Embed Size (px)
Citation preview

Using DERIVE 6.1 To Generate
DNA Fractal Representations
Joel Jeffrey’s Chaos Game
Representation, Genomic
Signatures, and Aligning Sequences
L. Carl Leinbach
Gettysburg College
Gettysburg, PA USA

The Chaos Game
The Chaos Game (Jeffrey’s name) is basically an Iterated Function System
dependent upon points arranged in some geometric configuration. The best
known example is that of Sierpinski’s Triangle.
In this case, the point configuration is the vertices of a triangle located in the
plane, say at vertices, v1 = (0,0), v2 = (1/2, 1), and v3 = (1,0). The sequence of
points (xi, yi) is then chosen with (x0, y0 ) = (1/2, 1/ 2) and
𝑥𝑖 = 𝑥𝑖−1 + 𝑣𝑟𝑖+ 𝑥𝑖−1
2
for I = 1, 2, . . .
𝑦𝑖= 𝑦𝑖−1 + 𝑣𝑟2 +𝑦𝑖−1
2
At each step vr is randomly chosen from among the vertices v1, v2, and v3.
This process is easily programmed in DERIVE and the result graphed.

Sierpinski(T, n, i, j, k, X, Y) ≔ Prog i ≔ 1 P ≔ [1/2, 1/2] X ≔ [P↓1] Y ≔ [P↓2] Loop i ≔ i + 1 If i > n RETURN VECTOR([X↓j, Y↓j], j, 1, n) k ≔ RANDOM(3) + 1 P ≔ [(P↓1 + T↓k↓1)/2, (P↓2 + T↓k↓2)/2] X ≔ INSERT(P↓1, X, i) Y ≔ INSERT(P↓2, Y, i)
Below is an example of a typical program for constructing the Sierpinski
Triangle and the result of the program after 10,000 iterations. The result is
always the same, independent of the choice of the starting point and, remarkably,
the quality of the pseudo-random number generator picking the target vertex.
Sierpinski(T, 10000)

The Birth of the Chaos Game Representation
In his 1990 article in Nucleic Acids Research , Jeffrey noted that if an iterated function
system using the same “Chaos Game” strategy is applied to figures with 5, 6, 7 as well
as three vertices, the result is the same independent of the quality of the random number
generator. However, with 8 vertices, and to a lesser extent 4 vertices, the result was
dramatically different.
Using a good random number generator with 8 vertices, the result of the Chaos Game
was an almost uniformly filled octagon. When the game was played using the random
number generator from Turbo Pascal 3.0 (a notoriously bad generator) the result was
an elaborate pattern generating a circle within a circle and the circles connected by 8
spidery lines. The DOS Basic v2.1 function RND produced no visible pattern at all for
8 vertices.
This experience, according to Jeffrey’s account, led him to the conclusion that the
Chaos Game can display certain kinds of non randomness visually. In particular,
patterns in a four vertex Chaos Game having the vertices labeled starting at the bottom
left and proceeding clockwise with the letters, “A”, “C” , “G”, and “T” could be
observed by controlling the moves with successive letters in a particular DNA sequence.
He called the resulting pattern the “Chaos Game Representation” of the sequence.

An All Too Brrief
Introduction to DNA
Much work preceded the spectacular 1953 announcement of the
double helical structure of DNA by Watson and Crick. Nucleic
Acid was known prior to 1820. DNA was first identified in 1869
and, simultaneously Mendel was doing his pioneering research in
genetics. In the 1940’s Linus Pauling predicted a three
dimensional structure for the DNA molecule. The X-ray
diffraction work of Rosalind Franklin and Maurice Wilkins
played a major role in the Watson–Crick discovery.
DNA consists chains made up from four nucleotides,
two Purines:
Adenine - A
Guanine - G
and two Pyrimidines:
Thymine – T
Cytosine - C

What Watson and Crick Discovered
Erwin Chargaff, an Austrian born biochemist ,who
immigrated to the United States during the Nazi
era, discovered two rules in 1950 that were
fundamental to the discovery of the double helical
structure of DNA.
1. In natural DNA the number of guanine units
equals the number of cytozine units and the
number of adenine units equals the number of
thymine units
2. The composition of DNA varies from one
species to another.
However he did not grasp the connection of the first
rule to the structure of DNA.
Using Chargaff’s first ‘rule’, the results from Roslind Franklin and Maurice
Wilkins’ Xray diffraction work, and Linus Pauling’s work on the three dimensional
structure of molecules, Watson an Crick painstakingly constructed a cardboard
model of the DNA molecule showing the correct nucleic acid bonds and bonding
distances. Thus, they became the first to accurately formulate the molecule’s
structure.

The Chaos Game Representation (CGR) of DNA
Since A always bonds with T and C always bods with G, one strand of the double helix
is a reflection of the other. Thus, if we know the sequence for one strand , we know the
other; When we see a listing of the nucleic acid content of DNA, we see a single
sequence. These sequences are the inputs for the CGR.
The DERIVE program for obtaining the CGR is very similar to that for generating
Sierpinski’s Triangle. Other than the fact that we are using the corners of a square as
the reference points, the main difference is that there is no need for a random number
generator. The following code is the heart of the program:
L ≔ D↓j If L = "A" R ≔ A If L = "C" R ≔ C If L = "G" R ≔ G R ≔ T P ≔ [(P↓1 + R↓1)/2, (P↓2 + R↓2)/2]
NOTE: A, C, G, and T are the locations of the four corners of the unit square.

C
A T
G
Below is the CGR for Human Hemoglobin B: (HUMHBB) on Chromozome 11 The sequence is 73308 base pairs (bp) long. The starting point was (1/2, 1/2). The location of the most recent ly calculated point was a global variable. The results were plotted after each 20000 bp’s and the end of the sequence. Accurate rational arithmetic was essential for correctly locating points

The Fractal Nature Of The CGR
The pattern exhibited by the entire Hemoglobin B shown has a
double scoop in the upper right hand or quadrant of the CGR.
This reflects a paucity of the CG dinucleotide in the sequence.
This double scoop is exhibited in the displays shown below.
Lower Right Quadrant Lower Right Octant

The Shape of the CGR
For Any Sequence Of Reasonable Length
If we choose any two base pairs that are at least a few thousand positions apart in the
sequence, we will see the same basic pattern exhibited. Shown below is the CGR of
the HUMHBB sequence between bp 53000 and 58271
According to Jeffrey the double scoop shape is has been found only in DNA sequences
for vertebrates and in the human and chimpanzee HIV virus. And ,it can be used to
classify these viruses.

Another Example of the Fractal Behavior
To illustrate that the phenomenon observed for the Human
Hemoglobin B is not a one time occurrence, The CGR was applied
to Gordonia otitidis, a pathogen that can cause blood infections. On
the left is the complete sequence for the Gordonia and on the right is
a partial sequence involving 2500 base pairs. Note that we do not
have the double scoops in the CG region; however there is a paucity
in the AT region.
Godonia otitdis – 9421 bp CGR for bp 3241 to 5678

Jeffrey’s Observations
1. The k-th point on the CGR of a sequence corresponds to the first k-long initial
subsequence and no other subsequence. Thus there is a one-to-one correspondence
between the subsequences of a gene (anchored at a start) and the points of the CGR.
2. Visible patterns in the CGR correspond to some pattern in the sequence of bases..
3. The resolution of the screen limits the amount of detail that may be shown in the CGR.
However, the fractal nature of the CGR allows for for any portion of the CGR to be
magnified to show fine structure.
4. Adjacent bases in the sequence are not plotted adjacent to each other. Euclidean distance
in the CGR therefore implies a new metric on subsequences, or bases.
5. Generally, two close points in a CGR correspond to different sequences, however two
points that are close, but in different quadrants may belong to the same sequence.
6. Points 4 and 5 imply that the visible patterns in a CGR represent global as well as local
patterns. A density (or paucity) of points in a region corresponds to a large (or small)
number of sequences with suffixes in that region.
7. Any mathematical description of the CGR of a sequence is a characterization of the
underlying sequence.

Jeffrey’s Theorem
Theorem 1. In a CGR whose side is of length 1, two sequences with suffix of length
k are contained within the square with side of length 2-k. Further, the center of the
square is give by the following recursive definition:
(a) The center of the suffix of 0 length is (1/2, 1/2)
(b) If the center of the square containing sequences with suffix w is at (x, y),
then
i. the center of the square containing sequences with suffix wa is
(x/2, y/2);
ii. the center of the square containing sequences with suffix wc is
(x/2, (y+1)/2);
iii. the center of the square containing sequences with suffix wg is at
((x+1)/2, (y+1)2);
iv. the center of the square containing sequences with suffix wt (or wu) is
at ((x+1)/2, y/2).
Conversely, all points within this square correspond to sequences with this suffix.

Illustrating Jeffrey’s Theorem
Starting at the center of the square with coordinates A = [0,0], C = [0,1], G = [1,1]
and T = [1,0], trace each successive nucleotide base pair in th e string. Each
point in the plot represents the entire sequence to that point. Also note the
position of the G term is closer to the first T term than it is to C.
G GC GCT GCTT
GCTTA
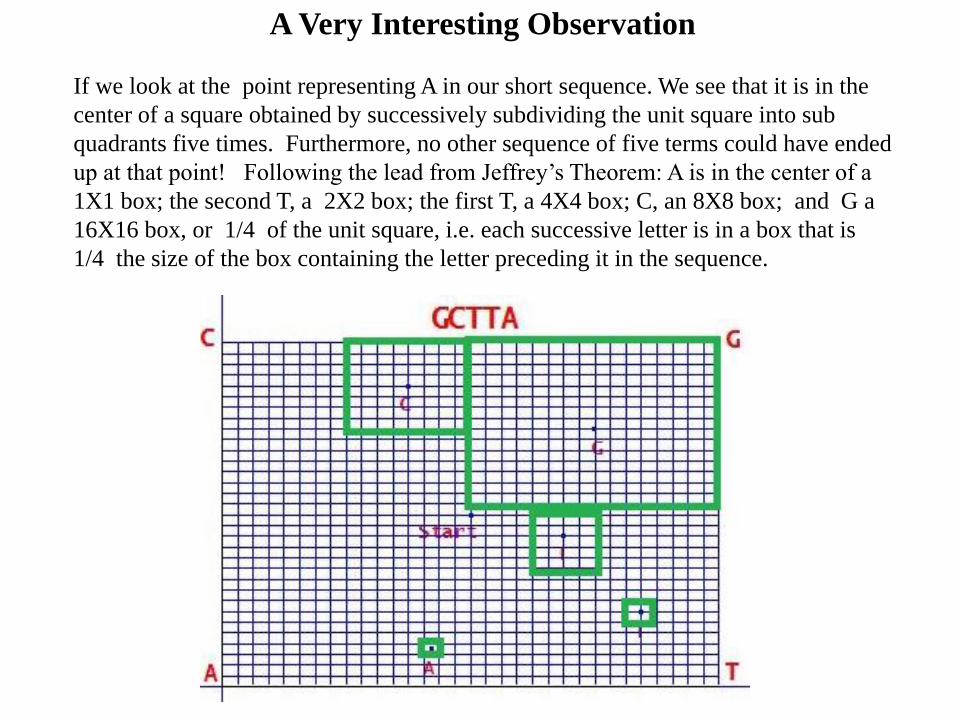
A Very Interesting Observation
If we look at the point representing A in our short sequence. We see that it is in the
center of a square obtained by successively subdividing the unit square into sub
quadrants five times. Furthermore, no other sequence of five terms could have ended
up at that point! Following the lead from Jeffrey’s Theorem: A is in the center of a
1X1 box; the second T, a 2X2 box; the first T, a 4X4 box; C, an 8X8 box; and G a
16X16 box, or 1/4 of the unit square, i.e. each successive letter is in a box that is
1/4 the size of the box containing the letter preceding it in the sequence.

An Illustration of The
Independence of Starting Point
On the CGR Graph below the sequence GCTTA was plotted from three different
initial points: [1/2, 1/2] (black), [2/3, 5/9] (red), and [9/13, 13/15] (blue). Note that
all three ending points ended in the same 1/32 X 1/32 region of the graph.

Towards Constructing a Genomic Signature
The location of a point of the CGR can allow us to reconstruct the last n terms of a
sequence where n is the size of n-th sub-sub- ٠٠٠-sub quadrant of the unit square on
which the CGR is constructed. Consequently, any sequence ending in GCTTA will end
up in this 5-th sub quadrant. The example sequence is in the middle since it started at
the middle of the unit square. This can result in a further refinement for identifying the
genomic source of the sequence..

Deschavanne’s Genomic Signature
Suppose that we number each of the possible 5-th order sub quadrants according to the
following scheme:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 ٠٠٠ 32 33 34 35 ٠ ٠ ٠ 64
٠٠
٠٠ ٠٠ ٠٠
993 ٠ ٠ ٠ 1024
The five letter sequences starting at the upper left and proceeding down to the lower right
Listed according to the folowing scheme:
CCCCC, GCCCC, CGCCC, GGCCC, ٠٠٠TTTTT
Note that CCCCC corresponds to cell 1, GGGGG to cell 32, AAAAA to cell 993 and
TTTTT to cell 1024.
In 1999, Patrick J. Deschavanne and his team of researchers saw that by comparing the
distribution of position s in the plane of the CGR of a sequence could be used to identify
sequences and also to differentiate between sequences having similar appearing CGR’s , but
being from different sources.

Creating a Genomic Signature
The heart of the “Genomic Signature” is that the number of points of the CGR that lie in
each of the specified sub regions of the CGR normalize across the entire region will
disclose the concentration of each of the possible oligonucleotides of a certain length
that are in the sequence.
Below are CGR’s of sub sequences of Beta Hemoglobin from a Human, a Whale , and a
Chimpanzee.
HUMAN WHALE CHIMPANZEE
816 bp 640 bp 1729 bp
At first glace these CGR’s appear to be very similar and one might be hard pressed to
tell which CGR came from which sequence

The Genomic Sequence Revealed Given the matrix, M, showing the normalized concentrations in each block of the CGR,
construct a 3-D point (x,y,z), where (x,y) is the center f the block and z is the number of
points lying in that block divided by the maximum number of points in a block of M,
the infinity norm of M. The following graphs were derived from the three CGRs on the
previous slide.
Human Whale Chimpanzee
A natural concern is that the difference in these graphs may be a function of the
subsequence and its size that is chosen for each species. In his paper Deschvanne and
his team considered the distance between several samples from several species and
several samples of reasonable size within each species and showed that the distances
between interspecies samples are much more tightly grouped about their mean than
the distances between the means of different species.
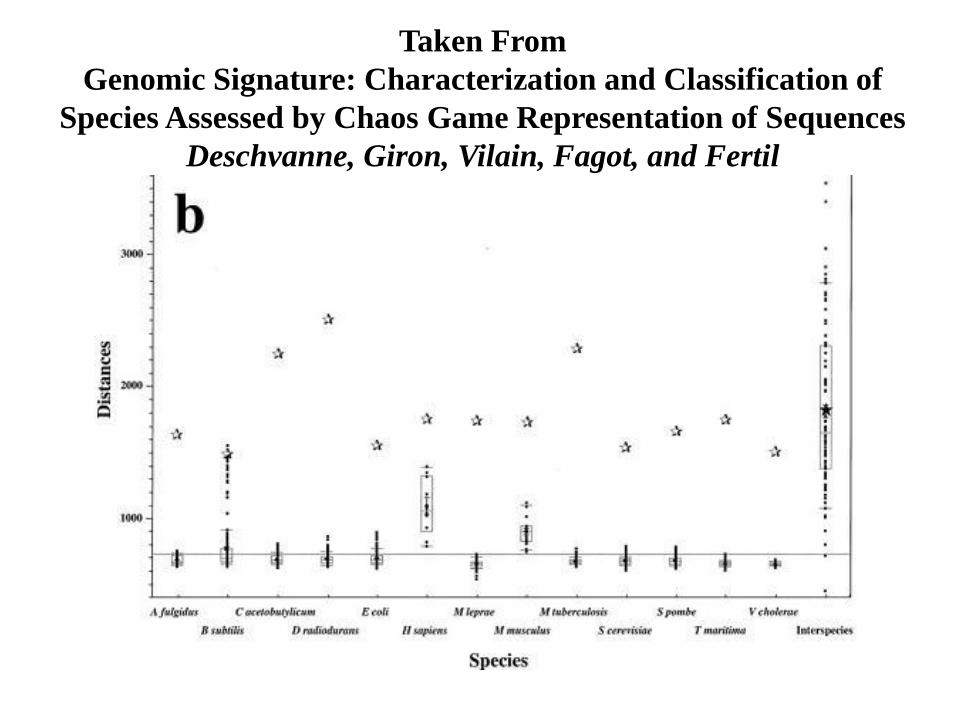
Taken From
Genomic Signature: Characterization and Classification of
Species Assessed by Chaos Game Representation of Sequences
Deschvanne, Giron, Vilain, Fagot, and Fertil

Sequence Homology Why Sequence Alignment is Important
DNA sequences develop from pre-existing sequences instead of being invented by nature
from scratch . This fact makes sequence alignment an important part of biological
research. If the DNA is taken from an area which has a function or structure from a
known sequence, then , if the sequences exhibit regions of similarity, then it is possible
that the surce f the new sequence will exhibit similar structure or function. When a match
occurs, we say the old and the new sequence are homologous.
At this point we will look for sequence similarity, a first step towards sequence homology.
Several tools exist and are publically available to researchers. The most commonly used
both for DNA and protein sequences is called BLAST (Basic Linear Alignment Tool)
developed by Stephan Altschul, Warren Gish, and David Lipman of the National Center for Biotechnical Information.. It looks for short portions of each sequence that are identical and then stretches in each direction and looks for a good alignment.
The identical sub sequences are called “conserved sequences.” We will show how some
practitioners in the biotechnical field are uing Jeffrey’a CGR as a means of finding
conserved sequences.

A DERIVE Program
For Finding Conserved Sequences
It has been noted that if two CGR points reside in the same block of the 1024 block of a
32 X 32 grid imposed upon the CGR they are the result of mapping the exact same six
letter sequence of the DNA alphabet. The problem is one of finding a strategy for
quickly finding the box in which a point of the CGR resides. We circumvent this point
by saying that since the box is 1
64 X
1
64 , their l1norm of the distance between the two
points in the box is at most 1
32 . We will use a much stricter criterion of considering
only points that lie within 1
256 units of each other. This criterion is not “fool proof”, but
gives a good start for
the search:

An Example
We will consider the following two sequences:
Seq1 ≔ TCCCAGTTATGTCAGGGGACACGAGCATGCAGAGAGAC
Seq2 ≔ AATTGCCGTCGTTTTCAGCAGTTATGTCAGATC
The result of the previous program is: [12, 27]
This means that our search will begin at the 12-th letter of sequence 1 and the 27-th of
sequence 2 If, as is hoped, the points are in the same grid box, the five letters preceding
these positions will match and we extend to the right until our criterion on the CGR
points fails. The result
For the two sequences
above is:
[TTATGTCAG, [17, 32],
TTATGTCAG]
Indicating that the conserved
sequence extends from
character 7 = 12 -5 to 17 in
seequence 1 and from 22 to
32 in sequence 2.

Shot Gunning The Genome
The DNA making up the human genome is estimated to be about 3.2 Mb in
size. Only about 1.5% of this total is protein coding DNA The rest is made
up of other components; non coding RNA genes, regulatory sequences,
introns, non coding DNA (aka junk DNA).
To fully sequence the human genome in one shot would be an impossible
task. In the late 1940’s , before DNA structure was even known, Sanger was
awarded a Nobel Prize for determining the amino acid sequence of insulin.
He digested insulin in enzymes that broke up the sequence and was able to
sequence the short sequences rather easily. Then since the fragments
overlapped, he was able (no small task) to put the puzzle back together. In
1974, Russian scientist, Andrey Mirzabekov, whiole visiting Harvard found a
way to selectively break DNA at A and G. Later Allan Maxam and Walter
Gilbert found a method to break DNA at every C and T. The short sequences
could be sequenced and then using combinatorics algorithms, be put back
together. This is known as shotgunning.

A Small Example
Of Shot Gunning
The following three sequences were shot gunned from a protein sequnce.
sg1 ≔ GGATCGTCGTCG
sg2 ≔ CGTCGTCGAACAATG
sg3 ≔ CGAACAATGTCGGA
Using the Derive program we are able to solve this very simple example.
First we try sequence 1 and sequence 2
ConsSeq(sg1, sg2)
[CGTCG, [14, 10], CGTCG]
Indeed there is overlap and it occurs at the end of sequence 1 and beginning of sequence
2. However, we still need to check if there is also overlap between seqquence 1 and 3.
ConsSeq(sg1, sg3)
No Sequence Found
So now what about 2 and 3?
ConsSeq(sg3, sg2)
[AACAA, [10, 15], AACAA]
Here we see one of the weaknesses of the heuristic in determining the same block of the
CGR and also the fact that we are dealing with the end of one sequence and the
beginning of another. The program missed th e beginning CG and final TG. There is
still work to be done! – Next time.

Why CAS? Recovery:
Each point of the CGR is the history of the sequence up to that point. Given the
simple nature of the IFS (Iterrated Function System) used to generate the CGR, the
sequence can be completely recovered from the position of the point and the position of
the starting point. A finite word length decimal approximation will quickly loose this
accuracy. Rational arithmetic of the CAS does not have this restriction.
Quoting from Almeida: “The nucleotide sequence can (also) be recovered from the
CGR coordinate, where the the number of the bases resolved is a function of the
resolution of the CGR coordinates.
Sequence Comparison:
We use a grid of size 2𝑛 𝑋 2𝑛 and store the location of the n-length oligonucleotide
sequence represented by the corresponding grid box containing the CGR points from
the sequences under comparison and then extend then extend the sequence as far as
possible within each sequence. Thus, the CGR jump starts the comparison procedure.
In this paper oligonucleotide of length 6 were used.
Quoting from Joseph: “For long identical sequence segments, the minimum value of
the distance value may go below the minimum possible floating point variable… in
double precision the variable becomes zero when the length of identical segments is
greater than 64.”

References
1. Almeida J. S., Carrico J. A., Maretzek A., Nobel P. A. and Fletcher M., “Analysis of
Genomic Sequences by Chaos Game Representation, Bioinformatics, v. 17, no. 4,
2001, pp 429 – 437
2. Deschavanne P. J., Giron P., Vilain J., Fagot G., and Fertil B., “Genomic Signature:
Characterization and Classification by Chaos Game Representation of Sequences” ,
Molecular Biology and Evolution, http://mbe.oxfordjournals.org/ Stanley Sawer,
reviewing editor, June 1999
3. Isaev, A., Introduction to Mathematical Methods in Bioinformatics, Springer, Berlin,
2004
4. Jeffrey, H. J., “Chaos game Representation of Gene Structure”, Nucleic Acids
Research,1 v 18, no, 8, 1990
5. Jones N. C. and Pevsner P. A., An Introduction to Bioinformatics Algorithms, The
MIT Press, Cambridge, Mass., 2004
6. Joseph J. and Sasikumar, R., “Chaos Game Representation of Whole Genomes”,
BioMed Central (Open Access), 10 pages, May, 2006
7. Pevsner, P., Computational Molecular Biology, An Algoritmic Approach, The MIT
Press. Cambridge, Mass., 2000, (Second Printing), 2001





























