Embed Size (px)
Citation preview

Journal of Catalysis 330 (2015) 6–18
Contents lists available at ScienceDirect
Journal of Catalysis
journal homepage: www.elsevier .com/locate / jcat
Understanding the selectivity of methanol steam reforming on the (111)surfaces of NiZn, PdZn and PtZn: Insights from DFT
http://dx.doi.org/10.1016/j.jcat.2015.06.0200021-9517/� 2015 Elsevier Inc. All rights reserved.
⇑ Corresponding author.
M. Krajcí a,⇑, A.-P. Tsai b, J. Hafner c
a Institute of Physics, Slovak Academy of Sciences, Dúbravská cesta 9, SK-84511 Bratislava, Slovak Republicb Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai 980-8577, Japanc Fakultät für Physik and Center for Computational Materials Science, Universität Wien, Sensengasse 8/12, A-1090 Wien, Austria
a r t i c l e i n f o
Article history:Received 14 January 2015Revised 28 April 2015Accepted 25 June 2015Available online 17 July 2015
Keywords:Methanol steam reformingSelectivityIntermetallic compoundsNiZnPdZnPtZnDensity-functional theory
a b s t r a c t
This work is focused on better understanding of the origin of CO2/CO selectivity of methanol steamreforming (MSR) catalyzed by three isostructural (L10-type) intermetallic compounds NiZn, PdZn, andPtZn. Following earlier suggestions we assume that the selectivity of MSR arises from the competitionbetween two possible transformations of the reaction intermediate formaldehyde: the reaction offormaldehyde with hydroxyl leading ultimately to the formation of CO2, and the direct dehydrogenationof formaldehyde leading to the production of CO. We have investigated whether the differences in theactivation energies for these steps explain the experimentally observed trends in selectivity. Usingdensity-functional theory detailed atomistic scenarios for both reactions have been developed. As in vac-uum the reaction of formaldehyde with hydroxyl is non-activated, it does not need the support of a cat-alyst to proceed. There is a clear correlation between high CO2 selectivities and weak adsorption energiesof formaldehyde. On the L10(111) surfaces we have to consider the existence of four inequivalent adsorp-tion sites, caused by the monoclinic shift of subsurface atomic planes resulting from tetragonal deforma-tion of the L10 structure. At PtZn(111) this shift could explain the observed mixed CO2/CO selectivity ofthe PtZn catalyst. The proposed mechanism of selectivity can qualitatively explain the experimentallyobserved selectivities of the investigated intermetallic compounds. Our results can contribute also tounderstanding the influence of the ZnO support and the Zn-enriched PdZn surface on the selectivity ofthe MSR process. The possibilities how to improve the CO2 selectivity of the catalyst are also presented.
� 2015 Elsevier Inc. All rights reserved.
1. Introduction
Fuel cells are a promising technology for the production of elec-trical power by small scale devices and in automotive transportapplications [1]. As fuel cells require hydrogen as a fuel and gas-eous hydrogen is extremely difficult to store and to transport,many alternative hydrogen carriers are being considered. The useof methanol as hydrogen carrier has a number of advantages[2,3]. Methanol is liquid at atmospheric conditions and has a highH/C ratio. Although methanol is toxic, it is biodegradable and incontrast to conventional fuels it does not contain sulfur or nitro-gen. With only one carbon atom it is the simplest of all alcohols.While the conversion of ethanol to hydrogen requires a rather hightemperature of about 400�, due to the absence of a strong carbon–carbon bond the conversion of methanol can be performed at lowertemperatures of 200–300� [3].
There are three ways to obtain hydrogen from methanol: metha-nol decomposition, partial oxidation, and methanol steam reform-ing (MSR). For in-situ hydrogen generation MSR is the mostfavorable of these alternatives. MSR produces in addition to hydro-gen CO2 and CO. The formation of CO must be avoided since it poi-sons the anode of fuel cells. For MSR a number of different catalystshave been proposed [3–5]. The traditional catalyst is copper dis-persed on an oxide support. The Cu-based catalysts are efficientand highly selective toward CO2, avoiding the production of CO.However, Cu-based catalysts suffer from pyrophoricity, insufficientthermal stability, and metal sintering resulting in a rapiddegradation of the catalyst. Palladium is known to be an efficientand thermally stable catalyst for various hydrogenation anddehydrogenation processes. Unfortunately, for MSR Pd catalystsare almost 100% CO selective. If Pd is dispersed on a ZnO supportthe catalyst has the required CO2 selectivity and maintains a highefficiency and thermal stability [6,7]. The change in selectivity hasbeen attributed to the formation of the intermetallic compoundPdZn due to heating of Pd/ZnO under reaction conditions [8].

M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18 7
Much effort has been directed toward a better understanding ofMSR over Pd/ZnO catalysts. The MSR reaction CH3OH + H2O ? 3H2 + CO2 on the surface of a catalyst is a very complex process.Despite a large number of investigations [4–28] the reaction mech-anism of MSR remains controversial. Very recently the state of theart has been summarized in a comprehensive review [29]. Manyexperimental investigations have been accompanied by theoreticalstudies. Ab-initio density-functional theory (DFT) studies havehelped to unravel many aspects of the reaction. It is now generallyaccepted [29] that MSR is initiated by O–H bond cleavage leadingto the formation of methoxyl (CH3O), CH3OH ? CH3O + H. The ratecontrolling step is the following dehydrogenation of chemisorbedmethoxyl to formaldehyde (CH2O), CH3O ? CH2O + H, as demon-strated by both experimental [11,19] and theoretical[13,20,22,23] studies. The adsorbed formaldehyde seems to be animportant intermediate of the MSR process [6,7] serving as abranching point for different product channels. A possible reactionnetwork of MSR has been graphically presented, e.g., in the worksof Sá et al. [3] and Frank et al. [17]. Formaldehyde can react withhydroxyl OH (formed by the dissociative adsorption of water) toform hydroxymethoxy CH2OOH (reaction R1),
R1 : CH2Oþ OH! CH2OOH; ð1Þ
it can dehydrogenate to formyl CHO (reaction R2),
R2 : CH2O! CHOþH: ð2Þ
In an alternative reaction channel formaldehyde can react witha methoxyl molecule (CH3O) forming methyl formate (CHOOCH3)as intermediate [16,17,24]. At high-temperatures methanol candecompose and side reactions can appear [3,22,23]. The importantrole of the formaldehyde intermediate has been demonstrated bythe observation [7] that if steam reforming starts with formalde-hyde (formaldehyde steam reforming, FSR) the reaction productsremain essentially the same as for MSR.
As the reaction mechanism for MSR is not yet completelyunderstood, the conditions determining selectivity to CO2 or COare still discussed. Recent DFT studies have confirmed that on Cuand PdZn catalysts the reaction of formaldehyde with hydroxyldominates over the dehydrogenation of formaldehyde, its desorp-tion or other reaction channels [22,24,29]. These studies suggestthat the reaction of formaldehyde with hydroxyl is the key forthe observed CO2 selectivity of MSR on Cu and PdZn catalysts[22,24], while on Pd the dehydrogenation to formyl is favored,leading to selective CO production. The alternative reaction chan-nel via methyl formate [17] plays a minor role on Cu or PdZn sur-faces according to a recent DFT study [25]. If the reaction offormaldehyde with hydroxyl is preferred, CH2OOH can furtherdecompose via formic acid CHOOH (or alternatively viadioxymethlene CH2OO) to formate CHOO and finally to CO2 [24].If direct dehydrogenation is energetically more favorable, than inaddition to hydrogen the undesired CO is produced.
The influence of the oxide support on the selectivity of MSR isalso not completely clear. From the observation that supportedand unsupported catalysts show comparable selectivities [30,31]one could conclude that the role of the ZnO support on the selec-tivity is not significant. However, recent studies demonstrated[23,26] that the ZnO support and perhaps also oxidized surfaceZn can play an important role in the activity and selectivity ofthe PdZn catalyst. The ZnO support itself has been identified [26]as an almost 100% CO2 selective catalyst for both MSR and FSR. Itwas also suggested that a metallic catalyst alone cannot achievea high CO2 selectivity. These authors also assume that the reactionon the metallic catalyst is only part of the story and that the cat-alytic reaction could proceed near the interface between thePdZn compound and the ZnO phase [23,27].
The similarity of the catalytic properties of Cu and PdZn ishighly intriguing. If its origin is clarified, this would permit thedesign of improved catalysts based on intermetallic compounds[30]. The catalytic properties depend on details of the atomic andelectronic structures of the surfaces and their interactions withreactants and intermediates. Interestingly, Cu has the same elec-tron per atom ratio and a similar electronic density of states(DOS) as PdZn, as shown by DFT calculations and X-ray photoelec-tron spectroscopy (XPS) [30]. With the aim of understanding theinfluence of the electronic structure on the catalytic properties ofCu and PdZn the MSR reaction on the three isostructural com-pounds NiZn, PdZn, and PtZn has been investigated [30,31]. Allthree compounds crystallize in the L10 (AuCu-type) structurewhich can be considered as a tetragonally deformed cubic B2(bcc) structure. The catalyst samples were prepared in the formof powders obtained from bulk crystals [30] (unsupported cata-lysts) and also in the form of supported catalysts where Ni, Pd,and Pt nanoparticles were dispersed on a ZnO support [31].
While PdZn catalysts are almost completely CO2 selective, PtZnexhibits a mixed CO2/CO selectivity and NiZn is CO selective. Thecatalytic activity as well as the selectivity for CO2 decrease in theorder PdZn > PtZn > NiZn. XPS measurements accompanied byDOS calculations found a correlation between position of the d-bandof the transition metal and the observed selectivities [30,31].
The present work focuses on a better understanding of themicroscopic mechanism determining the selectivity of the MSRprocess. Starting from the DFT results [24,25] for MSR catalyzedby PdZn and Cu we have investigated whether the observed trendsin the CO2 selectivity of the isostructural and isoelectronic NiZn,PdZn, and PtZn catalysts can be understood on the basis of thereaction of formaldehyde with hydroxyl. We have used DFT calcu-lations to investigate the adsorption geometries and the diffusionof the reactants on the (111) surfaces of the three compounds.We have constructed atomistic scenarios for the two competingreactions determining the selectivity: the reaction of formaldehydewith hydroxyl (R1) and the dehydrogenation of formaldehyde (R2).We have found that the tetragonal distortion of the L10 structure isresponsible for the formation of inequivalent adsorption sites onthe (111) surfaces. In vacuum the reaction R1 between formalde-hyde and hydroxyl is non-activated. For reactants adsorbed on thecatalyst the reaction is activated and the transition state corre-sponds to the breaking of the bond between the hydroxyl groupand the surface, enabled by the formation of a new bond betweenthe oxygen atom of hydroxyl and the carbon atom of formalde-hyde. Our results also contribute to a better understanding of theselectivity of the MSR process. Possibilities how to improve CO2
selectivity are discussed.
2. Theory
2.1. Computational methodology
Total-energy and electronic structure calculations have beenperformed using the Vienna Ab-initio Simulation Package (VASP)[32,33]. VASP performs an iterative solution of the Kohn–Shamequations of Density Functional Theory (DFT) within aplane-wave basis. The electron–ion interaction is described withinthe projector-augmented wave (PAW) method [34]. We used thesemilocal PBE exchange–correlation functional in the generalizedgradient approximation proposed by Perdew et al. [35,36]. Thebasis set contains plane waves with a kinetic energy up toEcut�off ¼ 700 eV. The selfconsistency iterations were stopped whentotal energies are converged to within 10�6 eV. The atomic struc-ture of the surfaces has been optimized by static relaxations usinga quasi-Newton method and the Hellmann–Feynman forces acting

8 M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18
on the atoms. Transition-states were determined using theclimbing-image nudged-elastic band method [37]. If necessary,transition states have been further optimized by the dimer method[38,39]. For the structural optimizations and transition-statesearches convergence criteria of 10�4 eV for total energies and0.05 eV/Å for forces acting on the atoms have been applied.
Surface calculations have been performed for slab models cutfrom the bulk structure of the L10 compounds. For the calculationsof adsorption energies and reaction paths we have used a slab con-sisting of 4 atomic layers with fixed coordinates of the atoms in thebottom layer, altogether 32 atoms. Periodically repeated slabs areseparated by a vacuum layer of 13 Å. Because of the tetragonaldeformation of the L10 structure (see below) the computationalcell representing the (111) surface has a monoclinic shape. Thesurface area of the computational cell contains 2 � 2 surface cells.More structural data on the computational cell are compiled inTable 1. Total energies of adsorption complexes and of states alongthe reaction path were calculated using a 4 � 6 � 1 k-point meshfor Brillouin-zone integration. Zero-point-energy (ZPE) correctedvalues of the adsorption energies are also presented. We note thatthe calculated binding and activation energies depend on the cov-erage. To compare the activities and selectivities of different sur-faces it is thus important to perform the calculations at the sameor at very similar coverages and with the same computational set-ting. To compare our results for the L10(111) surfaces with theknown results for the Cu(111) and Pd(111) surfaces we recalcu-lated the latter ones at the comparable coverages.
2.2. Structure of L10(111) surfaces
The intermetallic compounds NiZn, PdZn and PtZn crystallize inthe body-centered tetragonal (bct) L10 (AuCu-type) structure,Pearson symbol tP4. The space group is P4/mmm (No 123). TheL10 structure can be understood as a tetragonally deformed cubicB2 (CsCl) structure. It is well known that the bct lattice can bedescribed alternatively as a face-centered tetragonal (fct) lattice.Hence the L10 structure can also be considered as a tetragonallydeformed cubic B1 (NaCl) structure, see Fig. 2 in Ref. [31]. The rela-tion between the lattice parameters of both descriptions is simple:afct ¼ abct
ffiffiffi2p
, cfct ¼ cbct ¼ c. The tetragonal deformation can beexpressed by the c=abct parameter. The L10 structure is identicalto B2 for c=abct ¼ 1. The calculated lattice parameters of all threecompounds together with their experimental values [31] are pre-sented in Table 1. Although the fct lattice is not a Bravais lattice,the description in terms of fct Miller indices is most frequentlyused in the literature. Therefore here we also adopt thisconvention.
Previous DFT calculations [40] for bulk PdZn have shown thatthe (111) surfaces (in the fct notation) exposing zinc and palla-dium in equimolar ratio are more stable than the (001) or (100)surfaces terminated by alternating Zn and Pd layers. Therefore
Table 1Calculated lattice parameters of the L10 compounds. Experimental values [31] aregiven in brackets. Structural data of the studied (111) surface: dx � dy are dimensionsof the surface cell of the (1 11) surface, dz is interlayer spacing of atomic planes, / isthe monoclinic angle.
NiZn PdZn PtZn
abct 2.722(2.74) 2.930(2.90) 2.886(2.88)afct 3.849(3.87) 4.144(4.10) 4.081(4.07)c 3.185(3.24) 3.321(3.36) 3.409(3.53)c=abct 1.170(1.182) 1.133(1.159) 1.181(1.226)c=afct 0.827(0.837) 0.801(0.820) 0.835(0.867)
dx � dy 4.26 � 2.72 4.48 � 2.93 4.56 � 2.89dz 2.095 2.215 2.233/ 81.03� 82.84� 80.50�
our investigations concentrate on the (111) surfaces. In the fccstructure the most densely packed atomic planes are perpendicularto the [111] direction. In the fct reference the L10 structure can bedescribed as a stacking of flat, densely packed atomic planes alongthe [111] direction. The interlayer distances dz between the atomicplanes are given in Table 1. Because of the tetragonal deformationthe stacking sequence is monoclinically tilted in the ½11 �2� direc-tion. The monoclinic angle / is given by / ¼ p� 2 arctanðc=abctÞ.Although the tilt p=2� / of the stacking direction is only 7–10�,in Section 3.1 we demonstrate that it can have a significant influ-ence on the energetics of the adsorption sites. The surface cell con-tains one transition metal (TM) atom (Ni, Pd, Pt) and one Zn atom;
the rectangular dimensions dx � dy (dx ¼ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffic2 þ a2
bct
q, dy ¼ abct) are
also listed in Table 1. We note that in the bct framework the stack-ing sequence of the atomic planes corresponds to the [110] direc-tion. The L10 surface is thus also closely related to the (110)surfaces of B2 compounds. In the following the tetragonal defor-mation of the L10 structure is measured relative to the cubic B2structure. Fig. 1 shows a view of the clean L10(111) surface. Theshift of the subsurface atomic planes in the ½11 �2� direction (hori-zontal) is clearly recognizable. The perpendicular (vertical) direc-tion expressed in fct Miller indices is ½�110�. The reasons for thetetragonal distortion of the L10-type structure of PdZn have beendiscussed by Friedrich et al. [41] and shown to be related to astrong, partially covalent bonding between the Pd atoms formingfour-coordinated nets parallel to the (100) planes. This also sug-gests that the increasing covalent character of the bondingbetween the transition-metal atoms causes the increase of thetetragonal distortion from NiZn to PdZn and further to PtZn.
The structural relaxation of the surface reduces the monoclinicshift of the atomic planes. The tilt of the stacking direction is smal-ler than in the bulk, approximately 7�, 3�, and 8� for the NiZn, PdZn,and PtZn surfaces, respectively. On the relaxed PdZn(111) surfacethe shift of the atomic planes is quite small, while it is largest onPtZn(111). The relaxed (111) surfaces are slightly corrugated. Onthe NiZn, PdZn, and PtZn surfaces the Zn atoms are shifted outwardby Dh = 0.26 Å, 0.21 Å, and 0.20 Å relative to the transition metalatoms.
The preferred adsorption sites for both reactants – CH2O mole-cules and OH radicals – are atomic triplets. We use the followingnotation: A triplet consisting of two transition metal atoms and aZn atom is denoted as PPZ, P stands for any TM atom (Ni, Pd, orPt), and Z denotes a Zn atom. Because of the horizontal shift ofthe subsurface layers the ZPP triplet with the opposite orientationin the ½11 �2� direction (see Fig. 1) is not equivalent to a PPZ triplet.Likewise, the PZZ and ZZP triplets are not equivalent.
The calculated lattice parameters of PdZn are in reasonableagreement with the results of Lin et al. [24] calculated using the
Pd
Zn
PPZ ZPP
PZZ ZZP
Fig. 1. Top view of the clean L10(111) surface. Because of the tetragonaldeformation of the lattice the subsurface atomic planes are shifted in the ½11 �2�direction (horizontal). The preferred adsorption sites for CH2O and OH moleculesare atomic triplets. The PPZ triplet and the ZPP triplet consist both of one Zn atomand two Pd(Ni, Pt) atoms differ in the coordination to subsurface atoms. LikewisePZZ and ZZP triplets are also inequivalent.

M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18 9
same code, but a lower cut-off energy and a coarser k-space grid. Intheir surface calculations, however, Lin et al. neglected surfacerelaxation and the inequivalency of surface triplets due to thetetragonal distortion. We shall come back to this point when wediscuss the results for adsorption and chemical reactions.
3. Results
3.1. Adsorption and diffusion of OH and CH2O
In this section we summarize our results for the adsorption anddiffusion of hydroxyl groups and formaldehyde molecules on the(111) surfaces. Adsorption energies are determined as the differ-ence in the total energies of the relaxed adsorbate/substrate com-plex and the energies of the clean substrate plus the energy of theadsorbed species in the gas-phase.
3.1.1. Adsorption of hydroxyl groupsOH groups are adsorbed in a vertical orientation, bound through
the oxygen atom in the hollows of the atomic triplets. The adsorp-tion energies Eb of OH in the ZZP, PZZ, ZPP, and PPZ triplets aresummarized in Table 2. On the NiZn(111) surface the adsorptionenergies of OH range between �322 kJ/mol to �308 kJ/mol, onthe PdZn(111) surface between �307 kJ/mol and �272 kJ/mol,and on PtZn(111) between �301 kJ/mol and �224 kJ/mol. Hencethe adsorption strength of OH groups decreases in the sequenceNiZn > PdZn > PtZn.
The difference in the adsorption energies on different triplets iscorrelated with their chemical composition and with the tetrago-nal deformation of the L10 lattice. The distortion from cubic B2symmetry and thus the difference between inequivalent tripletsis smallest for PdZn. The adsorption energies in triplets of oppositeorientations (ZZP, PZZ) and (ZPP, PPZ) differ only by 6 kJ/mol and7 kJ/mol, respectively. Adsorption energies in the Zn-rich triplets(ZZP and PZZ) are by 31 kJ/mol more exothermic than on thePd-rich (ZPP and PPZ) triplets. On the NiZn(111) where the distor-tion is stronger the differences in the adsorption energies oninequivalent triplets are 13 kJ/mol and 12 kJ/mol, but the differ-ence between Zn- and Ni-rich triplets is smaller. On thePtZn(111) surface the hydroxyl group is most strongly adsorbedin the ZZP triplet. The PZZ and ZPP adsorption sites are not stableand upon relaxation the OH group shifts to the most stable ZZPsite. Adsorption in the PPZ triplet is locally stable, but energeticallyby 77 kJ/mol less favorable than adsorption on ZZP. On the
Table 2Adsorption energies Eb of OH (in kJ/mol) and distances between the oxygen atom andsurface atoms, d(O–P), d(O–Z) (in Å) for the four inequivalent adsorption sites ZZP,PZZ, ZPP, and PPZ on the (11 1) surfaces of NiZn, PdZn, and PtZn. Entries in theparentheses are ZPE-corrected values. Two values of binding distances correspond tobridge sites.
Site Property NiZn(111) PdZn(111) PtZn(111)
ZZP Eb �320 (�318) �307 (�297) �301 (�288)d(O–P) 2.15 2.52 2.51d(O–Z) 2.05, 2.05 2.15, 2.15 2.01, 2.01
PZZ Eb �308 (�298) �301 (�291) ?ZZPd(O–P) 1.87 2.26d(O–Z) 2.21, 2.21 2.11, 2.11
ZPP Eb �322 (�319) �278 (�269) ?ZZPd(O–P) 2.14, 2.14 2.48, 2.48d(O–Z) 1.87 1.82
PPZ Eb �310 (�307) �272 (�263) �224 (�216)d(O–P) 1.96,1.96 2.28, 2.30 2.23, 2.36d(O–Z) 2.20 2.10 2.15
PtZn(111) surface the difference between the adsorption sitescaused by the tetragonal deformation is thus the largest.
The inequivalency of the surface triplets is also reflected in theadsorption geometries. For example, on NiZn(111) the O–Zn dis-tance is smaller than d(O–Ni) on ZZP triplets and vice versa onPZZ. On all both types of triplets adsorption is more exothermicif the O–Zn distance is shorter. On PdZn(111) the chemical compo-sition is most important (as expected, because the tetragonal dis-tortion is small), with the triplets with two Zn atoms binding OHmore strongly. On the Pd-rich triplets, OH is more stronglyadsorbed on ZPP where the Zn–O distance is smallest. OnPtZn(111) binding is strongest on ZZP where the O–Zn distanceis smallest and d(O–Pt) largest.
3.1.2. Adsorption of formaldehydeFormaldehyde binds to the surface via both carbon and oxygen
atoms, with the C–O axis essentially parallel to the surface. Thestable adsorption geometries of the CH2O molecule on the fourinequivalent triplets are shown in Figs. 2–4. The calculated adsorp-tion energies of CH2O and binding distances are presented inTable 3.
On the Zn-rich ZZP and PZZ triplets the oxygen atom is locatedclose to the center of the threefold hollow, while on ZPP and PPZ itoccupies an off-symmetry position in the P–Z bridge. On all ZZPand ZPP triplets the oxygen atom is closer to the Zn atoms, whileon PZZ and PPZ this holds only for PdZn and PtZn while on NiZnthe O–P distances are shorter. This demonstrates the dominantstrength of the O–Zn bonds for the adsorption of formaldehydeon PdZn and PtZn, while on NiZn the bonding to both species isof comparable strength.
The carbon atom of the CH2 group adsorbed on ZZP and PZZ tri-plets is located on-top of the transition-metal atom, shifted towardthe hollow on PdZn and PtZn. On ZPP and PPZ triplets C is close tothe Ni–Zn bridge site on NiZn(111), but slightly off from the top onthe transition-metal atom on PdZn(111) and PtZn(111). A remark-able fact is that on NiZn(111) one of the C–H bonds of the mole-cule bound in ZPP and PPZ triplets is considerably stretched (to1.18 Å and 1.20 Å, respectively, compared to 1.12 Å in thegas-phase, we shall return to this point in Section 3.2.2). Alladsorption geometries are different variants of di-r configurations.On the carbon atom the hybridization of the atomic eigenstates ischanged upon adsorption from sp2 in the gas-phase to sp3. Forexample, for adsorption on PdZn the H–C–H angle is 117�(110�),the H–C–O angles are 112�(118�), the H–C–Pd angles 106�(108�)for molecules adsorbed on a ZZP(PZZ) triplet. In both cases theangles are close to a tetrahedral configuration.
The adsorption energies of formaldehyde do not show the sametrend as found for hydroxyl groups. Adsorption of CH2O is weakeston PdZn(111) surfaces. Adsorption energies range between�25 kJ/mol and �18 kJ/mol, and they are less exothermic onPd-rich triplets. NiZn(111) adsorbs CH2O most strongly. The bind-ing energies range from �60 kJ/mol to �37 kJ/mol, and adsorptionis less exothermic on the Zn-rich triplets. On PtZn(111) adsorptionenergies vary between �45 kJ/mol and �17 kJ/mol, the differencesare largest between triplets of the same composition, but differentorientation. It is also remarkable that the most stable adsorptionsite is different for each compound: ZPP on NiZn, stabilized bystrong Ni–C, Ni–O and Zn–O bonds. PZZ on PdZn, promoted bystrong Pd–C and Zn–O bonds. ZZP (energetically almost degeneratewith ZPP) on PtZn where the strong O–Zn bonds dominate. The dif-ference in the adsorption energies on triplets of opposite orienta-tion is largest on the PtZn(111) surface where the difference inthe adsorption energies on ZPP and PPZ triplets reaches 28 kJ/mol.
Adsorption for both reactants on PdZn(111) was studied by Linet al. [24] and Neyman et al. [18]. For OH an adsorption energies of�292 and �294 kJ/mol on a PdZn2 triplet and for CH2O binding

Fig. 2. Adsorption geometries of CH2O on the four inequivalent atomic triplets on the NiZn(111) surface (a) ZZP, (b) PZZ, (c) ZPP, and (d) PPZ. Carbon atoms are shown inblack, oxygen in blue, and hydrogen in green. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3. Adsorption geometries of CH2O on the four inequivalent atomic triplets on the PdZn(111) surface are different from those on NiZn(111). (a) ZZP, (b) PZZ, (c) ZPP, and(d) PPZ. Cf. Fig. 2.
Fig. 4. Adsorption geometries of CH2O on the four inequivalent atomic triplets on the PtZn(111) surface are similar to those on PdZn(111). The main difference is in thearrangement of subsurface atoms. (a) ZZP, (b) PZZ, (c) ZPP, and (d) PPZ. Cf. Fig. 3.
Table 3Adsorption energies Eb of CH2O (in kJ/mol) and binding distances d(C–P), d(O–P), andd(O–Z) (in Å) between carbon, oxygen and the neighboring atoms for fourinequivalent adsorption sites ZZP, PZZ, ZPP, and PPZ on the (111) surfaces of NiZn,PdZn, and PtZn. Entries in the parentheses are ZPE-corrected values. Two values ofbinding distances correspond to bridge sites.
Site Property NiZn(111) PdZn(111) PtZn(111)
ZZP Eb �38 (�32) �23 (�18) �45 (�35)d(C–P) 2.05 2.25 2.29d(O–P) 2.28 2.87 2.90d(O–Z) 2.12, 2.12 2.12, 2.12 2.05, 2.05
PZZ Eb �37 (�31) �25 (�20) �36 (�27)d(C–P) 2.03 2.07 1.99d(O–P) 1.94 2.52 2.49d(O–Z) 2.38, 2.38 2.22, 2.22 2.19, 2.19
ZPP Eb �60 (�54) �19 (�14) �44 (�34)d(C–P) 2.09, 2.13 2.28 2.27d(O–P) 2.19 2.62 2.45d(O–Z) 1.99 1.93 1.89
PPZ Eb �48 (�45) �18 (�14) �17 (�10)d(C–P) 2.12, 2.19 2.16 2.07d(O–P) 1.89 2.48 2.21d(O–Z) 2.44 2.24 2.29
Table 4Activation energies for diffusion of OH (in kJ/mol). Diffusion is strongly anisotropic.
Direction NiZn(111) PdZn(111) PtZn(111)
½11 �2� 42 80 87
½�110� 17 31 48
10 M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18
energies between �20 and �23 kJ/mol on a Pd2Zn triplet werereported. The inequivalency of the surface triplets arising fromthe tetragonal distortion (which is even more pronounced sincesurface relaxation was neglected) was disregarded. Nevertheless,their values are comparable with the average adsorption energiescalculated here.
3.1.3. Mobility of adsorbatesA necessary condition for catalytic activity is a sufficient mobil-
ity of the reactants on the surface of the catalyst. The highest acti-vation energies for diffusion jumps of hydroxyl groups between thestable adsorption sites are summarized in Table 4. The mobility ofOH groups is anisotropic: the activation energies for diffusion par-allel to the rows of TM atoms in the ½�110� direction (crossing a PZbridge site) are much lower than for diffusion in the perpendicular½11 �2� direction where PP, PZ, ZZ, and ZP bridge sites have to bepassed before returning to an equivalent adsorption site. On theNiZn surface the highest barrier for diffusion in the ½11 �2� directionis 42 kJ/mol across the ZZ bridge. On the PdZn surface the highestbarrier of 80 kJ/mol is across a PP bridge. Diffusion on the PtZn sur-face follows a different scenario: As diffusion across a PP bridge siterequires a large activation energy of 98 kJ/mol, a direct jump over aPt atom with a barrier of 87 kJ/mol is more favorable. From theinstable PZZ hollow the OH group drifts directly to the stable ZZPsite. In general the mobility of hydroxyl groups decreases withincreasing corrugation of the potential energy surface, i.e. fromNiZn to PdZn (see Table 2).
While the diffusion of OH groups proceeds essentially via trans-lational movements, the diffusion of CH2O can proceed also by
12 32
11
13
31
33
21 41
23 43
22 42
10
14
20
24
30
34
40
44
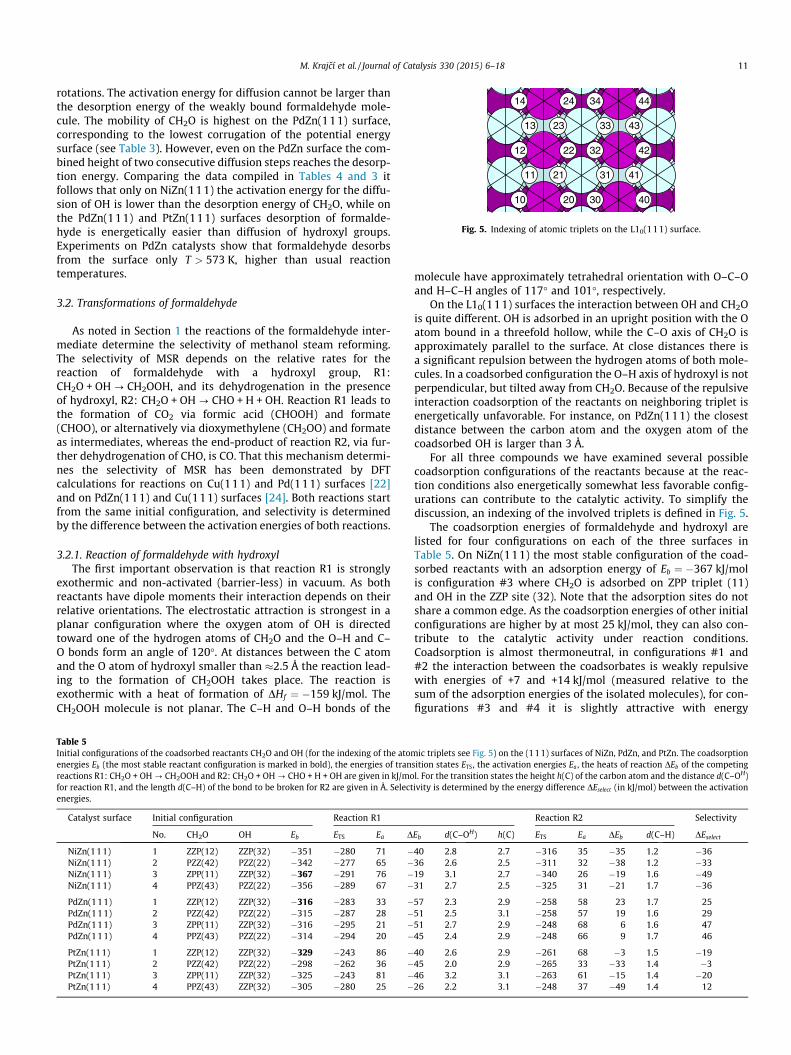
Fig. 5. Indexing of atomic triplets on the L10(111) surface.
M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18 11
rotations. The activation energy for diffusion cannot be larger thanthe desorption energy of the weakly bound formaldehyde mole-cule. The mobility of CH2O is highest on the PdZn(111) surface,corresponding to the lowest corrugation of the potential energysurface (see Table 3). However, even on the PdZn surface the com-bined height of two consecutive diffusion steps reaches the desorp-tion energy. Comparing the data compiled in Tables 4 and 3 itfollows that only on NiZn(111) the activation energy for the diffu-sion of OH is lower than the desorption energy of CH2O, while onthe PdZn(111) and PtZn(111) surfaces desorption of formalde-hyde is energetically easier than diffusion of hydroxyl groups.Experiments on PdZn catalysts show that formaldehyde desorbsfrom the surface only T > 573 K, higher than usual reactiontemperatures.
3.2. Transformations of formaldehyde
As noted in Section 1 the reactions of the formaldehyde inter-mediate determine the selectivity of methanol steam reforming.The selectivity of MSR depends on the relative rates for thereaction of formaldehyde with a hydroxyl group, R1:CH2O + OH ? CH2OOH, and its dehydrogenation in the presenceof hydroxyl, R2: CH2O + OH ? CHO + H + OH. Reaction R1 leads tothe formation of CO2 via formic acid (CHOOH) and formate(CHOO), or alternatively via dioxymethylene (CH2OO) and formateas intermediates, whereas the end-product of reaction R2, via fur-ther dehydrogenation of CHO, is CO. That this mechanism determi-nes the selectivity of MSR has been demonstrated by DFTcalculations for reactions on Cu(111) and Pd(111) surfaces [22]and on PdZn(111) and Cu(111) surfaces [24]. Both reactions startfrom the same initial configuration, and selectivity is determinedby the difference between the activation energies of both reactions.
3.2.1. Reaction of formaldehyde with hydroxylThe first important observation is that reaction R1 is strongly
exothermic and non-activated (barrier-less) in vacuum. As bothreactants have dipole moments their interaction depends on theirrelative orientations. The electrostatic attraction is strongest in aplanar configuration where the oxygen atom of OH is directedtoward one of the hydrogen atoms of CH2O and the O–H and C–O bonds form an angle of 120�. At distances between the C atomand the O atom of hydroxyl smaller than �2.5 Å the reaction lead-ing to the formation of CH2OOH takes place. The reaction isexothermic with a heat of formation of DHf ¼ �159 kJ/mol. TheCH2OOH molecule is not planar. The C–H and O–H bonds of the
Table 5Initial configurations of the coadsorbed reactants CH2O and OH (for the indexing of the atomenergies Eb (the most stable reactant configuration is marked in bold), the energies of tranreactions R1: CH2O + OH ? CH2OOH and R2: CH2O + OH ? CHO + H + OH are given in kJ/mofor reaction R1, and the length d(C–H) of the bond to be broken for R2 are given in Å. Selectenergies.
Catalyst surface Initial configuration Reaction R1
No. CH2O OH Eb ETS Ea D
NiZn(111) 1 ZZP(12) ZZP(32) �351 �280 71 �NiZn(111) 2 PZZ(42) PZZ(22) �342 �277 65 �NiZn(111) 3 ZPP(11) ZZP(32) �367 �291 76 �NiZn(111) 4 PPZ(43) PZZ(22) �356 �289 67 �
PdZn(111) 1 ZZP(12) ZZP(32) �316 �283 33 �PdZn(111) 2 PZZ(42) PZZ(22) �315 �287 28 �PdZn(111) 3 ZPP(11) ZZP(32) �316 �295 21 �PdZn(111) 4 PPZ(43) PZZ(22) �314 �294 20 �
PtZn(111) 1 ZZP(12) ZZP(32) �329 �243 86 �PtZn(111) 2 PZZ(42) PZZ(22) �298 �262 36 �PtZn(111) 3 ZPP(11) ZZP(32) �325 �243 81 �PtZn(111) 4 PPZ(43) ZZP(32) �305 �280 25 �
molecule have approximately tetrahedral orientation with O–C–Oand H–C–H angles of 117� and 101�, respectively.
On the L10(111) surfaces the interaction between OH and CH2Ois quite different. OH is adsorbed in an upright position with the Oatom bound in a threefold hollow, while the C–O axis of CH2O isapproximately parallel to the surface. At close distances there isa significant repulsion between the hydrogen atoms of both mole-cules. In a coadsorbed configuration the O–H axis of hydroxyl is notperpendicular, but tilted away from CH2O. Because of the repulsiveinteraction coadsorption of the reactants on neighboring triplet isenergetically unfavorable. For instance, on PdZn(111) the closestdistance between the carbon atom and the oxygen atom of thecoadsorbed OH is larger than 3 Å.
For all three compounds we have examined several possiblecoadsorption configurations of the reactants because at the reac-tion conditions also energetically somewhat less favorable config-urations can contribute to the catalytic activity. To simplify thediscussion, an indexing of the involved triplets is defined in Fig. 5.
The coadsorption energies of formaldehyde and hydroxyl arelisted for four configurations on each of the three surfaces inTable 5. On NiZn(111) the most stable configuration of the coad-sorbed reactants with an adsorption energy of Eb ¼ �367 kJ/molis configuration #3 where CH2O is adsorbed on ZPP triplet (11)and OH in the ZZP site (32). Note that the adsorption sites do notshare a common edge. As the coadsorption energies of other initialconfigurations are higher by at most 25 kJ/mol, they can also con-tribute to the catalytic activity under reaction conditions.Coadsorption is almost thermoneutral, in configurations #1 and#2 the interaction between the coadsorbates is weakly repulsivewith energies of +7 and +14 kJ/mol (measured relative to thesum of the adsorption energies of the isolated molecules), for con-figurations #3 and #4 it is slightly attractive with energy
ic triplets see Fig. 5) on the (1 11) surfaces of NiZn, PdZn, and PtZn. The coadsorptionsition states ETS, the activation energies Ea , the heats of reaction DEb of the competingl. For the transition states the height h(C) of the carbon atom and the distance d(C–OH)ivity is determined by the energy difference DEselect (in kJ/mol) between the activation
Reaction R2 Selectivity
Eb d(C–OH) h(C) ETS Ea DEb d(C–H) DEselect
40 2.8 2.7 �316 35 �35 1.2 �3636 2.6 2.5 �311 32 �38 1.2 �3319 3.1 2.7 �340 26 �19 1.6 �4931 2.7 2.5 �325 31 �21 1.7 �36
57 2.3 2.9 �258 58 23 1.7 2551 2.5 3.1 �258 57 19 1.6 2951 2.7 2.9 �248 68 6 1.6 4745 2.4 2.9 �248 66 9 1.7 46
40 2.6 2.9 �261 68 �3 1.5 �1945 2.0 2.9 �265 33 �33 1.4 �346 3.2 3.1 �263 61 �15 1.4 �2026 2.2 3.1 �248 37 �49 1.4 12

OC
OH
Fig. 7. Schematic presentation of the bonding situation in the transition state of theR1 reaction of formaldehyde with hydroxyl. At the start of the reaction the bondbetween the C atom and the surface is broken and CH2O remains attached to thesurface by the OC atom only. The transition state corresponds to the breaking thebond between the OH atom of the adsorbed hydroxyl and the surface (red dottedline). The energy of the transition state is substantially reduced if the OH atomsimultaneously forms a bond with the C atom of co-adsorbed formaldehyde (greendashed line). Carbon atom is shown in black, oxygen in blue, and hydrogen in green.(For interpretation of the references to color in this figure legend, the reader isreferred to the web version of this article.)
12 M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18
differences of �7 and �9 kJ/mol. On PdZn(111) the co-adsorptionenergies of all four configurations agree with ±1 kJ/mol, again theinteraction is repulsive for configurations #1 and #2, and attractivefor configurations #3 and #4, with interaction energies varyingbetween �17 and +14 kJ/mol. On PtZn(111) the interactionbetween the coadsorbates is much stronger. It is attractive onlyfor configuration #2 where hydroxyl occupies the energeticallymuch less favorable PPZ triplet, but repulsive for the other threeconfigurations.
The product of reaction R1 is hydroxymethoxy CH2OOH, and theadsorption energies are collected in Table 6. CH2OOH binds ratherstrongly via the oxygen atom in the triplet hollows, with an almostvertical O–C axis and the OH group high above a neighboringtransition-metal atom. As an example schematic coadsorptionand product configurations are shown in Fig. 6.
During the formation of CH2OOH the bonds between the CH2
group of formaldehyde and the surface, and between the O atomof the hydroxyl group and the surface have to be broken and anew C–OH bond has to be formed, see Fig. 7. The transition stateof R1 corresponds to breaking the bond between the OH atom ofhydroxyl and the surface (labeled OH to avoid confusion with theOC atom of the formaldehyde molecule). The energy necessaryfor breaking the strong bond of the OH group to the surface arisesfrom the pronounced exothermicity of the reaction.
We have studied the bond-breaking/bond-formation process indetail for all twelve initial configurations. The OH atom is stronglybound in a triplet hollow at a height of 1.3–1.6 Å, in the coadsorp-tion geometry the OH–H axis is tilted away from formaldehyde.The C atom of formaldehyde is located nearly on-top of atransition-metal atom at a height of approximately 2.0 Å. To initi-ate the reaction, the bond between the CH2 group of formaldehydeand the surface must be broken. The height h(C) of the C atomincreases by up to 1.1 Å such that the C–OC axis forms an angleof about 45� with respect to the surface plane (Fig. 7). The valuesof h(C) at the transition state are also presented in Table 5. OnPdZn and PtZn surfaces h(C) � 3.0 Å, on NiZn h(C) � 2.6 Å. Thisreflects the stronger adsorption on the NiZn surface. Althoughthe bond between the CH2 group and the surface is broken, themolecule is still attached to the surface through the OC atom. OnNiZn(111) the height of OC increases only slightly during the reac-tion from �1.7 Å to �2.0 Å, but simultaneously with the formationof the new C–OH bond the OC atom moves again closer to the sur-face. On PdZn(111) and PtZn(111) the enhancement of the height
Table 6Adsorption energies Eb (in kJ/mol) of CH2OOH molecules on the (1 11) surfaces ofNiZn, PdZn, and PtZn. Entries in the parentheses are ZPE-corrected values.
Site NiZn(1 11) PdZn(111) PtZn(111)
ZZP �231 (�215) �213 (�198) �209 (�191)PZZ �219 (�204) �206 (�192) �184 (�168)ZPP �227 (�212) �208 (�193) �212 (�195)PPZ �228 (�213) �202 (�188) �172 (�156)
Fig. 6. Atomic configurations of coadsorbed formaldehyde and hydroxyl on the L10(111OH forming CH2OOH [reaction R1 – (b)] or dehydrogenate to CHO [reaction R2 – (c)]. On Pon PtZn(111) the preferred product depends on the configuration of the reactants, see
of the OC atoms is much more pronounced, and it increases from�1.7 Å to �2.5 Å and decreases again to �1.5 Å simultaneouslywith the formation of the C–OH bond. For a short part of the reac-tion path the bond between the OC atom and the surface is weak-ened such that formaldehyde can adopt a position facilitating theformation of the new C–OH bond. This is essential for achieving alow activation energy. In their DFT study of MSR on Cu(111) Guand Li [22] observed that CH2O is lifted from the substrate to per-mit the reaction with hydroxyl.
The important reaction coordinate is also the distance d(C–OH)between the carbon atom and the oxygen atom of the hydroxylgroup. To form the new C–OH bond the OH atom has to get out ofthe hollow, it attacks the C atom from below. The energy to over-come the binding of the hydroxyl group to the surfaces comes fromthe strong exothermicity of its reaction with formaldehyde. In thetransition state the attractive interaction between the reactants isstrong enough to break the strong bond of the OH atom to the sur-face. On the reference catalysts Pd(111) and Cu(111) the C–OH dis-tances at the transition state are 3.0 Å and 2.7 Å respectively. Thisis much longer than the C–OH bond length of 1.45 Å in freeCH2OOH. The value of C–OH distance at the transition state is cor-related with the activation energy. On the NiZn(111) andPdZn(111) surfaces the average C–OH distances are 2.8 Å and2.5 Å corresponding to average activation energies of 70 kJ/moland 25 kJ/mol, respectively. Detailed values of the reaction coordi-nates and activation energies are given in Table 5, and the transi-tion states for reactions starting from the energetically mostfavorable coadsorption states are depicted in panels (a, c) and (e)of Fig. 8. The correlation between the C–OH distance and the acti-vation energy holds also for the different reaction channels. Thisis most evident for PtZn(111) where the C–OH distances are largerfor configurations #1 and #3 with a high activation energy above80 kJ/mol, but smaller for configurations #2 and #4 with muchlower activation energies around 30 kJ/mol.
) surface (a) and possible products (b, c). Formaldehyde CH2O can either react withdZn(111) reaction R1 is favored, on NiZn(111) reaction R2 is more favorable, while
Table 5.

Fig. 8. Transition states of the reactions R1 (a, c, e) and R2 (b, d, f) starting from theconfigurations #3, #4, and #1 of the reactants on the NiZn(111) (a, b), PdZn(111)(c, d), and PtZn(111) (e, f) surfaces, respectively.
Table 8Adsorption energies Eb (in kJ/mol) of atomic hydrogen on the (111) surfaces of NiZn,PdZn, and PtZn. The energy of 1=2H2(g) is subtracted from the adsorption energies.Entries in the parentheses are ZPE-corrected values. On Zn-rich triplets H is adsorbedclose to top of the P atom, on TM rich triplets the position of hydrogen atom is close tothe PP bridge.
Site NiZn(111) PdZn(111) PtZn(111)
ZZP �24 (�12) +1 (+12) �37 (�20)PZZ �9 (+3) +6 (+17) ?ZZPZPP �54 (�38) �19 (�6) �37 (�21)PPZ ?ZPP �18 (�5) ?ZPP
M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18 13
The reaction between formaldehyde and hydroxyl onPdZn(111) was also considered by Lin et al. [24]. Starting fromCH2O and OH coadsorbed on neighboring ZZP and PZZ triplets anactivation energy of Ea ¼ 16 kJ/mol and a heat of reaction ofDEb ¼ �48 kJ/mol were calculated. The product CH2OOH is boundthrough the oxygen atom in a PdZn2 hollow. While the adsorptionenergy and the heat of reaction are in good agreement with ourresults, the somewhat lower activation energy is due to the factthat the interaction between the reactants is already repulsivewhen coadsorbed on neighboring triplets.
3.2.2. Dehydrogenation of formaldehydeDehydrogenation of formaldehyde (reaction R2) produces for-
myl CHO. On the PdZn and PtZn surfaces CHO is bound throughthe oxygen atom in a bridge position between Zn and a Pd or Ptatom with the carbon atom nearly on top of the transition-metalatom [see Fig. 6(c)] and with O weakly bound to Zn. Binding isstronger in the ZP than in the PZ bridge. On the NiZn surface formylis adsorbed on a ZPP or PPZ triplet, the CH group binding in theNi–Ni bridge and oxygen to Zn. The binding energies collected inTable 7 demonstrate that formyl, similarly as formaldehyde,adsorbs on the PdZn surface substantially weaker (by 30–50 kJ/mol) than on the other two compounds. Table 8 summarizesthe adsorption energies of atomic hydrogen relative to molecularhydrogen in the gas-phase (the energy of 1=2H2(g) is subtractedfrom the adsorption energies). On Zn-rich triplets ZZP and PZZhydrogen is adsorbed close to top of the P atom, and on the TM richtriplets ZPP and PPZ the position of H is close to the PP bridge.
For dehydrogenation the relevant reaction coordinate is thelength of the C–H bond to be broken. In the gas-phase
Table 7Adsorption energies Eb of CHO molecules (in kJ/mol) on the (111) surfaces of NiZn,PdZn, and PtZn. Entries in the parentheses are ZPE-corrected values.
Site NiZn(111) PdZn(111) PtZn(111)
ZPP �183 (�173)PPZ �181 (�172)ZP �154 (�143) �206 (�192)PZ �150 (�139) �192 (�179)
dehydrogenation of formaldehyde is strongly endothermic with aheat of reaction of 380 kJ/mol. For adsorbed CH2O the energyrequired to break the one of the C–H bonds arises from the strongbonding of the products to the surface. To achieve a good CO2
selectivity the catalyst should not favor this reaction too much.During a reaction on the NiZn(111) surface the adsorption geom-etry of the CHO group remains essentially unchanged, the dissoci-ated hydrogen atom moves to a Ni–Ni bridge position on the otherside of the Ni atom. As mentioned above in Section 3.1.2, one of theC–H bonds of formaldehyde is elongated already in the initial state.For the energetically most favorable adsorption on the ZPP tripletthis leads to a low activation energy for reaction R2 of 26 kJ/moland a modest exothermic heat of reaction of �19 kJ/mol. The cor-responding transition state is shown in Fig. 8(b). For the otherstarting configurations, the activation energies, and heats of reac-tion see Table 5.
On the PdZn(111) surface where formaldehyde is adsorbed inan atomic triplet dehydrogenation has to be accompanied by arearrangement of the CHO product. The transition state reachedfrom configuration #2 with formaldehyde initially bound in aPZZ triplet [see Fig. 3(c)] is shown in Fig. 8(d). The O atom movesfrom the Zn–Zn bridge to the Pd–Zn bridge, while the CH groupremains bound to the Pd atom and the dissociated H atom is onthe other side of the Pd atom. The reaction is weakly endothermicand because of the change in the bonding of the CHO molecule theactivation energy of 57 kJ/mol is much higher than on NiZn. Forthe other starting configurations the reaction is also endothermic,the activation energies range from 58 kJ/mol to 68 kJ/mol,see Table 5.
On PtZn(111) the initial configurations #1 and #3, withformaldehyde bound in the energetically more favorable ZZP andZPP triplets the activation energies of 68 kJ/mol and 61 kJ/molare about twice as large as for configurations #2 and #4, againbecause the reaction requires a substantial rearrangement of theCHO binding. See again Figs. 3(a) and 8(d) for the initial and tran-sition states. For the more weakly bound initial states activationenergies are only 35 ± 2 kJ/mol, see Table 5 for more details.
The dehydrogenation of formaldehyde on bulk-terminatedPdZn(111) was studied by Lim et al. [16], Neyman et al. [18] andLin et al. [24]. In reasonable agreement with our results activationenergies ranging between 78 and 86 kJ/mol and an essentiallythermoneutral reaction were reported.
For MSR over Cu(111) and PdZn(111) Gu and Li [22] havepointed out that the transition-state energies for dehydrogenationfollow a Brønsted–Evans–Polanyi correlation with the adsorptionenergies of the dissociation products. The adsorption energiesof hydrogen atoms in the most stable PP sites are �54 kJ/molon NiZn(111), �37 kJ/mol on PtZn(111) and �19 kJ/mol onPdZn(111), see Table 8. Together with the adsorption energiesfor CHO collected in Table 7 this shows the difference betweenthe transition state energies on NiZn on one side and on PdZnand PtZn on the other side. The weak adsorption of atomic hydro-gen on the catalyst surface thus contributes to higher CO2
selectivity.
14 M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18
On PdZn(111) the detached H atom is preferably adsorbed in anearby PP bridge site with a binding energy of Eb ¼ �19 kJ/mol.This is an important observation. Our previous experiencewith the investigation of the selectivity of acetylene semi-hydrogenation on surfaces of the Ga–Pd compounds [42–44]shows that (de)hydrogenation activity of the catalyst can besignificantly suppressed [45] if the active TM atom is surroundedby simple metal atoms only. Hence the dehydrogenation activitycan be reduced if the surface is enriched by Zn atoms. This leadsto a lower number of PP bridge adsorption sites that are favoredby the hydrogen atoms. Hence it is conceivable that onZn-enriched surfaces dehydrogenation will be even more difficultthan on stoichiometric PdZn surfaces, in agreement with theexperimental observations of Friedrich et al. [46]. On the otherside, this simultaneously reduces the activity of the other reactionsteps of the MSR process.
On all three surfaces the coadsorbed hydroxyl group has only anegligible influence on the dehydrogenation reaction.
3.2.3. SelectivitySelectivity is determined by the difference in the activation
energies of the two competing reactions
DEselect ¼ EaðR2Þ � EaðR1Þ: ð3Þ
If DEselect < 0 dehydrogenation, resulting in the undesired produc-tion of CO is preferred, while the required selectivity to the CO2 pro-duction is obtained for DEselect > 0. The values of DEselect are compiledin the last column of Table 5. The selectivities calculated for differ-ent initial configurations exhibit a rather large scatter. The differ-ences between the highest and lowest values of DEselect are31 kJ/mol on PtZn(111), 17 kJ/mol on PdZn(111), and about15 kJ/mol on NiZn(111). However, only for PtZn(111) the differ-ence is large enough to lead to a qualitatively different predictionof selectivity for different initial configurations.
On NiZn(111) direct dehydrogenation is favored for all fourinitial configurations. It is most pronounced for the most stablestarting configuration #3. We note that already in the initialconfiguration one of the C–H bonds of the CH2 group is stretched,formaldehyde is activated for dehydrogenation.
On PdZn(111) reaction R1 is more exothermic and has loweractivation energies than on NiZn, whereas reaction R2 is endother-mic, with much larger activation energies. Hence on PdZn(111)MSR is strongly selective for CO2. This agrees with the combined
-380 -360 -340 -320 -300 -280
Eb(CH2O+OH) [kJ/mol]
-60
-40
-20
0
20
40
60
Sele
ctiv
ity Δ
Ese
lect
[kJ/
mol
]
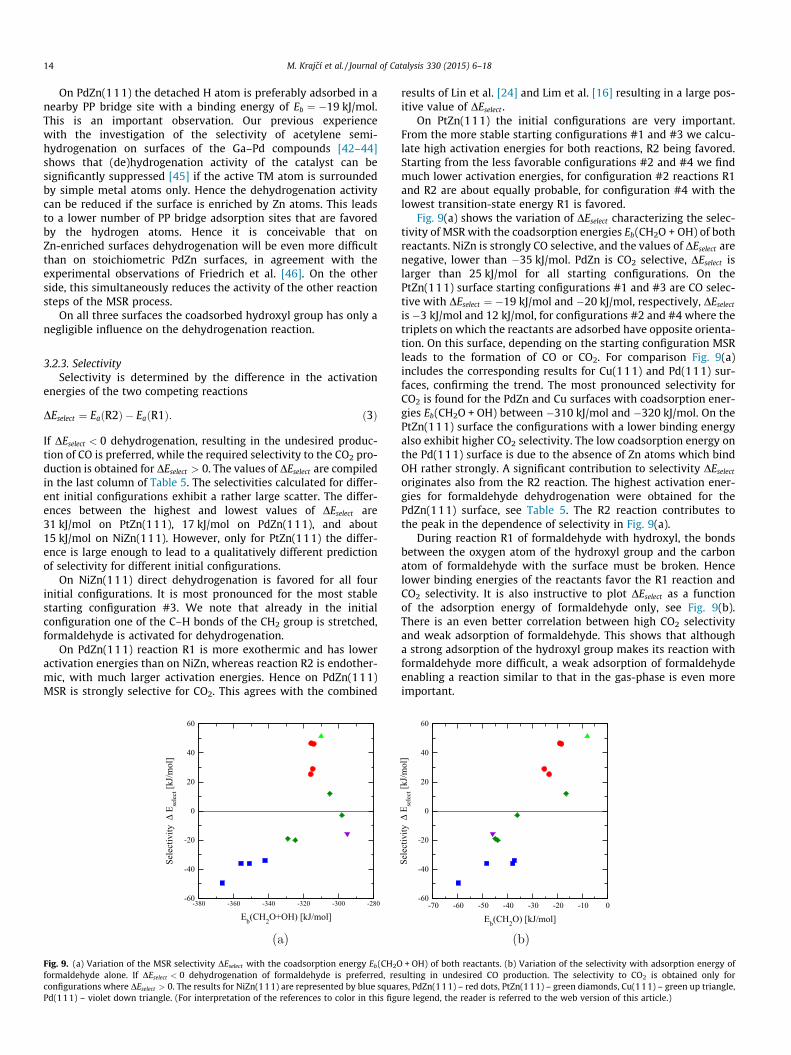
Fig. 9. (a) Variation of the MSR selectivity DEselect with the coadsorption energy Eb(CH2Oformaldehyde alone. If DEselect < 0 dehydrogenation of formaldehyde is preferred, reconfigurations where DEselect > 0. The results for NiZn(111) are represented by blue squarPd(111) – violet down triangle. (For interpretation of the references to color in this figu
results of Lin et al. [24] and Lim et al. [16] resulting in a large pos-itive value of DEselect .
On PtZn(111) the initial configurations are very important.From the more stable starting configurations #1 and #3 we calcu-late high activation energies for both reactions, R2 being favored.Starting from the less favorable configurations #2 and #4 we findmuch lower activation energies, for configuration #2 reactions R1and R2 are about equally probable, for configuration #4 with thelowest transition-state energy R1 is favored.
Fig. 9(a) shows the variation of DEselect characterizing the selec-tivity of MSR with the coadsorption energies Eb(CH2O + OH) of bothreactants. NiZn is strongly CO selective, and the values of DEselect arenegative, lower than �35 kJ/mol. PdZn is CO2 selective, DEselect islarger than 25 kJ/mol for all starting configurations. On thePtZn(111) surface starting configurations #1 and #3 are CO selec-tive with DEselect ¼ �19 kJ/mol and �20 kJ/mol, respectively, DEselect
is �3 kJ/mol and 12 kJ/mol, for configurations #2 and #4 where thetriplets on which the reactants are adsorbed have opposite orienta-tion. On this surface, depending on the starting configuration MSRleads to the formation of CO or CO2. For comparison Fig. 9(a)includes the corresponding results for Cu(111) and Pd(111) sur-faces, confirming the trend. The most pronounced selectivity forCO2 is found for the PdZn and Cu surfaces with coadsorption ener-gies Eb(CH2O + OH) between �310 kJ/mol and �320 kJ/mol. On thePtZn(111) surface the configurations with a lower binding energyalso exhibit higher CO2 selectivity. The low coadsorption energy onthe Pd(111) surface is due to the absence of Zn atoms which bindOH rather strongly. A significant contribution to selectivity DEselect
originates also from the R2 reaction. The highest activation ener-gies for formaldehyde dehydrogenation were obtained for thePdZn(111) surface, see Table 5. The R2 reaction contributes tothe peak in the dependence of selectivity in Fig. 9(a).
During reaction R1 of formaldehyde with hydroxyl, the bondsbetween the oxygen atom of the hydroxyl group and the carbonatom of formaldehyde with the surface must be broken. Hencelower binding energies of the reactants favor the R1 reaction andCO2 selectivity. It is also instructive to plot DEselect as a functionof the adsorption energy of formaldehyde only, see Fig. 9(b).There is an even better correlation between high CO2 selectivityand weak adsorption of formaldehyde. This shows that althougha strong adsorption of the hydroxyl group makes its reaction withformaldehyde more difficult, a weak adsorption of formaldehydeenabling a reaction similar to that in the gas-phase is even moreimportant.
-70 -60 -50 -40 -30 -20 -10 0
Eb(CH2O) [kJ/mol]
-60
-40
-20
0
20
40
60
Sele
ctiv
ity Δ
Ese
lect
[kJ/
mol
]
+ OH) of both reactants. (b) Variation of the selectivity with adsorption energy ofsulting in undesired CO production. The selectivity to CO2 is obtained only fores, PdZn(111) – red dots, PtZn(111) – green diamonds, Cu(111) – green up triangle,re legend, the reader is referred to the web version of this article.)
M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18 15
3.2.4. Reaction pathways to carbon dioxideIn the previous subsections it was demonstrated that on the
PdZn(111) surface and partially also on the PtZn(111) surfacethe R1 reaction of formaldehyde with hydroxyl is preferred toformaldehyde dehydrogenation. To prove the required CO2 selec-tivity of these surfaces it still remains to show that the followingreaction pathway to carbon dioxide is not energetically prohibitive.In a recent DFT study [24] it has been explicitly shown that on thePdZn(111) surface the endproduct of the R1 reaction is CO2, fol-lowing a pathway where hydroxymethoxy (CH2OOH) first dehy-drogenates to formic acid (CHOOH) which reacts with hydroxylto form formyl (CHOO) that finally dehydrogenates to CO2. In analternative reaction path CH2OOH first reacts with OH to formCH2OO which in two dehydrogenation steps produces CO2. Thereactions of the intermediates with further hydroxyl moleculesare very easy, with very low reaction barriers. Such reactions arecertainly favorable from the viewpoint of the catalytic activity.However, as a low concentration of OH on the catalyst surfacecan reduce the selectivity (see also Section 4.5), the consumptionof further water molecules in the MSR reaction is not desirablefrom the viewpoint of CO2 selectivity. We have therefore consid-ered a possible reaction path from hydroxymethoxy to carbondioxide proceeding by dehydrogenation steps only.
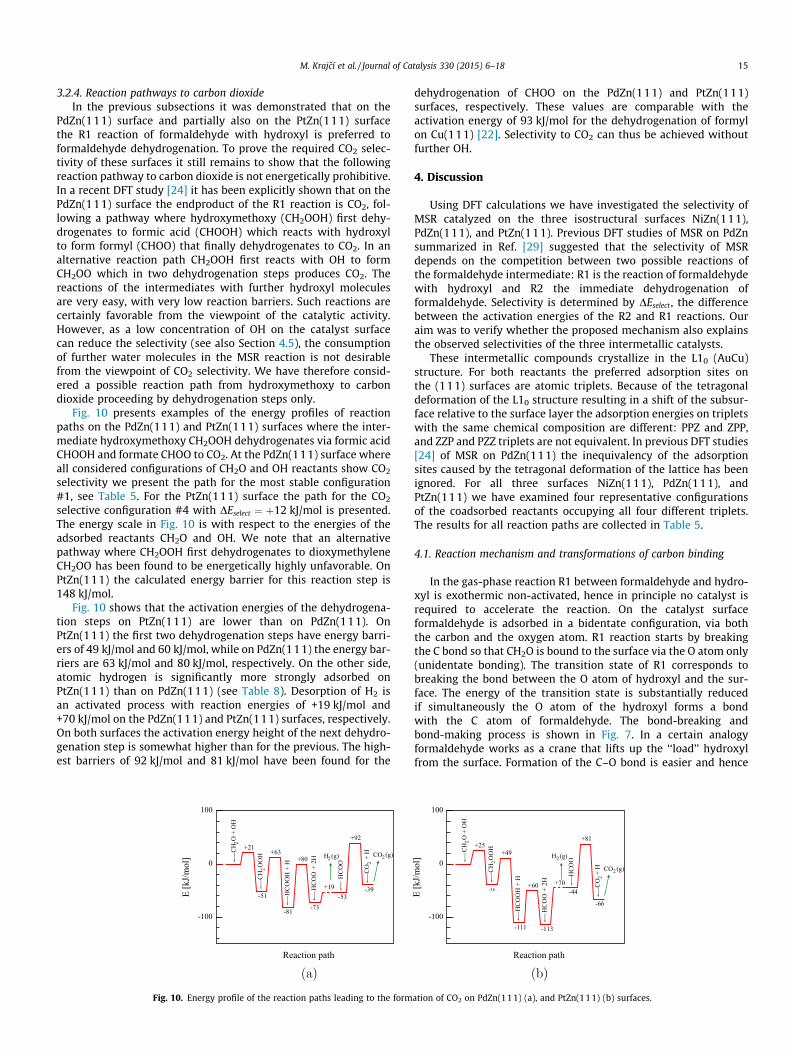
Fig. 10 presents examples of the energy profiles of reactionpaths on the PdZn(111) and PtZn(111) surfaces where the inter-mediate hydroxymethoxy CH2OOH dehydrogenates via formic acidCHOOH and formate CHOO to CO2. At the PdZn(111) surface whereall considered configurations of CH2O and OH reactants show CO2
selectivity we present the path for the most stable configuration#1, see Table 5. For the PtZn(111) surface the path for the CO2
selective configuration #4 with DEselect ¼ þ12 kJ/mol is presented.The energy scale in Fig. 10 is with respect to the energies of theadsorbed reactants CH2O and OH. We note that an alternativepathway where CH2OOH first dehydrogenates to dioxymethyleneCH2OO has been found to be energetically highly unfavorable. OnPtZn(111) the calculated energy barrier for this reaction step is148 kJ/mol.
Fig. 10 shows that the activation energies of the dehydrogena-tion steps on PtZn(111) are lower than on PdZn(111). OnPtZn(111) the first two dehydrogenation steps have energy barri-ers of 49 kJ/mol and 60 kJ/mol, while on PdZn(111) the energy bar-riers are 63 kJ/mol and 80 kJ/mol, respectively. On the other side,atomic hydrogen is significantly more strongly adsorbed onPtZn(111) than on PdZn(111) (see Table 8). Desorption of H2 isan activated process with reaction energies of +19 kJ/mol and+70 kJ/mol on the PdZn(111) and PtZn(111) surfaces, respectively.On both surfaces the activation energy height of the next dehydro-genation step is somewhat higher than for the previous. The high-est barriers of 92 kJ/mol and 81 kJ/mol have been found for the
-100
0
100
Reaction path
E [k
J/m
ol]
CH 2
O +
OH
+21
-51
CH
2OO
H +63
-81
HC
OO
H +
H
+80
-73
HC
OO
+ 2
H
+19-53
HC
OO
+92
-39
CO
2 +
H CO2(g)H2(g)
Fig. 10. Energy profile of the reaction paths leading to the form
dehydrogenation of CHOO on the PdZn(111) and PtZn(111)surfaces, respectively. These values are comparable with theactivation energy of 93 kJ/mol for the dehydrogenation of formylon Cu(111) [22]. Selectivity to CO2 can thus be achieved withoutfurther OH.
4. Discussion
Using DFT calculations we have investigated the selectivity ofMSR catalyzed on the three isostructural surfaces NiZn(111),PdZn(111), and PtZn(111). Previous DFT studies of MSR on PdZnsummarized in Ref. [29] suggested that the selectivity of MSRdepends on the competition between two possible reactions ofthe formaldehyde intermediate: R1 is the reaction of formaldehydewith hydroxyl and R2 the immediate dehydrogenation offormaldehyde. Selectivity is determined by DEselect , the differencebetween the activation energies of the R2 and R1 reactions. Ouraim was to verify whether the proposed mechanism also explainsthe observed selectivities of the three intermetallic catalysts.
These intermetallic compounds crystallize in the L10 (AuCu)structure. For both reactants the preferred adsorption sites onthe (111) surfaces are atomic triplets. Because of the tetragonaldeformation of the L10 structure resulting in a shift of the subsur-face relative to the surface layer the adsorption energies on tripletswith the same chemical composition are different: PPZ and ZPP,and ZZP and PZZ triplets are not equivalent. In previous DFT studies[24] of MSR on PdZn(111) the inequivalency of the adsorptionsites caused by the tetragonal deformation of the lattice has beenignored. For all three surfaces NiZn(111), PdZn(111), andPtZn(111) we have examined four representative configurationsof the coadsorbed reactants occupying all four different triplets.The results for all reaction paths are collected in Table 5.
4.1. Reaction mechanism and transformations of carbon binding
In the gas-phase reaction R1 between formaldehyde and hydro-xyl is exothermic non-activated, hence in principle no catalyst isrequired to accelerate the reaction. On the catalyst surfaceformaldehyde is adsorbed in a bidentate configuration, via boththe carbon and the oxygen atom. R1 reaction starts by breakingthe C bond so that CH2O is bound to the surface via the O atom only(unidentate bonding). The transition state of R1 corresponds tobreaking the bond between the O atom of hydroxyl and the sur-face. The energy of the transition state is substantially reducedif simultaneously the O atom of the hydroxyl forms a bondwith the C atom of formaldehyde. The bond-breaking andbond-making process is shown in Fig. 7. In a certain analogyformaldehyde works as a crane that lifts up the ‘‘load’’ hydroxylfrom the surface. Formation of the C–O bond is easier and hence
-100
0
100
Reaction path
E [k
J/m
ol]
CH
2O +
OH
+25
-38
CH
2OO
H +49
-111
HC
OO
H +
H
+60
-113
HC
OO
+ 2
H +70-44
HC
OO
+81
-66
CO
2 + H CO2(g)
H2(g)
ation of CO2 on PdZn(111) (a), and PtZn(111) (b) surfaces.

16 M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18
the energy of the transition state is lower if formaldehyde isadsorbed only weakly so that it can easily approach and accommo-date its position to the strongly adsorbed hydroxyl molecule.
The easy change of the formaldehyde-surface bonding configu-ration from bidentate to unidentate is a prerequisite for CO2 selec-tivity. We note that the classification of catalysts with respect tothe CO2/CO selectivity on the basis of unidentate/bidentate bond-ing of the aldehyde intermediates on the catalyst surface has beenproposed already by Takezawa and Iwasa in 1997 [6].
In the gas-phase R2 is strongly endothermic. Many reactionsteps of MSR require a good dehydrogenation catalyst. A catalystmust reverse the preference at the gas-phase reaction. However,too strong dehydrogenation activity of catalyst leads to undesiredCO selectivity. Weak adsorption of atomic hydrogen on the catalystsurface contributes to higher CO2 selectivity. At dehydrogenationof formaldehyde the relevant reaction coordinate is the length ofthe C–H bond to be broken. The detached H atom is preferablyadsorbed in a nearby PP bridge site. Dehydrogenation activity ofthe catalyst can be reduced if the surface is enriched by Zn atoms.This leads to a lower number of PP bridge adsorption sites thatattract the dissociating hydrogen atoms.
4.2. Comparison with experiment
The predicted selectivities can be compared with availableexperimental data [30,31]. PtZn shows mixed CO2/CO selectivity.An unsupported PtZn catalyst prepared in the form of a powderexhibited �50% selectivity [30] in the temperature range from513 K to 573 K. For a supported Pt/ZnO catalyst the selectivityis larger, �80% [31]. The increased selectivity could indicate apositive effect of the ZnO support (as discussed below). In ourrecent studies of selectivity of Ga–Pd catalysts for acetylenesemi-hydrogenation [42,44] we have demonstrated differentpreparation techniques can affect the relative stabilities and thestoichiometries of low-index surfaces, with consequent changesin the selectivities. Our present results suggest that the observedmixed selectivity of PtZn catalysts is due to the presence ofinequivalent adsorption sites on the same (111) surface. Out ofthe four investigated configurations of coadsorbed reactants twoexhibit CO selectivity with DEselect � �19 kJ/mol, one shows essen-tially no selectivity, DEselect � �3 kJ/mol, and one CO2 selectivity,DEselect � 12 kJ/mol. These results provide a qualitative explanationof the experimentally observed [30] mixed CO2/CO selectivity ofthe PtZn catalyst.
Many studies have been devoted to the catalytic properties ofPdZn. For all reaction paths DEselect has positive values between25 kJ/mol and 47 kJ/mol, clearly favoring CO2 selectivity. On thePdZn(111) surface both R1 and R2 reactions proceed like onCu(111). The predicted high CO2 selectivity agrees with the workof Tsai et al. [30] and Nozawa et al. [31] who reported for bothunsupported and supported samples almost 100% CO2 selectivity.Our results also agree at least semiquantitatively with the resultsof previous DFT calculations, see Ref. [29] and references therein.
For the NiZn(111) surface our calculations predict a strong COselectivity with all values of DEselect smaller than �34 kJ/mol, inagreement with experimental observations. For supportedNi/ZnO samples essentially zero CO2 selectivity was observed[6,7]. For unsupported NiZn catalysts poor selectivity for CO2 for-mation, decreasing from 35% at 513 K to 7% at 573 K was reported[30]. Very recently Friedrich et al. [47] reported that unsupportedsingle-phase NiZn catalysts are unstable under MSR conditionsand decompose, forming ZnO and a Ni-enriched substitutionalNi–Zn alloy. Parallel to the decomposition selectivity for CO2
increases from very low values for the initial NiZn compound tolarge values for the mixed ZnO/Ni–Zn samples.
4.3. Selectivity and d-band position
Nozawa et al. [31] presented an extensive theoretical andexperimental study of the electronic structure of the NiZn, PdZn,and PtZn compounds. They observed a correlation between theposition of d-band and selectivity of the catalysts at MSR. InPdZn the top of the d-band coincides is located �1.8 eV below EF
as in Cu. In PtZn and NiZn the d-bands shifted to slightly lowerbinding energies, parallel to a decreased CO2 selectivity. In our pre-sent study we have observed a clear correlation between a highCO2 selectivity and a weak adsorption of the formaldehyde onthe surface of the catalysts, see Fig. 9(b). With decreasing adsorp-tion energy CO2 selectivity increases.
A strong correlation between the d-band position and theadsorption energy has been reported for many molecules [48]. Alower binding energy of the d-states facilitates the donation ofelectrons to empty antibonding molecular eigenstates. Here wehave found that the adsorption energy of formaldehyde (averagedover the four possible adsorption sites) becomes more exothermicin the sequence PdZn (Eb ¼ �21 kJ/mol) ? PtZn (Eb ¼�35 kJ/mol) ? NiZn (Eb ¼ �46 kJ/mol), parallel to the decrease ofthe energy at the upper edge of the d-band of the transition metalfrom �1.8 eV to �1.2 eV and �0.9 eV, respectively. A similardependence of the adsorption energy on the d-band position hasalso been reported for ethylene (which is isoelectronic to CH2Oand also adsorbed in a flat configuration) [49,50].
4.4. Correlation with the tetragonal distortion of the L10 structure
Our investigation is the first to account for inequivalence of thehollow sites in surface triplets caused by the tetragonal distortionof the crystal structure. On NiZn(111) and PdZn(111) surfaces theinequivalence of the surface sites manifests itself in differences inthe adsorption energies of the reactants of up to 35 kJ/mol (seeTables 4 and 3). However, these differences are not large enoughto affect the ordering of the activation energies of the competingreactions of formaldehyde determining the selectivity of MSR.Only for PtZn with the strongest tetragonal distortion certain sitesare destabilized for OH adsorption (see Table 4) and these sitesshow also a weaker adsorption of formaldehyde. As a result itdepends on the initial coadsorption configuration whether thedehydrogenation of formaldehyde or its reaction with hydroxyl isenergetically favored (see Table 5).
4.5. Comments on recent results
Recently several investigations [23,27,51] of the influence of theZnO support on the selectivity of MSR have appeared. They suggestthat the MSR reaction on PdZn is only a part of the story and theinterface between PdZn and the ZnO support plays an importantrole. It is known that ZnO is an efficient catalyst for producinghydroxyl molecules. Smith et al. [23] reported that the activationenergy for splitting water is much lower on ZnO(0001)(Ea ¼ 0:42 eV) than on PdZn(111) (Ea ¼ 0:92 eV). Lorenz et al.[26] have shown that ZnO itself is an almost completely CO2 selec-tive catalyst (although with a very low activity) for both MSR andFSR. Friedrich et al. [27] demonstrated that the high CO2 selectivityis obtained by cooperative action of both PdZn and ZnO. ZnO canalso be formed by the oxidation of Zn surface atoms [21].
Our present results show that CO2 selectivity increases with thedecreasing adsorption energies of the reactants, see Fig. 9. It is alsoknown that adsorption energies of reactants decrease with theincreasing coverage [52]. The presence of the ZnO phase as supplyof split water molecules to pre-cover the surface of the catalyst cancontribute to the increased coverage and enhance CO2 selectivity ofthe catalyst. Our results could thus explain the observations

M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18 17
[27,51] suggesting that a higher CO2 selectivity is obtained ina teamwork of PdZn and ZnO phases. On the other side thecalculated activation energy of 0.92 eV for water splitting onPdZn(111) [23] is comparable with that of 0.89 eV for dehydro-genation of methoxyl to formaldehyde [23] which is consideredto be the rate-controlling step of the MSR reaction. MSR can thusproceed with rather high CO2 selectivity also without the ZnOphase as reported in other experimental studies [30,31,46].
CO2 selectivity is higher at reaction conditions with very highcoverages of both reactants. The obvious possibility to reachhigh coverage could be also achieved if the MSR proceeds underhigh pressure.
Very recently we have performed a study of MSR selectivity onthree compounds Ni5Ga3, Pd5Ga3, and Pt5Ga3 (oC16). The com-pounds have a layered atomic structure consisting of two alternat-ing flat atomic planes. We have considered the (001) surfaceformed by the atomic plane with the equal stoichiometric compo-sition of TM and Ga atoms. It is remarkable that the structure ofthis surface can be understood as a laterally deformed B2 (110)surface. Preliminary calculations of MSR selectivity based on com-petition of the R1 and R2 reactions indicate that while the Pd5Ga3
surface is CO2 selective the surfaces of Ni5Ga3 and Pt5Ga3 com-pounds exhibit CO selectivity.
4.6. Comments on used models and methods
Catalysts in the form of nanoparticles expose a much richervariety of active sites than the bulk-terminated (111) surfaces con-sidered as model catalysts in the present work. Nanoparticlesexpose various edge and kink sites with lower-coordinated atoms[53]. New kind of active sites can be formed also on reconstructedsurfaces [54]. It is therefore legitimate to ask whether the resultsfor (111) surfaces are relevant for real catalysts.
Steps, edges and kinks can significantly contribute to the activ-ity of the catalyst. In a recent review [29] activation energies andreaction enthalpies of the steps of MSR on PdZn(111) are com-pared with data for PdZn(100) and the Pd-, resp. Zn-terminatedsurfaces PdZn(221)Pd and PdZn(221)Zn. The activation energiesof some reaction steps on PdZn(221)Pd and PdZn(221)Zn are signif-icantly lower than on PdZn(111). However, from our results it fol-lows that for CO2 selectivity the situation is different. From theobservation that CO2 selectivity is improved with decreasingadsorption energy of formaldehyde (Fig. 9) one can expect that astronger binding of formaldehyde (or hydroxyl) on more reactivesites will make reaction R1 more difficult and hence lower CO2
selectivity. The nanostructured catalysts will thus exhibit higheractivity, but lower CO2 selectivity than it can be expected fromour calculations.
It could also be questioned whether the description of adsorp-tion at the level of the gradient-corrected PBE functional is ade-quate. It is well known that DFT calculations generally tend tooverestimate adsorption energies, but also do not include the effectof dispersion forces. As the adsorption energies of formaldehydeare very weak (see Table 3) it is important to verify whether moreaccurate calculations lead to significant changes in the adsorptionenergies. We have recalculated the adsorption energies offormaldehyde using the DFT-D3 functional (Becke–Jonson) [55]that includes also dispersion forces. On the PdZn(111) surfacethe adsorption energies for all four configurations (see Table 3)are by ��37 kJ/mol more exothermic. The stronger adsorptionhelps to prevent desorption at reaction temperatures. We also notethat DEselect characterizing the selectivity is determined by the dif-ference of the transition-state energies of the R1 and R2 reactionsand the dispersion forces enhance the adsorbate–surface bindingfor all configurations, the influence of the choice of a functionalis at least partially compensated.
5. Conclusions
The origin of MSR selectivity catalyzed by the L10-type inter-metallic compounds NiZn, PdZn, and PtZn, based on the competi-tion between the dehydrogenation of the formaldehydeintermediate (reaction R2) and its reaction with co-adsorbedhydroxyl (reaction R1), has been investigated using DFT.
NiZn(111) is an efficient dehydrogenation catalyst and bindsboth formaldehyde and hydroxyl rather strongly. The high energyrequired to break the bond of the C atom of formaldehyde with thesurface leads to a preference for direct dehydrogenation and to ahigh CO selectivity. On PdZn(111) the adsorption of formaldehydeis weaker, but strong enough to prevent desorption. The reactantremains bound to the surface through the O atom, leading to a con-figuration favoring a reaction with hydroxyl and a high CO2 selec-tivity, similarly as on Cu(111) surfaces. On the L10(111) surfacesthe adsorption properties in threefold hollows are different dueto the shift of the subsurface relative to the surface layer. On thePtZn(111) surface where this shift is largest, on some triplets thebinding of formaldehyde is nearly as strong as on NiZn while onothers it is as weak as on PdZn. Hence some configurations of thereactants show a preference for the R2 reaction, while for othersthe R1 reaction has a lower activation energy. This explains theobserved mixed CO2/CO selectivity of the PtZn catalyst. Our resultsconfirm that formaldehyde is the key intermediate whose reac-tions determine the selectivity of the MSR reaction.
There are two ways to improve the CO2-selectivity of MSR: (i)To make the R1 reaction easier. The MSR reaction requires a gooddehydrogenation catalyst. The R1 reaction is exothermic andnon-activated in the gas-phase, hence in principle no catalyticactivity is required. The surface of the catalyst should bindformaldehyde weakly, just strong enough to prevent desorption.As increasing surface coverage leads to a reduction of the adsorp-tion energy MSR should proceed at high coverages. Selectivitycan be improved if hydroxyl molecules which are easier split fromwater molecules on an oxidic support (such as ZnO) increase thecoverage on the intermetallic surface. Another possible way toachieve the high coverage is to perform the MSR reaction underhigh pressure. (ii) To make the R2 reaction more difficult. Weakadsorption of atomic hydrogen on the catalyst surface contributesto higher selectivity. The (de)hydrogenation activity of the catalystcan be reduced by isolation of the catalytically active transitionmetal atoms by simple metal atoms (Zn, Al, Ga, etc.). It seems thatthis is the way to get a high CO2 selectivity of MSR for Ga–Pd cat-alysts [56–58]. Our preliminary results for the MSR selectivity ofthese compounds show that the Ga3Pd5(001) surface and Gaenrichment of the surface can play an important role. The obviousdisadvantage of this way is that simultaneously with the increasedCO2 selectivity the activity of the catalyst (as measured by themethanol conversion rate) decreases.
Acknowledgments
This work has been supported by the Austrian Ministry forScience through the Center for Computational Materials Science.M.K. thanks also for support from the Grant Agency for Scienceof Slovakia (No. 2/0189/14), from CEX FUN-MAT, and from theSlovak Research and Development Agency (Grants Nos.APVV-0647-10, APVV-0076-11, APVV-0492-11). Part of the calcu-lations were performed in the Computing Centre of the SlovakAcademy of Sciences using the supercomputing infrastructureacquired in project ITMS 26230120002 and 26210120002 (Slovakinfrastructure for high-performance computing) supported by theResearch and Development Operational Programme funded bythe ERDF.

18 M. Krajcí et al. / Journal of Catalysis 330 (2015) 6–18
References
[1] A. Iulianelli, P. Ribeirinha, A. Mendes, A. Basile, Renew. Sustain. Energy Rev. 29(2014) 355.
[2] D.R. Palo, R.A. Dagle, J.D. Holladay, Chem Rev. 107 (2007) 3992.[3] S. Sá, H. Silva, L. Brandão, J.M. Sousa, A. Mendes, Appl. Catal. B: Environ. 99
(2010) 43.[4] N. Iwasa, S. Masuda, N. Takegawa, React. Kinet. Catal. Lett. 55 (1995) 349.[5] N. Iwasa, T. Mayanagi, N. Ogawa, K. Sakata, N. Takegawa, Catal. Lett. 54 (1998)
119.[6] N. Takezawa, N. Iwasa, Catal. Today 36 (1997) 45.[7] N. Iwasa, N. Takezawa, Top. Catal. 22 (2003) 215.[8] N. Iwasa, S. Masuda, N. Ogawa, N. Takegawa, Appl. Catal. 125 (1995) 145.[9] J.R.B. Gomes, J.A.N.F. Gomes, Surf. Sci. 471 (2001) 59.
[10] J. Greeley, M. Mavrikakis, J. Catal. 208 (2002) 291.[11] J.K. Lee, J.B. Ko, D.H. Kim, Appl. Catal. A 278 (2004) 25.[12] Z.-X. Chen, K.M. Neyman, K.H. Lim, N. Rösch, Langmuir 20 (2004) 8068.[13] Z.-X. Chen, K.H. Lim, K.M. Neyman, N. Rösch, Phys. Chem. Chem. Phys. 6 (2004)
4499.[14] N. Iwasa, M. Yoshikawa, W. Nomura, M. Arai, Appl. Catal. A 292 (2005) 215.[15] Z.-X. Chen, K.H. Lim, K.M. Neyman, N. Rösch, J. Phys. Chem. B 109 (2005) 4568.[16] K.H. Lim, Z.-X. Chen, K.M. Neyman, N. Rösch, J. Phys. Chem. B 110 (2006)
14890.[17] B. Frank, F.C. Jentoft, H. Soerijanto, J. Kröhnert, R. Schögl, R. Schomäcker, J.
Catal. 246 (2007) 177.[18] K.M. Neyman, K.H. Lim, Z.-X. Chen, L.V. Moskaleva, A. Bayer, A. Reindl, D.
Borgmann, R. Deneke, H.-P. Steinrück, N. Rösch, Phys. Chem. Chem. Phys. 9(2007) 3470.
[19] D.K. Kim, E. Iglesia, J. Phys. Chem. C 112 (2008) 17235.[20] D.H. Mei, L.J. Xu, G. Henkelman, J. Phys. Chem. C 113 (2009) 4522.[21] C. Rameshan, C. Weilach, W. Stadlmayr, S. Penner, H. Lorenz, M. Hävecker, R.
Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlögl, D. Zemlyanov, N.Memmel, G. Rupprechter, B. Klötzer, J. Catal. 276 (2010) 101.
[22] X.-K. Gu, W.-X. Li, J. Phys. Chem. C 114 (2010) 21539.[23] G.K. Smith, S.W. Lin, W. Lai, A. Datye, D. Xie, H. Guo, Surf. Sci. 605 (2011) 750.[24] S. Lin, D. Xie, H. Guo, J. Phys. Chem. C 115 (2011) 20583.[25] S. Lin, D. Xie, H. Guo, J. Mol. Catal. A 356 (2012) 165.[26] H. Lorenz, M. Friedrich, M. Armbrüster, B. Klötzer, S. Penner, J. Catal. 297
(2013) 151.[27] M. Friedrich, S. Penner, M. Heggen, M. Armbrüster, Angew. Chem. Int. Ed. 52
(2013) 4389.[28] Z.-J. Zuo, L. Wang, P.-D. Han, W. Huang, Int. J. Hydrogen Energy 39 (2014)
1664.
[29] M. Armbrüster, M. Behrens, K. Föttinger, M. Friedrich, E. Gaudry, S.K. Matam,H.R. Sharma, Catal. Rev.: Sci. Eng. 55 (2013) 289.
[30] A.P. Tsai, S. Kameoka, Y. Ishii, J. Phys. Soc. Jpn. 73 (2004) 3270.[31] K. Nozawa, N. Endo, S. Kameoka, A.P. Tsai, Y. Ishii, J. Phys. Soc. Jpn. 80 (2011)
064801.[32] G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169.[33] G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758.[34] P.E. Blöchl, Phys. Rev. B 50 (1994) 17953.[35] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.[36] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 78 (1997) 1396.[37] D. Sheppard, R. Terrel, G. Henkelman, J. Chem. Phys. 128 (2008) 134106.[38] G. Henkelman, H. Jónsson, J. Chem. Phys. 111 (1999) 7010.[39] A. Heyden, A.T. Bell, F.J. Keil, J. Chem. Phys. 123 (2005) 224101.[40] Z.-X. Chen, K.M. Neyman, A.B. Gordienko, N. Rösch, Phys. Rev. B 68 (2003)
075417.[41] M. Friedrich, A. Ormeci, Y. Grin, M. Armbrüster, Z. Anorg. Algg. Chem. 636
(2010) 1735.[42] M. Krajcí, J. Hafner, J. Catal. 295 (2012) 70.[43] M. Krajcí, J. Hafner, J. Chem. Phys. 138 (2013) 124703.[44] M. Krajcí, J. Hafner, J. Catal. 312 (2014) 232.[45] M. Krajcí, J. Hafner, J. Phys. Chem. C 118 (2014) 12285.[46] M. Friedrich, D. Teschner, A. Knop-Gericke, M. Armbrüster, J. Catal. 285 (2012)
41.[47] M. Friedrich, D. Teschner, A. Knop-Gericke, M. Armbrüster, J. Phys. Chem. C 116
(2012) 14930.[48] A. Nilsson, L.G.M. Pettersson, B. Hammer, T. Bligaard, C.H. Christensen, J.K.
Norskov, Catal. Lett. 100 (2005) 111.[49] Q. Ge, M. Neurock, Chem. Phys. Lett. 358 (2002) 377.[50] C.G.P.M. Bernardo, J.A.N.F. Gomes, J. Mol. Struct. (Theochem) 542 (2001) 263.[51] M. Armbrüster, R. Schlögl, Yu. Grin, Sci. Techol. Adv. Mater. 15 (2014) 034803.
17pp.[52] P.A. Sheth, M. Neurock, C.M. Smith, J. Phys. Chem. B 107 (2003) 2009.[53] K.M. Neyman, R. Sahnoun, C. Inntam, S. Hengrasmee, N. Rösch, J. Phys. Chem. B
108 (2004) 5424.[54] C. Weilach, S.M. Kozlov, H.H. Holzapfel, K. Föttinger, K.M. Neyman, G.
Rupprechter, J. Phys. Chem. C 116 (2012) 18768.[55] S. Grimme, S. Ehrlich, L. Goerigk, J. Comput. Chem. 32 (2011) 1456.[56] L. Li, B. Zhang, E. Kunkes, K. Föttinger, M. Armbrüster, D.S. Su, W. Wei, R.
Schlögl, M. Behrens, Chem. Cat. Chem. 4 (2012) 1764.[57] A. Haghofer, K. Föttinger, F. Girgsdies, D. Teschner, A. Knop-Gericke, R. Schlögl,
G. Rupprechter, J. Catal. 286 (2012) 13.[58] A. Ota, E.L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R.M. Navarro
Yerga, M. Behrens, J. Catal. 293 (2012) 27.



![[XLS]ictmumbai.edu.inictmumbai.edu.in/ScrollingFiles/NIRF information for... · Web viewM/s. Indian Oil Corporation Ltd. To study aqueous phase reforming of methanol and bio-oil for](https://img.pdfslide.us/doc/110x75/5ae290167f8b9a0d7d8c8397/xls-information-forweb-viewms-indian-oil-corporation-ltd-to-study-aqueous.jpg)

![Influence of methanol reformate injection strategy on ... · engine fueled with methanol reforming products mainly consisting of H 2 and CO 2. Li et al. [31] as well as Shimada and](https://img.pdfslide.us/doc/110x75/6062eb425f9a6a352b0bc8c9/influence-of-methanol-reformate-injection-strategy-on-engine-fueled-with-methanol.jpg)















![Methanol Steam Reforming Performance Optimisation of ... · were used as optimisation objects by utilising the coupling method and artificial neural networks [23]. Although some research](https://img.pdfslide.us/doc/110x75/5f1acffd2be672440c330099/methanol-steam-reforming-performance-optimisation-of-were-used-as-optimisation.jpg)







