Embed Size (px)
Citation preview

Unusual Complete Reduction of Cu2+ Species in Cu-ZSM-5 Zeolitesunder Vacuum Treatment at High TemperatureManlio Occhiuzzi,*,† Giuseppe Fierro,‡ Giovanni Ferraris,‡ and Giuliano Moretti†
†Dipartimento di Chimica, “SAPIENZA”Universita di Roma, Piazzale A. Moro 5, 00185 Roma, Italy‡Consiglio Nazionale delle Ricerche (CNR)Istituto per lo Studio dei Materiali Nanostrutturati (ISMN),c/o Dipartimento di Chimica, “SAPIENZA”Universita di Roma, Piazzale A. Moro 5, 00185 Roma, Italy
*S Supporting Information
ABSTRACT: By electron paramagnetic resonance (EPR),coupled to elemental and thermogravimetric analysis anddiffuse reflectance spectroscopy (DRS), we investigated theunusual quasi-complete reduction of Cu2+ species in Cu-ZSM-5 zeolites (Si/Al = 80 and 25) at different copper loadingsinduced by high-temperature vacuum treatments. It has beenfound that at high temperatures (673−773 K) in vacuum aquasi-complete reduction of Cu2+ unexpectedly occurred in allthe “as-prepared Cu-ZSM-5 samples. Evidence is given thatsuch extensive reduction of Cu2+ species is caused bycarbonaceous deposits. In order to completely remove anyresidual carbonaceous species, the “as-prepared” Cu-ZSM-5materials must be heated in air (or O2) at high temperature(673−773 K) for prolonged time. Differently, some carbon residue, as in particular originated from the organic template used inthe zeolite synthesis, can be left within the channels of the ZSM-5 structure. We show also that such a carbon residue, even whenescapes the detection accuracy of elemental analysis, can be checked by a careful EPR characterization.
KEYWORDS: Cu-ZSM-5 zeolites, reduction of Cu2+ species by carbonaceous deposits, EPR evidence of organic template residue,DRS characterization, EPR characterization
1. INTRODUCTIONThe transition-metal-ion-containing zeolites represent animportant class of materials showing a rather complexchemistry that is particularly evidenced when these solids areused as catalysts. In particular, the Cu-ZSM-5 catalysts haveattracted great attention, because they are active and selectivefor both the NO decomposition and the reduction of NOxby hydrocarbons in the presence of O2,
1a−e and, more recently,as a biomimetic inorganic model for methane oxidation.1f
Important chemical modification can occur in these solidsunder practical conditions,2 as aggregation of Cu2+ species withthe formation of CuO-like particles within the channels or onthe external surface of the zeolite,3 and dealumination.4 Indeed,the nature of the active sites both for NO decomposition and forthe selective catalytic reduction of NOx by hydrocarbons in thepresence of O2 is still debated.
1a−e The chemical and structuralproperties of the Cu-ZSM-5 catalysts as well as the nature of thecopper species are affected by many parameters such as the Si/Alatomic ratio of the starting ZSM-5 matrix, the preparationmethod, the copper loading, the activation procedure.5 Such anintriguing and complex chemistry, leading to specific localarrangement of the copper species at the extra-framework site ofthe zeolite, is testified by the fact that the Cu-ZSM-5 catalyst isthe only one to be remarkably active for NO decompositionamong all the transition-metal-ion-containing zeolites.1a−e,2−10
A crucial aspect of the chemistry of these materials is relatedto the reduction of Cu2+ species to Cu+ species, also becausethis phenomenon has been suggested to play a key role in theNO decomposition mechanism.6−10 We reported that a specificconfiguration of dimeric Cu+ species at the intersection bet-ween the straight and sinusoidal channels of the Cu-ZSM-5zeolites are able to irreversibly adsorb N2 at low temperature(273−325 K) and could represent the active site for the NOdecomposition.6 Also, monomeric Cu+7 or others dimeric(Cu+···Cu+) species,8−10 that must be anyway close toAl−−O−Si framework sites,11,12 have been reported asactive sites for this reaction. On the other hand, some authorsstated that the Cu2+ species in Cu-ZSM-5 were stable under thereaction conditions.13−15 These different conclusions are notsurprising, because a truly coherent description of the NOdecomposition catalytic process on Cu-ZSM-5 has proven to beelusive. On the other hand, the reduction of copper species inCu-ZSM-5 catalysts in vacuum or under a flow of inert gas is amatter of fact. However, the extent of such a reduction and thechemistry behind this process is still lively debated. The extentof reduction is mainly depending on temperature, as well as on
Received: December 21, 2011Revised: April 27, 2012Published: May 3, 2012
Article
pubs.acs.org/cm
© 2012 American Chemical Society 2022 dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−2031

the sample history. For instance, an almost complete reductionof the Cu2+ species occurs when “as prepared” Cu-ZSM-5materials are first treated at high temperature under vacuumor inert gas flow. On the other hand, a much less extentof reduction of the Cu2+ species was observed when theCu-ZSM-5 samples were initially treated in O2 at hightemperature before the treatment under vacuum or inert gasflow at high temperature. Hall and co-workers,8 on the basis ofthermobalance experiments, found out that only a small fraction(maximum 20%) of the initial Cu2+ is reduced to Cu+ fromswitching the gas flow from O2 to pure helium in repeatedcycles at 773 K.8c Liu and Robota16 observed by XANESan almost complete reduction of Cu2+ ions to Cu+ after atreatment of as-prepared Cu-ZSM-5 samples in helium at 773 K.It may be worth to recall that, in 1977, Kasai and Bishop17
investigated, by EPR, the reduction of Cu2+ ions in mordeniteafter activation under vacuum at temperatures of >573 K. Theseauthors evidenced that strongly absorbed water molecules in thecoordination sphere of Cu2+ ions played a role either in thereduction process at high temperature, coupled to O2 desorption,and in the reoxidation of Cu+ to Cu2+ at room temperature.When the sample activated under vacuum was exposed at roomtemperature to degassed water or dry oxygen alone, the Cu2+
signal remained unchanged and increased only in the presence ofboth O2 and H2O. More recently Zecchina and co-workers haveconfirmed these results in both “as prepared” Cu-ZSM-5 andCu-mordenite zeolites concluding that a quasi-completereduction of Cu2+ to Cu+ ions occurred after a thermal treatmentin vacuum in the temperature range of 470−670 K.18a,b
If the extent of reduction involves only a partial amount ofthe total copper, the process has been usually reported inliterature as a “self-reduction” process. According to Hall andco-workers,8 a “self-reduction” involves a decrease of the sampleweight coupled to O2 desorption with the simultaneous releaseof four electrons to the solid and consequent reduction of4 Cu2+ to 4 Cu+ (no metal copper formation was reported byHall’s group). However, the oxygen source for the “self-reduction” process is questioned. While it has been reportedthat the “self-reduction” in CuY zeolites occurs only throughoxygen atoms of the zeolitic framework,19 in the case ofCu-ZSM-5 materials, Hall8 and Iwamoto9 groups suggestedthat in the “self-reduction” only extra-framework oxygen speciesare involved. This idea was also supported by Sachtler5 and Bell20
groups who, in turn, have suggested different mechanismsthrough which the “self-reduction” process is accomplished,namely, oxygen evolution from [Cu−O−Cu]2+ species or waterelimination from [CuOH]+ species, respectively.In the case of a complete reduction of the Cu2+ species in
copper-containing zeolites, the situation is much less clear andis still lively debated. It seems to be very unlike that a totalreduction of Cu2+ to Cu+ species could be ascribed only to an‘intrisic’ process, like the “self-reduction”. In this respect, it hasbeen reported that traces of hydrocarbons from the environ-ment may play a role in presence of ionizing radiation, as underXPS measurements.19 However, much more relevant can bethe effect of carbonaceous deposits left in the Cu-ZSM-5zeolites along the preparation. This aspect, to the best of ourknowledge, has never been investigated in detail and seems tobe missing either in literature dealing with, in general, zeolite-based materials or, specifically, NOx abatement over Cu-ZSM-5catalysts. The formation of carbonaceous species is well-documented in relation to acid-catalyzed hydrocarbon reac-tions.21 In this work, the role of carbonaceous deposits in the
Cu-ZSM-5 zeolites has been investigated by EPR and DRSspectroscopy, coupled to elemental and thermogravimetricanalyses, adding new insights to our previous partial findings.22,23
In particular, the reduction of Cu2+ species was studied fortwo set of Cu-ZSM-5 samples having different copper loadings.They were prepared from ZSM-5 parent materials characterizedby a remarkable difference in the Si/Al ratio (80 and 25) andobtained from two different preparation methods (i.e., with andwithout the use of the organic template).
2. EXPERIMENTAL SECTION2.1. Catalysts Preparation. The H-ZSM-5 zeolite with Si/Al = 80
was prepared in three different batches using tetraethylsilicate,tetrapropylammonium hydroxide, and a solution of Al3+ nitrate inethanol. This mixture was kept under stirring at 333 K for 3 h and thenheated, with no stirring, under autogenous pressure in a stainlessautoclave at 448 K for 24 h.2,3,6 Then the crystalline product wasseparated from the mother liquor by centrifugation, washed severaltimes with distilled water and dried for 2 h at 383 K. The driedpowder was finally heated in air, with the temperature gradually rising(8.5 K/min) from room temperature up to 823 K and kept at thistemperature in air for 5 h, that is the usual standard procedureemployed to remove the organic ammine template from the MFIstructure.24 At the end of this step, the “as-prepared” H-ZSM-5-80parent material was obtained. For the sake of comparison, a portion ofone of the ZSM-5-80 preparations was not treated in air at hightemperature in order to leave the tetrapropylammonium templateinside the MFI structure. This solid will be labeled as “as-synthesized”ZSM-5-80.
The H-ZSM-5 zeolite with Si/Al = 25 was prepared starting froma commercial product, NH4-ZSM-5 by PQ Corp. (No. CBV5020).The NH4−ZSM-5 was first treated in air at increasing temperature(8.5 K/min from room temperature up to 773 K) and then kept in airat 773 K for 5 h. At the end of this step the “as-prepared” H-ZSM-5-25parent material was obtained. The Cu-ZSM-5 samples were preparedthrough a standard ion exchange procedure starting from the “as-prepared” H-ZSM-5 parent materials and using diluted cupric acetate,or nitrate, aqueous solutions. In detail, 2.0 g of the “as-prepared”H-ZSM-5 zeolites were added under continuous stirring to 250 mL ofthe salt solution containing copper in suitable concentration. This ion-exchange step was made at 323 K for 2 h. The exchanged samples werewashed several time with distilled water, dried for 2 h at 383 K andfinally stored in a glass vessel under a controlled relative humidity(ca. 79%). These solids are labeled and hereafter reported like “as-prepared” Cu-ZSM-5 samples. In purposely devoted experiments, wefound out that if the exchange process is made for longer times this hasno effect on the final copper loading. Copper content was determinedby atomic adsorption spectroscopy (Varian Model SpectrAA-30).According to a standard nomenclature adopted in literature for thesematerials, the parent materials and the catalysts will be reported asH-ZSM-5-a and Cu-ZSM-5-a-b where the “a” indicate the Si/Al atomratio (80 or 25) and “b” the nominal extent of Cu2+ exchange. Theextent of Cu2+ exchange was calculated from the Cu/Al atomic ratio[extent of Cu2+ exchange (%) = 2 × (Cu/Al) × 100], assuming that, inany H-ZSM-5 sample, the number of Al3+ ions is equal to the numberof H+ ions and that 2 H+ ions can be, in principle, replaced by oneCu2+ ion. Accordingly, an atomic ratio Cu/Al = 0.5 corresponds to100% of the basic exchange capacity (stoichiometric preparations),while for atomic ratios Cu/Al > 0.5 an overexchanging occurs(overexchanged preparations). Details about the H-ZSM-5 unit cellformula and the calculation of the extent of Cu2+ exchange arereported in the Supporting Information section (Part 1 and 2).
2.2. Catalysts Characterization. 2.2.1. Textural and ElementalAnalyses. Textural analysis was performed by N2 adsorption−desorption isotherm at 77 K using a Micromeritics ASAP 2010analyzer. Before measurements, all the “as-prepared” samples under-went a three-step pretreatment under vacuum: (i) at 423 K for 1 h,(ii) at 523 K for 1 h, and, finally, (iii) at 623 K for 4 h. The“as-synthesized” ZSM-5-80 sample was outgassed at 523 K for 4 h before
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312023
measurements in order to preserve the integrity of the tetrapropylammo-nium template within the MFI framework. The Brunauer−Emmett−Teller (BET) specific surface area (St) was calculated using adsorptiondata in the relative pressure range 0 < P/P0 < 0.1. The microporevolume (Vμ) (i.e., the empty volume in the MFI framework) and theexternal surface area (Se) (i.e., the area not belonging to microporesin the MFI framework), were determined by t-test25 in the t-rangefrom 5 Å to 15 Å for which the Harkins and Jura reference isothermequation was used.26
Elemental analyses of carbon, hydrogen, and nitrogen (whichshould belong to carbonaceous deposits present in the samples) weremeasured with a Model EA CHNS-O 1110 Carlo Erba analyzer whoseaccuracy is ±0.01 wt %, ±0.02 wt %, and ±0.02 wt % for C, H and N,respectively.Thermogravimetric analysis (TGA) was performed by a Stanton
Redcroft Model STA-781 analyzer under flowing N2 (20 mL/min),increasing the temperature from room temperature up to 993 K at aheating rate of 5 K/min.2.2.2. Electron Paramagnetic Resonance (EPR) and Diffuse
Reflectance Spectroscopy (DRS). The EPR spectra were recorded atroom temperature on a Varian E-9 spectrometer (X-band), equippedwith a TE102 cavity and an online computer for data processing.Spin-Hamiltonian parameters (g and A values) were obtained fromspectra calculated with the program SIM14 A.27 The g-values werecomputed taking as reference the sharp peak at g = 2.0008 of the E1′center formed by the UV irradiation of one of the two silica dewarsused as sample holders28 (one was irradiated and the other was notirradiated). The EPR spectra were collected on the “as-prepared”catalysts which, after treatment in a vacuum line under differentconditions (vide infra), were transferred to the spectrometer in apurposely designed sealed reactor. The treatments in the vacuum linefollowed this sequence: (a) vacuum at room temperature for 1 h,followed by (b) heating in vacuum at increasing temperatures, from393 to 773 K for 1 h, followed by (c) treatment at room temperaturefor 1 h in the presence of H2O (∼20 Torr), followed by (d) treatmentat 773 K for 1 h in dry O2 (∼80 Torr).The concentration, as absolute values, of Cu2+ species (i.e., spins/g,
±10%) was calculated from the integrated area of the spectra, taking asstandard the Varian strong pitch (5 × 1015 spin/cm). This secondarystandard was accurately calibrated by a series of primary standardsincluding Cu(acac)2 (where acac = acetylacetonate) and CuSO4·5H2Oin polycrystalline state.29 The specimen, as powder, was placed in theEPR silica tube (i.d. = 3 mm) in a weighted amount to fill the resonantcavity completely in order to have the so-called “full length geometry”.Under these conditions, the numbers of spins/cm (N) are given by thefollowing equation:
=++
⎡⎣⎢⎢
⎤⎦⎥⎥⎛⎝⎜
⎞⎠⎟N N
g S S
g S SAA
( 1)
( 1)a bb b b
a a a
a
b (1)
where, for the two samples (a and b), Aa and Ab values are therespective integrated areas normalized for the instrumental conditions,ga and gb are the respective average g-values, and Sa and Sb are the
respective spins. Na was then divided by the linear density (g/cm) ofthe sample in the EPR tube to yield the concentration of paramagneticspecies (spins/g).
In situ DRS spectra were recorded at room temperature on a CaryModel 5 spectrometer in the wavelength range of 200−2500 nm.A purposely designed cell with a silica window23 permitted heatingtreatments under vacuum and in O2 (350 Torr) from roomtemperature up to 790 K.
3. RESULTS AND DISCUSSIONFor a more comprehensive understanding of the results, a briefreference to the MFI structure30 is given in the SupportingInformation (Part 1).
3.1. Chemistry of Base Exchange and TexturalProperties. Results concerning the “as-prepared” Cu-ZSM-5zeolites together with their copper loadings are reported inTable 1.Since the number of H+ ions available for exchange should, in
principle, be equal to that of Al3+ ions, it would be expected ahigher exchange capacity of H-ZSM-5-25 with respect to thehomologous H-ZSM-5-80 matrix. Surprisingly, the data inTable 1 indicate that the opposite occurs. Indeed, regardless thecupric salt (nitrate or acetate) used in the exchange procedure,compared to the H-ZSM-5-25, the H-ZSM-5-80 parent materialcan be loaded to higher extent of Cu2+ exchange under thesame experimental conditions and using a copper acetatesolution of the same molarity: the Cu-ZSM-5-80-540 (over-exchanged) appears to be loaded to a much higher coppercontent than the Cu-ZSM-5-25-102 (stoichiometric) sample(see Table 1). These findings can be explained if it is taken intoaccount that single positively charged hydroxy cupric species,Cu(OH)+, are very likely to be involved in the exchangingprocess. Indeed Cu(OH)+ species have been reported to be themost abundant in copper acetate solutions.3,31,32 Moreover, ithas been found that the exchange properties of the H-ZSM-5zeolites with high Si/Al ratio can also be remarkably influencedby the number of internal silanols, which are defects associatedwith Si(IV) vacancies in the zeolite framework.30b,c,33 The lossof one framework Si4+ is counterbalanced by four Si−OHgroups located around the silicon vacancy. These Si−OHgroups are weak Bronsted acid sites with protons (in principle)available for exchange. The number of Si vacancies increases asthe Si/Al ratio increases.30b,c,33 It follows that the H-ZSM-5-80parent material can contain, in its framework, a higher numberof silanol defects, compared to the homologous H-ZSM-5-25matrix, which, in principle, should be free of silanol defects.This can explain why, regardless of the copper salt used (nitrateor acetate), no overexchanged Cu-ZSM-5-25 samples can beprepared while the H-ZSM-5-80 matrix can easily leads to
Table 1. Concentration of the Copper Nitrate or Acetate Solutions Used in the Exchange, Copper Content, Cu/Al Ratio, andCopper Exchange Extent for “As-Prepared” H-ZSM-5 and Cu-ZSM-5 Samples
sample Al−−O−Si per MFI unit cell exchange solutions [Cu2+]a (M) Cu (wt %) Cu/Al extent of Cu2+ exchange (%)
H-ZSM-5-25 3.7Cu-ZSM-5-25-55 3.7 0.10 (n) 1.02 0.28 55Cu-ZSM-5-25-102 3.7 0.10 (a) 1.89 0.51 102
H-ZSM-5-80 1.2Cu-ZSM-5-80-84 1.2 0.10 (n) 0.50 0.42 84Cu-ZSM-5-80-225 1.2 0.010 (a) 1.35 1.13 225Cu-ZSM-5-80-540 1.2 0.10 (a) 3.24 2.70 540
aIn this table, “n” indicates that the sample was prepared using nitrate solution (pH ≅4), and “a” indicates that the sample was prepared using acetatesolution (pH ≅5.5).
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312024
overexchanged Cu-ZSM-5-80 samples (see Table 1). Moreover,our data clearly show that the cation exchange capacity of theinternal silanols is strongly dependent on the solution pH.Indeed, at pH ∼4, i.e., using copper nitrate solutions, it is nolonger possible to obtain overexchanged Cu-ZSM-5-80 samples,confirming the weak Bronsted acidity of internal silanols.30c
The results of the textural characterization (Table 2) showthat the internal and external specific surface area, as well as themicropore volume of both parent H-ZSM-5 zeolites, are onlyslightly modified by the presence of copper species in all the“as-prepared” Cu-ZSM-5 samples at lower copper content(Cu-ZSM-5-25-102 and Cu-ZSM-5-80-84; see Table 2).By contrast, in the case of the overexchanged catalysts asCu-ZSM-5-80-225 and especially Cu-ZSM-5-80-540, a decreaseof the total specific surface area and of the micropore volumewith respect to their H-ZSM-5-80 matrix occurred, thissuggesting that CuO-like nanoparticles are entrapped withinthe zeolite structure.3
It should be noted that, after the high-temperaturetreatments of overexchanged samples, especially in the presenceof water, the CuO nanoclusters tend to move to the externalsurface of the ZSM-5 zeolite and increase their size.2,3,5,32
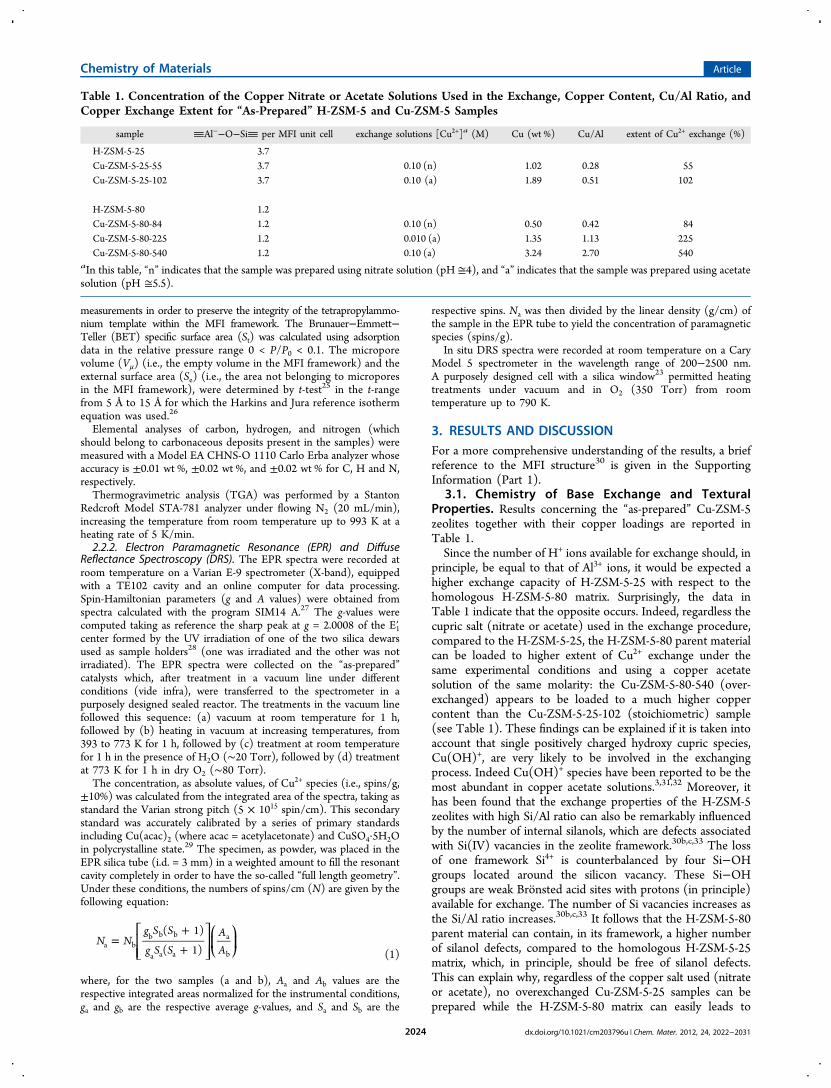
3.2. Local Symmetry of Cu2+-Exchanged Species. TheEPR spectra of “as-prepared” Cu-ZSM-5-25-102 (Cu = 1.89 wt %)and Cu-ZSM-5-80-225 (Cu = 1.35 wt %), after vacuum treat-ment at room temperature are reported in Figure 1. The spectraexhibit an axial, broad, and scarcely resolved signalwith hyperfine structure in the parallel region. The spectrasimulation gave for both samples similar spin-Hamiltonianparameters, which are reported in Table 3. These parametersare typical of isolated Cu2+ hydrated complexes, i.e.,[Cu(H2O)6]
2+ and [Cu(H2O)5(OH)]+, in a distorted octahe-
dral symmetry (Table 3: A species, 6-coordinate Cu2+ ions,Cu2+6c).
18a,20,32,34−39 These results suggest that, regardlessthe Si/Al and Cu/Al ratios, the spectra of the “as-prepared”Cu-ZSM-5-25-102 and Cu-ZSM-5-80-225 samples are charac-terized by almost-identical features.
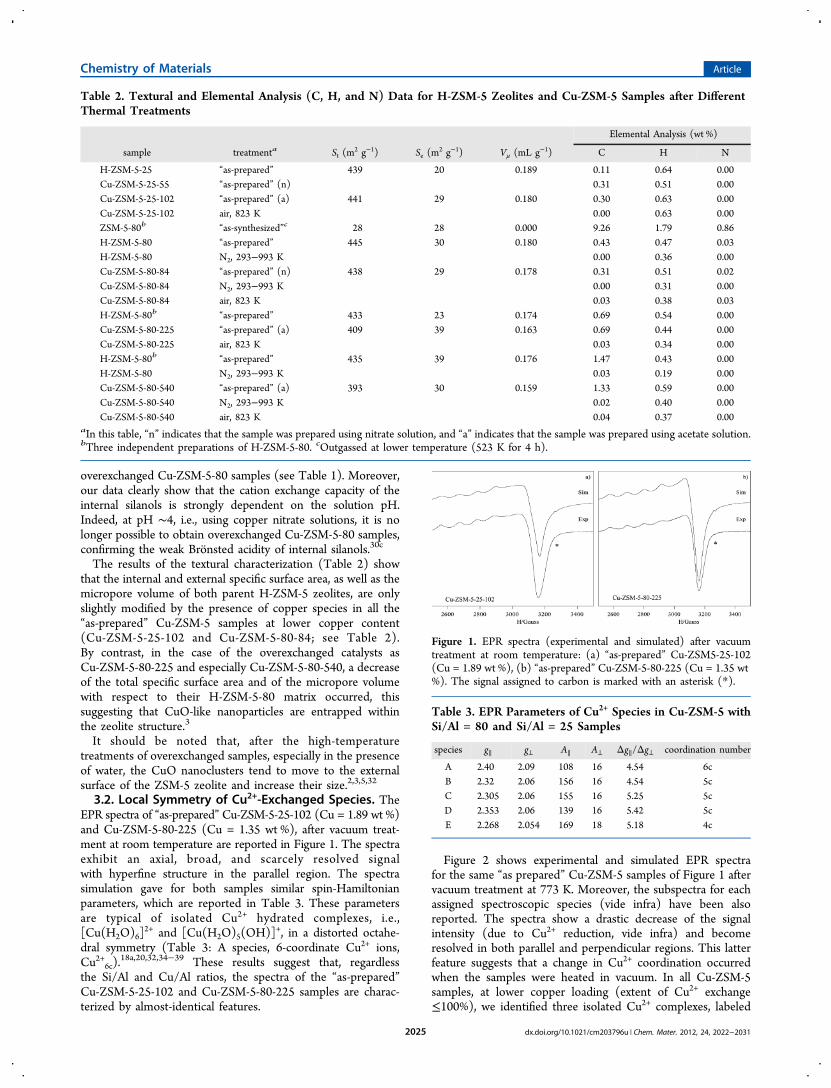
Figure 2 shows experimental and simulated EPR spectrafor the same “as prepared” Cu-ZSM-5 samples of Figure 1 aftervacuum treatment at 773 K. Moreover, the subspectra for eachassigned spectroscopic species (vide infra) have been alsoreported. The spectra show a drastic decrease of the signalintensity (due to Cu2+ reduction, vide infra) and becomeresolved in both parallel and perpendicular regions. This latterfeature suggests that a change in Cu2+ coordination occurredwhen the samples were heated in vacuum. In all Cu-ZSM-5samples, at lower copper loading (extent of Cu2+ exchange≤100%), we identified three isolated Cu2+ complexes, labeled
Table 2. Textural and Elemental Analysis (C, H, and N) Data for H-ZSM-5 Zeolites and Cu-ZSM-5 Samples after DifferentThermal Treatments
Elemental Analysis (wt %)
sample treatmenta St (m2 g−1) Se (m
2 g−1) Vμ (mL g−1) C H N
H-ZSM-5-25 “as-prepared” 439 20 0.189 0.11 0.64 0.00Cu-ZSM-5-25-55 “as-prepared” (n) 0.31 0.51 0.00Cu-ZSM-5-25-102 “as-prepared” (a) 441 29 0.180 0.30 0.63 0.00Cu-ZSM-5-25-102 air, 823 K 0.00 0.63 0.00ZSM-5-80b “as-synthesized”c 28 28 0.000 9.26 1.79 0.86H-ZSM-5-80 “as-prepared” 445 30 0.180 0.43 0.47 0.03H-ZSM-5-80 N2, 293−993 K 0.00 0.36 0.00Cu-ZSM-5-80-84 “as-prepared” (n) 438 29 0.178 0.31 0.51 0.02Cu-ZSM-5-80-84 N2, 293−993 K 0.00 0.31 0.00Cu-ZSM-5-80-84 air, 823 K 0.03 0.38 0.03H-ZSM-5-80b “as-prepared” 433 23 0.174 0.69 0.54 0.00Cu-ZSM-5-80-225 “as-prepared” (a) 409 39 0.163 0.69 0.44 0.00Cu-ZSM-5-80-225 air, 823 K 0.03 0.34 0.00H-ZSM-5-80b “as-prepared” 435 39 0.176 1.47 0.43 0.00H-ZSM-5-80 N2, 293−993 K 0.03 0.19 0.00Cu-ZSM-5-80-540 “as-prepared” (a) 393 30 0.159 1.33 0.59 0.00Cu-ZSM-5-80-540 N2, 293−993 K 0.02 0.40 0.00Cu-ZSM-5-80-540 air, 823 K 0.04 0.37 0.00
aIn this table, “n” indicates that the sample was prepared using nitrate solution, and “a” indicates that the sample was prepared using acetate solution.bThree independent preparations of H-ZSM-5-80. cOutgassed at lower temperature (523 K for 4 h).
Figure 1. EPR spectra (experimental and simulated) after vacuumtreatment at room temperature: (a) “as-prepared” Cu-ZSM5-25-102(Cu = 1.89 wt %), (b) “as-prepared” Cu-ZSM-5-80-225 (Cu = 1.35 wt%). The signal assigned to carbon is marked with an asterisk (*).
Table 3. EPR Parameters of Cu2+ Species in Cu-ZSM-5 withSi/Al = 80 and Si/Al = 25 Samples
species g∥ g⊥ A∥ A⊥ Δg∥/Δg⊥ coordination number
A 2.40 2.09 108 16 4.54 6cB 2.32 2.06 156 16 4.54 5cC 2.305 2.06 155 16 5.25 5cD 2.353 2.06 139 16 5.42 5cE 2.268 2.054 169 18 5.18 4c
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312025

as species B, C, and E (see Figure 2a and parameters inTable 3). In the overexchanged catalysts (the extent of Cuexchange is≫100%) besides species B and C, one more species(labeled as species D) was detected (see Figure 2b andparameters in Table 3). A further refinement of the assignmentof the Cu2+ signals was obtained by investigating how the EPRspectra change when the Cu-ZSM-5 samples, after treatmentsat 773 K under vacuum or in O2 (vide infra), are contacted withwater. In particular, to this purpose, the Cu2+ coordination andthe crystal-field axial component have been correlated to twospecific spectral features, namely, the Δg∥/Δg⊥ ratio and the A∥value [ref 40 and references therein]. The 6-coordinatedCu2+ complex (species A) shows the lowest value of both theΔg∥/Δg⊥ ratio and the A∥ parameters, indicating that the otherspecies (B, C, D, and E) have a higher crystal-field axialcomponent and a coordination number lower than 6 (Table 3).Such changes in the symmetry of the Cu2+ ions are notspecific for the ZSM-5 zeolite: they also occur in otherzeolites.14,15,17,18a,b,20,34−39,41−44 We assign species B, C, and Dto isolated copper complexes in square-pyramidal configuration,i.e., Cu2+5c, (possibly differing in the crystal-field symmetry),and the species E to isolated copper complexes in square-planarconfiguration (Cu2+4c). Accordingly, the signals with g∥ = 2.30−2.33 were assigned to square-pyramidal 5-coordinated Cu2+
species, while the signals with g∥ = 2.26−2.28 were assigned tosquare-planar 4-coordinated Cu2+ species.14,15,41,42 Therefore,the EPR investigation confirmed that, as the temperatureincreases, water ligands are gradually replaced by frameworkoxygen. This process leads to a decrease in the Cu2+
coordination, which changes from octahedral into squarepyramidal for the overexchanged Cu-ZSM-5 samples andfrom octahedral into square planar symmetry for the Cu-ZSM-5samples at lower extent of Cu2+ exchange. It should be noted thatwhen H2O was added to the Cu-ZSM-5-25 and Cu-ZSM-5-80samples treated under vacuum, species B, C, D, and E changedto species A, namely, the 6-coordinated Cu2+ ion. The aboveassignments are in agreement with the “Blumberg-Peisachcorrelation plot approach”, which was used by Larsen and co-workers for the analysis of their EPR spectra of Cu2+-exchanged
zeolites.44 On the other hand, it is worth noting that, in all ofour EPR spectra, and regardless of any treatment,no signal at half field due to Cu2+ ion pairs has been detected.This cannot exclude the presence of coupled Cu2+ ions, which,however, if present in our overexchanged Cu-ZSM-5 pre-parations, are certainly EPR silent. For instance, Cu2+ ion pairswere identified by EPR in CuO45 and in Cu−Y.46 Evidently, thegeometry of the Cu2+−O(H)−Cu2+ bonds is unique within theMFI structure giving rise, through the extra-framework oxygen,to strong antiferromagnetic exchange interaction between thecoupled Cu2+ spins, which change to EPR-silent species.
3.3. Carbonaceous Residues in H-ZSM-5 and Cu-ZSM-5Materials and the Reduction of Cu2+ Species. The resultsof the elemental analyses show that all the “as-prepared”H-ZSM-5 and Cu-ZSM-5 samples have no nitrogen but docontain a significant amount of carbon (see Table 2). Moreover,these data also clearly prove that copper acetate solutionscannot be the source of the carbon contamination, because,for the “as-prepared” Cu-ZSM-5-80 samples obtained via ionexchange in the copper acetate solution, there is no increase ofcarbon with respect to their “as-prepared” H-ZSM-5-80 parentmaterial. This suggests that the acetate species have been easilyeliminated during the final washing of the samples with water.On the other hand, the tetrapropylammonium used as anorganic template for the H-ZSM-5-80 preparation, by itself,can be a large reservoir of carbon in the solid. Indeed, in the“as-synthesized” ZSM-5-80 sample, still containing the organictemplate, carbon amounts to 9.26 wt % and nitrogen to0.86 wt % (Table 2), data almost perfectly matching thetheoretical values of C and N when the ZSM-5 structure is filledwith 4 tetrapropylammonium ions per unit cell (u.c.), with 4being the maximum number allowed by the MFI structure inwhich 4 channels intersections are present per u.c.30 In order toremove the carbon residue, the “as-synthesized” ZSM-5-80sample was heated in air at 823 K for 5 h. It should be recalledthat such a treatment usually represents the last step in thesynthesis of “as-prepared” H-ZSM-5 zeolites.24 Despite heatingin air at 823 for 5 h, a significant amount of carbon stillremains in the solid (see Table 2), namely, the “as-prepared”
Figure 2. EPR spectra (experimental and simulated) after vacuum treatment at 773 K of (a) Cu-ZSM-5-25-102 (Cu =1.89 wt %) and (b) Cu-ZSM-5-80-225 (Cu = 1.35 wt %). The subspectra of the species used in the simulation are sketched below both the simulated spectra. Magnification of thelow field hyperfine structure is presented in the inset. The signal assigned to carbon is marked with an asterisk (*).
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312026
H-ZSM-5-80 parent materials. This finding clearly proves thatcarbonaceous deposits can be left in the “as-prepared” samplesessentially as a consequence of a incomplete burning of thetetrapropylammonium template used in the synthesis in air athigh temperature. This is indeed confirmed by the “as-prepared”Cu-ZSM-5-80 samples that, originating from the rich-in-carbonH-ZSM-5-80 parent material, still contain about the sameamount of carbon (see Table 2). Also the “as-prepared”H-ZSM-25 and Cu-ZSM-5-25 samples are characterized bythe presence of carbon, although in a much lesser amount(see Table 2). In any case, we recall that EPR, because of itshigh sensitivity, is a very powerful tool for checking traces ofcarbonaceous deposits that almost always contain paramagneticcarbon species (vide infra).47
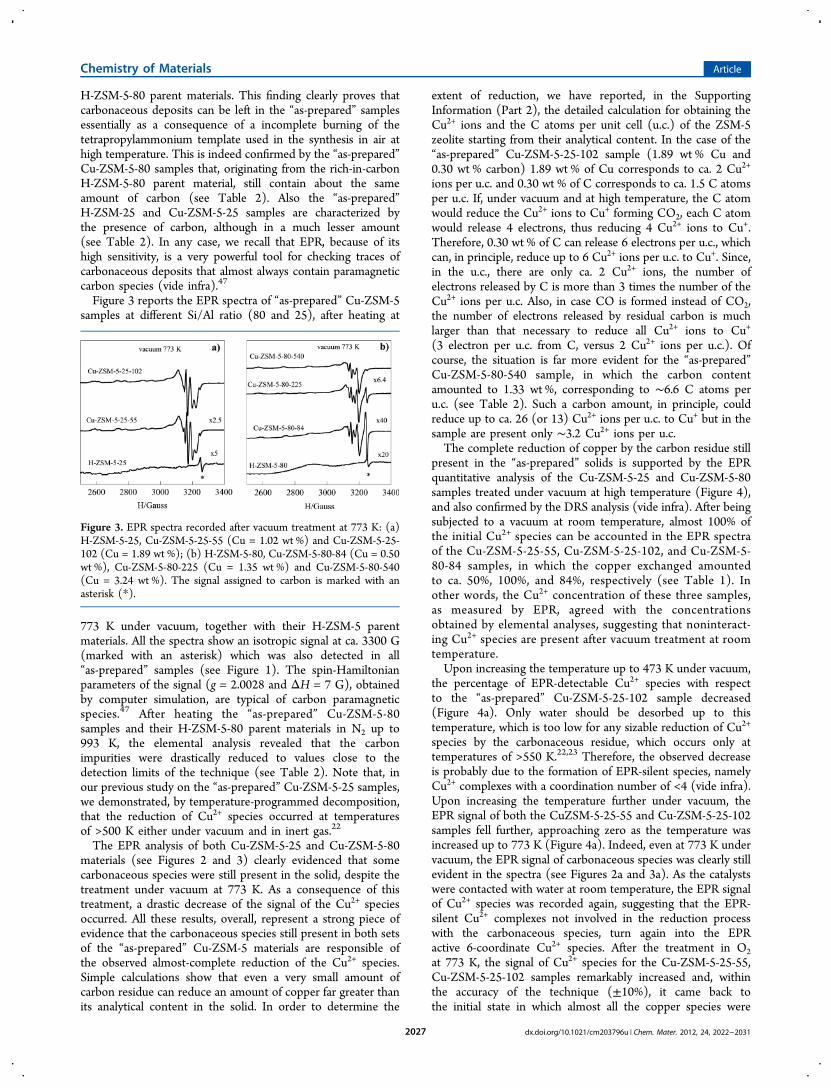
Figure 3 reports the EPR spectra of “as-prepared” Cu-ZSM-5samples at different Si/Al ratio (80 and 25), after heating at
773 K under vacuum, together with their H-ZSM-5 parentmaterials. All the spectra show an isotropic signal at ca. 3300 G(marked with an asterisk) which was also detected in all“as-prepared” samples (see Figure 1). The spin-Hamiltonianparameters of the signal (g = 2.0028 and ΔH = 7 G), obtainedby computer simulation, are typical of carbon paramagneticspecies.47 After heating the “as-prepared” Cu-ZSM-5-80samples and their H-ZSM-5-80 parent materials in N2 up to993 K, the elemental analysis revealed that the carbonimpurities were drastically reduced to values close to thedetection limits of the technique (see Table 2). Note that, inour previous study on the “as-prepared” Cu-ZSM-5-25 samples,we demonstrated, by temperature-programmed decomposition,that the reduction of Cu2+ species occurred at temperaturesof >500 K either under vacuum and in inert gas.22
The EPR analysis of both Cu-ZSM-5-25 and Cu-ZSM-5-80materials (see Figures 2 and 3) clearly evidenced that somecarbonaceous species were still present in the solid, despite thetreatment under vacuum at 773 K. As a consequence of thistreatment, a drastic decrease of the signal of the Cu2+ speciesoccurred. All these results, overall, represent a strong piece ofevidence that the carbonaceous species still present in both setsof the “as-prepared” Cu-ZSM-5 materials are responsible ofthe observed almost-complete reduction of the Cu2+ species.Simple calculations show that even a very small amount ofcarbon residue can reduce an amount of copper far greater thanits analytical content in the solid. In order to determine the
extent of reduction, we have reported, in the SupportingInformation (Part 2), the detailed calculation for obtaining theCu2+ ions and the C atoms per unit cell (u.c.) of the ZSM-5zeolite starting from their analytical content. In the case of the“as-prepared” Cu-ZSM-5-25-102 sample (1.89 wt % Cu and0.30 wt % carbon) 1.89 wt % of Cu corresponds to ca. 2 Cu2+
ions per u.c. and 0.30 wt % of C corresponds to ca. 1.5 C atomsper u.c. If, under vacuum and at high temperature, the C atomwould reduce the Cu2+ ions to Cu+ forming CO2, each C atomwould release 4 electrons, thus reducing 4 Cu2+ ions to Cu+.Therefore, 0.30 wt % of C can release 6 electrons per u.c., whichcan, in principle, reduce up to 6 Cu2+ ions per u.c. to Cu+. Since,in the u.c., there are only ca. 2 Cu2+ ions, the number ofelectrons released by C is more than 3 times the number of theCu2+ ions per u.c. Also, in case CO is formed instead of CO2,the number of electrons released by residual carbon is muchlarger than that necessary to reduce all Cu2+ ions to Cu+
(3 electron per u.c. from C, versus 2 Cu2+ ions per u.c.). Ofcourse, the situation is far more evident for the “as-prepared”Cu-ZSM-5-80-540 sample, in which the carbon contentamounted to 1.33 wt %, corresponding to ∼6.6 C atoms peru.c. (see Table 2). Such a carbon amount, in principle, couldreduce up to ca. 26 (or 13) Cu2+ ions per u.c. to Cu+ but in thesample are present only ∼3.2 Cu2+ ions per u.c.The complete reduction of copper by the carbon residue still
present in the “as-prepared” solids is supported by the EPRquantitative analysis of the Cu-ZSM-5-25 and Cu-ZSM-5-80samples treated under vacuum at high temperature (Figure 4),and also confirmed by the DRS analysis (vide infra). After beingsubjected to a vacuum at room temperature, almost 100% ofthe initial Cu2+ species can be accounted in the EPR spectraof the Cu-ZSM-5-25-55, Cu-ZSM-5-25-102, and Cu-ZSM-5-80-84 samples, in which the copper exchanged amountedto ca. 50%, 100%, and 84%, respectively (see Table 1). Inother words, the Cu2+ concentration of these three samples,as measured by EPR, agreed with the concentrationsobtained by elemental analyses, suggesting that noninteract-ing Cu2+ species are present after vacuum treatment at roomtemperature.Upon increasing the temperature up to 473 K under vacuum,
the percentage of EPR-detectable Cu2+ species with respectto the “as-prepared” Cu-ZSM-5-25-102 sample decreased(Figure 4a). Only water should be desorbed up to thistemperature, which is too low for any sizable reduction of Cu2+
species by the carbonaceous residue, which occurs only attemperatures of >550 K.22,23 Therefore, the observed decreaseis probably due to the formation of EPR-silent species, namelyCu2+ complexes with a coordination number of <4 (vide infra).Upon increasing the temperature further under vacuum, theEPR signal of both the CuZSM-5-25-55 and Cu-ZSM-5-25-102samples fell further, approaching zero as the temperature wasincreased up to 773 K (Figure 4a). Indeed, even at 773 K undervacuum, the EPR signal of carbonaceous species was clearly stillevident in the spectra (see Figures 2a and 3a). As the catalystswere contacted with water at room temperature, the EPR signalof Cu2+ species was recorded again, suggesting that the EPR-silent Cu2+ complexes not involved in the reduction processwith the carbonaceous species, turn again into the EPRactive 6-coordinate Cu2+ species. After the treatment in O2at 773 K, the signal of Cu2+ species for the Cu-ZSM-5-25-55,Cu-ZSM-5-25-102 samples remarkably increased and, withinthe accuracy of the technique (±10%), it came back tothe initial state in which almost all the copper species were
Figure 3. EPR spectra recorded after vacuum treatment at 773 K: (a)H-ZSM-5-25, Cu-ZSM-5-25-55 (Cu = 1.02 wt %) and Cu-ZSM-5-25-102 (Cu = 1.89 wt %); (b) H-ZSM-5-80, Cu-ZSM-5-80-84 (Cu = 0.50wt %), Cu-ZSM-5-80-225 (Cu = 1.35 wt %) and Cu-ZSM-5-80-540(Cu = 3.24 wt %). The signal assigned to carbon is marked with anasterisk (*).
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312027
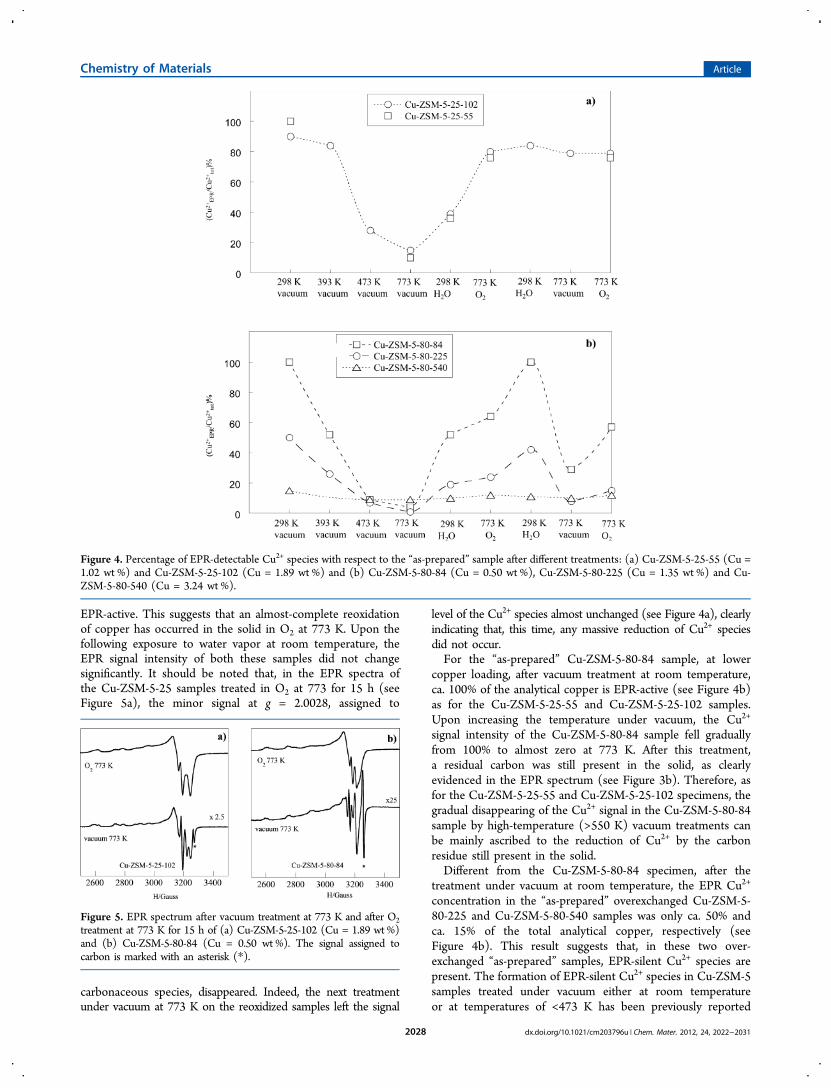
EPR-active. This suggests that an almost-complete reoxidationof copper has occurred in the solid in O2 at 773 K. Upon thefollowing exposure to water vapor at room temperature, theEPR signal intensity of both these samples did not changesignificantly. It should be noted that, in the EPR spectra ofthe Cu-ZSM-5-25 samples treated in O2 at 773 for 15 h (seeFigure 5a), the minor signal at g = 2.0028, assigned to
carbonaceous species, disappeared. Indeed, the next treatmentunder vacuum at 773 K on the reoxidized samples left the signal
level of the Cu2+ species almost unchanged (see Figure 4a), clearlyindicating that, this time, any massive reduction of Cu2+ speciesdid not occur.For the “as-prepared” Cu-ZSM-5-80-84 sample, at lower
copper loading, after vacuum treatment at room temperature,ca. 100% of the analytical copper is EPR-active (see Figure 4b)as for the Cu-ZSM-5-25-55 and Cu-ZSM-5-25-102 samples.Upon increasing the temperature under vacuum, the Cu2+
signal intensity of the Cu-ZSM-5-80-84 sample fell graduallyfrom 100% to almost zero at 773 K. After this treatment,a residual carbon was still present in the solid, as clearlyevidenced in the EPR spectrum (see Figure 3b). Therefore, asfor the Cu-ZSM-5-25-55 and Cu-ZSM-5-25-102 specimens, thegradual disappearing of the Cu2+ signal in the Cu-ZSM-5-80-84sample by high-temperature (>550 K) vacuum treatments canbe mainly ascribed to the reduction of Cu2+ by the carbonresidue still present in the solid.Different from the Cu-ZSM-5-80-84 specimen, after the
treatment under vacuum at room temperature, the EPR Cu2+
concentration in the “as-prepared” overexchanged Cu-ZSM-5-80-225 and Cu-ZSM-5-80-540 samples was only ca. 50% andca. 15% of the total analytical copper, respectively (seeFigure 4b). This result suggests that, in these two over-exchanged “as-prepared” samples, EPR-silent Cu2+ species arepresent. The formation of EPR-silent Cu2+ species in Cu-ZSM-5samples treated under vacuum either at room temperatureor at temperatures of <473 K has been previously reported
Figure 4. Percentage of EPR-detectable Cu2+ species with respect to the “as-prepared” sample after different treatments: (a) Cu-ZSM-5-25-55 (Cu =1.02 wt %) and Cu-ZSM-5-25-102 (Cu = 1.89 wt %) and (b) Cu-ZSM-5-80-84 (Cu = 0.50 wt %), Cu-ZSM-5-80-225 (Cu = 1.35 wt %) and Cu-ZSM-5-80-540 (Cu = 3.24 wt %).
Figure 5. EPR spectrum after vacuum treatment at 773 K and after O2treatment at 773 K for 15 h of (a) Cu-ZSM-5-25-102 (Cu = 1.89 wt %)and (b) Cu-ZSM-5-80-84 (Cu = 0.50 wt %). The signal assigned tocarbon is marked with an asterisk (*).
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312028
by us,32 Turnes Palomino et al.,18a and Soria et al.36 In theoverexchanged Cu-ZSM-5-80-540 sample, most of the copperspecies are in the form of superparamagnetic CuO nano-particles, as suggested by the textural analysis (vide supra), aswell as also by our recent magnetic susceptibility results.3 TheseCuO superparamagnetic particles in the Cu-ZSM-5-80-540sample are EPR-silent, thus explaining the very low intensity ofthe EPR signal, which remains almost unaffected by any treat-ment under vacuum or in O2, even at 773 K.The EPR spectra of the overexchanged Cu-ZSM-5-80-225
and Cu-ZSM-5-80-540 samples after the treatment under vacuumat 773 K (see Figure 3b) still show the signal typical of carbonspecies. Since EPR cannot quantitatively assay the Cu2+concentra-tion, in order to prove that the carbon residue is responsible forthe almost-complete reduction of copper, a specific DRS studywas made on the overexchanged Cu-ZSM-5-80-540 sample bycarrying out “in situ” treatments under vacuum or in O2 atdifferent temperatures (vide infra).However, it should be noted that, when a Cu-ZSM-5 sample
is treated under vacuum at high temperature, a “self-reduction”process can also very likely to occur. However, the “self-reduction” process cannot justify the observed quasi-completereduction of Cu2+ ions, because it involves the reduction ofonly a small fraction (5%−10%) of the total copper, as alsoconfirmed by our DRS study (vide infra) and in agreement withprevious literature data.6,8,32
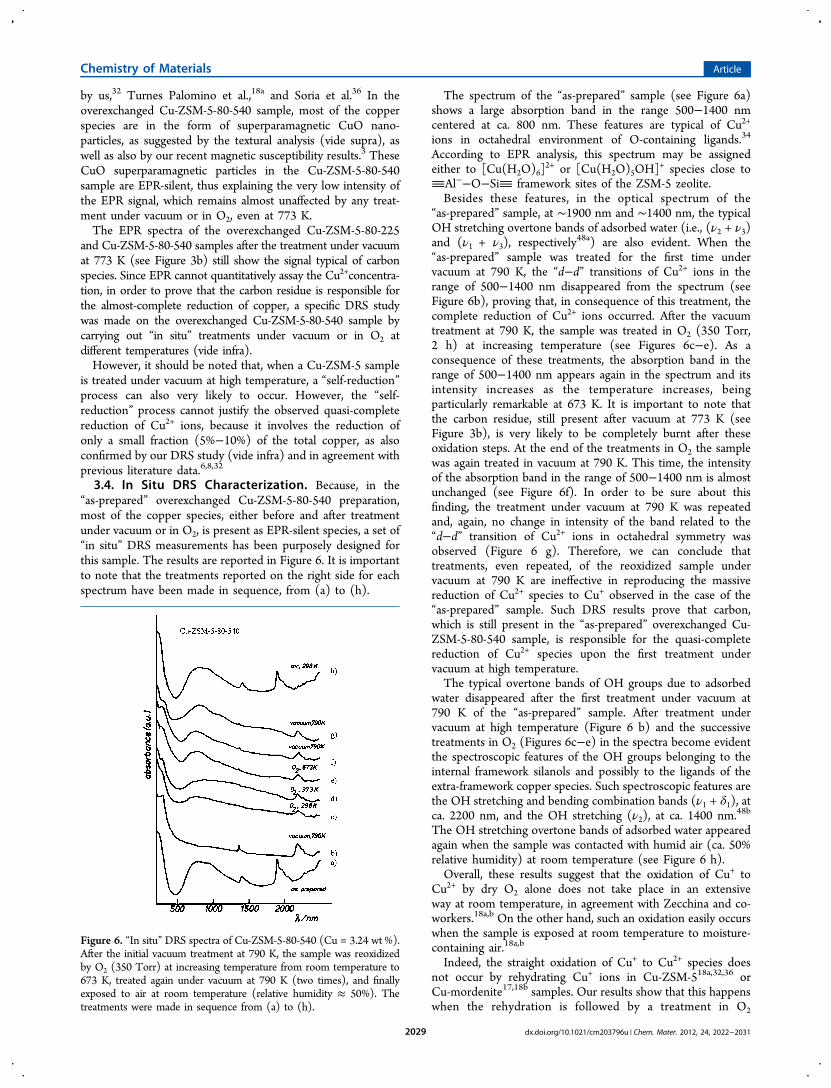
3.4. In Situ DRS Characterization. Because, in the“as-prepared” overexchanged Cu-ZSM-5-80-540 preparation,most of the copper species, either before and after treatmentunder vacuum or in O2, is present as EPR-silent species, a set of“in situ” DRS measurements has been purposely designed forthis sample. The results are reported in Figure 6. It is importantto note that the treatments reported on the right side for eachspectrum have been made in sequence, from (a) to (h).
The spectrum of the “as-prepared” sample (see Figure 6a)shows a large absorption band in the range 500−1400 nmcentered at ca. 800 nm. These features are typical of Cu2+
ions in octahedral environment of O-containing ligands.34
According to EPR analysis, this spectrum may be assignedeither to [Cu(H2O)6]
2+ or [Cu(H2O)5OH]+ species close to
Al−−O−Si framework sites of the ZSM-5 zeolite.Besides these features, in the optical spectrum of the
“as-prepared” sample, at ∼1900 nm and ∼1400 nm, the typicalOH stretching overtone bands of adsorbed water (i.e., (ν2 + ν3)and (ν1 + ν3), respectively
48a) are also evident. When the“as-prepared” sample was treated for the first time undervacuum at 790 K, the “d−d” transitions of Cu2+ ions in therange of 500−1400 nm disappeared from the spectrum (seeFigure 6b), proving that, in consequence of this treatment, thecomplete reduction of Cu2+ ions occurred. After the vacuumtreatment at 790 K, the sample was treated in O2 (350 Torr,2 h) at increasing temperature (see Figures 6c−e). As aconsequence of these treatments, the absorption band in therange of 500−1400 nm appears again in the spectrum and itsintensity increases as the temperature increases, beingparticularly remarkable at 673 K. It is important to note thatthe carbon residue, still present after vacuum at 773 K (seeFigure 3b), is very likely to be completely burnt after theseoxidation steps. At the end of the treatments in O2 the samplewas again treated in vacuum at 790 K. This time, the intensityof the absorption band in the range of 500−1400 nm is almostunchanged (see Figure 6f). In order to be sure about thisfinding, the treatment under vacuum at 790 K was repeatedand, again, no change in intensity of the band related to the“d−d” transition of Cu2+ ions in octahedral symmetry wasobserved (Figure 6 g). Therefore, we can conclude thattreatments, even repeated, of the reoxidized sample undervacuum at 790 K are ineffective in reproducing the massivereduction of Cu2+ species to Cu+ observed in the case of the“as-prepared” sample. Such DRS results prove that carbon,which is still present in the “as-prepared” overexchanged Cu-ZSM-5-80-540 sample, is responsible for the quasi-completereduction of Cu2+ species upon the first treatment undervacuum at high temperature.The typical overtone bands of OH groups due to adsorbed
water disappeared after the first treatment under vacuum at790 K of the “as-prepared” sample. After treatment undervacuum at high temperature (Figure 6 b) and the successivetreatments in O2 (Figures 6c−e) in the spectra become evidentthe spectroscopic features of the OH groups belonging to theinternal framework silanols and possibly to the ligands of theextra-framework copper species. Such spectroscopic features arethe OH stretching and bending combination bands (ν1 + δ1), atca. 2200 nm, and the OH stretching (ν2), at ca. 1400 nm.48b
The OH stretching overtone bands of adsorbed water appearedagain when the sample was contacted with humid air (ca. 50%relative humidity) at room temperature (see Figure 6 h).Overall, these results suggest that the oxidation of Cu+ to
Cu2+ by dry O2 alone does not take place in an extensiveway at room temperature, in agreement with Zecchina and co-workers.18a,b On the other hand, such an oxidation easily occurswhen the sample is exposed at room temperature to moisture-containing air.18a,b
Indeed, the straight oxidation of Cu+ to Cu2+ species doesnot occur by rehydrating Cu+ ions in Cu-ZSM-518a,32,36 orCu-mordenite17,18b samples. Our results show that this happenswhen the rehydration is followed by a treatment in O2
Figure 6. “In situ” DRS spectra of Cu-ZSM-5-80-540 (Cu = 3.24 wt %).After the initial vacuum treatment at 790 K, the sample was reoxidizedby O2 (350 Torr) at increasing temperature from room temperature to673 K, treated again under vacuum at 790 K (two times), and finallyexposed to air at room temperature (relative humidity ≈ 50%). Thetreatments were made in sequence from (a) to (h).
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312029

(see Figures 6g and 6h). Moreover, even after treatment of theoverexchanged Cu-ZSM-5-80-540 sample in O2 at hightemperature, there is no evidence of the typical absorptionedge at ca. 890 nm due to the energy gap of bulk CuO(ca. 1.4 eV), confirming that no large CuO particles are formedon the external surface of the zeolite.3 In the overexchangedsamples, the copper species are very likely to be present in theform of CuO nanoparticles, behaving as superparamagneticspecies, as suggested by EPR and our previous magneticsusceptibility study.3
Finally, it should be noted that when the reoxidized“carbon-free”sample is treated under vacuum at hightemperature, a “self-reduction” process is very likely to occur.However, the intensity of the band due to “d−d” transitions ofCu2+ ions remains almost unchanged (see Figures 6e and 6f andFigures 6f and 6g), confirming that the “self-reduction” processinvolves the reduction of only a rather small fraction of the totalcopper, as previously suggested in the literature.6,8,32
4. CONCLUSIONSEither in a commercial H-ZSM-5-25 zeolite and in a laboratory-made H-ZSM-5-80 zeolite, small amounts of carbon were foundby elemental analysis. The carbon amount in H-ZSM-5-80 wasmuch higher than in H-ZSM-5-25 zeolite, possibly due to aresidue of the organic template used for the synthesis and notcompletely removed after the standard treatment in air at hightemperature at the end of the preparation. Upon treatmentsof the “as-prepared” Cu-ZSM-5-25 and Cu-ZSM-5-80 samplesunder inert gas (He, N2) flow or under vacuum at hightemperatures, a complete reduction of Cu2+ ions occurs. Theresults coming from a combined use of elemental analysis,EPR and DR spectroscopy have given strong evidence thatthis unexpected quasi-complete reduction of Cu2+ ions in the“as-prepared’ Cu-ZSM-5 samples is caused by the carbonresidue that is still remaining in the H-ZSM-5 parent materials.These carbonaceous impurities, even when escape the detectionaccuracy of the elemental analysis, can be checked by a carefulEPR analysis. A complete removal of the carbon residue isobtained only by repeating or prolonging the treatment in O2,or air, at high temperature. As a general advice, the zeolite-basedmaterials, especially those prepared starting from an organictemplate, should be adequately pretreated in order to avoid thatcarbonaceous species still present in the solid can give rise tounexpected chemistry.
■ ASSOCIATED CONTENT*S Supporting InformationSupplemental material is provided in two parts. Part 1 describesthe MFI structure, and Part 2 describes the calulation of howmany Cu2+ and C ions are present per unit cell. This material isavailable free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*Tel.: +39-06-49913382. Fax: +39-06-490324. E-mail: [email protected] authors declare no competing financial interest.
■ DEDICATIONAll the authors dedicate this work to the memory of Prof. PieroPorta (1933−2012).
■ REFERENCES(1) (a) Shelef, M. Chem. Rev. 1995, 95, 209−225. (b) Parvulescu, V.I.; Grange, P.; Delmon, B. Catal. Today 1998, 46, 233−316. (c) Garin,F. Appl. Catal. A 2001, 222, 183−219. (d) Yashnik, S. A.; Ismagilov, Z.R.; Anufrienko, V. F. Catal. Today 2005, 110, 310−322. (e) Olsson, L.;Sjovall, H.; Blint, R. J. Appl. Catal. B 2009, 87, 200−210.(f) Vanelderen, P.; Hadt, R. G.; Smeets, P. J.; Solomon, E. I.;Schoonheydt, R. A.; Sels, B. F. J. Catal. 2011, 284, 157−164.(2) (a) Ciambelli, P.; Corbo, P.; Gambino, M.; Minelli, G.; Moretti,G.; Porta, P. Catal. Today 1995, 26, 33−39. (b) Moretti, G.; Minelli,G.; Porta, P.; Ciambelli, P.; Corbo, P.; Gambino, M.; Migliardini, F.;Iacoponi, S. Stud. Surf. Sci. Catal. 1997, 105, 1525−1532.(3) Fierro, G.; Ferraris, G.; Moretti, G. Appl. Catal. B 2009, 91, 499−506.(4) (a) Sano, T.; Yamashita, N.; Iwami, Y.; Takeda, K.; Kawakami, Y.Zeolites 1996, 16, 258−264. (b) Sano, T.; Ikeda, H.; Kasuno, T.;Wang, Z. B.; Kawakami, Y.; Soga, K. Zeolites 1997, 19, 80−86.(5) (a) Sarkany, J.; D’Itri, J. L.; Sachtler, W. M. H. Catal. Lett. 1992,16, 241−249. (b) Beutel, T.; Sarkany, J.; Lei, G.-D.; Yan, J. Y.; Sachtler,W. M. H. J. Phys. Chem. 1996, 100, 845−851.(6) Moretti, G.; Ferraris, G.; Fierro, G.; Lo Jacono, M.; Morpurgo, S.;Faticanti, M. J. Catal. 2005, 232, 476−487.(7) (a) Spoto, G.; Bordiga, S.; Scarano, D.; Zecchina, A. Catal. Lett.1992, 13, 39−44. (b) Spoto, G.; Zecchina, A.; Bordiga, S.; Ricchiardi,G.; Martra, G.; Leofanti, G.; Petrini, G. Appl. Catal. B 1994, 3, 151−172.(8) (a) Li, Y.; Hall, W. K. J. Catal. 1991, 129, 202−215; and J. Phys.Chem. 1990, 94, 6145−6148. (b) Hall, W. K.; Valyon, J. Catal. Lett.1992, 15, 311−315. (c) Jang, H.-J.; Hall, W. K.; d’Itri, J. L. J. Phys.Chem. 1996, 100, 9416−9420. (d) Valyon, J.; Hall, W. K. In NewFrontiers in Catalysis, Proceedings of the 10th International Congress onCatalysis; Guczi, L. et al., Eds.; Elsevier: Amsterdam, 1993; pp 1339−1350.(9) (a) Iwamoto, M.; Yahiro, H.; Mizuno, N.; Zang, W.; Mine, Y.;Furukawa, H.; Kagawa, S. J. Phys. Chem. 1992, 96, 9360−9366.(b) Iwamoto, M.; Yahiro, H.; Mizuno, N.; Tanda, K.; Mine, Y.;Kagawa, S. J. Phys. Chem. 1991, 95, 3727−3730. (c) Iwamoto, M.;Furukawa, H.; Mine, Y.; Uemura, F.; Mikuriya, S.; Kagawa, S. J. Chem.Soc., Chem. Commun. 1986, 1272−1273. (d) Iwamoto, M.; Furukawa,H.; Kagawa, S. In New Developments in Zeolite Science and Technology,Proceedings of the 7th International Congress on Zeolites; Murakami, Y.et al., Eds.; Elsevier: Amsterdam, 1986; pp 943−949.(10) (a) Groothaert, M. H.; Van Bokhoven, J. A.; Battiston, A. A.;Weckhuysen, B. M.; Schoonheydt, R. A. J. Am. Chem. Soc. 2003, 125,7629−7640. (b) Groothaert, M. H.; Lievens, K.; Leeman, H.;Weckhuysen, B. M.; Schoonheydt, R. A. J. Catal. 2003, 220, 500−512. (c) Groothaert, M. H.; Lievens, K.; van Bokhoven, J. A.;Battiston, A. A.; Weckhuysen, B. M.; Pierloot, K.; Schoonheydt, R. A.ChemPhysChem 2003, 4, 626−630.(11) Parrillo, D. J.; Dolenec, D.; Gorte, R. J.; McCabe, R. W J. Catal.1993, 142, 708−718.(12) Moretti, G.; Dossi, C.; Fusi, A.; Recchia, S.; Psaro, R. Appl.Catal. B 1999, 20, 67−73.(13) Shelef, M. Catal. Lett. 1992, 15, 305−310.(14) Kucherov, A. V.; Slinkin, A. A.; Goryashenko, S. S; Slovetskaja,K. I. J. Catal. 1989, 118, 459−465.(15) Kucherov, A. V.; Gerlock, J. L; Jen, H.-W.; Shelef, M. J. Phys.Chem. 1994, 98, 4892−4894.(16) Liu, D.-J.; Robota, H. Catal. Lett. 1993, 21, 291−301.(17) Kasai, P. H.; Bishop, R. J., Jr. J. Phys. Chem. 1977, 81, 1527−1529.(18) (a) Turnes Palomino, G.; Fisicaro, P.; Bordiga, S.; Zecchina, A.;Giamello, E.; Lamberti, C. J. Phys. Chem B 2000, 104, 4064−4073.(b) Llabres i Xamena, F. X.; Fisicaro, P.; Berlier, G.; Zecchina, A.;Turnes Palomino, G.; Prestipino, C.; Bordiga, S.; Giamello, E.;Lamberti, C. J. Phys. Chem. B 2003, 107, 7036−7044.(19) Jacobs, P. A.; De Wilde, W.; Schoonheydt, R. A.; Uytterhoeven,J. B.; Beyer, H. J. Chem. Soc. Faraday Trans. 1 1976, 72, 1221−1230.
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312030

(20) (a) Larsen, S. C.; Aylor, A.; Bell, A. T.; Reimer, J. A. J. Phys.Chem. 1994, 98, 11533−11540. (b) Trout, B. L.; Chakraborty, A. K.;Bell, A. T. J. Phys. Chem. 1996, 100, 4173−4179. (c) Hu, S.; Reimer, J.A.; Bell, A. T. J. Phys. Chem. B 1997, 101, 1869−1871.(21) (a) Guisnet, M.; Magnoux, P. Appl. Catal. 1989, 54, 1−27.(b) Karge, H. G. Stud. Surf. Sci. Catal. 1991, 58, 531−570.(22) Dossi, C.; Fusi, A.; Moretti, G.; Recchia, S.; Psaro, R. Appl.Catal. A 1999, 188, 107−119.(23) Moretti, G.; Ferraris, G.; Galli, P. Stud. Surf. Sci. Catal. 2001,135, 5020−5028.(24) (a) Argauer, R. J.; Landolt, G. R. Crystalline zeolite ZSM-5 andmethod of preparing the same, U.S. Patent 3,702,886, 1972. (b) Bellussi,G.; Carati, A.; Clerici, M. G.; Esposito, A. Stud. Surf. Sci. Catal. 1991,63, 421−429.(25) Gregg, S. J.; Sing, K. S. W. Adsorption, Surface Area and Porosity;Academic Press: London, 1982; p 94.(26) Harkins, W. D.; Jura, G. J. Am. Chem. Soc. 1944, 66, 1362−1366.(27) Lozos, G. P.; Hoffmann, B. M.; Franz, C. G. Quantum ChemistryProgram Exchange (QCPE); Northwestern University, Evanston, IL,1974; Vol. 11, pp 265−269.(28) Griscom, D. L. J. Ceram. Soc. Jpn. 1991, 99, 923−942.(29) Cordischi, D.; Occhiuzzi, M.; Dragone, R. Appl. Magn. Reson.1999, 16, 427−445.(30) (a) Baerlocher, Ch.; McCusker, L. B.; Olson, D. H. Atlas ofZeolite Framework Types, Sixth Revised ed.; Elsevier: Amsterdam, 2007.(b) Zecchina, A.; Bordiga, S.; Spoto, G.; Marchese, L.; Petrini, G.;Leofanti, L.; Padovan, M. J. Phys. Chem. 1992, 96, 4985−4990 and J.Phys. Chem. 1992, 96, 4991−4997. (c) Artioli, G.; Lamberti, C.;Marra, G. L. Acta Crystallogr., Sect. B: Struct. Sci. 2000, 56, 2−10.(31) Jolivet, J.-P. Metal Oxide Chemistry and Synthesis; J. Wiley &Sons, Ltd.: Chichester, U.K., 2000; pp 78−80.(32) Lo Jacono, M.; Fierro, G.; Dragone, R.; Feng, X.; D’Itri, J.; Hall,W. K. J. Phys. Chem. B 1997, 101, 1979−1984.(33) Woolery, G. L.; Alemany, L. B.; Dessau, R. M.; Chester, A. W.Zeolites 1986, 6, 14−16.(34) Schoonheydt, R. A. Catal. Rev.-Sci. Eng. 1993, 35, 129−168.(35) Giamello, E.; Murphy, D.; Magnacca, G.; Morterra, C.; Shioya,Y.; Nomura, T.; Anpo, M. J. Catal. 1992, 136, 510−520.(36) Soria, J.; Martínez-Arias, A.; Martínez-Chaparro, A.; Conesa, J.C.; Schay, Z. J. Catal. 2000, 190, 352−363.(37) Yashnik, A. S.; Ismagilov, Z. R.; Anufrienko, V. F. Catal. Today2005, 110, 310−322.(38) (a) Sojka, Z.; Che, M.; Giamello, E. J. Phys. Chem. B 1997, 101,4831−4838. (b) Gil, B.; Datka, J.; Witkowski, S.; Sojka, Z.; Broclawik,E. Stud. Surf. Sci. Catal. 2000, 130, 3249−3254. (c) Pierzyk, P.; Gil, B.;Sojka, Z. Catal. Today 2007, 126, 103−111.(39) Sass, C. E.; Kevan, L. J. Phys. Chem. 1989, 93, 7856−7859.(40) (a) Cordischi, D.; Campa, M. C.; Indovina, V.; Occhiuzzi, M. J.Chem. Soc., Faraday Trans. 1994, 9, 207−212. (b) Occhiuzzi, M.; Tuti,S.; Cordischi, D.; Dragone, R.; Indovina, V. J. Chem. Soc., FaradayTrans. 1996, 92, 4337−4345.(41) Dedecek, J.; Sobalik, Z.; Tvaruzkova, Z.; Kaucky, D.;Wichterlova, B. J. Phys. Chem. 1995, 99, 6327−16337.(42) (a) Groothaert, M. H.; Pierloot, K.; Delabie, A.; Schoonheydt,R. A. Phys. Chem. Chem. Phys. 2003, 5, 2135−2144. (b) Smeets, P. J.;Groothaert, M. H. R.; van Teeffelen, M.; Leeman, H.; Hensen, E. J.M.; Schoonheydt, R. A. J. Catal. 2007, 245, 358−368.(43) Conesa, J. C.; Soria, J. J. Phys. Chem. 1978, 82, 1847−1850.(44) (a) Carl, P. J.; Larsen, S. C. J. Phys. Chem. B 2000, 104, 6568−6575. (b) Ames, W. M.; Larsen, S. C. J. Phys. Chem. A 2010, 114,6568−6575.(45) Punnoose, A.; Maurya, B. P.; Mathew, J.; Umar, M.; Haque, M.I.; Singh, R. J. Solid State Commun. 1993, 88, 195−198.(46) Chao, C.-C.; Lunsford, J. H. J. Chem. Phys. 1972, 57, 2890−2897.(47) Keeble, D. J.; Robb, K. M.; Smith, G. M.; El Mkami, H.; Rodil,S. E.; Robertson, J. J. Phys.: Condens. Matter 2003, 15, 7463−7468.
(48) (a) Braun, C. L.; Smirnov, S. N. J. Chem. Educ. 1993, 70, 612−614. (b) Kustov, L. M.; Borovkov, V.Yu; Kazansky, V. B. J. Catal. 1981,72, 149−159.
Chemistry of Materials Article
dx.doi.org/10.1021/cm203796u | Chem. Mater. 2012, 24, 2022−20312031









![Nitrous Oxide for Space Propulsion Applications · Ru/Al2O3, Co3O4, FexCe1–xO2, LaCoO3, Fe-ZSM-5, and Cu-ZSM-5 [5–10]. Recently, spinel ... Accordingly, the BET surface area decreased](https://img.pdfslide.us/doc/110x75/5adf78cc7f8b9a1c248c0f1c/nitrous-oxide-for-space-propulsion-applications-co3o4-fexce1xo2-lacoo3-fe-zsm-5.jpg)

![A [Cu O]2 core in Cu-ZSM-5, the active site 2 in the ... [Cu 2O]2 core in Cu-ZSM-5, the active site in the oxidation of methane to methanol Julia S. Woertinka, Pieter J. Smeetsa,b,](https://img.pdfslide.us/doc/110x75/5b3418b17f8b9a7e4b8bb5a1/a-cu-o2-core-in-cu-zsm-5-the-active-site-2-in-the-cu-2o2-core-in-cu-zsm-5.jpg)

















