Embed Size (px)
Citation preview

Unstable fluorescent proteins used as UPS reporters
1
CMV promoter up-regulation is the major cause of increased protein levels of unstable reporter proteins after treatment of living cells with proteasome inhibitors
Beatriz Alvarez-Castelao1, Idoia Martín-Guerrero2, África García-Orad2 and José G. Castaño1 *
From 1 Departamento de Bioquímica, Instituto de Investigaciones Biomédicas "Alberto Sols". UAM-CSIC y Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas
(CIBERNED). Facultad de Medicina UAM. 28029 Madrid. 2Departamento de Genética, Antropología Física y Fisiología Animal. Facultad de Medicina. Universidad del País Vasco, UPV-
EHU. 48940 Leioa. Spain * Address correspondence to: José G. Castaño. Departamento de Bioquímica e Instituto de Investigaciones Biomédicas "Alberto Sols". UAM-CSIC. Facultad de Medicina de la Universidad Autónoma de Madrid. 28029 Madrid. Spain. FAX: 34-91-585-4401. E-mail: [email protected] Fluorescent unstable proteins obtained by the fusion of a fluorescent protein coding sequence with specific amino acid sequences that promote its fast degradation have become popular to gauge the activity of the ubiquitin/proteasome system in living cells. The steady-state levels of expression of those unstable proteins is low, in agreement with their short half-lives and accumulate in the cell upon treatment with proteasome inhibitors. We show here that this accumulation is mainly due to transcriptional up-regulation of the CMV promoter by proteasome inhibitors and mediated, at least in part, by AP-1 transactivation. Those simple facts put under quarantine conclusions reached about the activity of the ubiquitin/proteasome pathway in animal cells in culture, or in transgenic mice, where popular CMV-driven constructs are used, as transcriptional regulation of the expression of the reporter protein construct, and not degradation of the unstable protein by the ubiquitin/proteasome system, may contribute significantly to the interpretation of the results observed.
The ubiquitin/proteasome system (UPS) plays a central role in the degradation of nuclear and cytoplasmic proteins and constitutes a basic control mechanism of many cell functions, i.e.: DNA replication, transcription, translation, transport, etc.. The UPS is a multistep pathway that ends up in
the degradation of the selected and targeted protein by the proteolytic activity of the proteasome. The 20S proteasome is a multicatalytic proteinase, its structure can be described as an heterodimeric cylinder composed of two heptameric outer α-rings and two inner β-rings. The proteolytic activity of the proteasome is due to the N-terminal threonine of three of the β-subunits (β1, β2 and β5) and can be assayed in vitro with synthetic fluorogenic peptides that seem to freely diffuse into the catalytic chamber formed by the inner β-subunit rings (1). The main and consolidated pathway for protein degradation requires the post-translational modification of the targeted protein by ubiquitin. First, ubiquitin needs to be activated by the E1 activation enzyme. Second, active ubiquitin is transferred to one of the several UBC or E2 ubiquitin-conjugating enzymes. Then, ubiquitin is usually transferred to a member of the E3 ubiquitin-ligase family, which covalently links ubiquitin to the protein substrate. This is followed by poly-ubiquitylation of the substrate, generated via multi-isopeptide linkages between a lysine residue of the protein-attached ubiquitin and the carboxyl-terminal glycine of the next ubiquitin molecule to be added (2). The multiubiquitylated protein is not a direct substrate of the 20S proteasome. The recognition, unfolding and translocation of the poly-ubiquitylated proteins to the inner catalytic chamber of the 20S proteasome is performed with the help of a 19S/PA700 protein complex that associates to both ends
http://www.jbc.org/cgi/doi/10.1074/jbc.M109.004101The latest version is at JBC Papers in Press. Published on August 13, 2009 as Manuscript M109.004101
Copyright 2009 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
2
of the 20S proteasome core cylinder. This 26S proteasome complex performs the degradation of polyubiquitylated proteins requiring the concomitant hydrolysis of ATP and recovering intact ubiquitin that can be reused for future ubiquitylation reactions (1). A critical observation for protein degradation is that in some proteins a limited contiguous amino acid sequence (the simplest is the N-terminal aminoacid) is responsible of the covalent linkage of ubiquitin by E3 enzymes (3). Those modular sequences, or degrons, can often be transferred in-frame to otherwise stable proteins and promote the degradation of the fusion protein by the ubiquitin-26S proteasome system. In the recent past years several groups have developed protein constructs for measuring UPS activity in cells and animals. Those constructs can be easily detected by direct fluorescence, chemioluminiscence, or color development. Examples of those reporter proteins are: green fluorescent protein and their derivatives, luciferase. lactamase, β-galactosidase (4). Each of those reporter proteins are stable proteins and in-frame ligation with specific degrons from unstable proteins produces shortening of the half-life of the fusion proteins. One type of such constructs is based on the removal of ubiquitin from ubiquitin N-terminal fusion proteins rendering different N-terminal residues (4), or linking a modified ubiquitin (G76V) in the N-terminal of the green flourescent proteins (5) or luciferase (6) creating a signal for Ubiquitin Fusion Degradation pathway (UFD) In those cases the resulting proteíns, either after ubiqutin removal and N-terminal recognition or directly by UFD, are degraded after ubiqutilytation by the 26S proteasome (3) (4). More general UPS substrates have been designed by the fusion of the GFP to a degron found in unrelated proteins. Two main degrons have been used for fusion to the C-terminal end of GFP, the Cl1 degron (ACKNWFSSLSHFVIHL) found in a yeast screening for sequences targeting to the endoplasmic reticulum degradation pathway (7), and the ornithine decarboxylase C-terminal (a.a.422-461) region of the protein
(8). While GFP-Cl1, also called GFPu, likely requires ubiquitylation prior to its degradation (9); the second type, commercially named EGFPd2 (Becton and Dickinson), seems to be directly degraded by 26S proteasome without requirement of prior ubiquitylation (8;10). Other specific substrates have been generated like IκBα-GFP (11) and IκBα-luciferase (6) based on the short-half life of IκBα, that is unstable due to the targeting of IκBα by stress or cytokines to degradation by the UPS. A general property of all those constructs in cells, or in transgenic animals, is the low steady-state level of expression of the reporter fusion protein, attributed to the short-half-life of the protein and its rapid decay to almost undetectable levels when protein synthesis is inhibited because of its rapid degradation by the UPS. In agreement with the above conclusion, treatment of the transfected cells (or animals) with proteasome inhibitors produces a variable 2-10 fold increase in the amount of the reporter unstable protein (see references cited above). We have recently identified a putative C-terminal degron in the study of the degradation of Cot/Tpl2 kinase (12). Therefore, we decided to characterize EYFP-C-cot (a.a. 390-435) as another reporter that can be used in living cells to gauge UPS activity. In the course of our studies we found a similar situation to that described above, low levels of expression in transfected cells and a very significant increase in the amounts of the unstable protein in cells treated with different proteasome inhibitors. Further experiments demonstrate that the increase in reporter protein abundance in the cells after treatment with proteasome inhibitors is mainly due to an increase of the transcription of the reporter gene due to up-regulation of the CMV promoter by proteasome inhibitors, and the accumulation of the unstable fusion protein can be completely prevented by co-treatment with transcriptional inhibitors. We have generalized those results with other unstable fusion proteins and actually found that the up-regulation of the CMV promoter by proteasome inhibitors, mainly by AP-1 transcription factor, and not inhibition of the degradation of the reporter protein, is the
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
3
main cause of the accumulation of the unstable reporter protein in the cell.
Experimental Procedures Cell lines, DNA constructs and transfections. Rat pheochromocytoma PC12 cells were cultured in Dulbecco Modified Eagle’s medium containing 10% horse serum and 5% fetal bovine serum. NIH 3T3 (mouse) and N2A (mouse) and Hela (human) cells were cultured in the same medium containing 10% fetal bovine serum. The reporter fluorescent constructs used in this study are: EYFP C-ter Cot/Tpl2, that contains the EYFP coding sequence fused with the C-terminal of Cot/Tpl2, a.a. 390-435, (12); EGFPd2, that contains the EGFP coding sequence fused with the C-terminal sequence of ornithine decarboxylase (a.a. 422-461) obtained from Becton and Dickinson; GFP-Cl1, that contains the GFP coding sequence followed by the Cl1 sequence (ACKNWFSSLSHFVIHL) that was obtained from ATCC (8-10). All the above constructs are under the transcriptional control of the CMV promoter, as well as the corresponding EYFP and EGFP vectors (Clontech) used as controls. Firefly luciferase transcriptional reporters for AP1 (3 x AP-1 binding sites) (13), CREB (3 x CREB binding sites) (14) and NFΚB (3 x κB sites) (15) used in competition experiments have been previously described. The empty vectors pcDNA3.1 (Invitrogen) and pEF-BOS (16) were used as DNA carriers. Stable cell transfectants of all these protein constructs in PC12, N2A and NIH3T3 cells were obtained with Lipofectamine (Invitrogen) and selection with G418 at 800 µg/ml. Either pool of selected cells or individual cloned cell lines (obtained by dilution and single colony picking) have been used in the present studies with no significant differences in the results. Transient transfections were also performed with Lipofectamine and assayed within 48-72h after transfection. Cell assays. The basic cell assay was performed by seeding appropriate number of cells, either in 35mm dishes or in 24-well plates from Falcon. Cells were allowed to
attach to the bottom of the plastic plate and then subjected to the different treatments for different periods of times (regular assays, 12-14h), unless otherwise indicated. Proteasome inhibitors: MG132 and epoxomycin were from Calbiochem, and Lactacystin was kindly provided by Dr. E. J. Corey (Harvard, USA) or Sigma. Other protease inhibitors: Pefabloc (Roche), E64-d and leupeptin (Sigma) were also used to ascertain the specificity of the effects observed with proteasome inhibitors, Actinomycin D (Amersham Biosciences), cycloheximide and α-amanitin (Sigma) were used at the following concentrations: 10-500 ng/ml, 10-100 µg/ml, 1-20 µg/ml, respectively. Flow citometry and fluorescence microscopy. Cells were detached from the plates, washed once in cold PBS by centrifugation at 1000 x g for 5 min and resuspended in PBS. Cell viability was routinely assessed by staining with propidium iodine (1µg/ml) and was always below 10% in all experimental conditions tested in this study. Fluorescence was determined in a FACsort flow cytometer (Becton and Dickinson) and analyzed with Cellquest software. Gating was initially performed in FSC vs SSC dot-plots. For fluorescence microscopy cells were plated over glass-cover slips, subjected to different treatments, fixed with paraformaldehyde, mounted and examined with a Leica confocal microscope. Protein analysis. Expression of fluorescent proteins was analyzed by western immunoblotting of total cell extracts prepared by direct lysis of cells in SDS-loading buffer and separation on 12% SDS-PAGE. Anti-EGFP monoclonal antibodies were obtained from BD Biosciences (clone JL-8) and used at 1/5000 dilution. Anti c-Jun (H-79) polyclonal antibody and anti-phospho c-Jun (KM-1) monoclonal antibody were obtained from Santa Cruz Biotechnology and used at 1/1000 dilution. The rabbit antisera used against Fos family (RR26/8) was kindly provided by Dr. Juan Miguel Redondo (17) and used at 1/1000 dilution. Blots were developed with peroxidase-labeled goat anti-mouse (or anti-rabbit) from BioRad at 1/4000 dilution by chemiluminiscence method.
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
4
Controls for the amounts of loaded proteins were performed with anti-tubulin antibodies (Sigma). Blots were analyzed by quantitative densitometry using Quantity-one software (Bio-Rad). Luciferase activity was assayed with Dual-Luciferase Reporter Assay System from Promega as per manufacturer’s protocol. Results are expressed as the means ± standard errors of the means for the quotient of firefly luciferase and Renilla luciferase activity. RNA analysis. Total RNA was extracted by the method of Chomczynski and Sacchi (18) using Tryzol (Gibco–BRL). Isolated RNA was treated for 45 minutes at R.T. with DNase I Amplification Grade from Invitrogen, in order to eliminate traces for plasmidic and genomic DNA. The RNA integrity was assessed by RNA chips using RNA 6000 Nano Kit. 1-2 µg of total RNA was used for cDNA synthesis using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Semi-quantitative estimation of GFP RNA levels was performed by RT-PCR using the following oligonucleotides: forward, 5’ GAACGGCATCAAGGTGAACT 3’; reverse 5’ GAACTCCAGC AGGACCATGT 3’, control RT-PCR was performed with the inclusion in the same RT-PCR reaction of actin oligonucleotides: forward, 5’ AGCCATGTACGTAGCCATCC 3’; reverse, 5’ CTCTCAGCTGTGGTGGTGAA 3’. After cDNA synthesis, PCR was performed with Imolase (Bioline) with an initial cycle at 95ºC during 7 minutes to activate the polymerase. This initial cycle was followed by 30 cycles: 94ºC for 30 seconds, 60ºC for 30 seconds and 72ºC for 30 seconds. Final extension consisted of one cycle at 72ºC for 10 min. The PCR products were analyzed by 1.5% agarose gels and staining with ethidium bromide. For direct visualization and quantification of mRNAs, Northern blot analysis was performed, 5-10µg of total RNA was separated on 1% agarose-denaturing gel and transferred onto charged Nylon membranes (Zeta-probe GT, Bio-Rad), the membranes were UV cross-linked, and stained with methylene blue for ribosomal 28S and 18S visualization. DNA probes were generated from the purified inserts of the following
plasmid constructs after restriction endonuclease digestion (enzymes indicated between brackets): pEYFP C-1 (NheI/HindIII) from Clontech; pCMV Sport6 mouse GAPDH (I.M.A.G.E. Clone ID; 6494145) (SalI/NotI) and pGEM rat PDI (PstI) as described (19). The purified inserts (50ng) were labelled with 50µCi [α-32P]dCTP using the Rediprime™ II DNA Labeling System (GE Healthcare), and purified using Illustra NICK Columns (GE Healthcare), according to manufacturer's instructions. The same filter was probed, washed and hybridized successively with the above indicated cDNA probes. For developing, the blots were exposed to a storage phosphor screen (2-to 4h) and scan in a Typhoon Trio Imager (GE Healthcare), quantification was performed with ImageQuant TL or Quantity–one Software from Bio-Rad. Quantitative real-time PCR (qPCR) was performed using a 7900 HT Fast Real-Time PCR System, with Fast Sybr Green Master Mix (Applied Biosystems) using the standard protocol for PCR Fast. For the target gen (GFP) the following oligonucleotides forward, 5’ GGGCACAAGCTGGAGTACAACT 3’; reverse 5’ TCTGCTTGTCGGCCATGATA 3’ were used. The oligonucleotides of the genes used for normalization were; TATA binding protein, forward, 5’ TCATGAGAATAAGAGAGCCACGAA-3’; reverse ;5’TTCTTCACTCTTGGCTCCTGTG-3’; glyceraldehyde-3-Phosphate dehydrogenase, forward, 5’ TGCCAGCCTCGTCTCATAGA 3’; reverse, 5’ GTCCGATACGGCCAAATCC 3’ and eukaryotic elongation factor 1 alpha, forward, 5’ TCTGGTTGGAATGGTGACAACA 3’; and reverse, 5’ CCCTTGAACCACGGCATATT 3’. After validation of similar amplification efficiencies for target and reference genes (100%±5%), the relative expression levels were analyzed by the ∆∆Ct comparative method (20) from three different experiments. The Ct ranges for the analyzed genes were between 15-23 cycles. Results are expressed as fold changes, taking the control (no treatment) as a reference value of 1 plus minus the corresponding range.
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
5
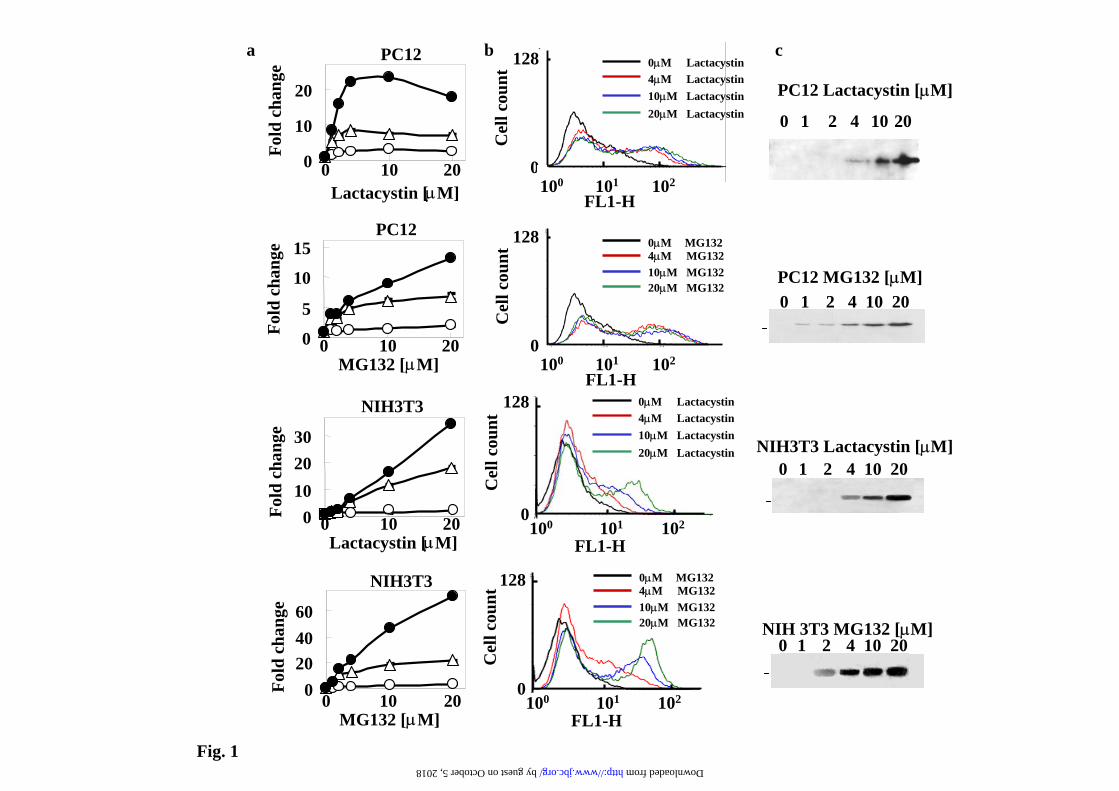
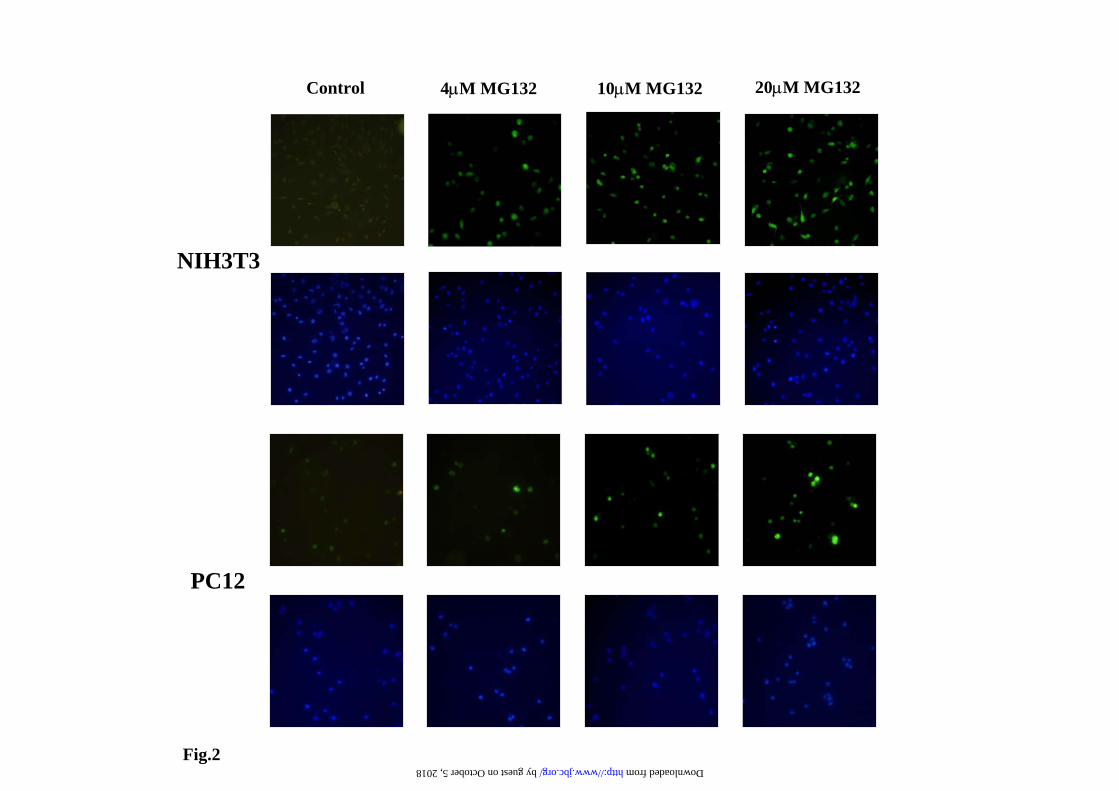
Results EYFP C-terminal Cot/Tpl2 UPS reporter. We have reported that the C-terminal of Cot/Tpl2 kinase when transferred to EYFP renders the fusion protein unstable and degraded by the proteasome (12). We used this construct (EYFP-Ct-Cot) as a possible new fluorescent reporter to monitor proteasome activity in living cells. Transient cell transfectants of this reporter construct were obtained in PC12 and NIH3T3, our aim was to study the possible differential regulation of proteasome activity in cells of neural and non-neural origin. Untreated cells show little fluorescence and after addition of proteasome inhibitors, a dose- dependent increase in the amount of the reporter fusion protein was invariably obtained, in parallel with increase levels of the reported protein as demonstrated by Western blot analysis (Fig.1). These results were also confirmed by direct fluorescent microscopy observation of the cells (Fig. 2). The results presented are similar to those reported by other authors with different unstable fluorescent proteins (8) (5) (9) (10). Other proteases inhibitors, E64-d (50 µM); Pefablock (100 µM) and leupeptin (200 µM) have no effect (Supplementary Figure 1), indicating the specificity of the effect of proteasome inhibitors. In general, three main cellular processes: transcription, translation and degradation, are responsible for the steady state levels of proteins in the cell. Total protein translation was not significantly affected by treatment of the cells with proteasome inhibitors, as measured by incorporation of 35S-Met/Cys into total proteins, identical results were obtained by other authors under experimental conditions similar to the ones used in this study (21). Then we check transcription, to our surprise the addition of actinomycin D (ActD), blocked the increase in the amount of the EYFP-Ct-Cot reporter protein produced by proteasome inhibitors (data not shown). Similar results were obtained with stable transfected cell lines, either transfected pools
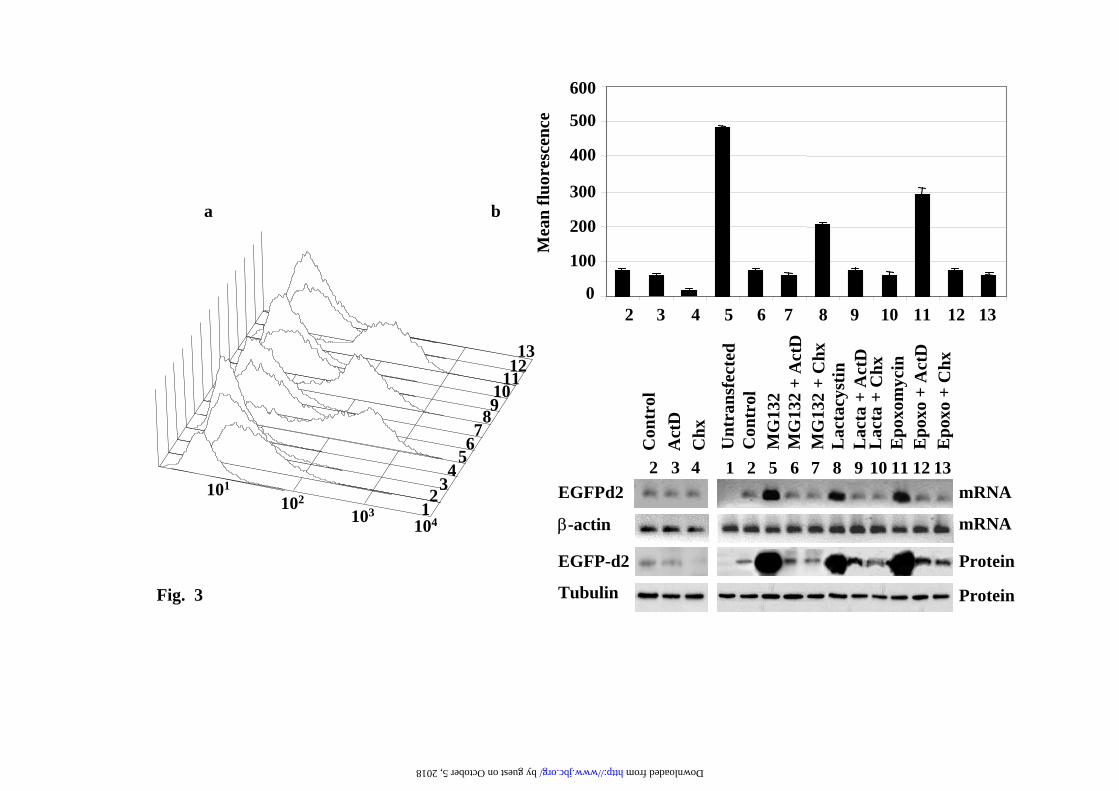
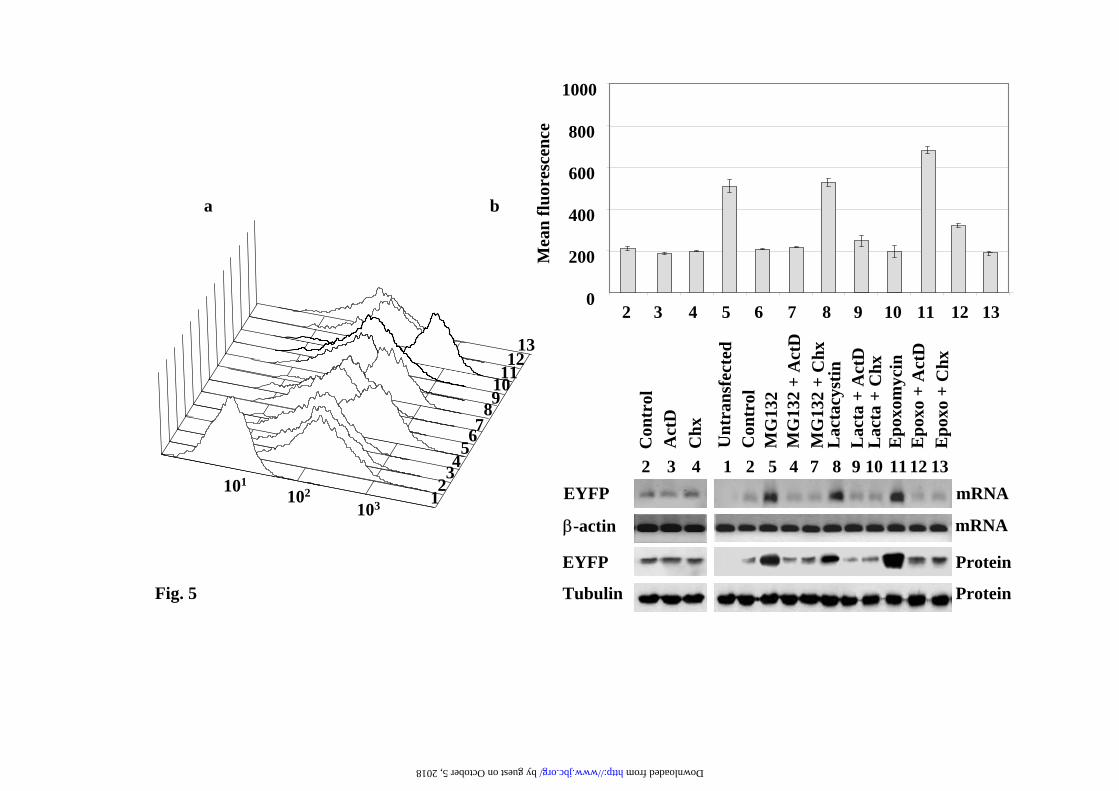
or clonal cell lines of PC12 and NIH3T3 cells. Transcriptional regulation of CMV-driven fluorescence reporter constructs. The above results could be a particular property of our reporter EYFP-Ct-Cot construct, then we decided to analyze other GFP reporters, EGFPd2 (EGFP-Ct- fused with a.a. 422-461 from ornithine decarboxylase), a direct substrate of the 26S proteasome (not requiring ubiquitylation for its degradation), GFPu (GFP-C1 construct, ACKNWFSSLSHFVIHL). To that end, PC12, NIH3T3 and N2A stable transfectants of the above reporters were isolated. Pools of selected cells and several individual clones were analyzed and the results obtained were similar to those presented above for EYFP-C-Cot, and in agreement with previous reports using those constructs (8-10). As an example, the results for the EGFPd2 in PC12 cells are presented. FACS-scan profiles are presented in Fig. 3a and its quantification in Fig. 3b, upper panel. The corresponding amounts of EGFd2 mRNA and protein (Fig.3b) were in parallel with the cell fluorescence results. The addition of ActD to control cells (Figure 3, lane 3) produce a small reduction in the basal expression of the protein, while cycloheximide (CHX, Figure 3, lane 4), as expected, substantially reduced the steady-state levels of the fluorescent reporter protein. These results demonstrate that inhibition of protein synthesis produces the rapid degradation of the EGFPd2 protein. Treatment with proteasome inhibitors (MG132, lactacystin or epoxomicin) produces a strong increase (Fig. 3b), in cell mean fluorescence intensity, in parallel with the increase in the amount of mRNA and protein levels (fig. 3 lanes 5, 8 and 11), as determined by semi-quantitative RT-PCR and immunoblotting, respectively. The parallel increase of fluorescence, mRNA and protein levels produced by treatment with proteasome inhibitors was abolished (or strongly reduced) when either ActD or CHX was added together with proteasome inhibitors (Figure 3, lanes 6, 7, 9, 10, 12 and 13). Time-course experiments demonstrate that the effects are clearly observed after 4h of treatment and
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
6
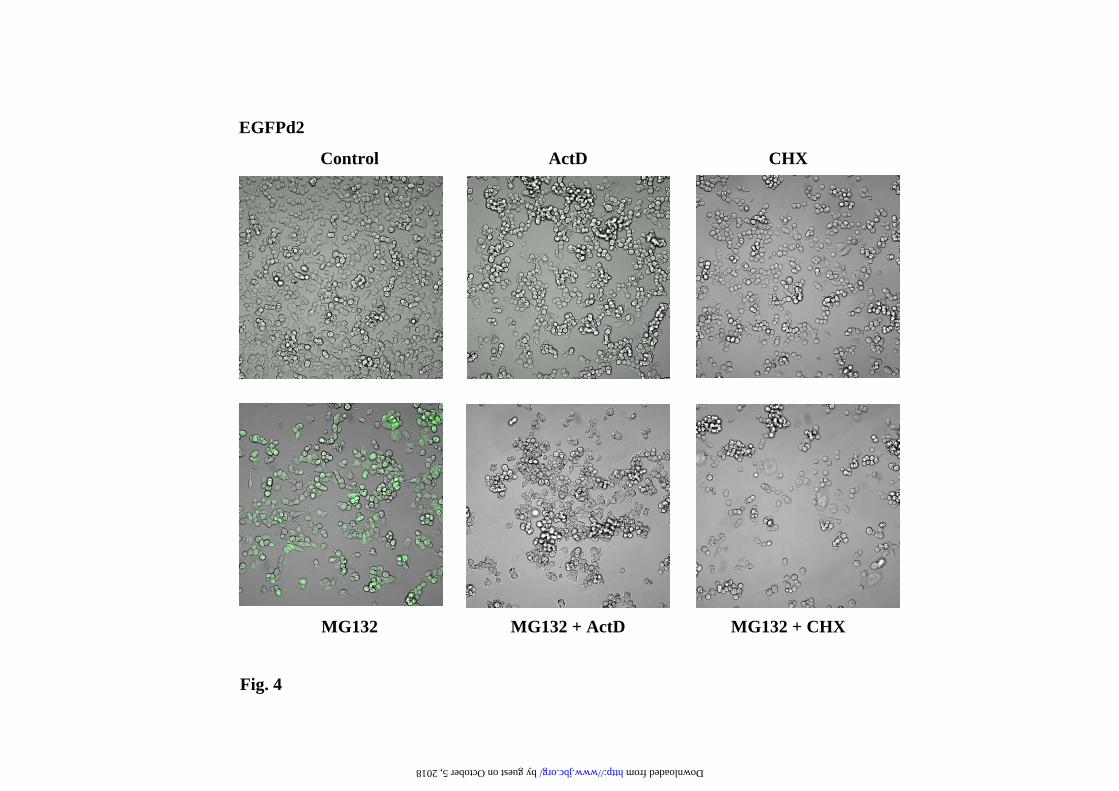
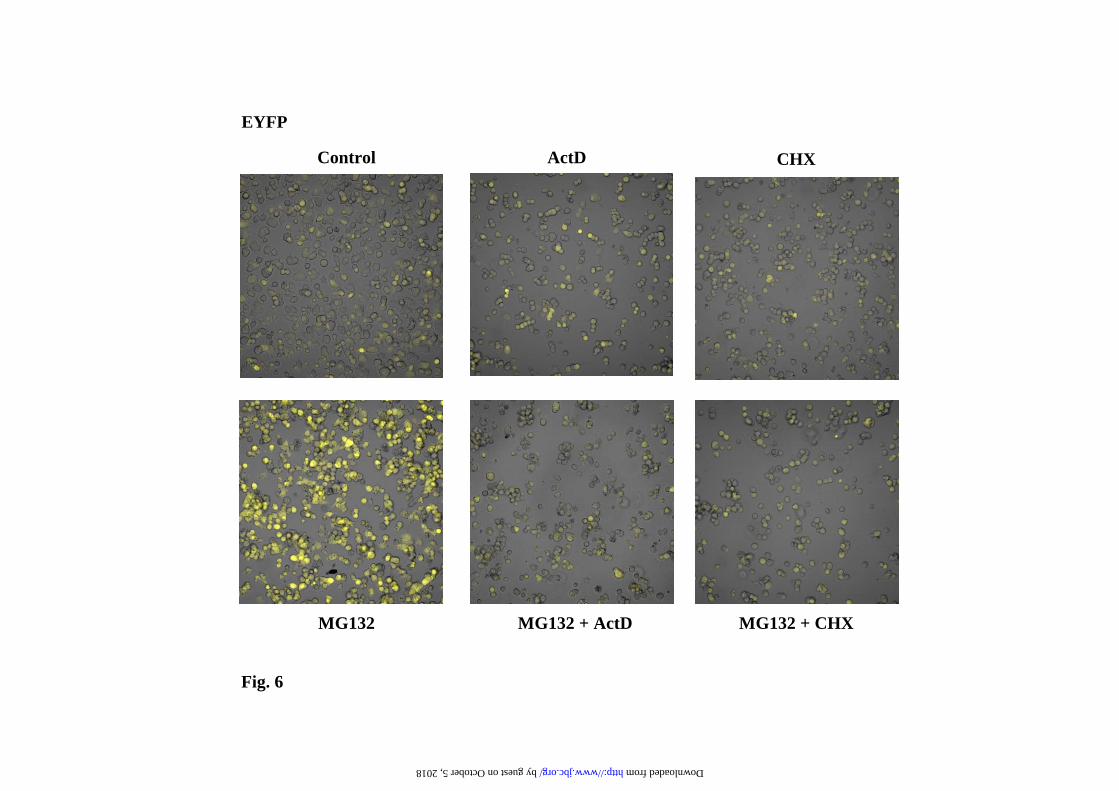
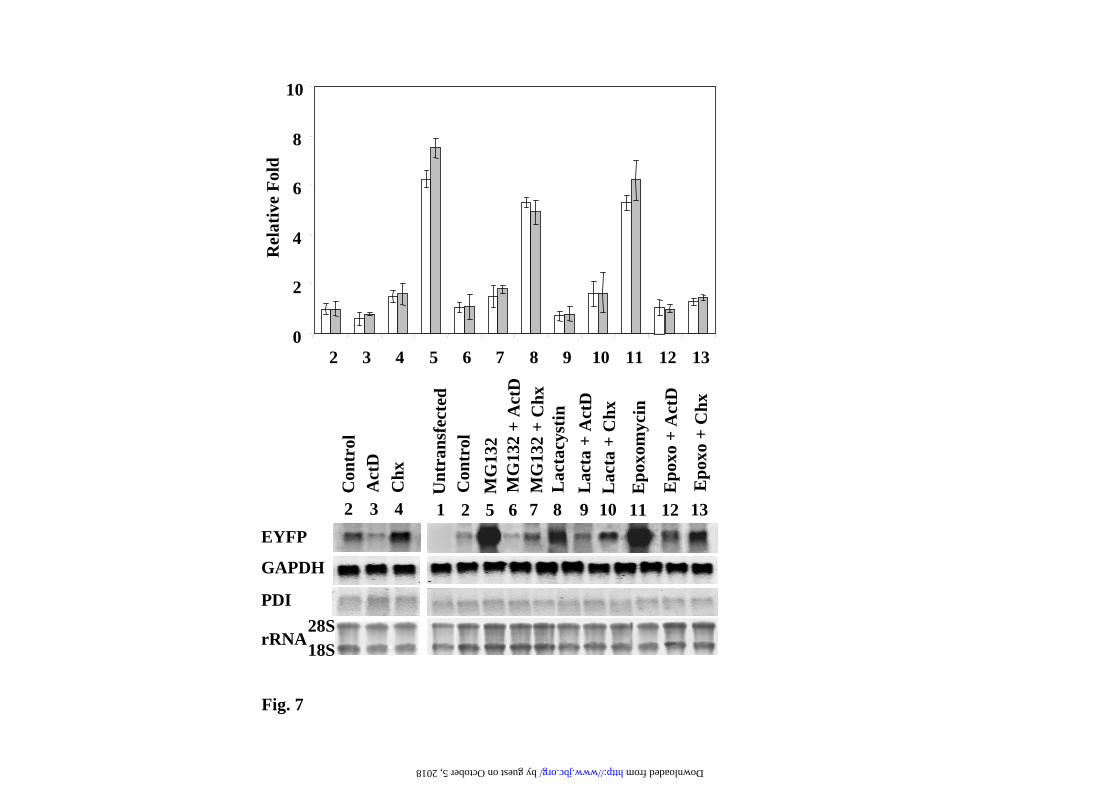
plateau in a dose-dependent manner after 12h of treatment. Those results are also evident by direct confocal cell fluorescence analysis, as illustrated in Fig. 4 for the experiments with MG132. The interpretation of the results presented in Fig. 3 can be misleading due to the instability of the EGFPd2 protein, but they predict that similar effects should be observed with the stable EGFP (or EYFP). The results for EYFP are presented in Fig. 5 and 6, clearly demonstrating similar results to those obtained with the unstable EGFPd2. In order to get a more direct demonstration of the effect of proteasome inhibitors on mRNA, mRNA levels of EYFP were analyzed by Northern blot, and the results are presented in Fig. 7. GAPDH (glyceraldehyde-3-phosphate dehydrogenase, a cytoplasmic and nuclear localized glycolytic enzyme) and PDI (Protein Disulfide Isomerase, a lumenal endoplasmic reticulum enzyme) mRNA levels were used as internal controls. Proteasome inhibitors produce (Fig. 7) a clear and significant increase in the mRNA expression levels of EYFP, suppressed by co-treatment with transcriptional (ActD) and translational (CHX) inhibitors. As expected, similar results were also obtained with EGFPd2 construct (as shown for MG132 in Supplementary Figure 2). Those changes in the mRNA levels for EGFPd2 and EYFP were also confirmed by qPCR analysis (Supplementary Fig. 3). The conclusion from these experiments is that proteasome inhibitors are able to activate the transcription of the reporter gene constructs and therefore producing an increase in the amount of mRNA and protein that is the main cause of the increase of the cell fluorescence and protein amounts of unstable (short-half life), as well as stable (long half-life), fluorescent proteins. Similar results were obtained with the EGFPu (as shown for MG132 in Supplementary Figs. 4 and 5). Alternative transcriptional (α-amanitin) and translational (emetine) inhibitors also produce similar results (Supplementary Fig. 6). While we have analyzed several different clones (and pools) of stable cell transfectants and the response is similar to that shown in Figures 3
and 5, it could be that proteasome inhibitors are able to release the transcriptional silencing of the cell integrated DNA constructs in the stable cell lines, because silencing has been reported to occur for the CMV promoter in stable transfectants (22;23). Therefore, the same type of experiments were performed in transiently transfected cells (PC12, N2A and Hela cells), unlikely to show those possible epigenetic modifications, since it has been demonstrated that the CMV-GFP plasmid remains unmethylated for a period up to 96 h in transiently transfected cells (22;23), and the results were similar to those presented in Fig. 3 (data not shown), as it would be expected by the results obtained with the transiently transfected EYFP-Ct-Cot construct shown above. Transcription factors responsible of CMV promoter up-regulation by proteasome inhibitors. The CMV promoter is a popular promoter because of its pan-active behavior in many cell lines and transgenic mice (24). The human CMV promoter allows high-level of “constitutive” gene expression, nevertheless its expression is modulated by a number of regulatory transcription factors that are cell specific and responds to different stimuli and stress: NFκB/Rel (25-27), AP-1(28), ATF/CREB (26;26;29;30;30), YY1 and NF-1(31;32;32;33;33). To explore the possible transcription factors responsible of CMV up-regulation by proteasome inhibitors, we used competition experiments in transiently transfected Hela cells. First, we performed similar experiments to those described above for murine cells with transiently transfected Hela cells (Supplementary Fig. 7) for both stable EYFP and unstable EGFPu. Treatment of transfected Hela cells with proteasome inhibitor (MG132) increases the amount of mRNAs (as measured by qPCR) and protein levels, and again, as shown above with murine cells, co-treatment with ActD and CHX suppress the increases observed (Supplementary Fig. 7). Once established that human cells behave similarly to murine cells, the DNA dose-dependence of both unstable (GFPu) and stable (EYFP) and its
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
7
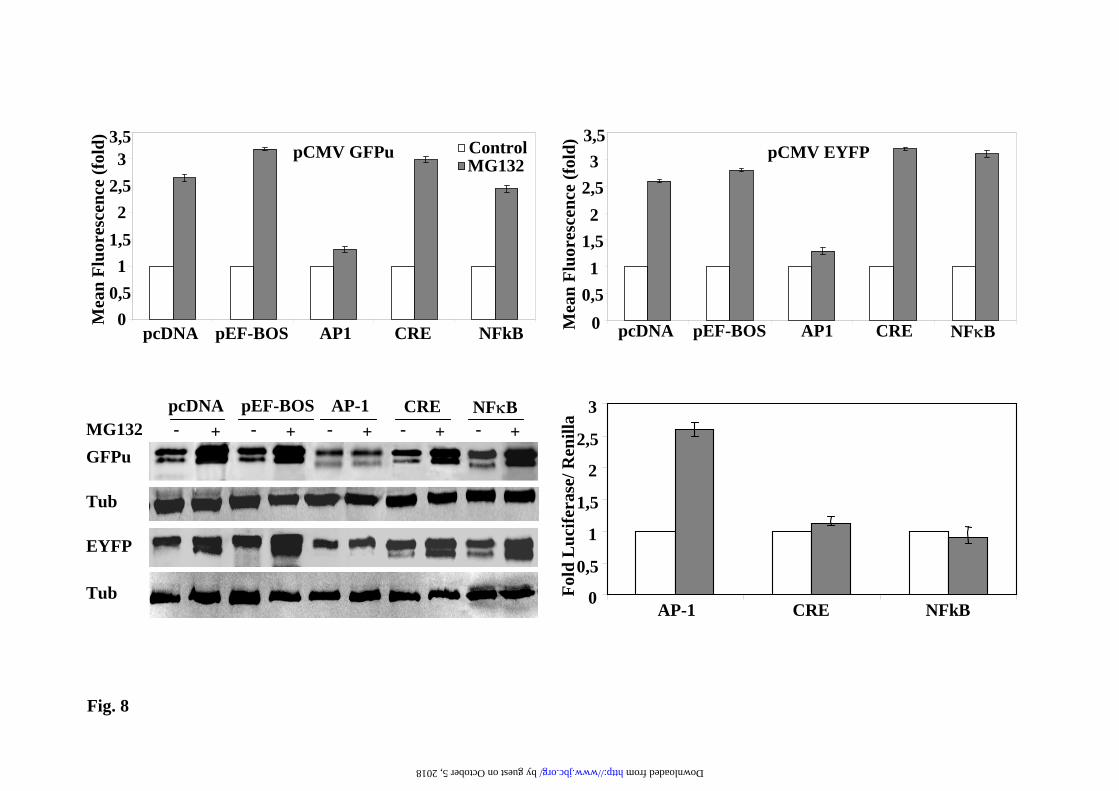
response to proteasome inhibition with MG132 were evaluated in transiently transfected Hela cells. Results are presented in Supplementary Fig. 8. MG132 treatment increases GFPu cell contents, measured both by cell fluorescence and immunobloting in an almost linear manner. In contrast, MG132 effect on EYFP cell contents levels off (both by cell fluorescence and immunoblotting), indicating saturation of the transfected cells for EYFP expression, accordingly the degree of MG132 activation decreases as expression saturation is reached. Once we have defined the experimental setting in Hela cells, three different luciferase reporters driven by AP1, CREB and NFκB binding sites were selected to perform competition experiments. In the absence of proteasome inhibitors, the expression levels of the GFPu and EYFP proteins from their respective plasmids was not affected by co-transfection with those luciferase reporters plasmids, behaving similarly to pcDNA 3.1 or pEF-Bos (Supplementary Fig. 9). Therefore, the study of competition for the activation in the presence of proteasome inhibitors was done by transfecting a fixed amount of DNA of GFPu and EYFP plasmids (50 ng) and 950 ng of the AP1, CREB and NFκB luciferase reporters plasmids (and pcDNA3.1 empty vector as control) . As shown in Fig. 8, cell fluorescence and protein immunoblot analysis demonstrate that only the co-transfection with AP-1 luciferase reporter prevents the activation of the expression of both unstable (GFPu) and stable (EYFP) proteins by MG132. These results indicate that AP-1 transcription factors (fos/jun) are implicated in the transcriptional activation of CMV promoter by proteasome inhibitors and are in perfect agreement with the results presented in Fig. 8d, showing that MG132 treatment only increases firefly luciferase expression in the case of the AP1 luciferase reporter. These results also suggest that effective transcriptional competition under these experimental conditions requires a high amount of AP-1 binding sites, as a 10 fold excess of empty pcDNA3.1 (one AP-1 binding site) vector is inefficient as competitor (see Fig. 8 and Supplementary
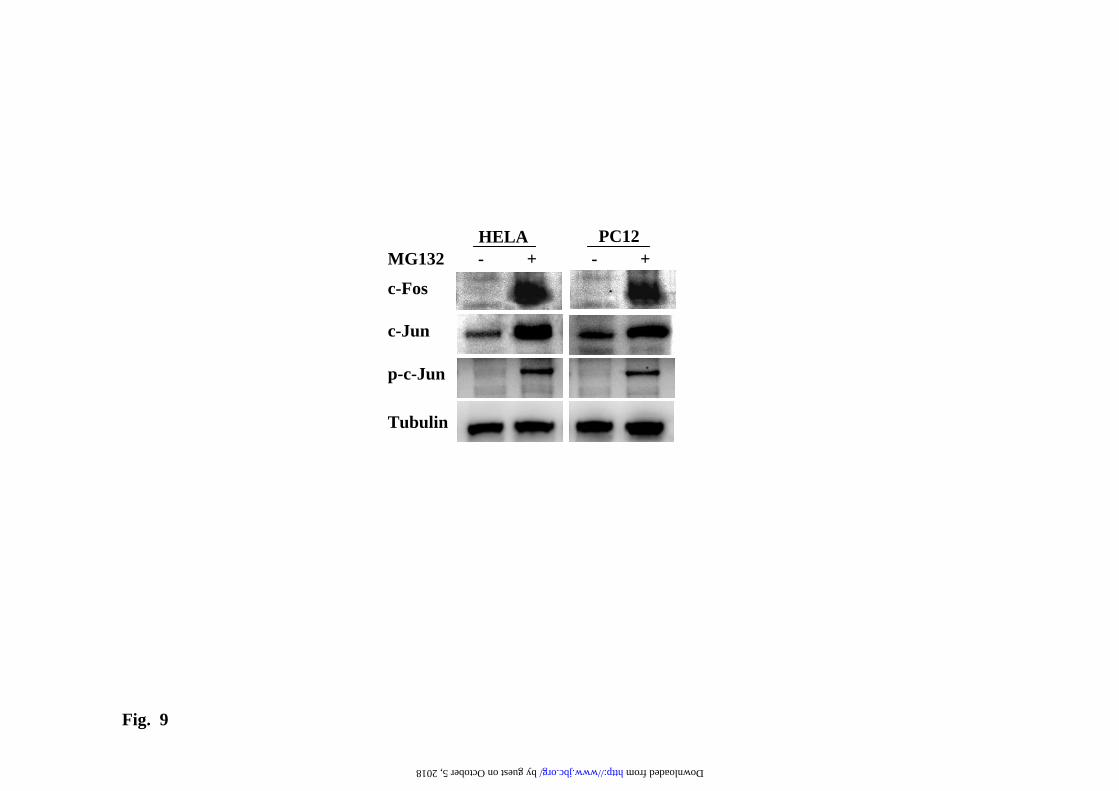
Figs. 8) to prevent the up-regulation of GFP reporter by proteasome inhibitors or the basal expression (Supplementary Fig. 9). In contrast, a 30-fold excess of AP-1 binding sites, as provided by 10-fold higher amounts of the AP-1 luciferase reporter with 3 AP-1 binding sites in tandem, can efficiently prevent the transcriptional activation of the GFP reporter. In agreement with the above results, treatment of cells with proteasome inhibitors produce an increase of c-jun and c-fos protein levels due to the stabilization of these transcription factors (Fig. 9) and increase in the phosphorylation of c-jun by activated stress-kinases, as it has already been demonstrated by other groups (34;35;35;36). These data set clearly support that the AP-1 is directly involved in the trans-activation of the CMV promoter by treatment with proteasome inhibitors. Avoiding transcriptional regulation in the study of protein degradation by UPS. The above results point clearly to the need of experimental conditions where the RNA of the reporter protein is not transcriptionally up-regulated by the use of proteasome inhibitors in order to measure protein degradation. To that end, and based on the results shown above, it is clear that an increase in the amounts of unstable GFP proteins are needed to accurately measure its degradation, because of the low steady-sate levels of the unstable proteins. The simplest way is to add proteasome inhibitors for a short period of time and get enough amount of the reporter protein to easily follow its degradation. For this type of experiments we have to use a reversible proteasome inhibitor, like MG132, that can be washed-out from the cell. Then the addition of CHX will produce protein synthesis inhibition, allowing to see the degradation of the unstable reporter protein, and because CHX is present, the addition of new proteasome inhibitor in the presence of CHX will not produce an up-regulation of the CMV promoter (as it has been shown above), as a consequence we can safely determined the half-life of the unstable protein without interference of transcriptional and translational input to the unstable protein pool in the cell. One such experiment is
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
8
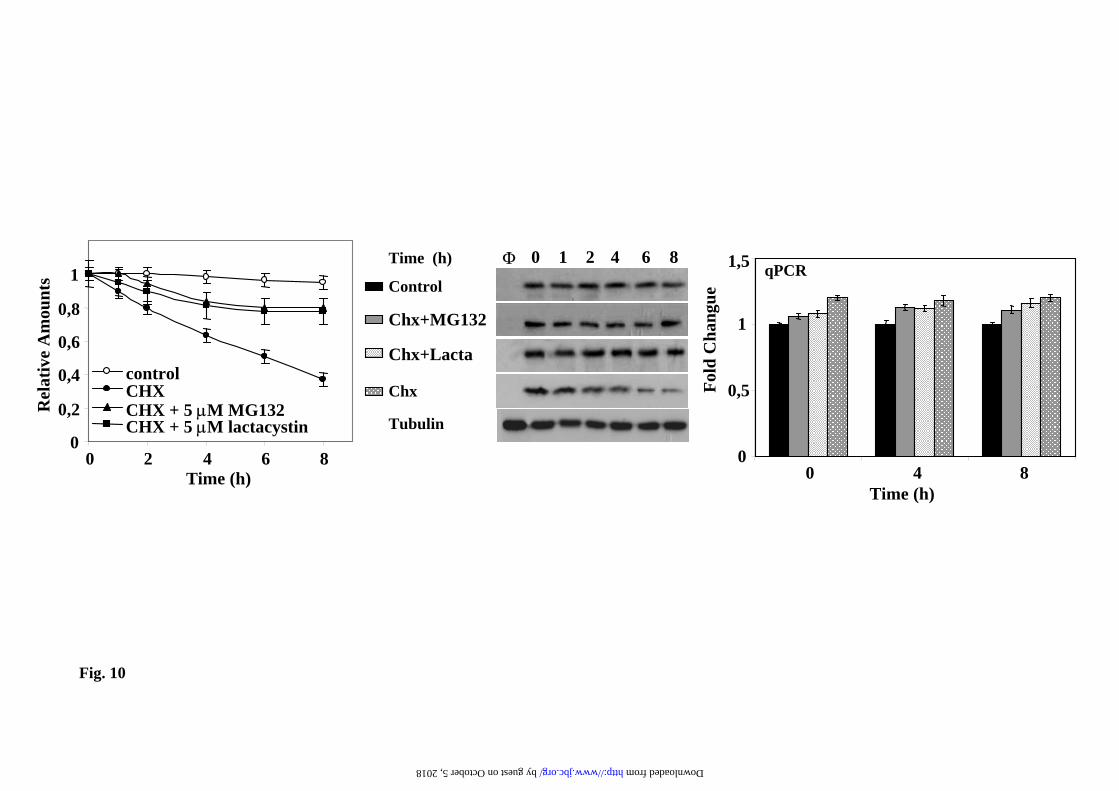
presented in Fig. 10 for the EGFPd2 unstable protein. The EGFPd2 protein is degraded with an estimated half-life of 4-5h in the presence of CHX, and proteasome inhibitors prevents its degradation without affecting the expression of the corresponding mRNA, as judged by qPCR (Fig. 10). Under these experimental conditions any compound, or gene product, can be tested for its effect on proteasome activity in the cell without the inconvenience of transcriptional regulation of the CMV promoter.
Discussion
We have presented experimental evidence showing that transcriptional up-regulation of the CMV promoter by proteasome inhibitors is mainly responsible of the increased levels of unstable GFP in treated cells, and not inhibition of the degradation of the reporter protein by the UPS. The ubiquitin/proteasome pathway plays a substantial role in transcriptional regulation, not only by degradation of transcription factors limiting the transcriptional output, but also in recycling of transcriptional complexes on chromatin to facilitate multiple rounds of transcription (37-39). Proteasome inhibitors, as a consequence, have been reported to have multiple effects, depending on the context of the specific promoter under study. Inhibition of proteasomal degradation increases transcriptional activity of some, but not all, steroid hormone receptors (40), produces a heat-shock response with an increase of cytoplasmic and endoplasmic reticulum chaperones (41) mediated by HSF1-3 (42) and favoring proteostasis (43), and promote an increase in the global levels of trimethyl histone H3K4 and phosphorylated RNA polymerase II forms that affect global gene transcription (44). In the context of the CMV promoter it would be expected that proteasome inhibitors could prevent NFkB/Rel activation by inhibition of Ikappab degradation (45), activate AP-1 by stabilization of both jun and fos transcription factors and direct activation of stress kinases (34;35;35;46), promote the activation of CREB by inhibition of its proteasomal degradation (47) and stabilizeYY1 that may
behave as an activator, or as a repressor, depending on the cell context (48). Our experimental strategy based on transcriptional competition experiments with specific transcriptional reporter constructs, instead of using stress kinase inhibitors or other interference treatments that are always subjected to non-specific effects, gave unequivocal experimental evidence that the AP1 transcription factor is the main transcription factor responsible of the observed increase in protein levels both of unstable and stable GFP fluorescent proteins after treatment with proteasome inhibitors, due to the stabilization of c-jun and c-fos transcription factors and the phosphorylation of c-jun, still other transcription factors that bind to the CMV promoter can also participate. Actually, the transcriptional activation of CMV-promoter by proteasome inhibitors have already been described by other groups (49;50). The fact that the increase of mRNA expression of CMV-driven constructs by proteasome inhibitors is blocked by ActD (and α-amanitin) is not surprising; more striking could be the results with CHX, a protein synthesis inhibitor, which also blocks the transcriptional up-regulation of the CMV-driven protein constructs. While CHX is an inhibitor of protein synthesis, one should remember that it can affect gene transcription, by facilitating the degradation of labile transcription factors, either activators or repressors, or by preventing its synthesis. In fact, Li et al. (50) also reported that CHX blocks the transcriptional activation of CMV-promoter by proteasome inhibitors, similarly to what is reported here. As a consequence, the up-regulation of CMV promoter by proteasome inhibitors may require protein synthesis directly or indirectly, through the post-translational modification of the transcriptional machinery involved in CMV transcription. The main message of the present work is that the measurement of UPS activity in cells, or in vivo in transgenic animals, with unstable proteins requires the compulsory demonstration that the level of the mRNAs for those protein constructs is not affected by
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
9
the experimental treatments made to cells or animals. Simple conclusions based on fluorescence and/or immunoblots of the reporter protein can give rise to unjustified and misleading conclusions about proteasome activity. The misleading conclusion may look even substantiated by the experiments. Under saturating amounts of transfected DNA (see Supplementary Fig. 8) proteasome inhibitor treatment will not affect the expression levels of the stable GFP constructs measured by protein amounts or fluorescence (as expected for an stable protein), because the cell expression system is saturated, while at the same DNA doses of the unstable GFP construct, the levels of the unstable GFP will be nicely increased by proteasome inhibitors (as expected for an unstable protein). This general statement is perfectly exemplified in the present case for the CMV promoter, where the results of proteasome inhibition on unstable (and stable) fluorescent protein levels can mainly be explained by transcriptional up-regulation and not by the inhibition of the proteasomal degradation of the unstable protein. The results presented here put into quarantine the interpretation of some published works using fluorescent, chromogenic or chemiluminescencent unstable reporter proteins, as in many cases the gene constructs used the human CMV promoter to drive transcription. Furthermore, the use of CMV-driven unstable protein
constructs in the study of protein degradation in neurodegenerative process, leading to the conclusion of UPS impairment by the expression of pathogenic proteins implicated in neurodegeneration, needs also to be re-evaluated by the results presented here and the reported increase by neuronal depolarization of CMV-EGFP expression constructs in neurons by CREB-mediated transactivation (51). Using other promoters, instead of the CMV, to drive the expression of unstable fluorescent proteins will also require to study the possible effects of proteasome inhibitors on the transcription of the particular promoter under use, due to the general role of proteasome activity in several aspects of transcriptional regulation (52)..Last, but not least, one has also to be cautious not only with the interpretation of experiments in which unstable (or stable) GFP reporters are used to gauge UPS activity in cells or in vivo, but also with the reported EGFP inhibitory effects on the ubiquitylation process itself (53). Nevertheless, as always in experimental science, with the appropriate controls and under well defined conditions (for example, as described in Fig. 10), the unstable protein constructs can still be used to gauge the activity of the UPS system in animal and human cells.
Acknowledgments
We want to express our thanks to Ana Belen Sánchez López and Esperanza Martin for their help with some of the experiments presented and Dr. Juan Miguel Redondo, CNIO, Madrid, for the c-fos antibodies This work was supported by Grants from SAF-2008-00766, CM SAL-0202, FMM, Fundación Mutua Madrileña, & CIBERNED to JGC and by RETICS G3/179 and Basque Government SAIOTEK to AGO.
Figure Legends
Legend to Fig.1 Dose-dependent accumulation of EYFP-C-cot in transiently transfected PC12 and NIH3T3 cells treated with proteasome inhibitors. Cells were treated with the indicated dose of proteasome inhibitors for 12h. a) Graphs of the relative change in fluorescence of the cell population in response to different doses of proteasome inhibitors: , mean fluorescence; , percent of positive cells; , product of mean fluorescence multiply by the percent positive cells. b)
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
10
Representative cell fluorescence profiles of the experiments presented in a). c) Representative immunoblots with anti-EGFP antibodies of the changes in protein levels at different doses of proteasome inhibitors. Legend to Fig. 2. Fluorescence microscopy analysis of EYFP-C-cot transiently transfected PC12 and NIH 3T3 cells treated with different doses of proteasome inhibitors. Cells were transiently transfected with EYFP-C-cot and treated for 12h with the indicated doses of proteasome inhibitors. Upper panels for each cell type show EYFP fluorescence and lower panels DAPI fluorescence (DNA staining). Note that, as presented in a quantitative way in Fig.1, both the percent of positive cells and the fluorescence intensity per cell increase in a dose-dependent manner. Low magnification confocal images, x200. All pictures were taken with the same settings of the confocal microscope. Legend to Fig. 3. Effect of proteasome, transcription and translation inhibitors on cell fluorescence mRNA and protein abundance of PC12 cells stably transfected with EGFPd2. a) Representative experiment of cell fluorescence profiles of stably EGFPd2 PC12 cells treated with proteasome, transcription and translation inhibitors. b), Upper panel, graph of cell fluorescence mean intensity analyzed by flow cytometry and expressed as mean ± S.E.M from three different experiments, each run in triplicate. Lower panels show representative experiments of the levels of mRNA (EGFP and actin, used as control) analyzed by 1.5% agarose gel and ethidium bromide staining of RT-PCRs products obtained from the respective total cell mRNAs samples; and protein (EGFPd2 and tubulin, used as control) by immunoblotting with anti-EGFP and anti-tubulin antibodies. PC12 cells untransfected (1) or stably transfected cell lines (2-13) where untreated (control, 2) or treated for 12h with proteasome inhibitors (10µM MG132, 10µM lactacystin, or 100nM epoxomycin) in the absence or in the presence of 500ng/ml actinomycin D (ActD), or 20 µg/ml cycloheximide (CHX), as indicated (3-13). Legend to Fig. 4. Fluorescence microscopy analysis of EGFPd2 stably transfected PC12 in response to proteasome, transcriptional and translational inhibitors. Cells stably expressing EGFPd2 were untreated (controls) or treated as indicated for 12h with MG132, ActD and CHX or the combinations indicated (see Legend to Fig.3 for doses). Living cells were examined under confocal microscopy with low magnification (x200), fluorescence images are superimposed over phase-contrast images of the cells. All fluorescent images were captured with the same settings of the confocal microscope. Legend to Fig. 5. Effect of proteasome, transcription and translation inhibitors on cell fluorescence, mRNA and protein abundance of PC12 cells stably transfected with EGFP. a), Representative experiment of cell fluorescence profiles of stably EGFP PC12 cells treated with proteasome, transcription and translation inhibitors. b), Upper panel, graph of cell fluorescence mean intensity analyzed by flow cytometry and expressed as mean ± S.E.M from three different experiments, each done by triplicate. Lower panels show representative experiments of the levels of mRNA (EGFP and actin, used as control) analyzed by 1.5% agarose gel and ethidium bromide staining of RT-PCRs products obtained from the respective total cell mRNAs samples; and protein (EGFP and tubulin, used as control) by immunoblotting with anti-EGFP and anti-tubulin antibodies. PC12 cells untransfected (1) or stably transfected cell lines (2-13) where untreated (control, 2) or treated for 12h with proteasome inhibitors (10µM MG132, 10µM lactacystin, or 100nM epoxomycin) in the absence or in the presence of 500ng/ml actinomycin D (ActD), or 20 µg/ml cycloheximide (CHX), as indicated (3-13). Legend to Fig. 6. Fluorescence microscopy analysis of EYFP stably transfected PC12 in response to proteasome, transcriptional and translational inhibitors. Cells stably expressing EYFP were untreated (controls) or treated as indicated for 12h with MG132, ActD and CHX or the
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
11
combinations indicated (see Legend to Fig.3 for doses). Living cells were examined under confocal microscopy with low magnification (x200), fluorescence images are superimposed over phase-contrast images of the cells. All fluorescent images were captured with the same settings of the confocal microscope. Legend to Fig. 7. Northern blot analysis of the effect of proteasome, transcription and translation inhibitors on the mRNA expression of stable EYFP. Experimental conditions are identical to those described in the legend to Fig. 3. Total RNA was isolated from cells and analyzed by Northern blot. Membranes of transferred RNAs were stained with methylene blue (28S and 18S RNAs) and successively hybridized with 32P-labeled probes for EYFP, GAPDH and PDI. A representative experiment is presented. Graph shows the relative changes in the amount of mRNA for EYFP quantitated using either GAPDH (white bars) or PDI (grey bars) mRNAs as controls, mean ± S.E.M from three experiments. Legend to Fig. 8. Transcription factors implicated in the activation of CMV fluorescent reporters by proteasome inhibitors. Hela cells were transiently transfected with 50ng of the corresponding pCMV GFPu (a) and pCMV EYFP (b) together with 950 ng of the indicated plasmids: empty pCDNA3.1, empty pEF BOS and AP-1, CREB, or NFκB luciferase reporters. Transfected cells where untreated (control) or treated with MG132 (10µM) for 12h. a) and b) Show fold changes in total cell fluorescence intensity analyzed by flow cytometry. c) Shows a representative western immunoblot of the expression of the fluorescent proteins, d), Hela cells where transiently transfected with a mixture of DNAs containing: 100ng of the indicated firefly luciferase reporters, 50ng of basal Renilla luciferase plasmid and 850 ng of empty pcDNA3.1 vector. Transfected cells were untreated (control) or treated with MG132 (10µM) for 12h and luciferase was assayed as described under “Experimental Procedures”. Graph shows fold changes in the quotient of firefly/renilla luciferase. Data are expressed as mean ± S.E.M. from three different experiments. Legend to Fig. 9. Effect of MG132 treatment of Hela and PC12 cells on c-jun and c-fos expression. Hela or PC12 cells were untreated (-) or treated (+) with 10 µM MG132 for 12h. Total cells extracts were analyzed by immunoblots with the indicated antibodies to detect total c-fos and c-jun and the phosphorylation of c-jun, as described under “Experimental Procedures”. Legend to Fig. 10. Studying protein degradation by proteasome under controlled cellular conditions. PC12 stably transfected with EGFPd2 were treated MG132 (5µM) for 4h, then washed 4 times with complete medium to remove the excess of proteasome inhibitor and plated again, 1h after plating medium was changed and fresh media containing: no addition (control), 20 µg/ml cycloheximide (CHX), or CHX and proteasome inhibitors as indicated in the figure. Left panel show the quantification of relative fluorescence and protein levels as determined by flow citometry and Western immunoblot, and expressed as mean ± S.E.M from three different experiments. Middle panel show a representative experiment of Western and immunoblots protein (EGFPd2 and tubulin, used as control) by immunoblotting with anti-EGFP and anti-tubulin antibodies, φ, untrasnfected PC12. Right panel, total RNA was isolated and analyzed by qPCR, as described under “Experimental Procedures”. Graphs show the relative fold change and the corresponding ranges (upper and lower bars) as calculated from three different experiments from –∆∆Ct values.
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
12
Reference List
1. Voges, D., Zwickl, P., and Baumeister, W. (1999) Annu.Rev.Biochem. 68, 1015-1068
2. Hershko, A. and Ciechanover, A. (1998) Annu.Rev.Biochem 67, 425-479
3. Varshavsky, A. (1995) Cold.Spring.Harb.Symp.Quant.B. 60, 461-478
4. Neefjes, J. and Dantuma, N. P. (2004) Nat.Rev.Drug Discov. 3, 58-69
5. Dantuma, N. P., Lindsten, K., Glas, R., Jellne, M., and Masucci, M. G. (2000) Nat.Biotechnol. 18, 538-543
6. Luker, G. D., Pica, C. M., Song, J., Luker, K. E., and Piwnica-Worms, D. (2003) Nat.Med. 9, 969-973
7. Gilon, T., Chomsky, O., and Kulka, R. G. (1998) EMBO J. 17, 2759-2766
8. Corish, P. and Tyler-Smith, C. (1999) Protein Eng 12, 1035-1040
9. Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001) Science 292, 1552-1555
10. Zhang, M., Pickart, C. M., and Coffino, P. (2003) EMBO J. 22, 1488-1496
11. Li, X., Fang, Y., Zhao, X., Jiang, X., Duong, T., and Kain, S. R. (1999) J.Biol.Chem. 274, 21244-21250
12. Gandara, M. L., Lopez, P., Hernando, R., Castano, J. G., and Alemany, S. (2003) Mol.Cell Biol. 23, 7377-7390
13. Angel, P., Imagawa, M., Chiu, R., Stein, B., Imbra, R. J., Rahmsdorf, H. J., Jonat, C., Herrlich, P., and Karin, M. (1987) Cell 49, 729-739
14. Fernandez, M., Sanchez-Franco, F., Palacios, N., Sanchez, I., and Cacicedo, L. (2005) J.Mol.Endocrinol. 34, 699-712
15. Castrillo, A., Diaz-Guerra, M. J., Hortelano, S., Martin-Sanz, P., and Bosca, L. (2000) Mol.Cell Biol. 20, 1692-1698
16. Mizushima, S. and Nagata, S. (1990) Nucleic Acids Res. 18, 5322
17. Aragones, J., Lopez-Rodriguez, C., Corbi, A., del Arco, P. G., Lopez-Cabrera, M., de Landazuri, M. O., and Redondo, J. M. (1996) J.Biol.Chem. 271, 10924-10931
18. Chomczynski, P. and Sacchi, N. (1987) Anal.Biochem. 162, 156-159
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
13
19. Nieto, A., Mira, E., and Castano, J. G. (1990) Biochem.J. 267, 317-323
20. Schmittgen, T. D. and Livak, K. J. (2008) Nat.Protoc. 3, 1101-1108
21. Qian, S. B., Princiotta, M. F., Bennink, J. R., and Yewdell, J. W. (2006) J.Biol.Chem. 281, 392-400
22. Detich, N., Hamm, S., Just, G., Knox, J. D., and Szyf, M. (2003) J.Biol.Chem. 278, 20812-20820
23. Grassi, G., Maccaroni, P., Meyer, R., Kaiser, H., D'Ambrosio, E., Pascale, E., Grassi, M., Kuhn, A., Di Nardo, P., Kandolf, R., and Kupper, J. H. (2003) Carcinogenesis 24, 1625-1635
24. Schmidt, E. V., Christoph, G., Zeller, R., and Leder, P. (1990) Mol.Cell Biol. 10, 4406-4411
25. Ramanathan, M., Hasko, G., and Leibovich, S. J. (2005) Inflammation 29, 94-102
26. Prosch, S., Heine, A. K., Volk, H. D., and Kruger, D. H. (2001) J.Biol.Chem. 276, 40712-40720
27. Sambucetti, L. C., Cherrington, J. M., Wilkinson, G. W., and Mocarski, E. S. (1989) EMBO J. 8, 4251-4258
28. Lembo, D., Angeretti, A., Foresta, P., Gribaudo, G., Gariglio, M., and Landolfo, S. (1994) J.Gen.Virol. 75 ( Pt 7), 1685-1692
29. Stamminger, T., Fickenscher, H., and Fleckenstein, B. (1990) J.Gen.Virol. 71 ( Pt 1), 105-113
30. Sun, B., Harrowe, G., Reinhard, C., Yoshihara, C., Chu, K., and Zhuo, S. (2001) J.Cell Biochem. 83, 563-573
31. Niller, H. H. and Hennighausen, L. (1991) Nucleic Acids Res. 19, 3715-3721
32. Hennighausen, L. and Fleckenstein, B. (1986) EMBO J. 5, 1367-1371
33. Meier, J. L. and Stinski, M. F. (1996) Intervirology 39, 331-342
34. Chan, Y. J., Chiou, C. J., Huang, Q., and Hayward, G. S. (1996) J.Virol. 70, 8590-8605
35. Bruening, W., Giasson, B., Mushynski, W., and Durham, H. D. (1998) Nucleic Acids Res. 26, 486-489
36. Sandbo, N., Qin, Y., Taurin, S., Hogarth, D. K., Kreutz, B., and Dulin, N. O. (2005) Mol.Pharmacol. 67, 789-797
37. Lipford, J. R. and Deshaies, R. J. (2003) Nat.Cell Biol. 5, 845-850
38. Auld, K. L., Brown, C. R., Casolari, J. M., Komili, S., and Silver, P. A. (2006) Mol.Cell 21, 861-871
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Unstable fluorescent proteins used as UPS reporters
14
39. Collins, G. A. and Tansey, W. P. (2006) Curr.Opin.Genet.Dev. 16, 197-202
40. Nawaz, Z. and O'Malley, B. W. (2004) Mol.Endocrinol. 18, 493-499
41. Bush, K. T., Goldberg, A. L., and Nigam, S. K. (1997) J.Biol.Chem. 272, 9086-9092
42. Kawazoe, Y., Nakai, A., Tanabe, M., and Nagata, K. (1998) Eur.J.Biochem. 255, 356-362
43. Mu, T. W., Ong, D. S., Wang, Y. J., Balch, W. E., Yates, J. R., III, Segatori, L., and Kelly, J. W. (2008) Cell 134, 769-781
44. Kinyamu, H. K. and Archer, T. K. (2007) Mol.Cell Biol. 27, 4891-4904
45. DiDonato, J. A., Mercurio, F., and Karin, M. (1995) Mol.Cell Biol. 15, 1302-1311
46. Sandbo, N., Qin, Y., Taurin, S., Hogarth, D. K., Kreutz, B., and Dulin, N. O. (2005) Mol.Pharmacol. 67, 789-797
47. Taylor, C. T., Furuta, G. T., Synnestvedt, K., and Colgan, S. P. (2000) Proc.Natl.Acad.Sci.U.S.A 97, 12091-12096
48. Walowitz, J. L., Bradley, M. E., Chen, S., and Lee, T. (1998) J.Biol.Chem. 273, 6656-6661
49. Biasini, E., Fioriti, L., Ceglia, I., Invernizzi, R., Bertoli, A., Chiesa, R., and Forloni, G. (2004) J.Neurochem. 88, 545-553
50. Li, X., Chen, D., Yin, S., Meng, Y., Yang, H., Landis-Piwowar, K. R., Li, Y., Sarkar, F. H., Reddy, G. P., Dou, Q. P., and Sheng, S. (2007) J.Cell Physiol 212, 298-306
51. Wheeler, D. G. and Cooper, E. (2001) J.Biol.Chem. 276, 31978-31985
52. Collins, G. A. and Tansey, W. P. (2006) Curr.Opin.Genet.Dev. 16, 197-202
53. Baens, M., Noels, H., Broeckx, V., Hagens, S., Fevery, S., Billiau, A. D., Vankelecom, H., and Marynen, P. (2006) PLoS.ONE. 1, e54
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from
PC12
0
10
20
0 10 20Lactacystin [µM]
Fold
chan
ge
100 101 1020
128
Cel
lcou
nt
FL1-H
0µM Lactacystin4µM Lactacystin10µM Lactacystin20µM Lactacystin
PC12
0
5
10
15
0 10 20MG132 [µM]
Fold
chan
ge
100 101 1020
128
Cel
lcou
nt
FL1-H
0µM MG1324µM MG13210µM MG13220µM MG132
0 2 4 10 20PC12 MG132 [µM]
1
NIH3T3 Lactacystin [µM]
NIH3T3
0102030
0 10 20Lactacystin [µM]
Fold
chan
ge
100 101 1020
128
Cel
lcou
nt
FL1-H
0µM Lactacystin4µM Lactacystin10µM Lactacystin20µM Lactacystin
0 2 4 10 201
NIH3T3
0204060
0 20MG132 [µM]
Fold
chan
ge
100 101 1020
128
Cel
lcou
nt
FL1-H
0µM MG1324µM MG13210µM MG13220µM MG132 NIH 3T3 MG132 [µM]
0 2 4 10 201
10
Fig. 1
a
PC12 Lactacystin [µM]
0 2 4 10 201
b c
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
NIH3T3
PC12
Control 4µM MG132 10µM MG132 20µM MG132
Fig.2
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
0
100
200
300
400
500
600
Mea
n flu
ores
cenc
e
2 43 5 6 7 8 9 10 11 12 13
1312
a b
EGFPd2
β-actin
mRNA
mRNA
Protein
Protein
EGFP-d2
Tubulin
2
Con
trol
5
MG
132
6
MG
132
+ A
ctD
7
MG
132
+ C
hx
8
Lac
tacy
stin
9
Lac
ta +
Act
D
10
Lac
ta +
Chx
11
Epo
xom
ycin
12
Epo
xo+
Act
D
13
Epo
xo+
Chx
1
Unt
rans
fect
ed
Con
trol
Act
DC
hx
2 3 4
12
34
567
910
11
8
101
102103
104
Fig. 3
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
Control
MG132 MG132 + ActD MG132 + CHX
ActD CHX
Fig. 4
EGFPd2
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
EYFP
β-actin
EYFP
Tubulin
mRNA
mRNA
Protein
Protein
4
MG
132
+ A
ctD
7
MG
132
+ C
hx
8
Lac
tacy
stin
9
Lac
ta +
Act
D
10
Lac
ta +
Chx
11
Epo
xom
ycin
12
Epo
xo+
Act
D
13
Epo
xo+
Chx
1
Unt
rans
fect
ed
2
Con
trol
5
MG
132
0
200
400
600
800
1000
2 43 5 6 7 8 9 10 11 12 13
Mea
n flu
ores
cenc
e
Fig. 5
b
1213
1103
234
567
891011
101102
a
Act
DC
hx
2
Con
trol
3 4
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
Control
MG132 MG132 + ActD MG132 + CHX
ActD CHX
Fig. 6
EYFP
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
2
Con
trol
5M
G13
26
MG
132
+ A
ctD
7M
G13
2 +
Chx
1
Unt
rans
fect
ed
EYFP
GAPDH
PDI
8
Lac
tacy
stin
9
Lac
ta +
Act
D
10
Lac
ta +
Chx
11
Epo
xom
ycin
12
Epo
xo+
Act
D
13
Epo
xo+
Chx
rRNA18S28S
0
2
4
6
8
10
2 3 4 5 6 7 8 9 10 11 12 13
Rel
ativ
eFo
ld
Act
DC
hx
2
Con
trol
3 4
Fig. 7
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
0
0,5
1
1,5
2
2,5
3
AP-1 NFkBCRE
Fold
Luc
ifera
se/ R
enill
a
pCMV GFPu
00,51
1,52
2,53
3,5
pcDNA pEF-BOS AP1 CRE NFkB
Mea
n Fl
uore
scen
ce(f
old) Control
MG132pCMV EYFP
00,51
1,52
2,53
3,5
pcDNA pEF-BOS AP1 CRE NFκBMea
n Fl
uore
scen
ce(f
old)
Fig. 8
NFκB
EYFP
GFPuMG132 - + - -+ + - + - +
pcDNA pEF-BOS AP-1 CRE
Tub
Tub
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
MG132
c-Fos
c-Jun
p-c-Jun
Tubulin
Fig. 9
- +HELA
- +PC12
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from
Time (h) 0 1 2 4 6 8Φ
Chx
Chx+MG132
Chx+Lacta
Control
Tubulin
Fold
Cha
ngue
0
0,5
1
1,5
0 4 8Time (h)
Rel
ativ
eA
mou
nts
0
0,2
0,4
0,6
0,8
1
0 2 4 6 8Time (h)
controlCHXCHX + 5 µM MG132CHX + 5 µM lactacystin
Fig. 10
qPCR
by guest on October 5, 2018 http://www.jbc.org/ Downloaded from

CastañoBeatriz Alvarez-Castelao, Idoia Martín-Guerrero, África García-Orad and José G.
inhibitorsunstable reporter proteins after treatment of living cells with proteasome CMV promoter up-regulation is the major cause of increased protein levels of
published online August 13, 2009J. Biol. Chem.
10.1074/jbc.M109.004101Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2009/08/13/M109.004101.DC1
by guest on October 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from