical and fluorescent in situ hybridization study. Mod Pathol, 17: 895904, 2004.
177. Yamanaka, Y., Friess, H., Kobrin, M. S., Buchler, M., Kunz, J., Beger, H. G., and Korc, M.
Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Hum Pathol, 24:
11271134, 1993.
178. Perez, S. A., Sotiropoulou, P. A., Sotiriadou, N. N., Mamalaki, A., Gritzapis, A. D., Echner, H.,
Voelter, W., Pawelec, G., Papamichail, M., and Baxevanis, C. N. HER2/neuderived peptide
884899 is expressed by human breast, colorectal and pancreatic adenocarcinomas and is
recognized by invitroinduced specific CD4(+) T cell clones. Cancer Immunol Immunother,
50: 615624, 2002.
179. Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J. A., Wong, S. G., Keith, D. E., Levin, W. J.,
Stuart, S. G., Udove, J., Ullrich, A., and et al. Studies of the HER2/neu protooncogene in
human breast and ovarian cancer. Science, 244: 707712, 1989.
180. Kono, K., Rongcun, Y., Charo, J., Ichihara, F., Celis, E., Sette, A., Appella, E., Sekikawa, T.,
Matsumoto, Y., and Kiessling, R. Identification of HER2/neuderived peptide epitopes recog
nized by gastric cancerspecific cytotoxic T lymphocytes. Int J Cancer, 78: 202208, 1998.
181. Rongcun, Y., SalazarOnfray, F., Charo, J., Malmberg, K. J., Evrin, K., Maes, H., Kono, K.,
Hising, C., Petersson, M., Larsson, O., Lan, L., Appella, E., Sette, A., Celis, E., and Kiessling, R.
Identification of new HER2/neuderived peptide epitopes that can elicit specific CTL against
autologous and allogeneic carcinomas and melanomas. J Immunol, 163: 10371044, 1999.
182. Kono, K., Takahashi, A., Sugai, H., Fujii, H., Choudhury, A. R., Kiessling, R., and Matsumoto,
Y. Dendritic cells pulsed with HER2/neuderived peptides can induce specific Tcell respon
ses in patients with gastric cancer. Clin Cancer Res, 8: 33943400, 2002.
183. Peoples, G. E., Gurney, J. M., Hueman, M. T., Woll, M. M., Ryan, G. B., Storrer, C. E., Fisher, C.,
Shriver, C. D., Ioannides, C. G., and Ponniah, S. Clinical trial results of a HER2/neu (E75) vac
cine to prevent recurrence in highrisk breast cancer patients. J Clin Oncol, 23: 75367545, 2005.
184. Gritzapis, A. D., Mahaira, L. G., Perez, S. A., Cacoullos, N. T., Papamichail, M., and Baxevanis,
C. N. Vaccination with human HER2/neu (435443) CTL peptide induces effective antitumor
immunity against HER2/neuexpressing tumor cells in vivo. Cancer Res, 66: 54525460, 2006.
185. Mimura, K., Kono, K., Hanawa, M., Kanzaki, M., Nakao, A., Ooi, A., and Fujii, H. Trastuzu
mabmediated antibodydependent cellular cytotoxicity against esophageal squamous cell
carcinoma. Clin Cancer Res, 11: 48984904, 2005.
186. Slamon, D. J., LeylandJones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T.,
Eiermann, W., Wolter, J., Pegram, M., Baselga, J., and Norton, L. Use of chemotherapy plus a
Milano_opm_v5.indd 39 16-08-2007 15:42:24

Chapter 1
40
monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N
Engl J Med, 344: 783792, 2001.
187. Baselga, J., Tripathy, D., Mendelsohn, J., Baughman, S., Benz, C. C., Dantis, L., Sklarin, N.
T., Seidman, A. D., Hudis, C. A., Moore, J., Rosen, P. P., Twaddell, T., Henderson, I. C., and
Norton, L. Phase II study of weekly intravenous recombinant humanized antip185HER2
monoclonal antibody in patients with HER2/neuoverexpressing metastatic breast cancer. J
Clin Oncol, 14: 737744, 1996.
188. Montemurro, F., Valabrega, G., and Aglietta, M. Trastuzumabbased combination therapy for
breast cancer. Expert Opin Pharmacother, 5: 8196, 2004.
189. Valabrega, G., Montemurro, F., and Aglietta, M. Trastuzumab: mechanism of action, resistance
and future perspectives in HER2overexpressing breast cancer. Ann Oncol, 2007.
190. Delord, J. P., Allal, C., Canal, M., Mery, E., Rochaix, P., Hennebelle, I., Pradines, A., Chatelut,
E., Bugat, R., Guichard, S., and Canal, P. Selective inhibition of HER2 inhibits AKT signal
transduction and prolongs diseasefree survival in a micrometastasis model of ovarian carci
noma. Ann Oncol, 16: 18891897, 2005.
191. Klos, K. S., Zhou, X., Lee, S., Zhang, L., Yang, W., Nagata, Y., and Yu, D. Combined trastu
zumab and paclitaxel treatment better inhibits ErbB2mediated angiogenesis in breast
carcinoma through a more effective inhibition of Akt than either treatment alone. Cancer, 98:
13771385, 2003.
192. Molina, M. A., CodonyServat, J., Albanell, J., Rojo, F., Arribas, J., and Baselga, J. Trastuzumab
(herceptin), a humanized antiHer2 receptor monoclonal antibody, inhibits basal and activa
ted Her2 ectodomain cleavage in breast cancer cells. Cancer Res, 61: 47444749, 2001.
193. Clynes, R. A., Towers, T. L., Presta, L. G., and Ravetch, J. V. Inhibitory Fc receptors modulate
in vivo cytoxicity against tumor targets. Nat Med, 6: 443446, 2000.
194. Gennari, R., Menard, S., Fagnoni, F., Ponchio, L., Scelsi, M., Tagliabue, E., Castiglioni, F., Vil
lani, L., Magalotti, C., Gibelli, N., Oliviero, B., Ballardini, B., Da Prada, G., Zambelli, A., and
Costa, A. Pilot study of the mechanism of action of preoperative trastuzumab in patients with
primary operable breast tumors overexpressing HER2. Clin Cancer Res, 10: 56505655, 2004.
Milano_opm_v5.indd 40 16-08-2007 15:42:25

Chapter 2
An improved protocol for generation of
immuno-potent Dendritic Cells through
direct electroporation of CD14+ monocytes
Francesca Milano, Jantine W.P.M. van Baal, Agnieszka M. Rygiel, Jacques J.G.H.M. Bergman,
Sander J.H.van Deventer, Martien L. Kapsenberg, Maikel P. Peppelenbosch, Kausilia K. Krishnadath
J Immunol Methods. 2007 Apr 10;321(12):94106
Milano_opm_v5.indd 41 16-08-2007 15:42:25

Chapter 2
42
ABSTRACT
In this study we demonstrate a novel protocol showing that electroporation of CD14+ monocytes directly isolated from blood with Green Fluorescent Protein (GFP) RNA results in a 3fold higher yield of antigen presenting Dendritic Cells (DCs) when compared to conventional methods employing immature DCs for electroporation. We further show a stable electroporation efficacy resulting in 60% of GFP positive cells. Expression of costimulatory molecules and maturation markers such as CD80, CD86, CD83 as well of the chemokine receptor 7 (CCR7) was found in 90% of the mature DCs. Importantly, production of IL12p70 was 10 times higher in cells electroporated at the monocyte stage compared to cells electroporated at the immature DC stage. Stimulation of autologous naïve lymphocytes by DCs electroporated at monocytes stage elicited proliferation of CD8+ Tcell with 7 fold increase in IFNγ release. Blocking of the MHCClass I molecules significantly inhibited the IFNγ release, indicating that antigen presentation was MHCClass I mediated. In summary, electroporation of CD14+ monocytes with RNA results in a high yield of antigen presenting DCs with high immunostimulatory capacity and antigen presentation on MHC class I molecules. This improved method may represent an attractive approach for RNA based DC immunotherapy.
Milano_opm_v5.indd 42 16-08-2007 15:42:25

Chapter 2
43
INTRODUCTION
Cancer immunotherapy is a novel approach which has been investigated in the last few decades as an alternative therapy against oncogenic disease. Dendritic Cells (DCs) are professional antigen presenting cells (APCs) with a unique ability to capture and process tumor associated antigens for inducing Tcell mediated immune responses. In the last few years, the possibility to generate functional DCs from human peripheral blood monocytes that can be loaded with tumor antigens using various protocols has been explored extensively.1
5 In the past, most antigenbased vaccines employing either known tumor antigens or wholecell tumor lysates to initiate a specific Tcell response exhibited limited immunogenicity, raising concern that human cancer cells might possess inherently poor antigenicity, or that antigenpresentation and Tcell activation may be suppressed in cancer patients.6 Moreover, there is raising concern that the tumor itself is surrounded by an unfavorable environment that can induce immunotolerance and allow the tumor to escape from immune surveillance inducing deleterious effect on Tcells.7,8 For these reasons, several approaches have been employed to obtain high yields of immunopotent antigen presenting DCs. In the development of DCs vaccines, one of the first concerns is to obtain sufficient amounts of DCs that fully mature with preserved immunostimulatory capacity.9 Secondly, the DCs have to be loaded with tumor associated antigens able to induce a specific Tcell response against cancer cells. DCs have been successfully loaded with antigens using different methodology, such as lipofection, passive pulse, or transfected through electroporation with DNA, total RNA or mRNA of defined tumor antigens.1012 In general most assays employ blood or bone marrow monocytes that are first stimulated to become immature or mature DCs before cells are loaded with the antigens.13
Electroporation proves to be a more efficient and frequently applied method for antigen loading of DCs compared to other methods.10 This procedure, however, is associated with a significant loss of cells. It appears that in vivo the capacity to interiorize and process antigens is a constitutive property of immature DCs when present in nonlymphoid organs.14 Therefore most researchers rely on employing immature DCs for electroporation. Yet, electroporation of monocytes directly isolated from blood has been described as well.15 Electroporating monocytes directly isolated from peripheral blood without any preincubation, would give the possibility to avoid several laborious cell culturing and harvesting steps leading to a more efficient protocol for obtaining antigen loaded DCs. Moreover, monocytes may be more resistant against the electroporation procedure itself that may lead to a significant higher recovery of cells after the electroporation technique. It is, however, not known whether electroporated monocytes would efficiently present the electroporated antigens and mature into potent APCs. In
Milano_opm_v5.indd 43 16-08-2007 15:42:25

Chapter 2
44
this study we present a novel efficient protocol for obtaining high yields of potent DCs through electroporation of monocytes directly isolated from peripheral blood. We first examined whether we could achieve stabile electroporation of monocytes with Green Fluorescent Protein (GFP) RNA and maintain a persistent expression of the antigens during the maturation process into DCs. We measured the cell recovery rate of monocytes after the electroporation procedure and compared it to that of immature DCs. We tested whether the electroporated monocytes subsequently matured into effective immunopotent DCs, expressing costimulatory molecules and exhibiting a mature phenotype. We looked into the production of pro and antiinflammatory cytokines such as IL12p70 and of IL10 and compared our protocol with the more conventional method, in which immature DCs are employed for electroporation. Finally, we studied the immunostimulatory ability of the mature DCs for inducing a CD8+ Tcell response, measuring the CD4+/CD8+ Tcell ratio, Tcell viability and Tcell proliferation and looked into IFNγ production. This study demonstrates that electroporation of monocytes directly isolated from blood is an efficient procedure that results in a 3 times higher yield of immunopotent DCs when compared to the more conventional electroporation of immature DCs. The presented protocol may considerably facilitate the manufacturing of DCs and may greatly enhance further development of future DCs RNAbased vaccines.
MATERIALS AND METHODS
Isolation of monocytes and lymphocytes from peripheral blood Human monocytes were isolated from peripheral blood of healthy volunteers using the FicollPercoll gradient separation method. Sixty four ml of blood was drawn from each volunteer. A first separation of peripheral blood mononuclear cells (PBMCs) was done using FicollHypaque solution (Amersham, Pharmacia, Piscataway, NJ, USA). A further separation of monocytes from lymphocytes was done using a Percoll (Amersham Biosciences Europe Freiburg, Germany) gradient separation, following manufacture’s protocol: briefly, PBMCs were washed in RPMI 1640 medium (BioWhittakerCambrex Bioscience, Walkersville, MD, USA) at 1500 rpm for 5 minutes twice. At the mean time, Percoll was mixed with 10X Phosphate Buffer Saline (PBS) to obtain Standard Isotone Percoll solution (SIP), and then with IMDM medium (BioWhittaker), to obtain three solutions at different concentration (60, 47.5 and 34% SIP). PBMCs were then resuspended in 2.5 ml 60% SIP, then 5 ml 47.5% SIP and finally 2 ml 34% SIP were added to the 60%SIP cell suspension and a centrifugation at 3100 rpm for 45 minutes was performed. Once monocytes were separated from lymphocytes, both populations were resuspended in IMDM and washed twice. Monocytes
Milano_opm_v5.indd 44 16-08-2007 15:42:26

Chapter 2
45
and lymphocytes were counted and either used immediately or cryopreserved in a DMSO/FCS (2:8) solution (Merck, Darmstadt, Germany; GIBCO BRL, Grand Island, NY, USA, respectively).
Electroporation of monocytes with In Vitro Transcribed (IVT) GFP-RNA Monocytes characterized as CD14+, CD45+, CD80, CD86, CD83 and CD209 (Fig.2A) were used for electroporation with IVT GFPRNA as follows: Monocytes freshly isolated from blood were washed twice in IMDM and electroporated using the Amaxa cell line Nucleofector Kit V (Amaxa GmBh, Cologne, Germany) : 2x106 cells were mixed with Nucleofector transfection solution V, and 5 μg/ml of IVT GFPRNA or 1 μg/ml of autologous normal RNA (control); the program U16 of the Amaxa transfection machine was used. Likewise, immature DCs, harvested after six days of culturing and characterized as CD14 and CD80+, CD83+, CD86+ and CD209+, were electroporated with IVT GFPRNA using the same procedure as described above.
Maturation of electroporated monocytes into DCs.For maturing the monocytes into DCs the protocol as described by Braat et al.16 was used. Upon electroporation monocytes characterized as CD14+, CD45+, CD80, CD86, CD83 and CD209 (Fig.2A) were immediately cultured in IMDM medium with 10% FCS and 2% antibiotics (GIBCO) in 24 wells plate in a concentration of 5x105 cells/ml and left for 1 hour at 37˚C and 5%CO2 for the adherence step. After one hour from the electroporation, cells were washed with IMDM, than 1000 U/ml of IL4 and 800 U/ml GMCSF were added to initiate the maturation process. After six days at the stage of immature DCs, the proinflammation cytokines IL1β and TNFα and LPS (10 ng/ml, 25 ng/ml, 0.02 ng/ml respectively) were added to further enhance the maturation process. After 2 more days of maturation, cells proved to have a mature phenotype characterized as: CD14, CD45, CD80+, CD86+, CD83+ and CD209+ (Fig 2B).
Detection of markers in monocytes and DCs Detection of CD14, CD45, CD80, CD83, CD86, CD209 and CCR7 in monocytes, immature and mature DCs was as follows: cells were harvested, washed and resuspended in a concentration of 1x106 in FACS buffer (5 g BSA, 0.1 g NaN3, 100 mM EDTA in 1 L PBS), then incubated with various fluorochromeconjugated antibodies on ice for 30 minutes in the dark, then washed again and analyzed with the FACSCALIBUR apparatus (BectonDickinson, Franklin Lakes, NJ, USA); antibodies PE conjugated, specific for CD14, CD45, CD86, CD80 ,CD83, CD209 and CCR7 (BD, San Jose, CA, USA) were used, in a concentration of 1:25.
Milano_opm_v5.indd 45 16-08-2007 15:42:26

Chapter 2
46
Electroporation efficacy in monocytes as measured by GFP expression during maturation into DCs The electroporation efficacy of GFPRNA was assessed through measuring of GFP expression by FACS analysis. Briefly, the procedure was as follows: cells were harvested and spun down at 1500 rpm for 10 minutes, then resuspended in FACS buffer in a concentration of 1x106 cells/ml and sorted by the FACS apparatus for direct measurement of GFP expression. GFP expression was measured in monocytes before electroporation (day one) and daily on eight subsequent days after electroporation, during the maturation of the monocytes into immature and mature DCs. For each time point GFP expression was measured in three different samples.
Cell loss and recovery due to culturing and electroporation of monocytes and immature DCsFor assessing cell loss due to the process of culturing and electroporation of cells the Trypan Blue viability test was used following the manufacture’s procedure. Two millions monocytes freshly isolated from blood were directly electroporated and immediately plated in 24 well plates in a concentration of 5x105 cells/well, at 37˚C and 5%CO2. After 8 days of culturing, at the mature DC stage, cells were harvested and counted to assess the recovery rate after electroporation. To obtain DCs at the immature stage, monocytes freshly isolated were directly plated in 24 well plates at the concentration of 5x105 cell/well, and stimulated for six days to differentiate into immature DCs (see above). After six days of culturing, at the immature DC stage, cells were electroporated and immediately plated in 24 well plates in a concentration of 5x105 cell/well, at 37˚C and 5%CO2. After 48 hours, cells were harvested and counted to assess the recovery rate of immature DCs after electroporation.
Stimulations of autologous lymphocytes with mature DCs. For stimulation of the autologous lymphocytes, lymphocytes and DCs were incubated in a proportion of 1:4 (5x105 DCs and 2x106 lymphocytes) in 24 wells plate in IMDM medium with 5% FCS and 2% penicillinstreptomycin (Gibco). Cells were left for 7 days in the autologous mixed cellular reaction. A second stimulation was performed after 7 days and cells were again cocultured for one week. Before and after the first and second stimulations aliquots of lymphocytes were taken to measure changes in the CD4+/CD8+ ratio by FACS analysis.
Cytometric Bead Array (CBA) Multiplex AssaysMonocytes were matured into DCs, and at day 8 supernatants of the cultured cells were collected and used for measuring cytokine production using the Cytometric
Milano_opm_v5.indd 46 16-08-2007 15:42:26

Chapter 2
47
Bead Array (CBA). The analysis was done using the CBA Inflammation kit (BD), and the standard procedure was followed, as previously described.17 Briefly the principle of the technique is the following: different particles are labelled with a fluorescent dye which has an emission wavelength of 650 nm. Each different group of beads is labelled with such a level of fluorescent dye so that it can be distinguished by its Mean Fluorescent Intensity (MFI) by flow cytometric analysis. Beads are also coupled with antibodies that specifically recognize the molecule of interest present in the supernatant. The captured molecule is then detected through addition of a secondary fluorescent antibody. For each supernatant sample and cytokine standard mixture, 10 μl of sample or standard was added to 10 μl of the solution of the mixed bead and 10 μl of PE detection reagent. The mixture was incubated for 3 hours at RT in the dark, and afterwards data was acquired by flow cytometry using the Plate Manager ® software.
MHC-Class I restriction and IFN-γ releaseTo asses whether the Tcell response was MHCClass I mediated, DCs were incubated with a MHCClass I blocking antibody (W6/32 clone, Acris, Hiddenhausen, Germany) a method which has been previously described by Liu et al.18 DCs were incubated with the antiMHCClass I antibody for 1 hour at 37˚C before the addition of lymphocytes. Lymphocytes were incubated for 5 hours at 37˚C with the DCs in a 24 wells plate. The supernatants of each sample were immediately collected and used for detecting the IFNγ release using an ELISA kit (Biosource, Camarillo, CA, USA). IFNγ release was measured with and without inhibition of the MHCClass I molecules by using multiwell scanning spectrophotometer (ELISA reader).
CD4/CD8 ratios before and after stimulation of lymphocytes with DCsBefore and after the first and the second incubation of lymphocytes with the electroporated DCs, lymphocytes were harvested, spun down at 1250 rpm for 5 minutes and resuspended in FACS buffer in a concentration of 1x106 cells/ml. Cells were then stained in the dark for 30 minutes with the following antibodies: antihuman CD3 PE conjugated, antihuman CD4 FITC conjugated, antihuman CD8 PerCyp conjugated (R&D systems, Minneapolis, MN, USA); the antibodies were used in a concentration of 1:25.
Mixed Leucocytes Reaction (MLR)The capacity of DCs to present antigens and evoke a lymphocytes response was tested in an autologous Mixed Leucocytes Reaction (MLR). Briefly, mature DCs electroporated either at monocytes or at immature DCs stage were cocultured in flatbottom 96well plates (Nunc, Roskilde, Denmark) in IMDM culture medium at different targeteffector ratios, starting from 4x103/well, down to 1.25x102 DCs
Milano_opm_v5.indd 47 16-08-2007 15:42:27

Chapter 2
48
(APC) per well, while each well contained a fixed number of 5x104 lymphocytes.
After 5 days in culture, 0.2 μCi [3H] thymidine (Amersham Biosciences, Amersham, UK) was added to each well and the incorporation of radioactivity was measured after 16 h using liquid scintillation counting.
MTT assayBefore and after stimulation of the lymphocytes by the DCs electroporated either by IVT GFPRNA or normal autologous RNA (control), an aliquot of Tcells was examined for assessing the viability rate by an MTT assay. MTT[3(4,5dimethylthiazol2yl)2,5diphenyltetrazolium bromide] assay, first described by Mosmann et al.19 is based on the ability of a mitochondrial dehydrogenase enzyme from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form a dark blue formazan crystals which is largely impermeable to cell membranes, thus resulting in its accumulation within healthy cells. Addition of a detergent results in the liberation of the crystals which are solubilized. The number of surviving cells is directly proportional to the level of the formazan product created. The color can then be quantified using a simple colorimetric assay. The results were read on a multiwell scanning spectrophotometer (ELISA reader).
RESULTS
Lymphocyte and monocytes yields from peripheral bloodTwelve volunteers were included, 8 male and 4 female, with a mean age of 30 (Range 2438). Sixty four ml of blood was taken from each volunteer. The average yield of monocytes per volunteer was 20x106 (Range: 10 to 30x106). On average 60x106 (Range: 40 to 90x106) lymphocytes were obtained.
Electroporation of monocytes with GFP-RNA and expression of GFP du-ring the maturation of monocytes into DCsIn Vitro Transcribed GFPRNA was electroporated in 2x106 monocytes.Twenty four hours after electroporation GFP expression was seen in 62% of the gated monocytes (Fig.1B). GFP expression was measured in the GFPRNA electroporated monocytes during eight consecutive days of culturing and maturation of the monocytes into mature DCs. GFP expression was seen at day four in 57% of the gated cells (Fig.1C), at day six, the immature DCs stage, GFP expression was seen in 55% of the gated cells (Fig.1D), and at day eight, the mature DCs stage, GFP expression was still found in 51% of the gated DCs. (Fig.1E) The maturation of monocytes into DCs was further marked by
Milano_opm_v5.indd 48 16-08-2007 15:42:27
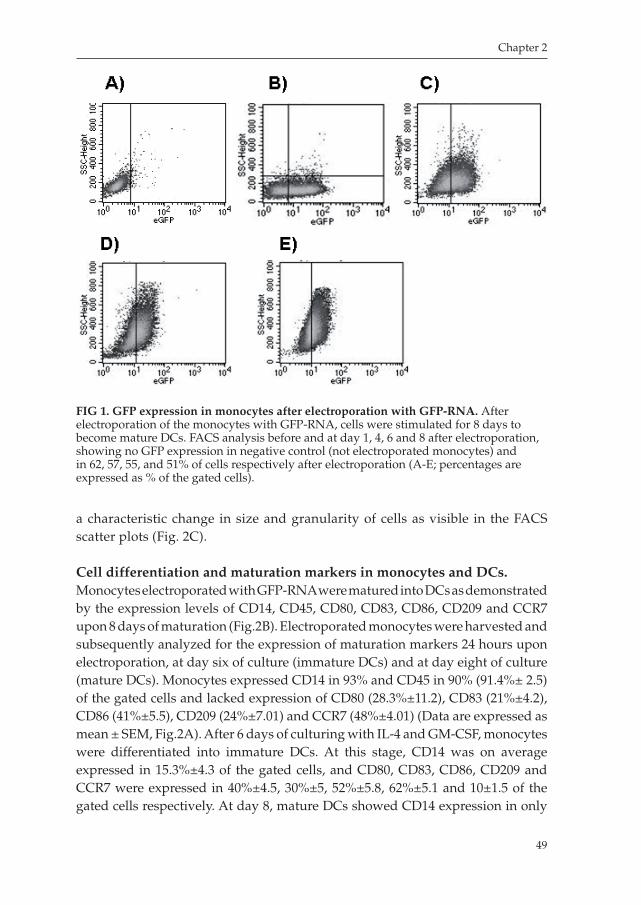
Chapter 2
49
a characteristic change in size and granularity of cells as visible in the FACS scatter plots (Fig. 2C).
Cell differentiation and maturation markers in monocytes and DCs.Monocytes electroporated with GFPRNA were matured into DCs as demonstrated by the expression levels of CD14, CD45, CD80, CD83, CD86, CD209 and CCR7 upon 8 days of maturation (Fig.2B). Electroporated monocytes were harvested and subsequently analyzed for the expression of maturation markers 24 hours upon electroporation, at day six of culture (immature DCs) and at day eight of culture (mature DCs). Monocytes expressed CD14 in 93% and CD45 in 90% (91.4%± 2.5) of the gated cells and lacked expression of CD80 (28.3%±11.2), CD83 (21%±4.2), CD86 (41%±5.5), CD209 (24%±7.01) and CCR7 (48%±4.01) (Data are expressed as mean ± SEM, Fig.2A). After 6 days of culturing with IL4 and GMCSF, monocytes were differentiated into immature DCs. At this stage, CD14 was on average expressed in 15.3%±4.3 of the gated cells, and CD80, CD83, CD86, CD209 and CCR7 were expressed in 40%±4.5, 30%±5, 52%±5.8, 62%±5.1 and 10±1.5 of the gated cells respectively. At day 8, mature DCs showed CD14 expression in only
FIG 1. GFP expression in monocytes after electroporation with GFP-RNA. After electroporation of the monocytes with GFPRNA, cells were stimulated for 8 days to become mature DCs. FACS analysis before and at day 1, 4, 6 and 8 after electroporation, showing no GFP expression in negative control (not electroporated monocytes) and in 62, 57, 55, and 51% of cells respectively after electroporation (AE; percentages are expressed as % of the gated cells).
Milano_opm_v5.indd 49 16-08-2007 15:42:28
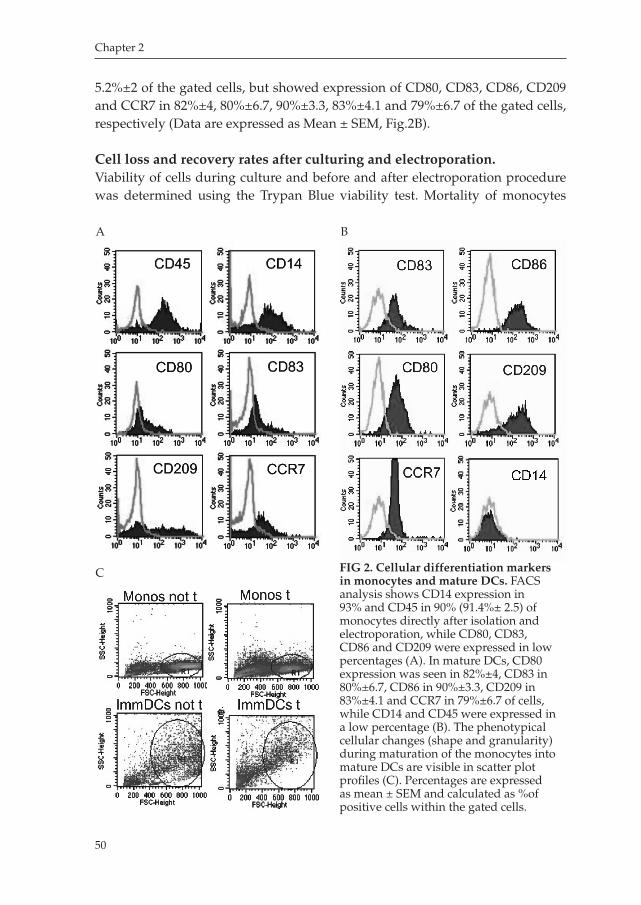
Chapter 2
50
FIG 2. Cellular differentiation markers in monocytes and mature DCs. FACS analysis shows CD14 expression in 93% and CD45 in 90% (91.4%± 2.5) of monocytes directly after isolation and electroporation, while CD80, CD83, CD86 and CD209 were expressed in low percentages (A). In mature DCs, CD80 expression was seen in 82%±4, CD83 in 80%±6.7, CD86 in 90%±3.3, CD209 in 83%±4.1 and CCR7 in 79%±6.7 of cells, while CD14 and CD45 were expressed in a low percentage (B). The phenotypical cellular changes (shape and granularity) during maturation of the monocytes into mature DCs are visible in scatter plot profiles (C). Percentages are expressed as mean ± SEM and calculated as %of positive cells within the gated cells.
5.2%±2 of the gated cells, but showed expression of CD80, CD83, CD86, CD209 and CCR7 in 82%±4, 80%±6.7, 90%±3.3, 83%±4.1 and 79%±6.7 of the gated cells, respectively (Data are expressed as Mean ± SEM, Fig.2B).
Cell loss and recovery rates after culturing and electroporation.Viability of cells during culture and before and after electroporation procedure was determined using the Trypan Blue viability test. Mortality of monocytes
A B
C
Milano_opm_v5.indd 50 16-08-2007 15:42:29
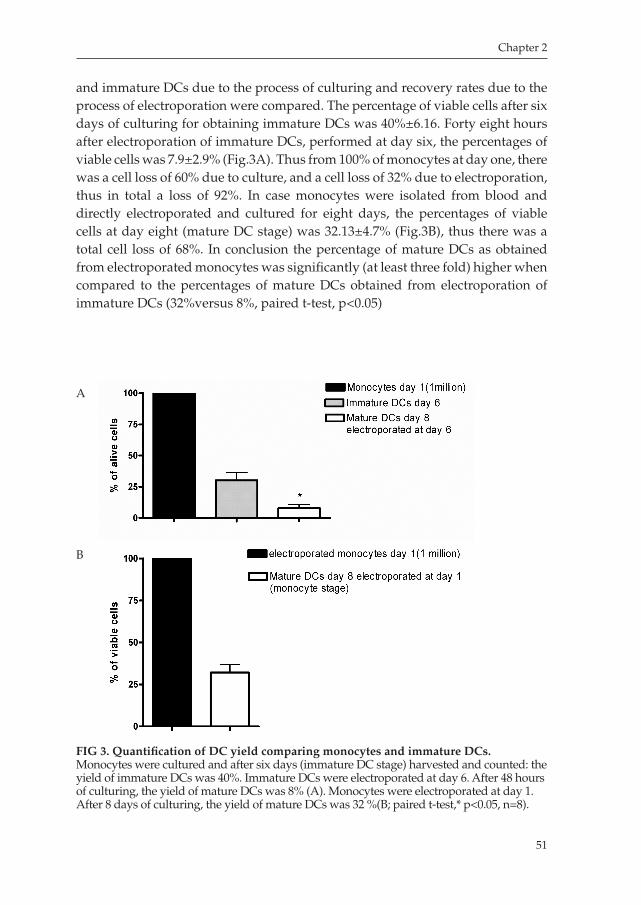
Chapter 2
51
and immature DCs due to the process of culturing and recovery rates due to the process of electroporation were compared. The percentage of viable cells after six days of culturing for obtaining immature DCs was 40%±6.16. Forty eight hours after electroporation of immature DCs, performed at day six, the percentages of viable cells was 7.9±2.9% (Fig.3A). Thus from 100% of monocytes at day one, there was a cell loss of 60% due to culture, and a cell loss of 32% due to electroporation, thus in total a loss of 92%. In case monocytes were isolated from blood and directly electroporated and cultured for eight days, the percentages of viable cells at day eight (mature DC stage) was 32.13±4.7% (Fig.3B), thus there was a total cell loss of 68%. In conclusion the percentage of mature DCs as obtained from electroporated monocytes was significantly (at least three fold) higher when compared to the percentages of mature DCs obtained from electroporation of immature DCs (32%versus 8%, paired ttest, p<0.05)
FIG 3. Quantification of DC yield comparing monocytes and immature DCs. Monocytes were cultured and after six days (immature DC stage) harvested and counted: the yield of immature DCs was 40%. Immature DCs were electroporated at day 6. After 48 hours of culturing, the yield of mature DCs was 8% (A). Monocytes were electroporated at day 1. After 8 days of culturing, the yield of mature DCs was 32 %(B; paired ttest,* p<0.05, n=8).
A
B
Milano_opm_v5.indd 51 16-08-2007 15:42:29
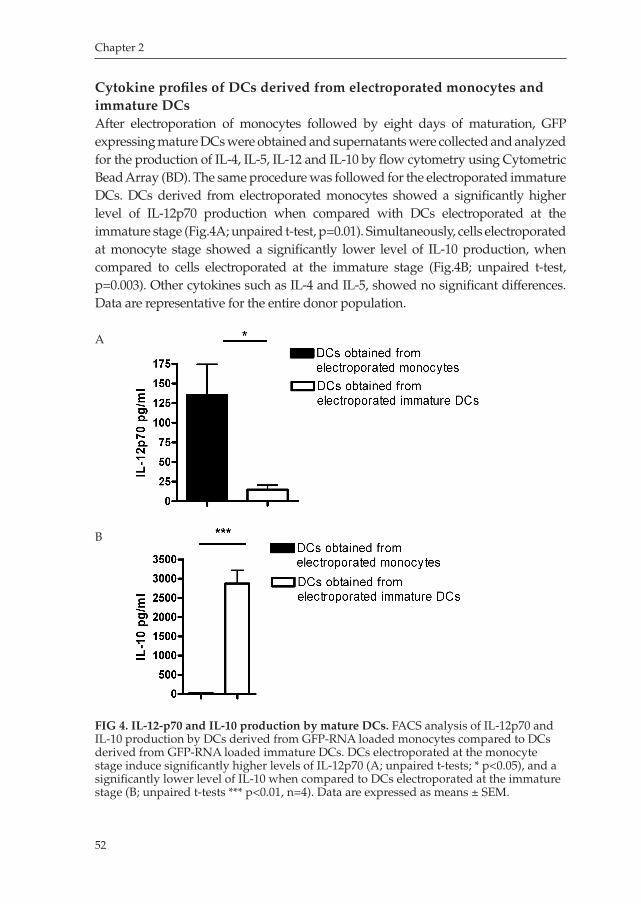
Chapter 2
52
Cytokine profiles of DCs derived from electroporated monocytes and immature DCs After electroporation of monocytes followed by eight days of maturation, GFP expressing mature DCs were obtained and supernatants were collected and analyzed for the production of IL4, IL5, IL12 and IL10 by flow cytometry using Cytometric Bead Array (BD). The same procedure was followed for the electroporated immature DCs. DCs derived from electroporated monocytes showed a significantly higher level of IL12p70 production when compared with DCs electroporated at the immature stage (Fig.4A; unpaired ttest, p=0.01). Simultaneously, cells electroporated at monocyte stage showed a significantly lower level of IL10 production, when compared to cells electroporated at the immature stage (Fig.4B; unpaired ttest, p=0.003). Other cytokines such as IL4 and IL5, showed no significant differences. Data are representative for the entire donor population.
FIG 4. IL-12-p70 and IL-10 production by mature DCs. FACS analysis of IL12p70 and IL10 production by DCs derived from GFPRNA loaded monocytes compared to DCs derived from GFPRNA loaded immature DCs. DCs electroporated at the monocyte stage induce significantly higher levels of IL12p70 (A; unpaired ttests; * p<0.05), and a significantly lower level of IL10 when compared to DCs electroporated at the immature stage (B; unpaired ttests *** p<0.01, n=4). Data are expressed as means ± SEM.
A
B
Milano_opm_v5.indd 52 16-08-2007 15:42:30
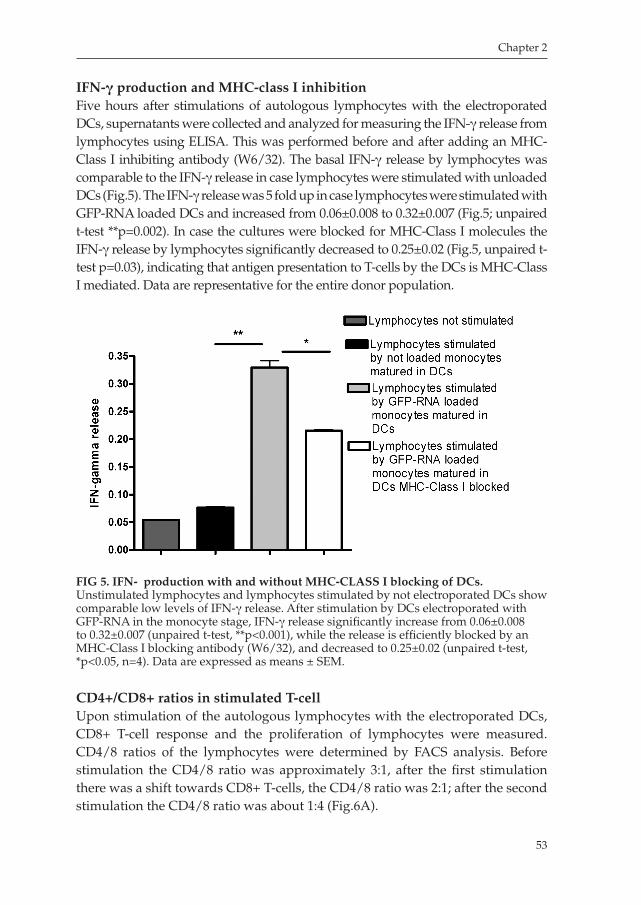
Chapter 2
53
IFN-γ production and MHC-class I inhibitionFive hours after stimulations of autologous lymphocytes with the electroporated DCs, supernatants were collected and analyzed for measuring the IFNγ release from lymphocytes using ELISA. This was performed before and after adding an MHCClass I inhibiting antibody (W6/32). The basal IFNγ release by lymphocytes was comparable to the IFNγ release in case lymphocytes were stimulated with unloaded DCs (Fig.5). The IFNγ release was 5 fold up in case lymphocytes were stimulated with GFPRNA loaded DCs and increased from 0.06±0.008 to 0.32±0.007 (Fig.5; unpaired ttest **p=0.002). In case the cultures were blocked for MHCClass I molecules the IFNγ release by lymphocytes significantly decreased to 0.25±0.02 (Fig.5, unpaired ttest p=0.03), indicating that antigen presentation to Tcells by the DCs is MHCClass I mediated. Data are representative for the entire donor population.
CD4+/CD8+ ratios in stimulated T-cell Upon stimulation of the autologous lymphocytes with the electroporated DCs, CD8+ Tcell response and the proliferation of lymphocytes were measured. CD4/8 ratios of the lymphocytes were determined by FACS analysis. Before stimulation the CD4/8 ratio was approximately 3:1, after the first stimulation there was a shift towards CD8+ Tcells, the CD4/8 ratio was 2:1; after the second stimulation the CD4/8 ratio was about 1:4 (Fig.6A).
FIG 5. IFN-γ production with and without MHC-CLASS I blocking of DCs. Unstimulated lymphocytes and lymphocytes stimulated by not electroporated DCs show comparable low levels of IFNγ release. After stimulation by DCs electroporated with GFPRNA in the monocyte stage, IFNγ release significantly increase from 0.06±0.008 to 0.32±0.007 (unpaired ttest, **p<0.001), while the release is efficiently blocked by an MHCClass I blocking antibody (W6/32), and decreased to 0.25±0.02 (unpaired ttest, *p<0.05, n=4). Data are expressed as means ± SEM.
Milano_opm_v5.indd 53 16-08-2007 15:42:30
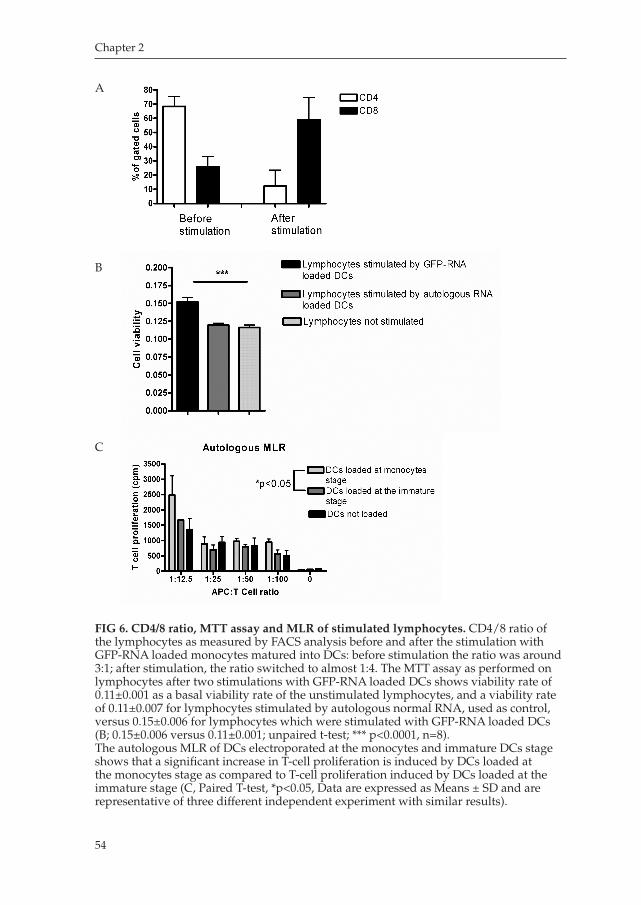
Chapter 2
54
FIG 6. CD4/8 ratio, MTT assay and MLR of stimulated lymphocytes. CD4/8 ratio of the lymphocytes as measured by FACS analysis before and after the stimulation with GFPRNA loaded monocytes matured into DCs: before stimulation the ratio was around 3:1; after stimulation, the ratio switched to almost 1:4. The MTT assay as performed on lymphocytes after two stimulations with GFPRNA loaded DCs shows viability rate of 0.11±0.001 as a basal viability rate of the unstimulated lymphocytes, and a viability rate of 0.11±0.007 for lymphocytes stimulated by autologous normal RNA, used as control, versus 0.15±0.006 for lymphocytes which were stimulated with GFPRNA loaded DCs (B; 0.15±0.006 versus 0.11±0.001; unpaired ttest; *** p<0.0001, n=8).The autologous MLR of DCs electroporated at the monocytes and immature DCs stage shows that a significant increase in Tcell proliferation is induced by DCs loaded at the monocytes stage as compared to Tcell proliferation induced by DCs loaded at the immature stage (C, Paired Ttest, *p<0.05, Data are expressed as Means ± SD and are representative of three different independent experiment with similar results).
A
B
C
Milano_opm_v5.indd 54 16-08-2007 15:42:31

Chapter 2
55
MTT assay and Mixed Leukocytes Reaction (MLR) Tcell viability was measured by MTT Assay. Lymphocytes stimulated with GFPRNA electroporated DCs showed a viability rate of 0.15±0.006. This was 1.4 fold higher when compared to unstimulated lymphocytes which showed a viability rate of 0.11±0.001 and lymphocytes stimulated by DCs loaded with autologous normal RNA (control) which showed a viability rate of 0.11±0.007 (Fig.6B; paired ttest, p<0.0001). Simultaneously, the ability of the electroporated DCs as APC was measured performing an autologous MLR. Mature DCs electroporated with IVT GFPRNA electroporated at the monocyte stage (day 1) induced higher Tcell proliferation as compared to mature DCs electroporated at the immature DCs stage (day 6), (Paired ttest, p<0.05, Fig. 6C). Mature DCs that were not electroporated were taken as control. Data are expressed as Mean ±SD of three independent experiments with similar results.
DISCUSSION
The possibility to isolate monocytes from peripheral blood and transform these exvivo into DCs, that in turn can evoke a primary immune response, has strongly enhanced the use of peripheral blood monocytes for DCs immunotherapy, particularly against several types of cancers.2024 Although many functions of monocytes and DCs are already well known, several aspects of their complex biology still are unclear. It is known for instance that the ability of presenting antigens highly depends on the maturation state of the DCs: mature DCs are significantly more able to present antigens on their surface in order to stimulate Tcells in comparison to the immature or monocyte state, when their main ability is to process the captured antigens.6 A possible explanation for this phenomenon is the fact that, upon maturation, the expression level of MHCClass I molecules increase up to a 100 fold.1113, 2529 This theory, supported by the work of Kalady et al.2, suggests that DCs at best can be electroporated in their immature state when they develop the ability to process antigens. Upon maturation these DCs will be able to efficiently present the antigens to induce an immune response, for instance, via activation of cytotoxic Tcells through the MHCClass I pathway.18 Therefore, the majority of studies that employed DNA or RNA for the loading of DCs, electroporated the APCs in the immature or mature state.3037 This means that CD14+ monocytes, that are isolated from PBMCs, are first stimulated to become (CD14low) immature DCs mostly with low expression of the maturation markers such as CD80 and CD83 before performing electroporation with antigens.15,38,39 This two step approach in which (CD14+) monocytes first have to be cultured in a growth medium and stimulated exvivo to become (CD14 low) immature DCs, and than electroporated and recultured to be stimulated to become mature
Milano_opm_v5.indd 55 16-08-2007 15:42:31

Chapter 2
56
DCs, is a laborious method, since significant amounts of cells are lost during culturing, harvesting and reculturing. In addition, it has been observed that a considerable amount of the immature and mature DCs may be lost during the process of electroporation, although a recent study demonstrated a more efficient protocol in which fully matured DCs are more efficiently pulsed with antigens.40 Another general assumption is that antigens presented by monocytes may induce tolerance through stimulation of regulatory Tcells.41 For these reasons transfection of CD14+ monocytes has been regarded as unsuitable for inducing a cytotoxic Tcell response. Interestingly, it has been reported that monocytes directly isolated from peripheral blood and electroporated with RNA or DNA have the ability to differentiate into DCs with a mature phenotype.15 Yet, the immunopotency and yield of electroporated monocytes as compared to conventional electroporation of immature DCs, and the ability to present antigens on MHCClass I molecules in order to stimulate CD8+ Tcell clones are hardly known. Here we demonstrate an improved and efficient one step approach that results in an at least three fold higher yield of immunocompetent DCs compared to conventional methodology. In the present protocol we show that CD14+ monocytes directly isolated from peripheral blood, have significantly higher recovery after electroporation compared to immature DCs. This confirms that the tolerance of monocytes to the electroporation procedure is superior to that of immature DCs. Moreover, employing monocytes rather than immature DCs for electroporation prevents unnecessary cell loss due to culturing and harvesting of cells (Fig.3). However, it could be possible that electroporation of monocytes would change the nature of the monocytes to an extent that they would loose the capacity to become fully mature DCs. Here we demonstrated that the electroporated monocytes develop into a consistent mature DC phenotype with high expression of costimulatory molecules and maturation markers such as CD80, CD83 and CD86 (Fig.2B). The DCs have a high production of the important lymphocyte homing receptor CCR7 (Fig.2B), demonstrating the capability of these cells to respond to chemokine stimulation and attraction. We showed that these cells produce sufficient amounts of the proinflammatory cytokine IL12p70 (Fig.4A), and, simultaneously, the release of the immunosuppressive IL10 was significantly lower than the IL10 release of immature electroporated DCs (Fig.4B). We further demonstrate that the mature DCs were able to elicit a cytotoxic Tcell response. The observed Tcell response went along with sufficient IFNγ production. Furthermore, upon stimulation of the lymphocytes with GFPRNA electroporated DCs, the CD4/CD8 ratio shift in favour of the CD8+ Tcell clones with simultaneous increased metabolic activity, as indicated by the Tcell viability value observed in the MTT assay, which was significantly different in lymphocytes stimulated by GFPRNA loaded DCs as compared to lymphocytes stimulated by DCs loaded by autologous normal RNA and unstimulated lymphocytes (controls), and by the induced Tcell
Milano_opm_v5.indd 56 16-08-2007 15:42:32

Chapter 2
57
proliferation in the autologous MLR, where induction of Tcell proliferation was significantly higher when DCs where loaded at the monocytes stage, as compared to DCs loaded at the immature stage (Paired ttest, p<0.05). We showed a high electroporation efficacy for GFPRNA that sustained during and after the eight days of maturation of the electroporated monocytes into mature DCs (Fig.1BE). The persistent expression of GFP antigen during and after the maturation (Fig.1CE) indicated that the electroporated monocytes are able to efficiently translate RNA into proteins, process antigens and express these antigens on their surface. To prove that the expression of antigens is via MHCClass I molecules, we demonstrate that specific blocking of the MHCClass I molecules, resulted in inhibition of IFNγ production (Fig.5). In summary we have developed an improved and simplified method for exvivo manufacturing of DCs through electroporation of RNA into CD14+ monocytes directly isolated from blood. We have proved that these cells can be stimulated to differentiate into potent, fully mature DCs, highly expressing costimulatory molecules, DC maturation markers and CCR7 receptor, and are able to produce high levels of proinflammatory cytokines. Furthermore the obtained mature DCs were able to induce an MHCClass I mediated cytotoxic Tcell response and sustained Tcell proliferation. The currently described methodology results in an at least three fold higher yield of potent mature DCs, while redundant laborious culturing steps are eliminated. Future application of this simplified methodology may greatly enhance further developments and applications of DC RNAbased immunotherapy.
ACKNOWLEDGMENTS
We would like to kindly thank all the volunteers who accepted to participate to the study and a special thanks to Dr. H. Braat for his support for the laboratory work.
This work was supported by a grant of the Dutch National Cancer Foundation (KWF).
Milano_opm_v5.indd 57 16-08-2007 15:42:32

Chapter 2
58
REFERENCES
1. Banchereau, J. and Steinman, R.M. Dendritic cells and the control of immunity. Nature 392, 24552,
1998
2. Kalady, M.F., Onaitis, M.W., Padilla, K.M., Emani, S., Tyler, D.S. and Pruitt, S.K. Enhanced
dendritic cell antigen presentation in RNAbased immunotherapy. J Surg Res 105, 1724.,
2002.
3. Gilboa, E., Nair, S.K. and Lyerly, H.K. Immunotherapy of cancer with dendriticcellbased
vaccines. Cancer Immunol Immunother 46, 827, 1998.
4. Steinman, R.M. and Dhodapkar, M. Active immunization against cancer with dendritic cells:
the near future. Int J Cancer 94, 45973, 2001.
5. Fong, L. and Engleman, E.G. Dendritic cells in cancer immunotherapy. Annu Rev Immunol
18, 24573, 2000.
6. Steinman, R.M. and Pope, M. Exploiting dendritic cells to improve vaccine efficacy. J Clin
Invest 109, 151926, 2002.
7. Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic
relevance. Nat Rev Cancer 5, 26374, 2005.
8. Tada, T., Ohzeki, S., Utsumi, K., Takiuchi, H., Muramatsu, M., Li, X.F., Shimizu, J., Fujiwara,
H. and Hamaoka, T. Transforming growth factorbetainduced inhibition of T cell function.
Susceptibility difference in T cells of various phenotypes and functions and its relevance to
immunosuppression in the tumorbearing state. J Immunol 146, 107782, 1991.
9. Mailliard, R.B., WankowiczKalinska, A., Cai, Q., Wesa, A., Hilkens, C.M., Kapsenberg, M.L.,
Kirkwood, J.M., Storkus, W.J. and Kalinski, P. alphatype1 polarized dendritic cells: a novel
immunization tool with optimized CTLinducing activity. Cancer Res 64, 59347, 2004.
10. Van Tendeloo, V.F., Ponsaerts, P., Lardon, F., Nijs, G., Lenjou, M., Van Broeckhoven, C., Van
Bockstaele, D.R. and Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation
in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to
electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 98, 4956,
2001.
11. O’Neill, D.W., Adams, S. and Bhardwaj, N. Manipulating dendritic cell biology for the active
immunotherapy of cancer. Blood 104, 223546, 2004.
12. Brossart, P., Wirths, S., Brugger, W. and Kanz, L. Dendritic cells in cancer vaccines. Exp Hema
tol 29, 124755, 2001.
13. Porgador, A., Snyder, D. and Gilboa, E. Induction of antitumor immunity using bone marrow
generated dendritic cells. J Immunol 156, 291826, 1996.
14. Sallusto, F., Cella, M., Danieli, C. and Lanzavecchia, A. Dendritic cells use macropinocytosis
and the mannose receptor to concentrate macromolecules in the major histocompatibility
complex class II compartment: downregulation by cytokines and bacterial products. J Exp
Med 182, 389400, 1995.
15. Ponsaerts, P., Van den Bosch, G., Cools, N., Van Driessche, A., Nijs, G., Lenjou, M., Lardon,
F., Van Broeckhoven, C., Van Bockstaele, D.R., Berneman, Z.N. and Van Tendeloo, V.F. Mes
Milano_opm_v5.indd 58 16-08-2007 15:42:32

Chapter 2
59
senger RNA electroporation of human monocytes, followed by rapid in vitro differentiation,
leads to highly stimulatory antigenloaded mature dendritic cells. J Immunol 169, 166975,
2002.
16. Braat, H., de Jong, E.C., van den Brande, J.M., Kapsenberg, M.L., Peppelenbosch, M.P., van
Tol, E.A. and van Deventer, S.J. Dichotomy between Lactobacillus rhamnosus and Klebsiella
pneumoniae on dendritic cell phenotype and function. J Mol Med 82, 197205, 2004.
17. Chen, R., Lowe, L., Wilson, J.D., Crowther, E., Tzeggai, K., Bishop, J.E. and Varro, R. Simulta
neous Quantification of Six Human Cytokines in a Single Sample Using Microparticlebased
Flow Cytometric Technology. Clin Chem 45, 16931694, 1999.
18. Liu, G., Ying, H., Zeng, G., Wheeler, C.J., Black, K.L. and Yu, J.S. HER2, gp100, and MAGE
1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res 64,
49806, 2004.
19. Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to prolife
ration and cytotoxicity assays. J Immunol Methods 65, 5563, 1983.
20. Coughlin, C.M., Vance, B.A., Grupp, S.A. and Vonderheide, R.H. RNAtransfected CD40
activated B cells induce functional Tcell responses against viral and tumor antigen targets:
implications for pediatric immunotherapy. Blood 103, 204654, 2004.
21. Heiser, A., Dahm, P., Yancey, D.R., Maurice, M.A., Boczkowski, D., Nair, S.K., Gilboa, E. and
Vieweg, J. Human dendritic cells transfected with RNA encoding prostatespecific antigen
stimulate prostatespecific CTL responses in vitro. J Immunol 164, 550814, 2000.
22. Gilboa, E. and Vieweg, J. Cancer immunotherapy with mRNAtransfected dendritic cells. Im
munol Rev 199, 25163, 2004.
23. Panelli, M.C., Wunderlich, J., Jeffries, J., Wang, E., Mixon, A., Rosenberg, S.A. and Marincola,
F.M. Phase 1 study in patients with metastatic melanoma of immunization with dendritic
cells presenting epitopes derived from the melanomaassociated antigens MART1 and gp100.
J Immunother 23, 48798, 2000.
24. Moll, H. Antigen delivery by dendritic cells. Int J Med Microbiol 294, 33744, 2004.
25. Koido, S., Kashiwaba, M., Chen, D., Gendler, S., Kufe, D. and Gong, J. Induction of antitumor
immunity by vaccination of dendritic cells transfected with MUC1 RNA. J Immunol 165,
57139, 2000.
26. Schadendorf, D. and Nestle, F.O. Autologous dendritic cells for treatment of advanced cancer
an update. Recent Results Cancer Res 158, 23648, 2001.
27. Gilliet, M., Kleinhans, M., Lantelme, E., Schadendorf, D., Burg, G. and Nestle, F.O. Intranodal
injection of semimature monocytederived dendritic cells induces T helper type 1 responses
to protein neoantigen. Blood 102, 3642, 2003.
28. Maier, T., TunKyi, A., Tassis, A., Jungius, K.P., Burg, G., Dummer, R. and Nestle, F.O. Vac
cination of patients with cutaneous Tcell lymphoma using intranodal injection of autologous
tumorlysatepulsed dendritic cells. Blood 102, 233844, 2003.
29. Mule, J.J. Tumor vaccine strategies that employ dendritic cells and tumor lysates: experimen
tal and clinical studies. Immunol Invest 29, 1279, 2000.
30. Muller, M.R., Tsakou, G., Grunebach, F., Schmidt, S.M. and Brossart, P. Induction of chronic
Milano_opm_v5.indd 59 16-08-2007 15:42:33

Chapter 2
60
lymphocytic leukemia (CLL)specific CD4 and CD8mediated Tcell responses using RNA
transfected dendritic cells. Blood 103, 17639, 2004.
31. Takahashi, M., Narita, M., Ayres, F., Satoh, N., Abe, T., Yanao, T., Furukawa, T., Toba, K.,
Hirohashi, T. and Aizawa, Y. (2003) Cytoplasmic expression of EGFP in dendritic cells trans
fected with in vitro transcribed mRNA or cellular total RNA extracted from EGFP expressing
leukemia cells. Med Oncol 20, 33548, 2003.
32. Ashley, D.M., Faiola, B., Nair, S., Hale, L.P., Bigner, D.D. and Gilboa, E. Bone marrow
generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immu
nity against central nervous system tumors. J Exp Med 186, 117782, 1997.
33. Nair, S.K., Heiser, A., Boczkowski, D., Majumdar, A., Naoe, M., Lebkowski, J.S., Vieweg, J. and
Gilboa, E. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors
using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 6, 10117, 2000.
34. Strobel, I., Berchtold, S., Gotze, A., Schulze, U., Schuler, G. and Steinkasserer, A. Human
dendritic cells transfectedwith either RNA or DNA encoding influenza matrix protein M1
differ in their ability to stimulate cytotoxic T lymphocytes. Gene Ther 7, 202835, 2000.
35. Su, Z., Dannull, J., Heiser, A., Yancey, D., Pruitt, S., Madden, J., Coleman, D., Niedzwiecki,
D., Gilboa, E. and Vieweg, J. Immunological and clinical responses in metastatic renal cancer
patients vaccinated with tumor RNAtransfected dendritic cells. Cancer Res 63, 212733, 2003.
36. Milazzo, C., Reichardt, V.L., Muller, M.R., Grunebach, F. and Brossart, P. Induction of my
elomaspecific cytotoxic T cells using dendritic cells transfected with tumorderived RNA.
Blood 101, 97782, 2003.
37. Grunebach, F., Muller, M.R., Nencioni, A. and Brossart, P. Delivery of tumorderived RNA for
the induction of cytotoxic Tlymphocytes. Gene Ther 10, 36774, 2003.
38. Kalady, M.F., Onaitis, M.W., Emani, S., AbdulWahab, Z., Pruitt, S.K. and Tyler, D.S. Dendritic
cells pulsed with pancreatic cancer total tumor RNA generate specific antipancreatic cancer T
cells. J Gastrointest Surg 8, 17581; discussion 1812, 2004.
39. Nair, S.K., Morse, M., Boczkowski, D., Cumming, R.I., Vasovic, L., Gilboa, E. and Lyerly, H.K.
Induction of tumorspecific cytotoxic T lymphocytes in cancer patients by autologous tumor
RNAtransfected dendritic cells. Ann Surg 235, 5409, 2002.
40. Grunebach, F., Muller, M.R. and Brossart, P. New developments in dendritic cellbased vac
cinations: RNA translated into clinics. Cancer Immunol Immunother 54, 51725, 2005.
41. Beyth, S., Borovsky, Z., Mevorach, D., Liebergall, M., Gazit, Z., Aslan, H., Galun, E. and
Rachmilewitz, J. Human mesenchymal stem cells alter antigenpresenting cell maturation and
induce Tcell unresponsiveness. Blood 105, 22149, 2005.
Milano_opm_v5.indd 60 16-08-2007 15:42:33

Chapter 3
An ex-vivo read out for evaluation of
Dendritic Cells induced autologous CTL
responses against esophageal cancer
Francesca Milano, Agnieszka M. Rygiel, Navtej Buttar, Jacques J.G.H.M. Bergman, Carine Sondermeijer,
Jantine W. P.M. van Baal, Anja ten Brinke, Martien Kapsenberg, Marieke S. van Ham,
Maikel P. Peppelenbosch, Kausilia K. Krishnadath
Cancer Immunol Immunother. 2007 Jun 13
Milano_opm_v5.indd 61 16-08-2007 15:42:33

Chapter 3
62
ABSTRACT
Esophageal cancer is a highly malignant disease that despite surgery and adjuvant therapies has an extremely poor outcome. Dendritic cell (DC) immunotherapy as a novel promising strategy could be an alternative for treating this malignancy. Effective DC mediated immune responses can be achieved by raising cytotoxic T lymphocyte (CTL) response against multiple antigens, through loading DCs with total tumor RNA. However, the efficacy of this strategy first needs to be evaluated in a preclinical setting. The aim of the study was to set up an ex-vivo autologous human read out assay for assessing the effects of DCmediated cytotoxic responses, using total tumor RNA as an antigen load. Biopsy specimens of seven esophageal cancer patients were used to establish primary cultures of normal and cancer cells, and to obtain autologous RNA for loading DCs. Mature DCs loaded with either normal or tumor RNA were obtained and subsequently used to raise various lymphocytes populations. Apoptosis levels of the autologous cultures were measured before and after incubating the cultures with the different lymphocytes populations. The mean apoptosis levels in the tumor cell cultures induced by lymphocytes instructed by DCs loaded with tumor RNA, significantly increased with 15.6% ±2.9 SEM (range: 3.4 to 24.5%, ttest, p<0.05). Incubation of the normal cultures with the lymphocytes populations showed a mean non significant increase in apoptosis of 0.4% ±3.4 SEM (range: 13.9 to 9.8%, ttest, p=0.7). Here we introduce a practical patient specific autologous read out assay for preclinical testing of DC mediated cytotoxic responses. Additionally, we demonstrated that the use of autologous tumor RNA as a strategy for raising cytotoxic responses against multiple tumor antigens could be effective for treating esophageal cancer.
Milano_opm_v5.indd 62 16-08-2007 15:42:33

Chapter 3
63
INTRODUCTION
There are two major types of esophageal cancer: esophageal squamous cell carcinoma, and esophageal adenocarcinoma. For decades, the incidence of esophageal squamous cell carcinoma has been unchanged and is approximately 1 per 100.000 cases per year. Of major concern is the steadily increasing incidence of esophageal adenocarcinoma which has become an important health problem1,2. While esophageal squamous cell carcinoma is associated with poor socioeconomy status, smoking habits and alcohol intake3, esophageal adenocarcinoma has a strong association with Barrett’s esophagus4. Barrett’s esophagus is a metaplastic premalignant transformation of the esophageal epithelium associated with gastroesophageal reflux disease (GERD)57. Although the two types of esophageal cancer have different pathophysiology, the clinical outcomes of both are poor. Even after surgical resection, the overall 5 years survival rate of these patients is less than 15%, and adjuvant treatments such as chemo and radiotherapy have only little effect on patient outcome811.
Dendritic Cell (DC) therapy, as a promising strategy to treat cancer, has been intensively investigated in the last few years. Dendritic cells (DCs) are specialized antigenpresenting cells involved in innate and adaptive immune responses1213. Functional DCs can be generated from human peripheral blood monocytes and be further matured into DCs that in turn can be used as vaccines for treating malignancies1417. To generate a cytotoxic Tcell (CTL) response against tumor cells, specific tumor antigens have to be presented to Tlymphocytes by immunoactivatory DCs. Therefore, the immunogenicity of the tumor associated antigens that are used for loading the DCs is crucial. Different antigens have been used and tested for their immunopotency. These include synthetic peptides1820, tumor lysates21, and cDNA or RNA encoding for specific tumorassociated antigens as well as total tumor mRNA2226. The introduction of autologous total tumor RNA as an antigen source for loading DCs has several advantages. Such a strategy will not restrict DC vaccination therapy to patients with certain HLA haplotypes. Moreover, it is most suitable to treat cancers with heterogeneous phenotypes, such as esophageal cancers and other solid malignancies that have variable expression of diverse tumor antigens. It is reasonable to assume that normal RNA that will be cotransferred with the total tumor RNA into the DCs may not result in immune responses, since there is tolerance towards self proteins through depletion of selfspecific T cells. Nevertheless, when using total RNA, it is of importance to evaluate whether this strategy would induce a break in the tolerance against selfantigens. A valid preclinical method, which would enable us to test the efficacy of immunoactivatory DCs loaded with total tumor RNA to induce T cell reactivity in an autologous system could be of use to monitor direct adverse effects but as well to predict potential clinical responses.
Milano_opm_v5.indd 63 16-08-2007 15:42:34

Chapter 3
64
The aim of our study was to create an effective autologus ex-vivo read out system to evaluate cytotoxic responses induced by DCs based on total tumor RNA as an antigen load. To this aim primary cultures were established from biopsies of normal and tumor tissues taken during endoscopy from esophageal cancer patients. DCs were generated from peripheral blood monocytes of the patients and loaded with either autologous normal or total tumor RNA, and subsequently studied for their immunostimulatory capacity. Hereupon, the DCs were used to stimulate the patient’s lymphocytes to obtain several lymphocytes populations that were subsequently tested for their cytolytic responses against the autologous primary cell cultures.
In this study we introduce a patient tailored approach using an ex vivo cell culture read out system for evaluating autologous DC induced cytotoxic responses. We were able to generate fully mature DCs loaded with total tumor RNA that in the autologous ex-vivo cultures were able to elicit cytotoxic responses specifically against the autologous cancer cells, while no significant direct adverse effect were seen against the normal cells.
MATERIALS AND METHODS
Patient’s materialThe study was approved by the Academic Medical Center (Amsterdam, The Netherlands) Hospital’s Medical Ethical Committee. After informed consent and written permission, twelve patients who met the inclusion criteria (see supplementary data), were included. Before the endoscopic procedure, 64 ml of blood was drawn and collected in heparinized vials for extraction of peripheral blood mononuclear cells (PBMCs). Patients underwent endoscopic procedures for classifying, staging and grading of the esophageal cancer. During this procedure, 12 extra biopsies of each patient were taken to be used for culturing purposes and for RNA isolation; biopsies were obtained from both normal squamous epithelium taken at least 3 cm above the mass, and from the malignancy. Matching biopsies from the same spots were taken for histopathological diagnosis.
Primary cell cultures of esophageal normal squamous epithelium and esophageal cancer epitheliumFor establishing the autologous ex-vivo test model, biopsies from normal esophageal epithelium and from esophageal cancer were used to establish primary cell cultures. Histological examination of the matching biopsies of the tumors showed that on estimate the biopsy specimens contained at least 50% (range 50 to 80%) of tumor cells. The culture medium MCDB 153 (Sigma) was
Milano_opm_v5.indd 64 16-08-2007 15:42:34

Chapter 3
65
modified by the adding 5% fetal bovine serum, 0.4 μg/mL hydrocortisone, (Sigma), 20 ng/ml epidermal growth factor (GIBCO, Grand Island, NY), 1010 mol/L cholera toxin (Sigma), 140 μg/mL bovine pituitary extract (Sigma), 20 μg/mL adenine (Sigma), 100 U/mL penicillin (GIBCO), 100 μg/mL streptomycin (GIBCO), 0.25 μg/mL amphotericin B (GIBCO), 5 μg/mL insulintransferrin (GIBCO) and 4 mmol/L glutamine. The explant method was used as described before27. Briefly, biopsies from normal and tumor esophageal mucosa were collected aseptically into MCDB153 modified medium during routine endoscopy of patient with esophageal cancer. Specimens were processed within half an hour of procurement as follows: biopsy specimens were minced into fragments of 12 mm3 in size. The pieces of tissue were placed in a 24 wells plate and anchored by a sterile glass microscope slide before adding of growth medium. One ml/well MCDB 153 modified medium was added and cultures were placed at 37ºC and 5% CO2. Fresh medium was replaced every three days for three weeks until measurement of autologous cytotoxic responses.
RNA isolationBiopsies from normal and cancer tissues were collected in Trizol Reagent (Life Technologies Inc, Invitrogen, Breda, The Netherlands) and processed according to the manufacturer’s instructions. Briefly, tissues were lysed by adding 200 μl Trizol. After phenol/chloroform extraction, RNA was precipitated with isopropanol, washed with 70% ethanol and airdried. The RNA was then dissolved in RNasefree H2O and stored at 80 °C until required. One μl of total RNA was used to quantitate the RNA by spectrophotometry using the Nanodrop apparatus (type ND1000, Wilmington, USA).
Isolation of lymphocytes and monocytes from peripheral blood of the patient Lymphocytes and monocytes were isolated from 64 ml of peripheral blood collected from patients in heparinized vials, using the FicollPercoll gradient separation method13. A first separation of peripheral blood mononuclear cells (PBMCs) was done using FicollHypaque solution (Amersham, Pharmacia, Piscataway, NJ, USA). A further separation of monocytes from lymphocytes was done using a Percoll (Amersham Biosciences Europe Freiburg, Germany) density gradient separation, as described previously29. Briefly, PBMCs were washed in Roswell Park Memorial Institute (RPMI) 1640 medium (BioWhittakerCambrex Bioscience, Walkersville, MD, USA) at 1500 rpm for 5 minutes twice. At the mean time, 19.8 ml of Percoll were mixed with 2.2 ml 10x Phosphate Buffered Saline (PBS) to obtain Standard Isotonic Percoll solution (SIP), and then with Iscove’s Modified Dulbecco’s Medium (IMDM) medium (BioWhittaker), to obtain three solutions at different concentrations (60, 47.5 and 34% SIP). PBMCs were then resuspended in 2.5 ml of 60% SIP, then 5 ml of 47.5% SIP and 2 ml of 34% SIP
Milano_opm_v5.indd 65 16-08-2007 15:42:35

Chapter 3
66
were added and a centrifugation at 3100 rpm for 45 minutes was performed. After centrifugation, the upper layer (monocytes) was collected as well as the lower layer (lymphocytes). Once separated, monocytes and lymphocytes were washed twice in IMDM, and either used immediately or cryopreserved in a Dimethylsulfoxide (DMSO)/ Fetal calf serum(FCS) (2:8) solution (Merck, Darmstadt, Germany; GIBCO BRL, Grand Island, NY, USA, respectively).
Electroporation of monocytes with normal and tumor autologous total RNA Monocytes characterized as CD14+, CD83, CD86, CD209 were used for electroporation with autologous total normal or tumor RNA following the procedure recently described by Milano et al.54. To monitor for the electroporation efficacy, In Vitro Transcribed (IVT) GFPRNA was used. The procedure was as follows: monocytes freshly isolated from blood were washed twice in IMDM and electroporated using the Amaxa cell line Nucleofector Kit V (Amaxa GmbH, Cologne, Germany): 0.5 up to 1x106 cells were mixed with Cell Line Nucleofector solution V, and 5 µg/ml of RNA was added to the cuvette to be electroporated using the Nucleofector program U16 of the Amaxa Nucleofector device. After electroporation, 1 million/ml monocytes were cultured in 24 wells plate using IMDM with 10%FCS and 2% penicillin/streptomycin (GIBCO) and cells were placed at 37ºC and 5% CO2 for one hour, then cells were washed twice with IMDM, and 1000 U/ml of IL4 and 800 U/ml GMCSF were added to initiate the maturation process. After six days at the stage of immature DCs, the cytokines IL1β, TNFα and LPS (10 ng/ml, 25 ng/ml, 0.02 ng/ml respectively) were added to further enhance the maturation process. Cells were analysed for the expression of maturation markers at day six and eight by FACS.
Detection of markers and MHC class I and II in monocytes and DCs Monocytes were harvested by pipetting, washed and resuspended in FACS buffer (5 g BSA, 0.1 g NaN3, 100 mM EDTA in 1 L PBS) in a concentration of 1 million cells/ml, then incubated with various fluorochromeconjugated antibodies on ice for 30 minutes in the dark, then washed again and analyzed using the FACSCALIBUR apparatus (BectonDickinson, Franklin Lakes, NJ, USA) and BD CellQuest Pro software. PE or FITC conjugated antibodies, specific for CD14, CD83, CD86, CD209, CCR7, HLAA,B,C and HLADR (BD, San Jose, CA, USA) were used as appropriate, in a concentration of 1:25. Sample stained for FITC and PEconjugated IgG2a/IgG1 Isotype controls (BD Biosciences) were included in the staining procedure. For all cases markers and MHCClass I and II expression were measured during maturation from monocytes, to immature and mature DCs. Measurements were performed of both electroporated and not electroporated (control) cells.
Milano_opm_v5.indd 66 16-08-2007 15:42:35

Chapter 3
67
Stimulation of autologous lymphocytes with electroporated DCsAfter eight days from the isolation of monocytes, the resulting mature DCs were twice coincubated for seven days with autologous lymphocytes in a proportion of 1:4 (5x105 DCs and 2x106 lymphocytes) in 24 wells plate in IMDM medium with 5% FCS and 2% penicillinstreptomycin (GIBCO). Before and after the first and second stimulation (day 1, day 7 and day 14 of the coculture) aliquots of lymphocytes were taken to measure changes in the CD4/8 ratio by flow cytometry and the supernatants were collected to measure production of inflammatory cytokines. Finally, per patient two populations of lymphocytes were obtained: lymphocytes stimulated by DCs electroporated with tumor RNA and a second population of lymphocytes stimulated by DCs electroporated with normal RNA.
Measuring of the cytotoxic responses on the ex-vivo cell culturesAfter stimulation with the normal and tumor RNA electroporated DCs, the two different populations of lymphocytes mentioned above were washed in IMDM and added to the autologous cultured normal and tumor epithelial cells at a target/effector ratio of 2x104/4x105cells for 5 hours. For each patient autologous tumor and normal epithelial cell cultures were incubated with lymphocytes either stimulated by DCs loaded with autologous normal RNA or with autologous tumor RNA (see supplementary picture). The epithelial normal and tumor cells were washed with cold PBS and detached by adding 0.5ml trypsin (GIBCO, Auckland, NZ) for five minutes at 37°C; cells were then collected in 1 ml MCDB 153 modified medium, spun down and resuspended in Annexin V buffer (2.38g HEPES, 8.8g NaCl, 0.38g KCl2 , 0.2g CaCl2, 0.20 g MgCl2) in a concentration of 1 million cells/ml. Apoptosis of the normal and cancer cells was measured by using the following antibodies: antihuman AnnexinV APC conjugated (ICQ, Groningen, The Netherlands); Viaprobe 7AAD (necrosis marker; R&D System); antihuman EpCam FITC conjugated (epithelial specific marker; Miltenyi Biotech, Auburn, CA); antihuman CD3 PE conjugated (Tcells marker; R&D System). Data were acquired using BD Cell Quest Pro Software. Apoptotic epithelial cells were gated as double positive for AnnexinV and EpCam, and negative for CD3 and 7AAD.
Cytometric Bead Array (CBA) Multiplex AssaysDuring stimulation of lymphocytes with normal and tumor RNA electroporated DCs supernatants were collected of the samples of the patients at day 7 (after the first stimulation) and at day 14 (after the second stimulation) and analysed for cytokine contents using the Cytometric Bead Array (CBA). Experiments were performed using the CBA Inflammation kit (BD) following the manufacturer’s instructions, and the standard procedure was followed as previously described30. Acquired data were analyzed using the BD calibration and analysis software.
Milano_opm_v5.indd 67 16-08-2007 15:42:35
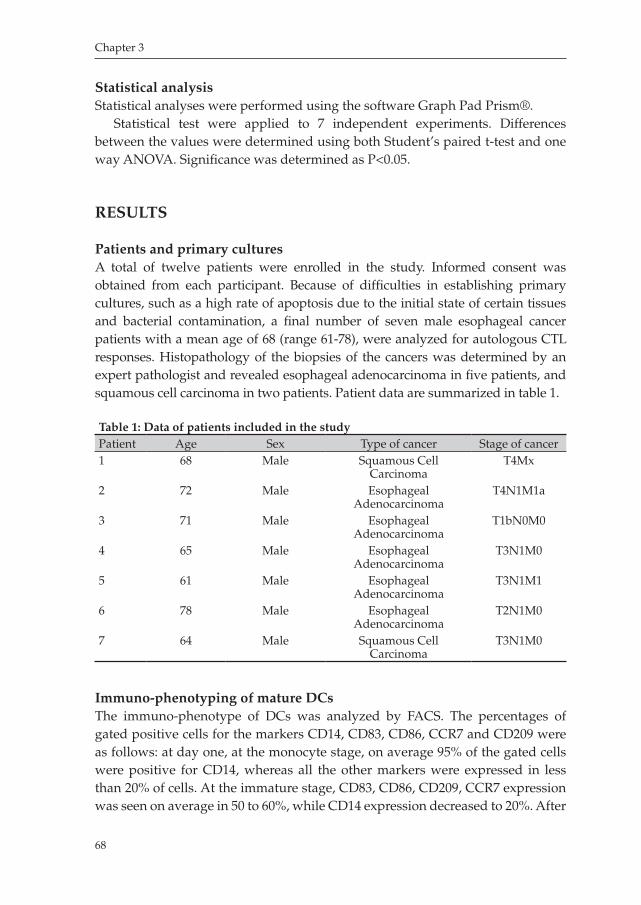
Chapter 3
68
Statistical analysisStatistical analyses were performed using the software Graph Pad Prism®.
Statistical test were applied to 7 independent experiments. Differences between the values were determined using both Student’s paired ttest and one way ANOVA. Significance was determined as P<0.05.
RESULTS
Patients and primary culturesA total of twelve patients were enrolled in the study. Informed consent was obtained from each participant. Because of difficulties in establishing primary cultures, such as a high rate of apoptosis due to the initial state of certain tissues and bacterial contamination, a final number of seven male esophageal cancer patients with a mean age of 68 (range 6178), were analyzed for autologous CTL responses. Histopathology of the biopsies of the cancers was determined by an expert pathologist and revealed esophageal adenocarcinoma in five patients, and squamous cell carcinoma in two patients. Patient data are summarized in table 1.
Table 1: Data of patients included in the studyPatient Age Sex Type of cancer Stage of cancer1 68 Male Squamous Cell
CarcinomaT4Mx
2 72 Male Esophageal Adenocarcinoma
T4N1M1a
3 71 Male Esophageal Adenocarcinoma
T1bN0M0
4 65 Male Esophageal Adenocarcinoma
T3N1M0
5 61 Male Esophageal Adenocarcinoma
T3N1M1
6 78 Male Esophageal Adenocarcinoma
T2N1M0
7 64 Male Squamous Cell Carcinoma
T3N1M0
Immuno-phenotyping of mature DCsThe immunophenotype of DCs was analyzed by FACS. The percentages of gated positive cells for the markers CD14, CD83, CD86, CCR7 and CD209 were as follows: at day one, at the monocyte stage, on average 95% of the gated cells were positive for CD14, whereas all the other markers were expressed in less than 20% of cells. At the immature stage, CD83, CD86, CD209, CCR7 expression was seen on average in 50 to 60%, while CD14 expression decreased to 20%. After
Milano_opm_v5.indd 68 16-08-2007 15:42:36
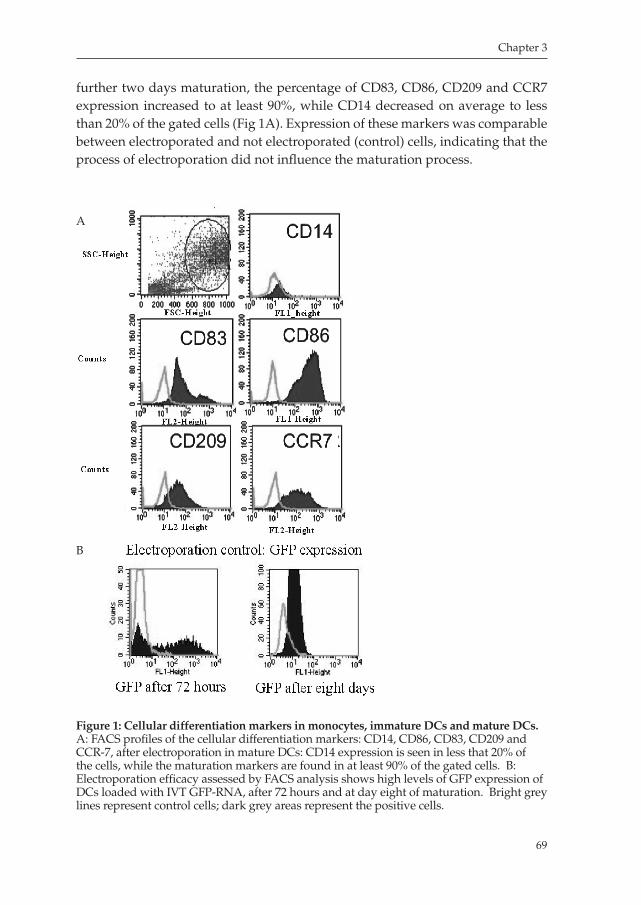
Chapter 3
69
further two days maturation, the percentage of CD83, CD86, CD209 and CCR7 expression increased to at least 90%, while CD14 decreased on average to less than 20% of the gated cells (Fig 1A). Expression of these markers was comparable between electroporated and not electroporated (control) cells, indicating that the process of electroporation did not influence the maturation process.
Figure 1: Cellular differentiation markers in monocytes, immature DCs and mature DCs. A: FACS profiles of the cellular differentiation markers: CD14, CD86, CD83, CD209 and CCR7, after electroporation in mature DCs: CD14 expression is seen in less that 20% of the cells, while the maturation markers are found in at least 90% of the gated cells. B: Electroporation efficacy assessed by FACS analysis shows high levels of GFP expression of DCs loaded with IVT GFPRNA, after 72 hours and at day eight of maturation. Bright grey lines represent control cells; dark grey areas represent the positive cells.
A
B
Milano_opm_v5.indd 69 16-08-2007 15:42:37
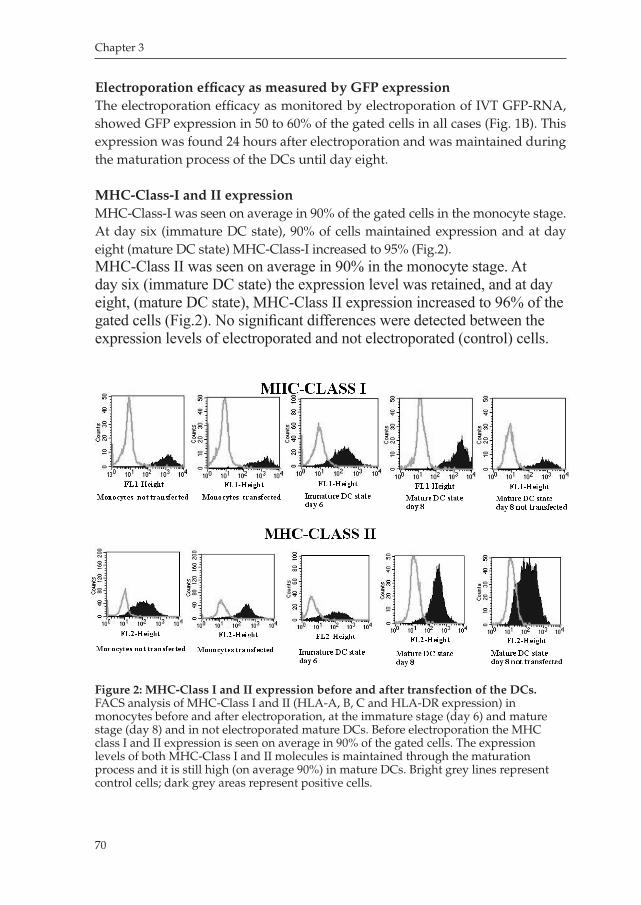
Chapter 3
70
Electroporation efficacy as measured by GFP expressionThe electroporation efficacy as monitored by electroporation of IVT GFPRNA, showed GFP expression in 50 to 60% of the gated cells in all cases (Fig. 1B). This expression was found 24 hours after electroporation and was maintained during the maturation process of the DCs until day eight.
MHC-Class-I and II expression MHCClassI was seen on average in 90% of the gated cells in the monocyte stage. At day six (immature DC state), 90% of cells maintained expression and at day eight (mature DC state) MHCClassI increased to 95% (Fig.2). MHC-Class II was seen on average in 90% in the monocyte stage. At day six (immature DC state) the expression level was retained, and at day eight, (mature DC state), MHC-Class II expression increased to 96% of the gated cells (Fig.2). No significant differences were detected between the expression levels of electroporated and not electroporated (control) cells.
Figure 2: MHC-Class I and II expression before and after transfection of the DCs. FACS analysis of MHCClass I and II (HLAA, B, C and HLADR expression) in monocytes before and after electroporation, at the immature stage (day 6) and mature stage (day 8) and in not electroporated mature DCs. Before electroporation the MHC class I and II expression is seen on average in 90% of the gated cells. The expression levels of both MHCClass I and II molecules is maintained through the maturation process and it is still high (on average 90%) in mature DCs. Bright grey lines represent control cells; dark grey areas represent positive cells.
Milano_opm_v5.indd 70 16-08-2007 15:42:37
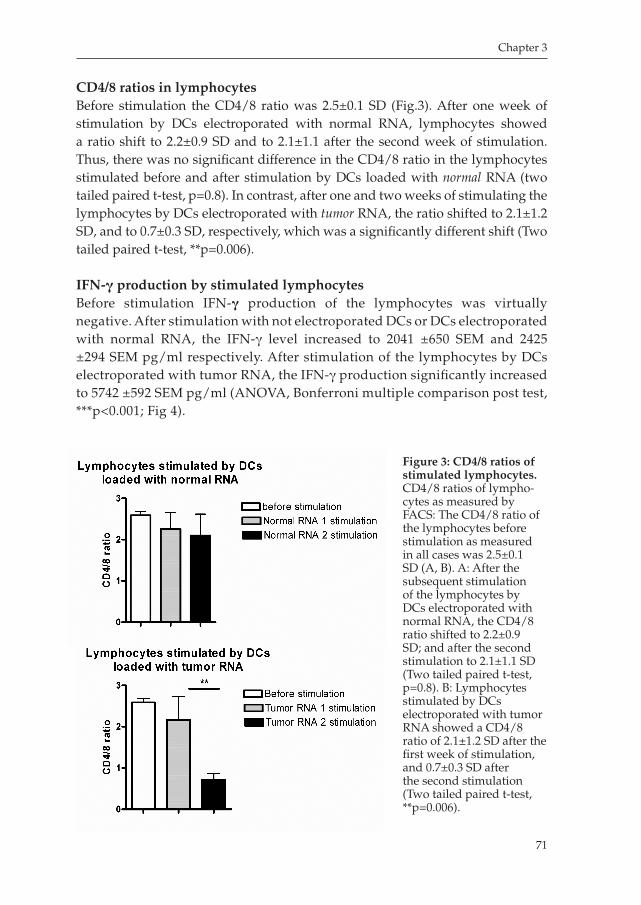
Chapter 3
71
CD4/8 ratios in lymphocytes Before stimulation the CD4/8 ratio was 2.5±0.1 SD (Fig.3). After one week of stimulation by DCs electroporated with normal RNA, lymphocytes showed a ratio shift to 2.2±0.9 SD and to 2.1±1.1 after the second week of stimulation. Thus, there was no significant difference in the CD4/8 ratio in the lymphocytes stimulated before and after stimulation by DCs loaded with normal RNA (two tailed paired ttest, p=0.8). In contrast, after one and two weeks of stimulating the lymphocytes by DCs electroporated with tumor RNA, the ratio shifted to 2.1±1.2 SD, and to 0.7±0.3 SD, respectively, which was a significantly different shift (Two tailed paired ttest, **p=0.006).
IFN-γ production by stimulated lymphocytesBefore stimulation IFNγ production of the lymphocytes was virtually negative. After stimulation with not electroporated DCs or DCs electroporated with normal RNA, the IFNγ level increased to 2041 ±650 SEM and 2425 ±294 SEM pg/ml respectively. After stimulation of the lymphocytes by DCs electroporated with tumor RNA, the IFNγ production significantly increased to 5742 ±592 SEM pg/ml (ANOVA, Bonferroni multiple comparison post test, ***p<0.001; Fig 4).
Figure 3: CD4/8 ratios of stimulated lymphocytes. CD4/8 ratios of lymphocytes as measured by FACS: The CD4/8 ratio of the lymphocytes before stimulation as measured in all cases was 2.5±0.1 SD (A, B). A: After the subsequent stimulation of the lymphocytes by DCs electroporated with normal RNA, the CD4/8 ratio shifted to 2.2±0.9 SD; and after the second stimulation to 2.1±1.1 SD (Two tailed paired ttest, p=0.8). B: Lymphocytes stimulated by DCs electroporated with tumor RNA showed a CD4/8 ratio of 2.1±1.2 SD after the first week of stimulation, and 0.7±0.3 SD after the second stimulation (Two tailed paired ttest, **p=0.006).
Milano_opm_v5.indd 71 16-08-2007 15:42:38
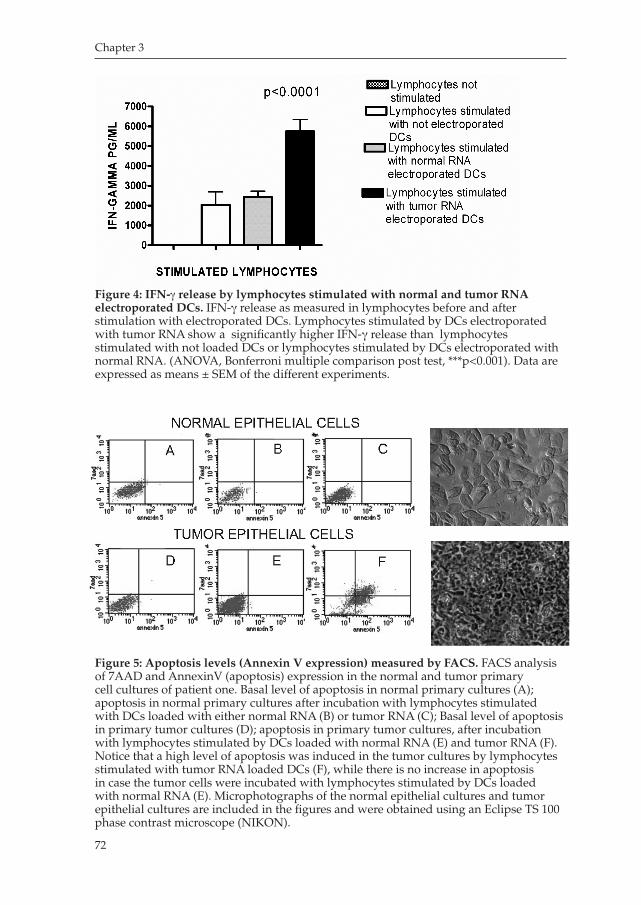
Chapter 3
72
Figure 4: IFN-γ release by lymphocytes stimulated with normal and tumor RNA electroporated DCs. IFNγ release as measured in lymphocytes before and after stimulation with electroporated DCs. Lymphocytes stimulated by DCs electroporated with tumor RNA show a significantly higher IFNγ release than lymphocytes stimulated with not loaded DCs or lymphocytes stimulated by DCs electroporated with normal RNA. (ANOVA, Bonferroni multiple comparison post test, ***p<0.001). Data are expressed as means ± SEM of the different experiments.
Figure 5: Apoptosis levels (Annexin V expression) measured by FACS. FACS analysis of 7AAD and AnnexinV (apoptosis) expression in the normal and tumor primary cell cultures of patient one. Basal level of apoptosis in normal primary cultures (A); apoptosis in normal primary cultures after incubation with lymphocytes stimulated with DCs loaded with either normal RNA (B) or tumor RNA (C); Basal level of apoptosis in primary tumor cultures (D); apoptosis in primary tumor cultures, after incubation with lymphocytes stimulated by DCs loaded with normal RNA (E) and tumor RNA (F). Notice that a high level of apoptosis was induced in the tumor cultures by lymphocytes stimulated with tumor RNA loaded DCs (F), while there is no increase in apoptosis in case the tumor cells were incubated with lymphocytes stimulated by DCs loaded with normal RNA (E). Microphotographs of the normal epithelial cultures and tumor epithelial cultures are included in the figures and were obtained using an Eclipse TS 100 phase contrast microscope (NIKON).
Milano_opm_v5.indd 72 16-08-2007 15:42:39
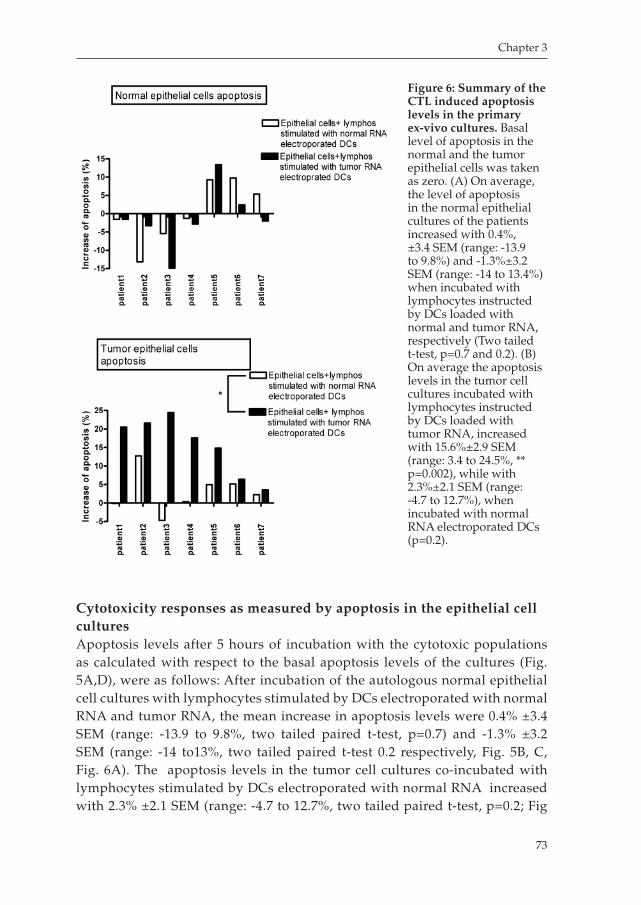
Chapter 3
73
Cytotoxicity responses as measured by apoptosis in the epithelial cell culturesApoptosis levels after 5 hours of incubation with the cytotoxic populations as calculated with respect to the basal apoptosis levels of the cultures (Fig. 5A,D), were as follows: After incubation of the autologous normal epithelial cell cultures with lymphocytes stimulated by DCs electroporated with normal RNA and tumor RNA, the mean increase in apoptosis levels were 0.4% ±3.4 SEM (range: 13.9 to 9.8%, two tailed paired ttest, p=0.7) and 1.3% ±3.2 SEM (range: 14 to13%, two tailed paired ttest 0.2 respectively, Fig. 5B, C, Fig. 6A). The apoptosis levels in the tumor cell cultures coincubated with lymphocytes stimulated by DCs electroporated with normal RNA increased with 2.3% ±2.1 SEM (range: 4.7 to 12.7%, two tailed paired ttest, p=0.2; Fig
Figure 6: Summary of the CTL induced apoptosis levels in the primary ex-vivo cultures. Basal level of apoptosis in the normal and the tumor epithelial cells was taken as zero. (A) On average, the level of apoptosis in the normal epithelial cultures of the patients increased with 0.4%, ±3.4 SEM (range: 13.9 to 9.8%) and 1.3%±3.2 SEM (range: 14 to 13.4%) when incubated with lymphocytes instructed by DCs loaded with normal and tumor RNA, respectively (Two tailed ttest, p=0.7 and 0.2). (B) On average the apoptosis levels in the tumor cell cultures incubated with lymphocytes instructed by DCs loaded with tumor RNA, increased with 15.6%±2.9 SEM (range: 3.4 to 24.5%, ** p=0.002), while with 2.3%±2.1 SEM (range: 4.7 to 12.7%), when incubated with normal RNA electroporated DCs (p=0.2).
Milano_opm_v5.indd 73 16-08-2007 15:42:43

Chapter 3
74
5E, Fig.6). The mean level of apoptosis in the tumor cell cultures incubated with lymphocytes instructed by tumor RNA electroporated DCs increased significantly with 15.6% ±2.9 SEM (range: 3.4 to 24.5%, Two tailed paired ttest, p<0.05 Fig.5F, Fig.6B).
DISCUSSION
Clinical trials using antigenpulsed DCs have been conducted in patients with various types of cancer, including myeloid leukemia, glioblastoma, metastatic melanoma, pancreas, colorectal cancer, and many others3135. To our knowledge, no reports have been yet published regarding DC therapy on esophageal cancer. Genotypically and phenotypically, esophageal cancers are highly heterogeneous36, which makes it difficult to choose a single effective antigenic target for treatment of these type of malignancy. Using total tumor RNA as an antigen load for DCs overcomes the need to identify specific tumor antigens, but as well provides the opportunity to evoke more effective anticancer cytotoxic T cell responses against tumors without knowing the exact nature of the targeted antigens37. RNAbased DC immunotherapy with the use of total tumor RNA provides the potential to generate a polyclonal immune response to multiple known and unknown tumor antigens without the limitation to specific HLA types. However, when using total tumor RNA for loading DCs there is a major concern for inducing direct adverse effects through breaking tolerance to self antigens resulting in autoimmune responses. Therefore, a preclinical ex-vivo evaluation in an autologous set up to estimate potential adverse effects of this strategy could be useful to predict inauspicious effects in a clinical setting.
In our study we analyzed the feasibility of an autologous read out system to evaluate cytotoxicity responses against esophageal cancer cells. Furthermore, in this study, the concern of breaking tolerance to selfantigens, when immunizing with total tumor RNA, was addressed by monitoring lysis activity against the patient’s normal epithelial cells in the ex-vivo read out system. In general we found that DCs electroporated with tumor RNA elicited cytotoxic lymphocytes which were able to recognize and induce significantly high levels of apoptosis in the autologous cultured cancer cells (p<0.05, Fig.6), while these lymphocytes did not induce a cytotoxic reaction against the autologous normal epithelial cells (p=0.2, Fig.6). In addition, in the control experiments with lymphocytes stimulated by DCs loaded with normal RNA, in general no significant increase in apoptosis levels in the autologous normal epithelial cells was detected. This observation corresponds with a previous study, in which DCs electroporated with renal tumor RNA, did not show a cytotoxic response against benign renal parenchyma38. It needs to be pointed out, however, that the increase in level of
Milano_opm_v5.indd 74 16-08-2007 15:42:43

Chapter 3
75
apoptosis in the normal cultures ranged from 13.9 to 12%. Thus, in certain cases this could mean that the DCs can mediate adverse effects on normal tissues. In one particular case, for instance, the DCs loaded with tumor RNA induced a cytotoxic lymphocytes mediated increase in apoptosis level of 12% in the normal cells and, although this was significantly lower when compared to the apoptosis level of 22% as induced in the tumor cells (p<0.05, Fig.6), it would be doubtful whether or not these DCs would be suitable as a vaccine for patient therapy.
Despite the strong potency of DCs to present tumor antigens, the efficacy of therapeutic DC vaccination against cancer is questioned. So far only a limited rate of objective tumor regressions have been observed in clinical studies21,32,3943. One reason for the discrepancy between the outcomes as seen in the preclinical feasibility assays with respect to the true clinical responses could be that most preclinical in vitro tumor models to evaluate direct cytotoxicity of DCs have been performed on either cancer cell lines or mice models4448. Because of an absent or different immune system, tumor cell lines and animal models in general will exhibit remarkable anticancer responses, while these models are not suitable to predict direct adverse immune responses in humans. Indirectly, the potency of DCs is measured through their ability to induce proinflammatory cytokines such as IL12, for their migratory ability, their potency to induce allogenic Tcell responses and tumor antigenspecific CD8+ T cells39,4951. Therefore, another potential application of the test model is its application for predicting clinical anti tumor responses. In our seven cases cytotoxic lymphocytes induced apoptosis was on average 15% in the autologous tumor cultures, but the range of lysis was as low as 4% and as high as 24%. It is possible that these ex-vivo responses would correlate with clinical responses, and as such this set up might be used as a prognostic clinical tool. Unquestionably, this should be first evaluated in patient trials, during which as well late adverse responses can be monitored and long term safety of total tumor RNA based DCs can be studied. Of interest is a similar recent study, which demonstrated that DC induced preclinical ex-vivo CTLs responses on autologous tumor cell cultures of melanoma patients do correlate with clinical tumor responses53.
The main difficulty of our set up is the loss of the cultures due to bacterial overgrowth and cell death. Five of twelve cultures were lost because of these reasons. This is not surprising since esophageal cancers are highly contaminated and may contain necrotic areas. The majority of cultures were lost in the initiation phase of the study. Optimizing the culturing media, targeting biopsies from non necrotic tumor areas and minimizing the delay between collection of the tissues and setting up of the cultures highly diminished the number of losses.
In this study loading of the DCs was performed through direct electroporation of the monocytes with total RNA. This method that has been recently described was found to be particularly efficient with a high yield of fully mature immunopotent
Milano_opm_v5.indd 75 16-08-2007 15:42:43

Chapter 3
76
DCs54. Furthermore, the method used did not interfere with the expression of the most important maturation markers, such as CD83, CD86, CD80, CD209, the expression of MHCClass I, which is of pivotal importance for a proper CD8+ T cell priming, and MHCClass II, of central importance for inducing CD4+ T helper response (Fig.2).
Another important finding in the present study is that the immunopotency of the DCs loaded with tumor RNA was significantly higher compared to those loaded with normal RNA. This was demonstrated by a significantly more profound shift of the CD4/8 ratio towards CD8+ T cells (Fig. 3), and the 3 fold increase in the level of IFNγ (Fig. 4). The more significant shift towards a CD8+ T cells subpopulation induced by tumor RNA loaded DCs on autologous lymphocytes as compared by the shift induced by normal RNA loaded DCs, indicates that lymphocytes stimulated by tumor RNA loaded DCs were primed towards a cytotoxic population, whereas lymphocytes stimulated by normal RNA loaded DCs maintained tolerance for self antigens and as consequence the shift towards CD8+ T cells was not significant. This hypothesis is further sustained by the additional shift towards CD8+ T cells after the second stimulation of lymphocytes that was only seen in case DCs were electroporated with tumor RNA. This suggests that lymphocytes that were primed in the first stimulation, further expanded after the additional stimulation with tumor antigen presenting DCs.
In summary, here we present a human autologous ex-vivo read out system that appears to be an effective and useful read out for testing the efficacy of DC induced cytotoxic responses against esophageal cancer cells, and to monitor for direct adverse effects on autologous normal tissues of esophageal cancer patients. In addition, the minimal lytic effects on the normal cell cultures encourages the use of total tumor RNA based DC vaccines. This important ex-vivo validation study sets the stage to proceed with establishment of this novel model for preclinical testing of CTLs and antigen presenting cells, and to further explore the strategy of total tumor RNA based DC therapy for treatment of esophageal cancer.
Milano_opm_v5.indd 76 16-08-2007 15:42:44

Chapter 3
77
REFERENCES
1. Pera, M. Trends in incidence and prevalence of specialized intestinal metaplasia, barrett’s
esophagus, and adenocarcinoma of the gastroesophageal junction. World J Surg, 27: 9991008;
discussion 10061008, 2003.
2. O’Connor, J. B., Falk, G. W., and Richter, J. E. The incidence of adenocarcinoma and dysplasia
in Barrett’s esophagus: report on the Cleveland Clinic Barrett’s Esophagus Registry. Am J
Gastroenterol, 94: 20372042, 1999.
3. Hashibe, M., Boffetta, P., Janout, V., Zaridze, D., Shangina, O., Mates, D., SzeszeniaDabrows
ka, N., Bencko, V., and Brennan, P. Esophageal cancer in Central and Eastern Europe: Tobacco
and alcohol. Int J Cancer, 2007.
4. Reid, B. J., Barrett, M. T., Galipeau, P. C., Sanchez, C. A., Neshat, K., Cowan, D. S., and Levine, D. S.
Barrett’s esophagus: ordering the events that lead to cancer. Eur J Cancer Prev, 5 Suppl 2: 5765, 1996.
5. Cossentino, M. J. and Wong, R. K. Barrett’s esophagus and risk of esophageal adenocarci
noma. Semin Gastrointest Dis, 14: 128135, 2003.
6. Spechler, S. J., Zeroogian, J. M., Antonioli, D. A., Wang, H. H., and Goyal, R. K. Prevalence of
metaplasia at the gastrooesophageal junction. Lancet, 344: 15331536, 1994.
7. Demeester, S. R., Peters, J. H., and Demeester, T. R. Barrett’s esophagus. Curr Probl Surg, 38:
558640, 2001.
8. Peracchia, A., Bonavina, L., Via, A., and Incarbone, R. Current trends in the surgical treatment
of esophageal and cardia adenocarcinoma. J Exp Clin Cancer Res, 18: 289294, 1999.
9. Bosset, J. F., Gignoux, M., Triboulet, J. P., Tiret, E., Mantion, G., Elias, D., Lozach, P., Ollier, J. C.,
Pavy, J. J., Mercier, M., and Sahmoud, T. Chemoradiotherapy followed by surgery compared
with surgery alone in squamouscell cancer of the esophagus. N Engl J Med, 337: 161167, 1997.
10. Pulkkinen, J., Sipila, J., Hujala, K., and Grenman, R. Intraluminal radiotherapy in esophageal
cancer. An update. Otolaryngol Pol, 58: 191195, 2004.
11. Di Fiore, F., Lecleire, S., Galais, M. P., Rigal, O., Vie, B., David, I., Hamidou, H., Paillot, B., Jacob,
J. H., and Michel, P. Impact of radiation schedule and chemotherapy duration in definitive
chemoradiotherapy regimen for esophageal cancer. Gastroenterol Clin Biol, 30: 845851, 2006.
12. Banchereau, J. and Steinman, R. M. Dendritic cells and the control of immunity. Nature, 392:
245252, 1998.
13. Lanzavecchia, A. and Sallusto, F. Regulation of T cell immunity by dendritic cells. Cell, 106:
263266, 2001.
14. Ardavin, C., Amigorena, S., and Reis e Sousa, C. Dendritic cells: immunobiology and cancer
immunotherapy. Immunity, 20: 1723, 2004.
15. Stift, A., Friedl, J., Dubsky, P., BachleitnerHofmann, T., Schueller, G., Zontsich, T., Benkoe,
T., Radelbauer, K., Brostjan, C., Jakesz, R., and Gnant, M. Dendritic cellbased vaccination in
solid cancer. J Clin Oncol, 21: 135142, 2003.
16. Berger, T. G., Feuerstein, B., Strasser, E., Hirsch, U., Schreiner, D., Schuler, G., and Schuler
Thurner, B. Largescale generation of mature monocytederived dendritic cells for clinical
application in cell factories. J Immunol Methods, 268: 131140, 2002.
Milano_opm_v5.indd 77 16-08-2007 15:42:44

Chapter 3
78
17. Schultz, E. S., SchulerThurner, B., Stroobant, V., Jenne, L., Berger, T. G., Thielemanns, K., van der
Bruggen, P., and Schuler, G. Functional analysis of tumorspecific Th cell responses detected in
melanoma patients after dendritic cellbased immunotherapy. J Immunol, 172: 13041310, 2004.
18. Nestle, F. O., Filgueira, L., Nickoloff, B. J., and Burg, G. Human dermal dendritic cells process
and present soluble protein antigens. J Invest Dermatol, 110: 762766, 1998.
19. SchulerThurner, B., Schultz, E. S., Berger, T. G., Weinlich, G., Ebner, S., Woerl, P., Bender, A.,
Feuerstein, B., Fritsch, P. O., Romani, N., and Schuler, G. Rapid induction of tumorspecific
type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopre
served, peptideloaded monocytederived dendritic cells. J Exp Med, 195: 12791288, 2002.
20. Dhodapkar, M. V., Krasovsky, J., Steinman, R. M., and Bhardwaj, N. Mature dendritic cells
boost functionally superior CD8(+) Tcell in humans without foreign helper epitopes. J Clin
Invest, 105: R9R14, 2000.
21. Maier, T., TunKyi, A., Tassis, A., Jungius, K. P., Burg, G., Dummer, R., and Nestle, F. O. Vac
cination of patients with cutaneous Tcell lymphoma using intranodal injection of autologous
tumorlysatepulsed dendritic cells. Blood, 102: 23382344, 2003.
22. Strobel, I., Berchtold, S., Gotze, A., Schulze, U., Schuler, G., and Steinkasserer, A. Human den
dritic cells transfected with either RNA or DNA encoding influenza matrix protein M1 differ
in their ability to stimulate cytotoxic T lymphocytes. Gene Ther, 7: 20282035, 2000.
23. Van Tendeloo, V. F., Ponsaerts, P., Lardon, F., Nijs, G., Lenjou, M., Van Broeckhoven, C., Van
Bockstaele, D. R., and Berneman, Z. N. Highly efficient gene delivery by mRNA electroporation in
human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to elec
troporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood, 98: 4956, 2001.
24. Nair, S. K., Morse, M., Boczkowski, D., Cumming, R. I., Vasovic, L., Gilboa, E., and Lyerly,
H. K. Induction of tumorspecific cytotoxic T lymphocytes in cancer patients by autologous
tumor RNAtransfected dendritic cells. Ann Surg, 235: 540549, 2002.
25. Heiser, A., Maurice, M. A., Yancey, D. R., Wu, N. Z., Dahm, P., Pruitt, S. K., Boczkowski, D.,
Nair, S. K., Ballo, M. S., Gilboa, E., and Vieweg, J. Induction of polyclonal prostate cancer
specific CTL using dendritic cells transfected with amplified tumor RNA. J Immunol, 166:
29532960, 2001.
26. Muller, M. R., Grunebach, F., Kayser, K., Vogel, W., Nencioni, A., Brugger, W., Kanz, L., and
Brossart, P. Expression of her2/neu on acute lymphoblastic leukemias: implications for the
development of immunotherapeutic approaches. Clin Cancer Res, 9: 34483453, 2003.
27. PalancaWessels, M. C., Barrett, M. T., Galipeau, P. C., Rohrer, K. L., Reid, B. J., and Rabino
vitch, P. S. Genetic analysis of longterm Barrett’s esophagus epithelial cultures exhibiting
cytogenetic and ploidy abnormalities. Gastroenterology, 114: 295304, 1998.
28. Cranmer, L. D., Trevor, K. T., and Hersh, E. M. Clinical applications of dendritic cell vaccina
tion in the treatment of cancer. Cancer Immunol Immunother, 53: 275306, 2004.
29. Braat, H., van den Brande, J., van Tol, E., Hommes, D., Peppelenbosch, M., and van Deventer,
S. Lactobacillus rhamnosus induces peripheral hyporesponsiveness in stimulated CD4+ T
cells via modulation of dendritic cell function. Am J Clin Nutr, 80: 16181625, 2004.
Milano_opm_v5.indd 78 16-08-2007 15:42:44

Chapter 3
79
30. Cook, E. B., Stahl, J. L., Lowe, L., Chen, R., Morgan, E., Wilson, J., Varro, R., Chan, A., Grazi
ano, F. M., and Barney, N. P. Simultaneous measurement of six cytokines in a single sample of
human tears using microparticlebased flow cytometry: allergics vs. nonallergics. J Immunol
Methods, 254: 109118, 2001.
31. Anichini, A., Molla, A., Mortarini, R., Tragni, G., Bersani, I., Di Nicola, M., Gianni, A. M.,
Pilotti, S., Dunbar, R., Cerundolo, V., and Parmiani, G. An expanded peripheral T cell popu
lation to a cytotoxic T lymphocyte (CTL)defined, melanocytespecific antigen in metastatic
melanoma patients impacts on generation of peptidespecific CTLs but does not overcome
tumor escape from immune surveillance in metastatic lesions. J Exp Med, 190: 651667, 1999.
32. Burgdorf, S. K., Fischer, A., Claesson, M. H., Kirkin, A. F., Dzhandzhugazyan, K. N., and Ro
senberg, J. Vaccination with melanoma lysatepulsed dendritic cells, of patients with advanced
colorectal carcinoma: report from a phase I study. J Exp Clin Cancer Res, 25: 201206, 2006.
33. Fong, L. and Engleman, E. G. Dendritic cells in cancer immunotherapy. Annu Rev Immunol,
18: 245273, 2000.
34. Liu, G., Ying, H., Zeng, G., Wheeler, C. J., Black, K. L., and Yu, J. S. HER2, gp100, and MAGE
1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res, 64:
49804986, 2004.
35. Morse, M. A., Nair, S. K., Boczkowski, D., Tyler, D., Hurwitz, H. I., Proia, A., Clay, T. M.,
Schlom, J., Gilboa, E., and Lyerly, H. K. The feasibility and safety of immunotherapy with
dendritic cells loaded with CEA mRNA following neoadjuvant chemoradiotherapy and resec
tion of pancreatic cancer. Int J Gastrointest Cancer, 32: 16, 2002.
36. Krishnadath, K. K., Wang, K. K., Taniguchi, K., Sebo, T. J., Buttar, N. S., Anderson, M. A.,
Lutzke, L. S., and Liu, W. Persistent genetic abnormalities in Barrett’s esophagus after photo
dynamic therapy. Gastroenterology, 119: 624630, 2000.
37. Kalady, M. F., Onaitis, M. W., Padilla, K. M., Emani, S., Tyler, D. S., and Pruitt, S. K. Enhanced
dendritic cell antigen presentation in RNAbased immunotherapy. J Surg Res, 105: 1724, 2002.
38. Su, Z., Dannull, J., Heiser, A., Yancey, D., Pruitt, S., Madden, J., Coleman, D., Niedzwiecki,
D., Gilboa, E., and Vieweg, J. Immunological and clinical responses in metastatic renal cancer
patients vaccinated with tumor RNAtransfected dendritic cells. Cancer Res, 63: 21272133, 2003.
39. Banchereau, J. and Palucka, A. K. Dendritic cells as therapeutic vaccines against cancer. Nat
Rev Immunol, 5: 296306, 2005.
40. Babatz, J., Rollig, C., Lobel, B., Folprecht, G., Haack, M., Gunther, H., Kohne, C. H., Ehninger,
G., Schmitz, M., and Bornhauser, M. Induction of cellular immune responses against carcin
oembryonic antigen in patients with metastatic tumors after vaccination with altered peptide
ligandloaded dendritic cells. Cancer Immunol Immunother, 55: 268276, 2006.
41. Dees, E. C., McKinnon, K. P., Kuhns, J. J., Chwastiak, K. A., Sparks, S., Myers, M., Collins, E. J.,
Frelinger, J. A., Van Deventer, H., Collichio, F., Carey, L. A., Brecher, M. E., Graham, M., Earp,
H. S., and Serody, J. S. Dendritic cells can be rapidly expanded ex vivo and safely administered
in patients with metastatic breast cancer. Cancer Immunol Immunother, 53: 777785, 2004.
42. Lee, W. C., Wang, H. C., Hung, C. F., Huang, P. F., Lia, C. R., and Chen, M. F. Vaccination of
Milano_opm_v5.indd 79 16-08-2007 15:42:45

Chapter 3
80
advanced hepatocellular carcinoma patients with tumor lysatepulsed dendritic cells: a clini
cal trial. J Immunother, 28: 496504, 2005.
43. Roddie, H., Klammer, M., Thomas, C., Thomson, R., Atkinson, A., Sproul, A., Waterfall, M.,
Samuel, K., Yin, J., Johnson, P., and Turner, M. Phase I/II study of vaccination with dendritic
like leukaemia cells for the immunotherapy of acute myeloid leukaemia. Br J Haematol, 133:
152157, 2006.
44. Muller, M. R., Grunebach, F., Nencioni, A., and Brossart, P. Transfection of dendritic cells with
RNA induces CD4and CD8mediated T cell immunity against breast carcinomas and reveals
the immunodominance of presented T cell epitopes. J Immunol, 170: 58925896, 2003.
45. Kalady, M. F., Onaitis, M. W., Emani, S., AbdulWahab, Z., Pruitt, S. K., and Tyler, D. S. Den
dritic cells pulsed with pancreatic cancer total tumor RNA generate specific antipancreatic
cancer T cells. J Gastrointest Surg, 8: 175181; discussion 181172, 2004.
46. PilonThomas, S., Li, W., Briggs, J. J., Djeu, J., Mule, J. J., and Riker, A. I. Immunostimulatory
effects of CpGODN upon dendritic cellbased immunotherapy in a murine melanoma mo
del. J Immunother, 29: 381387, 2006.
47. Warncke, M., Dodero, A., Dierbach, H., Follo, M., and Veelken, H. Murine dendritic cells ge
nerated under serumfree conditions have a mature phenotype and efficiently induce primary
immune responses. J Immunol Methods, 310: 111, 2006.
48. Nair, S. K., Snyder, D., Rouse, B. T., and Gilboa, E. Regression of tumors in mice vaccinated with
professional antigenpresenting cells pulsed with tumor extracts. Int J Cancer, 70: 706715, 1997.
49. Osada, T., Nagawa, H., Takahashi, T., Tsuno, N. H., Kitayama, J., and Shibata, Y. Dendritic
cells cultured in antiCD40 antibodyimmobilized plates elicit a highly efficient peptidespeci
fic Tcell response. J Immunother, 25: 176184, 2002.
50. Nair, S. K., Boczkowski, D., Morse, M., Cumming, R. I., Lyerly, H. K., and Gilboa, E. Induction
of primary carcinoembryonic antigen (CEA)specific cytotoxic T lymphocytes in vitro using
human dendritic cells transfected with RNA. Nat Biotechnol, 16: 364369, 1998.
51. Dannull, J., Nair, S., Su, Z., Boczkowski, D., DeBeck, C., Yang, B., Gilboa, E., and Vieweg,
J. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA
encoding OX40 ligand. Blood, 105: 32063213, 2005.
52. Warncke M., Dodero A., Dierbach H., Follo M., Veelken H., Murine dendritic cells generated
under serumfree conditions have a mature phenotype and efficiently induce primary im
mune responses. J Immunol Methods 310: 111, 2006.
53. Schmidt C.W., Martinez N.R., Neller M., Lopez J.A., Lanagan C.M., O’Connor L.E., O’Rourke
M.G.E., Johnson M.K., See J.L., Slater G.J., Ellem K.A.O., An immunological insight into effec
tive dendritic cell immunotherapy of metastatic melanoma. Abstract, 9th International Confer
ence Dendritic Cells, Edimburgh, Scotland, 1620 sept. 2006.
54. Milano F., van Baal J.W., Rygiel A.M., Bergman J.J., van Deventer S.J., Kapsenberg M., Pep
pelenbosch M.P., Krishnadath K.K., An improved Protocol for generation of immunopotent
DC through direct electroporation Of CD14+ monocytes. Accepted for publication, Journal of
Immunological Methods, 2007.
Milano_opm_v5.indd 80 16-08-2007 15:42:45

Chapter 4
Characterization of esophageal cancer
microenvironment: a balance towards tumor
or towards anticancer immune-response?
Francesca Milano, Tineke Jorritsma, Agnieszka M. Rygiel, Jacques J.G.H.M. Bergman, Carine Sondermeijer,
Maikel P.Peppelenbosch, Anja Ten Brinke, Marieke S. van Ham, Kausilia K. Krishnadath
Submitted
Milano_opm_v5.indd 81 16-08-2007 15:42:46

Chapter 4
82
ABSTRACT
OBJECTIVE: Immunotherapy of esophageal adenocarcinoma (EAC) remains disappointing, possibly because of a therapyunfavorable tumor microenvironment, prompting investigations as the nature of this environment. DESIGN AND PATIENTS: Biopsies of tumor and normal tissues were collected from 17 EAC patients, and investigated using fluorescent immunohistochemistry (IHC) for COX2, VEGF, TGFβ, IDO, CXCL3 and the receptor CXCR1 expression, or for measuring cytokine levels by cytometric bead array (CBA) and quantitative PCR (QPCR). RESULTS: IHC of normal and tumor samples showed that COX2, VEGF, TGF β, CXCL3, CXCR1, and IDO expression is upregulated in varying degrees in the tumor biopsies in 80% to 93% of the patients, whereas QPCR revealed that the cytokine IL8 is significantly upregulated in 58% of the EAC cases. CBA confirmed high IL8 expression in 80% of the tumors (MannWhitney test, p<0.05), and high IL1β in 86% of the tumor tissues as compared to normal tissues (p<0.05). IL6 was only detectable in 10% of tumors, and IL10, IL12, IL4 and IFNgamma were detected neither in the tumor nor in the normal tissues. CONCLUSIONS: The EAC microenvironment is characterized by a lack of cytokines that normally would enhance anticancer responses, such as IFNγ and IL12, and by a high expression of several immunosuppressive cytokines, and a wide range of tumor growth promoting factors, such as COX2, VEGF, TGFβ and IL8. For future treatment of patients with immunotherapies it will be important to specifically target these factors and combine immunotherapy with immune modulating agents in order to improve treatment efficacy.
Milano_opm_v5.indd 82 16-08-2007 15:42:46

Chapter 4
83
INTRODUCTION
Dendritic Cell (DC) therapy for various types of cancer has received considerable attention the last decades, and proof of principle that DC vaccination is an appealing strategy to treat cancer has been obtained on multiple occasions. Nevertheless, the limited rate of objective tumor regressions observed in clinical studies, have raised questions as to the actual efficacy of therapeutic vaccination against cancer in a practical setting13. The reasons underlying the relatively poor efficacy of DC vaccination remain only partially understood4.
Among the oftencited factors hampering effective anticancer immunotherapy, the immunosuppressive network created as a consequence of pathological interactions between cancer cells and host immune cells features prominently57. It has become clear that it involves a multitude of factors whose production is under control of a complex interplay between the normal host epithelial cells, invading tumor cells, stromal fibroblasts, inflammatory cells, proliferating endothelial cells, an altered extracellular matrix, and growth factors activating oncogenic signaling pathways8. This altered tumor environment will result in altered production of for instance costimulatory and coinhibitory molecules, such as the cytokines IL6, IL8, IL1β, TNFα, IFNγ, IL12, IL4 and many others, and an imbalance of effector T and regulatory Tcells (Tregs)9 finally resulting in tumor immune evasion10,11.
Although in vivo data are still largely lacking, a number of interesting factors, which may participate in tumorpermissive microenvironments have now been identified. Maintaining tumor growth requires an efficient supply of nutrients and oxygen, therefore proangiogenic factors, like vascular endothelial growth factor (VEGF)12 and the CXCR/CXCR ligand axis including IL8 (CXCL8)13,14 seem important factors, and may exert immunosuppressive actions as well. In addition, it has been demonstrated that production of VEGF profoundly inhibits maturation of DCs, which are major inducers of effector Tcell proliferation15. Generally considered as a potential more important factor is the expression of COX2, which provokes strong immunesuppression through the production of PGE216, and subsequent diminished IL12 release by mature DCs, in turn impairing Th1 responses17. Several studies have demonstrated that COX2 expression is strongly correlated with poor prognosis18 and it is assumed that inhibition of this protein would induce downregulation of important oncogenic pathways19,20. In addition, another factor, which can contribute to the immunesuppression and inhibition of Tcell proliferation and attracts substantial attention is Indoleamine 23 dioxygenase (IDO), a tryptophancatabolizing enzyme, which inhibits Tcell proliferation. Munn et al. have previously described a subset of human monocytederived DCs that can induce Tcell inhibition by excretion of IDO21,22. Finally, some authors note that IL1β, together with TNFα are known to induce production of IL6, which in turn mediates the inhibition of an appropriate
Milano_opm_v5.indd 83 16-08-2007 15:42:46

Chapter 4
84
DC differentiation2325 and may also be involved in providing the tumor with a favorable environment.
Apart from the active expression of immunosuppressive factors, inhibition of production of cytokines that induce DC differentiation, such as IL12 and IFNgamma, may also induce tumor immunetolerance6. It may be predicted that an impaired balance in immunestimulatory and immunesuppressive factors leads to defects in adaptive immune responses, and in particular may impair antitumor DC functioning6, partly explaining the limited success obtained hitherto with DC vaccination studies of solid cancer and prompting investigation to clarify whether such a disturbed balance is present in patients. It is important to stress, however, that a clear demonstration that such a tumor microenvironment exists in esophageal adenocarcinomas (EAC) patients has not been clearly provided yet.
Since EAC are highly malignant and seem to be resistant to conventional therapies such as chemo and radiotherapy, we anticipated that EAC patients may highly benefit from DC immunotherapies in the future. EAC is associated with Barrett’s esophagus (BE), a metaplastic condition of the distal esophagus, in which after gastroesophageal reflux injury, the normal squamous epithelium is replaced by columnar epithelium26,27. These cancers have an extremely poor prognosis, with an overall five years survival rate of less than 15%28,29. The aim of our study was to gain further insight in the EAC tumor microenvironment, in order to find novel targets for developing immunotherapies and for more effective combinatorial treatment strategies. In order to achieve this, we performed immunohistochemistry (IHC) on normal and tumor EAC biopsies, staining for several factors, such as COX2, VEGF, TGFbeta, IDO, and CXCL3 and its receptor CXCR1. Furthermore we isolated RNA from biopsies and perform quantitative PCR to establish the balance between proinflammatory and antiinflammatory cytokines, such as IL6 and IL8. To confirm the presence of the cytokines analyzed by PCR, we obtained lysates of biopsies and measured the cytokine levels by Cytometric Bead Array.
Our results show that the EAC microenvironment is characterized by a disturbed immunological background specifically characterized by high levels of IL8 and IL1β, while other immunosuppressive factors such as IL6 and IL10, are not found in EAC. Definition of the immunological microenvironment in EAC provides us with rational clues for the future treatment of this disease.
MATERIALS AND METHODS
Patient material and characteristicsAfter informed consent and written permission, 17 EAC patients were included. Patients underwent endoscopic procedures for classifying, staging and grading of the esophageal cancer. During this procedure, 12 extra biopsies, 6 from normal
Milano_opm_v5.indd 84 16-08-2007 15:42:47
Chapter 4
85
and 6 from tumor tissues, of each patient were collected and immediately stored at 80˚C to be used for RNA isolation, IHC, and for obtaining lysates for protein measurements; biopsies were obtained from normal squamous epithelium, taken at least 3 cm above the visible mass, and from the malignancy. Matching biopsies from the same spots were taken for histopathological diagnosis.
Table 1
Patients Age Sex Type of cancer Stage
1 51 male EAC G2T3N1M1b
2 75 male EAC T3N0
3 50 male EAC T1N0
4 71 male EAC T3N1
5 53 male EAC T1N0M0
6 73 male EAC T3N0M0
7 80 male EAC T3N0M0
8 76 female EAC T1N0
9 58 male EAC T1N0
10 68 male EAC T4N1
11 76 female EAC T1N0
12 60 male EAC T1N0M0
13 82 male EAC T3N1M0
14 85 male EAC T3N1
15 78 male EAC T3/4N1
16 80 male EAC T1N0
17 77 male EAC T3N1M0
RNA isolationTotal RNA for PCR purposes was isolated from normal squamous and tumor biopsies. For each biopsy, 300 μl of Trifast (Peqlab Biotech, GmbH, Erlangen, Germany) were mixed with 3μl of glycogen, samples were vortex for 10 seconds, then 60 μl chloroform was added and again the samples were vortex for 30 seconds. A centrifugation step of 10 minutes at 13000 rpm at RT followed, then the supernatants were collected into a fresh tube and 180μl isopropanol was added and samples vortex for 30 seconds. The RNA was precipitated overnight at 20ºC. The next day, a centrifugation step of 30 minutes at 13000 rpm at 4ºC was performed, then the supernatant was discarded and the RNA pellet was washed with 75% ethanol. The pellet was airdryed, and then dissolved in 16.5 μl DEPC water (Ambion, Nieuwerkerk a/d, Nijssel, The Netherlands). In order to dissolve the RNA completely, the tubes were placed at 5560ºC for 3 minutes,
Milano_opm_v5.indd 85 16-08-2007 15:42:47

Chapter 4
86
vortexed and spun for two times. Upon isolation, 1.5μl of RNA solution was used to determine purity and concentration using Nanodrop (type ND1000). Assessment of the quality was performed with the RNA 2100 Pico Labchip kit (Agilent Technologies, Amstelveen, The Netherlands) using 1µl of total RNA.
Quantitative PCR (Taqman)Preparation of cDNA: to each RNA sample, 2μl of random Hexamers were added, and then incubated for 10 minutes at 70ºC. Tubes were cooled on ice, then 15μl of RNA was mixed with the following: 10μl 5X RTbuffer, 5μl 0.1M DTT, 10μl 10mM dNTPs, 0.5μl RNAse inhibitor, and 0.5μl of Superscript RT (200U/μl). Samples were incubated for 10 minutes at RT, then 50 minutes at 42ºC, and 15 minutes at 70ºC. cDNA was obtained for quantitative RT PCR using Taqman 7900HT Fast RealTime PCR System (Applied Biosystems, Foster City, CA, USA). Real time PCR was performed using primers for selected genes which were developed, such as for IL8, IL6 and 18S RNA as control (Table 2). Reactions were performed in 20μl total volume, consisting of 5μl of sample diluted 1:50, and 15μl Master Mix constituted by PCR Master Mix SYBR Green PCR Master Mix (Applied Biosystem, Foster City, CA, USA) , and DEPC water (Ambion). Each sample was analysed in duplicate. The thermal Cycle conditions were: Cycle Time Temperature Repeat Ramp Time, 2’at 50ºC, hold 10’ at 95 ºC, [(cycle 15” at 95 ºC, 1’at 60 ºC )x 40], [(15”20 ºC , 1’ at 95 ºC )x1]. The level of gene expression within each sample was adjusted to levels of an internal control (human ribosomal 18S RNA) before expression was calculated as a percentage of the level of gene expression using the control samples.
Table 2Cytokines PrimersIL8 F: TTGGCAGCCTTCCTGATTTC
R: AACTTCTCCACAACCCTCTGIL6 F: GTACATCCTCGACGGCATC
R: CCAGGCAAGTCTCCTCATTG18SrRNA F: CGGCTACCACATCCAAGGAA
R: GCTGGAATTACCGCGGCT
Fluorescent immuno-histochemistry of normal and tumor biopsiesTissue sections from squamous and EAC patients were embedded in TissueTek®, Optimum Cutting Temperature compound (Sakura Finetek, USA, Torrance, CA) at 20°C, and sectioned using a cryomicrotome (Microm HM 550) in serial cryostat sections of 6μm, placed on Superfrost + (Menzel Glaser, Braunschweig, Germany) glass slides and airdried overnight. For routine histological examination, of each block one slide was routinely stained by Hematoxylin and Eosin (H&E). Slide preparations were fixed for 20 minutes
Milano_opm_v5.indd 86 16-08-2007 15:42:48
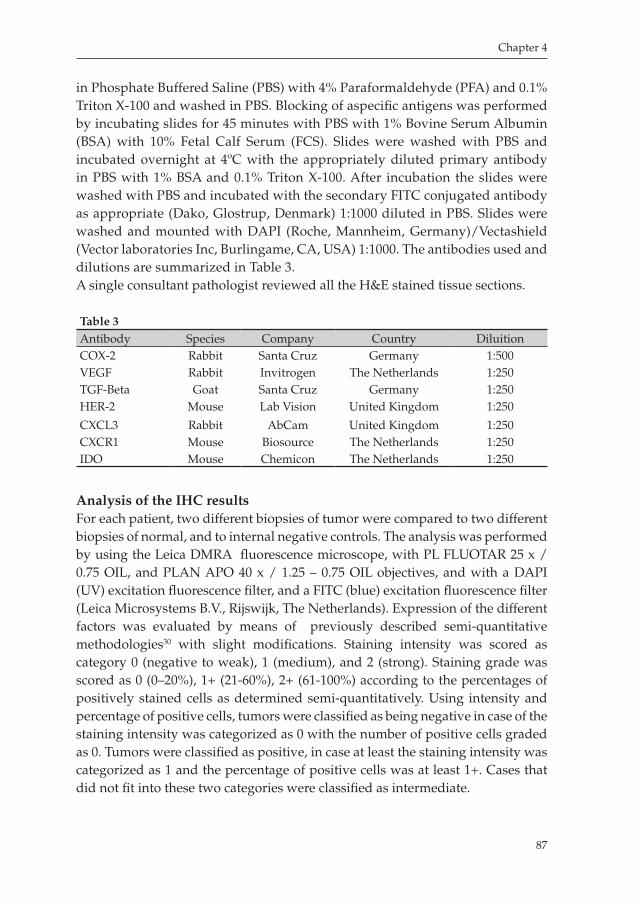
Chapter 4
87
in Phosphate Buffered Saline (PBS) with 4% Paraformaldehyde (PFA) and 0.1% Triton X100 and washed in PBS. Blocking of aspecific antigens was performed by incubating slides for 45 minutes with PBS with 1% Bovine Serum Albumin (BSA) with 10% Fetal Calf Serum (FCS). Slides were washed with PBS and incubated overnight at 4ºC with the appropriately diluted primary antibody in PBS with 1% BSA and 0.1% Triton X100. After incubation the slides were washed with PBS and incubated with the secondary FITC conjugated antibody as appropriate (Dako, Glostrup, Denmark) 1:1000 diluted in PBS. Slides were washed and mounted with DAPI (Roche, Mannheim, Germany)/Vectashield (Vector laboratories Inc, Burlingame, CA, USA) 1:1000. The antibodies used and dilutions are summarized in Table 3. A single consultant pathologist reviewed all the H&E stained tissue sections.
Table 3Antibody Species Company Country DiluitionCOX2 Rabbit Santa Cruz Germany 1:500VEGF Rabbit Invitrogen The Netherlands 1:250TGFBeta Goat Santa Cruz Germany 1:250HER2 Mouse Lab Vision United Kingdom 1:250
CXCL3 Rabbit AbCam United Kingdom 1:250CXCR1 Mouse Biosource The Netherlands 1:250IDO Mouse Chemicon The Netherlands 1:250
Analysis of the IHC results For each patient, two different biopsies of tumor were compared to two different biopsies of normal, and to internal negative controls. The analysis was performed by using the Leica DMRA fluorescence microscope, with PL FLUOTAR 25 x / 0.75 OIL, and PLAN APO 40 x / 1.25 – 0.75 OIL objectives, and with a DAPI (UV) excitation fluorescence filter, and a FITC (blue) excitation fluorescence filter (Leica Microsystems B.V., Rijswijk, The Netherlands). Expression of the different factors was evaluated by means of previously described semiquantitative methodologies30 with slight modifications. Staining intensity was scored as category 0 (negative to weak), 1 (medium), and 2 (strong). Staining grade was scored as 0 (0–20%), 1+ (2160%), 2+ (61100%) according to the percentages of positively stained cells as determined semiquantitatively. Using intensity and percentage of positive cells, tumors were classified as being negative in case of the staining intensity was categorized as 0 with the number of positive cells graded as 0. Tumors were classified as positive, in case at least the staining intensity was categorized as 1 and the percentage of positive cells was at least 1+. Cases that did not fit into these two categories were classified as intermediate.
Milano_opm_v5.indd 87 16-08-2007 15:42:48

Chapter 4
88
Cytometric Bead Array (CBA) Multiplex AssaysBiopsies from both normal and tumor tissues were used to obtain lysates by dissolving them in Greenberger Lysis Buffer (GLB). Briefly, the procedure was as follows: tissues were collected in liquid nitrogen and subsequently frozen down and suspended in GLB (150mM NaCl, 15mM Tris 1mM MgCl.H2O, 1mM CaCl2, 1% Triton X100, Protease Inhibitor) diluited 1:1 with PBS. Biopsies were homogenised on ice and then placed on ice for 30 minutes. The disrupted tissues were spun down at 4000 rpm for 7 minutes, and the supernatant was collected and spun down at 1400 rpm for 10 minutes. One aliquot was taken to measure protein with the BCA protein assay kit (Pierce chemical co. Rockford, IL, USA). Lysates were used to perform Cytometric Bead Array (CBA) using the CBA Inflammation kit (BD) following the manufacturer’s instructions as previously described31. Acquired data were analyzed using BD CBD calibration and analysis software.
BCA Protein assayTo determine the protein concentration in the analyzed samples, the bicinchoninic acid (BCA) protein assay kit was used. Briefly, 5μl of the lysates obtained as described above were used and incubated in a 96 wells plate together with protein assay reagents A and B following the manufacturer’s instructions, and the protein content was measured spectrophotometrically using Plate Manager ® software.
Statistical analysisStatistical analyses were performed using the software Graph Pad Prism®.Statistical tests were applied to17 independent experiments. Differences between the values were determined using both Students’ paired ttest, one way ANOVA and MannWhitney test, as appropiate. Significance was determined as p<0.05.
RESULTS
Patient’s characteristics and routine histologyThe study was approved by the Academic Medical Center (Amsterdam, The Netherlands) Hospital’s Medical Ethical Committee. All 17 patients enrolled in the study obtained informed consent and gave written permission. The population constituted of 15 males and 2 females, the mean age was 74 (range 5087), and cancers were at variable stages of differentiation. Histopathology of the H&E staining of the biopsies as evaluated by an expert pathologist showed EAC in all the 17 patients (Fig.3A). Patient data are summarized in table 1.
Milano_opm_v5.indd 88 16-08-2007 15:42:49
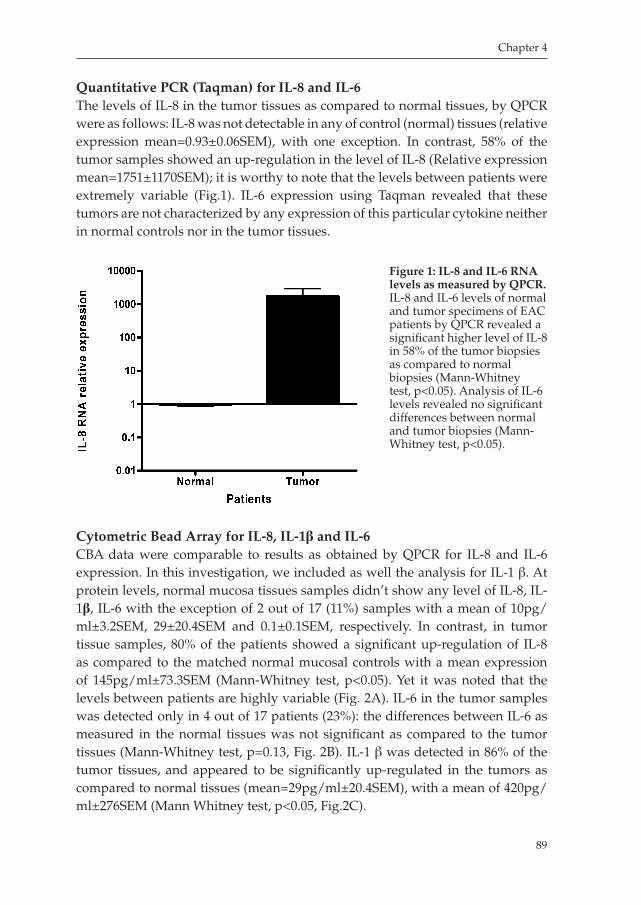
Chapter 4
89
Quantitative PCR (Taqman) for IL-8 and IL-6The levels of IL8 in the tumor tissues as compared to normal tissues, by QPCR were as follows: IL8 was not detectable in any of control (normal) tissues (relative expression mean=0.93±0.06SEM), with one exception. In contrast, 58% of the tumor samples showed an upregulation in the level of IL8 (Relative expression mean=1751±1170SEM); it is worthy to note that the levels between patients were extremely variable (Fig.1). IL6 expression using Taqman revealed that these tumors are not characterized by any expression of this particular cytokine neither in normal controls nor in the tumor tissues.
Cytometric Bead Array for IL-8, IL-1β and IL-6CBA data were comparable to results as obtained by QPCR for IL8 and IL6 expression. In this investigation, we included as well the analysis for IL1 β. At protein levels, normal mucosa tissues samples didn’t show any level of IL8, IL1β, IL6 with the exception of 2 out of 17 (11%) samples with a mean of 10pg/ml±3.2SEM, 29±20.4SEM and 0.1±0.1SEM, respectively. In contrast, in tumor tissue samples, 80% of the patients showed a significant upregulation of IL8 as compared to the matched normal mucosal controls with a mean expression of 145pg/ml±73.3SEM (MannWhitney test, p<0.05). Yet it was noted that the levels between patients are highly variable (Fig. 2A). IL6 in the tumor samples was detected only in 4 out of 17 patients (23%): the differences between IL6 as measured in the normal tissues was not significant as compared to the tumor tissues (MannWhitney test, p=0.13, Fig. 2B). IL1 β was detected in 86% of the tumor tissues, and appeared to be significantly upregulated in the tumors as compared to normal tissues (mean=29pg/ml±20.4SEM), with a mean of 420pg/ml±276SEM (Mann Whitney test, p<0.05, Fig.2C).
Figure 1: IL-8 and IL-6 RNA levels as measured by QPCR. IL8 and IL6 levels of normal and tumor specimens of EAC patients by QPCR revealed a significant higher level of IL8 in 58% of the tumor biopsies as compared to normal biopsies (MannWhitney test, p<0.05). Analysis of IL6 levels revealed no significant differences between normal and tumor biopsies (MannWhitney test, p<0.05).
Milano_opm_v5.indd 89 16-08-2007 15:42:50
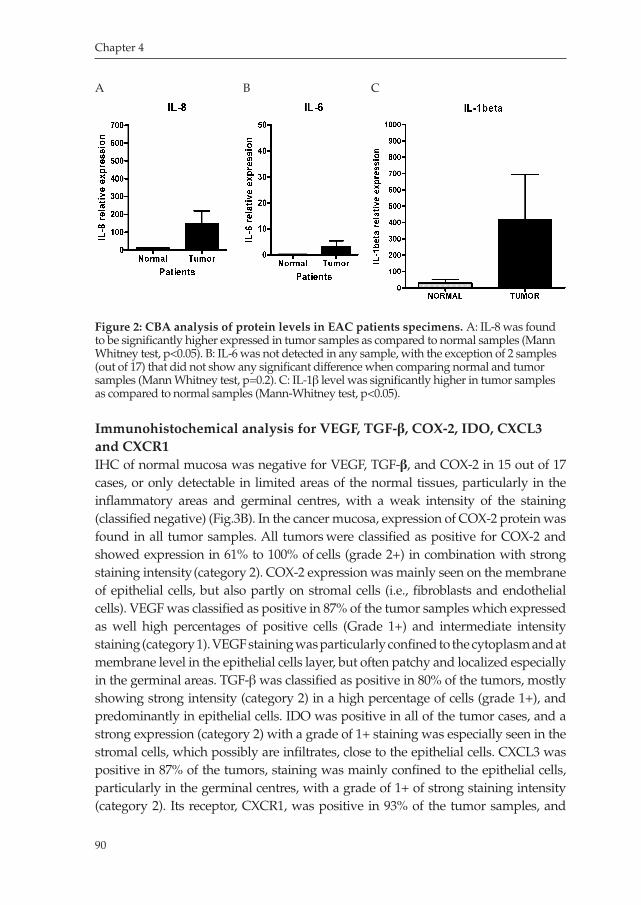
Chapter 4
90
Immunohistochemical analysis for VEGF, TGF-β, COX-2, IDO, CXCL3 and CXCR1IHC of normal mucosa was negative for VEGF, TGFβ, and COX2 in 15 out of 17 cases, or only detectable in limited areas of the normal tissues, particularly in the inflammatory areas and germinal centres, with a weak intensity of the staining (classified negative) (Fig.3B). In the cancer mucosa, expression of COX2 protein was found in all tumor samples. All tumors were classified as positive for COX2 and showed expression in 61% to 100% of cells (grade 2+) in combination with strong staining intensity (category 2). COX2 expression was mainly seen on the membrane of epithelial cells, but also partly on stromal cells (i.e., fibroblasts and endothelial cells). VEGF was classified as positive in 87% of the tumor samples which expressed as well high percentages of positive cells (Grade 1+) and intermediate intensity staining (category 1). VEGF staining was particularly confined to the cytoplasm and at membrane level in the epithelial cells layer, but often patchy and localized especially in the germinal areas. TGFβ was classified as positive in 80% of the tumors, mostly showing strong intensity (category 2) in a high percentage of cells (grade 1+), and predominantly in epithelial cells. IDO was positive in all of the tumor cases, and a strong expression (category 2) with a grade of 1+ staining was especially seen in the stromal cells, which possibly are infiltrates, close to the epithelial cells. CXCL3 was positive in 87% of the tumors, staining was mainly confined to the epithelial cells, particularly in the germinal centres, with a grade of 1+ of strong staining intensity (category 2). Its receptor, CXCR1, was positive in 93% of the tumor samples, and
Figure 2: CBA analysis of protein levels in EAC patients specimens. A: IL8 was found to be significantly higher expressed in tumor samples as compared to normal samples (Mann Whitney test, p<0.05). B: IL6 was not detected in any sample, with the exception of 2 samples (out of 17) that did not show any significant difference when comparing normal and tumor samples (Mann Whitney test, p=0.2). C: IL1β level was significantly higher in tumor samples as compared to normal samples (MannWhitney test, p<0.05).
A B C
Milano_opm_v5.indd 90 16-08-2007 15:42:51
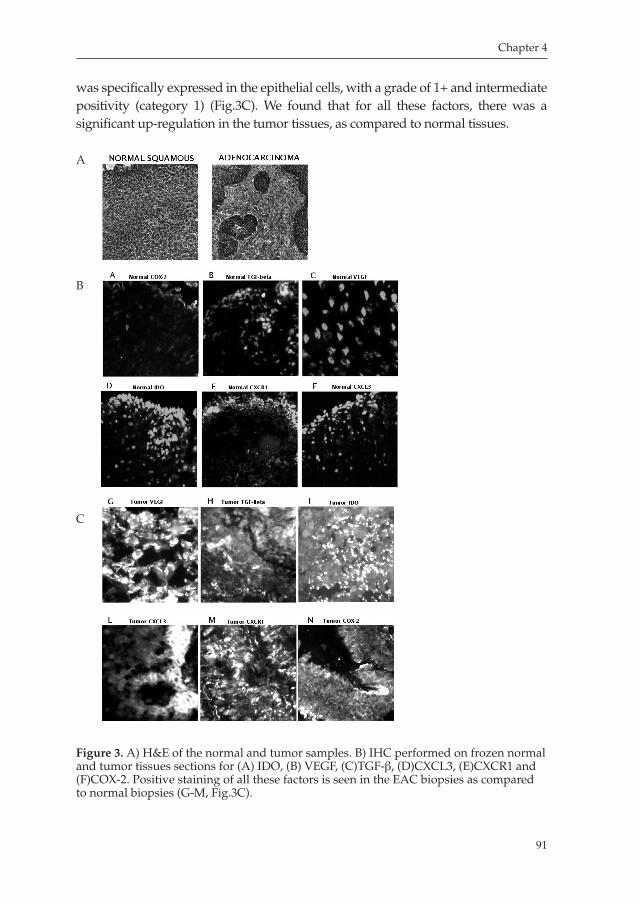
Chapter 4
91
was specifically expressed in the epithelial cells, with a grade of 1+ and intermediate positivity (category 1) (Fig.3C). We found that for all these factors, there was a significant upregulation in the tumor tissues, as compared to normal tissues.
Figure 3. A) H&E of the normal and tumor samples. B) IHC performed on frozen normal and tumor tissues sections for (A) IDO, (B) VEGF, (C)TGFβ, (D)CXCL3, (E)CXCR1 and (F)COX2. Positive staining of all these factors is seen in the EAC biopsies as compared to normal biopsies (GM, Fig.3C).
A
B
C
Milano_opm_v5.indd 91 16-08-2007 15:42:52

Chapter 4
92
DISCUSSION
Tumor resistance to immunological attack is thought to be a result of alteration in the production of specific factors, for instance costimulatory and coinhibitory molecules, or an imbalanced activation of effector Tcells and regulatory Tcells. Such an essential balance is especially altered in patients with advanced stage cancers. These malignancies for instance present high levels of inflammatory molecules, cytokines, chemokines, and tumor infiltrating immunosuppressive cells32,33. Only few factors had been systematically studied in the immunological microenvironment of EAC. It has been broadly demonstrated that factors, such as cyclin D1, EGFR, Her2/Neu, APC, TGFbeta, P53, Bcl2, NFkB, COX2, Ecadherin, βcatenin, are highly expressed at RNA and protein levels, and correlate with poor prognosis and tumor progression in several cancers, including EAC34
40.Our study was aimed at gaining insight in the EAC microenvironment to
study the balance of a panel of immunomodulating substances and factors, which characterize these cancers. Our results show that EAC features a particular pattern of immuneinhibiting factors. Significant high levels of VEGF were seen in 90% of the patient’s samples. Such a high expression was as well previously observed in EAC39. Several studies have confirmed that VEGF is known to enhance and allow the process of metastasis41,42. This factor has been previously described in EAC as a molecule, which plays a role in lymphatic metastasis via lymphangiogenesis and angiogenesis, indicating that pharmacological inhibition of VEGF might be useful to hinder progression of these cancers12,43,44. VEGF is further described as an inhibitor of DC differentiation and maturation32,45. For instance, in vitro data have shown that antiVEGF neutralizing antibodies block the negative effects of tumor cell supernatants on DC maturation15. These results are even more interesting when looking at the simultaneous expression with COX2 in the tumors. We observed that COX2 is highly expressed by the tumor epithelial cells and stroma in significant levels as compared to normal tissues. Similar results were seen in previous studies, where it was shown that the expression of COX2 and VEGF correlated with metastatic stage, and may be used as important prognostic factors18,30,34,35,46,49. As COX2 inhibition is clinically very feasible and has limited side effects, our results may be interpreted that such inhibition is an attractive adjuvant strategy in EAC. Another key player in the mechanism of tumor immuneescape is TGFβ. Many tumors, including EAC, overexpress TGFβ, and high circulating levels of TGFβ1 in cancer patients are frequently associated with poor prognosis34,50,51. Von Radhen et al, showed that high expression of TGFβ, is an important promoter of tumor growth in a series of primary resected esophageal adenocarcinomas36. It has been previously shown that TGFβ mediates hyporesponsiveness of the memory T cells in human lung
Milano_opm_v5.indd 92 16-08-2007 15:42:52

Chapter 4
93
tumor microenvironment in situ by blocking TCR signalling52. Furthermore, adoptive transfer of tumorreactive, TGFβinsensitive CD8+ T cells into immunocompetent mice was able to eradicate lung metastasis of mouse prostate cancer53. In addition, there is evidence that TGFβ in general reduces Tcell functions54. In our study, we could detect positive or intermediate positivity for TGFβ in the majority of the population, confirming the results of previous studies55, suggesting that this factor may be of critical importance in mediating immunoevasion is EAC and that inhibition of its signaling might be beneficial in patient treatment.
Next to these factors, we selected molecules which are thought to be important in the process of tumor immuneescape, but which expression has never been investigated in EAC. These include the enzyme IDO, the chemokine CXCL3 and its receptor CXCR1. Several studies in the last few years have been supporting the idea about the pivotal role of IDO in the inhibition of Tcells50,5658. In vitro and in vivo, and in different models, this enzyme, responsible for the process of tryptophan catabolism, has attracted the interest of immunologists because of its potent inhibitory effect on Tcells. It has been observed that tryptophan depletion consequently induces apoptosis of Tcells. IDO and its enzymatic products have been found in many tumors, and, as proved by functional studies, Tcell responses can be greatly enhanced by inhibiting its action. Therefore, it was of outmost importance in our study to investigate whether or not this factor is expressed in esophageal cancers. Our results show that this molecule is highly upregulated in the stroma of EAC tumor tissues, as shown by IHC, and we therefore speculate that IDO as a potent inhibitor of the immunedefence is one of the active mechanisms in these cancers leading to immuneescape. Another important factor is the CXC ligand CXCL3, a chemokine found previously to be upregulated in EAC patients biopsies by using Serial Analysis of Gene Expression (SAGE) in our laboratory (data not shown). It has been demonstrated that CXCR receptors and related ligands, are profoundly associated to angiogenesis and tumorigenesis13,59. Our results show that this molecule is extensively overexpressed in EAC, especially with regard to epithelial cells. This pattern correlates with the expression of the receptor CXCR1, which was also found to be extremely high expressed in tumor tissues as compared to normal tissues. Surprisingly, the levels of IL8, another important factor known to interact with VEGF and therefore, also responsible for metastatic behaviour of cancers5960, was found to be significantly upregulated in tumors both at RNA and protein level6164. Significantly high levels of IL8 were detectable in human melanoma cell lines, and it has been demonstrated that reduction of the protein level of human IL8 in human melanoma cell lines is associated with a substantial inhibition of their colony formation and proliferative rate14. High secretion of IL8 has as well been described in human colon carcinoma cell lines and in colon carcinoma65. This may confirm the hypothesis that there
Milano_opm_v5.indd 93 16-08-2007 15:42:53

Chapter 4
94
is an important crosstalk between these two factors, which can be responsible for the aggressiveness of EAC. Based on previous studies, and analysing these results, we may speculate that, as shown for other cancers, targeting either IL8 or VEGF and inhibiting this intense crosstalk, can be an attractive strategy to decrease tumor immuneescape in EAC patients.
Analyzing the results for other cytokines, such as IL1β, we could observe that the level of this proinflammatory cytokine was significantly increased in the tumor tissues. These results confirm a previous study where it was shown that expression of IL8 and IL1β are significantly upregulated not only in EAC, but as well in its precursor lesions, Barrett’s esophagus and even in esophagitis6667. The high levels observed in the tumor tissue samples, confirmed what previously has been reported on the IL1β role in cancer progression and its correlation with poor prognosis62,68. This can be explained as a role of this molecule in maintaining an inflammation status in cancer tissues, promoting nonfavourable impaired interactions between the different cell populations that characterize the tumor tissue.
As expected, IFNγ levels in lysates of tumor biopsies did not show any significant difference as compared to the normal tissues, indicating that effector Tcell responses are not initiated in these cancers, or have been inhibited. We observed a significant correlation between the results as obtained for the cytokines at RNA levels and those as measured at protein level. Our results are similar to a previous study where low levels of IFNγ were detected in patients with EAC69.
Surprisingly, the levels of IL6 observed in the tumor tissues as compared to normal, both at RNA and protein levels, did not show a significant difference, indicating that probably this well known immunosuppressive factors, in this particular cancer does not exert a main role in terms of decreasing patient immuneresponses. This confirms previous observations, which have reported on the role of IL6 in the chronic inflammation status in gastroesophageal cancers62.
In summary, in this study we have analyzed the esophageal cancer tumor microenvironment, in order to define the balance of factors that characterize its immunosurroundings. We selected several factors known to be involved in the mechanism of tumor immuneescape in order to establish the pattern of expression of these factors both at RNA and protein level. Here we describe a specific tumor microenvironment, in which significant upregulation of particular immunosuppressive factors are observed, while several important immunogenic factors are lacking. We hypothesize that these molecules are involved in the inhibition of an appropriate and efficient immuneresponse, which lead to escape from immune surveillance. These factors may potentially contribute to resistance to DCs vaccination and other immunotherapeutic approaches. Targeting these molecules in the future may lead to significant improvement of the efficacy of immunotherapies.
Milano_opm_v5.indd 94 16-08-2007 15:42:53

Chapter 4
95
REFERENCES
1. Lee, W. C., Wang, H. C., Hung, C. F., Huang, P. F., Lia, C. R., and Chen, M. F. Vaccination of
advanced hepatocellular carcinoma patients with tumor lysatepulsed dendritic cells: a clini
cal trial. J Immunother, 28: 496504, 2005.
2. Burgdorf, S. K., Fischer, A., Claesson, M. H., Kirkin, A. F., Dzhandzhugazyan, K. N., and Ro
senberg, J. Vaccination with melanoma lysatepulsed dendritic cells, of patients with advanced
colorectal carcinoma: report from a phase I study. J Exp Clin Cancer Res, 25: 201206, 2006.
3. Babatz, J., Rollig, C., Lobel, B., Folprecht, G., Haack, M., Gunther, H., Kohne, C. H., Ehninger,
G., Schmitz, M., and Bornhauser, M. Induction of cellular immune responses against carcin
oembryonic antigen in patients with metastatic tumors after vaccination with altered peptide
ligandloaded dendritic cells. Cancer Immunol Immunother, 55: 268276, 2006.
4. Banchereau, J. and Palucka, A. K. Dendritic cells as therapeutic vaccines against cancer. Nat
Rev Immunol, 5: 296306, 2005.
5. Smyth, M. J., Godfrey, D. I., and Trapani, J. A. A fresh look at tumor immunosurveillance and
immunotherapy. Nat Immunol, 2: 293299, 2001.
6. Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic
relevance. Nat Rev Cancer, 5: 263274, 2005.
7. Rabinovich, G. A., Gabrilovich, D., and Sotomayor, E. M. Immunosuppressive Strategies that
are Mediated by Tumor Cells. Annu Rev Immunol, 25: 267296, 2007.
8. Mahadevan, D. and Von Hoff, D. D. Tumorstroma interactions in pancreatic ductal adenocar
cinoma. Mol Cancer Ther, 6: 11861197, 2007.
9. Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic selftolerance and
negative control of immune responses. Annu Rev Immunol, 22: 531562, 2004.
10. Fricke, I. and Gabrilovich, D. I. Dendritic cells and tumor microenvironment: a dangerous
liaison. Immunol Invest, 35: 459483, 2006.
11. Balkwill, F. and Mantovani, A. Inflammation and cancer: back to Virchow? Lancet, 357: 539
545, 2001.
12. Ding, M. X., Lin, X. Q., Fu, X. Y., Zhang, N., and Li, J. C. Expression of vascular endothelial
growth factorC and angiogenesis in esophageal squamous cell carcinoma. World J Gastroen
terol, 12: 45824585, 2006.
13. Mestas, J., Burdick, M. D., Reckamp, K., Pantuck, A., Figlin, R. A., and Strieter, R. M. The role of
CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. J Immunol, 175: 53515357, 2005.
14. Schadendorf, D., Moller, A., Algermissen, B., Worm, M., Sticherling, M., and Czarnetzki, B. M.
IL8 produced by human malignant melanoma cells in vitro is an essential autocrine growth
factor. J Immunol, 151: 26672675, 1993.
15. Gabrilovich, D. I., Chen, H. L., Girgis, K. R., Cunningham, H. T., Meny, G. M., Nadaf, S., Ka
vanaugh, D., and Carbone, D. P. Production of vascular endothelial growth factor by human
tumors inhibits the functional maturation of dendritic cells. Nat Med, 2: 10961103, 1996.
16. Sombroek, C. C., Stam, A. G., Masterson, A. J., Lougheed, S. M., Schakel, M. J., Meijer, C.
J., Pinedo, H. M., van den Eertwegh, A. J., Scheper, R. J., and de Gruijl, T. D. Prostanoids
Milano_opm_v5.indd 95 16-08-2007 15:42:54

Chapter 4
96
play a major role in the primary tumorinduced inhibition of dendritic cell differentiation. J
Immunol, 168: 43334343, 2002.
17. Jongmans, W., Tiemessen, D. M., van Vlodrop, I. J., Mulders, P. F., and Oosterwijk, E. Th1
polarizing capacity of clinicalgrade dendritic cells is triggered by Ribomunyl but is compro
mised by PGE2: the importance of maturation cocktails. J Immunother, 28: 480487, 2005.
18. Bhandari, P., Bateman, A. C., Mehta, R. L., Stacey, B. S., Johnson, P., Cree, I. A., Di Nicolantonio,
F., and Patel, P. Prognostic significance of cyclooxygenase2 (COX2) expression in patients with
surgically resectable adenocarcinoma of the oesophagus. BMC Cancer, 6: 134, 2006.
19. Tuynman, J. B., Buskens, C. J., Kemper, K., ten Kate, F. J., Offerhaus, G. J., Richel, D. J., and
van Lanschot, J. J. Neoadjuvant selective COX2 inhibition downregulates important oncoge
nic pathways in patients with esophageal adenocarcinoma. Ann Surg, 242: 840849, discus
sion 849850, 2005.
20. BagumaNibasheka, M., Barclay, C., Li, A. W., Geldenhuys, L., Porter, G. A., Blay, J., Casson,
A. G., and Murphy, P. R. Selective cyclooxygenase2 inhibition suppresses basic fibroblast
growth factor expression in human esophageal adenocarcinoma. Mol Carcinog, 2007.
21. Munn, D. H., Sharma, M. D., Lee, J. R., Jhaver, K. G., Johnson, T. S., Keskin, D. B., Marshall, B.,
Chandler, P., Antonia, S. J., Burgess, R., Slingluff, C. L., Jr., and Mellor, A. L. Potential regula
tory function of human dendritic cells expressing indoleamine 2,3dioxygenase. Science, 297:
18671870, 2002.
22. Munn, D. H., Shafizadeh, E., Attwood, J. T., Bondarev, I., Pashine, A., and Mellor, A. L. Inhibition
of T cell proliferation by macrophage tryptophan catabolism. J Exp Med, 189: 13631372, 1999.
23. MenetrierCaux, C., Montmain, G., Dieu, M. C., Bain, C., Favrot, M. C., Caux, C., and Blay, J.
Y. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells:
role of interleukin6 and macrophage colonystimulating factor. Blood, 92: 47784791, 1998.
24. Strassmann, G., Masui, Y., Chizzonite, R., and Fong, M. Mechanisms of experimental cancer
cachexia. Local involvement of IL1 in colon26 tumor. J Immunol, 150: 23412345, 1993.
25. O’Riordain, M. G., Falconer, J. S., Maingay, J., Fearon, K. C., and Ross, J. A. Peripheral blood
cells from weightlosing cancer patients control the hepatic acute phase response by a prima
rily interleukin6 dependent mechanism. Int J Oncol, 15: 823827, 1999.
26. Spechler, S. J., Zeroogian, J. M., Antonioli, D. A., Wang, H. H., and Goyal, R. K. Prevalence of
metaplasia at the gastrooesophageal junction. Lancet, 344: 15331536, 1994.
27. Ransford, R. A. and Jankowski, J. A. Genetic versus environmental interactions in the oeso
phagitismetaplasiadysplasiaadenocarcinoma sequence (MCS) of Barrett’s oesophagus. Acta
Gastroenterol Belg, 63: 1821, 2000.
28. Pera, M. Trends in incidence and prevalence of specialized intestinal metaplasia, barrett’s
esophagus, and adenocarcinoma of the gastroesophageal junction. World J Surg, 27: 9991008;
discussion 10061008, 2003.
29. O’Connor, J. B., Falk, G. W., and Richter, J. E. The incidence of adenocarcinoma and dysplasia
in Barrett’s esophagus: report on the Cleveland Clinic Barrett’s Esophagus Registry. Am J
Gastroenterol, 94: 20372042, 1999.
Milano_opm_v5.indd 96 16-08-2007 15:42:54

Chapter 4
97
30. Villanacci, V., Rossi, E., Zambelli, C., Galletti, A., Cestari, R., Missale, G., Casa, D. D., and Bas
sotti, G. COX2, CDX2, and CDC2 immunohistochemical assessment for dysplasiacarcinoma
progression in Barrett’s esophagus. Dig Liver Dis, 39: 305311, 2007.
31. Cook, E. B., Stahl, J. L., Lowe, L., Chen, R., Morgan, E., Wilson, J., Varro, R., Chan, A., Grazi
ano, F. M., and Barney, N. P. Simultaneous measurement of six cytokines in a single sample of
human tears using microparticlebased flow cytometry: allergics vs. nonallergics. J Immunol
Methods, 254: 109118, 2001.
32. Kryczek, I., Lange, A., Mottram, P., Alvarez, X., Cheng, P., Hogan, M., Moons, L., Wei, S., Zou,
L., Machelon, V., Emilie, D., Terrassa, M., Lackner, A., Curiel, T. J., Carmeliet, P., and Zou, W.
CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in
human ovarian cancers. Cancer Res, 65: 465472, 2005.
33. Gabrilovich, D. I., Bronte, V., Chen, S. H., Colombo, M. P., Ochoa, A., OstrandRosenberg, S.,
and Schreiber, H. The terminology issue for myeloidderived suppressor cells. Cancer Res, 67:
425; author reply 426, 2007.
34. Lagarde, S. M., ten Kate, F. J., Richel, D. J., Offerhaus, G. J., and van Lanschot, J. J. Molecular
prognostic factors in adenocarcinoma of the esophagus and gastroesophageal junction. Ann
Surg Oncol, 14: 977991, 2007.
35. Buskens, C. J., Van Rees, B. P., Sivula, A., Reitsma, J. B., Haglund, C., Bosma, P. J., Offerhaus, G. J.,
Van Lanschot, J. J., and Ristimaki, A. Prognostic significance of elevated cyclooxygenase 2 expres
sion in patients with adenocarcinoma of the esophagus. Gastroenterology, 122: 18001807, 2002.
36. von Rahden, B. H., Stein, H. J., Feith, M., Puhringer, F., Theisen, J., Siewert, J. R., and Sarbia,
M. Overexpression of TGFbeta1 in esophageal (Barrett’s) adenocarcinoma is associated with
advanced stage of disease and poor prognosis. Mol Carcinog, 45: 786794, 2006.
37. Buskens, C. J., Ristimaki, A., Offerhaus, G. J., Richel, D. J., and van Lanschot, J. J. Role of
cyclooxygenase2 in the development and treatment of oesophageal adenocarcinoma. Scand J
Gastroenterol Suppl 8793, 2003.
38. von Rahden, B. H., Stein, H. J., Puhringer, F., Koch, I., Langer, R., Piontek, G., Siewert, J. R.,
Hofler, H., and Sarbia, M. Coexpression of cyclooxygenases (COX1, COX2) and vascular
endothelial growth factors (VEGFA, VEGFC) in esophageal adenocarcinoma. Cancer Res,
65: 50385044, 2005.
39. Mobius, C., Freire, J., Becker, I., Feith, M., Brucher, B. L., Hennig, M., Siewert, J. R., and Stein,
H. J. VEGFC Expression in Squamous Cell Carcinoma and Adenocarcinoma of the Esopha
gus. World J Surg, 2007.
40. Lebman, D. A., Edmiston, J. S., Chung, T. D., and Snyder, S. R. Heterogeneity in the transfor
ming growth factor beta response of esophageal cancer cells. Int J Oncol, 20: 12411246, 2002.
41. Risau, W. and Flamme, I. Vasculogenesis. Annu Rev Cell Dev Biol, 11: 7391, 1995.
42. DiazRubio, E. Vascular endothelial growth factor inhibitors in colon cancer. Adv Exp Med
Biol, 587: 251275, 2006.
43. von Rahden, B. H., Stein, H. J., PuhringerOppermann, F., and Sarbia, M. cmyc amplification
is frequent in esophageal adenocarcinoma and correlated with the upregulation of VEGFA
expression. Neoplasia, 8: 702707, 2006.
Milano_opm_v5.indd 97 16-08-2007 15:42:54

Chapter 4
98
44. Shimizu, D., Vallbohmer, D., Kuramochi, H., Uchida, K., Schneider, S., Chandrasoma, P. T.,
Shimada, H., DeMeester, T. R., Danenberg, K. D., Peters, J. H., DeMeester, S. R., and Danen
berg, P. V. Increasing cyclooxygenase2 (cox2) gene expression in the progression of Barrett’s
esophagus to adenocarcinoma correlates with that of Bcl2. Int J Cancer, 119: 765770, 2006.
45. Carmeliet, P. and Jain, R. K. Angiogenesis in cancer and other diseases. Nature, 407: 249257, 2000.
46. Alici, S., Ugras, S., Bayram, I., and Izmirli, M. Prognostic factors and COX2 expression in
advanced stage esophageal squamous cell carcinoma. Adv Ther, 23: 672679, 2006.
47. Liu, J. F., Jamieson, G., Wu, T. C., Zhang, S. W., Wang, Q. Z., and Drew, P. Cyclooxygenase2
expression in squamous cell carcinoma of the esophagus. Dis Esophagus, 19: 350354, 2006.
48. Yu, H. P., Xu, S. Q., Liu, L., Shi, L. Y., Cai, X. K., Lu, W. H., Lu, B., Su, Y. H., and Li, Y. Y.
Cyclooxygenase2 expression in squamous dysplasia and squamous cell carcinoma of the
esophagus. Cancer Lett, 198: 193201, 2003.
49. Buskens, C. J., Sivula, A., van Rees, B. P., Haglund, C., Offerhaus, G. J., van Lanschot, J. J., and
Ristimaki, A. Comparison of cyclooxygenase 2 expression in adenocarcinomas of the gastric
cardia and distal oesophagus. Gut, 52: 16781683, 2003.
50. Bruna, A., Darken, R. S., Rojo, F., Ocana, A., Penuelas, S., Arias, A., Paris, R., Tortosa, A.,
Mora, J., Baselga, J., and Seoane, J. High TGFbetaSmad activity confers poor prognosis in
glioma patients and promotes cell proliferation depending on the methylation of the PDGFB
gene. Cancer Cell, 11: 147160, 2007.
51. Saunier, E. F. and Akhurst, R. J. TGF beta inhibition for cancer therapy. Curr Cancer Drug
Targets, 6: 565578, 2006.
52. Broderick, L. and Bankert, R. B. Membraneassociated TGFbeta1 inhibits human memory T
cell signaling in malignant and nonmalignant inflammatory microenvironments. J Immunol,
177: 30823088, 2006.
53. Zhang, Q., Yang, X. J., Kundu, S. D., Pins, M., Javonovic, B., Meyer, R., Kim, S. J., Greenberg,
N. M., Kuzel, T., Meagher, R., Guo, Y., and Lee, C. Blockade of transforming growth factor
{beta} signaling in tumorreactive CD8(+) T cells activates the antitumor immune response
cycle. Mol Cancer Ther, 5: 17331743, 2006.
54. Tada, T., Ohzeki, S., Utsumi, K., Takiuchi, H., Muramatsu, M., Li, X. F., Shimizu, J., Fujiwara,
H., and Hamaoka, T. Transforming growth factorbetainduced inhibition of T cell function.
Susceptibility difference in T cells of various phenotypes and functions and its relevance to
immunosuppression in the tumorbearing state. J Immunol, 146: 10771082, 1991.
55. Koliopanos, A., Friess, H., di Mola, F. F., Tang, W. H., Kubulus, D., Brigstock, D., Zimmer
mann, A., and Buchler, M. W. Connective tissue growth factor gene expression alters tumor
progression in esophageal cancer. World J Surg, 26: 420427, 2002.
56. Hwu, P., Du, M. X., Lapointe, R., Do, M., Taylor, M. W., and Young, H. A. Indoleamine 2,3di
oxygenase production by human dendritic cells results in the inhibition of T cell proliferation.
J Immunol, 164: 35963599, 2000.
57. Frumento, G., Rotondo, R., Tonetti, M., and Ferrara, G. B. T cell proliferation is blocked by
indoleamine 2,3dioxygenase. Transplant Proc, 33: 428430, 2001.
Milano_opm_v5.indd 98 16-08-2007 15:42:55

Chapter 4
99
58. Muller, A. J. and Scherle, P. A. Targeting the mechanisms of tumoral immune tolerance with
smallmolecule inhibitors. Nat Rev Cancer, 6: 613625, 2006.
59. Wente, M. N., Keane, M. P., Burdick, M. D., Friess, H., Buchler, M. W., Ceyhan, G. O., Reber,
H. A., Strieter, R. M., and Hines, O. J. Blockade of the chemokine receptor CXCR2 inhibits
pancreatic cancer cellinduced angiogenesis. Cancer Lett, 241: 221227, 2006.
60. Curiel, T. J., Cheng, P., Mottram, P., Alvarez, X., Moons, L., EvdemonHogan, M., Wei, S., Zou,
L., Kryczek, I., Hoyle, G., Lackner, A., Carmeliet, P., and Zou, W. Dendritic cell subsets dif
ferentially regulate angiogenesis in human ovarian cancer. Cancer Res, 64: 55355538, 2004.
61. Ren, Y., Cao, B., Law, S., Xie, Y., Lee, P. Y., Cheung, L., Chen, Y., Huang, X., Chan, H. M.,
Zhao, P., Luk, J., Vande Woude, G., and Wong, J. Hepatocyte growth factor promotes cancer
cell migration and angiogenic factors expression: a prognostic marker of human esophageal
squamous cell carcinomas. Clin Cancer Res, 11: 61906197, 2005.
62. Deans, D. A., Wigmore, S. J., Gilmour, H., PatersonBrown, S., Ross, J. A., and Fearon, K. C.
Elevated tumour interleukin1beta is associated with systemic inflammation: A marker of
reduced survival in gastrooesophageal cancer. Br J Cancer, 95: 15681575, 2006.
63. Savage, S. A., Abnet, C. C., Mark, S. D., Qiao, Y. L., Dong, Z. W., Dawsey, S. M., Taylor, P. R.,
and Chanock, S. J. Variants of the IL8 and IL8RB genes and risk for gastric cardia adenocar
cinoma and esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev, 13:
22512257, 2004.
64. Kitadai, Y., Onogawa, S., Kuwai, T., Matsumura, S., Hamada, H., Ito, M., Tanaka, S., Yoshiha
ra, M., and Chayama, K. Angiogenic switch occurs during the precancerous stage of human
esophageal squamous cell carcinoma. Oncol Rep, 11: 315319, 2004.
65. Brew, R., Erikson, J. S., West, D. C., Kinsella, A. R., Slavin, J., and Christmas, S. E. Interleukin8
as an autocrine growth factor for human colon carcinoma cells in vitro. Cytokine, 12: 7885,
2000.
66. O’Riordan, J. M., Abdellatif, M. M., Ravi, N., McNamara, D., Byrne, P. J., McDonald, G. S.,
Keeling, P. W., Kelleher, D., and Reynolds, J. V. Proinflammatory cytokine and nuclear factor
kappaB expression along the inflammationmetaplasiadysplasiaadenocarcinoma sequence
in the esophagus. Am J Gastroenterol, 100: 12571264, 2005.
67. Fitzgerald, R. C., Abdalla, S., Onwuegbusi, B. A., Sirieix, P., Saeed, I. T., Burnham, W. R., and
Farthing, M. J. Inflammatory gradient in Barrett’s oesophagus: implications for disease com
plications. Gut, 51: 316322, 2002.
68. Lewis, A. M., Varghese, S., Xu, H., and Alexander, H. R. Interleukin1 and cancer progression:
the emerging role of interleukin1 receptor antagonist as a novel therapeutic agent in cancer
treatment. J Transl Med, 4: 48, 2006.
69. Uenaka, A., Wada, H., Isobe, M., Saika, T., Tsuji, K., Sato, E., Sato, S., Noguchi, Y., Kawabata,
R., Yasuda, T., Doki, Y., Kumon, H., Iwatsuki, K., Shiku, H., Monden, M., Jungbluth, A. A.,
Ritter, G., Murphy, R., Hoffman, E., Old, L. J., and Nakayama, E. T cell immunomonitoring
and tumor responses in patients immunized with a complex of cholesterolbearing hydropho
bized pullulan (CHP) and NYESO1 protein. Cancer Immun, 7: 9, 2007.
Milano_opm_v5.indd 99 16-08-2007 15:42:55

Milano_opm_v5.indd 100 16-08-2007 15:42:55

Chapter 5
Trastuzumab synergistically acts with
HER-2 specific CTL to induce tumor cell lysis in
Esophageal Adenocarcinoma cell lines
Francesca Milano, Agnieszka M. Rygiel, Jacques J.G.H.M. Bergman, Paul Fockens, Carine Sondermeijer
Maikel P. Peppelenbosch, Kausilia K. Krishnadath
Manuscript in preparation
Milano_opm_v5.indd 101 16-08-2007 15:42:56

Chapter 5
102
ABSTRACT
Esophageal adenocarcinoma (EAC) is a highly aggressive disease that with the conventional treatment strategies has extremely poor prognosis. In a previous study we demonstrated that CTL mediated DC therapy is a promising novel treatment option for EAC. EACs are known to have high amplification of the HER2 gene, and therefore it is of interest to explore combinatorial treatment strategies employing DC immunotherapy and antiHER2 treatment for this patient group. Previous studies showed that HER2derived peptides are naturally processed as Cytotoxic Tcell (CTL) epitopes that can be recognized by tumorspecific CTLs. The humanized antibody against HER2, Trastuzumab, has potent inhibitory effects on HER2 overexpressing tumors. In addition, this antibody may induce an increase of HER2 epitope presentation via MHCClass I molecule on tumor cells that may mediate a HER2 specific CTL response and tumor cell lysis. In this study we first, determined the HER2 gene amplification and protein expression in EAC cell lines, and investigated the biological activity of Trastuzumab. Secondly, we exvivo evaluated whether Trastuzumab and HER2specific CTLs act synergistically for inducing tumor lysis, and then did the same using PBMCs of patients with and without HER2 overexpressing EAC. The HLAA2/HER2 positive cell lines, OE33 and OE19, and HLAA2 positive/HER2 negative cell line Bic1 were used to test the effect of Trastuzumab performing an MTT assay. In the same time, HLAA2 matched donor DCs were loaded with HER2 RNA to obtain HER2 specific CTLs. Hereupon, a cytotoxicity assay was performed, which revealed a synergistic effect of Trastuzumab and HER2 specific CTLs in lysing the HER2 overexpressing EAC cell line OE33, but not the HER2 negative Bic1. Most interestingly, PBMCs of HLAA2 matched EAC patients with high HER2 amplification induced significantly higher levels of lysis of the HER2 positive cell lines as compared to PBMCs of patients without HER2 amplification. This effect was further enhanced in case cells were pretreated with Trastuzumab. These results show that Trastuzumab treatment of HER2 expressing cell lines enhances the susceptibility to lysis by HER2specific CTLs. Furthermore, we provide evidence that EAC patients with HER2 overexpressing cancers might have circulating anti HER2 CTLs that upon treatment with Trastuzumab may lead to an improvement for a specific anti HER2 cytoloytic immune response.
Milano_opm_v5.indd 102 16-08-2007 15:42:56

Chapter 5
103
INTRODUCTION
HER2/neu is a 185 KDa transmembrane glycoprotein with tyrosine kinase activity and extensive homology to the epidermal growth factors receptor1.
HER2 is ubiquitously expressed in a large proportion of epithelial cancers, and it has been widely reported to be overexpressed, mostly via gene amplification, in several aggressive cancers2, such as in 2530% of ovarian and breast cancer3,4, 3545% of pancreatic carcinoma5, in a wide range of colorectal carcinomas6, and in 3080% of esophageal adenocarcinoma714,43. Since cytotoxic Tlymphocytes (CTL) response for several HER2 peptides have been observed in cancer patients, the HER2/neu protein appears to be immunogenic, and therefore is an ideal Tumor Associated Antigen that can be employed for anticancer immunotherapy1519. The fully humanized antibody Trastuzumab (Herceptin), which specifically targets the extracellular domain of the HER2 protein, exhibits potent growth inhibitory activity against HER2 overexpressing tumors2023. Trastuzumab has been used in clinical studies showing encouraging results, particularly when used as therapy for breast cancer2426. Applying Trastuzumab as an additional treatment option could be an attractive strategy to consider for tumors with a notoriously poor prognosis such as EAC. This cancer, in Western countries, is the major type of esophageal cancer; the incidence is the most rapidly increasing compared to other malignancies and has exceeded 5 per 100.000 per year27,28. Conventional treatment options for these patients can guarantee only low survival rate: the main treatment of EAC is surgical resection, yet, even after curative resection with or without neoadjuvant or adjuvant chemo and or radiotherapy, the median survival is less than 2 years29,30. In addition, several studies suggested that HER2 amplification/overexpression may be relevant for these tumor entities10. In an exvivo study, we previously showed that DC vaccines could be an advantageous approach for curing esophageal cancer44. For these reason, we questioned whether immunotherapy of EAC in combination with the use of Trastuzumab could be a valid approach as a more effective strategy to treat EAC patients. The underlying mechanisms of the action of Trastuzumab
include blockade of the signalling pathway as well as induction of apoptosis in tumor cells and inhibition of tumor cell growth through downregulation of the HER2 receptor and the enhancement of the immune system response such as Antibody Dependent Cellular Cytotoxicity (ADCC)31,32. It has been previously shown that HER2 overexpressing cell lines treated with Trastuzumab are more susceptible to killing by HER2 specific CTLs33,34. To date, only one report describes the possibility of applying Trastuzumab as a treatment for EAC patients showing an overall toxicity, which was not increased as compared to previous studies35.
Milano_opm_v5.indd 103 16-08-2007 15:42:56

Chapter 5
104
In this study, we examined the level of gene amplification and protein overexpression in diverse EAC cell lines by Fluorescence In Situ Hybridization (FISH) and immunocytochemistry (ICC). We first evaluated the effect of Trastuzumab against HER2 expressing EAC cell lines and showed that Trastuzumab induces cell growth arrest in HER2 overexpressing cell lines. We then evaluated whether Trastuzumab increases the capacity of HER2 specific CTLs to induce lysis of tumor cells and showed that in fact pretreatment with Trastuzumab can enhance CTLs lytic capacity for the HER2 positive cell line OE33, but not for the HER2 negative cell line Bic1. In addition, we showed that PBMCs isolated from patients with HER2 amplification induce lysis of Trastuzumab pretreated EAC cell lines, while this effect is significantly less when employing PBMCs of patients without HER2 amplification.
MATERIALS AND METHODS
Cell Lines and PBMCs from EAC patients.EAC cell lines OE19 and OE33 were obtained from ECACC (Porton Down, Wiltshire, SP4 DJG, UK), Bic1 was a gift from Dr. N.S. Buttar (Mayo Clinic, Rochester, MN, USA). OE19 and OE33 were maintained in RPMI 1640 medium with addition of Lglutamine, antibiotics and Fetal Calf Serum (FCS). Bic1 was kept in DMEM medium with addition of Lglutamine, antibiotics and FCS. All the cell lines were incubated at 37ºC and 5% CO2, and medium was refreshed every three days.
Peripheral blood mononuclear cells (PBMCs) were separated from blood obtained from healthy donors and EAC patients by density gradient centrifugation using FicollPaque (Amersham, Pharmacia, Piscataway, NJ, USA) and cryopreserved in a DMSO/FCS (2:8) solution (Merck, Darmstadt, Germany; GIBCO BRL, Grand Island, NY, USA, respectively) for further use.
Fluorescent in situ hybridization (FISH)EAC cell lines OE33, OE19 and Bic1 were trypsinized at a confluence of 7080%, spun down and collected in PBS. A Cytospin (Shandon Cytospin 4 Cytocentrifuge, Thermo, Waltham, MA) was used to generate a single layer of cells on a glass slide as described before45. The cytospin slides were dried overnight at RT, and then stored at 80ºC until FISH analysis was performed. We used directly labeled fluorescent chromosomal centromeric probes (CEP) for chromosome 17 and 17q11.2 q12 (Her2/neu), obtained from Vysis (Downers Grove, IL). DNAFISH was performed according to the manufacturer’s instructions provided by Vysis as described before by our group45. After the FISH procedure, 100 interphase nuclei of tumor cells were scored per slide by an experienced scorer (A.M Rygiel)
Milano_opm_v5.indd 104 16-08-2007 15:42:57

Chapter 5
105
using Olympus BX61 fluorescent microscope (Germany). The categories of Her2/neu gene abnormalities were determined by calculating the ratio of Her-2/neu locus signals (orange) to chromosome 17 centromere signals (green). The following categories were distinguished: ratio <2 were considered as having no amplification, ratios ≥2 and <5 were considered as low amplification and ratio ≥5 was considered as high amplification. More then two green signals (CEP 17) accompanied by the same number of orange signals (Her2/neu locus) was considered to be indicative of polysomy of chromosome 17 (ratio 1:1). Following these criteria the cases were classified as displaying a polysomy of chromosome 17 (cutoff ≥3 % of abnormal nuclei) and an amplification of Her2/neu locus (cutoff ≥5 % of abnormal nuclei) as established before.
Fluorescent immuno-cytochemistry of cultured cells. Cultured cells were plated in a 24 well plate at a concentration of 1x 104/ml and grown on coverslips. After 24 hours, cultured cells were fixed directly into the well plate for 20 minutes in Phosphate Buffered Saline (PBS) with 4% Paraformaldehyde (PFA) and 0.1% Triton X100 and then washed in PBS. Blocking of aspecific antigens was performed by incubating slides for 45 minutes with PBS containing 1% BSA and10% Fetal Calf Serum (FCS). Slides were washed with PBS and incubated overnight at 4ºC with the appropriately diluted primary HER2 antibody, cerbB2/HER2/neuAb17 (Lab Vision Corporation, Freemont, CA, USA) in PBS with 1% BSA and 0.1% Triton X100. After washing with PBS, cells were incubated with a mouse antihuman secondary FITC conjugated antibody (Dako, Denmark) 1:500 diluted in PBS. Cells attached on coverslips were placed on Superfrost + (Menzel Glaser, Braunschweig, Germany) glass slides and mounted with DAPI (Roche, Mannheim, Germany)/vectashield (Vector laboratories Inc, Burlingame, CA, USA) 1:1000.
Flow cytometric analysis for HLA-A2 typing.To assess the HLAA2 type of the EAC cell lines, cells were harvested, washed and resuspended in a concentration of 1x106 in FACS buffer (5 g BSA, 0.1 g NaN3, 100 mM EDTA in 1 L PBS), then incubated with antihuman HLAA2 antibody (BD) PE conjugated. Cells were stained for 30 minutes on ice in the dark, then spun down and resuspended in FACS buffer, and analyzed with the FACSCALIBUR apparatus (BectonDickinson, Franklin Lakes, NJ, USA). Analysis of the result was done using Cell Quest Pro Software (BD). Isotypes antibodies were used as control.
Isolation of monocytes and lymphocytes from peripheral blood Human monocytes were isolated from peripheral blood of healthy volunteers HLAA2 positive using the FicollPercoll gradient separation method,
Milano_opm_v5.indd 105 16-08-2007 15:42:57

Chapter 5
106
previously described by Milano et al.46. Once monocytes were separated from lymphocytes, both populations were resuspended in IMDM and washed twice. Monocytes and lymphocytes were counted and either used immediately or cryopreserved in a DMSO/FCS (2:8) solution (Merck, Darmstadt, Germany; GIBCO BRL, Grand Island, NY, USA, respectively). A further separation of monocytes from lymphocytes was done using a Percoll (Amersham Biosciences Europe Freiburg, Germany) gradient separation, following manufacture’s protocol. Briefly, PBMCs were washed in RPMI 1640 medium (BioWhittakerCambrex Bioscience, Walkersville, MD, USA) at 1500 rpm for 5 minutes twice. At the mean time, Percoll was mixed with 10X Phosphate Buffer Saline (PBS) to obtain Standard Isotone Percoll solution (SIP), and then with IMDM medium (BioWhittaker), to obtain three solutions at different concentration (60, 47.5 and 34% SIP). PBMCs were then resuspended in 2.5 ml 60% SIP, then 5 ml 47.5% SIP and finally 2 ml 34% SIP were added to the 60%SIP cell suspension and a centrifugation at 3100 rpm for 45 minutes was performed. Once monocytes were separated from lymphocytes, both populations were resuspended in IMDM and washed twice. Monocytes and lymphocytes were counted and either used immediately or cryopreserved in a DMSO/FCS (2:8) solution (Merck, Darmstadt, Germany; GIBCO BRL, Grand Island, NY, USA, respectively).
Generation of HER-2 mRNA by in vitro transcription.HER2 mRNA was made as previously described36. Briefly, to generate HER2 mRNA, plasmid pSPJC1 with an insert coding for HER2 (a gift from Prof. A.Ulrich, Martinsried, Germany) was used. This plasmid allows in vitro transcription under the control of an SP6 promoter. The plasmid was linearized using NdeI (MBI Fermentas, St. LeonRot, Germany) and in vitro transcription was subsequently performed using Ambion in vitro transcription kit with an SP6 promoter (Ambion, Nieuwerkerk a/d, IJssel, The Netherlands), and the manufactures’s procedure was followed. The in vitro transcribed mRNA was purified using RNeasy Midi Kit (Qiagen, Hilden, Germany) according to the manufacture’s instructions. The RNA was then dissolved in RNasefree H2O and stored at 80 °C until required. Spectrophotometry was performed with 1μl of total RNA to quantitate on the Nanodrop (Nanodrop Tecnologies, type ND1000, Wilmington, USA).
Electroporation of monocytes with HER-2 mRNA Monocytes characterized as CD14+, CD83, CD86, CD209 were used for electroporation with HER2 mRNA. The electroporation procedure was performed as previously described by F.Milano et al.46, using Amaxa cell lines Nucleofector procedure, kit V. After electroporation, 1x105 monocytes were
Milano_opm_v5.indd 106 16-08-2007 15:42:58

Chapter 5
107
plated in 24 well plates with 1 ml of Cell Gro Medium (Cellgenix, Freiburg, Germany) with the addition of 2% of Penicillin/Streptomycin (GIBCO, Breda, The Netherlands), and 800U/ml IL4 and 1000U/ml GMCSF for obtaining immature DCs. After six days, IL1β, TNFα and LPS (10 ng/ml, 25 ng/ml, 0.02 ng/ml respectively) were added to obtain mature DCs. The electroporation efficacy of HER2 mRNA was assessed through measuring HER2 expression by FACS analysis. Cells were harvested and spun down at 1500 rpm for 10 minutes, then resuspended in FACS buffer in a concentration of 1x106 cells/ml and sorted by the FACS apparatus for direct measurement of HER2 extra cellular domain using antihuman HER2 primary monoclonal antibody anti HER2 clone 9G6.10 (Lab Vision, Fremont, CA). Expression was measured in monocytes 24 hours after electroporation (day one) and daily on eight subsequent days after electroporation, during the maturation of the monocytes into mature DCs.
Induction of HER-2 specific CTLsAfter eight days from the isolation of monocytes, the obtained HER2 loaded mature DCs were twice coincubated for seven days with autologous lymphocytes in a proportion of 1:4 (5x105 DCs and 2x106 lymphocytes) in 24 wells plates in Cell Gro medium with 2% penicillinstreptomycin (GIBCO). As control population, lymphocytes were as well incubated with DCs which underwent the electroporation procedure but without adding HER2 RNA.
Mixed Leucocyte Reaction (MLR) using [3H] thymidine incorporation assayThe capacity of DCs to present antigens and evoke a lymphocytes response was tested in an autologous Mixed Leucocyte Reaction (MLR). Briefly, mature DCs electroporated either with HER2 RNA and DCs electroporated with no antigen source were cocultured in flatbottom 96well plates (Nunc, Roskilde, Denmark) with lymphocytes in IMDM medium at different targeteffector ratios, starting from 4x103/well, down to 1.25x102 DCs (APC) per well, while each well contained a fixed number of 5x104 lymphocytes. After 5 days in culture, 0.2 μCi [3H] thymidine (Amersham Biosciences, Amersham, UK) was added to each well and the incorporation of radioactivity was measured after 16 hours using liquid scintillation counting.
MTT assay of Trastuzumab treated cellsTo evaluate the effect of Trastuzumab on EAC cell lines, cultured cells were treated with increasing concentration of Trastuzumab, namely 2.5, 5 and 10 μg/ml, and left untreated for control purposes. Cells were harvested after 24 hours, 48 hours and 72 hours of treatment with Trastuzumab, transferred on an enzymatic 96 wells plate and coincubated with MTT[3(4,5dimethylthiazol
Milano_opm_v5.indd 107 16-08-2007 15:42:58

Chapter 5
108
2yl)2,5diphenyltetrazolium bromide], for 4 hours. Ethanol absolute (96%) was added to stop the reaction, and results were read on a multiwell scanning spectrophotometer (ELISA reader).
Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) assay.In this assay the Trastuzumab treated EAC cell lines were incubated with HLA matched HER2 specific CTLs or lymphocytes obtained by stimulation with DCs which underwent the electroporation procedure, but without the addition of HER2, and cell lysis was measured. The HLAA2 positive cell lines OE33 and OE19 that were as well HER2 positive, and the HLAA2 positive Bic1 cell line that was HER2 negative was incubated for 48 hours with 10μg/ml Trastuzumab. Hereupon, 1x104 treated and not treated cells (target) were coincubated with effector cells , i.e., the HER2 specific CTLs or non specific CTLs, and PBMCs isolated from patients with or without HER2 amplification, at various E:T ratios, namely 5:1, 2.5:1, 1.25:1 and 0.62:1, in 100 μl of RPMI 1640 in a 96well plates in triplicate for 4 hours at 37ºC. The percentage of cytotoxicity was measured by the CytoTox 96 NonRadioactive Cytotoxicity assay (Promega, Madison, USA). This is a colorimetric cytotoxicity assay, which quantitatively measures lactate dehydrogenase (LDH), a stable cytosolic enzyme that is released upon cell lysis. Released LDH in culture supernatant is measured with a 30minute coupled enzymatic assay, which results in the conversion of a tetrazolium salt (INT) into a red formazan product. The amount of colour will be proportional to the number of lysed cells. Wavelength absorbance data were collected using a multiwell scanning spectrophotometer (ELISA reader). The percentage of specific cytotoxicity was calculated according to the formula: % specific lysis= [(ExperimentalEffector SpontaneousTarget Spontaneous)/ (Target MaximumTarget Spontaneous)] x100.
Cytometric Bead Array Supernatant from the different samples was collected after performing the cytotoxicity assay and used for measuring IFNγ release from activated HER2 specific CTLs. The procedure was followed as described previously44, following the manufacture’s instruction, and samples were examined in triplo for each condition.
Apoptosis in esophageal adenocarcinoma cell linesEach cell line (1x 104 cells) was cultured in 2 ml of RPMI 1640 and DMEM as appropriate with or without Trastuzumab (10 μg/mL) at 37˚C for 48 hours in a sixwells plate. After incubation, apoptosis in each cell line was measured by staining with APCconjugated AnnexinV and 7aad via probe as previously described44 and a staining procedure was followed as described before.
Milano_opm_v5.indd 108 16-08-2007 15:42:58
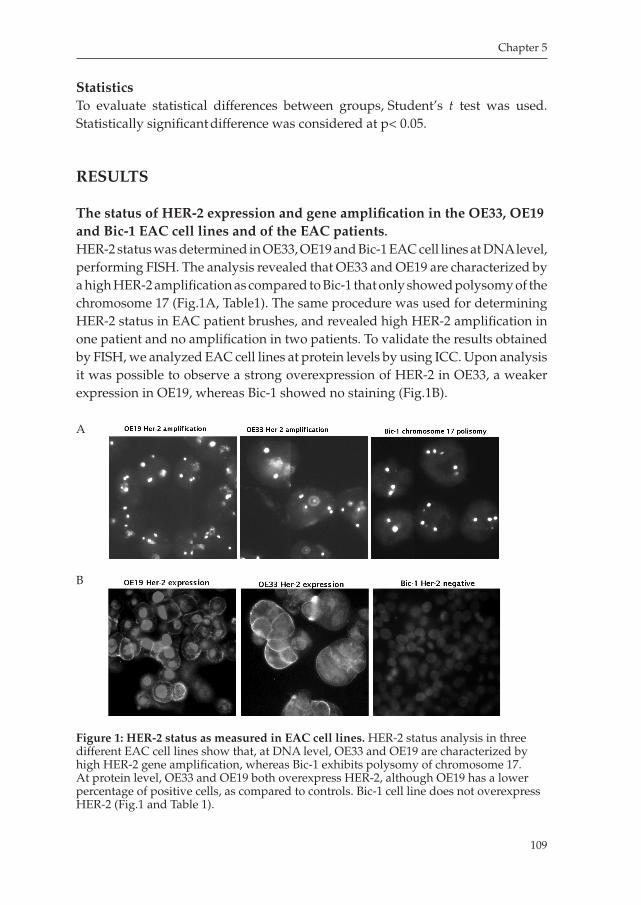
Chapter 5
109
StatisticsTo evaluate statistical differences between groups, Student’s t test was used. Statistically significant difference was considered at p< 0.05.
RESULTS
The status of HER-2 expression and gene amplification in the OE33, OE19 and Bic-1 EAC cell lines and of the EAC patients.
HER2 status was determined in OE33, OE19 and Bic1 EAC cell lines at DNA level, performing FISH. The analysis revealed that OE33 and OE19 are characterized by a high HER2 amplification as compared to Bic1 that only showed polysomy of the chromosome 17 (Fig.1A, Table1). The same procedure was used for determining HER2 status in EAC patient brushes, and revealed high HER2 amplification in one patient and no amplification in two patients. To validate the results obtained by FISH, we analyzed EAC cell lines at protein levels by using ICC. Upon analysis it was possible to observe a strong overexpression of HER2 in OE33, a weaker expression in OE19, whereas Bic1 showed no staining (Fig.1B).
Figure 1: HER-2 status as measured in EAC cell lines. HER2 status analysis in three different EAC cell lines show that, at DNA level, OE33 and OE19 are characterized by high HER2 gene amplification, whereas Bic1 exhibits polysomy of chromosome 17. At protein level, OE33 and OE19 both overexpress HER2, although OE19 has a lower percentage of positive cells, as compared to controls. Bic1 cell line does not overexpress HER2 (Fig.1 and Table 1).
A
B
Milano_opm_v5.indd 109 16-08-2007 15:42:59

Chapter 5
110
Table 1: HER-2 STATUSCELL LINES FISH ICCOE33 High amplification +++ positivityOE19 High amplification ++ positivityBic1 polysomy negative
Trastuzumab inhibits growth, but does not increase apoptosis, in HER-2 overexpressing EAC cell lines.
To evaluate the potential inhibitory activity of Trastuzumab on HER2 overexpressing cell lines, namely OE33 and OE19, cells treated at different time points and different concentrations of Trastuzumab were analyzed by MTT, and, further, stained by Annexin V and 7aad to assess the level of apoptosis comparing treated with not treated cells. Growth arrest in not treated cells was on average 0.21±0.02SEM. Inhibition of growth induced by Trastuzumab in OE33 at 24 hours using 2.5μg dose, was 0.1±0.01SEM (Paired Ttest, p<0.05). Increment of the Trastuzumab dosage at this time point did not further influence cell viability. After 48 hours, growth inhibition was not significantly different as compared to not treated cells (One way ANOVA, Bonferroni PostTest, p=0.7). After 72 hours, using 2.5μg dose, cell viability significantly decreased to 0.2±0.02SEM, Paired Ttest, p<0.05). Further increasing the doses of Trastuzumab at this time point did not result in enhanced sensitivity of the OE33 cell line (One way ANOVA, Bonferroni Post Test, p=0.05, Fig.2A). The basal cell viability before treatment of OE19 was on average 0.34±0.02SEM. After incubating the cells with a dose of 2.5μg of Trastuzumab for 24 hours OE19 cell viability significantly decreased as compared to not treated cells (0.2±0.03SEM, Paired Ttest, p<0.05). Significant inhibition was observed as well using a 5μg and 10μg dose (Mean 0.2±0.01SEM and 0.18±0.02SEM, respectively, paired Ttest, p<0.05). After 48 hours of treatment, using a 2.5μg dose showed significant growth inhibition as compared to the not treated cells (0.2±0.03, Paired Ttest, p<0.05). Using 5μg dose induced no further decrease in cell viability: 0.2±0.01 (Paired Ttest, p=0.08), but 10μg dose again induced a significant decrease in cell viability as compared to not treated cells (Paired Ttest, p<0.05, Fig.2B). After 72 hours of treatment, either using 2.5, 5, or 10μg dose, inhibition of growth was not significantly different as compared to not treated cells (One way ANOVA, Bonferroni Post Test, p=0.7). In Bic1 (EAC cell line HER2 negative, used as control), no differences were observed at different time point using different concentrations of Trastuzumab (Fig.2C).
Annexin V and 7aad staining as performed to assess the apoptosis level induced in EAC cell lines by the treatment with Trastuzumab showed no significant increase of apoptosis levels in the Trastuzumab treated cell lines as compared to not treated cell lines, either expressing or not expressing HER2 (Fig.2D).
Milano_opm_v5.indd 110 16-08-2007 15:42:59
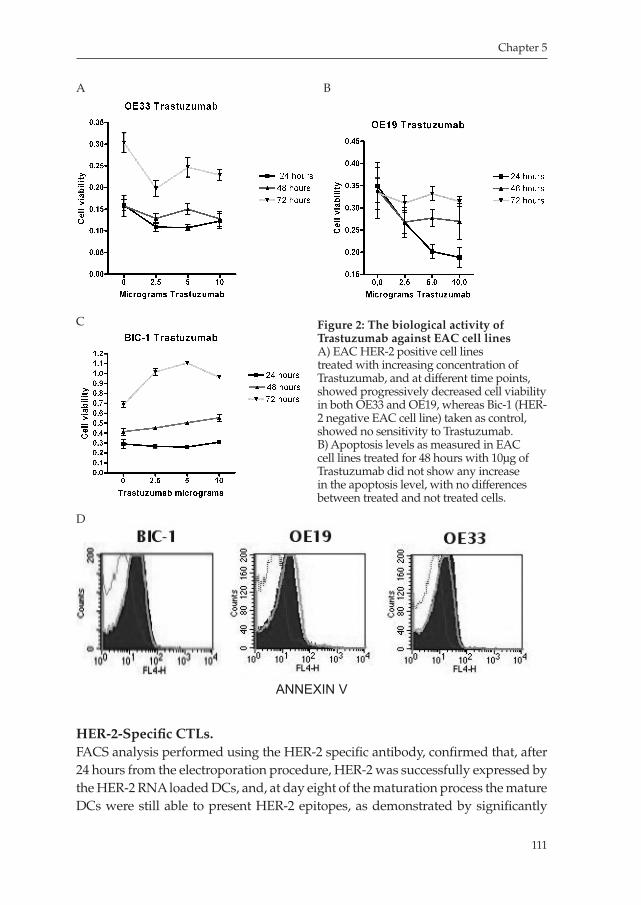
Chapter 5
111
HER-2-Specific CTLs.FACS analysis performed using the HER2 specific antibody, confirmed that, after 24 hours from the electroporation procedure, HER2 was successfully expressed by the HER2 RNA loaded DCs, and, at day eight of the maturation process the mature DCs were still able to present HER2 epitopes, as demonstrated by significantly
Figure 2: The biological activity of Trastuzumab against EAC cell linesA) EAC HER2 positive cell lines treated with increasing concentration of Trastuzumab, and at different time points, showed progressively decreased cell viability in both OE33 and OE19, whereas Bic1 (HER2 negative EAC cell line) taken as control, showed no sensitivity to Trastuzumab. B) Apoptosis levels as measured in EAC cell lines treated for 48 hours with 10μg of Trastuzumab did not show any increase in the apoptosis level, with no differences between treated and not treated cells.
A B
C
D
ANNEXIN V
Milano_opm_v5.indd 111 16-08-2007 15:43:01
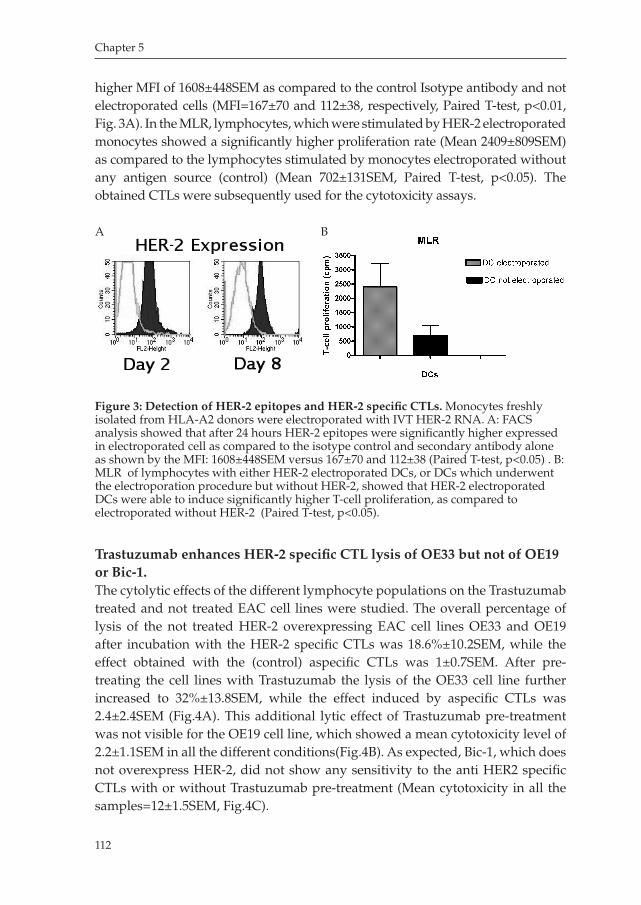
Chapter 5
112
higher MFI of 1608±448SEM as compared to the control Isotype antibody and not electroporated cells (MFI=167±70 and 112±38, respectively, Paired Ttest, p<0.01, Fig. 3A). In the MLR, lymphocytes, which were stimulated by HER2 electroporated monocytes showed a significantly higher proliferation rate (Mean 2409±809SEM) as compared to the lymphocytes stimulated by monocytes electroporated without any antigen source (control) (Mean 702±131SEM, Paired Ttest, p<0.05). The obtained CTLs were subsequently used for the cytotoxicity assays.
Trastuzumab enhances HER-2 specific CTL lysis of OE33 but not of OE19 or Bic-1.The cytolytic effects of the different lymphocyte populations on the Trastuzumab treated and not treated EAC cell lines were studied. The overall percentage of lysis of the not treated HER2 overexpressing EAC cell lines OE33 and OE19 after incubation with the HER2 specific CTLs was 18.6%±10.2SEM, while the effect obtained with the (control) aspecific CTLs was 1±0.7SEM. After pretreating the cell lines with Trastuzumab the lysis of the OE33 cell line further increased to 32%±13.8SEM, while the effect induced by aspecific CTLs was 2.4±2.4SEM (Fig.4A). This additional lytic effect of Trastuzumab pretreatment was not visible for the OE19 cell line, which showed a mean cytotoxicity level of 2.2±1.1SEM in all the different conditions(Fig.4B). As expected, Bic1, which does not overexpress HER2, did not show any sensitivity to the anti HER2 specific CTLs with or without Trastuzumab pretreatment (Mean cytotoxicity in all the samples=12±1.5SEM, Fig.4C).
Figure 3: Detection of HER-2 epitopes and HER-2 specific CTLs. Monocytes freshly isolated from HLAA2 donors were electroporated with IVT HER2 RNA. A: FACS analysis showed that after 24 hours HER2 epitopes were significantly higher expressed in electroporated cell as compared to the isotype control and secondary antibody alone as shown by the MFI: 1608±448SEM versus 167±70 and 112±38 (Paired Ttest, p<0.05) . B: MLR of lymphocytes with either HER2 electroporated DCs, or DCs which underwent the electroporation procedure but without HER2, showed that HER2 electroporated DCs were able to induce significantly higher Tcell proliferation, as compared to electroporated without HER2 (Paired Ttest, p<0.05).
A B
Milano_opm_v5.indd 112 16-08-2007 15:43:02
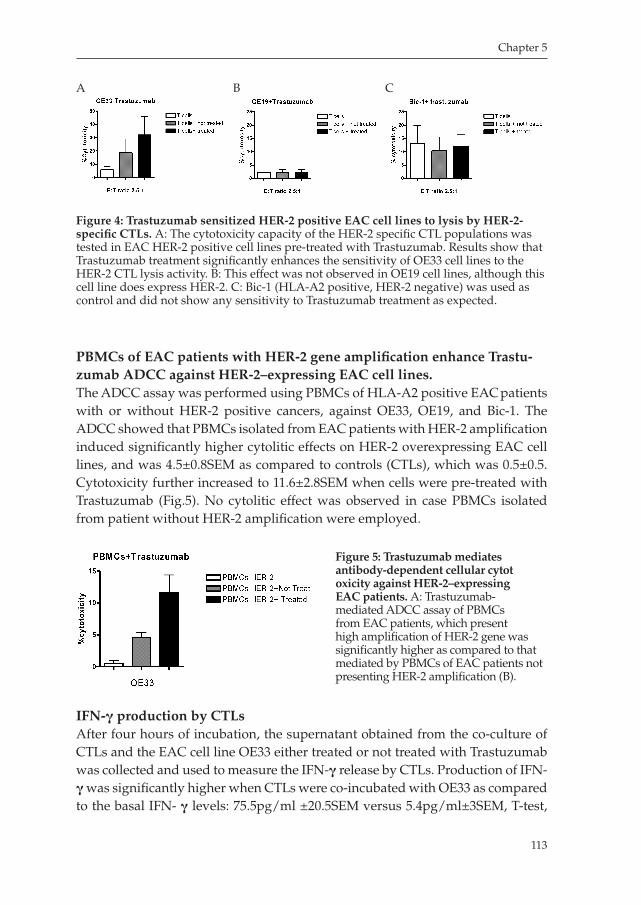
Chapter 5
113
PBMCs of EAC patients with HER-2 gene amplification enhance Trastu-zumab ADCC against HER-2–expressing EAC cell lines.The ADCC assay was performed using PBMCs of HLAA2 positive EAC patients with or without HER2 positive cancers, against OE33, OE19, and Bic1. The ADCC showed that PBMCs isolated from EAC patients with HER2 amplification induced significantly higher cytolitic effects on HER2 overexpressing EAC cell lines, and was 4.5±0.8SEM as compared to controls (CTLs), which was 0.5±0.5. Cytotoxicity further increased to 11.6±2.8SEM when cells were pretreated with Trastuzumab (Fig.5). No cytolitic effect was observed in case PBMCs isolated from patient without HER2 amplification were employed.
IFN-γ production by CTLsAfter four hours of incubation, the supernatant obtained from the coculture of CTLs and the EAC cell line OE33 either treated or not treated with Trastuzumab was collected and used to measure the IFNγ release by CTLs. Production of IFNγ was significantly higher when CTLs were coincubated with OE33 as compared to the basal IFN γ levels: 75.5pg/ml ±20.5SEM versus 5.4pg/ml±3SEM, Ttest,
Figure 4: Trastuzumab sensitized HER-2 positive EAC cell lines to lysis by HER-2-specific CTLs. A: The cytotoxicity capacity of the HER2 specific CTL populations was tested in EAC HER2 positive cell lines pretreated with Trastuzumab. Results show that Trastuzumab treatment significantly enhances the sensitivity of OE33 cell lines to the HER2 CTL lysis activity. B: This effect was not observed in OE19 cell lines, although this cell line does express HER2. C: Bic1 (HLAA2 positive, HER2 negative) was used as control and did not show any sensitivity to Trastuzumab treatment as expected.
A B C
Figure 5: Trastuzumab mediates antibody-dependent cellular cytot oxicity against HER-2–expressing EAC patients. A: Trastuzumabmediated ADCC assay of PBMCs from EAC patients, which present high amplification of HER2 gene was significantly higher as compared to that mediated by PBMCs of EAC patients not presenting HER2 amplification (B).
Milano_opm_v5.indd 113 16-08-2007 15:43:04
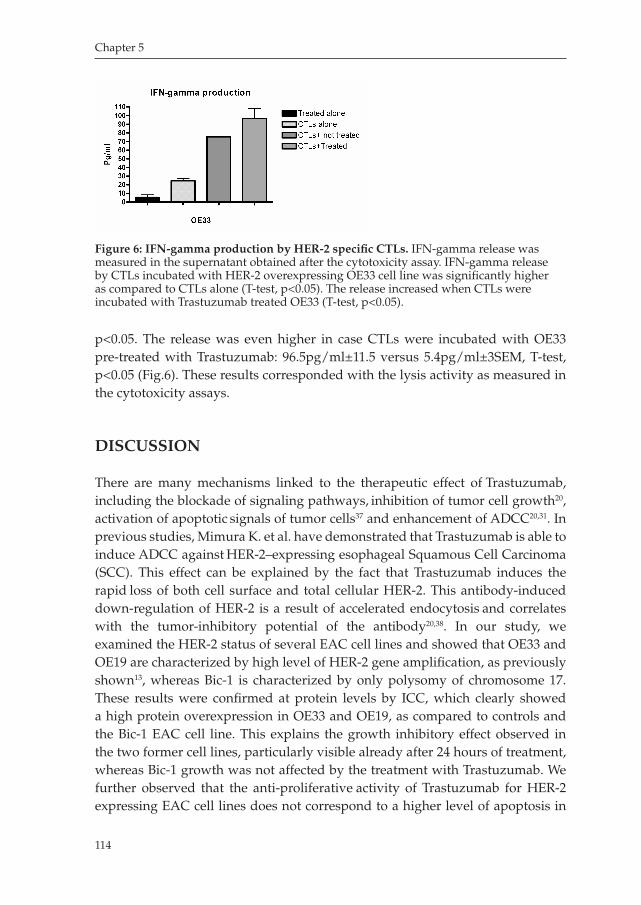
Chapter 5
114
p<0.05. The release was even higher in case CTLs were incubated with OE33 pretreated with Trastuzumab: 96.5pg/ml±11.5 versus 5.4pg/ml±3SEM, Ttest, p<0.05 (Fig.6). These results corresponded with the lysis activity as measured in the cytotoxicity assays.
DISCUSSION
There are many mechanisms linked to the therapeutic effect of Trastuzumab, including the blockade of signaling pathways, inhibition of tumor cell growth20, activation of apoptotic signals of tumor cells37 and enhancement of ADCC20,31. In previous studies, Mimura K. et al. have demonstrated that Trastuzumab is able to induce ADCC against HER2–expressing esophageal Squamous Cell Carcinoma (SCC). This effect can be explained by the fact that Trastuzumab induces the rapid loss of both cell surface and total cellular HER2. This antibodyinduced
downregulation of HER2 is a result of accelerated endocytosis and correlates with the tumorinhibitory potential of the antibody20,38. In our study, we examined the HER2 status of several EAC cell lines and showed that OE33 and OE19 are characterized by high level of HER2 gene amplification, as previously shown13, whereas Bic1 is characterized by only polysomy of chromosome 17. These results were confirmed at protein levels by ICC, which clearly showed a high protein overexpression in OE33 and OE19, as compared to controls and the Bic1 EAC cell line. This explains the growth inhibitory effect observed in the two former cell lines, particularly visible already after 24 hours of treatment, whereas Bic1 growth was not affected by the treatment with Trastuzumab. We further observed that the antiproliferative activity of Trastuzumab for HER2 expressing EAC cell lines does not correspond to a higher level of apoptosis in
Figure 6: IFN-gamma production by HER-2 specific CTLs. IFNgamma release was measured in the supernatant obtained after the cytotoxicity assay. IFNgamma release by CTLs incubated with HER2 overexpressing OE33 cell line was significantly higher as compared to CTLs alone (Ttest, p<0.05). The release increased when CTLs were incubated with Trastuzumab treated OE33 (Ttest, p<0.05).
Milano_opm_v5.indd 114 16-08-2007 15:43:05

Chapter 5
115
treated cells as compared to not treated cells, confirming prior observations33. One important mechanism that has been shown previously39, is that the tumorinhibitory antibodies to HER2 enhance ubiquitination of HER2. Protein tagged to ubiquitin is known to be targeted to the proteasome, which cleaves the protein into small peptides. These peptides are then transported into the endoplasmic reticulum, loaded onto MHC class I molecules, and transferred to the cell surface
for recognizing CTLs39. In addition, some recent studies demonstrated that HER2specific CTLs, generated from an HLAA2 positive patient with HER2 positive breast cancer, lysed the HER2 positive SKOV3tA2 breast cancer cells in an HLAA2 restricted manner34. These results suggest that Trastuzumab sensitized HER2overexpressing tumors inducing lysis in a MHC class Irestricted manner by HER2specific CTLs33. Based on these prior findings, we investigated whether this phenomenon can as well be observed in EAC cell lines using HER2 specific CTLs. Our findings confirmed that treatment with Trastuzumab significantly increased the capacity of HER2 specific CTLs to lyse the HER2 overexpressing EAC cell line OE33. Surprisingly, this effect was not observed in OE19 EAC cell line, although this cell line does overexpress HER2, but at a lower level (Fig 1B). The reason for this might be that the OE19 cell line has an impairment in the MHCClass I antigen presentation machinery, which is a subject for our future studies. Reasonably, this effect was not observed in Bic1 cell line, which does not overexpress HER2. Interestingly, performing the ADCC test using lymphocytes not specific for HER2, no differences were observed in OE33 cell line, as well as in OE19 and Bic1, providing evidence that in this particular circumstance, the lytic effect is rather mediated by anti HER2 specific CTLs than by NK cells. Kono et al. have shown both in gastric cancer and in esophageal squamous cell carcinoma that treatment with Trastuzumab induces ADCC using PBMCs from healthy donors, while PBMCs from patients showed a significantly lower ADCC, meaning that probably these patients may have impaired NK Tcell functions31. In our study, we first selected patients with advanced stage disease with or without HER2 gene amplification and compared the level of ADCC in these two different patient populations. The Trastuzumabmediated ADCC of PBMCs from EAC patients with HER2 amplification was significantly higher as compared to that observed in patients who did not present HER2 gene amplification. The result partly confirms the observations of Kono et al. In addition we show that there is an important difference in ADCC between PBMCs obtained from EAC patients with HER2 gene amplification compared to those without HER2 gene amplification. It is likely that patient with high HER2 levels are already sensitized and have developed HER2 specific CTLs, of which the lysis capacity may be enhanced by the action of Trastuzumab. This confirms other studies where it was shown that some breast cancer patients with HER2 positive tumors have preexistent T and Bcell–mediated immunity to the HER2 protein40. These results have been
Milano_opm_v5.indd 115 16-08-2007 15:43:05

Chapter 5
116
later confirmed by the same groups and by others41,42. It is important to note that cancer patients exhibiting natural antibody and Tcell immunity to HER2 do not develop autoimmune responses, suggesting that HER2 specific antibodies and T cells generated because of HER2 overexpression do not recognize basal HER2 expression on normal epithelial cells41. Our findings described here, confirming that Trastuzumab enhances the cytolitic activity of HER2 specific CTLs and showing that this antibody can mediate a significantly higher ADCC in HER2 positive EAC patients, encourages us to initiate combination therapies including both the antibody and CTLs directed against HER2. In summary, this study shows that the EAC cell line OE33 with high HER2 amplification is susceptible to Trastuzumab treatment. In addition, we found that specific anti HER2 CTL responses can be enhanced through pretreatment with Trastuzumab. We further provide data that EAC patients with HER2 expressing cancers may have specific anti HER2 CTLs, and that these patients may highly benefit from the combinatorial therapy by combining Trastuzumab with anti HER2 CTL mediated immunotherapies.
Milano_opm_v5.indd 116 16-08-2007 15:43:06

Chapter 5
117
REFERENCES
1. Hung, M. C. and Lau, Y. K. Basic science of HER2/neu: a review. Semin Oncol, 26: 5159, 1999.
2. Perez, S. A., Sotiropoulou, P. A., Sotiriadou, N. N., Mamalaki, A., Gritzapis, A. D., Echner, H.,
Voelter, W., Pawelec, G., Papamichail, M., and Baxevanis, C. N. HER2/neuderived peptide
884899 is expressed by human breast, colorectal and pancreatic adenocarcinomas and is
recognized by invitroinduced specific CD4(+) T cell clones. Cancer Immunol Immunother,
50: 615624, 2002.
3. Slamon, D. J., LeylandJones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T.,
Eiermann, W., Wolter, J., Pegram, M., Baselga, J., and Norton, L. Use of chemotherapy plus a
monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N
Engl J Med, 344: 783792, 2001.
4. Murray, J. L., Gillogly, M. E., Przepiorka, D., Brewer, H., Ibrahim, N. K., Booser, D. J., Horto
bagyi, G. N., Kudelka, A. P., Grabstein, K. H., Cheever, M. A., and Ioannides, C. G. Toxicity,
immunogenicity, and induction of E75specific tumorlytic CTLs by HER2 peptide E75 (369
377) combined with granulocyte macrophage colonystimulating factor in HLAA2+ patients
with metastatic breast and ovarian cancer. Clin Cancer Res, 8: 34073418, 2002.
5. Yamanaka, Y., Friess, H., Kobrin, M. S., Buchler, M., Kunz, J., Beger, H. G., and Korc, M.
Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Hum Pathol, 24:
11271134, 1993.
6. Ooi, A., Takehana, T., Li, X., Suzuki, S., Kunitomo, K., Iino, H., Fujii, H., Takeda, Y., and
Dobashi, Y. Protein overexpression and gene amplification of HER2 and EGFR in colorectal
cancers: an immunohistochemical and fluorescent in situ hybridization study. Mod Pathol, 17:
895904, 2004.
7. Jankowski, J., Coghill, G., Hopwood, D., and Wormsley, K. G. Oncogenes and oncosuppres
sor gene in adenocarcinoma of the oesophagus. Gut, 33: 10331038, 1992.
8. Geddert, H., Zeriouh, M., Wolter, M., Heise, J. W., Gabbert, H. E., and Sarbia, M. Gene ampli
fication and protein overexpression of cerbb2 in Barrett carcinoma and its precursor lesions.
Am J Clin Pathol, 118: 6066, 2002.
9. Walch, A., Bink, K., Gais, P., Stangl, S., Hutzler, P., Aubele, M., Mueller, J., Hofler, H., and Wer
ner, M. Evaluation of cerbB2 overexpression and Her2/neu gene copy number heterogen
eity in Barrett’s adenocarcinoma. Anal Cell Pathol, 20: 2532, 2000.
10. Reichelt, U., Duesedau, P., Tsourlakis, M., Quaas, A., Link, B. C., Schurr, P. G., Kaifi, J. T., Gros,
S. J., Yekebas, E. F., Marx, A., Simon, R., Izbicki, J. R., and Sauter, G. Frequent homogeneous
HER2 amplification in primary and metastatic adenocarcinoma of the esophagus. Mod
Pathol, 20: 120129, 2007.
11. Persons, D. L., Croughan, W. S., Borelli, K. A., and Cherian, R. Interphase cytogenetics of
esophageal adenocarcinoma and precursor lesions. Cancer Genet Cytogenet, 106: 1117, 1998.
12. Brien, T. P., Odze, R. D., Sheehan, C. E., McKenna, B. J., and Ross, J. S. HER2/neu gene am
plification by FISH predicts poor survival in Barrett’s esophagusassociated adenocarcinoma.
Hum Pathol, 31: 3539, 2000.
Milano_opm_v5.indd 117 16-08-2007 15:43:06

Chapter 5
118
13. Dahlberg, P. S., Jacobson, B. A., Dahal, G., Fink, J. M., Kratzke, R. A., Maddaus, M. A., and Ferrin,
L. J. ERBB2 amplifications in esophageal adenocarcinoma. Ann Thorac Surg, 78: 17901800, 2004.
14. Reichelt, U., Duesedau, P., Tsourlakis, M. C., Quaas, A., Link, B. C., Schurr, P. G., Kaifi, J. T., Gros,
S. J., Yekebas, E. F., Marx, A., Simon, R., Izbicki, J. R., and Sauter, G. Frequent homogeneous HER2
amplification in primary and metastatic adenocarcinoma of the esophagus. Mod Pathol, 2006.
15. Sotiropoulou, P. A., Perez, S. A., Voelter, V., Echner, H., Missitzis, I., Tsavaris, N. B., Papami
chail, M., and Baxevanis, C. N. Natural CD8+ Tcell responses against MHC class I epitopes
of the HER2/ neu oncoprotein in patients with epithelial tumors. Cancer Immunol Immu
nother, 52: 771779, 2003.
16. Gritzapis, A. D., Mahaira, L. G., Perez, S. A., Cacoullos, N. T., Papamichail, M., and Baxevanis,
C. N. Vaccination with human HER2/neu (435443) CTL peptide induces effective antitumor
immunity against HER2/neuexpressing tumor cells in vivo. Cancer Res, 66: 54525460, 2006.
17. Baxevanis, C. N., Sotiriadou, N. N., Gritzapis, A. D., Sotiropoulou, P. A., Perez, S. A., Cacoull
os, N. T., and Papamichail, M. Immunogenic HER2/neu peptides as tumor vaccines. Cancer
Immunol Immunother, 55: 8595, 2006.
18. Fisk, B., Blevins, T. L., Wharton, J. T., and Ioannides, C. G. Identification of an immunodomi
nant peptide of HER2/neu protooncogene recognized by ovarian tumorspecific cytotoxic T
lymphocyte lines. J Exp Med, 181: 21092117, 1995.
19. Kono, K., Takahashi, A., Sugai, H., Fujii, H., Choudhury, A. R., Kiessling, R., and Matsumoto,
Y. Dendritic cells pulsed with HER2/neuderived peptides can induce specific Tcell respon
ses in patients with gastric cancer. Clin Cancer Res, 8: 33943400, 2002.
20. Mimura, K., Kono, K., Hanawa, M., Kanzaki, M., Nakao, A., Ooi, A., and Fujii, H. Trastuzu
mabmediated antibodydependent cellular cytotoxicity against esophageal squamous cell
carcinoma. Clin Cancer Res, 11: 48984904, 2005.
21. Kimura, K., Sawada, T., Komatsu, M., Inoue, M., Muguruma, K., Nishihara, T., Yamashita,
Y., Yamada, N., Ohira, M., and Hirakawa, K. Antitumor effect of trastuzumab for pancreatic
cancer with high HER2 expression and enhancement of effect by combined therapy with
gemcitabine. Clin Cancer Res, 12: 49254932, 2006.
22. Zhang, Y., Banerjee, S., Wang, Z., Xu, H., Zhang, L., Mohammad, R., Aboukameel, A., Adsay,
N. V., Che, M., Abbruzzese, J. L., Majumdar, A. P., and Sarkar, F. H. Antitumor activity of epi
dermal growth factor receptorrelated protein is mediated by inactivation of ErbB receptors
and nuclear factorkappaB in pancreatic cancer. Cancer Res, 66: 10251032, 2006.
23. Diermeier, S., Horvath, G., KnuechelClarke, R., Hofstaedter, F., Szollosi, J., and Brockhoff,
G. Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in
HER2/neu overexpressing breast cancer cells via specific erbBreceptor interaction and acti
vation. Exp Cell Res, 304: 604619, 2005.
24. Baselga, J., Tripathy, D., Mendelsohn, J., Baughman, S., Benz, C. C., Dantis, L., Sklarin, N.
T., Seidman, A. D., Hudis, C. A., Moore, J., Rosen, P. P., Twaddell, T., Henderson, I. C., and
Norton, L. Phase II study of weekly intravenous recombinant humanized antip185HER2
monoclonal antibody in patients with HER2/neuoverexpressing metastatic breast cancer. J
Clin Oncol, 14: 737744, 1996.
Milano_opm_v5.indd 118 16-08-2007 15:43:06

Chapter 5
119
25. LeylandJones, B. Trastuzumab: hopes and realities. Lancet Oncol, 3: 137144, 2002.
26. Tripathy, D., Slamon, D. J., Cobleigh, M., Arnold, A., Saleh, M., Mortimer, J. E., Murphy, M.,
and Stewart, S. J. Safety of treatment of metastatic breast cancer with trastuzumab beyond
disease progression. J Clin Oncol, 22: 10631070, 2004.
27. Gamliel, Z. Incidence, epidemiology, and etiology of esophageal cancer. Chest Surg Clin N
Am, 10: 441450, 2000.
28. Pera, M. Trends in incidence and prevalence of specialized intestinal metaplasia, barrett’s
esophagus, and adenocarcinoma of the gastroesophageal junction. World J Surg, 27: 9991008;
discussion 10061008, 2003.
29. Donington, J. S., Miller, D. L., Allen, M. S., Deschamps, C., Nichols, F. C., 3rd, and Pairolero,
P. C. Preoperative chemoradiation therapy does not improve early survival after esophagec
tomy for patients with clinical stage III adenocarcinoma of the esophagus. Ann Thorac Surg,
77: 11931198; discussion 11981199, 2004.
30. Bosset, J. F., Gignoux, M., Triboulet, J. P., Tiret, E., Mantion, G., Elias, D., Lozach, P., Ollier, J. C.,
Pavy, J. J., Mercier, M., and Sahmoud, T. Chemoradiotherapy followed by surgery compared
with surgery alone in squamouscell cancer of the esophagus. N Engl J Med, 337: 161167, 1997.
31. Kono, K., Takahashi, A., Ichihara, F., Sugai, H., Fujii, H., and Matsumoto, Y. Impaired anti
bodydependent cellular cytotoxicity mediated by herceptin in patients with gastric cancer.
Cancer Res, 62: 58135817, 2002.
32. Clemenceau, B., Gallot, G., Vivien, R., Gaschet, J., Campone, M., and Vie, H. Longterm pres
ervation of antibodydependent cellular cytotoxicity (ADCC) of natural killer cells amplified
in vitro from the peripheral blood of breast cancer patients after chemotherapy. J Immunoth
er, 29: 5360, 2006.
33. Kono, K., Sato, E., Naganuma, H., Takahashi, A., Mimura, K., Nukui, H., and Fujii, H. Tras
tuzumab (Herceptin) enhances class Irestricted antigen presentation recognized by HER2/
neuspecific T cytotoxic lymphocytes. Clin Cancer Res, 10: 25382544, 2004.
34. zum Buschenfelde, C. M., Hermann, C., Schmidt, B., Peschel, C., and Bernhard, H. Antihu
man epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances
cytolytic activity of class Irestricted HER2specific T lymphocytes against HER2overexpress
ing tumor cells. Cancer Res, 62: 22442247, 2002.
35. Safran, H., Dipetrillo, T., Akerman, P., Ng, T., Evans, D., Steinhoff, M., Benton, D., Purviance,
J., Goldstein, L., Tantravahi, U., and Kennedy, T. Phase I/II study of trastuzumab, paclitaxel,
cisplatin and radiation for locally advanced, HER2 overexpressing, esophageal adenocarci
noma. Int J Radiat Oncol Biol Phys, 67: 405409, 2007.
36. Muller, M. R., Grunebach, F., Kayser, K., Vogel, W., Nencioni, A., Brugger, W., Kanz, L., and
Brossart, P. Expression of her2/neu on acute lymphoblastic leukemias: implications for the
development of immunotherapeutic approaches. Clin Cancer Res, 9: 34483453, 2003.
37. Cuello, M., Ettenberg, S. A., Clark, A. S., Keane, M. M., Posner, R. H., Nau, M. M., Dennis, P.
A., and Lipkowitz, S. Downregulation of the erbB2 receptor by trastuzumab (herceptin) en
hances tumor necrosis factorrelated apoptosisinducing ligandmediated apoptosis in breast
and ovarian cancer cell lines that overexpress erbB2. Cancer Res, 61: 48924900, 2001.
Milano_opm_v5.indd 119 16-08-2007 15:43:07

Chapter 5
120
38. Hurwitz, E., Stancovski, I., Sela, M., and Yarden, Y. Suppression and promotion of tumor
growth by monoclonal antibodies to ErbB2 differentially correlate with cellular uptake. Proc
Natl Acad Sci U S A, 92: 33533357, 1995.
39. Klapper, L. N., Waterman, H., Sela, M., and Yarden, Y. Tumorinhibitory antibodies to HER
2/ErbB2 may act by recruiting cCbl and enhancing ubiquitination of HER2. Cancer Res, 60:
33843388, 2000.
40. Disis, M. L., Calenoff, E., McLaughlin, G., Murphy, A. E., Chen, W., Groner, B., Jeschke, M.,
Lydon, N., McGlynn, E., Livingston, R. B., and et al. Existent Tcell and antibody immunity to
HER2/neu protein in patients with breast cancer. Cancer Res, 54: 1620, 1994.
41. Disis, M. L., Knutson, K. L., Schiffman, K., Rinn, K., and McNeel, D. G. Preexistent immunity
to the HER2/neu oncogenic protein in patients with HER2/neu overexpressing breast and
ovarian cancer. Breast Cancer Res Treat, 62: 245252, 2000.
42. Baxevanis, C. N., Sotiropoulou, P. A., Sotiriadou, N. N., and Papamichail, M. Immunobiology
of HER2/neu oncoprotein and its potential application in cancer immunotherapy. Cancer
Immunol Immunother, 53: 166175, 2004.
43. A.M Rygiel, F.Milano, F.J ten Kate, B. Elzer, P.W. Fockens, J.J.G.H.M Bergman M.P Peppelen
bosh, K.K.Krishnadath. Clonal evolution of chromosome 17 polysomy to Her-2/neu proto
oncogene amplification correlates with increasing stage of Barrett’s esophagus. (paper in
preparation).
44. F.Milano, A.M.Rygiel, J.W. P.M. van Baal, N.Buttar, J.J.G.H.M. Bergman, C. Sondermeijer,
A.ten Brinke, M.Kapsenberg, S.M. van Ham, M.P. Peppelenbosch, K.K. Krishnadath. An
exvivo read out for evaluation of Dendritic Cell induced autologous CTL responses against
esophageal cancer (Cancer Immunology and Immunotherapy, 2007, In Press).
45. A.M. Rygiel, J.W.P.M. van Baal, F.Milano, K.K. Wang, F.J. ten Kate, P. Fockens, W.D. Rosmo
len, J. J.G.H.M. Bergman, M.P. Peppelenbosch, K.K. Krishnadath Efficient automated assess
ment of genetic abnormalities detected by Fluorescent in situ hybridization on brush cytology
of a Barrett’s esophagus surveillance population. Cancer. 2007 May 15;109(10):19808.
46. F.Milano, J. W.P.M. van Baal, A.M. Rygiel, J.J.G.H.M. Bergman, S.J.H.Van Deventer,
M.L.Kapsenberg, M.P. Peppelenbosch, K.K. Krishnadath. An Improved Protocol for Genera
tion of ImmunoPotent Dendritic Cells through Direct Electroporation of CD14+ Monocytes. J
Immunol Methods. 2007 Apr 10;321(12):94106.
Milano_opm_v5.indd 120 16-08-2007 15:43:07

Chapter 6
Bone Morphogenetic Protein
(BMP)-4-expressed in esophagitis induces a
columnar phenotype in esophageal squamous cells
Francesca Milano, Jantine W.P.M. van Baal, Navtej S. Buttar, Agnieszka M. Rygiel, Floor de Kort
Wilda D. Rosmolen, Jacques J.G.H.M. Bergman, Jan van Marle, Kenneth K. Wang,
Maikel P. Peppelenbosch, Kausilia K. Krishnadath
Gastroenterology. 2007 Jun;132(7):24122421
Milano_opm_v5.indd 121 16-08-2007 15:43:07

Chapter 6
122
ABSTRACT
BACKGROUND &AIMS: Barrett Esophagus (BE) is a metaplastic condition in which normal squamous esophageal epithelium is replaced by columnar epithelium. It is proposed that one of the possible mechanisms is dedifferentiation of squamous epithelium into columnar epithelium. The pathophysiology through which this metaplasia occurs is unknown. A recent study by SAGE analysis showed that Bone Morphogenetic Protein 4 (BMP4) is uniquely expressed in BE. In this study the role of the BMP pathway in the metaplastic transformation of normal squamous cells into columnar cells was examined. METHODS: Tissues from esophagitisBE patients and in an esophagitisBE rat model were examined for the activation of the BMPpathway. Short term cultures of primary normal squamous esophageal cells were treated with BMP4, and cell biological changes were examined by Western blot analysis, immunohistochemistry and microarrays. RESULTS: In both human and rat tissues the BMP pathway proved to be activated in esophagitis and BE. Upon incubation of squamous cell cultures with BMP4, the cytokeratin (CK) expression pattern showed a shift which was consistent with columnar epithelium. Involvement of the BMP pathway was suggested by upregulation of PhosphorylatedSmad 1/5/8 (PSmad 1/5/8) that was effectively blocked by Noggin, a BMP antagonist. Comparison of the gene expression profiles of squamous cells, BMP4 treated squamous cells and BE cells, showed a significant shift in the profile of the BMP4 treated squamous cells towards that of the cultured BE cells. CONCLUSIONS: These results suggest that the BMP pathway could play a role in the transformation of normal esophageal squamous cells into columnar cells.
Milano_opm_v5.indd 122 16-08-2007 15:43:08

Chapter 6
123
INTRODUCTION
Barrett Esophagus (BE) is a premalignant condition of the distal esophagus, which is associated with an increased risk of developing esophageal adenocarcinoma. Esophageal adenocarcinoma is one of the most rapidly increasing, lethal cancers in Western countries and is becoming a major health concern.13 BE is believed to be a metaplastic change in which the normal stratified squamous epithelium in the distal esophagus is replaced by columnar epithelium.4 This process is assumed to be the result of longstanding gastroesophageal reflux disease (GERD). GERD is a common condition that can be found in 2030% of the general Western population.5 It has been estimated that BE can be found in 612% of patients who suffer from GERD and it is assumed that individuals with GERD and esophagitis subsequently may develop BE. 6, 7 Although the concept of metaplasia is largely accepted, the actual pathophysiology by which these changes take place is unknown. In order to identify genes specifically involved in the transition of normal squamous esophageal mucosa into metaplastic BE, we previously used serial analysis of gene expression (SAGE), a technique to compare the expression profile of BE with that of its surrounding epithelia, i.e. normal squamous esophageal and gastric cardia mucosa. BMP4 was found to be abundantly and uniquely expressed in the SAGE library of BE, but not in normal squamous or cardia epithelium. BMP4 is a protein belonging to the transforming growth factor beta (TGFβ) family. Members of the TGFβ family are involved in controlling cellular differentiation, migration and proliferation.8 BMPs are 3035 kD hetero or homodimeric proteins. They were originally shown to play a role in bone formation, but were also found to be essential during embryonic development.9, 10 BMPs induce the formation of a heterodimeric complex of the BMP receptor type I and type II. This receptor complex signals downstream by phosphorylating specific BMP receptor regulated Smads (Smad 1, 5 and 8). The PSmad 1/5/8 forms a heterocomplex with Smad 4, this complex translocates into the nucleus, where certain target genes, such as ID2, can be transcribed.11 Previous studies demonstrated that BMPs are induced during inflammation and injury.1215
In this study, we provide evidence that refluxinduced metaplastic transformation of inflamed squamous esophageal mucosa to columnar type mucosa is mediated by BMP4. Consequently, manipulation of this pathway could potentially prevent the development of BE and thus reduce the occurrence of esophageal adenocarcinoma.
MATERIALS AND METHODS
Patient and Biopsy SpecimensBiopsy specimens obtained during routine surveillance endoscopies from 28 nondysplastic BE patients were used. The median age was 67 years (range 3988
Milano_opm_v5.indd 123 16-08-2007 15:43:08

Chapter 6
124
years) and 13 patients were male. The median length of the BE segment measured endoscopically was 5.4 cm (range 213 cm). All patients were on long term proton pump inhibitor therapy. Paired biopsies were obtained from the Barrett segment as well as of normal squamous esophagus. The Barrett biopsies were taken at least 2 cm above the gastroesophageal junction. Normal squamous epithelium was taken at least 2 cm above the Barrett segment. Special care was taken to avoid sampling Barrett areas containing islands of quamous mucosa. Additional biopsies from 6 patients, who had at least had grade B (Los Angeles classification) esophagitis were collected. Four of these patients were male and the mean age was 54 years (range 2770 years). Paired biopsies were taken next to each other from the inflamed squamous mucosa and normal squamous segment of these patients. None of the esophagitis patients had a concomitant BE. The inflammation was histologically confirmed on the pairwise taken control biopsies.
Primary Cell CultureBiopsies specimens of 15 BE patients were used for tissue culture. Biopsies were collected during endoscopic procedure and immediately placed in MCDB 153 medium (Sigma Chemical Co., St Louis, MO, USA) on ice. Biopsy specimens were processed within 4 hours after endoscopy. The explant method described by K. Washington et al. was used.16 Primary cell cultures were initiated by maintaining the cells in Barrett plus media, as previously described.17, 18 Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C and the media was replaced twice weekly. After two to three weeks of culturing, squamous cells were incubated with 100 ng/ml rec h BMP4 (R&D Systems, Minneapolis, MN, USA) for several time points and/or 50 µg/ml recombinant Mouse noggin/Fc chimera (R&D) for several time points. For the preparation of cells on glass slides, cells were harvested and resuspended in medium, dropped on Superfrost + (Menzel Glaser, Braunschweig, Germany) glass slides and airdried overnight.
RNA isolationTotal RNA for Microarray was isolated from squamous, BMP4 treated squamous, and Barrett primary cell cultures derived from three different patients, using TRIzol Reagent (according to manufacturer’s instructions, Life Technologies Inc, Invitrogen, Breda, The Netherlands). Upon isolation, spectrophotometry was performed with 1 μl of total RNA to quantitate on the Nanodrop (type ND1000). Assessment of the quality was performed with the RNA 2100 Pico Labchip kit (Agilent Technologies, Amstelveen, The Netherlands) using 1 µl of total RNA.
Microarray analysis: RNA Amplification, Labeling and HybridizationMicroarray analysis was performed for three different patients. The mRNA was double amplified using the Amino Allyl MessageAmp kit (Ambion, Austin,
Milano_opm_v5.indd 124 16-08-2007 15:43:09

Chapter 6
125
USA). The labeling, hybridization and data extraction were performed at ServiceXS (Leiden, The Netherlands). Briefly, 20 ng total RNA was mixed with 1 µl of T7 Oligo(dT) primer in a total volume of 12 µl. Primer and template were denatured by incubating at 70°C for 10 minutes and annealed by putting the reaction tubes on ice. The First Strand Reaction was performed by adding 8 μl Reverse Transcription Master Mix (containing 10x First Strand buffer, Ribonuclease Inhibitor, dNTP Mix and Reverse Transcriptase) and incubating at 42°C for 2 hours. Second Strand cDNA Synthesis was done by adding 63 μl NucleaseFree Water, 10 μl 10x Second Strand Buffer, 4μl dNTP Mix, 2μl DNA polymerase and 1 μl RNase H and incubating at 16°C for 2 hours. cDNA purification was done according to the manufacturer’s protocol (Ambion). In vitro transcription was initiated by addition of 3 μl UTP solution (50mM), 12 μl ATP, CTP, GTP Mix (25mM), 4 μl T7 10x Reaction Buffer and 4 μl T7 Enzyme Mix and incubated at 37°C for 5 hours. Purification of aRNA was done according to manufacturers protocol (Ambion). The second round First Strand Synthesis was done using 2 μg purified aRNA, adding 2 μl 10x First Strand Buffer, 1 μl Ribonuclease Inhibitor, 4 μl dNTP Mix and 1 μl Reverse Transcriptase and incubating at 42°C for 2 hours. After adding 1 μl of RNase H, samples were incubated at 37°C for 30 minutes. Five μl of T7 Oligo (dT) primer was added and the sample was denatured by incubating at 70°C for 10 minutes and annealed by putting the samples on ice. The second round Second Strand cDNA Synthesis was done by adding 58 μl NucleaseFree Water, 10 μl 10x Second Strand Buffer, 4 μl dNTP Mix and 2 μl DNA polymerase and incubating at 16°C for 2 hours. cDNA purification was done according manufacturers protocol (Ambion). In vitro transcription was initiated by addition of 2 μl aaUTP Solution (50mM), 12 μl ATP, CTP, GTP mix (25mM), 3 μl UTP Solution (50mM), 4 μl T7 10x Reaction Buffer and 4 μl T7 Enzyme Mix and incubation at 37°C for 9 hours. Qiagen’s RNeasy mini spin columns (Qiagen, Benelux B.V., Venlo, The Netherlands) were used for purification of the cRNA as described in Agilent’s user manual. Dye Coupling Reaction was performed using 5 μg amino allyl aRNA, 9 μl Coupling Buffer and 11 μl NHS ester dye, prepared according manufacturers protocol (Amersham, Buckinghamshire, United Kingdom). After an incubation at room temperature for 30 minutes, 4.5 μl 4M Hydroxylamine was added and incubated at room temperature for 15 minutes. Dye labeled aRNA was purified according to the manufacturers protocol (Ambion) and the samples were checked on concentration and dye incorporation on the Nanodrop ND1000. The cRNA yield was between 91 µg and 123 µg. Hybridization was performed with 600 ng or 1 µg of each labeled target together with control targets, fragmentation and hybridization buffer at 60°C for 17 hours onto Agilent Human Whole Genome Oligo arrays (Amsterdam, the Netherlands) following the manufacturer’s protocol.
Milano_opm_v5.indd 125 16-08-2007 15:43:09

Chapter 6
126
Microarray Imaging, Data and Statistical Analysis The microarray slides were washed following the instructions in the user manual and scanned on the Agilent dual laser DNA microarray scanner. The microarray data was normalized using the Agilent feature extraction software (version 7.5) to correct for the background correction and analyzed using the Rosetta Resolver v5.0 Expression Data Analysis System (Rosetta Biosoftware, Seattle, USA). To compare the normal squamous cells with the BMP4 incubated squamous cells we hybridized every sample to the BE sample and as such used the BE cells as the standard. Statistical analysis was done using the Chi square test to test whether there was a significant difference in genes that were more than 2 fold up or downregulated in the normal squamous cells compared to the BE cells versus the BMP4 treated squamous cells compared to BE cells. To make a more strict statistical comparison between the non treated squamous cells and BMP4 treated squamous cells, with the expression levels of the BE cells as the reference, we build new ratios using Ratiosplit software of the Rosetta Resolver Package. Using this Ratiosplit, statistical analysis, ANOVA (on intensity data) was performed for the 3 different experiments (p<0.01).
Western blot analysisThe squamous cells treated with BMP4 were washed with ice cold PBS and lysed with 200 µl lysis buffer (Cell Signaling), 1 mM Pefablock (Sigma). Biopsy material from squamous, esophagitis and BE specimens were collected in liquid nitrogen and immediately placed at 80ºC. Samples were subsequently lysed with 200 μl lysis buffer 1 mM Pefablock for western blot analysis. The lysates were sonicated and then centrifuged at 20g for 10 minutes at 4ºC. The pellet were discarded and the protein concentration was measured with the BCA protein assay kit (Pierce chemical co. Rockford, IL, USA). Lysates were diluted 1:2 in protein sample buffer (125 mM Tris/HCl, pH 6,8; 4%SDS; 2% βmercapto ethanol ; 20% glycerol; 1 mg bromphenol blue) and incubated for 5 minutes at 95ºC. Twenty μg of protein per lane was loaded onto SDSPAGE and subsequently transferred onto PVDF membrane (Millipore, Amsterdam, The Netherlands). The blots were blocked with 2% BSA in Tris Buffered Saline supplemented with 0.1% Tween20 (TBST) for one hour at room temperature and washed in TBST before overnight incubation at 4ºC with primary antibody in 2% BSA in TBST. Blots were then washed with TBST and incubated for 1 hour at room temperature in 1:1000 Horse Radish Peroxidase (HRP) conjugated secondary antibody in 2% BSA in TBST. After a final wash with TBST, blots were incubated for 5 minutes in Lumilite plus (BoehringerMannheim, Mannheim, Germany) and then chemiluminescence detected using a Fuji LAS3000 illuminator (Fuji Film Medical Systems, Stamford, USA). The antibodies used and dilutions are summarized in Table 1.
Milano_opm_v5.indd 126 16-08-2007 15:43:09
Chapter 6
127
Table 1: Antibodies as used for Western blotting and immuno-histochemistry.Antibody Species Company Country DilutionCytokeratin 7 Mouse monoclonal Chemicon USA 1:500Cytokeratin 10/13 Mouse monoclonal Dako Denmark 1:500Cytokeratin 20 Mouse monoclonal Progen Germany 1:500BMP4 Mouse monoclonal R&D USA 1:1000PSmad 1/5/8 Rabbit monoclonal Cell Signaling USA 1:1000BMP RIA Goat polyclonal R&D USA 1:1000BMP RII Goat polyclonal Abcam USA 1:500ID 2 Rabbit polyclonal Santa Cruz USA 1:1000Smad 4 Mouse monoclonal Santa Cruz USA 1:1000Actin Goat polyclonal Santa Cruz USA 1:2000
Rat model of BESixweekold male SpragueDawley rats were purchased from Harlan (Indianapolis, IN, USA). A detailed description of the procedure and conditions of the preparation of the BE rat model has been described by Buttar et al. 19 Briefly, 8 rats underwent a midline laparotomy and Levrat’s esophagojejunostomy to induce enteroesophageal reflux.20 Four rats were kept under the same conditions but with sham procedure, for control purposes. Rats were euthanized between 20 to 22 weeks postoperatively using CO2 narcosis, intramuscular injection of 12 mg/kg xylazine hydrochloride and removal of 5 ml intracardiac blood. The whole body was cooled to 4ºC and a midline incision was made from the laryngopharynx to the lower abdomen. The site of anastomosis was identified by finding the polypropylene sutures. It was freed of any adhesions and then dissected free of surrounding tissue up to the laryngopharynx. The esophagus was then cut at the level of the larynx and 2 mm above the site of anastomosis and opened longitudinally. Finally esophageal tissues from 4 control rats and 4 rats that had both esophagitis and BE were collected. Tissue samples from inflamed esophagus and columnar lined esophagus were divided in two equal parts. Half of the tissue was formalin fixed and sectioned to confirm the diagnosis of esophagitis and BE, the remaining half was stored at 80˚C until processing for immunohistochemistry and H&E staining. The animal study was approved by the animal care committee (IACUC) at the Mayo Clinic (Rochester, MN, USA).
Fluorescent immuno-histochemistry of tissues and cultured cells Biopsies from squamous mucosa, esophagitis and BE specimens were embedded in TissueTek® Optimum Cutting Temperature compound (Sakura Finetek, USA, Torrance, Calif.) at 20°C and sectioned using a cryomicrotome (Microm HM 550) in serial cryostat sections of 6 μm, placed on Superfrost + (Menzel Glaser, Braunschweig, Germany) glass slides and airdried overnight. For routine histological examination, of each block one slide was stained by hematoxylin and eosin. Cultured cells were harvested and dropped on Superfrost + (Menzel
Milano_opm_v5.indd 127 16-08-2007 15:43:10

Chapter 6
128
Glaser, Braunschweig, Germany) glass slides and airdried overnight. Slide preparations were fixed for 20 minutes in Phosphate Buffered Saline (PBS) with 4% Paraformaldehyde (PFA) and 0.1% TritonX 100 and then washed in PBS. Blocking of aspecific antigens was performed by incubating slides for 45 minutes with PBS containing 1% BSA and10% Fetal Calf Serum (FCS). Slides were washed with PBS and incubated overnight at 4ºC with the appropriately diluted primary antibody in PBS with 1% BSA and 0.1% TritonX 100. After incubation the slides were washed with PBS and incubated with the secondary FITC conjugated antibody (Dako, Denmark) 1:500 diluted in PBS. Slides were washed and mounted with DAPI (Roche, Mannheim, Germany)/vectashield (Vector laboratories Inc, Burlingame, CA, USA) 1:1000. The antibodies used and dilutions are summarized in Table 1.
Confocal microscopy on rat esophagitis-BE animal modelRat tissues were embedded in TissueTek® Optimum Cutting Temperature compound (Sakura Finetek, USA, Torrance, Calif.) at 20°C and sectioned using a cryomicrotome (Microm HM 550) in serial cryostat sections of 6 μm, placed on Superfrost + (Menzel Glaser, Braunschweig, Germany) glass slides and airdried overnight. For routine histological examination, of each block one slide was stained by hematoxylin and eosin. Other slides were used to perform fluorescent immunohistochemistry for BMP4, Smad 1/5/8 and ID2, as described above and examined by confocal microscopy using a Leica TCSSP2 filterfree Spectral Confocal Microscope (Heidelberg GmbH, Molecular Imaging Center, Bergen, Norway). A 40X magnifying objective was used with the numerical aperture (NA) at 1.25, type HCX PL fluotar.
RESULTS
Expression of BMP-4 and downstream targets in BE, squamous and inflamed squamous epithelium of patients.In a recently published SAGE analysis we found that BMP4 is 19 fold upregulated in BE compared to normal squamous epithelium.8 Validation by Western blot analysis of BE and normal squamous esophagus biopsies confirmed the SAGE data and showed a high expression of BMP4 on protein level in BE and a low expression in normal squamous esophagus. Importantly, we now found a high expression of BMP4 in the inflamed squamous epithelium (Figure 1). Western blot analysis demonstrated that Smad4 as well as BMP receptors: BMP RIA and BMP RII, were expressed in BE, in normal squamous epithelium and in esophagitis (Figure 1), yet ID2, a downstream BMP4 target, and PSmad 1/5/8 were only detectable in BE and in esophagitis but not in normal noninflamed squamous epithelium (Figure 1). Immunohistochemistry of biopsy specimens demonstrated the expression of BMP4, PSmad1/5/8 and ID2 in esophagitis and BE while there was no expression in normal squamous epithelium
Milano_opm_v5.indd 128 16-08-2007 15:43:10
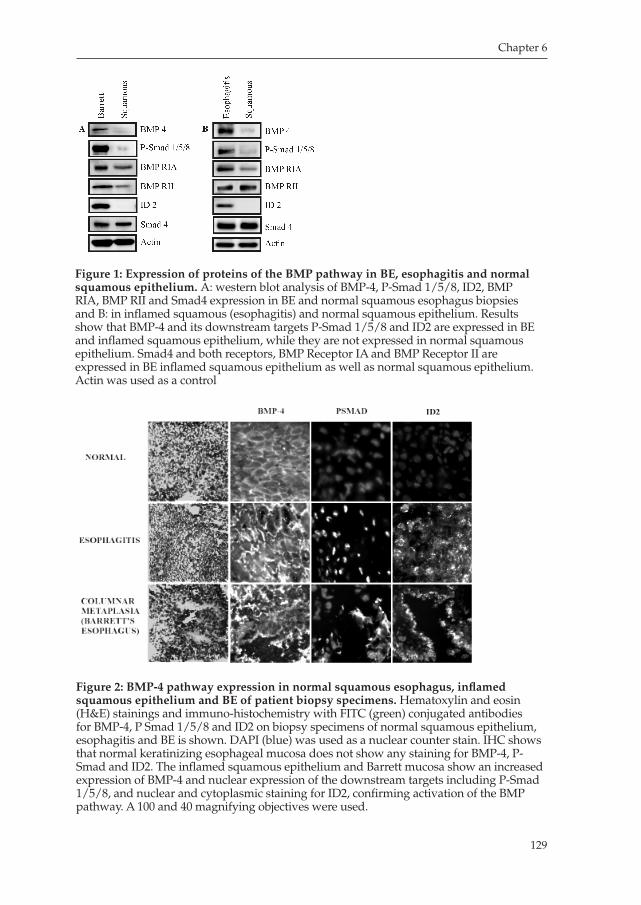
Chapter 6
129
Figure 1: Expression of proteins of the BMP pathway in BE, esophagitis and normal squamous epithelium. A: western blot analysis of BMP4, PSmad 1/5/8, ID2, BMP RIA, BMP RII and Smad4 expression in BE and normal squamous esophagus biopsies and B: in inflamed squamous (esophagitis) and normal squamous epithelium. Results show that BMP4 and its downstream targets PSmad 1/5/8 and ID2 are expressed in BE and inflamed squamous epithelium, while they are not expressed in normal squamous epithelium. Smad4 and both receptors, BMP Receptor IA and BMP Receptor II are expressed in BE inflamed squamous epithelium as well as normal squamous epithelium. Actin was used as a control
Figure 2: BMP-4 pathway expression in normal squamous esophagus, inflamed squamous epithelium and BE of patient biopsy specimens. Hematoxylin and eosin (H&E) stainings and immunohistochemistry with FITC (green) conjugated antibodies for BMP4, P Smad 1/5/8 and ID2 on biopsy specimens of normal squamous epithelium, esophagitis and BE is shown. DAPI (blue) was used as a nuclear counter stain. IHC shows that normal keratinizing esophageal mucosa does not show any staining for BMP4, PSmad and ID2. The inflamed squamous epithelium and Barrett mucosa show an increased expression of BMP4 and nuclear expression of the downstream targets including PSmad 1/5/8, and nuclear and cytoplasmic staining for ID2, confirming activation of the BMP pathway. A 100 and 40 magnifying objectives were used.
Milano_opm_v5.indd 129 16-08-2007 15:43:11

Chapter 6
130
(Figure 2). BMP4 expression in the inflamed esophagus and BE was typically localized in the stromal tissue, while PSmad 1/5/8 was localized in nuclei suggesting transcriptional activity (Figure 2). ID2, which is a transcription product of PSmad, is seen in the nuclei and cytoplasm of the epithelial cells (Figure 2).21
BMP-4 and downstream targets expression in BE, squamous and inflamed squamous epithelium in the BE rat model.Normal keratinizing squamous epithelium is found in the control rats (Figure 3). Reflux esophagitis and intestinal type of metaplasia of the mucosa, resembling BE, are found in the rats above and at the esophagojejunostomy site (Figure 3). Confocal analysis of immunohistochemical stainings of the fresh frozen material of the normal rat esophagus showed no expression of BMP4, PSmad 1/5/8 and ID2. BMP4 and the downstream BMP4 targets, PSmad 1/5/8 and ID2, were only seen in the inflamed esophagus and in the BE resembling epithelium (Figure 3). The pattern and localization of the expression of these factors were comparable to the patterns found in the human biopsy specimens as shown in Figure 2.
Figure 3: BMP-4 pathway expression in normal squamous esophagus, inflamed squamous epithelium and of the rat-BE model. The Hematoxylin and eosin (H&E) stainings and immunohistochemistry (IHC) using FITC (green) conjugated antibodies for BMP4, PSmad 1/5/8 and ID2 of the tissue from the rat esophagusBE model is shown. The left column shows normal nonkeratinizing squamous epithelium, inflamed squamous mucosa and columnar mucosa resembling BE. IHC shows keratinizing normal rat esophageal mucosa with negative staining for BMP4, and negative staining for the BMP4 downstream targets. The inflamed squamous epithelium and columnar epithelium show activation of the BMP4 pathway with increased expression of BMP4 and nuclear expression of the downstream targets PSmad 1/5/8 and ID2. DAPI (blue) was used as a nuclear counter stain. A 40X magnifying objective was used.
Milano_opm_v5.indd 130 16-08-2007 15:43:11

Chapter 6
131
BMP-4 activation in primary cultures of normal squamous esophageal cellsTimecourse incubation of primary cultured normal squamous cells with 100 ng/ml rec h BMP4 was performed. Western blot analysis showed that after 5 minutes of BMP4 incubation there are increased levels of PSmad 1/5/8 (Figure 4A). These phosphorylation levels were increased even more at 10 minutes and 20 minutes of incubation.
The untreated squamous cell cultures did not show any phosphorylation of Smad 1/5/8 (Figure 4A). Pretreatment of squamous cells with the BMP4 antagonist Noggin for 10 minutes decreased the phosphorylation level of Smad 1/5/8 to a basal level, indicating that the BMP pathway was blocked (Figure 4B).
Cytokeratin expression in cultured squamous cells before and after BMP-4 treatmentCK10 and CK13 are normally only expressed by squamous cells, while CK7 and CK20 are more specific for columnar types of cells such as BE.8 After 5 hours of treatment of squamous cells with BMP4, Western Blot analysis showed upregulation of expression of CK7 and CK20, while these cells were still expressing CK10/13 (Figure 5). Immunohistochemistry showed that the normal squamous cells do express CK10/13 but not CK7 or CK20, while the cultured Barrett cells express CK7 and CK20 but not CK10/13 (Figure 6). After 5 days of treatment of squamous epithelial cells with BMP4 the upregulation of CK7 and CK20 becomes clearly visible by immunohistochemistry. At this time point decreased expression of CK10/13 was also seen. (Figure 6).
Figure 4: P-Smad 1/5/8 expression in cell cultures stimulated with BMP-4. A: Western blot analysis of PSmad 1/5/8 in primary cultured Barrett and squamous esophageal cells. In cultured squamous cells there is no phosphorylation of Smad 1/5/8, while cultured Barrett cells show high levels of PSmad 1/5/8. Upon treatment of the squamous cell cultures with BMP4 for 5, 10 or 20 minutes PSmad 1/5/8 levels are upregulated. B: This upregulation in phosphorylation level of Smad 1/5/8 is effectively inhibited when the squamous cells are incubated with Noggin (a BMP antagonist). Actin was used as a control.
Milano_opm_v5.indd 131 16-08-2007 15:43:12
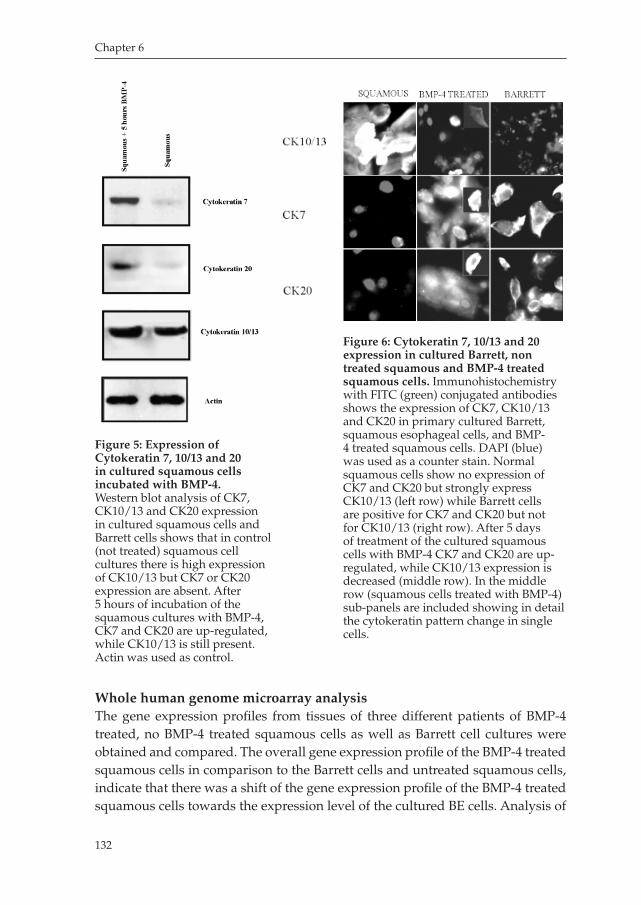
Chapter 6
132
Whole human genome microarray analysisThe gene expression profiles from tissues of three different patients of BMP4 treated, no BMP4 treated squamous cells as well as Barrett cell cultures were obtained and compared. The overall gene expression profile of the BMP4 treated squamous cells in comparison to the Barrett cells and untreated squamous cells, indicate that there was a shift of the gene expression profile of the BMP4 treated squamous cells towards the expression level of the cultured BE cells. Analysis of
Figure 5: Expression of Cytokeratin 7, 10/13 and 20 in cultured squamous cells incubated with BMP-4.Western blot analysis of CK7, CK10/13 and CK20 expression in cultured squamous cells and Barrett cells shows that in control (not treated) squamous cell cultures there is high expression of CK10/13 but CK7 or CK20 expression are absent. After 5 hours of incubation of the squamous cultures with BMP4, CK7 and CK20 are upregulated, while CK10/13 is still present. Actin was used as control.
Figure 6: Cytokeratin 7, 10/13 and 20 expression in cultured Barrett, non treated squamous and BMP-4 treated squamous cells. Immunohistochemistry with FITC (green) conjugated antibodies shows the expression of CK7, CK10/13 and CK20 in primary cultured Barrett, squamous esophageal cells, and BMP4 treated squamous cells. DAPI (blue) was used as a counter stain. Normal squamous cells show no expression of CK7 and CK20 but strongly express CK10/13 (left row) while Barrett cells are positive for CK7 and CK20 but not for CK10/13 (right row). After 5 days of treatment of the cultured squamous cells with BMP4 CK7 and CK20 are upregulated, while CK10/13 expression is decreased (middle row). In the middle row (squamous cells treated with BMP4) subpanels are included showing in detail the cytokeratin pattern change in single cells.
Milano_opm_v5.indd 132 16-08-2007 15:43:12
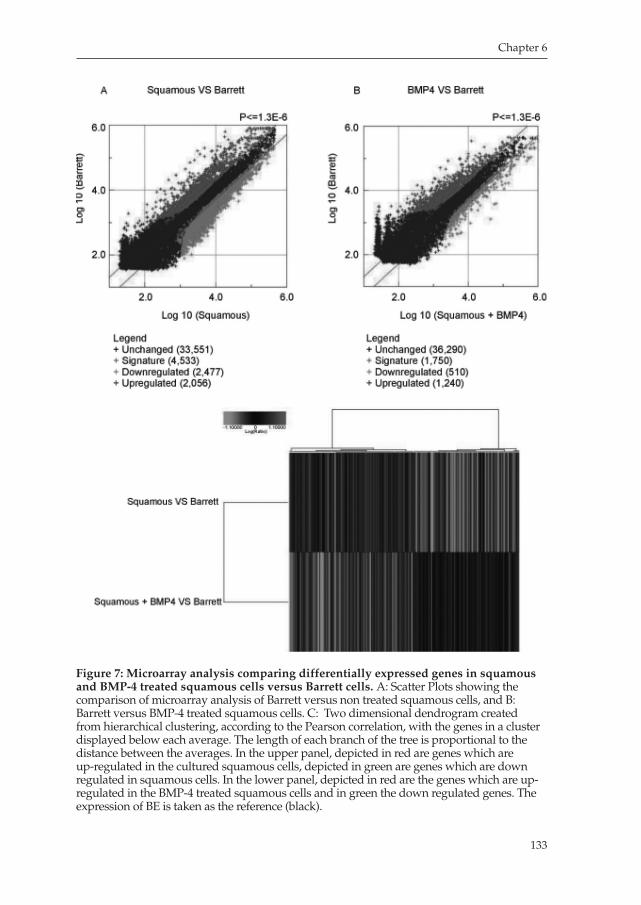
Chapter 6
133
Figure 7: Microarray analysis comparing differentially expressed genes in squamous and BMP-4 treated squamous cells versus Barrett cells. A: Scatter Plots showing the comparison of microarray analysis of Barrett versus non treated squamous cells, and B: Barrett versus BMP4 treated squamous cells. C: Two dimensional dendrogram created from hierarchical clustering, according to the Pearson correlation, with the genes in a cluster displayed below each average. The length of each branch of the tree is proportional to the distance between the averages. In the upper panel, depicted in red are genes which are upregulated in the cultured squamous cells, depicted in green are genes which are down regulated in squamous cells. In the lower panel, depicted in red are the genes which are upregulated in the BMP4 treated squamous cells and in green the down regulated genes. The expression of BE is taken as the reference (black).
Milano_opm_v5.indd 133 16-08-2007 15:43:13

Chapter 6
134
a subset of genes that are at least 2fold upor downregulated showed that there were on an average 11099 genes differentially expressed when comparing no BMP treated squamous versus Barrett cells, while on average 8226 differentially expressed genes were found when comparing BMP4 treated squamous versus BE cells. The decreased number of the at least 2fold differentially expressed genes as seen upon treatment of the squamous cells with BMP4 was statistically significant (Chi square test; p=0.0001). More strict statistical analysis of the 3 different microarray experiments showed that comparing the expression levels in the BE cells with non treated squamous cells revealed that 446 genes were significantly differently expressed, while the comparison of the BE cells with the BMP4 treated squamous cells showed that 392 genes were significantly differentially expressed, which was a decrease of 12% (ANOVA; p<0.01). Hierarchical clustering of the significantly differentially expressed genes, using Pearson’s correlation, showed that upon treatment of the squamous cells with BMP4, there was a shift towards the expression pattern which resembled BE cells. This is depicted in figure 7C that shows that genes highly expressed in the squamous cells (red in the upper panel) decrease in level of expression after BMP4 treatment (green in lower panel), while a set of genes which are not expressed in the squamous cells (green in the upper panel) gain expression in the BMP4 treated squamous cells (red/black, lower panel). Note that most of the induced genes in the BMP4 treated cells have a similar level (black) compared to BE cells.
DISCUSSION
In previous studies BMPs have been found to directly change the pathophysiology of certain inflammatory conditions.13, 14 Due to their chemotactic activity on inflammatory cells and fibroblasts, BMPs have been considered to influence inflammatory processes in adults.12 BMP4 is a protein belonging to the TGFβ protein family. So far, the involvement of members of the TGFβ family in BE has not been investigated in detail. With microarray analysis Barrett et al. found that a TGFβ superfamily protein was one of the genes that was upregulated in BE.22 With the use of SAGE, we recently found that BMP4 is exclusively expressed in BE and absent in normal squamous epithelium.8 Since BE is associated with chronic inflammation as a result of reflux of gastric contents damaging the esophageal mucosa, we hypothesized that these inflammatory changes could induce the production of BMP4, which subsequently triggers transdifferentiation of squamous epithelial cells into a columnar cell type, resembling metaplastic Barrett mucosal cells, that replaces the normal squamous mucosa.
Milano_opm_v5.indd 134 16-08-2007 15:43:13

Chapter 6
135
In this study, we provide evidence which supports that BMP4 might hasve an important role in the process of transformation of squamous epithelium towards a mucosa, which resembles columnar metaplasia and that this process is initiated in the inflamed squamous mucosa. Using Western blot and immunohistochemistry we found that BMP4 and its downstream targets, PSmad 1/5/8 and ID2 are present in patient biopsies from BE and squamous epithelium in the area of esophagitis, but not in normal non inflamed squamous esophageal mucosa (Figure 1 and 2). This indicates that indeed in inflamed esophageal mucosa, BMP4 is upregulated and its downstream pathway is activated. The finding of BMP receptors (RIA and RII) expression in normal squamous esophageal tissue supports the hypothesis that under certain conditions the BMP pathway can be activated in squamous epithelium. Analysis of the tissues obtained from a rat model in which reflux esophagitis and subsequently BE is induced through chronic reflux of gastroduodenal contents into the esophagus, confirmed that the BMP pathway is activated in both the inflamed esophageal mucosa and BE. This was illustrated by an increased expression of BMP4 and the downstream targets PSmad and ID2 (Figure 3). We further investigated in an ex-vivo model whether the BMP pathway was active in BE mucosa and whether BMP4 could induce dedifferentiation of cultured squamous cells into a columnar cell type that resembles BE. To this end we established short term primary cultures of normal squamous epithelial cells and Barrett cells from patient biopsies. Treatment of squamous epithelial cells with recombinant human BMP4 for 5, 10 or 20 minutes showed expression of PSmad 1/5/8, that increased in time, while the control squamous cell cultures did not show any expression level of activated Smad proteins (Figure 4A). Blocking the pathway by preincubating squamous cells with the BMP antagonist Noggin before treatment with BMP4, showed a decreased phosphorylation level of Smad 1/5/8 (Figure 4B). From these ex-vivo experiments we conclude that the BMP pathway can be activated in normal squamous cell cultures and that this effect can be inhibited by Noggin.
To further study the transdifferentiation process of the squamous epithelium into columnar metaplasia, we examined the CK expression profiles in BMP stimulated and non stimulated squamous cells. The CK expression is known to be specific depending on type, location and differentiation of the epithelium and is therefore important for characterizing certain epithelia, for instance characterizing the difference between BE and normal squamous cells.8, 2326 CK7 and CK20 are known to be expressed in BE and other columnar types of mucosa but not in normal squamous mucosa. In contrast CK10/13 is expressed in normal squamous mucosa but not in BE or other columnar types of mucosa .8, 27, 28 After 5 hours of incubation of squamous cell cultures with BMP4, Western blot analysis revealed that there was upregulation of CK7 and CK20, while CK10/13 was still present in these cells (Figure 5). After 5 days treatment of squamous epithelium with BMP
Milano_opm_v5.indd 135 16-08-2007 15:43:14

Chapter 6
136
4, CK7 and CK20 induction could be demonstrated by immunohistochemistry, whereas nontreated squamous cells did not express these CKs. At this point we noted a decreased CK10/13 expression in the BMP4 treated cells (Figure 6). From these results we can conclude that treatment with BMP4 induces a shift in the CK expression pattern towards a CK expression pattern that resembles that of BE. To further investigate to what extent a columnar type of mucosa is induced by BMP4, whole human genome arrays were used to compare the total gene expression profiles of primary BE cell cultures with the BMP4 treated and non treated squamous cell cultures. A remarkable change of the gene expression profile was seen of the BMP4 treated squamous cell cultures (Figure 7). When taking into account those genes that were more abundantly (> 2fold) up or downregulated, the overall number of differentially expressed genes diminished by 26%. Yet, using multivariate analysis for the three experiments and only take into account those genes that were significantly differentially expressed between the squamous cultured cells and the BMP4 treated squamous cells, with respect to expression levels in the BE cells, we observed a reduction of 12%. To further demonstrate that BMP4 down regulates a set of squamous specific genes and induces expression of a set of genes related to columnar metaplasia, we performed hierarchical clustering using Pearson’s correlation. From this analysis we could conclude that after BMP treatment of the squamous cells, within the significantly differentially expressed genes, there is a set of ‘squamous’ specific genes that is significantly reduced expressed while the expression of a set of ‘novel’ genes is induced. Most of the induced genes have expression levels that are similar to expression levels in the Barrett cells, but few genes have a higher expression than what is seen in the BE cells (Figure 7C). The latter phenomenon may be explained by the experimental conditions in which the level of BMP stimulation may be beyond physiological levels and counteracting effects and feed back mechanisms that normally may be present in the in vivo situation were lacking.
In summary, we made a novel observation that BMP4 plays an potentially important role in the process of metaplasia as occurs in the distal esophagus in which normal squamous epithelial cells are dedifferentiated into columnar type of cells. We demonstrated that the BMP pathway is activated in inflamed esophageal squamous epithelium to an extent, which is similar to what was seen in the columnar epithelium of both human and rat tissues and that the BMP4 pathway can be blocked effectively with Noggin, a specific BMP antagonist. In our ex-vivo experiments the dedifferentiation of normal squamous cells towards a columnar cell type as induced by BMP4, which was particularly illustrated by changes of the CK expression patterns. This was also illustrated by a shift in the gene expression profile of the BMP4 treated squamous cells that towards that of Barrett epithelial cells. Our findings suggest that BMP4 is involved in initiating the process of transformation of normal squamous esophageal mucosa into a
Milano_opm_v5.indd 136 16-08-2007 15:43:14

Chapter 6
137
columnar type of cells. Once we better understand the mechanisms involved in the process of dedifferentiation initiated by BMP4, manipulation of the BMP pathway may help us to prevent BE and subsequently the highly malignant BE associated esophageal adenocarcinoma.
ACKNOWLEDGEMENTS:
The authors would like to thank Arnold Spek (Laboratory of Experimental Internal Medicine, AMC, Amsterdam, the Netherlands) and Raymond Waaijer (Bioinformatics department, AMC, Amsterdam, the Netherlands) for their support with the microarray analysis.
Milano_opm_v5.indd 137 16-08-2007 15:43:14

Chapter 6
138
REFERENCES
1. Devesa SS, Blot WJ, Fraumeni JF, Jr. Changing patterns in the incidence of esophageal and
gastric carcinoma in the United States. Cancer 1998;83:204953.
2. Shaheen NJ. Advances in Barrett Esophagus and esophageal adenocarcinoma. Gastroentero
logy 2005;128:155466.
3. Bytzer P, Christensen PB, Damkier P, Vinding K, Seersholm N. Adenocarcinoma of the eso
phagus and Barrett Esophagus: a populationbased study. Am J Gastroenterol 1999;94:8691.
4. Ransford RA, Jankowski JA. Genetic versus environmental interactions in the oesophagitis
metaplasiadysplasiaadenocarcinoma sequence (MCS) of Barrett oesophagus. Acta Gastroen
terol Belg 2000;63:1821.
5. Locke GR, 3rd, Talley NJ, Fett SL, Zinsmeister AR, Melton LJ, 3rd. Prevalence and clinical
spectrum of gastroesophageal reflux: a populationbased study in Olmsted County, Minne
sota. Gastroenterology 1997;112:144856.
6. Winters C, Jr., Spurling TJ, Chobanian SJ, Curtis DJ, Esposito RL, Hacker JF, 3rd, Johnson DA,
Cruess DF, Cotelingam JD, Gurney MS, et al. Barrett Esophagus. A prevalent, occult complica
tion of gastroesophageal reflux disease. Gastroenterology 1987;92:11824.
7. Mann NS, Tsai MF, Nair PK. Barrett Esophagus in patients with symptomatic reflux esophagi
tis. Am J Gastroenterol 1989;84:14946.
8. van Baal JW, Milano F, Rygiel AM, Bergman JJ, Rosmolen WD, van Deventer SJ, Wang KK,
Peppelenbosch MP, Krishnadath KK. A comparative analysis by SAGE of gene expression
profiles of Barrett Esophagus, normal squamous esophagus, and gastric cardia. Gastroentero
logy 2005;129:127481.
9. Wall NA, Hogan BL. TGFbeta related genes in development. Curr Opin Genet Dev
1994;4:51722.
10. Kingsley DM, Bland AE, Grubber JM, Marker PC, Russell LB, Copeland NG, Jenkins NA. The
mouse short ear skeletal morphogenesis locus is associated with defects in a bone morphoge
netic member of the TGF beta superfamily. Cell 1992;71:399410.
11. Hollnagel A, Oehlmann V, Heymer J, Ruther U, Nordheim A. Id genes are direct targets of bone
morphogenetic protein induction in embryonic stem cells. J Biol Chem 1999;274:1983845.
12. Rosendahl A, Pardali E, Speletas M, Ten Dijke P, Heldin CH, Sideras P. Activation of bone
morphogenetic protein/Smad signaling in bronchial epithelial cells during airway inflamma
tion. Am J Respir Cell Mol Biol 2002;27:1609.
13. Kaiser S, Schirmacher P, Philipp A, Protschka M, Moll I, Nicol K, Blessing M. Induction of
bone morphogenetic protein6 in skin wounds. Delayed reepitheliazation and scar formation
in BMP6 overexpressing transgenic mice. J Invest Dermatol 1998;111:114552.
14. Frank S, Madlener M, Werner S. Transforming growth factors beta1, beta2, and beta3 and
their receptors are differentially regulated during normal and impaired wound healing. J Biol
Chem 1996;271:1018893.
15. Hubner G, Hu Q, Smola H, Werner S. Strong induction of activin expression after injury sug
Milano_opm_v5.indd 138 16-08-2007 15:43:14

Chapter 6
139
gests an important role of activin in wound repair. Dev Biol 1996;173:4908.
16. Washington K, Gottfried MR, Telen MJ. Tissue culture of epithelium derived from Barrett
oesophagus. Gut 1994;35:87983.
17. PalancaWessels MC, Barrett MT, Galipeau PC, Rohrer KL, Reid BJ, Rabinovitch PS. Genetic
analysis of longterm Barrett Esophagus epithelial cultures exhibiting cytogenetic and ploidy
abnormalities. Gastroenterology 1998;114:295304.
18. Buttar NS, Wang KK, Leontovich O, Westcott JY, Pacifico RJ, Anderson MA, Krishnadath KK,
Lutzke LS, Burgart LJ. Chemoprevention of esophageal adenocarcinoma by COX2 inhibitors
in an animal model of Barrett Esophagus. Gastroenterology 2002;122:110112.
19. Buttar NS, Wang KK, Anderson MA, Dierkhising RA, Pacifico RJ, Krishnadath KK, Lutzke
LS. The effect of selective cyclooxygenase2 inhibition in Barrett Esophagus epithelium: an in
vitro study. J Natl Cancer Inst 2002;94:4229.
20. Pera M, Trastek VF, Carpenter HA, Fernandez PL, Cardesa A, Mohr U, Pairolero PC. Influen
ce of pancreatic and biliary reflux on the development of esophageal carcinoma. Ann Thorac
Surg 1993;55:138692; discussion 13923.
21. Liu W, Oh SH, Kang Yk Y, Li G, Doan TM, Little M, Li L, Ahn K, Crenshaw EB, 3rd, Frenz
DA. Bone morphogenetic protein 4 (BMP4): a regulator of capsule chondrogenesis in the
developing mouse inner ear. Dev Dyn 2003;226:42738.
22. Barrett MT, Yeung KY, Ruzzo WL, Hsu L, Blount PL, Sullivan R, Zarbl H, Delrow J, Rabino
vitch PS, Reid BJ. Transcriptional analyses of Barrett metaplasia and normal upper GI muco
sae. Neoplasia 2002;4:1218.
23. Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: pat
terns of expression in normal epithelia, tumors and cultured cells. Cell 1982;31:1124.
24. Moll R. Cytokeratins in the histological diagnosis of malignant tumors. Int J Biol Markers
1994;9:639.
25. Quinlan RA, Schiller DL, Hatzfeld M, Achtstatter T, Moll R, Jorcano JL, Magin TM, Franke
WW. Patterns of expression and organization of cytokeratin intermediate filaments. Ann N Y
Acad Sci 1985;455:282306.
26. Southgate J, Harnden P, Trejdosiewicz LK. Cytokeratin expression patterns in normal and ma
lignant urothelium: a review of the biological and diagnostic implications. Histol Histopathol
1999;14:65764.
27. Gulmann C, Shaqaqi OA, Grace A, Leader M, Patchett S, Butler D, Kay E. Cytokeratin 7/20
and MUC1, 2, 5AC, and 6 expression patterns in Barrett Esophagus and intestinal metaplasia
of the stomach: intestinal metaplasia of the cardia is related to Barrett Esophagus. Appl Im
munohistochem Mol Morphol 2004;12:1427.
28. Ormsby AH, Goldblum JR, Rice TW, Richter JE, Falk GW, Vaezi MF, Gramlich TL. Cytoke
ratin subsets can reliably distinguish Barrett Esophagus from intestinal metaplasia of the
stomach. Hum Pathol 1999;30:28894.
Milano_opm_v5.indd 139 16-08-2007 15:43:15

Milano_opm_v5.indd 140 16-08-2007 15:43:15

Chapter 7
Summary, General discussion
and future perspective
Milano_opm_v5.indd 141 16-08-2007 15:43:15

Summary, general discussion and future perspective
142
Summary
Esophageal Adenocarcinoma (EA) is an extremely aggressive cancer, with the most rapidly rising incidence in the Western World. At presentation approximately half of the patients with EA have aggressive and incurable disease, and the other half will undergo surgery. Despite surgical with or without (neo) adjuvant treatment this patient group has a poor prognosis with a five years survival rate of less than 20%. For this reason, alternative and more efficient treatment strategies are urgently requested. Immunotherapy using Dendritic Cells (DC) has been used for treating several aggressive cancers, such as melanoma and prostate cancer. The principle of the therapy is similar to that of conventional antiviral vaccines, using DCs as the vehicles. These are the most potent antigen presenting cells (APC) of the immune system, with the unique ability to induce a primary immune response, thus permitting establishment of immunological memory. DCs are highly capable of inducing Tcell responses, and the interaction of these two cell types, is determinant for the type of immune response that will be raised. The capacity of the DCs to stimulate Tcells depends on many factors, such as surface expression of costimulatory molecules, and the cytokine microenvironment. Functional DCs can be generated from human peripheral blood monocytes and be further matured into DCs that in turn can be used as vaccines for treating malignancies. To generate a cytotoxic Tcell (CTL) response against tumor cells, specific tumor antigens have to be presented to Tlymphocytes by immunoactivatory DCs. Different antigens, such as synthetic peptides, tumor lysates, and cDNA or RNA encoding for specific tumorassociated antigens as well as total tumor mRNA, have been used and tested for their immunopotency. DC immunotherapy has been used in many clinical trials for treating cancer patients, particularly for melanoma and prostate cancer. Although DC therapy holds a promise for the future as a potent patient tailored approach for curing cancer, there are several caveats in this field. To further optimize DC therapy for treating EA patients, this thesis addressed several of these difficulties. Moreover, two important novel protocols aiming on more potent DCs and for developing an improved exvivo read out system have been established. In addition, more insight has been gained in the immunogeneic tumor environment, and finally an important combinatorial treatment regiment has been explored.
The first part of the thesis describes a novel method that was used to obtain mature antigen presenting immunostimulatory DCs (chapter 2). Here the novelty is the use of monocytes, freshly isolated from peripheral blood, as compared to immature DCs, classically used for the loading of antigens, for the preparation of the vaccines. Of interest is that the yield of mature DCs finally presenting antigens, still after 8 days by using monocytes, is significantly higher as compared to the use of immature DCs, which are as well less potent, as shown
Milano_opm_v5.indd 142 16-08-2007 15:43:15

Summary, general discussion and future perspective
143
in the paper. The electroporated monocytes that matured into DCs, were tested for their immunophenotype, antigen presentation capacity, migration properties, and Tcell stimulation ability, and resulted to be of higher quality as compared to immature DCs used for loading of antigens. This method that resulted to be valid to produce mature immunopotent DCs, was applied to patient material in chapter 3. In this chapter, the most important novelty is the establishment of a novel read out system for testing immune reponses (T cell response). The model consists of primary cultures established from fresh biopsies isolated from EAC patients. Here, monocytes freshly isolated from EAC patients, were loaded with total tumor RNA isolated from biopsies, as well as normal RNA (for control purposes), which were as well used for obtaining the primary cultures. These monocytes were stimulated by different cytokines to be matured into DCs, and subsequently cocultured with autologous lymphocytes for obtaining a population of lymphocytes containing several CTL populations against different tumor antigens. In the mean while the primary cultures were grown, and then cocultured with the lymphocytes obtained in the previous phase. The response of the lymphocytes stimulated by either normal or tumor RNA loaded DCs, was observed particularly against autologous tumor cells, but not against normal cells, demonstrating that, first, this strategy is valid to monitor CTL responses against tumor cells, that can be tested exvivo in primary cell cultures. Furthermore, this approach does not induce adverse undesirable effects. This system can be used as a valid read out for preclinical evaluation of tumor cells response and could be used to predict efficacy of the DC vaccines. In the next part of the thesis, chapter 4, another important issue has been remarked: so far, the outcomes of patient treated by DC immunotherapy are extremely disappointing and below expectations. One of the reasons, which has been addressed for this phenomenon, is the unfavorable tumor microenvironment, which is often characterized by the presence of molecules, such as tumor growth factors and immunosuppressive cytokines, which inhibits the immuneresponse raised against cancer. This concept prompted us to gain insight in the EAC microenvironment, to be able to find out whether in this cancer, as in many others, the immuneresponse can be hindered by the presence of factors, which are immunosuppressive. In this study, it was demonstrated that EAC, as many other cancer, is characterize by the expression of several molecules, such as COX2, VEGF, TGFβ, IL8, IL1β, and many others, which exert either immunosuppressive function or tumor growth functions. These findings are of interest to establish, for instance, combinatorial treatments regiments using DCs immunotherapy, and targeting one or more of these molecules, in order to enhance the efficacy of the DC approach, and to increase patient outcomes. One other approach which is interesting for enhancing the efficacy of DCs immunotherapy is the use of this approach in combination with drugs, such as
Milano_opm_v5.indd 143 16-08-2007 15:43:16

Summary, general discussion and future perspective
144
monoclonal antibodies, directed against tumor antigens. One very interesting example is the monoclonal antibody directed against the tumor antigen HER2, which is highly overexpressed, mainly via gene amplification, in several aggressive cancers. This antibody has already been used as treatment for breast cancer patients, and it has shown to have tumor growth inhibitory effects both in vitro and in vivo. EAC patients present HER2 overexpression in 30 to 80% of the cases. For this reason, the approach of combining DC immunotherapy and the use of Trastuzumab was used and this is described in chapter 5. In this chapter, three different EAC cell lines, OE33, OE19 and Bic1, were first characterized for the HER2 status by FISH and ICC, and used to study the biological effect of Trastuzumab by performing an MTT assay. Once established that OE33 and OE19 were HER2 positive, and Bic1 HER2 negative, and that Trastuzumab exerted highly inhibiting effects on OE33 and OE19 (HER2 positive), but not on Bic1 (HER2 negative), a cytotoxicity assay was performed to assess whether the use of Trastuzumab could enhance the cytotoxic response induced by HER2 specific CTLs against the two HER2 overexpressing EAC cell lines. It is known indeed that Trastuzumab exerts its function through different mechanisms, which are basically induction of cell growth, activation of pathway which are controlling cell apoptosis, but also mechanism which are enhancing the immunesystem, such as Antibody Depend Cellmediated Cytotoxicity. It is hypothesized that Trastuzumab induces receptor internalization and degradation, therefore, increasing the volume of antigens loaded onto the MHC class I complex. This would induce an enhancement of the epitopes presented to the CTLs. EAC cell lines were treated with Trastuzumab. At the same time, HLAA2 matched donors were used to isolate monocytes to load them with HER2 RNA, mature these into potent APCs to stimulate the autologous lymphocytes and obtain HER2 specific CTLs. These were subsequently coincubated with the three cell lines either pretreated or not treated with Trastuzumab. In addition, PBMCs from EAC patients with HER2 amplification were tested to investigate whether these patients could present self circulating HER2 specific CTLs able to respond to the Trastuzumab pretreated cancer cell lines. It was remarkable to note that the percentage of cytotoxicity mediated by the HER2 specific CTLs was significantly higher in the HER2 positive cell line OE33 as compared to the HER2 negative Bic1, and this effect was even more significantly different when cells were pretreated with Trastuzumab. In addition, PBMCs isolated form EAC patient, which present HER2 amplification in their cancer induced a significantly higher cytotoxicity effect on the cell line OE33 as compared to PBMCs isolated by EAC patients without HER2 amplification. In the last part of the thesis there is more focus on the genesis of EA. In chapter 6 the role of a factor, the Bone Morphogenetic Protein4 (BMP4) is described. This gene was previously found to be significantly higher overexpressed in BE as compared to normal tissues by
Milano_opm_v5.indd 144 16-08-2007 15:43:16

Summary, general discussion and future perspective
145
Serial Analysis of Gene Expression. BE is a premalignant condition of the distal esophagus and consist in a metaplastic change of the normal lined squamous epithelium into a columnar type of epithelium, as a result of chronic longstanding gastroesophageal reflux disease (GERD). Patients with BE have a significantly increased risk to develop EAC. In the last 3 decades, the incidence of BE and its associated EAC has increased and exceeds that of other malignancies. Therefore, when studying EAC it is as well of interest to gain further insight in the mechanism of development of BE which is the cause of EAC. In this chapter, we investigated whether BMP4 and BMP pathway plays a role in the metaplastic transformation of normal squamous epithelium towards a columnar type of mucosa. To this aim, tissues from patients and an esophagitisBE rat model were employed and investigated. Results showed that the BMP pathway is activated in esophagitis and BE. In addition, primary cell cultures of normal keratinocytes, obtained from biopsies, treated with BMP4 showed activation of the BMP pathway by upregulation of PhosphoSmad 1,5,8 and ID2 (BMP downstream targets). This effect could be blocked by pretreatment with Noggin, an antagonist of BMP. In addition, the cytokeratin expression patterns and gene expression profiles were examined. BMP4 treatment of keratinocytes resulted in shift of the cytokeratin expression pattern towards that of BE. In terms of gene expression, BMP4 treatment resulted in a shift in the gene expression pattern of the normal keratinocytes of 26% towards that of BE. These results suggested that BMP4 is involved in the process of transformation of the normal squamous epithelium into a columnar type of epithelium.
General discussion and future perspectives
The work described in this thesis focuses particularly on finding alternative and novel stratagems for treating esophageal adenocarcinoma patients and to improve the survival of this group of patients, which is considerably short and modest. Several approaches have been investigated in the past, including adjuvant and neoadjuvant therapies, such as chemotherapy and radiotherapy, and the combination of these with surgery. The major problem for these patients is that at presentation they often carry an already advanced disease, therefore leaving extremely low prospective of life. This is the reason why the achievement of this thesis was to explore innovative approaches to create different more effective strategy and improve the life expectancy for this patient population. One tremendously appealing strategy that has been applied as an attempt to cure several aggressive cancer is DC immunotherapy. Therefore this was studied in this thesis, and several findings have been accomplished. Important findings are reported about the establishment of an efficient method to obtain sufficient
Milano_opm_v5.indd 145 16-08-2007 15:43:17

Summary, general discussion and future perspective
146
amount of antigen presenting DC using total RNA as loading strategy. This method would overcome the enormous amount of cell lost which is determined by the laborious protocol that is currently used to prepare mature antigen presenting DCs evivo. Using our approach, in which monocyte are first electroporated and then further matured towards DCs, we were able to obtain higher yields immunestimulatory DCs as compared to the most used approach of loading immature DC. This is of importance since one of the difficulties correlated to this therapy is the possibility to obtain sufficient amount of DCs to be given to patients. This approach is effective as well when using esophageal cancer patients monocytes, which result in satisfactory amounts of immunopotent DC. The effectiveness of these cells to induce an immune response was tested exvivo using autologous primary cultures. This approach gives the remarkable possibility of testing the potential patient response in vivo, by using a novel autologous exvivo system. The feasibility of this autologous read out system to evaluate cytotoxicity responses against esophageal cancer cells is reported, next to the necessity to evaluate the concern of breaking tolerance to selfantigens, when immunizing with total tumor RNA. The use of non autologus systems can indeed induce to over evaluation of the possible results when looking at immune responses, therefore, an autologous read out might be of use, especially as preclinical tool to predict potential immuneresponse, or adverse effect, in a patient tailored approach. Numerous studies attempted at the development of very effective and safe DC vaccines, and this was partly successfully reported by several groups. However, patient who underwent this treatment did not show remarkable outcomes up to now, inducing the disappointment of the researcher and clinicians. The expectancy in terms of anticancer immune responses was significantly higher, considering the abundant highly promising results obtained in vitro, in terms of immuneresponse. Therefore it was required to address an explanation for this phenomenon. Finally few research groups emerged with a very essential issue: the tumor microenvironment, constituted by a complex network of cytokines, growth factors, and different cell types, is determinant for an appropriate immuneresponse in an immunocompetent host. These reports prompted to novel studies, based on a deeper and more profound understanding of the interactions, and the mutual influences, of cells, cytokines, and growth factors. In this thesis the answer to this issues, regarding particularly the esophageal adenocarcinoma, is given by the description of expression of several cytokines, growth factors, angiogenetic factors, and immunosuppressive factors which characterize the tumor surrounding in esophageal cancer patients. This part of the study provides an overview of the most important cytokines and tumor growth factors pattern of expression in esophageal cancer, and therefore can be of use for introducing novel approach based on targeting these molecules in order to overcome the immunosuppressive effect that they mediate. Despite
Milano_opm_v5.indd 146 16-08-2007 15:43:17

Summary, general discussion and future perspective
147
many efforts and several important achievements, the field of immunotherapy needs to be further explored. In particular, several issues has to be taken into account, like, for instance, the discrepancy between the in vitro data, highly promising, and the clinical outcomes, still below expectations, and limited to a small amount of patients. Further studies will have to be performed to establish to which extent the tumor microenvironment interferes with the functions of the immune system, permitting tumor escape from immunity. Therefore, more evaluation will be carried out. In future approaches, it is proposed to focus the attention not only towards one single strategy, but the combination of different stratagems, which could lead to more satisfactory results. Nowadays several approaches have been combined, like, for instance, surgery with chemo, and/or radiotherapy, and surgery with drugs, such as antibody and small molecules. To this point, we conducted a study in order to establish if such a strategy could be of interest, and, above all, applicable, to esophageal adenocarcinoma, therefore providing another alternative possibility of cure for this cancer. The use of antibodies in clinics is currently a common approach, based on targeting particular molecules which characterizes certain type of cancers. The HER2 gene is a very well know oncogene, which is already used as target for the therapy of HER2 overexpressing cancer, such as breast cancer. Esophageal cancer part of this group, with 50% of the cases bearing HER2 gene amplification, would result as a suitable candidate for such a strategy. Targeting HER2 with the use of a specific antibody of HER2, Trastuzumab, which represent a therapy for HER2 overexpressing breast cancers, was tested in vitro in esophageal cancer cells, used in combination with DC immunotherapy. Encouraging results elucidated the potential role of the antibody, in determining an improvement of Tcell responses against cancer cells mediating an Antibody Dependent Cellular Cytotoxicty mechanism. The mechanism of action of this particular antibody is not yet well understood; therefore further research is needed in order to achieve a more profound comprehension of this type of therapy, in terms of efficacy, but as well safety, for patients. Other similar approaches have been investigated, and these are the use of small molecules, such as tyrosine kinase inhibitors. Of interest is the fact that those have been already tested, with perhaps hopeful results, although one common features of this type of approach, is the heterogeneity of the patients response. Based on these observations, it is anticipated that in the future, curative treatment for cancer will become patient tailored, to ensure a more successful result. This concept is of high interest, because is taking into account the extreme diversity of patient even though belonging to the same group of disease.
Of outmost importance is certainly the exploration of earlier phases of this type of cancer development, which lead to the final aggressive adenocarcinoma, and this is the awareness of the Barrett’s metaplasia. It is still not know what the exact molecular mechanism behind the development of the Barrett’s metaplasia
Milano_opm_v5.indd 147 16-08-2007 15:43:18

Summary, general discussion and future perspective
148
is and the consequent progression to a malignant disease. The last part of this thesis is a “back to the origin” investigation, which focused on the role of Bone Morphogenetic Protein 4, a morphogen found to be overexpressed in Barrett’s esophagus as compared to normal controls tissues, in the transformation from the normal squamous to a columnar type of epithelium characteristic of Barrett’s eosphagus. It was ruled out that this molecule is certainly involved in this process of transformation towards a unspecificed columnar type of epithelium, certainly in the primary stages, but still a brick is missing in the wall: what determines the final transformation to a specialized columnar type of epithelium, such as the one characteristic of Barrett’s esophagus? This point needs to be clarified by performing additional studies in order to further delineate the molecular mechanism lying behind the development of Barrett’s esophagus.
This work provides a number of answers, and offer some advances, but consequently enlightens new questions, contributing to the intriguing neverending search of answers and progresses and prompting further investigations to improve patients outcomes when attempting to combat cancer diseases.
Milano_opm_v5.indd 148 16-08-2007 15:43:18

Appendices
Milano_opm_v5.indd 149 16-08-2007 15:43:18

Samenvatting
150
Samenvatting
Adenocarcinoma van de slokdarm is een extreem agressieve kanker en kent, in de Westerse wereld althans, de snelst stijgende incidentie van alle kankers. Op dit moment blijft in ongeveer de helft van alle patiënten met adenocarcinoom van de slokdarm de ziekte onbehandelbaar terwijl de andere helft operatief wordt behandeld. Desalniettemin kent ook dit laatste cohort een slechte prognose, met een vijfjaars overleving na diagnose van minder dan 20 % in deze patiëntengroep. Het moge duidelijk zijn dat we dringend behoefte hebben aan nieuwe meer efficiënte behandelwijzen.
Immunotherapie met dendritische cellen zou zo’n nieuwe behandelwijze kunnen vormen. In principe lijkt dergelijke therapie veel op conventionele vaccinatie. Dendritische cellen presenteren lichaamsvreemde componenten aan het immuunsysteem en hebben de unieke mogelijkheid om vervolgens een reactie van het immuunsysteem tegen deze lichaamsvreemde eiwitten op te wekken. Als dendritische cellen zo gek te krijgen zouden zijn dat ze componenten van de tumor aan het immuunsysteem zouden presenteren, dan zou het antwoord van het immuunsysteem zich vervolgens tegen de kanker richten en zou de ziekte door het lichaam zelf worden bestreden. Op deze manier zouden dendritische cellen als tumor vaccins kunnen dienen.
Het maken van dergelijke vaccins is echter niet zo simpel. Adenocarcinoma van de slokdarm verschilt van samenstelling van patiënt tot patiënt, wat het ontwikkelen van dergelijke vaccins niet helpt. Bovendien is nog slecht uitgezocht hoe men het effectiefst dendritische cellen aanzet om tumor materiaal aan het immuun systeem te presenteren. Ook is de lokale immunologische omgeving van adenocarcinoma in de slokdarm nog maar slecht in kaart gebracht, terwijl kennis hiervan noodzakelijk is om effectieve antikanker vaccins te ontwikkelen. Tenslotte weten we nog maar weinig hoe het combineren van verschillende behandelingen ons kan helpen bij het bestrijden van slokdarmkanker. Het onderzoek beschreven in dit proefschrift was opgezet deze barrières tegen het ontwikkelen van effectieve antikanker vaccinatie uit de weg te ruimen en heeft, nu aan het eind van de rit, er toe geleid dat de toekomst van een dergelijke strategie er rooskleurig uitziet.
Effectieve productie van verbeterde dendritische cellenEen eerste hindernis op weg naar effectieve vaccinatie tegen slokdarmkanker vormt het verkrijgen van effectieve dendritische cellen. In hoofdstuk 2 wordt deze barrière genomen. Dendritische cellen ontstaan uit de opeenvolgende ontwikkeling van monocyten (een celtype dat in grote aantallen in ons bloed aanwezig is) naar onvolgroeide dendritische cellen om uiteindelijk de volwassen dendritische immuunstimulatoire effector cellen te vormen. Klassiek werden
Milano_opm_v5.indd 150 16-08-2007 15:43:18

Samenvatting
151
voor het maken van kanker vaccins altijd onvolgroeide dendritische cellen opgeladen met tumorcomponenten, na doorgroei tot volwassen dendritische cellen zouden deze componenten dan aan het immuunsysteem gepresenteerd worden. In dit hoofdstuk laat ik zien dat het opladen van monocyten een veel effectievere strategie is, wat de basis vormt voor het verdere werk beschreven in dit proefschrift.
Dendritische cellen zijn in staat om in het immuunsysteem een anti-slok-darmkanker antwoord op te wekkenIn hoofdstuk 3 worden dendritische cellen gemaakt uit het bloed van slokdarmkanker patiënten en worden deze cellen opgeladen met tumor materiaal uit dezelfde patiënt, waarna buiten het lichaam het vermogen van deze cellen om een antikanker antwoord van het immuunsysteem op te wekken wordt getest (met behulp van immuun cellen en cultures van het adenocarcinoma van de patiënt). Het blijkt dat de dendritische cellen krachtige antitumor activiteit opwekken, de slokdarmkanker kweken worden efficiënt gedood door de met dendritische cellen gestimuleerde immuuuncellen. Kweken van normale slokdarm van deze patiënten worden echter met rust gelaten. Deze proeven vormen een bewijs dat antitumor vaccins in ieder geval onder laboratorium omstandigheden werken.
Hoe zit het met de tumor omgeving?Eerdere experimenten met antikanker vaccins waren teleurstellend. Vaak wordt gesuggereerd dat de tumor een omgeving voor zich zelf creëert die ongunstig zou zijn voor het immuunsysteem. Deze kwestie wordt onderzocht in hoofdstuk 4. Hiertoe werd de omgeving van het adenocarcinoma van de slokdarm nauwkeurig in kaart gebracht. Het bleek inderdaad dat verschillende immuunsysteem onderdrukkende factoren aanwezig waren, samen ook met groeifactoren voor deze kankers. Kennis van deze factoren stelt ons in staat om tumor vaccins te combineren met andere interventies speciaal gericht om de voor de slokdarmkanker zo gunstige omgeving minder aangenaam te maken.
Combinatietherapie lijkt meer effectiefDe bovengeschetste mogelijkheid wordt direct getest in hoofdstuk 5, waar het vermogen van de HER2 antagonist Trastuzumab werd getest. Inderdaad bleek dat in HER2positieve kankercellen Trastuzumab de effectiviteit van het immuunantwoord kan versterken. Ofschoon deze experimenten onder tamelijk kunstmatige omstandigheden werden uitgevoerd, lijken zij wel aan te tonen dat combinatietherapie de aangewezen weg is.
Milano_opm_v5.indd 151 16-08-2007 15:43:18

Samenvatting
152
Beter inzicht in het ontstaan van adenocarcinoma van de slokdarmHet ontwikkelen van nieuwe therapie is uiteraard ook gebaat bij beter inzicht van hoe deze gevaarlijke ziekte tot stand komt. Het is bekend dat de ziekte ontstaat uit zogenaamd metaplastisch epitheel: normaal is het slokdarmepitheel (de eerste barrière tussen darminhoud en het lichaam) een meerlagig gebeuren, waarin langgerekte cellen liggen. Gedurende het voorstadium van adenocarcinoma verandert deze laag in een enkelvoudig rechthoekig epitheel, iets wat we Barrett’s oesofagus noemen. In hoofdstuk 6 identificeren we het hormoon dat deze overgang bewerkstelligt. Uiteraard biedt dit ons nog aangrijpingspunt voor betere preventie en behandeling van het adenocarcinoma van de slokdarm.
Samenvattend…Dit proefschrift geeft nieuwe antwoorden op een aantal vragen waarmee reeds lang geworsteld werd m.b.t. het ontwerpen van nieuwe therapie voor het adenocarcinoma van de slokdarm. De volgende stap zal nu een fase 1 studie waarin onze methodologie direct op de patiënt getest zal worden.
Milano_opm_v5.indd 152 16-08-2007 15:43:19

153
Dankwoorden
And now comes my opportunity to thank all the people that were next to me during these years and made this experience not only possible but intense and full of emotions. I will start with the two persons that in these 4 years represented for me a guide, a support, a font of ideas and great supervisors: Sheila, you are a very special mentor, but above all you are a great person: thanks for giving me the amazing possibility to start and continue this beautiful project which I loved from the beginning and I still love, this is exactly what I have always dreamt to do and this would not be possible without your brilliant and smart ideas, your ability to create always something original and to build from this good works, a continuously renewed enthusiasm for science guides you next to all the million stuff you have to arrange, this amazed me and still does when we spend time discussing together, having brainstorms, I have the fortune to do it together with you to learn everyday something more and to increase curiosity and desire to know new things… thanks!!! My promotor, great Maikel: you are amazing!! Thanks for all, enthusiasm, creativity, and whish to dare in science: it will always be printed in my mind what I have learned from you and your spirit of being positive, and your way of coming up always with novel initiatives with enormous passion for this beautiful world which is biology!! Pulling these two together, you find a marvelous couple of supervisors perfectly combined, that I had the great fortune to have at the beginning of my PhD… I will never forget this!
In all these how could it be the same without Agni! You are a special person, I am so happy that in these years we shared our daily life and work, that we could live so many moments, and had the opportunity to know each other: I learned a lot from you, thanks for being my paranimf, colleague, friend! And when we speak about friends, there comes Liudmila! Luda you are a very sweat person and I am thankful that I had the possibility to share this experience with you, you taught me a lot about life, I think is not easy to find persons like you, thanks to you both for all the support always, for our nice moments together, coffies, lunches, fun, time spent outside!!
Jantine, to you also much thanks for the help and support that you gave me when I arrived in the lab, and for the nice trips together, like the one to Florida, that was fun! With this trip how to forget Carina… Thanks for all the fun and also for all your translation DutchEnglish when I was lost amongst Dutch letters and mails, and your smile everyday! My aio kamer genoten: Joppi!! It is very nice to arrive at work in the morning and hear “Ciao bella!” thanks to you and Willemijn and Inge Hui, you are all so shiny and you always smile, I like it a lot, that’s nice to have you around! And still Sylvia, the lunchfreezer organizer, thanks for not killing me after all my questions about how to arrange things for the thesis! And of course the new, Maarten and Marleen, thanks for always sharing stuff in the
Milano_opm_v5.indd 153 16-08-2007 15:43:19

Dankwoord
154
lab and for the long discussions, thanks to Wendy and the ones that left already, Sjoukje and Jan, and all the rest, thanks to you all.
Fred, you are such a sweet person, your enthusiasm and keenness to help, and especially your smile have accompanied me all the time, thanks for the many organizations and your wonderful guided tour through Vienna! To Greetje also many thanks for all the arrangements and our nice email correspondence. Many thanks to the people in the lab that helped me all the time I asked although they were very busy: to Hella for pleasantly remembering me that I have to follow all the rules in the lab and to Jennie for the help you gave me when I had the moments of crisis trying to understand what the FACS machine is and does, to Esther for answering me so quickly everytime I order primers and for walking always with head up and a nice smile across the lab! To Alex to share the love for Italian countryside and food, to Marcel to amuse the “culturers” of the MLI with singing, thanks a lot to Monique for arranging all the stuff, especially for filling up the empty spaces that I constantly forget when I fill in the orderingform, to Heleen and Suzanne, for arranging always things so nicely and always with enthusiasm, to all the MDLers, to Susan and Esmerji, very sweat and nice girls, is gezellig to have you in the lab and work next to you in the cell cultures room speaking about our little babies (dendritic cells and macrophages) and all the rest! Thanks to the computer guys, Piet, always so kind and helpful, and thanks also for your precious suggestions on how to save my turtles from starving! Eric for his patience and great help.
Many thanks to all the Lexor group people, I will not name you all because you are a lot: you are a very good group, I had the opportunity to know a little better some of you, and I think you are very nice persons. Thanks for being always very supportive, to be ready to share stuff and interesting discussions. I cannot forget of course Carla, Floor and Regina: you are all very lovely girls, I am very glad we had the opportunity to share some experiments and to speak so much about us during the long days in the MLII without music and the noise of the flow in the background… heel gezellig! Many thanks also to Arnold, very sweat guy, always ready to lend a hand, Henry and Zuzy (special dendriten genoten) thanks for shearing with me your knowledge and giving always support. Still to Andrea (Greece) Andrea (Brazil), Carmen, Maria, Luci (Troppo Carina) really great to know you during these years, and…of course thanks to all the lab people that I did not name, both oud and nieuw, for creating a good atmosphere and to accept my horrible dutch all the time! Lots of thanks to the Barrett group: Jacques, Carine, Wilda, Brenda, Annette, Femke, Wouter, Mo, Ineke… all my experiments would not be possible without the precious samples that some of you, with so much patience and spending their time, collected for me, thanks for also the nice moments spent together outside the lab, you are all so kind! I want to thank my big dendriten genoten of the ECDC group: Martien, Marieke and Anja, Tineke
Milano_opm_v5.indd 154 16-08-2007 15:43:19

Dankwoord
155
and Mjriam, for all our brainstorms, and to share ideas, for help, long meeting, thanks for all the tips and suggestions, and for the gezelligheid. Thanks to Jan and Kees from cell biology, you spent a lot of time to make difficult experiments possible and to teach me precious skills and notions. Last, but of course not least, all my friends, dutch and not, here in holland, Nicole, Virna, Andrea, Leo, Martine, Sandro, Valerie, Vasso, Harald, Joana, Rogier, Dani, Suro, Angela, Ubi, David, Tami, Giulia, Rocco, Galma, Japi, Giovanna and all the rest, for all the experience that we share and for being next to me and to be good friends, for the fun, for the dinners, for the parties!! To all my Italian friends in Italy, always in my mind and in my heart, especially the “magic five” Viviella, Gina, Rosso, Rosa, Alis… siete e sarete sempre le mie specialissime! And of course Amina, Alio, Benny e Giammi, Sandrina e Stefano, Marco Bof and all the rest, I miei amici di sempre!! My family, Mamma, Papa’ and my brother Valerio, thanks for always,and I say always support me and encouraging me, for being next to me all the time in all the steps of my life.…I could not have done this without you…siete meravigliosi e io vi adorooo!! And now Edo, my love, you are too special, thanks for being with me, to support, to understand, to listen, for having fun, for sharing our lives, and for teaching me everyday something new and to be always yourself and who you are…. Ti Amooooo!!
Francesca
Milano_opm_v5.indd 155 16-08-2007 15:43:20

Dankwoord
156
Milano_opm_v5.indd 156 16-08-2007 15:43:20

157
List of Publications
An improved Protocol for generation of immunopotent DC through direct electroporation Of
CD14+ monocytes.
Milano F, van Baal JW, Rygiel AM, Bergman JJ, van Deventer SJ, Kapsenberg M, Peppelenbosch
MP, Krishnadath KK. J Immunol Methods. 2007 Apr 10; 321 (12):9410 8
An exvivo read out for evaluation of Dendritic cell induced autologous CTL responses against
esophageal cancer.
Francesca Milano, Agnieszka M.Rygiel, Navtej Buttar, Jacques J.G.H.M. Bergman, Carine
Sondermeijer, Jantine W. P.M. van Baal, Anja ten Brinke, Martien Kapsenberg, S. Marieke van
Ham, Maikel P. Peppelenbosch, Kausilia K. Krishnadath. Cancer Immunol Immunother. 2007
Jun 13 (Epub ahead of print).
Bone Morphogenetic Protein 4 expressed in esophagitis induces a columnar phenotype inesopha
geal squamous cells. Francesca Milano, Jantine W.P.M. van Baal, Navtej S. Buttar, Agnieskza
M. Rygiel, Floor de Kort, Wilda D. Rosmolen, Jacques J.G.H.M. Bergman, Jan Van Marle,
Kenneth K. Wang, Maikel P. Peppelenbosch, Kausilia K Krishnadath. Gastroenterology. 2007
Jun; 132(7): 24122421.
A comparative analysis by SAGE of gene expression profiles of Barrett’s esophagus, normals qua
mous esophagus, and gastric cardia.
J.W.P.M. van Baal, F. Milano, A.M. Rygiel, J.J.G.H.M. Bergman, W.D. Rosmolen, S.J. vanDe
venter, K.K. Wang, M.P. Peppelenbosch, K.K. Krishnadath. Gastroenterology. 2005 Oct; 129(4):
127481.
RNABased Gene Therapy for Haemophilia B.Maikel P. Peppelenbosch, Francesca Milano, Carmen
V. Ferreira, Anna Knapinska, Sander H.Diks and C. Arnold Spek, Current Genomics, 2005, 6,
315318.
Efficient automated assessment of genetic abnormalities detected by Fluorescent In Situ Hybridiza
tion on brush cytology of a Barrett’s esophagus surveillance population.
A.M. Rygiel, J.W. Van Baal, F. Milano, K.K. Wang, F.J. Ten Kate, P. Fockens W.D. Rosmolen, J.J.
Bergman, M.P.Peppelenbosch, K.K. Krishnadath. Cancer. 2007 May 15; 109(10): 19808.
Comparison of kinome profiles of Barrett’s esophagus with normal squamousesophagus and nor
mal gastric cardia.
Jantine W.P.M. van Baal, Sander H. Diks, Ronald J.A. Wanders, Agnieszka M. Rygiel, Fran
cesca Milano, Jacques J.G.H.M.. Bergman, Maikel P. Peppelenbosch, Kausilia K. Krishnadath.
Cancer Res. 2006 Dec 15; 66(24): 1160512.
Milano_opm_v5.indd 157 16-08-2007 15:43:20

Milano_opm_v5.indd 158 16-08-2007 15:43:20

159
Curriculum Vitae
Francesca Milano was born on April 20, 1976 in Perugia, Italy. She graduated from highschool in 1995 at the Liceo Classico Mariotti in Perugia. In 1995 she started biology school at the University of Perugia. In 2002 Francesca graduated in medical biology in Perugia, and, in the same year, she was granted with a research scholarship at the Netherland Cancer Institute, in Amsterdam. Therefore she moved to Holland and, under the supervision of DR. D. Weinkove, she studied the role of Ppk2 and Daf16 in the oxidative stress in Caenorabditis Elegans. In 2003 Francesca started her PhD project at the Laboratory of Experimental Internal Medicine and Experimental Gastroenterology in the Academic Medical Center, in Amsterdam. Under the supervision of Dr. K.K. Krishnadath and Prof. M.P. Peppelenbosch, she conducted her research directed to find novel and more effective strategy to treat Esophageal Adenocarcinoma patients, particularly focusing on Dendritic Cell immunotherapy, and to study the molecular mechanism underlying the cause which is at the origin of this type of cancer, which is Barrett’s Esophagus. The results obtained so far are reported in this thesis. Currently she is continuing her studies on how to use Dendritic Cells immunotherapy in combination with other approaches, such as the use of small molecules inhibitors and blocking antibodies, in order to improve patient’s outcomes. Furthermore, she is further characterizing the dynamics of the development of Barrett’s esophagus.
Milano_opm_v5.indd 159 16-08-2007 15:43:20
![UvA-DARE (Digital Academic Repository) [Introductie] van ... · UvA-DARE is a service provided by the library of the University of Amsterdam () UvA-DARE (Digital Academic Repository)](https://img.pdfslide.us/doc/110x75/5fb83dbe0812d641087c115b/uva-dare-digital-academic-repository-introductie-van-uva-dare-is-a-service.jpg)




























