Embed Size (px)
Citation preview

Universidad de Sevilla
Analisis Genomico y Transcriptomico
de Zea mays basados en Datos de
Next Generation Sequencing
Autor:
Pablo Villegas
Miron
Trabajo Fin de Grado
26/08/2014

ÍNDICE
1. Resumen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
2. Introducción . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
3. Objetivos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
4. Materiales yMétodos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1. Materiales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1. Genoma y anotación de referencia . . . . . . . . . . . . . . . . . . . . . . . . . 2
2. Datos de RNA-seq . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
2. Métodos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .3
1. Procesamiento de los datos crudos y obtención de la expresión génica . . . . . . . 3
2. Selección de los genes diferencialmente expresados . . . . . . . . . . . . . . . . 5
3. Construcción de la red de co-expresión génica . . . . . . . . . . . . . . . . . . . .5
4. Análisis de la topología y técnicas de agrupamiento . . . . . . . . . . . . . . . . .7
5. Enriquecimiento de términos de ontología de genes (GO) . . . . . . . . . . . . . .7
6. Identificación de factores de transcripción . . . . . . . . . . . . . . . . . . . . . .8
7. Identificación de genes regulados por luz, genes fotoperiódicos y genes
fotomorfogénicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .8
8. Identificación de genes de interés biotecnológico y sus potenciales reguladores . . 9
9. Coste computacional . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
5. Resultados y Discusión . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .9
1. Generación de la red y estudio de la topología . . . . . . . . . . . . . . . . . . . . . .9
2. Análisis del agrupamiento general . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
3. Estudio de los factores de transcripción . . . . . . . . . . . . . . . . . . . . . . . . 16
4. Estudio de genes regulados por luz, genes fotoperiódicos y genes fotomorfogénicos . 20
5. Estudio de genes de interés biotecnológico . . . . . . . . . . . . . . . . . . . . . . . 24
6. Conclusiones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
7. Bibliografía . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
8. Agradecimientos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .28

1. RESUMEN
Se ha realizado un estudio integrativo global a nivel genómico y transcriptómico de la
planta superior Zea mays (Z. mays) o maíz. Este es un organismo modelo de gran
importancia, ya que no solo tiene relevancia en la investigación celular y molecular, sino que
también lo hace a nivel económico, siendo una de las plantas de cultivo de mayor producción
mundial.
Para hacer este estudio se han utilizado datos de Next Generation Sequencing (NGS)
basados en RNA-seq, los cuales abarcan un espectro más completo y sensible de información
que otras técnicas para análisis transcriptómicos como los microarrays. Se ha construido una
red de coexpresión génica, para la cual se han considerado 27 condiciones distintas de 7
experimentos. Esta red ha servido de base para estudios topológicos, funcionales y regulativos
del transcriptoma de Z. mays.
Se han empleado métodos de inteligencia artificial para revelar los distintos módulos
funcionales (agrupamiento) y potenciales reguladores (factores de transcripción) de la red.
Además se han buscado sitios de unión de factores de transcripción en los promotores de
genes para aportar mayor información sobre la regulación del transcriptoma. Debido a la
importancia de la luz en la fisiología y desarrollo vegetal, se han estudiado genes de Z. mays
potencialmente ortólogos a genes de respuesta a luz en Arabidopsis thaliana (A. thaliana). Por
último, al ser una de las plantas de mayor importancia agrícola, se han analizado genes de
interés biotecnológico que puedan sugerir posibles mejoras en el rendimiento de los cultivos.
2. INTRODUCCIÓN
El continuo avance en las tecnologías de secuenciación de nueva generación ha
llevado a un incremento masivo en la cantidad de datos de expresión génica, provocando el
avance paralelo de las técnicas de análisis en Biología Molecular de Sistemas. Esto nos
permite pasar de la tradicional perspectiva reduccionista a una perspectiva global e
integrativa, consiguiendo llegar a la caracterización y estudio riguroso de múltiples
propiedades biológicas en el contexto de los Sistemas Complejos.
Una técnica para estudios globales e integrativos del transcriptoma de un organismo
son las redes de coexpresión génica (Zhang et al, 2005). En éstas nos encontramos a los genes
representados como nodos unidos entre sí por aristas en función de cómo se encuentren
correlacionados sus perfiles de expresión, permitiéndonos analizar sus diferencias a través de
múltiples condiciones experimentales. Cuando se comparan dos condiciones distintas,
generalmente la expresión de determinados genes también es distinta, ya que el organismo
adapta su transcriptoma en función de las condiciones que se den. Lo mismo ocurre entre1

distintos tejidos o estadios de desarrollo, los patrones de expresión entre tejidos similares
generalmente van a mostrar perfiles de expresión similares. La relación entre estos perfiles de
expresión constituye la base para la construcción de redes de coexpresión génica..
Este tipo de redes no sólo nos permiten ver que genes se coexpresan entre sí en estas
condiciones, sino que su distribución en la red nos permite identificar distintas propiedades
topológicas que sugieren su participación en módulos funcionales. Estos módulos aparecen en
la red como grupos de genes coexpresados y potencialmente corregulados, lo cual aporta
evidencia sobre su participación en procesos biológicos específicos en el organismo. Sin
embargo, no sólo se pueden describir las propiedades funcionales de estas redes, sino también
cómo se regulan y que genes participan en esta modulación.
Uno de los organismos vegetales de mayor interés, sobre todo a nivel agrícola, es Z.
mays o maíz, una de las plantas de cultivo que más se utiliza actualmente. La producción
mundial de maíz llegó, en el periodo 2013/2014, a los 973 millones de toneladas, siendo
EEUU el mayor país productor (http://www.usda.gov/wps/portal/usda/usdahome). Es, por
tanto, un importante organismo modelo para su estudio a nivel molecular.
3. OBJETIVOS
Obtención de una red de coexpresión génica de Z. mays, basada en datos de RNA-seq.
Su estudio a nivel estructural, funcional y regulatorio, identificando los posibles sitios de
unión de factores de transcripción. Identificación y localización de genes regulados por luz y
genes de interés biotecnológico, así como el análisis de su regulación potencial a partir de las
relaciones con la red de coexpresión génica.
4. MATERIALES Y MÉTODOS
4.1. Materiales
4.1.1. Genoma y anotación de referencia
El genoma de Z. mays utilizado como referencia en este trabajo (Maize Golden Path
B73 RefGen_v2, Patrick et al, 2009) fue descargado de Gramene (Ware et al, 2002.
http://www.gramene.org/), una base de datos utilizada por la plataforma web Phytozome
(Goodstein et al, 2012. http://www.phytozome.net/) de recursos genómicos de plantas.
También fue descargado de la misma base de datos la correspondiente anotación del genoma
(versión 5b.60), utilizada como transcriptoma de referencia.
4.1.2. Datos de RNA-seq
En este análisis se han utilizado datos de secuenciación basados en RNA-seq de Z.2

mays, obtenidos de la base de datos Sequence Read Archive (SRA,
http://www.ncbi.nlm.nih.gov/Traces/sra/) y disponibles públicamente. SRA pertenece al
National Center for Biotechnology Information (NCBI), que almacena más de 500 TeraBases
de datos de Next Generation Sequencing.
Se han analizado más de 58 Gb de información sobre muestras a lo largo de 7
experimentos, los cuales recogen un total de 27 condiciones distintas que representan el
transcriptoma de Z. mays analizado en este estudio. Todos estos datos fueron obtenidos
utilizando la misma plataforma de secuenciación de nueva generación, Illumina Genome
Analyzer. La tabla suplementaria 1, que se encuentra en la web pública
“http://viridiplantae.ibvf.csic.es/tabla_condiciones1.png”, recoge los detalles de cada uno de
los experimentos de los cuales se han obtenido los datos de secuenciación.
4.2. Métodos
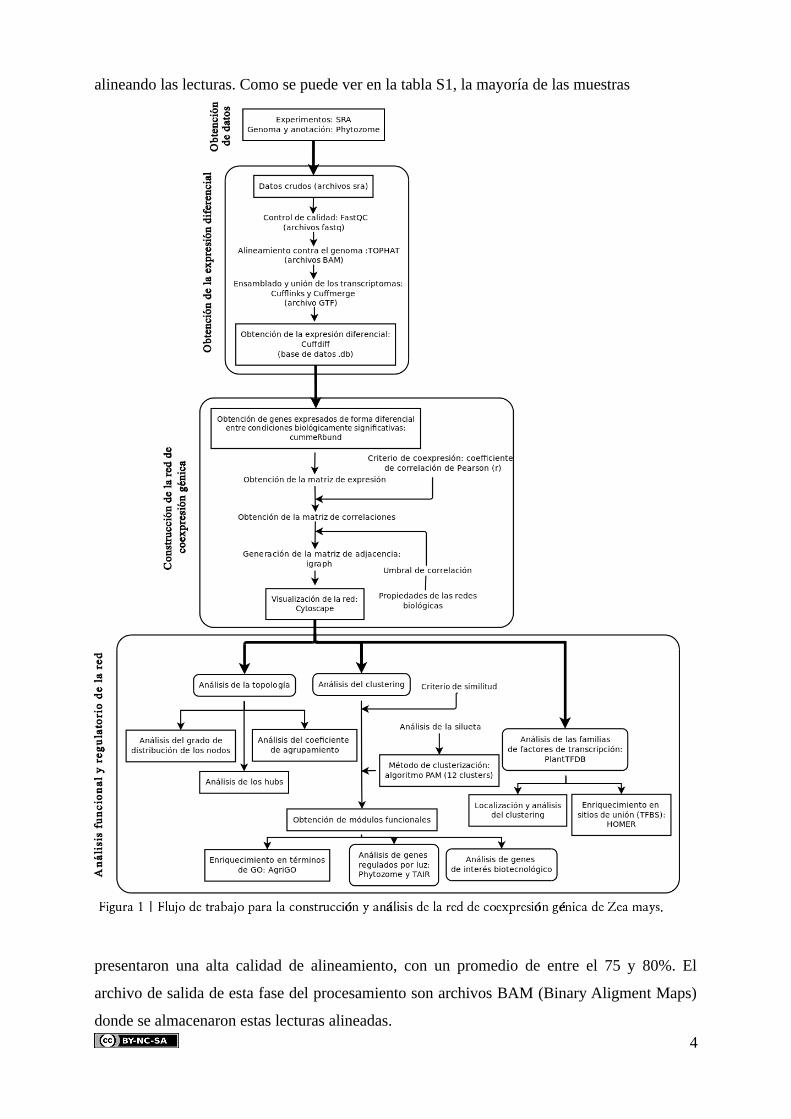
La metodología utilizada en la construcción y posterior análisis de la red de
coexpresión génica es una adaptación de la seguida en (Linyong et al, 2009;
Romero-Campero et al, 2013), una aproximación “top-down” en la que se parte de la red y se
avanza dividiendo su estructura en módulos e identificando funciones concretas. Todos los
pasos dados a lo largo del estudio se recogen en el flujo de trabajo de la figura 1, los cuales se
detallan a continuación.
4.2.1. Procesamiento de los datos crudos y obtención de la expresión génica
El flujo de trabajo para procesar los datos crudos, cuando hay un genoma y anotación
de referencia disponibles, sigue las siguientes fases: (i) eliminación de las muestras de baja
calidad y de bajo porcentaje de alineamiento, (ii) alineamiento de las lecturas al genoma de
referencia, (iii) ensamblado de los transcritos y (iv) estimación de los valores de expresión
diferencial (figura 1).
Esta metodología es similar a la seguida en (Trapnell et al, 2012) y adaptada a las
necesidades de este estudio. En un primer momento (i), las muestras crudas fueron filtradas
con el paquete de software FastQC (http://www.bioinformatics.babraham.ac.uk) en busca de
lecturas de mala calidad originadas en la secuenciación. También se consideraron de mala
calidad las que presentaban un mal alineamiento con el genoma. Finalmente, estas muestras se
eliminaron del conjunto total ya que, en caso contrario, distorsionarían los resultados.
Tras este filtrado, las lecturas se alinearon contra el genoma de referencia (ii) con el
programa Tophat (Trapnell et al, 2009), que hace uso del algoritmo de Burrows-Wheeler
implementado eficientemente en la herramienta Bowtie, que trabaja indexando el genoma y3
alineando las lecturas. Como se puede ver en la tabla S1, la mayoría de las muestras
presentaron una alta calidad de alineamiento, con un promedio de entre el 75 y 80%. El
archivo de salida de esta fase del procesamiento son archivos BAM (Binary Aligment Maps)
donde se almacenaron estas lecturas alineadas. 4

En la siguiente fase (iii), estas lecturas alineadas o transcritos son ensamblados en
transcriptomas específicos de cada muestra usando el paquete de software Cufflinks (Trapnell,
2010) junto con la anotación del genoma 5b.60. Tras esto se utilizó el programa Cuffmerge
del paquete Cufflinks para integrarlos en un transcriptoma general, almacenándose en un
archivo GTF (gene transfer format) donde quedaron reunidas todas las muestras.
Finalmente los niveles de expresión diferencial entre las distintas condiciones fueron
obtenidos con el programa CuffDiff, incluido en el paquete Cufflinks. La unidad de medida de
expresión que se utilizó fue fragmentos por kilobase de transcritos por millón de fragmentos
alineados (FPKM), la cual aplica una normalización en la que se eliminan los sesgos debidos
a las longitudes y al número de las lecturas obtenidos en la secuenciación (Garber et al, 2011).
Los resultados de este paso son recogidos en un archivo de texto en formato tabla, en el que se
recogen todas las comparaciones posibles entre las condiciones y los datos de expresión
diferencial.
El paquete de cummeRbund (Goff et al, 2011), obtenido de la plataforma
Bioconductor, ayuda a manejar este tipo de archivos a través del lenguaje de programación R,
facilitando los siguientes pasos de visualización de resultados y análisis estadísticos.
4.2.2. Selección de los genes diferencialmente expresados
El paso siguiente fue seleccionar los genes que presentaban una expresión diferencial
significativa entre las distintas condiciones biológicas. Para ello se eligió como criterio para
todos los experimentos un p-valor de 0.05, considerando a todos los genes cuyo p-valor se
encontraba por debajo de éste diferencialmente expresados.
También se consideró un factor de proporcionalidad como medida indicativa de
expresión diferencial. Este es una medida de cuánto cambia el valor de expresión de un gen
desde su mínimo hasta su máximo a lo largo de las comparaciones. Se aplicó un factor de 2
para los genes activados que, dado que se establece en función del logaritmo en base 2,
corresponde a un valor de logaritmo igual a 1. En el caso de los genes inhibidos, se escogió la
misma diferencia que en los activados. (Romero-Campero et al, 2013)
Tras haber obtenido los genes diferencialmente expresados, se identificaron según la
nomenclatura de la base de datos Gramene para los genes de Z. mays (“GRMZM”, Gramene
Zea mays). Esta se utiliza como sinónimo de la nomenclatura establecida en Marzo del 2013
por el Maize Nomenclature Committee (“ZEAMMB73”, Zea mays ssp. mays B73), estando
presente de igual modo en las bases de datos.
4.2.3. Construcción de la red de coexpresión génica. 5

Una red de coexpresión génica está formada por genes, representados como nodos y
conectados de acuerdo a una regla de coexpresión. Cada par de genes estarán unidos tan cerca
entre sí como mayores sean estos valores de coexpresión. Esta medida puede ser traducida
como el modo en que estos genes comparten información mutua, por ejemplo, interviniendo
en las mismas vías metabólicas (Linyong et al, 2009; Aoki et al, 2007).
Como medida del grado de coexpresión se ha utilizado el coeficiente de correlación de
Pearson (r), ya que es el más comúnmente utilizado, junto con su valor absoluto y el valor
absoluto elevado a un exponente (Ma et al, 2013). Tras su aplicación sobre los genes
diferencialmente expresados, se obtuvo una matriz donde se representan los valores de
correlación entre los distintos genes.
Para determinar que genes se encuentran coexpresados de forma significativa para la
construcción de la red, se estableció un umbral de correlación en base al compromiso entre
dos propiedades de las redes biológicas: la libre escala (Barabasi et al, 1999) y la alta
conectividad (Aoki et al, 2007). La primera surge del hecho de que las redes biológicas que se
encuentran en la Naturaleza presentan muy pocos nodos con alta conectividad, denominados
hubs. Como consecuencia, la información que viaja a través de la red lo hace atravesando
siempre estos nodos, adquiriendo éstos una gran importancia para su funcionamiento. Esta
propiedad también le confiere robustez a la red, ya que ante ataques fortuitos (no dirigidos),
como mutaciones, va a haber una baja probabilidad de que estos genes se vean afectados y por
tanto dañen la funcionalidad de la red.
La segunda propiedad se basa en la alta conectividad presente entre los nodos, la
información que fluye desde un nodo A a uno B lo va a hacer a través del camino mínimo que
se pueda establecer entre estos dos, resultando en una alta velocidad en la comunicación
dentro de la red. A estas redes de alta conectividad también se las denomina “redes de mundo
pequeño”.
Por tanto, a la hora de elegir el umbral de correlación, se procedió atendiendo a estas
propiedades. En el caso de la libre escala, se debe estudiar el ajuste de la distribución del
grado de los nodos a una exponencial negativa. El grado de los nodos es el número de vecinos
que se encuentran directamente unidos a cada uno de ellos. Por lo tanto, esta exponencial
negativa indicará que la mayoría de los nodos tiene muy pocos vecinos, pero habrá un
reducido número de ellos con una alta conectividad (hubs). Esto se hizo convirtiendo esta
distribución a escala logarítmica y calculando el ajuste de estos valores a una recta según su
R2 (Romero-Campero et al, 2013).
Para la propiedad de mundo pequeño simplemente se obtuvo la conectividad entre los
nodos de la red. Esta conectividad se estudió a partir del coeficiente de agrupamiento de los6

nodos, el cual es una medida del número de conexiones que existe entre los vecinos de cada
nodo. (Romero-Campero et al, 2013; Aoki et al, 2007).
Este umbral se utilizó para filtrar los valores de correlación, utilizándose para la
construcción de la red solo aquellos obtenidos para estas propiedades de libre escala y mundo
pequeño. Esto se hizo utilizando el paquete de R “igraph” (Csardi et al, 2006), con diversas
funciones específicas de construcción de redes. Éste también se utilizó para obtener la matriz
de adyacencia que define la red, que finalmente se visualizó mediante el paquete de software
Cytoscape (Cline et al, 2007), utilizándose el algoritmo de estructuración “Organic” para
representar la estructura.
4.2.4. Análisis de la topología y técnicas de agrupamiento
A partir de los valores del grado de los nodos y coeficiente de agrupamiento se hizo un
estudio de la topología de la red. Se utilizó la herramienta Cytoscape para visualizar estos
datos, mostrando cómo varía la conectividad entre las distintas regiones y definiendo posibles
módulos de interés (Aoki et al, 2007).
El análisis del agrupamiento nos muestra la organización funcional que presentan los
nodos dentro de la red. Está basado en la similitud entre los patrones de expresión de los
genes, que lleva a los nodos más similares a organizarse en módulos funcionales involucrados
en procesos biológicos relacionados. Estos nodos presentan una alta conectividad dentro del
módulo, pero limitada con los nodos que se encuentran fuera del conjunto (Ma et al, 2013).
Para obtener esta medida de similitud se partió de la matriz de correlaciones, sobre la
que se aplicó un criterio basado en el método de correlación de Pearson. En un primer
momento, estos valores de similitud se deben analizar para determinar que método de
agrupamiento resulta más adecuado para la red. Este estudio se basa en buscar el máximo
valor de la silueta del agrupamiento, la cual es una medida que nos dice que método nos da
una máxima similitud intra-grupo y una máxima disimilitud inter-grupo dentro de la red.
Por tanto, se determinó el valor de esta medida para dos métodos de agrupamiento en
un rango de 1 a 20 clústeres. Los métodos examinados fueron la técnica de agrupamiento
jerárquico y la técnica de agrupamiento por partición entorno a centroides (PAM). El primero
procede con la construcción de un dendrograma que representa las relaciones de similitud
entre los nodos, y el segundo consiste en organizar los nodos entorno a un centroide,
buscando la máxima cercanía de los nodos a este elemento central en un proceso iterativo.
4.2.5. Enriquecimiento de términos de ontología de genes
Tras la división de la estructura de la red en módulos funcionales, se procedió a7

identificar los procesos en los que estos grupos estaban enriquecidos. Para ello se hizo uso de
la herramienta web de análisis de ontología de genes y base de datos agriGO (Du et al, 2010).
En ella se utilizó el algoritmo de análisis de enriquecimiento singular (SEA) de genes, el cual
es el método tradicional y de mayor uso. Este método utiliza el test estadístico de Fisher con
el que se aplicó el p-valor por defecto de 0.05.
4.2.6. Identificación de factores de transcripción
El siguiente paso fue el análisis de la regulación de las funciones encontradas. Para
ello se procedió a identificar en la red todos los factores de transcripción disponibles,
utilizando la colección de familias de factores de transcripción para Z. mays de la base de
datos PlantTFDB (Jin et al, 2014).
Una vez localizados en la red se procedió a obtener el agrupamiento de los factores
según se ha descrito anteriormente, usando la silueta como método de elección del algoritmo
óptimo. Esto sirvió para delimitar los grupos de factores que intervienen en la regulación de
las distintas funciones en la red, ya que la organización de los genes en módulos funcionales
sugiere que se encuentran regulados por los mismos factores de transcripción (Ma et al,
2013).
Para poder estudiar la potencial regulación de los genes de cada uno de los módulos,
se siguió con el análisis de la vecindad de estos factores de transcripción y su enriquecimiento
en sitios de unión de factores de transcripción (TFBS). Esto se hizo por el hecho de que la
presencia significativa de sitios de unión en promotores de genes de esta vecindad podría
sugerir que estos genes y, por tanto, el papel que juegan en el módulo funcional, se encuentran
regulados por estos factores de transcripción. Con este fin se hizo uso del paquete de
herramientas HOMER v4.6 (Hipergeometric Optimization of Motif Enrichment) para el
descubrimiento de sitios de unión y análisis de datos de NGS (Heinz et al, 2010).
4.2.7. Identificación de genes regulados por luz, genes fotoperiódicos y genes
fotomorfogénicos
Para la búsqueda de genes regulados por luz se procedió identificando los potenciales
ortólogos de los genes conocidos de A. thaliana en el genoma del maíz. En esta búsqueda se
hizo uso de la base de datos Phytozome, en la que se consideró un umbral de similitud de
secuencia del 40%, por encima del cual se tomaron los genes como potenciales ortólogos,
además de la conservación relativa de los dominios proteicos.
Estos ortólogos podrían sugerir la función que llevan a cabo estos genes regulados por
luz en la red, por lo que se identificó su posible función a partir de la base de datos TAIR, la8

cual guarda datos genómicos y de biología molecular de la planta superior A. thaliana (The
Arabidopsis Information Resource. www.arabidopsis.org). Tras esto se estudió su localización
en la red, su función en los grupos de genes y su potencial regulación a partir de los factores
de transcripción encontrados anteriormente.
4.2.8. Identificación de genes de interés biotecnológico y sus potenciales reguladores
Se ha buscado en la bibliografía varios genes que podrían tener interés biotecnológico
actual para Z. mays, aquellos que se podrían aplicar a mejoras agronómicas (Kenneth et al,
2013). Tras esto se procedió a localizarlos en la red, analizar su función, su vecindad y
potencial regulación por los factores de transcripción identificados anteriormente.
4.2.9. Coste computacional
Debido al alto coste computacional que conllevan las operaciones de este flujo de
trabajo, se ha tenido que recurrir al uso de máquinas de supercomputación en las que ejecutar
todos los scripts y programas. Se han utilizado los servicios del CICA, quienes
proporcionaron las máquinas sesamo y bigfoot (550 cores de cálculo y sistema de colas SGE)
para el alineamiento de las lecturas con el genoma de referencia, ensamblado del
transcriptoma y obtención de la expresión génica diferencial.
El tiempo requerido para la ejecución de la mayoría de los scripts de alineamiento y
ensamblado fue de varias semanas cada uno, destacando la estimación de la expresión
diferencial con Cuffdiff con un tiempo total aproximado de un mes.
También se ha utilizado la máquina viridiplantae proporcionada por el Grupo en
Desarrollo Molecular y Metabolismo de Plantas perteneciente al Instituto de Bioquímica
Vegetal y Fotosíntesis (IBVF) y cuya capacidad es de 16 procesadores con 4 cores cada uno
(64 cores en total).
5. RESULTADOS Y DISCUSIÓN
5.1. Generación de la red y estudio de la topología
El estudio de los umbrales de correlación para el ajuste a una red de libre escala y
mundo pequeño muestra la distribución de la figura 2A. Como se puede ver, uno de los
mejores ajustes a estas propiedades corresponde con un valor de R² de 0.782, que coincide
con un valor de conectividad media de aproximadamente 10 nodos.
Estos valores reflejan las propiedades de las redes biológicas que se estaban buscando,
como se indica en Materiales y Métodos. En la figura 2B se muestra la distribución del grado
de los nodos como una exponencial negativa, como se esperaba, y el coeficiente de9

agrupamiento refleja una clara desviación hacia la derecha en la figura 2C, demostrando una
alta conectividad de los nodos.
Esta conectividad también se observa en los caminos mínimos entre los nodos en la
figura 2D, donde vemos una distribución trimodal con una alta presencia de caminos cortos.
Por tanto estos valores se consideraron la elección óptima para obtener una red con
propiedades de las redes biológicas y de la que extraer información estructural y funcional.
Estos valores corresponden con un umbral de correlación de 0.98 (figura 2A), el cual se
utilizó para generar la matriz de adyacencia y la red de coexpresión final.
10

En la figura 3A se puede ver la red final representada con la herramienta Cystoscape,
compuesta por 26.194 nodos y 115.055 aristas, sobre la cual se aplicaron los posteriores
análisis. La red construida se puede considerar muy restrictiva, ya que se ha elegido un valor
de umbral de correlación muy exigente. Como se verá más adelante, esto va a afectar tanto al
número de nodos unidos como a su estructuración. Una muestra de esta restricción se puede
ver en la parte inferior de la figura 3A, donde se encuentra la mayoría de los genes de forma
aislada, sin pertenecer a la componente principal de la red. Estos no resultan de interés para su
estudio y son solo los que se muestran los que potencialmente tienen significado biológico y
los que se han utilizado en posteriores análisis.
El grado de los nodos se utilizó para localizar las regiones de mayor conectividad de la
red (Materiales y Métodos). Como resultado, en la figura 3B vemos la representación de los
1000 nodos de mayor grado y, por tanto, con mayor probabilidad de constituir hubs de la red.
Se puede observar su localización específica en regiones modulares, donde los nodos se
encuentran aglomerados en grupos que sugieren la presencia de regiones con funciones
relevantes.
Como se explicó en Materiales y Métodos, uno de los elementos de mayor importancia
en una red son los hubs, representados en la figura 3C. Vemos que su localización es
mayoritaria en la región central, lo cual sugiere que esta es la región de mayor conectividad y11

que toda la información que viaja por la red fluye a través de ella. La figura 3D muestra la
distribución del coeficiente de agrupamiento, el cual, como medida de conectividad, también
sugiere la presencia de regiones modulares que desempeñan funciones específicas.
5.2. Análisis del agrupamiento general
Una vez estudiada la topología, se pasó a analizar la estructura interna de la red a partir
de su agrupamiento. Como ya se indicó en Materiales y Métodos, este análisis comienza con
el estudio de la silueta, que mostró que la mejor alternativa para agrupar la red es el algoritmo
PAM con 12 grupos. Estos se identificaron en la red, como vemos en la figura 4, mostrando
las mismas regiones funcionales que sugería el análisis de la topología.
Como se ha indicado en Materiales y Métodos, la pertenencia de estos nodos a
regiones modulares definidas sugiere su participación en procesos biológicos similares. Para
descubrir las funciones específicas de cada módulo, se llevó a cabo un enriquecimiento de
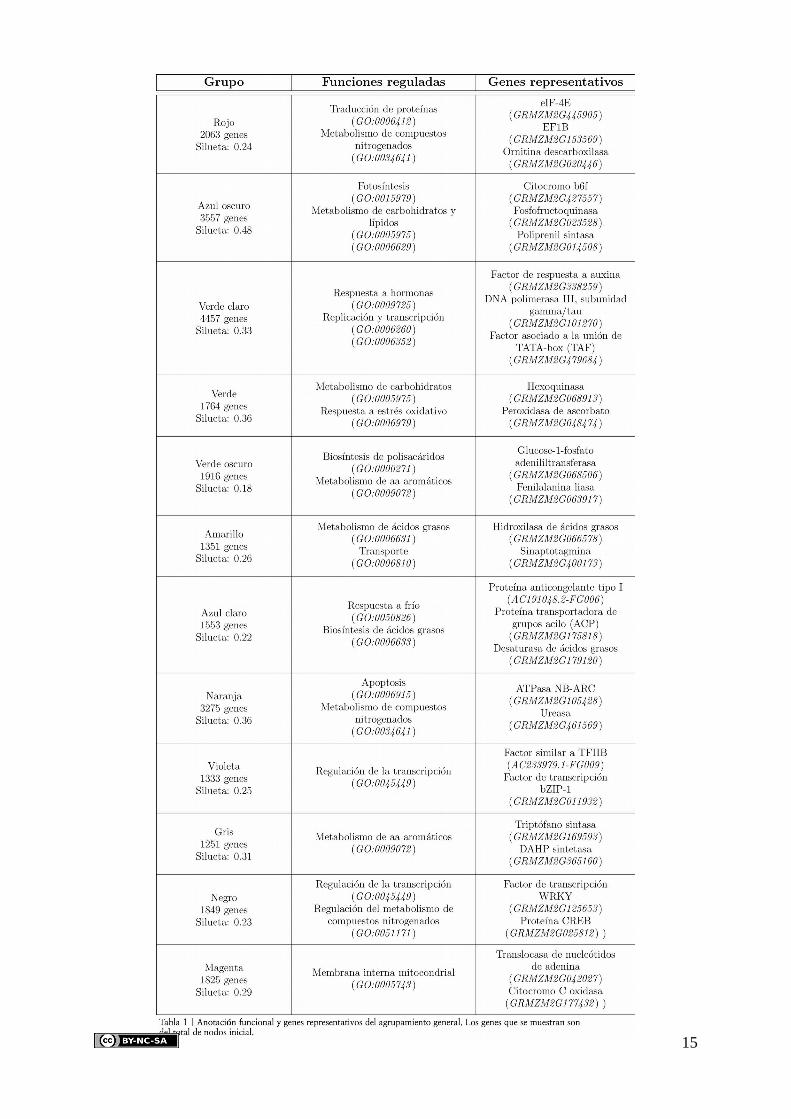
términos de GO de los genes con la herramienta agriGO, el cual se recoge en la tabla 1. Para
una aproximación más detallada a este estudio funcional, a continuación se describen cada
uno de los grupos representados en la figura 4:
• Grupo Azul oscuro: En el centro de la red se puede ver el grupo que ocupa la mayor
parte de la estructura, con 3557 nodos y una densidad de 0.48, por lo que es el que
presenta mayor cohesión. Como se empieza a intuir en las redes de organismos
fotosintéticos, el grupo de mayor tamaño se ocupa de las funciones principales de
12

metabolismo y fotosíntesis, con genes como la fosfofructoquinasa
(GRMZM2G023528, vía glucolítica) y el citocromo b6f (GRMZM2G427557,
transferencia de electrones entre los fotosistemas I y II). Por tanto, resulta lógico que
este grupo abarque los principales hubs, ya que es una región de alto tránsito de
información.
• Grupo Rojo: En rojo se muestra, con 2063 genes y cohesión de 0.24, el grupo
dedicado a las funciones de traducción y metabolismo de compuestos nitrogenados, lo
cual sugiere que estos genes se complementan recíprocamente en estas funciones.
Como ejemplos de estos genes vemos el factor eIF-4E (GRMZM2G445905), que actúa
formando el complejo de pre-iniciación de la traducción y la ornitina descarboxilasa
(GRMZM2G020446), componente del ciclo de la urea.
• Grupo Verde claro: Este grupo es el que presenta el mayor número de nodos, con
4457 y una silueta de 0.33. A pesar de ello, este grupo presenta la mayoría de sus
nodos aislados, resultando en una componente pequeña aislada de la principal. Sin
embargo, resulta lógico que ocupe un gran número de nodos, ya que sus funciones de
respuesta a hormonas (factor de respuesta a auxina, GRMZM2G338259) y regulación
de la replicación y transcripción (TAF, GRMZM2G479684) implica la participación de
una parte importante del genoma de las plantas.
• Grupo Verde: Este grupo se sitúa muy cerca de los citados anteriormente,
presentando 1764 genes con una cohesión de 0.36. Su ubicación en la región central
de la red es significativa, ya que, al igual que sus vecinos, está enriquecido en
funciones de metabolismo con genes como la hexoquinasa (GRMZM2G068913),
primer componente de la vía glucolítica. Por tanto se podría decir que éste, junto con
los grupos azul y rojo, conforman el núcleo metabólico de la red.
• Grupo Verde oscuro: Este grupo, con 1916 genes y una silueta de 0.18, también está
involucrado en procesos metabólicos (fenilalanina liasa, GRMZM2G063917), sin
embargo se encuentra más separado del núcleo metabólico de la red. De hecho se
puede apreciar que este grupo se ha abierto hacia la periferia, desde una hipotética
posición central, lo cual puede deberse a la alta restricción de la red.
• Grupo Amarillo: La anterior situación se da también en el grupo amarillo, el cual
presenta 1351 genes, una cohesión de 0.26 y funciones de transporte y metabolismo de
ácidos grasos. Éste conforma un grupo aislado de la componente principal, lo cual
indica que no presenta grandes valores de coexpresión con esta componente. Esto
también explica el hecho de que no forme parte del núcleo metabólico, ya que sus
13

funciones se encuentran más representadas para los procesos de transporte que para el
metabolismo de ácidos grasos, con genes como la sinaptotagmina
(GRMZM2G400173), involucrada en el tráfico vesicular.
• Grupo Azul claro: Este grupo presenta 1553 genes y una cohesión de 0.22. El análisis
funcional reveló que se encuentra enriquecido en los procesos de respuesta a frío y
biosíntesis de ácidos grasos, con genes como la proteína ACP (GRMZM2G17581),
proteína transportadora de grupos acilo. Estos procesos se encuentran
complementados, como en el caso del grupo rojo, ya que la adaptación de la
membrana celular a condiciones de frío depende de su composición en fosfolípidos.
• Grupo Naranja: Este grupo, con 3275 genes y una cohesión de 0.36, está enriquecido
principalmente en funciones de apoptosis, representada con la ATPasa NB-ARC
(GRMZM2G105428), dominio que forma parte del factor Apaf-1 del apoptosoma. Es
un grupo que pertenece a la región central, envolviendo al núcleo metabólico de la red,
lo cual sugiere que sus funciones metabólicas sobre los compuestos nitrogenados
tienen cierta relevancia, con genes como la ureasa (GRMZM2G461569), componente
principal del ciclo de la urea.
• Grupo Violeta: Este grupo se encuentra, como otros mencionados, en una
componente aislada de la principal. Tiene 1333 nodos y una cohesión de 0.25.
También presenta funciones de regulación de la transcripción, algo que puede sugerir
que ante condiciones menos restrictivas podría estar unido al grupo negro descrito más
abajo. Un ejemplo para este proceso es el gen bZIP-1 (GRMZM2G011932), factor de
transcripción envuelto en la homeostasis de nutrientes.
• Grupo Gris: El grupo gris es el de menor tamaño, con 1251 genes y una silueta de
0.31, presentando funciones de metabolismo de aminoácidos aromáticos, con genes
como la triptófano sintasa (GRMZM2G169593). Sin embargo, como el amarillo, se
encuentra aislado de la componente principal, lo cual puede ser un reflejo tanto de la
alta restricción de la red como del bajo número de genes.
• Grupo Negro: Como ya se ha comentado anteriormente, el grupo negro, con 1849
genes y cohesión de 0.23, presenta funciones de regulación de la transcripción. Sin
embargo esta función la comparte con la regulación del metabolismo de compuestos
nitrogenados, algo que puede explicar su cercanía al núcleo metabólico. Para este
proceso se han encontrado ejemplos como la proteína CREB (GRMZM2G025812),
importante regulador de, por ejemplo, la tirosina hidroxilasa.
14
15

• Grupo Magenta: Por último, este grupo es el que presenta funciones más dudosas.
Está compuesto por 1825 genes y presenta una cohesión de 0.29. Se encuentra
enriquecido en procesos que actúan a nivel de la membrana interna mitocondrial, lo
cual puede explicar su cercanía al núcleo metabólico, debido a los altos requerimientos
energéticos que estos procesos necesitan. Un ejemplo de este grupo es el citocromo C
oxidasa (GRMZM2G177432), componente de la cadena de transporte de electrones.
5.3. Estudio de los factores de transcripción
Una vez terminado el análisis funcional de la red, se pasó a estudiar la regulación de estos
módulos o grupos funcionales a partir de factores de transcripción.
Como se indica en Materiales y Métodos, se utilizaron todas las familias de factores de
transcripción disponibles en la base de datos PlantTFDB para Z. mays y se localizaron en la red,
como se muestra en la figura 5A. En ella se puede ver cómo la región central, altamente
conectada, es la que presenta mayor densidad de factores de transcripción, lo cual sugiere una
alta regulación.
Posteriormente se realizó el análisis del agrupamiento (figura 5B) de estos factores con el
fin de identificar los módulos funcionales en los que participan, los procesos que regulan y los
sitios de unión (TFBS) representativos en su vecindad (tabla 2).
Para una aproximación más detallada al papel de estas familias de factores como
potenciales reguladores, a continuación se describe cada uno de los grupos encontrados (Jin et al,
2014):
• Grupo Rojo (círculo): La región promotora E-box es el motivo de reconocimiento más
significativo para este grupo. Es el sitio de unión de las proteínas con dominios bHLH
(helix-loop-helix) que, como se puede ver, es una de las familias más representadas. Su
vecindad no presenta términos de GO significativos, sin embargo vemos que este grupo
de factores pertenece al módulo con funciones de regulación de la transcripción en el
agrupamiento general (grupo violeta, figura 4). Esto coincide con la función de estos
factores, ya que las proteínas bHLH es una superfamilia de factores muy involucrada en
la regulación transcripcional de múltiples procesos celulares.
• Grupo Verde (círculo): Al igual que en el anterior, en este grupo se ha encontrado a la
superfamilia de proteínas bHLH como una de las más representadas. De nuevo, la
región promotora E-box se ha visto representada significativamente en su vecindad,
sugiriendo que existe una regulación por estos factores. Enriquecida su vecindad en
funciones de fotosíntesis y metabolismo de carbohidratos, también concuerda con las
funciones del agrupamiento general (grupo azul oscuro, figura 4), así como con las
16

funciones de la familia bHLH, dada la amplia gama de procesos en los que actúa.
• Grupo Azul (círculo): En este caso, no se han encontrado motivos de unión
significativos. A pesar de ello, se ha encontrado en un alto porcentaje el sitio de unión
similar a ERF1 que, efectivamente, concuerda con el motivo de unión de la familia ERF
(ethylene response factor), ampliamente representada en este grupo. En cuanto a las
funciones de la vecindad, se puede ver que están involucradas en la regulación
transcripcional, estando esta última representada por la actividad metiltransferasa
17
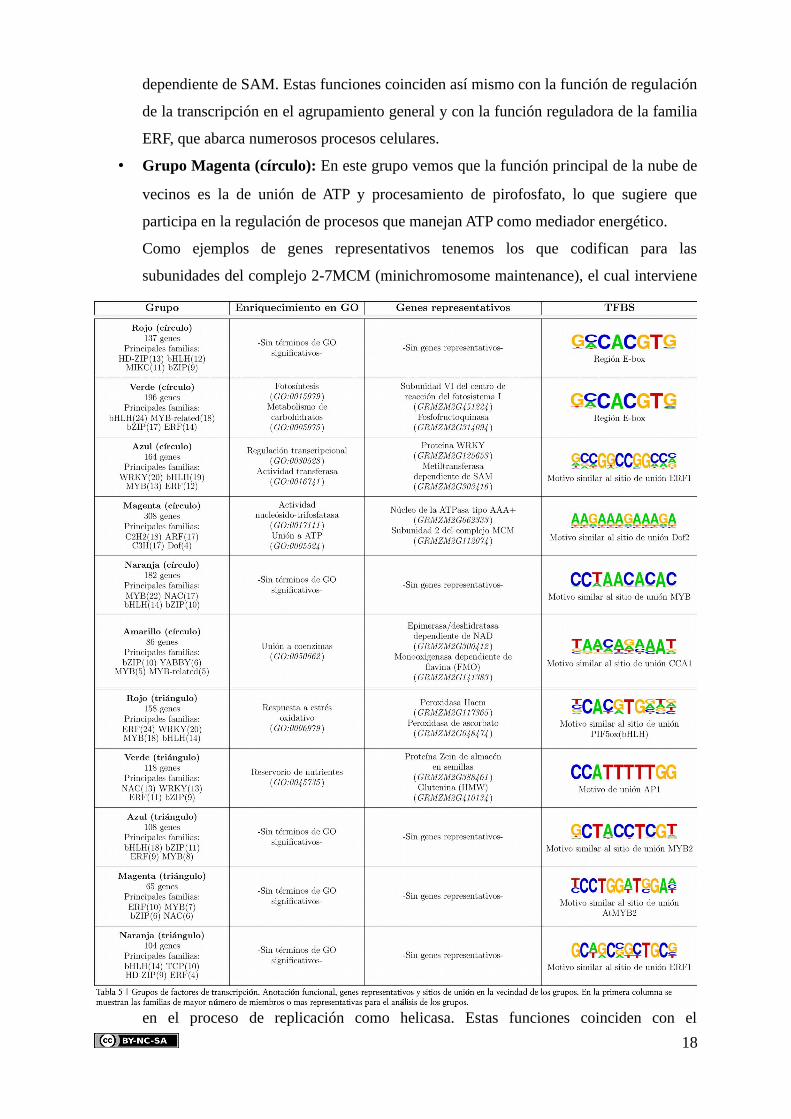
dependiente de SAM. Estas funciones coinciden así mismo con la función de regulación
de la transcripción en el agrupamiento general y con la función reguladora de la familia
ERF, que abarca numerosos procesos celulares.
• Grupo Magenta (círculo): En este grupo vemos que la función principal de la nube de
vecinos es la de unión de ATP y procesamiento de pirofosfato, lo que sugiere que
participa en la regulación de procesos que manejan ATP como mediador energético.
Como ejemplos de genes representativos tenemos los que codifican para las
subunidades del complejo 2-7MCM (minichromosome maintenance), el cual interviene
en el proceso de replicación como helicasa. Estas funciones coinciden con el
18

agrupamiento general, donde el grupo verde claro (figura 4) está involucrado en la
replicación y transcripción. En cuanto al sitio de unión enriquecido, ha resultado que es
similar al sitio de unión de dominios Dof 2 de A. thaliana, para el cual vemos algunos
miembros en este grupo. Se ha visto que las proteínas Dof se encuentran involucradas
en respuestas a hormonas, algo que concuerda con la función del agrupamiento general
y con procesos que implican la replicación celular, como el crecimiento.
• Grupo Naranja (círculo): Para este grupo no se han encontrado términos de GO
significativos, pero sí un enriquecimiento en motivos similares a sitios de unión MYB
en la vecindad, lo que concuerda con la presencia de un gran número de miembros de la
familia MYB en el grupo. Se sabe que la familia MYB está altamente relacionada con
procesos de diferenciación celular, procesos que pueden involucrar a la apoptosis, la
cual es la función enriquecida en el agrupamiento general (grupo naranja, figura 4).
• Grupo Amarillo (círculo): Este grupo vemos que está involucrado en la regulación de
procesos que incluyen funciones de unión a coenzimas. Como ejemplos representativos
vemos a enzimas dependientes de NAD (epimerasa/deshidratasa) y de flavina (FMO).
Estos coinciden en las funciones del agrupamiento general (grupo rojo, figura 4) con
procesos de traducción y metabolismo de compuestos nitrogenados, ya que, por
ejemplo, la monooxigenasa dependiente de flavina (FMO) es una enzima involucrada
en procesos de oxidación de heteroátomos, como el nitrógeno. En cuanto al sitio de
unión, vemos que la vecindad está enriquecida en motivos similares a sitios de unión de
CCA1. Este motivo de unión se ha visto que actúa como diana de factores de
transcripción de la famila MYB-related, de los cuales vemos algunos miembros en este
grupo. En cuanto a su función, también se ha visto que este motivo está presente en
genes regulados por luz, lo cual está relacionado con múltiples procesos que involucra a
la actividad de unión a coenzimas que regula el grupo.
• Grupo Rojo (triángulo): En este caso, los factores de transcripción encontrados están
involucrados en la regulación de procesos de respuesta a estrés oxidativo. Esto coincide
con la función en el agrupamiento general (grupo verde, figura 4). Como ejemplos de
genes de la vecindad se pueden ver hemo peroxidasas y la peroxidasa de ascorbato
(Vitamina C), la cual participa en el ciclo glutatión-ascorbato. En cuanto a los miembros
del grupo, vemos que una de las familias más representadas es la bHLH, la cual
presenta el sitio de unión PIF5ox en la vecindad.
• Grupo Verde (triángulo): En este grupo vemos que la vecindad se encuentra
enriquecida en la función de reserva de nutrientes. Esta es de especial importancia en el
mantenimiento de la semilla, para la cual se han encontrado ejemplos de genes
19

representativos, como la proteína de almacenaje Zein, específica del maíz. Dado que
esta función consiste en el uso de proteínas como método de almacenaje de nutrientes,
resulta lógico que este grupo coincida con la función de metabolismo de aminoácidos
aromáticos en el agrupamiento general (grupo verde oscuro, figura 4). En cuanto a los
sitios de unión enriquecidos, vemos altamente significativa la presencia del sitio de
unión AP1, diana de factores de transcripción como Jun y Fos, pertenecientes a la
familia bZIP.
• Grupo Azul (triángulo): Este grupo tampoco tiene términos de GO enriquecidos en su
vecindad, mientras que en el agrupamiento general (grupo magenta, figura 4)
corresponde con el componente celular de la membrana interna mitocondrial, por lo que
no se puede asociar a ninguna función específica. Por otro lado, uno de los sitios de
unión enriquecidos que se ha encontrado es el similar a MYB 2, el cual es diana de la
familia MYB, una de las principales familias representadas en el grupo.
• Grupo Magenta (triángulo): Este es otro grupo que no presenta términos de GO
significativos. En cuanto a los sitios de unión enriquecidos en la nube de vecinos,
vemos que uno de ellos son los motivos similares a sitios de unión MYB, diana de una
de las familias más representadas en el grupo (MYB). Estos factores regulan vías
metabólicas requeridas para mantener las vías de señalización y estructuras celulares,
como el metabolismo de aminoácidos aromáticos, función representada en el
agrupamiento general (grupo gris, figura 4).
• Grupo Naranja (triángulo): Para este último grupo tampoco se han encontrado
términos de GO significativos en la nube de vecinos. Sin embargo, sí que se han
encontrado motivos similares a sitios de unión ERF, diana de los factores de la familia
ERF, de los cuales encontramos ejemplos en este grupo. Se sabe que el etileno controla
múltiples procesos fisiológicos en la planta, uno de ellos son las respuestas a estrés
abiótico, como la respuesta a frío, función representada en el agrupamiento general
(grupo azul claro, figura 4).
5.4. Estudio de genes regulados por luz, genes fotoperiódicos y genes fotomorfogénicos
Se seleccionaron los ortólogos regulados por luz de A. thaliana en Z. mays como se
indicó en Materiales y Métodos, identificando sus potenciales funciones en la base de datos TAIR
(The Arabidopsis Information Resource) (tabla 3). Se lograron localizar ocho de entre todos los
genes estudiados, identificándose después los potenciales factores de transcripción en su
vecindad (figura 6). A continuación se describe con más detalle cada uno de los genes y su
potencial regulación.
20

• Gen AC207722.2_FG009: Este gen se encuentra en el grupo general (grupo azul oscuro,
figura 4) encargado del metabolismo y fotosíntesis. Es un gen prácticamente aislado en el
grupo, por lo que no se han encontrado potenciales factores de transcripción ni funciones
significativas. Es un gen cuyo ortólogo en A. thaliana codifica para una subunidad del
complejo LHCII (light harvesting complex II), involucrado en el proceso de fotosíntesis
junto con el fotosistema II (PSII), función que coincide con la función del grupo general.
• Gen GRMZM2G014902: Este gen se encuentra en el centro de la red, coincidiendo con
el grupo general de metabolismo y fotosíntesis (grupo azul oscuro, figura 4). Es un gen
altamente conectado, formando parte de la región de los principales hubs de la red que se
mostraban en el análisis topológico. En A. thaliana vemos que este gen presenta dos
ortólogos (At1G01060 y At2G46830). El primero de ellos corresponde con un factor de
transcripción de la familia MYB-related (LHY) involucrado en la regulación del ritmo
circadiano. El segundo corresponde con un represor transcripcional asociado al bucle de
regulación de LHY (CCA1), participando también en la regulación del oscilador
circadiano de A. thaliana. Ambos genes también se encuentran involucrados en funciones
de floración dependiente de día largo y en las respuestas a distintos factores ambientales
y hormonas (Jiao et al, 2007). Este gen se encuentra unido a una nube de vecinos
involucrada en funciones de metabolismo de carbohidratos, fotosíntesis y generación de
precursores metabólicos y energía (tabla 4), funciones que actúan acorde con la función
del grupo general. Son funciones que implican la generación de componentes
21

estructurales y energéticos necesarios para los múltiples procesos controlados por el
ritmo circadiano, como la floración y fotomorfogénesis. Por tanto resulta lógico que este
gen coordine a su vecindad en el contexto de la regulación circadiana. También se pueden
ver distintos factores de transcripción, pertenecientes a distintas familias (MYB-related,
CO-like, bHLH, bZIP y EIL), implicados en la regulación por luz del gen.
• Gen GRMZM2G032351: Este gen se encuentra también en una región altamente
conectada, por lo que su función debe ser importante para la regulación de la red. Como
ortólogos vemos que presenta dos en A. thaliana, uno de ellos corresponde con el
receptor de luz azul PHOT1 (At3G45780), involucrado en múltiples procesos, como el
fototropismo y movimiento de los estomas. El otro ortólogo encontrado corresponde a su
vez con el receptor de luz azul PHOT2 (At5G58140), que actúa junto con PHOT1. Al
igual que el primero, este actúa en procesos de fototropismo así como en procesos de
respuesta a distintos estímulos, como a citoquininas y sacarosa (Jiao et al, 2007). Este gen
se encuentra en el grupo general (grupo azul oscuro, figura 4) involucrado en la
fotosíntesis, dependiente de la luz azul a través de procesos como el fototropismo y la
distribución de los cloroplastos en la célula. Estos permiten adaptar a la planta a las
situaciones cambiantes de iluminación, consiguiendo una captación de luz óptima. La
22

vecindad de este gen no presenta procesos significativos, pero si se han encontrado
factores de transcripción coexpresados. Son factores de las familias NF-YB y NF-YC
(nuclear factor Y), las cuales están implicadas en la floración, embriogénesis y
fotomorfogénesis, procesos altamente dependientes de la luz.
• Gen GRMZM2G081474: Este gen presenta como ortólogo a una deacetilasa de histonas
(At4G38130, HDA1) involucrada en la regulación de la transcripción. Se presenta en la
mayoría de los tejidos y participa en la resistencia sistémica a patógenos dependiente de
jasmonato y etileno. Este gen se encuentra en el grupo general con funciones de respuesta
a hormonas y transcripción (grupo verde claro, figura 4), lo cual responde a la función del
ortólogo. Sin embargo, no se han encontrado términos significativos de GO en la
vecindad ni factores de transcripción que sugieran su regulación.
• Gen GRMZM2G104549: Este gen presenta dos ortólogos en A. thaliana, involucrados
en la fotosíntesis. Uno de ellos como proteína de unión a clorofila a/b (At1G29910,
CAB3) y el otro como un componente del LHCIIb, asociado al fotosistema II
(At1G29920, CAB2). En el agrupamiento general vemos que este gen pertenece al grupo
encargado de la fotosíntesis, lo cual coincide con su función. Sin embargo, no se han
encontrado términos de GO significativos ni factores de transcripción coexpresados aun
estudiándolos a distancia 3.
• Gen GRMZM2G120619: Para este gen encontramos dos ortólogos en A. thaliana, uno
de ellos (At1G29930, CAB1) codifica para una subunidad del complejo antena II
(LHCII) y el otro (At1G29920, CAB2) vemos que también codifica para un componente
del LHCIIb, asociado al fotosistema II y por tanto también con funciones fotosintéticas
de captación de luz (Jiao et al, 2007). El grupo general (grupo negro, figura 4) en el que
se encuentra este gen cumple funciones muy generales de regulación de la transcripción,
algo que involucra a múltiples procesos, entre ellos la fotosíntesis. A pesar de no
presentar procesos significativos en su vecindad, se han encontrado tres factores de
transcripción de la familia WRKY y uno de la bHLH coexpresados con este gen. Los
factores de transcripción WRKY se sabe que son reguladores muy importantes de las
respuestas a estrés biótico y abiótico, sin embargo también se han encontrado que están
involucrados en procesos de desarrollo, los cuales pueden depender de la luz, como es el
caso de la fotomorfogénesis. En el caso de los bHLH ya se ha visto que son factores muy
importantes en la regulación de las redes transcripcionales.
• Gen GRMZM2G181028: Este es un gen cuyo ortólogo en A. thaliana codifica para
un fotorreceptor de luz roja/infrarroja (At1G09570, PHYA), involucrado
principalmente en procesos fotomorfogénicos. Este es un receptor cuya actuación
23

depende de la conversión entre dos formas, Pr y Pfr, siendo esta última la más activa
(Jiao et al, 2007). Vemos que este gen se encuentra en el grupo general encargado del
metabolismo de ácidos grasos (grupo amarillo, figura 4), lo cual sugiere su
participación aportando lípidos como componentes estructurales en muchos procesos
fotomorfogénicos. Este gen también está relacionado con el transporte (grupo
amarillo, figura 4), ya que la transducción de la señal de luz roja/infrarroja implica el
paso del fotorreceptor al núcleo, actuando después sobre factores de transcripción.
Aun sin un enriquecimiento significativo en términos de GO, en su vecindad se han
encontrado hasta 3 factores de transcripción de las familias MYB-related, SRS y
NAC, lo que, como en casos anteriores, podrían sugerir vías de regulación
“downstream” de este ortólogo de PHYA.
• Gen GRMZM2G351977: En el caso de este gen, en A. thaliana presenta tres
ortólogos, todos ellos involucrados en la fotosíntesis y presentando distintos papeles.
Este gen se encuentra en el grupo general negro (figura 4) con funciones de regulación
de la transcripción, un papel muy general que abarca múltiples procesos. Sin embargo,
no se puede especificar una regulación potencial del gen al no haber encontrado
factores de transcripción en su vecindad ni términos de GO significativos.
5.5. Estudio de genes de interés biotecnológico
De los distintos genes de potencial interés biotecnológico que se han estudiado, en la
red se han encontrado tres, que a continuación se describen. Sobre ellos se ha estudiado su
función, su vecindad (Figura 7A y B) y su relación con la función del grupo general en el que
se encuentran. También se han estudiado a nivel regulatorio, identificando los posibles
factores de transcripción que potencialmente regulan su expresión y su función de acuerdo
con el agrupamiento de factores de transcripción anteriormente estudiado (tabla 4).
• Gen GRMZM2G024993 (Waxy): El gen waxy (Wx) codifica para una almidón sintasa
soluble asociada a gránulos (Cao et al, 1999), el cual se encuentra mutado (Fan et al,
2008) en la variedad de maíz waxy, procedente de Asia y caracterizada por presentar una
alta proporción de amilopectina en sus granos. Este gen, por tanto, esta involucrado en la
síntesis de carbohidratos, lo cual coincide con la función del grupo general en el que se
encuentra (grupo verde oscuro, figura 4). Su vecindad (figura 7A y B) se ha visto que
está enriquecida en la actividad de reservorio de nutrientes (tabla 4), con genes
representativos como la proteína Zein de almacenamiento en semillas. A nivel
regulatorio se han localizado en la vecindad dos factores de transcripción de la familia
NAC (grupo verde triángulo, figura 5B), familia principalmente involucrada en procesos
24
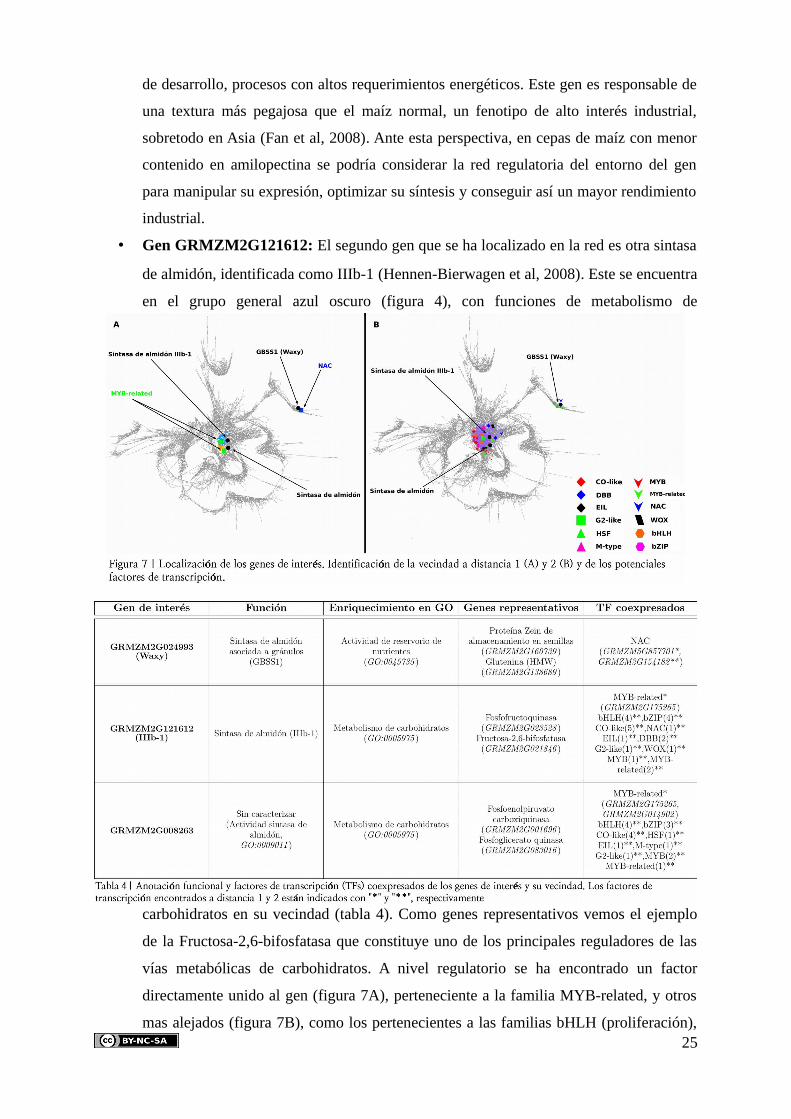
de desarrollo, procesos con altos requerimientos energéticos. Este gen es responsable de
una textura más pegajosa que el maíz normal, un fenotipo de alto interés industrial,
sobretodo en Asia (Fan et al, 2008). Ante esta perspectiva, en cepas de maíz con menor
contenido en amilopectina se podría considerar la red regulatoria del entorno del gen
para manipular su expresión, optimizar su síntesis y conseguir así un mayor rendimiento
industrial.
• Gen GRMZM2G121612: El segundo gen que se ha localizado en la red es otra sintasa
de almidón, identificada como IIIb-1 (Hennen-Bierwagen et al, 2008). Este se encuentra
en el grupo general azul oscuro (figura 4), con funciones de metabolismo de
carbohidratos en su vecindad (tabla 4). Como genes representativos vemos el ejemplo
de la Fructosa-2,6-bifosfatasa que constituye uno de los principales reguladores de las
vías metabólicas de carbohidratos. A nivel regulatorio se ha encontrado un factor
directamente unido al gen (figura 7A), perteneciente a la familia MYB-related, y otros
mas alejados (figura 7B), como los pertenecientes a las familias bHLH (proliferación),25

CO-like (floración) y bZIP, involucrados en procesos con alta demanda energética.
• Gen GRMZM2G008263: El tercer gen encontrado en la red presenta de nuevo
actividad sintasa de almidón. Sin embargo, en este caso no se encuentra caracterizado, y
solo presenta funciones en su vecindad similares a la de IIIb-1 y coexpresado con
factores de transcripción de la familia MYB-related a distancia 1, y otros muchos a
distancia 2 (figuras 7A y B), como miembros de las familias bZIP y bHLH.
6. CONCLUSIONES
1) A partir de la metodología seguida se ha construido una red de coexpresión génica
basada en datos de RNA-seq de Zea mays, constituida por 26.194 nodos y 115.055
aristas y definida por las propiedades de libre escala y mundo pequeño de las redes
biológicas.
2) El análisis de la red de coexpresión génica ha permitido descubrir la organización de los
genes en módulos funcionales, revelando 12 grupos de genes con baja inter-coexpresión
y alta intra-coexpresión. De entre todos ellos, cabe destacar la existencia de un grupo
central donde se encuentran la mayoría de hubs autoritativos de la red y que se
encuentra involucrado en procesos de metabolismo de carbohidratos y fotosíntesis.
3) Se han identificado todos los factores de transcripción de la red y analizado los procesos
biológicos que potencialmente regulan en Zea mays junto con sitios de unión
significativos en los promotores de genes dianas potenciales. Cabe destacar las
existencia de un grupo donde aparecen 24 factores de transcripción de la familia bHLH,
localizados en el centro de la red y que posiblemente reconocen el motivo E-box en
genes involucrados en metabolismo de carbohidratos y fotosíntesis.
4) Se ha estudiado la función y regulación de genes individuales, como genes regulados
por luz y con potencialidad biotecnológica. Se han identificado potenciales genes de
respuesta a luz como posibles ortólogos de los genes LHY y CCA1 de A. thaliana, cuyo
análisis sugiere que la respuesta a luz en maíz regula procesos metabólicos de
carbohidratos y fotosíntesis. Un ejemplo de gen de interés biotecnológico es el gen
waxy, cuya alta coexpresión con un factor de transcripción NAC sugiere que podría ser
una diana para la mejora de la producción de amilopectina en maíz.
7. BIBLIOGRAFÍA
• Patrick S. Schable, Ware D., Fulton R. S., Stein C. J., Wei F. et al. (2009) The B73 Maize
26

Genome: Complexity, Diversity, and Dynamics Science 326,1112. doi10.1126/science.
1178534.
• Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten
U, Putnam N, S RD. (2012) Phytozome: a comparative platform for green plant genomics.
Nucleic Acids Research, 40:D1178–D1186.
• Ware D, Jaiswal P, Ni J, Pan X, Chang K, Clark K, Teytelman L, Schmidt S, Zhao W,
Cartinhour S, McCouch S, Stein L (2002). Gramene: a resource for comparative grass
genomics. Nucleic Acids Research, 30, 103-105.
• Trapnell, C., Pachter, L., and Salzberg, S. L. (2009) TopHat: discovering splice junctions with
RNA-Seq. Bioinformatics 25 (9):1105-1111. doi:10.1093/bioinformatics/btp120
• Trapnell C. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated
transcripts and isoform switching during cell differentiation. Nat. Biotechnol, 28:511–515. doi:
10.1038/nbt.1621.
• Garber M, Grabherr M, Guttman M, C T. (2011). Computational methods for transcriptome
annotation and quantification using RNA-seq. Nature Methods, 8(6):469–477.
doi:10.1038/nmeth.1613.
• Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg S, Rinn J,
Lior P. (2012). Differential gene and transcript expression analysis of RNA-seq experiments
with TopHat and Cufflinks. Nature Protocols, 7(3):562–578. 75. doi: 10.1038/nprot.2012.016.
• Goff L, Trapnell C. (2011). cummeRbund: Analysis, exploration, manipulation, and
visualization of Cufflinks high-throughput sequencing data. [R package version 1.2.0].
• Du Z., Zhou X., Ling Y., Zhang Z., and Su Z. (2010). agriGO: a GO analysis toolkit for the
agricultural community. Nucl. Acids Res 38: W64-W70. DOI 10.1093/nar/gkq310.
• Jin JP, Zhang H, Kong L, Gao G and Luo JC. (2014). PlantTFDB 3.0: a portal for the
functional and evolutionary study of plant transcription factors. Nucleic Acids Research,
42(D1):D1182-D1187. doi: 10. 1093/nar/gkt1016
• The Arabidopsis Information Resource (TAIR), on www.arabidopsis.org.
• Ma S, Shah S, Bohnert HJ, Snyder M, Dinesh-Kumar SP. (2013) Incorporating Motif Analysis
into Gene Co-expression Networks Reveals Novel Modular Expression Pattern and New
Signaling Pathways. PLoS Genet 9(10):e1003840 doi:10.1371/journal.pgen.1003840
• Zhang B., Horvath S. (2005). A General Framework for Weighted Gene Co-Expression
Network Analysis. Statistical Applications in Genetics and Molecular Biology. Volume 4,
Issue 1, ISSN (Online) 1544-6115, DOI: 10.2202/1544-6115.1128.
• Mao L., Van Hemert J. L., Dash S. and Dickerson J. A. (2009). Arabidopsis gene
co-expression network and its functional modules. BMC Bioinformatics, 10:346
doi:10.1186/1471-2105-10-346.
• Romero-Campero F., Lucas-Reina E., Said F. E., Romero J. M., and Valverde F. (2013) A
27

contribution to the study of plant development evolution based on gene co-expression
networks. Front. Plant. Sci, Volume 4, number 291 doi: 10.3389/fpls.2013.00291
• Aoki K., Ogata Y., and Shibata D. (2007). Approaches for Extracting Practical
Information from Gene Co-expression Networks in Plant Biology. Plant Cell Physiol.
48(3): 381–390 doi:10.1093/pcp/pcm013.
• Barabasi A., and Albert R. (1999). Emergence of Scaling in Random Networks. Science
286, 509-512. doi: 10.1126/science.286.5439.509.
• Olsen K. M. and Wendel J. F. (2013). Crop plants as models for understanding plant
adaptation and diversification. Front. Plant. Sci, 4: 290. doi: 10.3389/fpls.2013.00290.
• Csardi G, Nepusz T (2006). The igraph software package for complex network research,
InterJournal, Complex Systems 1695. http://igraph.org
• Cline S. M, et al. (2007). Integration of biological networks and gene expression data
using Cytoscape. Nature Protocols 2, 2366 - 2382. doi:10.1038/nprot.2007.32.
• Heinz S, Benner C, Spann N, Bertolino E, Lin Y. C, Laslo P, Cheng J. X, Murre C., Singh
H, Glass C. K (2010). Simple combinations of lineage-determining transcription factors
prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell.
38(4):576-89. doi: 10.1016/j.molcel.2010.05.004.
• Jiao Y., Lau O. S. and Deng X. W (2007).. Light-regulated transcriptional networks in
higher plants. Nature Reviews Genetics 8, 217-230 doi:10.1038/nrg2049
• Fan L., Quan L., Leng X., Guo X., Hu W., Ruan S., Ma H., Zeng M. (2008). Molecular
evidence for post-domestication selection in the Waxy gene of Chinese waxy maize. Mol
Breeding 22:329–338. doi: 10.1007/s11032-008-9178-2
• Cao H., Imparl-Radosevich J., Guan H., Keeling P. L., James M. G. and Myers A. L.
(1999). Identification of the Soluble Starch Synthase Activities of Maize Endosperm.
Plant Physiology vol. 120 no. 1 205-216. doi: http://dx.doi.org/10.1104/pp.120.1.205
• Hennen-Bierwagen T. A. et al. (2008). Starch Biosynthetic Enzymes from Developing
Maize Endosperm Associate in Multisubunit Complexes. Plant Physiology vol. 146 no. 4
1892-1908. doi:http://dx.doi.org/10.1104/pp.108.116285
8. Agradecimientos
Este trabajo ha sido realizado con el apoyo de mi tutor Francisco J. Romero-Campero a
nivel de teoría de redes y técnicas bioinformáticas, con el soporte computacional de las máquinas
sesamo y bigfoot del Centro Informático Científico de Andalucía (CICA) y la máquina
viridiplantae del Grupo en Desarrollo Molecular y Metabolismo de Plantas perteneciente al
Instituto de Bioquímica Vegetal y Fotosíntesis (IBVF), ubicado en Isla de Cartuja.
28

Esta obra está sujeta a la licencia
Reconocimiento-NoComercial-Co
mpartirIgual 4.0 Internacional de
Creative Commons. Para ver una
copia de esta licencia, visite
http://creativecommons.org/license
s/by-nc-sa/4.0/.
29





























![Escalonamento multidimensional baseado em redes de arvore ...each.uspnet.usp.br/digiampietri/BraSNAM/2017/p05.pdf · [Yang et al. 2014, Fiedor 2014], e que pode revelar estruturas](https://img.pdfslide.us/doc/110x75/5c78c30209d3f2fb438bd921/escalonamento-multidimensional-baseado-em-redes-de-arvore-each-yang-et.jpg)