Embed Size (px)
Citation preview

Isolation and characterization of SOSconstitutive mutations in Escherichia coli.
Item Type text; Dissertation-Reproduction (electronic)
Authors Ossanna, Nina.
Publisher The University of Arizona.
Rights Copyright © is held by the author. Digital access to this materialis made possible by the University Libraries, University of Arizona.Further transmission, reproduction or presentation (such aspublic display or performance) of protected items is prohibitedexcept with permission of the author.
Download date 07/07/2018 02:38:28
Link to Item http://hdl.handle.net/10150/184398

INFORMATION TO USERS
The most advanced technology has been used to photograph and reproduce this manuscript from the microfilm master. UMI films the original text directly from the copy submitted. Thus, some dissertation copies are in typewriter face, while others may be from a computer printer.
In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyrighted material had to be removed, a note will indicate the deletion.
Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. Each oversize page is available as one exposure on a standard 35 mm slide or as a 17" x 23" black and white photographic print for an additional charge.
Photographs included in the original manuscript have been reproduced xerographically in this copy. 35 mm slides or 6" x 9" black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order.
I' ,U·M·I Accessing the World's Information since 1938
300 North Zeeb Road, Ann Arbor, M148106-1346 USA


Order Number 8814266
Isolation and characterization of SOS constitutive mutations in Escherichia coli
Ossanna, Nina, Ph.D.
The University of Arizona, 1988
U·M·I 300 N. Zeeb Rd. Ann Arbor, MI 48106

ISOLATION AND CHARACTERIZATION OF SOS CONSTITUTIVE
MUTATIONS IN ESCHERICHIA COLI
by
Nina Ossanna
A Dissertation Submitted to the Faculty of the
DEPARTMENT OF MOLECULAR AND CELLULAR BIOLOGY
In Partial Fulfillment of the Requirements For the Degree of
DOCTOR OF PHILOSOPHY WITH A MAJOR IN MOLECULAR BIOLOGY
In the Graduate College
THE 'UNIVERSITY OF ARIZONA
1 988
1

THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE
2
As members of the Final Examination Committee, we certify that we have read
the dissertation prepared by ---------------------------------------------Nina Ossanna
entitled Isolation and Characterization of SOS Constitutive -------------------------------------------------------------Mutations in Escherichia coli
and recommend that it be accepted as fulfilling the dissertation requirement
for the Degree of __ ~Ph~.D~. __ ~(~M_o~l~e_c_u_la_r __ B_i_o_l_o~g~y~) ________________________ _
Date
))f. John Little Date I I 17, /If??
Date
Date Dr. Marty
O~ Date Dr. Danny Brower
Final approval and acceptance of this dissertation is contingent upon the candidate's submission of the final copy of the dissertation to the Graduate College.
I hereby certify that I have read this dissertation prepared under my direction and r~commend that it be accepted as fulfilling the dissertation requirement.
Dissertation Director Date

3
STATEMENT BY AUTHOR
This dissertation has been submitted in partial fulfillment of requirements for an advanced degree at The University of Arizona and is deposited in the University Library to be made available to borrowers under rules of the Library.
Brief quotations from this dissertation are allowable without special permission, provided that accurate acknowledgment of source is made. Requests for permission for extended quotation from or reproduction of this manuscript in whole or in part may be granted by the head of the major department or the Dean of the Graduate College when in his or her judgment the proposed use of the material is in the interests of scholarship. In all other instances, however, permission must be obtained from the author.

4
AKNOWLEDGEMENTS
I would like to thank my committee members for their help during
this project and particularly for their assistance in writing this
dissertation. My committee included Dr. Danny Brower, Dr. Marty
Hewlett, Dr. June Ito and Dr. John Little.
I would like to thank my dissertation director, Dr. David Mount,
for the opportunity to work in his lab and his guidance to help me
become (in his words) a "true geneticist".
I have been fortunate during my time in David's lab to work with
fine people. The work contained in this dissertation would not have
been possible without their moral and scientific support, and I am
grateful. Particularly I wish to thank Mark Dubnick, Belva Fisher,
Nancy Istock, Ken Peterson, Andy Thliveris and Ken Wertman.
I would like to thank Cindy Parrill (fellow tech-nerd) for the
computer-generated graphics in this dissertation. My typist gets no
thanks--she could have done a better job.
Friends and family have helped enormously and I am grateful.
I am going to miss Tuesday lunches at McDonald's with Val. Particularly
I want to thank Anne Ryan for her support and encouragement throughout
this entire Ph.D. project. Also, Marcy, Phoebe and Willa for their
constant companionship even when there wasn't a milk bone in it for
them.

LIST OF ILLUSTRATIONS
LIST OF TABLES
ABSTRACT
1. INTRODUCTION
TABLE OF CONTENTS
..
Escherichia coli SOS response
The SOS inducing signal
Role of RecA protein
Inducing signals for RecA activation
Objective and rationale of this study
2. MATERIALS AND METHODS
Media ..
Materials
Plasmids, bacteriophage and strains
Strain used in mutant isolation
MNNG mutagenesis
STS test
Mapping mutant alleles
B-galactosidase assays
Spontaneous mutagenesis
UV survival assays
Recombination proficiency
5
page
8
9
11
12
12
13
14
15
19
21
21
21
22
29
29
30
31
32
32
33
33

6
TABLE OF CONTENTS--continued
3. RESULTS . . . . . . . . . . . . . . . . . . . . . . . . . . 34
4.
Results of mutagenesis . . . . . . . . . . . . . lOa mutant (lig252) . . . . . . . . . . . . . . . . . .
Mapping · . . . . . . . . . . . . . . . . . . . . SOS expression . . . . . . . . . . . . . . . . . . 1ig mutant phenotypes . . . . . . . . . . . . . .
9a mutant • • · . . . . . . . . . . . . . · . . . . . . 3e. 41 and 8a mutants (uvrD242. 243. 244) · . . . . . .
Mapping mutations in 3e, 41 and 8a · . . . . . . UV sensitivity of uvrD mutants . . . . . . . . . SOS expression in uvrD mutants . . . . . . . . . Spontaneous mutagenesis in uvrD mutants . . . . . uvrD spontaneous mutagenesis when SOS
induction is blocked •••••• . . . . uvrD mutagenesis in mismatch repair deficient
strains •• •• • • • • • • • • •
· . · .
Viability of uvrD244 rep5 double mutants . . . . . recA mutants · . . . . . . . . . . . . . . . . . . . .
SOS expression . . . . . . . . . . . . . . . . .
34
35
36
37
37
39
41
41
43
45
48
51
53
55
56
56
recA mutant recombination proficiency ••• • •• 56
Spontaneous mutagenesis in ~mutants •• 56
Viable and inviable mutations with recA81 and recAB 5 • • • • • • • • • • • • • • • • • • · . 60
DISCUSSION • • . . . . . . . . . . . . . . . . . . . . . . . 62
Isolation of SOS constitutive mutations . . . . . . . . 62
lig252 mutant • • • • • • • • • • • • • • • • • • • •• 66

7 .' r
TABLE OF CONTENTS--continued
dam17 mutant • • • • 67
uvrD mutants • • • • • • 69
~ mutants • • • • • • • 73
7. APPENDIX A Definitions of genetic terms • • • 76
6. REFERENCES . • • • · 77

LIST OF ILLUSTRATIONS
Figure
1. B-ga1actosidase expression from su1A::MuQ fusion in lig and dam strains . . . . . .
2. UV sensitivity of ~ mutants
3. B-galactosid~se expression from sulA::MuQ fusion in uvrD mutants . . . . . . . . . . .
4. Mitomycin C induced B-galactosidase expression from
8
page
38
44
46
sulA::MuQ fusion in uvrD mutants 49
5. Spontaneous mutagenesis in uvrD strains 50
6. B-galactosidase levels from sulA::MuQ fusion in mutant recA strains .... . . . . . 57
7. Spontaneous mutagenesis levels in recA mutants 59

LIST OF TABLES
Table
1. Bacteria, phage and plasmids used in this study
2. PI transductional mapping of mutant 10a(lig252)
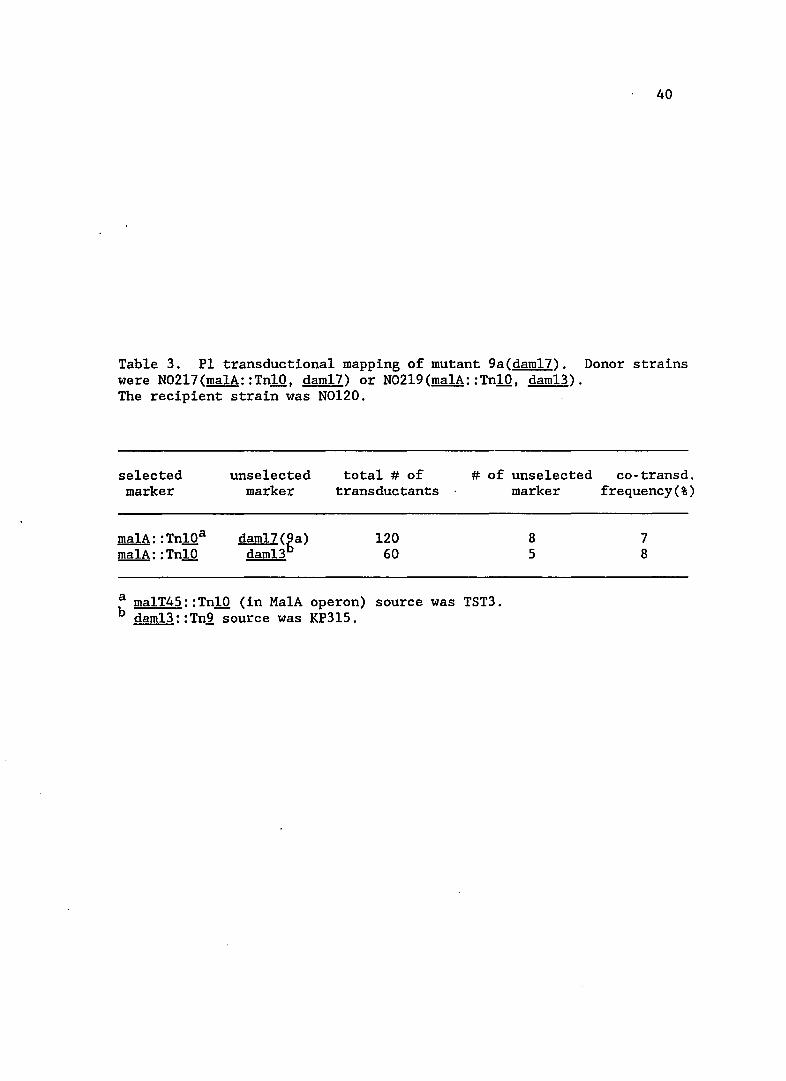
3. PI transductional mapping of mutant 9a(daml7) .
4. Results of three factor crosses to map uvrD alleles
5. B-galactosidase expression from sulA::MuQ fusion in previously isolated mutants . . . . . . . . . . . .
6. Spontaneous mutagenesis in uvrD244 strain when SOS response blocked . . . . . . . . . . . . . .
7. Spontaneous mutagensis in uvrD strains when mismatch repair blocked . . . . . . . . . . . . . . . . .
8. Recombination proficiency in mutant recA strains
9
page
23
36
40
42
47
52
54
58

10
ABSTRACT
Early events occuring during induction of the SOS response in
Escherichia coli are poorly understood. In order to understand the
early steps in SOS induction more fully, we have isolated several
mutations which constitutively express the SOS regulon.
Using a Mug(ApR,lac) fusion to the SOS regulated gene sulA, we
isolated Lac+ colonies as mutants in which RecA protein is
constitutively activated for repressor cleavage. The mutations map to
four loci: darn, lig, uvrD and recA. The extent of constitutive SOS
induction in these mutants varied greatly, indicating different levels
or types of signal in the cell.
The mutations isolated demonstrate two early steps in SOS
induction. The first step in SOS induction is signal generation and
includes mutations found in darn, lig and uvrD genes. The. mutant gene
products presumably alter DNA metabolism to produce an inducing signal.
These non-lethal mutations lead to sub-induction and probably generate
very specific signals, such as abnormally unwound DNA in the case of DNA
helicase II mutants or unsealed DNA nicks that result from deficient
ligation in l!g mutants. Greater induction may require quantitatively
more signal or different types of signal generated by severe defects
leading to cell death. These mutations also show that signal is a
variable quantity, allowing the cell to fine tune the levels of SOS
repair activity according to the amount or type of signal (damage)
perceived. In some cases (such as darn mutations), blocking the SOS
response by lexA(Ind-) alleles leads to cell death. In this type of
constitutively activated strain, the increased level of repair from SOS

11
induction is required to allow the cell to tolerate potentially lethal
DNA structures generated by the mutant gene product. The second step in
induction is the interaction of signal with RecA protein and is shown by
isolating 8 recA mutants. Mutant recA alleles caused the strongest SOS
induction of any mutants obtained, similar to the level found in strains
lacking repressor (lexA(Def) mutants). This full induction in the
absence of lethal DNA damage underscores the pivotal role of RecA
protein in regulating the SOS response.

12
CHAPTER 1
INTRODUCTION
Escherichia coli SOS response
The SOS response is normally induced when Escherichia coli
suffers DNA damage from physical agents such as ultra-violet (UV) light
or mutagenic chemicals such as Mitomycin C (Witkin, 1976). Induction of
SOS functions results in a diverse set of responses such as an increased
capacity for DNA repair, enhanced mutagenesis (SOS mutagenesis),
inhibition of cell division, cessation of respiration, alleviation of
host-controlled restriction, induction of stable DNA replication and
prophage induction (Little and Mount, 1982).
Many of the approximately twenty genes induced during the SOS
response that participate in repair phenomena have been identified. The
major repair pathways induced are excision repair and RecF
recombinational repair (also known as daughter-strand gap repair,
Peterson et al., 1987). Excision repair requires the SOS-regulated
uvrA, uvrB, uvrC (whether uvrC is SOS regulated is unclear) and uvrD
gene products. Inducible RecF recombinational repair requires the
products of the SOS regulated recA, recN, recO, uvrD and ~ genes, as
well as recF (not SOS regulated), and recO and recJ whose regulation is
unknown. Inhibition of cell division, or filamentation, requires the
SOS regulated sulA gene. SOS mutagenesis requires the products of the
recA and umuC.D genes. Induction of prophage requires the recA gene

13
product. Induction of stable DNA replication (replication in the
absence of protein synthesis) requires a functional RecA protein (Witkin
and Kogoma, 1984; Kogoma et a1., 1985). Other genes induced by the SOS
response, called din (for damage inducible) genes, have been identified
but their functions are unknown (Kenyon and Walker, 1980). For
additional reviews concerning the SOS response see Walker (1984) and
Ossanna et a1. (1986).
At the molecular level, the SOS response is regulated by the
gene products of the recA and lexA genes. When DNA damage occurs, an
inducing signal is produced that alters RecA protein to an activated
form. This activated RecA promotes cleavage of LexA protein, a
repressor of the various genes of the SOS regu1on. Cleaved LexA protein
is unable to act as a repressor and as a result, the genes of the SOS
regu10n are expressed, enhancing the ability of the cell to recover from
DNA damage.
The SOS induction process is reversible. As DNA damage is
repaired the level of inducing signal drops and RecA protein is no
longer activated. LexA repressor can again accumulate and repression of
the SOS genes is reestablished. The reversibility of SOS induction is
crucial to cell survival since one SOS protein, SulA, causes the cell to
lethally filament if continually expressed (George et a1., 1975).
The SOS inducing signal
The nature of signal(s) and cellular alterations that generate
inducing signal(s) to activate RecA protein after DNA damage is unclear.
Many suggestions have been made, and all involve aspects of DNA

14
metabolism or structure. Biochemical studies (discussed below) suggest
that a possible inducing signal is single-stranded regions of DNA.
These regions may arise from blocks in DNA synthesis due to damage, or
gaps in double-stranded DNA opposite damaged regions (Rupp and Howard-'
Flanders, 1968; Roberts and Devoret, 1982). DNA degradation products,
such as oligonucleotides, resulting from DNase action on damaged DNA
have also been suggested as inducing signals (Smith and Oishi, 1978).
Alternatively, induction may result from alterations in the nucleotide
pools after DNA damage occurs (Das and Loeb, 1984), changes in
superhe1icity of DNA (Little and Mount, 1982), or a combination of any
of the above.
Early events in the cell that generate signal may not give rise
to the same types of signal. Genetic evidence supports different types
of signal generation that may be dependent upon the nature of the
cellular damage. Cells with mutations in recB or recC (Exonuclease V)
are not inducible with nalidixic acid, but are inducible by UV
irradiation. Strains deficient in recF are poorly inducible by UV light
and show normal induction when treated with nalidixic acid (McPartland
et a1., 1980; Little and Hanawalt, 1977).
Role of RecA protein
Extensive genetic evidence exists to support a central role for
RecA protein in SOS regulation as the target for the inducing signal.
The recA430 mutation drastically reduces the ability of RecA protein to
participate in LexA repressor cleavage without affecting its
recombinationa1 properties (Morand et a1., 1977). Another recA allele,

15
recA441 (formerly tif-1), makes a protein which is constitutively
activated when grown at 41°C in the absence of inducing treatment. The
presence of adenine further stimulates activation of RecA441 protein,
while a mixture of cytidine and guanosine inhibit its activity (George
et a1., 1975). Many RecA mutations, termed PrtC , have now been isolated
which show constitutive SOS induction at all temperatures of growth
(Witkin et a1., 1982; Wang and Tessman, 1986). Additionally, deletions
or null alleles of recA prevent induction of any SOS functions. These
observations indicate a central role for RecA protein as a positive
regulator of the SOS response.
As well as being the target for inducing signal(s), it is clear
that RecA protein must be altered to an active form in order to induce
the SOS response. Mutations that result in the overproduction of RecA
protein (recAoc , operator-constitutive mutations) do not lead to SOS
induction (Quillardet et al., 1982). Conversely, cleavage of LexA
repressor can take place if RecA protein is activated but amplification
does not occur (Little, 1983; Baluch et a1., 1980).
Further support for the regulatory role of RecA protein in SOS
induction comes from biochemical studies. The earliest measured event
after UV irradiation is the appearance of LexA protein cleavage
fragments. These fragments appear 1-2 minutes after irradiation and are
dependent upon RecA protein function (Little, 1983). It is believed
that signal interacts directly with RecA protein to effect activation,
leading to LexA cleavage.

16
Inducing signals for RecA activation
Biochemical studies have provided insight into the molecules
that interact with RecA protein and may be involved in activation
during SOS induction. The proteolytic function of RecA protein was
demonstrated in vitro by Craig and Roberts (1980). RecA protein was
shown to cleave lambda £1 repressor in a reaction requiring ATP and a
polynucleotide. Further studies showed that ATP1S (a weakly-hydrolyzed
ATP analog) satisfied the ATP requirement, indicating that hydrolysis of
the nucleotide was not necessary for cleavage activity, and that a
ternary complex of RecA protein, DNA and ATP was active for cleavage of
£1 repressor (Craig and Roberts, 1981). A comparison of RecA protein
and the more easily activated RecA441 protein demonstrated that the
RecA441 protein interacts with DNA and ATP more efficiently than wi1d
type RecA protein (Phizicky and Roberts, 1981; McEntee and Weinstock,
1981). RecA protein was also shown to cleave purified LexA protein in
vitro, with requirements similar to £1 repressor of ATP and DNA for
activation (Little et a1., 1980; Horii et a1., 1981). These studies
suggest that a nucleotide cofactor and DNA may be important for
activation of RecA protein in inducing the SOS response.
An approach utilized to study factors effecting RecA protein
activation in vivo was to supply an inducing signal exogenously to
permeabi1ized cells (Irbe et a1., 1981). In this system, prophage ~80
induction was followed in the presence of different oligonucleotides
that mimick products of DNA degradation. The study was designed to test
the idea that recBC Dnase action produces oligonucleotides and is
responsible for generating the SOS inducing signal (Smith and Oishi,

17
1978). Induction occured in the presence of d(A-G), d(G-G), d(A-G-G)
and d(pG)8' while other similar molecules did not lead to ~80 induction.
While these studies indicated that some oligonucleotides may be signals
for SOS induction, the significance of the oligonucleotide specificity
is unclear.
Indirect induction results when UV irradiated replicons are
introduced into unirradiated cells. These types of experiments have
shed light on the relationship between replication of damaged DNA and
SOS induction. Introducing UV irradiated replicons such as F or F'
factors into cells induced SOS functions (George et al., 1974), as did
infection with irradiated Pl, lambda or M13 phages (D'Ari and Huisman,
1982). Irradiated lambda or Pl phage unable to replicate were
introduced into ~ coli. The ability of these non-replicating phage to
induce SOS functions was reduced, but not abolished, leading the authors
to suggest that two pathways or two signals exist to induce SOS
functions (D'Ari and Huisman, 1982). A possible signal for activation
in non-replicating phage may be the damaged DNA template. Lu et al.
(1986) found that RecA protein was better activated for LexA cleavage in
vitro by UV irradiated double-stranded DNA than unirradiated double
stranded DNA.
Certain mutations are also capable of generating an SOS inducing
signal leading to prophage induction. These mutations lie in genes
whose products are involved in replication of DNA and provide insight
into what perturbations of DNA synthesis induce SOS functions. In
strains with a temperature sensitive DNA ligase activity, prophage
induction occurred when cells were grown at the non-permissive

18
temperature (Gottesman et al., 1973). Strains with temperature
sensitive alleles of two DNA polymerase genes, dnaE (e subunit of
polymerase III) and polA (polymerase I), induced prophage when grown at
non-permissive temperatures (Schuster et al., 1973; Blanco, and Pomes,
1977).
Casaregola et al. (1982) surveyed the effect of several dna
mutants on the induction of a recA::lacZ fusion and found that not all
mutations affecting DNA replication are capable of inducing the SOS
response. They showed that strains with a temperature sensitive allele
of dnaB (involved in initiation and elongation during DNA synthesis)
induced SOS functions when grown at the non-permissive temperature
(dnaB(Ts) strains also induced prophage, Schuster et al., 1973).
Strains with a dnaG(Ts) allele, which is the RNA primase for lagging
strand DNA synthesis, did not induce the ~::lac fusion when grown at
the non-permissive temperature (but induced prophage, see Schuster et
al., 1973). This mutation did increase the basal level of recA
expression at all temperatures, however. Strains with dnaA and dnaC
alleles, which are required for initiation of new rounds of DNA
replication, did not induce recA::lacZ when grown at elevated
temperature. Mitomycin C was capable of inducing these strains during
the residual replication occuring after a temperature shift, but when
this replication ceased the dnaC strain was no longer inducible.
Another gene involved in replication, ssb (single-stranded binding
protein), does not induce prophage when mutants are UV irradiated at the
non-permissive temperature (Baluch et al., 1980a).
Several features about the induction of the SOS response emerge

19
from the studies presented. The SOS system is induced in response to
DNA damage or perturbations of DNA replication. During induction, a
signal (or signals) is generated, possibly by different pathways. The
signal(s) interacts with RecA protein, altering it to an activated form.
This activated form of RecA protein is required for the in vivo cleavage
of LexA repressor, resulting in SOS induction. What actually
constitutes the signal(s) that interacts with RecA protein in vivo, how
it is generated and its relationship to DNA replication is not clear.
Objective and rationale of this study
The work undertaken for this dissertation was designed to
provide additional information about the early events surrounding
induction of the SOS response in ~ coli. I have taken a genetic
approach to this problem by isolating mutants in which RecA protein is
constitutively activated for the SOS response. These mutants are
altered in functions that are associated with generating signal during
induction of the SOS response.
Using a screen described below, I isolated and characterized
mutants in which an SOS-regulated gene is derepressed without ~ priori
knowledge of specific genes that may be involved in SOS induction.
These mutants constitutively expressed SOS functions in the absence of
an inducing treatment. Unlike previously identified SOS inducing
mutants (except for those in the known SOS regulatory genes recA and
lexA) , these were viable at all temperatures, causing induction under
non-lethal conditions. Because these mutants induced the SOS response
under non-lethal conditions, I anticipated that the mutations would have

20
more specific defects leading to SOS induction with fewer secondary or
unforseeable effects (such as would be expected in conditionally lethal
mutants that stall DNA replication).
In order to identify SOS constitutive mutants, I utilized a MUQ
fusion to the SOS regulated sulA gene. The MUQ fusion placed
promoterless Lac structural genes (lacZYA) under control of the sulA
promoter. This fusion was useful for a number of reasons. First, since
the sulA gene product is responsible for lethal filamentation, I needed
a sulA(Null) mutation so that SOS inducing mutants isolated would not
be lethal. Second, the sulA gene has one of the highest induction
ratios of the SOS regulated genes. This provided me with a large window
of sulA expression levels. Third, this operon fusion made it possible
to easily follow SOS induction by scoring Lac phenotype on MacConkey
lactose plates. Strains induced for the SOS response had a Lac+
phenotype (red colonies), while uninduced strains were Lac- (white
colonies). An additional advantage of this fusion was the ability to
quantitate expression of the sulA fusion colorimetrically by B
galactosidase assays.

21
CHAPTER 2
MATERIALS AND METHODS
A glossary of genetic terms and definitions is in the Appendix.
Media
Strains were routinely grown in L broth containing 10 g Bacto
tryptone (Difco), 5 g yeast extract (Difco) and 10 g NaCl per liter.
Solid media also contained 15 g Difco agar. Cells to be infected with
lambda were grown in Lambda broth, which contained 10 g tryptone (BBL) ,
5 g NaCl and' 2 g maltose per liter. Lambda plates also contained 10 g
Difco agar per liter. lXA minimal medium for B-galactosidase assays was
prepared according to Miller (1972) and supplemented with 0.5% casamino
acids (Difco) for strains with auxotrophic markers. LC media for PI
transductions was L media supplemented with 25 mM CaC12 , 0.005%
thymidine and 0.2% glucose. MacConkey agar (Difco) was prepared
according to the manufacturer's directions. Antibiotics, when required,
were used in the following concentrations: 20 ug/ml chloramphenicol, 25
ug/ml tetracycline, 80 ug/ml kanamycin, 100 ug/ml streptomycin, 300
ug/ml 2-aminopurine, 100 ug/ml ampicillin for plasmid selection or 25
ug/ml ampicillin for chromosomal markers and 100 ug/ml spectinomycin.
Unless otherwise specified Mitomycin C was used at 0.25 ug/ml.
Materials
N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) and o-nitrophenyl-B
D-galactopyranoside (ONPG) were purchased from Sigma Chemical.

22
Plasmids, Bacteriophage and Strains
Table 1 lists plasmids,oscteriophage and ~ coli strains used
in this work. All bacterial genetic nomenclature and map positions for
alleles are in accordance with the current version of Bachmann's ~ coli
genetic map (Bachmann, 1983). Strains were constructed by standard
Plvir transductions (Miller, 1972). Transductants were selected for on
the appropriate antibiotic media, which also contained 5 glliter sodium
citrate, or minimal media (plus citrate) when selecting an amino acid
marker. Selection for TnlO excision was by the Bochner method (Davis et
al., 1980), while precise excisions (TnlO and Tn2) were selected for on
the appropriate minimal medium or screened for on MacConkey indicator
media. Strains bearing MUQ(Amp,Lac) gene fusions were temperature
stabilized to the XCam derivative (Baker et al., 1983). The MuQXCam
phage contained a Tn2 insertion in the Mu h gene which prevents
replication (and therefore transposition). This stabilized the
MUQ(Amp,Lac) fusion by allowing growth at elevated temperatures. The
XCam conversion also conferred chloramphenicol resistance on the strain.
Backcrosses, as noted in the strain list (Table 1), were done to
move transposon-linked mutant alleles into unmutagenized backgrounds. A
transposon (TnlO with a known map location) was crossed into the mutant
strains by PI transduction. If the transposon was closely linked to the
mutant allele, the allele would be crossed out by the wild-type gene and
the phenotype change to Lac- according to the co-transduction frequency.
An isolate that was still Lac+ was saved and used as a PI donor to cross
the transposon-linked mutant allele into a wild-type parent, either N056
or N0120, again saving a Lac+ transductant.

23
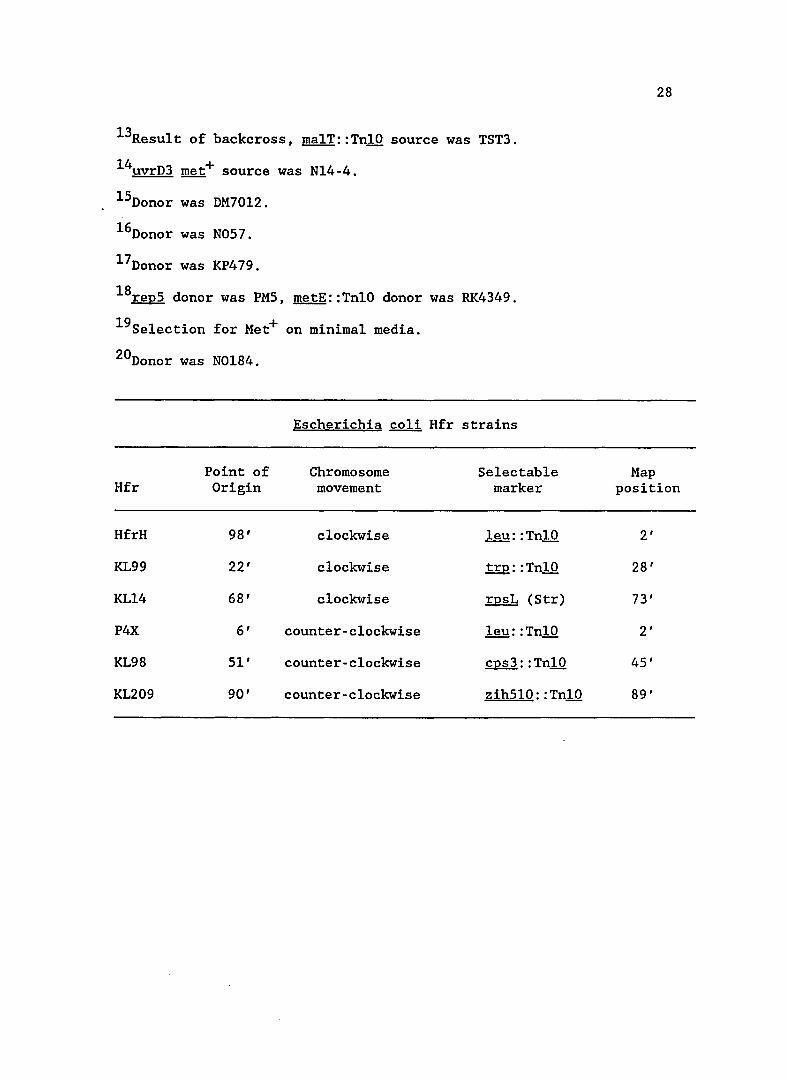
Table 1. Bacteria, phage and p1asmids used in this study
Plasmid
pNS-1
pVMK35
Phage
A38
A103
A120
Alnd-
AClear
Strain
594
DE309
Markers
Markers
bl453 nin5
i 21 cI- biol Q73 S7
i 434 cI- bio10 029
wild type phage
cI(Ind-)
cI(Inds )
P1asmids
Phage
Source
Samuel Sch1agman U of Rochester
Sidney Kushner U of Georgia, Athens
Source
This work
Don Ennis
Don Ennis
Don Ennis
D. Mount lab
D. Mount lab
D. Mount lab
D. Mount lab
Escherichia coli K-l2 strains
Genetic markers
ssb-l, ma1E::TnlO
Source or reference
D. Mount lab
Don Ennis

Strain
DE310
DM1901
DM7012
JIA07
KP281
KP315
KP419
KP479
KW88
N14-4
N2668
N056
N057
N060
N061
N062
N063
N064
N073
N074
N075
Table 1., continued
Genetic markers
ssb-113, rna1E::TnlO
HfrH lacU169 re1A1 thi-l spr55 malB: :Tn.2. sulA: : MUQ(Ap ,lac) (Mu)
lexA3 zja505::TnI0 malB45
srl- 300: : TnlO
as N0120 but 1exA71::Tn~
dam13 : : Tn.2. cysG: : Tn~
recB268: :TnlO
mutL: :TnlO
thil pyrD34 his68 trp45 mtl2 xyl7 ma1Al galk35 strAl18 recB21 argA81::TnlO
uvrD3
ligts-7
HfrH(?) lacU169 re1Al thi-l cps3 malF59::Tn~ su1A::MuQX(Cam) (Mu)
as N056 but de1(sr1R-recA)306::Tn10
as N056 but srl-300::TnI0 recA80(2a)
as N056 but srl-300::TnI0 recA81(2b)
as N056 but srl-300::TnlO recA730
as N056 but srl-300::TnlO recA441
as N056 but Xi434cI(Ind-)lexA+
as N056 but srl-300::TnI0 recA82(lf)
as N056 but srl-300::TnI0 recA83(8g)
as N056 but srl-300::TnI0 recA84(6i)
Source or reference
Don Ennis
(Huisman and D'Ari, 1981)
D. Mount lab
John Little
24
Kenneth Peterson
Kenneth Peterson
Kenneth Peterson
Kenneth Peterson
Kenneth Wertman
D. Mount lab
Martin Gellert
This work
This work
This work1
This workl
This work
This work
This work
This workl
This workl
This workl

25
Table 1., continued
Strain Genetic markers Source or reference
N076 as NOs6 but srl-300::TnlO recA8s(7f) This workl
N077 as NOs6 but srl-300::TnlO recA86(6j) This workl
N078 as NOs6 but srl-300::TnlO recA87(lh) This workl
NOll2 as N2668 but z£b::TnlO (ligts7) This workS
NOl20 as NOs6 but mal+ (Kans ) This work
NOl2l as N076 but TetS (recA8s) This work2
NOl22 as N06l but TetS (recA8l) This work2
NOl23 as NOl20 but ssb-l, malE::TnlO This work3
NOl24 as NOl20 but ssbll3, malE:: TnlO This work4
NOl34 as NOl23 but TetS (ssb-l) This work2
NOl3s as NOl24 but TetS (ssbll3) This work2
NOl37 as NOl20 but lig2s2(lOa) z£b: : TnlO This workS
NOl38 as NOl20 but z£b::TnlO This workS
NOl39 as NOl20 but uvrD244(8a) metE: :TnlO This work6
NOl40 as NOl20 but metE::TnlO This work6
NOls7 as NOl20 but lig2s2(lOa) nupC: : TnlO This work7
NOls8 as NOl20 but nupC::TnlO This work7
NOl62 as NOl20 but ligts-7 nupC::TnlO This work8
NOl73 as NOl20 but uvrD244(8a) fad7s1::TnlO This work9
NOl84. as NOl39 but met+ TetS (uvrD244) This work1O
NOl90 as NOl20 but dam13:: Tn.2, cysG:: Tn,2. This workll
NOl97 as NOl20 but uvrD242(3e) metE::TnlO This work6

26
Table 1., continued
Strain Genetic markers Source or reference
N0198 as N0120 but uvrD243(41) metE: :TnlO This work6
N0199 as N0120 but metE- Tets This work12
N0217 as N0120 but daml7(9a) malT::TnlO This work13
N0219 as N0120 but daml3: : Tn2 malT: : TnlO This work
N0220 as N0120 but malT: :TnlO This work13
N0221 as N0140 but uvrD3 met+ This work14
N0222 as N0140 but met+ This work14
N0223 N0139/pVMK35 (uvrD244/uvrD+) Transformation
N0224 N0140/pVMK35 (uvro+;uvrD+) Transformation
N0225 N0197/pVMK35 (uvrD242/uvrD+) Transformation
N0226 N0198/pVMK35 (uvrD243/uvrD+) Transformation
N0230 as N0137 but Tets (lig252) This work2
N0248 as N0184 but lexA3 zjaSOS::TnlO malB45 This work15
(uvrD244)
N0249 as N0184 but del(srlR-recA)306: :TnlO This work16
(uvrD244)
N0250 as N0197 but met+ Tets (uvrD242) This work10
N0251 as N0198 but met+ Tets (uvrD243) This work10
N0266 as N0120 but mutL: :TnlO This work17
N0267 as N0184 but mutL: :TnlO (uvrD244) This work17
N0268 as N0250 but mutL: :TnlO (uvrD242) This work17
N0269 as N0251 but mutL: :TnlO (uvrD243) This work17
N0276 as N0120 but repS metE: : TnlO This work18

Table 1., continued
Strain Genetic markers
N0278 as N0276 but rnet+ (repS)
N0280
Source or reference
This work19
This work20
27
N0282
as N0276 but uvrD244 (repS)
N0232/pNS-1 (dam17/dam+) Transformation
PMS
RK4349
RS3087
S~1024
YMC
repS re1Al spoT1 thil
metE163::Tn10 pro3 entA403 his2l8 metB1 xy1 rpsL109 de1(lac)6 supE44
fad-7Sl::TnlO relAl spoTl thil HfrH
HfrH relAl rnetC69 spoTl thi-l nupCS10: : TnlO
supFS8
Barbara Bachmann
Barbara Bachmann
Barbara Bachmann
Barbara Bachmann
D. Mount lab
1Resu1t of backcross (described in text), srl-300::TnlO source was JL407.
2TnlO excision by Bochner selection.
3ssb- l source DE309.
4ssbl13 sourc~ DE3l0.
SResult of backcross, zfb::TnlO generated in this study.
6Resu1t of backcross, metE: : TnlO source was RK4349.
7Resu1t of backcross, nupC: : TnlO source was S~1024.
8ligts-7 source was N2668, nupC::TnlO source was S~1024.
9Resu1t of backcross, fad7Sl::tnlO source was RS3087.
10Se1ection for Met+ on minimal media.
11Donor was KP31S.
12Bochner selection of N0140.
l3Result of backcross, malT::TnlO source was TST3.
l4uvrD3 met+ source was N14-4.
l5Donor was DM70l2.
l6Donor was N057.
l7Donor was KP479.
l8rep5 donor was PM5, metE::TnlO donor was RK4349.
19Selection for Met+ on minimal media.
20Donor was N0184.
Escherichia coli Hfr strains
Point of Chromosome Selectable Hfr Origin movement marker
HfrH 98' clockwise leu: :TnlO
KL99 22' clockwise trp: : TnlO
KL14 68' clockwise rpsL (Str)
P4X 6' counter-clockwise leu: : TnlO
KL98 51' counter-clockwise cps3: :TnlO
KL209 90' counter-clockwise zih5l0: :TnlO
28
Map position
2'
28'
73'
2'
45'
89'

29
Strain used in mutant isolation
The su1A::Mug fusion was constructed by Huisman and D'Ari (1981)
using the Mug(Lac,Amp) phage of Casadaban and Cohen (1979). This
strain, DM1901, was made 1exA+ via P1 transduction. The mucoid
phenotype of the original strain made it very difficult to lysogenize or
plate lambda phage; therefore I introduced a cps3::Tn10 allele which
relieved the mucoid phenotype. The Tn10 was removed by Bochner
selection (described above) to render the strain TetS and allow further
use of Tn10 in mutation mapping. The Mug fusion was temperature
stabilized as described above using MugxCam conversion, resulting in the
strain N056. Since the inactivation of 1exA was a likely mutation to
arise and result in derepression of su1A, a 1exA diploid of N056 (N064)
was constructed by lysogenizing N056 with ~i434cI(Ind-)lexA+. Using
the heteroimmune i 434 phage still allowed the use of ~ in further
screenings.
MNNG Mutagenesis
Mutagenesis by MNNG treatment was similar to previously
described methods (Adelberg et a1., 1965). N064 was grown to mid-log
phase and resuspended in 0.1 M sodium citrate (pH 5.5). MNNG, dissolved
in 0.1 M citrate, was added to the cells at a final concentration of 100
ug/m1. The culture was incubated for 20 minutes at 37oC. The
mutagenized culture was diluted 1 to 100 in L broth, dispensed into 10
culture tubes (to isolate independent mutants) and grown overnight at
32oC. Cells were spread onto MacConkey lactose plates to screen for
Lac+ colonies.

30
The Lac+ mutants to be studied further were ~creened for two
types of interfering mutations. Although the strain was a lexA diploid,
I checked for the unlikely possibility that a lexA (Null) mutation was
responsible for Lac expression. Lysogens of the mutants were made with
~lexA+ phage, an allele dominant to lexA (Null) mutations. If the
isolate became white on MacConkey lactose plates (Lac-), the mutation
may have been in lexA and not interesting to this study. The other type
of mutants to be screened out were sulA operator mutants or Mug(Ap,Lac)
phage transpositions. To detect this type of mutation, mutant strains
were lysogenized with ~lexA3 phage. This phage provided a non-cleavable
repressor to check that the sulA::Mug fusion was still repressable by
LexA. In this screen, white (Lac-) lysogens on MacConkey lactose agar
indicated an intact sulA::Mug fusion and were kept for further study.
STS Test
The STS test was a rapid screen to determine which mutants were
capable of constitutively inactivating £1 repressor (Mount, 1979).
Since £1 repressor is more resistant to cleavage than LexA, this enabled
me to determine which mutants were most strongly induced for the SOS
response. The mutant colonies were patched onto maltose-containing L
plates and incubated for four hours (until patch growth was barely
perceptible). These patch plates were replica plated sequentially onto
maltose-containing Lambda plates spread with ~cI(Ind-), ~cI(Ind+),
~cI(Inds) and finally ~cI-(Clear). This order of replica plating was
important so that phage with repressors less susceptible to cleavage
would not be carried over to a plate seeded with phage more likely to

31
grow 1ytica11y and interfere with the test results. Patch growth on any
of the phage seeded plates indicated lysogenization of the phage, not
constitutive cleavage of repressor. Lack of patch growth indicated
lytic growth of the phage (resulting in cell death) and constitutive
repressor cleavage. The Inds mutant produces a repressor that is more
easily cleaved than wild-type (Cohen et a1., 1981; Crowl et a1., 1981).
Ind- and clear plaque phage mutants were used as positive and negative
controls.
Mapping mutant alleles
Hfr matings for mapping mutations were performed according to
standard procedures (Miller, 1972). Hfr strains constructed for mapping
are shown at the end of Table 1. Since the recipient strain (mutant
N056) was an Hfr, these cultures were grown to saturation overnight at
320 C to discourage pilus formation. Donors were grown to mid-log phase
at 370 C and added to recipient cultures at one-tenth volume. Matings
were disrupted after 20 minutes by vortexing and an equal volume of L
broth containing 40 ug/m1 chloramphenicol was added and the cultures
allowed to grow out for one hour before plating. Recombinants were
selected on tetracycline chloramphenicol MacConkey lactose plates, since
donor strains mobilized an early Tn10 (Tet marker) and recipient strains
contained Tn2 (Cam marker). MacConkey plates allowed me to distinguish
between Lac+ and Lac- colonies and determine if the given Hfr crossed
out the mutant allele. This gave me a rough estimate of the map
location for a mutant allele.
Map location to the minute was determined by PI transductions.

P1 mapping is described with backcrosses in the section entitled
P1asmids, Bacteriophage and Strains.
B-ga1actosidase Assays
32
B-ga1actosidase assays were performed according to the method of
Miller (1972). Overnight cultures grown in the presence of chloramphen
icol were diluted back 1/200 in 1XA media and grown to early log phase
(Klett 15). The cultures were split and Mitomycin C was added (or not
added); cultures were allowed to grow for an additional two hours (to
mid-log), and the assays were performed. For each assay two samples
were taken and each experiment was done two or more times. Unless
specifically noted the cultures were grown at 37oC. The B-ga1actosidase
results are expressed as Miller units.
Spontaneous Mutagenesis
Single colonies were used to start overnight cultures in L
broth. The overnights were diluted 1/40 in L broth and grown to 1ate
log phase. The cells were concentrated by centrifugation, resuspended
in 0.85% saline and plated on streptomycin-containing L plates. Total
cell numbers were determined by plating appropriate dilutions on non
antibiotic L plates. All incubations were done at 37oC. Mutation
frequency was measured in at least three independent cultures. In two
cases jackpot cultures were found and the values not used.
UV Survival Assays
Cultures were grown to mid-log phase (3 X 108 ce11s/m1) in L
broth, diluted 1/100 in 0.85% saline and irradiated. Appropriate

33
dilutions were plated on L plates and incubated overnight at 370 C to
determine survival.
Recombination Proficiency
Recombination proficiency was assayed using lambda by lambda
. crosses. The phage used, Xl03 and X120, were deficient in recombination
due to red deletions. Also, they were unable to replicate in sup+
strains due to amber mutations. The test strains were co-infected with
both phage at an moi (multiplicity of infection) of 5 and incubated 1.5
hours to allow phage recombination and growth. The cells were killed
with chloroform and reSUlting phage titers determined on supF and sup+
strains. Only recombinant phage in which both amber markers were
crossed out plated on the sup+ strain. The percentage of recombinants
was determined by:
titer on sup+ strain titer on supF strain

34
CHAPTER 3
RESULTS
Results of mutagenesis
Approximately 40,000 colonies were screened for Lac+ phenotype
following MNNG mutagenesis of strain N064. I initially picked 116
strong Lac+ colonies as possible SOS constitutive mutants. Upon
restreaking these colonies, 84 had retained the Lac+ phenotype and the
remainder were discarded. The 84 Lac+ colonies were tested for the
ability to cleave h£I repressor, an in vivo indication of activated RecA
protein (STS test). Since the £1 repressor is more resistant to
cleavage than LexA, I had hoped that in conjunction with Lac+ phenotype
I could identify those mutants in which the SOS response was more
strongly induced. The STS screen identified 10 mutants with differing
abilities to cleave lambda repressor, while the remainder showed no
evidence of hcI cleavage by this test.
By P1 transduction analysis, eight of the ten mutants able to
cleave lambda repressor were found to reside in the recA gene (described
below). Strikingly, the two mutants that were not in recA had only a
marginal ability to cleave h repressor constitutively based upon plaque
morphology of hInd+ and the more easily cleaved hIndS. One of these
mutants mapped to the dam locus and is described in a later section. I
was not able to map the second mutation, and believe that there was more
than one mutation responsible for the Lac+ phenotype. Thus, the STS

test did unveil the most strongly induced mutants, and these were
exclusively recA mutations. Perhaps other mutations that altered
cellular metabolism to the extent that activated RecA could cleave
lambda repressor were lethal.
35
Several other mutants (of the 76/84 that did not cleave cI
repressor) were chosen for further study. These isolates had strong
Lac+ phenotypes and appeared stable. The mutants were screened for lexA
or suIA::MuQ fusion mutations as described in Materials and Methods.
Those mutants that checked out were roughly mapped on the ~ coli
chromosome using Hfrs (described in Materials and Methods). When a
given Hfr crossed out a mutant allele the strain phenotype changed from
Lac+ to Lac-. Once the mutations were roughly mapped, I mapped more
precisely using PI transduction of various known TnlO markers in that
area. Again, if the mutation was near the TnlO, a percentage
(transduction frequency) of transductants became phenotypically Lac- .
As well as providing map location to the minute, this provided a
selectable marker close to the mutation. Thus I could backcross the
mutation into a clean background (i.e., unmutagenized N056 or N0120) in
order to characterize the effect of the mutation. The mutants are each
discussed in detail below.
lOa mutant (lig252)
Mapping
Mapping data for the lOa mutant is shown in Table 2. This
mutation mapped to 52' on the ~ coli chromosome. When ligts7 was
crossed into lOa, no wild-type recombinants were recovered, indicating
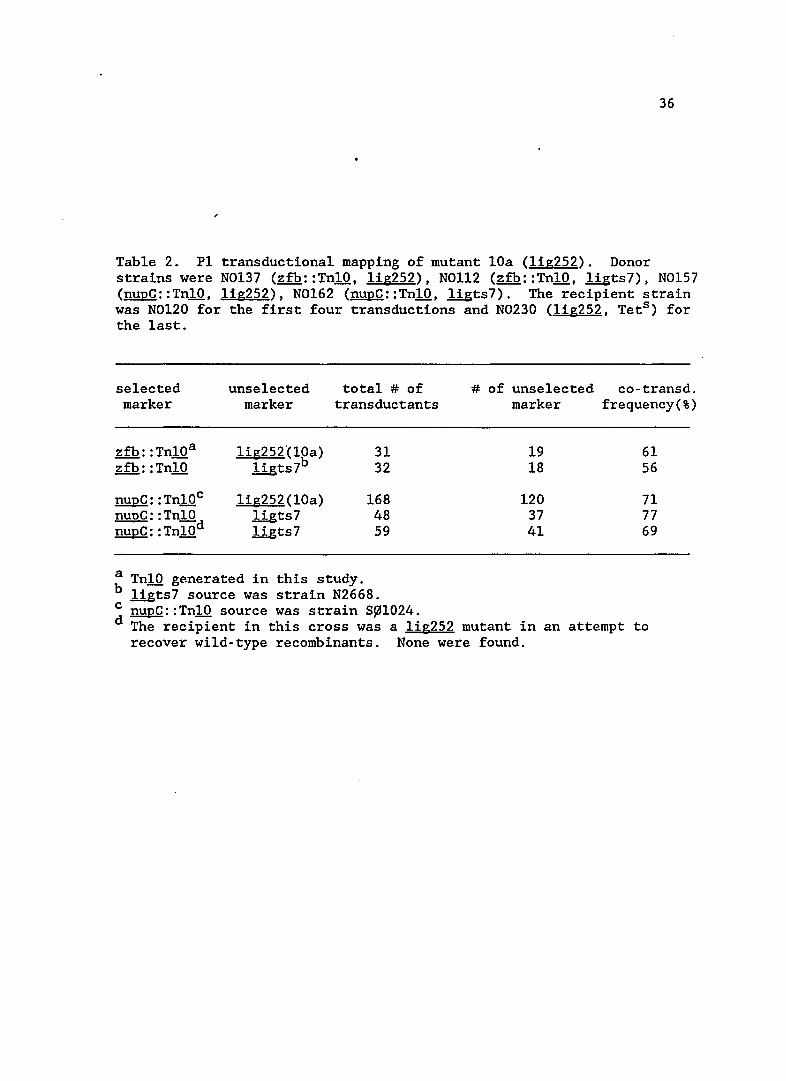
36
Table 2. P1 transductiona1 mapping of mutant lOa (lig252). Donor strains were N0137 (zfb::Tn10, 1ig252), NOl12 (zfb::Tn10, 1igts7), N0157 (nupC::Tn10, 1ig252), N0162 (nupC::Tn10, ligts7). The recipient strain was N0120 for the first four transductions and N0230 (lig252, Tets ) for the last.
selected unse1ected total # of marker marker transductants
zfb: :Tn10a lig252°(10a) 31 zfb: : Tn10 1igts7b 32
nupC: : Tn10c lig252(10a) 168 nupC: :Tn10 1igts7 48 nupC: : Tn10d llgts7 59
a Tn10 generated in this study. b llgts7 source was strain N2668.
# of unse1ected co-transd. marker frequency(%)
19 61 18 56
120 71 37 77 41 69
c nupC::Tn10 source was strain S~1024. d The recipient in this cross was a 1ig252 mutant in an attempt to
recover wild-type recombinants. None were found.

37
that these two mutations lie very close together. This observation,
along with the linkage frequencies, led me to conclude that the mutation
in lOa resided in the 1ig gene, which codes for DNA ligase. The allele
was designated 1ig252.
SOS expression
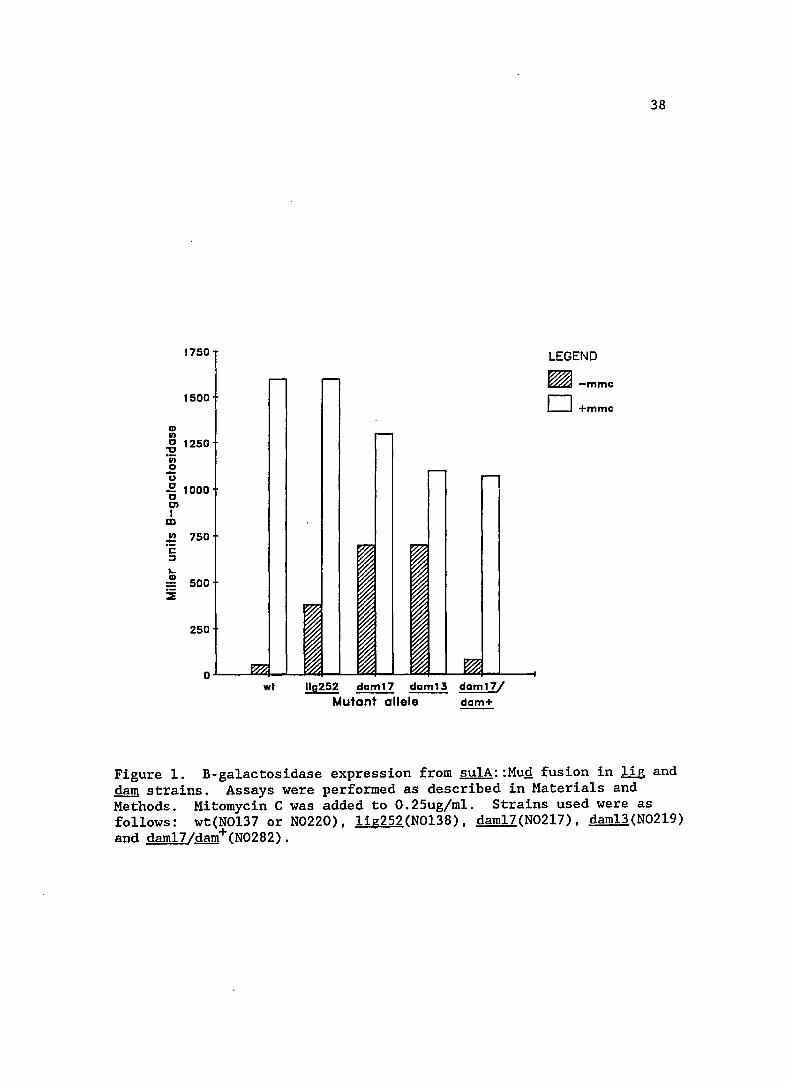
The extent of SOS induction in mutant lOa is shown in Figure 1.
This mutation resulted in a seven-fold increase of su1A expression,
indicating that this !ig mutation increases the basal level of SOS
expression. This increase is perhaps not surprising, since condition
ally lethal 1ig mutants induce prophage at non-permissive temperatures
(Gottesman et a1., 1973). This result does, however, show that 1ig
mutants can increase SOS expression under non-lethal conditions of cell
growth.
lig mutant phenotypes
Other observations indicate that mutant lOa is a lig mutation.
If lOa were a 1ig mutation, I expected Spi- lambda phage to grow very
poorly on the mutant host (Smith, 1983). Therefore, I did a burst size
experiment with mutant lOa. The phage us~a, ~38, has a b1453 deletion
which removes the red and gam genes and confers the Spi- phenotype.
After two hours of growth in isogenic lig+ and lig252 strains the burst
sizes were 64 and 4 phage, respectively, per input phage. This small
burst size indicates the growth of ~Spi- was much poorer in the strain
bearing the lig252 allele. characteristic of lig mutants.
Since recB mutations inhibit SOS induction by nalidixic acid
(Gudas and Pardee, 1975), I wanted to test whether recB would inhibit
CD 1/1
1750
1500
.g 1250 -;; o -u .g 1000 01 I
m !.! 750 C :::J
"-CD 500
SE
250
wt 110252 daml7 ~ daml7/ Mutant allele dam+
LEGEND
~-mmc D+mmc
38
Figure 1. B-ga1actosidase expression from sulA::MuQ fusion in lig and dam strains. Assays were performed as described in Materials and Methods. Mitomycin C was added to O.25ug/m1. Strains used were as follows: wt(N0137 or N0220), 1ig252(N0138), dam17(N0217), dam13(N0219) and dam17/dam+(N0282).

39
the high basal level of induction seen in the lOa mutant. I was unable
to construct the li&252 double mutant using recB21 or recB::TnlO as the
mutant donors. In the case of recB2l, ar&::TnlO (KW88 was the donor and
N0230 the li&252 recipient) 0/40 Tetr transductants screened were RecB- .
The normal co-transduction frequency for these markers is 65%. In
attempting to make the lig252 recB::TnlO double mutant (KP4l9 was the
recB::TnlO donor), 0 Tetr transductants were obtained in the N0230
recipient, while 300 Tetr transductants were obtained in a control
transduction into the wild-type N0120 strain. The 1i&252 recB
inviability may be characteristic of !1g mutants as an earlier study
showed that li&4 recB21 double mutants were barely viable at 420 C
(Gottesman et al., 1973).
9a mutant (dam17)
This mutant was one of those shown in the early screening to
have a slight ability to cleave £1 repressor, in addition to a strong
Lac+ phenotype. Subsequently, this mutation was mapped at 74' on the
~ coli linkage map (see Table 3) and had a similar linkage to
malA: :Tn10 as dam13: : Tn,2,.
Mutant 9a demonstrated several characteristics similar to those
of strains bearing dam mutations. First, when screening for su1A::MuQ
fusion mutations, I was unable to lysogenize 9a with ~lexA3, indicating
that the mutation was inviable with 1exA3. dam strains are a1'so
inviable with lexA3 mutations (Marinus and Morris, 1975). Second, the
9a strain was sensitive to 2-aminopurine, as are dam mutants. Third,
the basal level of SOS expression in this mutant was quite high and
40
Table 3. PI transductional mapping of mutant 9a(dam17). Donor strains were N02l7(malA::TnlO, dam17) or N02l9(malA::TnlO, dam13). The recipient strain was N0120.
selected marker
unselected marker
total # of transductants
# of unselected co-transd.
malA: :TnlOa
malA: : TnlO 120
60
a malT45::TnlO (in MalA operon) source was TST3. b dam13::Tn2 source was KP3l5.
marker frequency(%)
8 7 5 8

41
approximately l3-fold greater than wild-type (see Fi~ure 1). For
comparison, sulA expression in an isogenic dam13::Tn1 strain was
measured and shown to be nearly identical to 9a. Previous work had also
shown that da~ mutations increased the basal level of SOS expression
(Craig et al., 1983; Peterson et al., 1985). Finally, construction of a
partial diploid strain by introducing dam+ (on plasmid pNS-l) into 9a
restored sulA expression to near wild-type levels (see Figure 1) and
alleviated 2-aminopurine sensitivity. This confirmed that the 9a
mutation was a darn allele. The 9a mutation was designated dam17.
3e, 41. and 8a mutants (uvrD242, 243, 244)
Mapping mutations in 3e, 41 and 8a
These mutants were distinguished from the others by their
sensitivity to UV light. The mutations all mapped to the same region,
at 85' on the linkage map. Results of a three factor cross are shown in
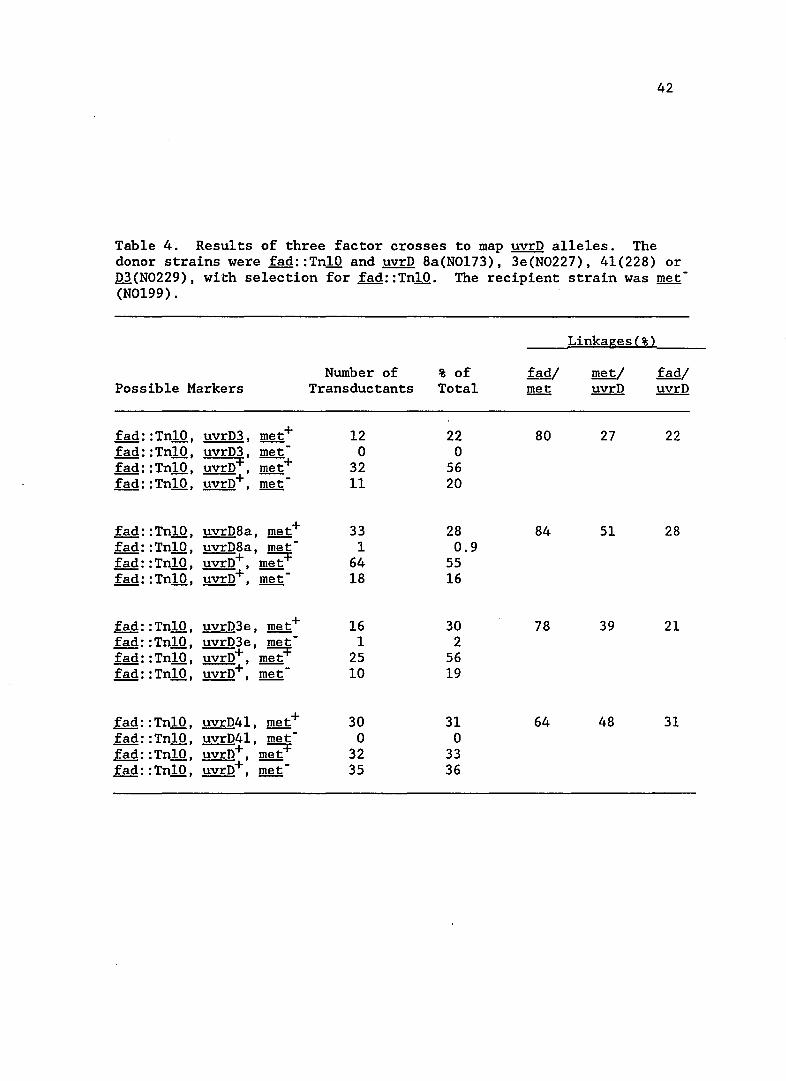
Table 4. A three factor cross was also included with a known uvrD
mutation, uvrD3, to compare linkages and map order. The order of the
markers on the ~ coli chromosome was shown to be fad met uvrD, in
agreement with the published genetic map (Bachmann, 1983). The UV
sensitivity of mutants 3e, 41 and 8a and their map position suggested
they were alleles of uvrD, the gene which encodes DNA helicase II.
Consequently the mutations were designated as uvrD242 for 3e, uvrD243
for 41 and uvrD244 for 8a.
Previously isolated mutants of uvrD have a variety of
phenotypes, differing from one allele to another, which were useful in
designing further experiments. The uvrD3 allele was isolated by Ogawa
42
Table 4. Results of three factor crosses to map uvrD alleles. The donor strains were fad::TnlO and uvrD 8a(N0173), 3e(N0227), 41(228) or D3(N0229), with selection for fad::TnlO. The recipient strain was met -(N0199).
Linkages(%)
Number of % of fad/ met/ fad/ Possible Markers Transductants Total met uvrD uvrD
fad: :TnlO, uvrD3, met+ 12 22 80 27 22 fad: :TnlO, uvrD3, met 0 0 fad: :TnlO, uvrD+, met+ 32 56 fad: :TnlO, uvrD+, met 11 20
fad: :TnlO, uvrD8a, met+ 33 28 84 51 28 fad: :TnlO, uvrD8a, met - 1 0.9 fad: :TnlO, uvrD+ met+ 64 55 --' --fad: :TnlO, uvrD+, met 18 16
fad: : TnlO , uvrD3e, met+ 16 30 78 39 21 fad: :TnlO, uvrD3e, met - 1 2 fad: :Tn10, uvrD+ met+ 25 56 --, --fad: :TnlO, uvrD+, met 10 19
fad: :TnlO, uvrD4l, met+ 30 31 64 48 31 fad: :TnlO, uvrD41, met - 0 0 fad: :TnlO, uvrD+ met+ 32 33 --, --fad: :TnlO, uvrD+, met- 35 36

43
(1968) as a mutant sensitive to UV light and methyl methanesulfonate. A
spontaneous mutator and UV sensitive mutation was isolated by Siegel
(1973) and designated mutU4. A similar mutant was isolated
independently as uvrE (Smirnov and Skavronskaya, 1971). While looking
for recombination deficient mutations in recBC sbcB strains Horii and
Clark (1973) isolated the recL152 mutant, which is not a spontaneous
mutator. Subsequently, these mutations have all been found to reside in
a single locus, uvrD, and share the common phenotype of UV sensitivity.
The product of the uvrD gene plays roles in several pathways of DNA
metabolism that include the SOS regulated repair functions excision
repair and RecF recombination repair, as well as methyl-directed
mismatch repair. In addition, uvrD is regulated by the SOS response
(Siegel, 1983). Identification of DNA helicase II as the product of the
uvrD gene uncovered its role in DNA replication as one of the known ~
coli DNA helicases that unwind duplex DNA (Maples and Kushner, 1982;
Oeda et al., 1982). The mutants were screened for some of these known
uvrD phenotypes in an attempt to understand which altered properties
were associated with SOS induction.
UV sensitivity of uvrD mutants
The UV sensitivity of strains bearing uvrD242, 243 and 244 is
shown in Figure 2. In order to show definitively that these mutations
did lie in uvrD, I constructed merodiploids of each mutant with a low
copy number uvrD+ plasmid, pVMK35. A low copy number plasmid was used
for these complementation studies because overproduction of the wild
type UvrD protein causes UV sensitivity (Map1es and Kushner, 1982).
44
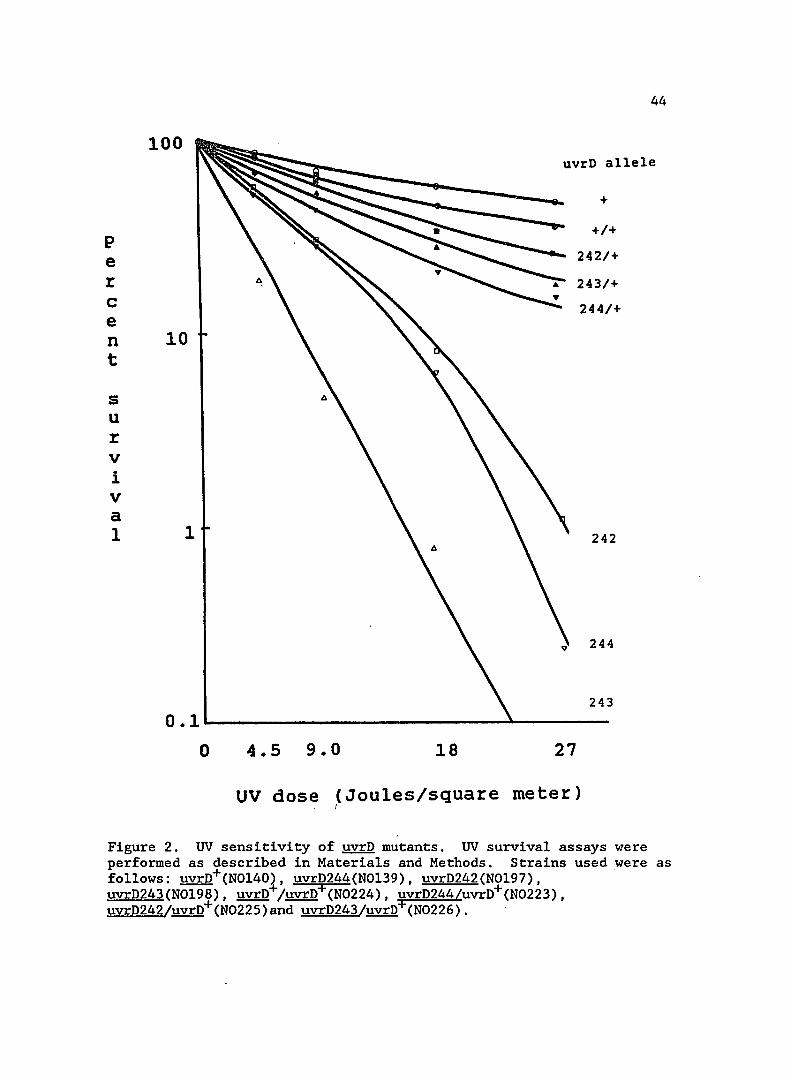
100 uvrD allele
+
+/+ P e 242/+
r 243/+
C 244/+ e n 10 t
s u r v i v a 1 1 242
244
243
0.1--------------------------~-------o 4.5 9.0 18 27
UV dose ~Joules/square meter)
Figure 2. UV sensitivity of ~ mutants. UV survival assays were performed as described in Materials and Methods. Strains used were as follows: uvrD+(N0140), uvrD244(N0139), uvrD242(N0197), uvrD243(N0198), uvrD+/uvrD+(N0224), uvrD244/uvrD+(N0223), uvrD242/uvrD+(N0225)and uvrD243/uvrD+(N0226).

45
As can be seen in Figure 2, uvrD+ largely complemented the UV
sensitivity in the mutant strains. This result indicates that the
mutations do lie in the uvrD gene and that the repair defect in these
strains is recessive and can be complemented.
SOS expression in uvrD mutants
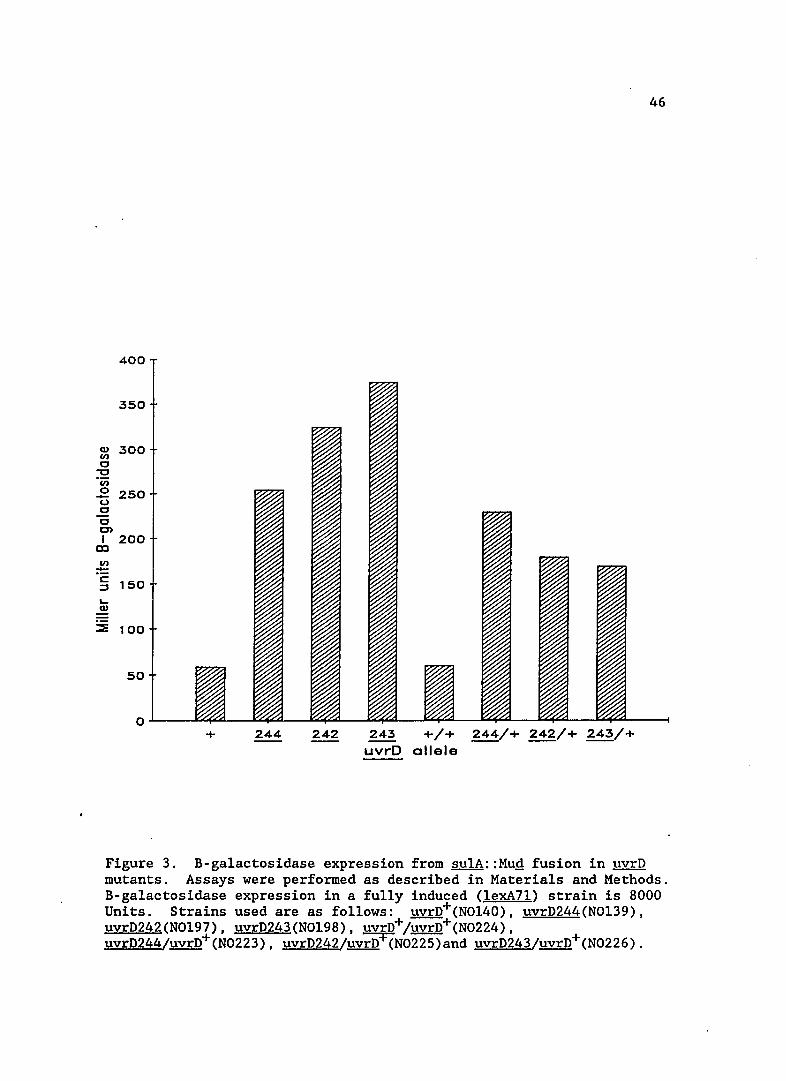
The basal level of SOS expression in the uvrD mutants is shown
in Figure 3. sulA expression is increased five to seven-fold in these
strains. The basal levels of two previously identified uvrD alleles,
uvrD3 and uvrDl56 (formerly mutU4) are shown in Table 5. The uvrD3
mutation did not lead to any increase in the basal level of SOS
expression, indicating that it is a different mutation from those
isolated in this study. This result also shows that a repair defect,
per se, does not increase sulA expression. In contrast, uvrDl56
increased sulA expression approximately ten-fold, indicating the new
uvrD mutations may be of a similar class.
Complementing the uvrD alleles isolated in this study with the
wild-type uvrD gene did not alleviate SOS induction, indicating there is
a dominant or incompletely dominant defect in the newly isolated
mutations. This dominance is interesting since the UV sensitivity of
the uvrD mutants was shown to be recessive (see previous section and
Figure 2). This result shows that the new uvrD alleles are not null
alleles and that the protein product still functions in some capacity.
The uvrD- strains containing the uvrD+ plasmids were unstable.
This was particularly true of uvrD243, strain N0226. Standard devia
tions in B-galactosidase units were approximately 5% in the uvrD strains
Q) UJ 0
-0 • iii 0 -(,) 0 0 Ol I
CD
UJ
== c ::J
~ Q)
:::::!!
400
350
300
250
200
150
100
50
0 + 244 242 243 +/+ 244/+ 242/+ 243/+
uvrD allele
46
Figure 3. B-galactosidase expression from sulA::Mug fusion in uvrD mutants. Assays were performed as described in Materials and Methods. B-galactosidase expression in a fully induced (lexA7l) strain is 8000 Units. Strains used are as follows: uvrD+(NOl40), uvrD244(NOl39), uvrD242(NOl97), uvrD243(NOl98), uvrD+/uvrD+(N0224), uvrD244/uvrD+(N0223), uvrD242/uvrD+(N0225)and uvrD243/uvrD+(N0226).
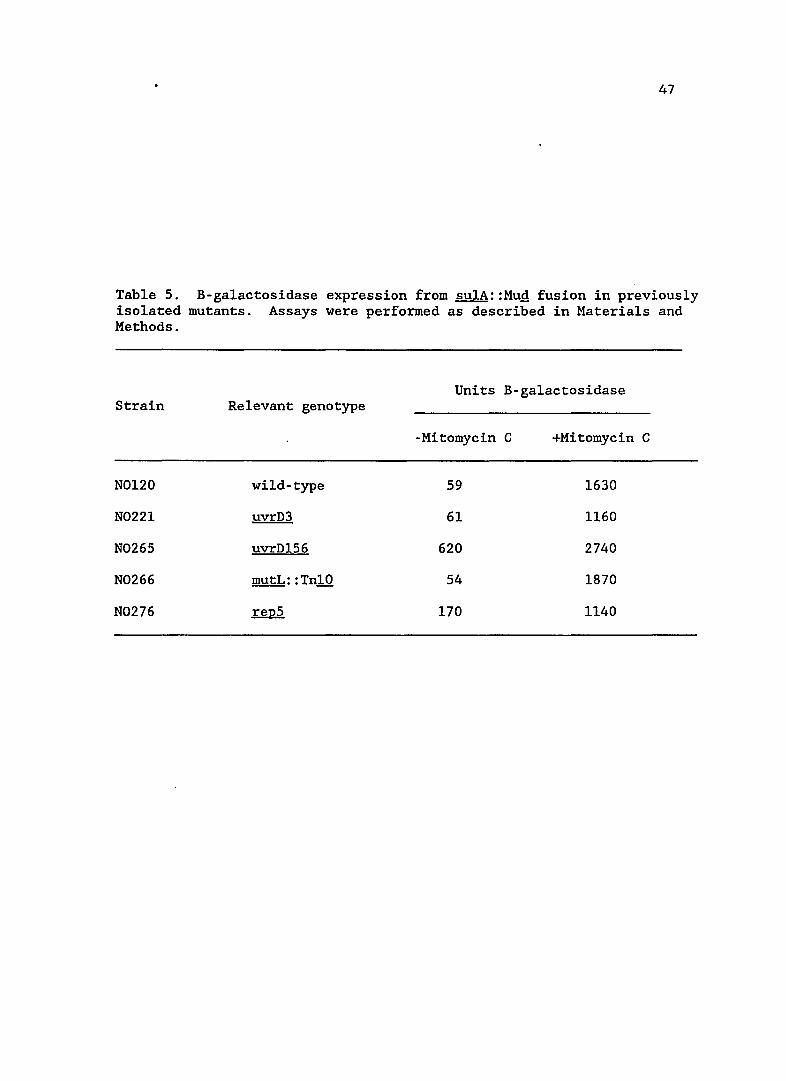
47
Table 5. B-ga1actosidase expression from su1A::MuQ fusion in previously isolated mutants. Assays were performed as described in Materials and Methods.
Units B-ga1actosidase Strain Relevant genotype
-Mitomycin C +Mitomycin C
N0120 wild-type 59 1630
N0221 uvrD3 61 1160
N0265 uvrD156 620 2740
N0266 mutL: :Tn10 54 1870
N0276 repS 170 1140

48
(not the merodip1oids). The standard deviation in B-ga1actosidase units
for the diploids were much greater, probably due to plasmid instability.
For example, in the most stable strain, N0223 (uvrD244), the standard
deviation in B-ga1actosidase units was 7%, for N0224 (uvrD242) the
standard deviation was 12%, and in N0226 (uvrD243) the standard
deviation was 20%.
Since uvrD plays a major role in excision repair, I considered
the possibility that a lack of excision repair results in an increased
level of SOS expression. I constructed a sulA::Mug fusion strain
containing uvrA6, a mutation which inactivates UvrABC excinuc1ease and
excision repair. While the strain was sensitive to UV irradiation, the .
basal level of sulA expression was not enhanced (data not shown),
suggesting that a defect in excision repair does not necessarily sub-
induce the SOS regu1on.
The levels of Mitomycin C induced SOS expression are presented
in Figure 4. The uvrD mutants were further inducible by the addition of
Mitomycin C, showing that the strains can respond to the addition of
further SOS inducing signal. This Mitomycin C induced expression was
enhanced above wild-type levels in strains bearing the new uvrD alleles.
This enhanced response to Mitomycin C induction was not complemented by
the uvrD+ plasmid.
Spontaneous mutagenesis in uvrD mutants
Since some alleles of uvrD confer a mutator phenotype, I
measured levels of spontaneous mutagenesis in uvrD242, 243 and 244. As
shown in Figure 5, the new uvrD alleles gave spontaneous mutator
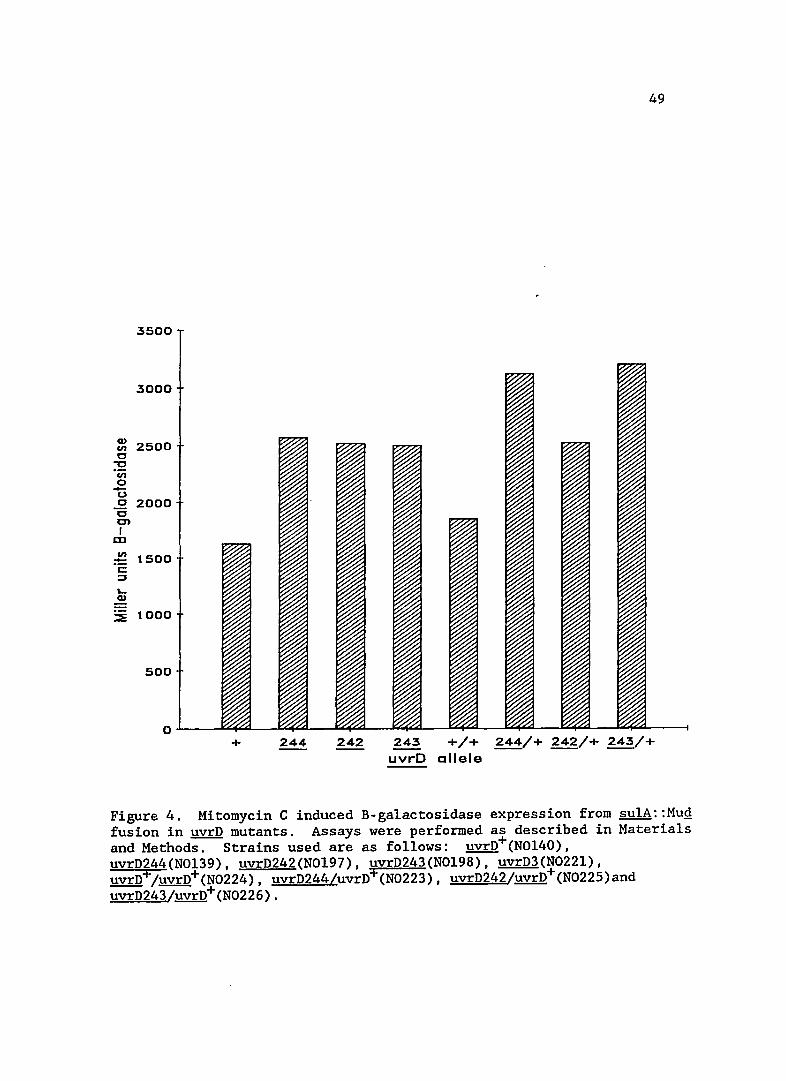
49
Q) 2500 en
c "'C ·m 0 -u c 2000 c C)
I CD
en 1500 := c: ::J
L-Q)
::E 1000
+ 244 242 243 +/+ 244/+ 242/+ 243/+
uvrD allele
Figure 4. Mitomycin C induced B-galactosidase expression from sulA::MuQ fusion in uvrD mutants. Assays were performed as described in Materials and Methods. Strains used are as follows: uvrD+(NOl40), uvrD244(NOl39), uvrD242(NOl97), uvrD243(NOl98), uvrD3(N022l), uvrD+/uvrD+(N0224), uvrD244/uvrD+(N0223), uvrD242/uvrD+(N0225)and uvrD243/uvrD+(N0226).
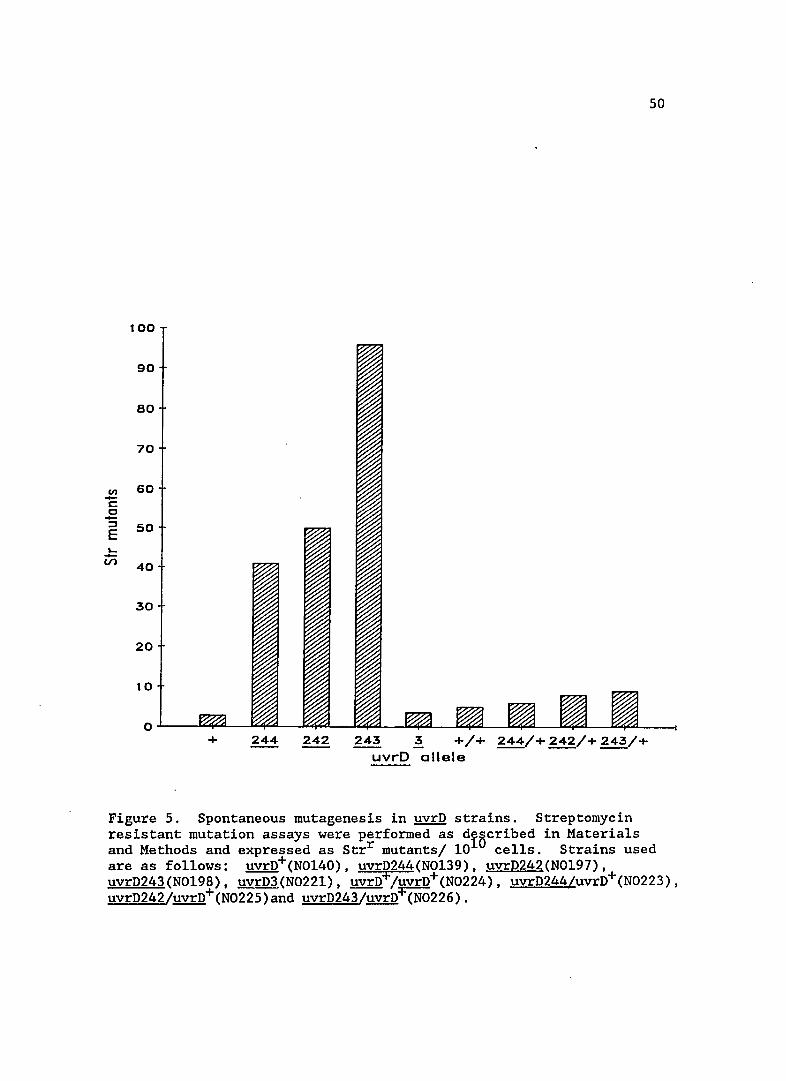
100
90
eo
70
UJ 60 -c: c -::l 50 E '--(/) 40
30
20
10
0 + 244 242 243 3 +/+ 244/+ 242/+ 243/+
uvrD allele
50
Figure 5. Spontaneous mutagenesis in uvrD strains. Streptomycin resistant mutation assays were performed as diBcribed in Materials and Methods and expressed as Strr mutants/ 10 cells. Strains used are as follows: uvrD+(N0140), uvrD244(N0139), uvrD242(N0197), uvrD243(N0198), uvrD3(N0221), uvrD+/uvrD+(N0224), uvrD244/uvrD+(N0223), uvrD242/uvrD+(N0225)and uvrD243/uvrn+(N0226).

51
activity, unlike uvrD3. The uvrD156 mutation, however, is known to
exhibit a mutator phenotype (Siegel, 1973), again indicating the new
alleles may be similar. Complementation studies with the newly isolated
uvrD mutants showed that increased spontaneous mutagenesis was recessive
to the wild-type allele, since mutagenesis was restored to near wi1d
type levels. Prior work has shown that uvrD156 spontaneous mutability
and UV sensitivity can be complemented by the uvrD+ allele (Siegel,
1981).
uvrD spontaneous mutagenesis when SOS induction is blocked
Since the new uvrD mutations resulted in an increase in SOS
expression, I wanted to see if this increase was responsible for the
elevated spontaneous mutation rate. SOS mutagenesis requires the
products of LexA regulated UmuC,D proteins as well as RecA protein.
Using the uvrD244 strain as a representative, I blocked SOS induction by
introducing a non-cleavable repressor, LexA3(Ind-). In another
strain, I introduced a deletion of recA, which not only prevented
derepression of SOS functions, but abolished SOS mutagenesis. As can be
seen in Table 6, the spontaneous mutation rate in the SOS blocked
uvrD244 strains was not reduced.
Construction of the uvrD244 de1recA or uvrD244 lexA3 double
mutant shows that a uvrD244 strain is viable when the induction of SOS
functions is blocked. su1A derepression is also blocked in the double
mutants as judged by Lac phenotype on MacConkey lactose indicator
plates. All of the new uvrD mutant strains were found to be viable with
1exA3 when I was screening for sulA fusion mutations early in this
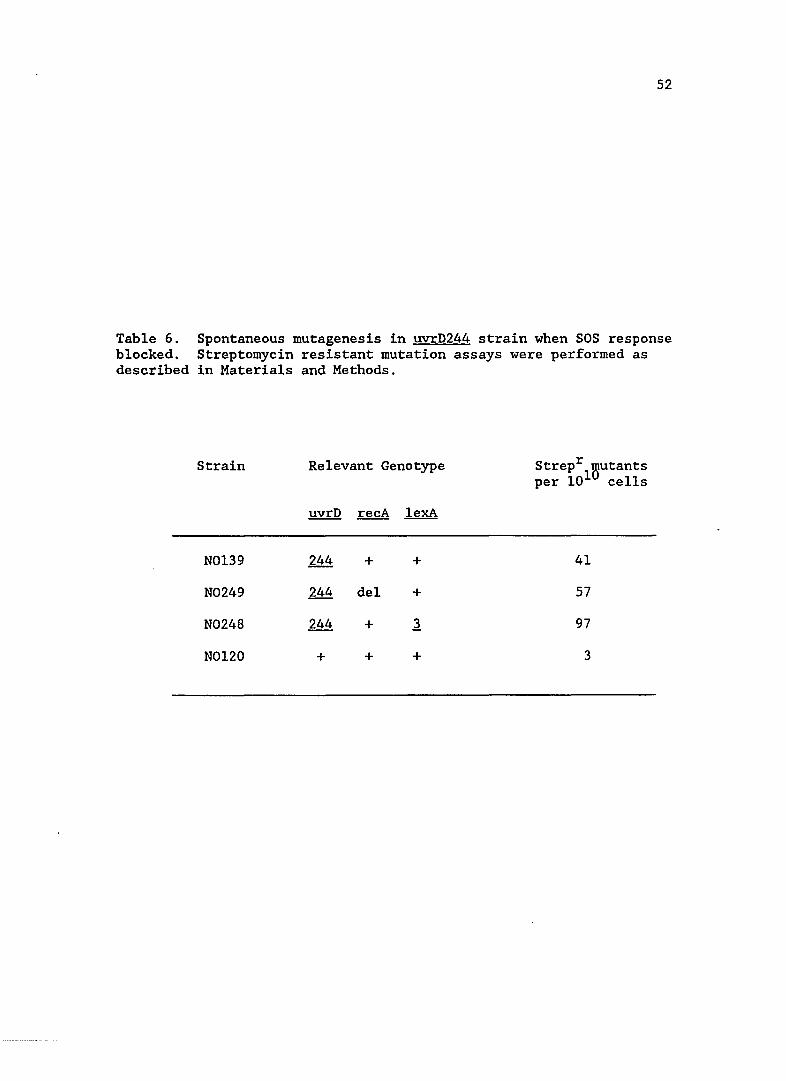
52
Table 6. Spontaneous mutagenesis in uvrD244 strain when SOS response blocked. Streptomycin resistant mutation assays were performed as described in Materials and Methods.
Strain Relevant Genotype Strepr 'Butants per 101 cells
!!Y.!:Q recA lexA
N0139 244 + + 41
N0249 244 del + 57
N0248 244 + .1 97
N0120 + + + 3

53
study. This result stands in contrast to darn mutant$ which require SOS
derepression for viability.
uvrD mutagenesis in mismatch repair deficient strains
Mutations in mutH, mutL or mutS abolish mismatch repair in ~
coli, and strains bearing these mutations have a high level of
spontaneous mutagenesis (Radman and Wagner, 1986). Since uvrD
participates in mismatch repair, I wanted to see if a lack of mismatch
repair is responsible for the increased level of mutagenesis seen in the
mutants I isolated. I reasoned that, if the lack of mismatch repair
caused the mutagenesis seen in these strains, double mutants with
mutL::TnlO would have a level of spontaneous mutagenesis similar to that
in a single (mutL or uvrD) mutant strain. If another deficiency of the
uvrD strains was responsible for mutagenesis, then spontaneous
mutagenesis in the double mutants would be greater than in strains
bearing either of the single mutant alleles.
The spontaneous mutability of mutL strains is shown in Table 7.
The levels of spontaneous mutagenesis in mutL uvrD strains is not
increased above that seen in mutL single mutation strains. This result
agrees with previous work (Glickman and Radman, 1980) which suggests that
the spontaneous mutagenesis seen in uvrD strains is due to a lack of
mismatch repair.
I also measured sulA expression in the mutL strain to check
whether a lack of mismatch repair by itself would elevate SOS
expression. The basal level of ~ulA expression in the mutL strain was
that of wild-type strains (shown in Table 5). These results indicate

54
Table 7. Spontaneous mutagenesis in uvrD strains when mismatch repair blocked. Streptomycin resistant mutation assays were performed as described in Materials and Methods.
Strain Relevant genotype
N0266
N0267 mutL uvrD242
N0268 mutL uvrD243
N0269 mutL uvrD244
Strepr mutants/ 1010 cells
134
132
83
140

55
that the lack of mismatch repair in the uvrD strains is unlikely to be
responsible for the increased expression of su1A in the uvrD mutants.
Viability of uvrD244 repS double mutants
The dominance of the new uvrQ alleles with respect to SOS
induction observed in the mutant strains indicates that they are not
null alleles. Previous studies showed that uvrD rep double mutants are
inviable or very unstable, which suggests that there is an interaction
between these two gene products during replication or that they perform
a similar essential function (Taucher-Scholz et a1., 1983; Siegel,
1973). Taucher-Scholz et a1. (1983) propose that UvrD and Rep proteins
act jointly during DNA replication to unwind duplex DNA, with each
protein acting on opposite arms of the replication fork. In the case of
Rep or UvrD mutations, they suggest that these proteins can substitute
for each other during replication. In order to determine if the uvrD244
strain was defective in replication functions, I attempted to construct
a repS uvrD244 double mutant. I was able to make this construction with
normal cotransduction frequencies and conclude that UvrD244 protein has
at least some function as a DNA helicase. It may be that an altered
helicase function in this mutant is responsible for the increased
expression of SOS functions.
Additionally, I measured B-galactosidase expression in the repS
strain. sulA expression was elevated 2-3 fold over wild-type levels,
demonstrating some sub-induction of sulA in this mutant (Table 5).

56
recA mutants
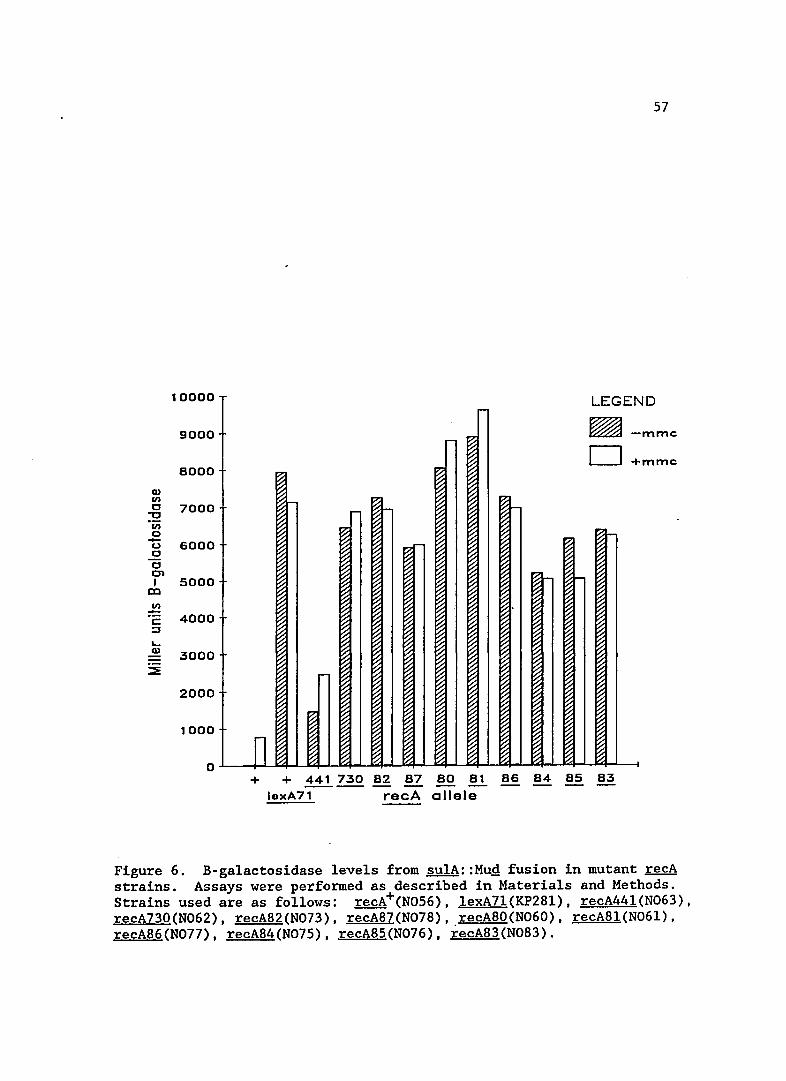
SOS expression
Expression levels from the sulA::MuQ fusion in the new recA
alleles is shown in Figure 6. For comparison, I included data for a
lexA7l(Def) mutant, the recA44l (formerly tif-l) allele and recA730
(Prtc ) which was isolated by Witkin et al. (1982). The expression of
sulA in these new recA mutants was very high, near that of a cell in
which LexA is non-functional. The high expression of SOS functions was
also seen in recA730, as would be expected in mutants of a similar
class. Standard deviations for the B-galactosidase values in the recA
mutant strains were approximately 5%. The addition of Mitomycin Chad
little effect on these strains, probably because they were nearly fully
induced.
recA mutant recombination proficieny
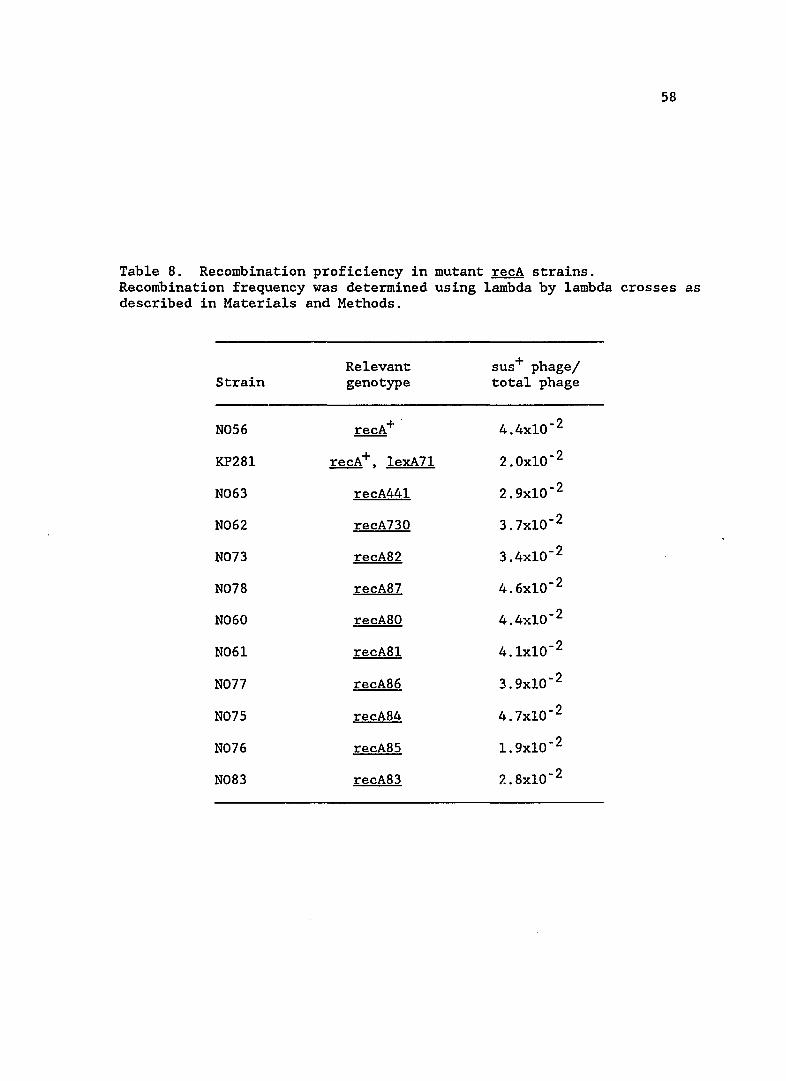
Since RecA protein has an additional function in promoting
homologous recombination, I assayed levels of recombination in the recA
mutant strains. Recombination efficiency is shown in Table 8. All
mutants appear to be proficient in homologous recombination of phage
lambda.
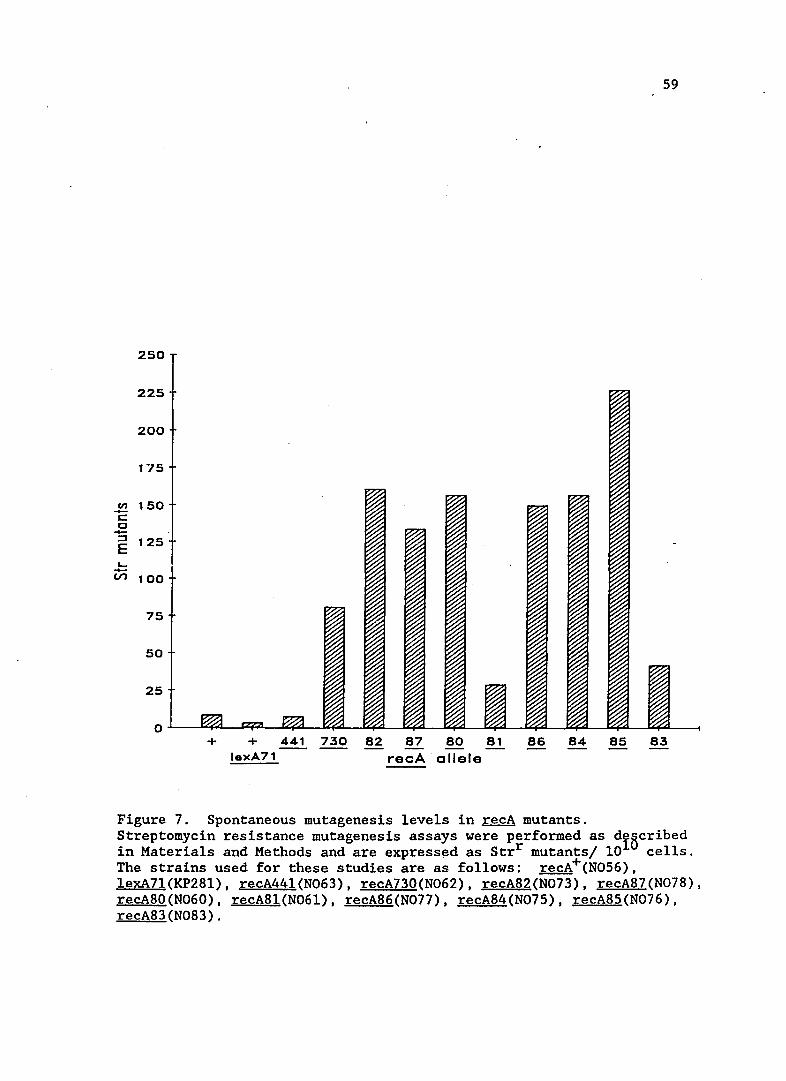
Spontaneous mutagenesis in recA mutants
Spontaneous mutagenesis in the new recA mutants is shown in
Figure 7. All strains have increased spontaneous mutator activity
compared to recA wild-type strains. recA44l strains had previously been
shown to have increased spontaneous mutations, but under conditions of
Q) VI C
'"C ·iii o -(,) c c en I
en
..... Q)
57
10000 LEGEND
9000 -mmc
+mmc 8000
7000
6000
5000
4000
3000
2000
~ O.L.-.......f..-n ~~~ ~~~~-'---I
1000
+ + 441 730 82 87 80 81 86 84 85 83 ----- - --lexA71 recA allele
Figure 6. B-galactosidase levels from sulA::Mug fusion in mutant recA strains. Assays were performed as described in Materials and Methods. Strains used are as follows: recA+(N056), lexA7l(KP28l), recA441(N063), recA730(N062), recA82(N073), recA87(N078), recA80(N060), recA81(N061), recA86(N077), recA84(N075), recA85(N076), recA83(N083).
58
Table 8. Recombination proficiency in mutant recA strains. Recombination frequency was determined using lambda by lambda crosses as described in Materials and Methods.
Relevant sus+ phage/ Strain genotype total phage
N056 recA+ . 4.4xlO- 2
KP28l recA+, lexA71 2.0xlO- 2
N063 recA441 2.9xlO- 2
N062 recA730 3.7xlO- 2
N073 recA82 3.4xlO- 2
N078 recA87 4.6xlO- 2
N060 recA80 4.4xlO- 2
N06l recA8l 4.lxlO- 2
N077 recA86 3.9xlO- 2
N075 recA84 4.7xlO- 2
N076 recA85 1. 9xlO- 2
N083 recA83 2.8xlO- 2
59
250
225
200
175
2 150 s:::: c -::l 125 E L. -Vl 100
75
50
25
a + + 441 730 82 87 80 81 8S 84 85 83
lexA71 recA allele
Figure 7. Spontaneous mutagenesis levels in recA mutants. Streptomycin resistance mutagenesis assays were performed as dIBcribed in Materials and Methods and are express~d as Strr mutants/ 10 cells. The strains used for these studies are as follows: recA+(N056). lexA7l(KP28l). recA441(N063). recA730(N062). recA82(N073). recA87(N078). recA80(N060). recA81(N06l). recA86(N077). recA84(N075). recA85(N076). recA83(N083).

60
growth different than used in this study. The growth conditions
conditions used in this study, 370 C versus 42oC, may account for the
relatively low level of mutagenesis for the recA44l strain shown in
Figure 7. Two mutants that exhibited extremes of mutagenesis and sulA
expression, recA8l and recA85 were chosen for further study.
Viable and inviable mutations with recA8l and recA85
I wanted to make double mutants of recA8l and recA85 with other
alleles whose mutant products affect SOS induction. These included
ssbl, ssbl13, recBC and recF. I successfully made double mutants of all
alleles in recA8l. The additional mutations did not affect constitutive
induction resulting from recA8l. This result indicates that the
mutations act at different stages in SOS induction. A similar result
was obtained by Lieberman and Witkin (1983) in finding that ssb
mutations did not affect SOS-constitutivity in recA730(prtc ) mutants.
In constructing the double mutants of ssb or recB with recA85 the
situation was very different. Pl transductions which attempted to move
ssbl or ssbl13 into a recA85 strain (N01.2l) yielded many colonies at the
expected transduct~on frequency; however, on patching or streaking,
those colonies which had the double mutant completely failed to grow.
In the case of recB, I was unable to construct recA85 recB2l or recA85
recB::TnlO double mutants. In attempting to construct recA85 recB2l
mutants (Pl KW88 --> NOl2l), 30 transductants failed to yield recB2l
mutations. In another Pl transduction to move recB::TnlO (donor KP4l9)
into recA85 strain NOl2l, no tetracycline resistant transductants were
obtained. A control transduction (recB::TnlO --> wild-type N0120

61
strain) run side-by-side yielded 300 transductants. recA85 recF142
double mutants were constructed without difficulty. The recA85 recF142
double mutant strain still expressed su1A constitutively as judged on
MacConkey plates. The difference in ability to construct double mutants
demonstrates that recA85 and recA81 are different" mutations that lead to
different effects within the cell.

62
CHAPTER 4
DISCUSSION
Isolation of SOS constitutive mutants
I have isolated, mapped and characterized several ~ coli
mutations which lead to constitutive induction of the SOS response.
These mutations include 8 recA (Prtc ) mutations, 3 uvrD mutations, and
single mutations in lig and dam. All of these mutations lead to
constitutive increased expression of the SOS response under conditions
of viable growth; that is, they are not conditionally lethal mutations.
The mutants isolated fall into two groups. The first group
consists of the uvrD, lig and dam mutations. These mutations subinduce
the SOS response, as shown by an increase in sulA expression (Figs. I
and 3). SOS induction in these mutants is most likely mediated through
RecA and LexA proteins, since LexA3(Ind-) blocks sulA expression in lig
and uvrD strains (dam mutants are inviable with lexA3 and require
expression of some SOS-regulated genes). This is in agreement with the
current model of SOS response regulation through RecA and LexA proteins
as discussed in the Introduction of this dissertation. The second type
of mutants, the recA mutations, arG those in which sulA is fully induced
to levels of expression similar to lexA(Null) mutants (Fig. 6). These
mutations are similar to others isolated previously and designated PrtC
for constitutive protease activity. SOS expression appears to be
mediated through LexA repressor destruction at least for RecA85, as the

63
LexA2(Ind-) hypocleavable repressor drastically reduces sulA expression
in a recA85 strain (Peterson et al., 1988a).
We interpret our results by proposing that the mutations
isolated represent two early steps in the induction of the SOS response.
The first step is generation of an SOS inducing signal mimicking (or
causing) DNA damage and is represented by the dam, lig and uvrD
mutations. The products of these genes all affect DNA metabolism and
the mutant proteins most likely alter DNA metabolism in such a way as to
produce an SOS inducing signal and activate RecA protein. The second
step of induction is the signal target and is represented by mutations
in recA, a key regulatory gene of the SOS response.
The alterations in DNA metabolism that induce the SOS response
are unclear. Finding mutations in !ig and uvrD which induce SOS is
consistent with other previously observed mutations causing induction,
such as !ig(Ts) , dnaB(Ts), dnaE(Ts) and polA(Ts). These mutations all
specifically affect synthesis of DNA indicating that the integrity of
DNA synthetic machinery plays a role in SOS induction. This appears to
be limited to the actual process of DNA synthesis, however, not
including re-initiation of new rounds of chromosomal replication.
Mutations that specifically halt initiation of new rounds of DNA
replication, such as dnaA(Ts) or dnaC(Ts), do not lead to spontaneous
SOS induction. Possible specific signals generated by each of the
mutations isolated in this study will be discussed later in this
chapter.
The uvrD, !ig and dam mutations lead to intermediate levels of
SOS induction, a condition sometimes refer.red to as sub-induction

64
(Bailone, et al., 1979; Little, 1983). These mutations all lead to
different basal levels of SOS expression, as shown in Figures land 3,
indicating that the level of inducing signal is a variable quantity.
The variable nature of signal is underscored by the observation that the
mutants isolated were capable of further induction by Mitomycin C
treatment. In control experiments, wild-type cells were induced with
Mitomycin C. The levels of SOS expression did not approach that of
strains which lacked LexA repressor. This difference indicates that
there was still intact LexA repressor remaining in the cell and may be
referred to as a sub-induced state as well. In the mutant strains, sub
induction may result from low levels of SOS inducing signal being
generated or incomplete signal if there are multiple' paths or types of
signal needed. My studies do not distinguish between these
possibilities.
The variable nature of signal probably allows the cell to fine
tune levels of SOS-associated repair proteins according to the amount of
DNA damage perceived. Based on the current model of SOS regulation,
signal most likely mediates this fine tuning by causing low levels of
RecA protein activation leading to cleavage of a fraction of the LexA
repressor population, partially derepressing SOS-regulated operons. A
steady state would be established producing a certain level of repair
proteins. Another factor infuencing the level of activated RecA protein
and SOS repair proteins is negative regulation of recA and lexA by the
lexA product. In the case of the darn mutation, the increased basal
level of SOS expression is required to accommodate some type of damage
generated by the defective protein, as dam strains are inviable when

65
lexA3(Ind-). The other mutations, li& and uvrD, do not generate damage
requiring SOS repair since they are viable when the SOS response is
blocked.
One problem with this type of study is trying to determine how
the mutant phenotype relates to signal generation. In these sub
induction mutants, one hopes that the mutant protein subtly perturbs a
specific and identifiable cellular process, allowing speculation as to
what constitutes an inducing signal. This direct type of perturbation
may be in DNA synthesis, causing a DNA structural alteration or an
immbalance in nucleotide pools for example. In reality, however, the
mutant protein may have wide-ranging effects that indirectly produce an
inducing signal by affecting other cellular processes. One possible
example would be of a mutation causing DNA damage that interferes with
DNA syt~thesis, subjecting DNA to exonuclease action or leading to DNA
repair intermediates that generate inducing signal. Mutations that
cause SOS induction under lethal conditions (such as Ts mutations) could
certainly cause such an effect. I will attempt to address this issue
when each of the specific mutations are discussed in detail.
The failure to find fully induced mutants (sulA levels analogous
to those found in lexA(Def) strains) in genes other than recA may be a
consequence of selecting non-lethal, SOS inducing mutations. The
mutants chosen for characterization in this study were those which
exhibited the strongest SOS induction, indicated by Lac+ phenotype.
Only mutants of recA, which represent the signal target, induced the SOS
system to the extetlt that lambda £1 repressor was cleaved constitutively
or that most (or all) LexA protein was cleaved. The ~ mutants

66
isolated were identified in the STS ~I repressor cleavage screen and
were shown to have levels of sulA expression similar to a lexA(Def)
strain. Mutations which caused this extent of SOS induction were not
found in any other genes--those that would be involved in the first step
of induction described above. It may be that severe defects in cellular
physiology (such as inhibition of DNA synthesis) generating an inducing
signal (or signals) strong enough to cleave most of the LexA in the cell
would be lethal. In fact, conditional mutations previously known to
induce prophage (cleave £1 repressor) do so at the non-permissive
temperature, as discussed in the Introduction. Since I was screening
for non-lethal SOS inducing mutants, this may explain why none of the
mutations I isolated (besides ~) lead to high levels of SOS
induction.
lig2S2 mutation
I have isolated a DNA ligase mutation which leads to sub
induction of the SOS response. This mutation is not conditionally
lethal, but leads to increased SOS expression under normal conditions
for growth. Since ligase is an essential gene, the lig2S2 mutation is
not a null allele but probably ligates replicating DNA and naturally
occuring nicks at a reduced efficiency.
We propose that the 1!g mutation generates an inducing signal by
the disrupting of DNA metabolism. One possibility is that unligated
nicks occurring in DNA during replication may serve directly as inducing
signals, being recognized by RecA protein. Alternatively, unligated
nicks may serve as a substrate for the action of further cellular

67
processes that generate SOS signals, thus inducing the SOS response
indirectly. These structures may be single-stranded regions of DNA or
oligonucleotides resulting from DNA degradation.
One clue bearing on SOS induction in ligase mutations is that
the mutant is inviable with recB mutations. This suggests that RecBCD
enzyme may act at DNA nicks to repair potentially lethal DNA damage
resulting from an over-abundance of nicks in DNA. This action most
likely involves recombination, since RecBCD is a major participant in
homologous recombination. The possible damage in the DNA ligase mutant
does not, however, require SOS repair functions to be expressed since
this mutant is viable when SOS is blocked (in lexA3 lig252 double
mutants).
daml7 mutant
The isolation of a dam mutation in this study comfirms earlier
observations that dam mutations increase the basal level of SOS
expression (Craig et al., 1984; Peterson et al., 1985). The allele
isolated in this study appears to be a null allele of dam since the
level of sulA expression in the mutant is similar to that found in an
isogenic strain bearing a known dam null allele, dam13::Tn2 (Figure 1).
Similar to other dam mutants, daml7 is sensitive to 2-aminopurine and
inviable in combination with lexA3.
The inviability of dam mutations with lexA3 indicates that
partial SOS derepression is required in strains lacking DNA-adenine
methylase and suggests that the lack of methylation produces a cellular
abnormality that needs to be repaired. Strains deficient in dam do not

68
simply lack mismatch repair, but are subject to indiscriminant mismatch
repair. Since neither DNA strand is methylated, MutHLS repair enzymes
attempt to repair both strands of DNA when mismatches occur (Lu et al,
1983; Pukkila et al., 1983). The lack of mismatch repair itself does
not lead to increased SOS expression, demonstrated in mutL strains
(Table 5) and in mutS strains (Craig et al., 1984). The results of
Peterson et ale (1985) show that strains bearing dam mutations require
SOS regulated recombination repair genes for viability. The required
genes are recA, ~ and recJ. The recB and Q genes, which are
associated with recombination but not under LexA repressor control, are
also required in dam strains (Marinus and Morris, 1974). This indicates
that in dam strains some type of DNA damage occurs that requires repair
for cell viability. Glickman and Radman (1980) suggest that
indiscriminant mismatch repair results in the formation of double
stranded breaks in DNA, which would require DNA repair processes to
correct these potentially lethal structures.
How dam mutations lead to SOS induction is not clear.
Certainly, DNA damage resulting from indiscriminant mismatch repair,
such as double- or single-strand DNA breaks, could produce an inducing
signal and lead to increased expression of sulA. Many of the phenotypes
associated with dam mutations are alleviated by second site mutations in
the mismatch repair enzymes (MutH, L or S). These phenotypes include 2-
aminopurine sensitivity and inviability with other allele combinations
such as recA(Def) and lexA(Ind-) (McGraw and Marinus, 1980). Other
evidence, however, shows that mutations in mutH, 1 or ~ do not suppress
SOS sub-induction in dam strains (Craig et al, 1984; K.R. Peterson,

69
personal communication). This result indicates that another de~ect,
which does not require SOS associated repair, sub-induces the SOS
response in dam mutants.
The extent of SOS induction in dam mutants as measured by su1A
expression may not be a true reflection of recA activation. The su1A
promoter contains a Dam methylation site: GATC (Cole, 1983). How the
lack of methylation affects promoter activity is unclear. In the case
of the trpR promoter, lack of methylation increases expression 2-3 times
(Marinus, 1985), however, lack of methylation in one of the dnaA
promoters decreases expression of the gene (Braun and Wright, 1986).
While the effect of methylation on su1A expression is not known, it is
still clear that dam mutations increase expression of other SOS
regulated genes (Craig et a1., 1984; Peterson et al., 1985).
uvrD mutants
The results of this study show that uvrD mutations can lead to
sub-induction of the SOS response (Figure 3 and Table 5). This includes
the new mutations isolated (uvrD242, 243 and 244), as well as a
previously isolated mutation, uvrD156. Not all uvrD mutations share
this property, however, since uvrD3 does not increase SOS expression.
All known uvrD mutations, including those isolated in this study, share
a UV sensitive phenotype (shown in Figure 2).
Several phenotypes are shared by uvrD242, 243, 244 and uvrD156,
suggesting that these may be of a similar class. These phenotypes
include SOS sub-induction, spontaneous mutagenesis and UV sensitivity.
Also, the spontanteous mutagenesis seen in all these uvrD alleles can be

70
complemented by uvrD+. Siegel (1983) found the spontaneous mutator
phenotype in a uvrD::Mu4 fusion strain, prompting him to suggest that
the mutator alleles of uvrD are the null phenotype. The uvrD mutants
isolated in this study, however, are not null alleles. This activity is
demonstrated by a lack nf uvrD+ complementation in SOS expression levels
of the mutant strains, shown in Figure 3. Also the viability of uvrD244
with the repS allele suggests at least some functioning of the mutant
protein. It is possible that the mutations isolated in this study may
not be as defective as uvrDls6 since the basal level of su1A expression
is higher in the uvrD1s6 strain. Alternatively, uvrD may be an
essential gene-- no known uvrD deletions have been constructed.
That the mutator alleles of uvrD sub-induce the SOS response may
affect what is currently thought about the levels of uvrD gene
expression. The uvrD gene is controlled by LexA repressor and is part
of the SOS regu1on. This SOS-mediated regulation was identified by
constructing Mu4 fusions to the uvrD gene and the resultant mutations
are UV sensitive and spontaneous mutators (Siegel, 1983). The level of
uvrD expression is increased 2.s-fo1d when induced with lug/ml Mitomycin
C and s-fold in 1exA71(Def) strains. If, however, the basal level of
SOS expression is increased due to the uvrD fusion mutation, the
induction ratio may actually be greater than what was observed in the
fusion strains.
This study demonstrates that certain defects in the uvrD mutants
can be complemented by a wild-type allele. These defects are UV
sensitivity and spontaneous mutator activity, shown in Figures 2 and 5.
Complementation of the UV sensitivity indicates that DNA repair

71
deficiencies associated with the uvrD mutation are complemented.
Experiments showed that spontaneous mutagenesis was not a result of SOS
mutagenesis (see Table 6), but may be due to a lack of mismatch repair
in the mutant strains (Table 7). This conclusion is in agreements with
previous studies that show the following: 1) mismatch repair deficient
strains (mutH, mutL, mutS or dam mutants) are spontaneous mutators
(Marinus, 1984), 2) evidence suggesting uvrE (now known to be uvrD) and
the mutHLS genes operate within the same pathway (Glickman and Radman,
1980), and 3) uvrD mutants and mutHLS and dam mutants all share the same
mutational spectrum (Cox, 1976; Glickman, 1979). I' conclude that one of
the defects in the newly isolated uvrD mutants is a repair defect which
can be complemented. This defect may be in the ability of UvrD protein
to recognize other repair proteins at DNA lesions. Pseudoreversion
analysis of these mutants by isolating UV resistant strains would
determine other proteins which interact with UvrD during DNA repair.
The mutant uvrD strains have another defect, possibly in DNA
replication, that leads to SOS induction and is not fully complemented
by uvrD+. Evidence presented in this study shows that a lack of DNA
repair functions is not sufficient to sub-induce the SOS response.
Excision repair defective uvrA6 strains and mismatch repair deficient
mutL strains were shown not to cause sub-induction of the sulA fusion.
Furthermore, uvrD3 strains which are deficient in DNA repair (UV
sensitive) do not cause increased expression of sulA.
The viability of uvrD244 in combination with repS suggests that
the mutant UvrD protein is proficient in DNA replication, since a uvrD
rep double mutant of other uvrD alleles cannot be made. I suggest that

72
an altered DNA helicase function during replication, rather than DNA
repair defects, is responsible for the SOS sub-induction seen in this
mutant. Furthermore, finding that the repS strain leads to a low level
of sulA expression suggests that improper helicase function during
replication can produce an inducing signal.
The specific role of DNA helicase II in DNA replication is not
clear. Taucher-Scholz et al. (1983) propose that during DNA replication
Rep and UvrD proteins act jointly on opposite strands to unwind duplex
DNA. This proposal was based on several observations. First, they were
unable to make double mutants of rep and uvrD (although the alleles used
in the attempted construction were not specified). Second, Rep helicase
unwinds DNA in the 3'--> 5' direction, the parental strand leading in
DNA synthesis (Yarranton and Gefter, 1979). Third, UvrD helicase was
thought at the time to unwind DNA on the parental strand for lagging DNA
synthesis, in the 5'--> 3' direction (Kuhn et al., 1979). In addition
to proposing joint action of Rep and UvrD helicases on opposite strands
at the replication fork, Taucher-Scholz et al. (1983) further suggested
that the two helicases could substitute for each other if necessary
(such as when one helicase is mutationally inactivated). More recent
studies, however, report that UvrD translocates on the 3'--> 5'strand,
the same direction as Rep (Matson, 1986). Perhaps UvrD and Rep can
substitute for one another on the 3'--> 5', leading strand during DNA
replication. Recently, DnaB protein has been shown to be a helicase,
unwinding DNA on the lagging strand template, or 5'--> 3' direction
(LeBowitz and McMacken, 1986). Rep, UvrD and DnaB may all function as
DNA helicases during replication.

73
DNA he1icases appear to play a role in signal metabolism and
the SOS response. rep and uvrD mutations have been shown to sub-induce
the SOS response and dnaB(Ts) mutations induce SOS functions at non
permissive temperatures. Another DNA unwinding protein, RecBCD, has
been shown to affect SOS induction as well (see Chapter I). Chaudhury
and Smith (1985) showed that the exonuclease activity of this enzyme was
not needed for SOS induction and suggested that RecBCD DNA unwinding
activity instead was involved in SOS signal generation. He1icases may
be important in regulating the amounts of single-stranded DNA in the
cell, an imbalance of which may be an inducing signal for RecA protein.
Alternatively, they may affect the rate of DNA replication. If
unwinding is impeded, the slowed replication fork movement may produce
an inducing signal, such as a nucleotide cofactor for RecA activation.
Biochemical studies of the mutant UvrD proteins may help to unravel the
alterations in DNA unwinding activity that produce an SOS inducing
signal.
recA mutants
The recA mutations isolated exhibited the strongest SOS
induction in any of the mutants screened. These mutations are of the
PrtC class because they are ~o~ease £onstitutive for assisting in
repressor cleavage. Additionally, the mutants all have increased
spontaneous mutability and are recombination proficient. Finding that
the only fully SOS-induced mutations isolated in this work map to the
recA gene underscores the central role of this gene product in
regulating the SOS response.

74
PrtC mutations have been isolated previously. These include the
initial temperature-sensitive allele, recA441 (tif-1)(Go1dthwait and
Jacob, 1964), the non-Ts PrtC allele, recA730 (Witkin et a1., 1982) and
several isolated by Tessman (Tessman and Peterson, 1985). All of these
alleles share phenotypes similar to the recA mutations I have isolated
including spontaneous phage induction, high levels of SOS expression,
recombination proficiency and spontaneous mutagenesis. It should be
noted that Tessman also isolated some recA(Prtc ) class alleles that were
recombination deficient; however, they cleaved LexA protein to a lesser
extent and did not spontaneously induce prophage (Tessman and Peterson,
1985a).
One of the recA mutations isolated in this study, recA85, had
unusual behavior. This mutation appeared to be inviable in combination
with ssbl, ssbll3 or recB mutations. This is in contrast to another of
the recA mutations isolated, recASl, in which the same double mutant
combinations could be made. In another study, a recA730 mutation was
viable in combination with ssbl or sbll3 mutations (Lieberman and
Witkin, 1983). These results demonstrate that the recA85 defect is
different from the defect in recASl or recA730, although other
phenotypes are similar. It is possible that these different mutations
define different sites on RecA protein that interact with inducing
signal(s) or show different abilities to bind ssDNA.
The relationship between RecA protein and Ssb (single-stranded
DNA binding) protein has been the subject of much investigation. In
vitro studies implicate Ssb protein in assisting strand exchange
reactions of RecA protein in homologous recombination (reviewed by

75
Radding, 1982). Repressor cleavage reactions, however, are enhanced at
optimal Ssb concentrations and inhibited by higher concentrations of Ssb
protein (Resnick and Sussman, 1982). Induction of the SOS response in
vivo is inhibited in ssb1 and ssb113 strains, indicating that this
protein plays a role in signal generation (Ba1uch et a1., 1980a).
Recent work suggests that there is no specific protein-protein
interaction between RecA and Ssb proteins, but that competitive binding
occurs on single-stranded DNA (Kowa1czykowski et a1., 1987). Based on
this evidence, a plausible explanation for the inviability of recA85 and
ssb mutations is that RecA85 protein may bind single-stranded regions of
DNA more tightly than wild-type RecA protein. In mutants with defective
Ssb, RecA85 may outcompete the protein for single-stranded DNA sites,
excluding Ssb. This competition could be lethal since Ssb is an
essential protein and its only known function is binding single-stranded
DNA. Tighter binding of RecA85 to single-stranded DNA could also
explain the constitutive activation seen in this protein.
Biochemical studies would provide more insight into the nature
of the RecA85 defects. Protein DNA binding studies could determine
whether this protein does bind single-stranded DNA more tightly.
Current collaboration with Dr. Patricia Weber (DuPont Corp.) to
investigate the biochemistry of purified RecA85 may provide this
information.

2-aminopurine
Mu.Q. fus ion
mutHLS
recBCD
SOS mutagenesis
uvrABC
76
APPENDIX A
Definitions of genetic terms
DNA base analogue that incorporates as adenine and causes transition mutations.
Gene coding for DNA adenine methylase, a protein involved in mismatch repair.
Gene encoding the repressor of the SOS regu10n.
Gene coding for DNA ligase.
Mu phage which inserts randomly into the chromosome and contains promoterless Lac structural genes (lac ZYA). The Lac genes are expressed when this phage inserts downstream from a promoter in the chromosome.
Genes encoding the mismatch repair proteins.
A gene encoding a key regulatory protein of the SOS response, also involved in strand exchange reactions during homologous recombination.
Genes encoding the multi-subunit protein Exonuclease V. As well as exonuclease activity, this protein has DNA unwinding ability and participates in homologous recombination.
Gene encoding replicase, an ATP-dependent DNA unwinding protein.
An increase in mutagenesis resulting from expression of the SOS genes recA and umuCD.
Gene encoding a cell division inhibitor, also known as sfiA. Mutations in sulA prevent lethal filamentation when the SOS response induced.
Genes encoding a protein complex that perfoms excision repair.
Gene encoding DNA helicese II, an ATP-dependent DNA unwinding protein. This protein also participates in excision repair, RecF recombination and mismatch repair.

77
REFERENCES
Ade1berg, E.A., M.Mande11 and G.Chein Chin Hem. 1965 Optimal conditions for mutagenesis by N-methy1-N-nitro-N-nitrosoguanidine in Escherichia coli K-12. Biochem. Biophys. Res. Commun. 18:788-795.
Bachmann, B.J. 1983 Linkage map of Escherichia coli K-12, Edition 7. Microbio1. Rev. 47:180-230.
Bai1one, A., A.Levine and R.Devoret. 1979 Inactivation of prophage lambda repressor in vivo. J. Mol. Bio. 131:553-572.
Baker, T.A., M.M.Howe and C.A.Gross. 1983 MudX, a derivative of Mug1(lac,Ap ) which makes stable 1acZ fusions at high temperature. J. Bacterio1 156:970-974.
Ba1uch, J., R.Sussman and J.W.Resnick. 1980 Induction of prophage lambda without amplification of recA protein. Mol. Gen. Genet. 178:317-323.
Ba1uch, J., J.W.Chase and R.Sussman. 1980a Synthesis of RecA protein and induction of bacteriophage lambda in single-strand deoxyribonucleic acid-binding protein mutants of Escherichia coli. J. Bacteriol. 144:489-498.
Blanco, M., and L.Pomes. 1977 Prophage induction in Escherichia coli K12 cells deficient in DNA polymerase I. Mol. Gen. Genet. 154:287-292.
Braun, R.E., and A.Wright. 1986 DNA methylation differentially enhances the expression of one of the two E. coli dnaA promoters in vivo and in vitro. Mol. Gen. Genet. 202:246-250.
Casadaban, M.J., and S.N.Cohen. 1979 Lactose genes fused to exogenous promoters in one step using a Mu-1ac bacteriophage: in vivo probe for transcriptional control sequences. Proc. Natl. Acad. Sci. USA 76:4530-4533.
Casaregola, S., R.D'Ari and O.Huisman. 1982 Role of DNA replication in the induction and turn-off of the SOS response in Escherichia coli. Mol. Gen. Genet. 185:440-444.
Chaudhury, A.M., and G.R.Smith. 1985 Role of Escherichia coli RecBC enzyme in SOS induction. Mol. Gen. Genet. 201:525-528.
Cohen, S., B.J.Knoll, J.W.Little and D.W.Mount. 1981 Preferential cleavage of phage lambda repressor monomers by RecA protease. Nature 294:182-184.

78
Cole, S.T. 1983 Characterization of the promoter for the lexA regulated sulA gene of Escherichia coli. Mol. Gen. Genet. 189:400-404.
Cox, E.C. 1976 Bacterial mutator genes and the'control of spontaneous mutation. Ann. Rev. Genet. 10:135-156.
Craig, N.L., and J.W.Roberts. 1980 ~ coli RecA protein-directed cleavage of phage lambda repressor requires polynucleotide. Nature 283:26-30.
Craig, N.L., and J.W.Roberts. 1981 Function of nucleoside triphosphate and polynucleotide in Escherichia coli RecA protein-directed cleavage of phage lambda repressor. J. BioI. Chem. 256:8039-8044.
Craig, R.L., J.A. Arraj and M.G. Marinus. 1984 Induction of damage inducible (SOS) repair in dam mutants of Escherichia coli exposed to 2-aminopurine. Mol. Gen. Genet. 194:539-540.
Crowl, R.M., R.P. Boyce, and H. Echols. 1981 Repressor cleavage as a prophage induction mechanism: hypersensitivity of a mutant AcI protein to RecA-mediated proteolysis. J. Mol. Bio. 152:815-820.
D'Ari, R., and O.Huisman. 1982 DNA replication and indirect induction of the SOS response in Escherichia coli. Biochimie 64:623-627.
Das, S.K., and L.A.Loeb. 1984 UV irradiation alters deoxynucleoside triphosphate pools in Escherichia coli. Mut. Res. 131:97-100.
Davis, R.W., D.Botstein and J.R.Roth. 1980 Advanced Bacterial Genetics. New York: Cold Spring Harbor.
Geider, K., and H.Hoffmann-Berling. 1981 Proteins controlling the helical structure of DNA. Ann. Rev. Biochem. 50:233-260.
George, J., R.Devoret and M.Radman. 1974 Indirect ultravioletreactivation of phage lambda. Proc. Natl. Acad. Sci. USA 71:144-147.
George, J., M.Castellazzi and G.Buttin. 1975 Prophage induction and cell division in ~ coli. III. Mutations sfiA and sfiB restore division in tif and Ion strains and permit the expression of mutator properties of tif. Mol. Gen. Genet. 140:309-332.
Glickman, B.W., and M.Radman. 1980 Escherichia coli mutator mutants deficient in methylation-instructed DNA mismatch correction. Proc. Natl. Acad. Sci. USA 77:1063-1067.
Glickman, B.W. 1979 Spontaneous mutagenesis in ~ coli strains lacking 6-methyladenine residues in their DNA. An altered mutational spectrum in dam mutants .. Mutat. Res. 61:153-162.

79
Goldthwait, D., and F.Jacob. 1964 Sur Ie mechanisme de l'induction du developpment du prophage chez les bacteries lysogenes. C.R. Acad. Sci. 259:661-664.
Gottesman, M.M., M.L.Hicks and M.Gellert. 1973 Genetics and function of DNA ligase in Escherichia coli. J. Mol. Bio. 77:531-547.
Gudas, L.J., and D.W.Mount. 1977 Identification of the recA (tif) gene product of Escherichia coli. Proc. Natl. Acad. Sci. USA 74:5280-5284.
Horii, Z.I., and A.J.Clark. 1971 Genetic analysis of the RecF pathway to genetic recombination in Escherichia coli: Isolation and characterization of mutants. J. Mol. Bio. 80:327-344.
Horii, T., T.Ogawa, T.Nakatani, T.Hase. H.Matsubara and H.Ogawa. 1981 Regulation of SOS functions: Purification of h coli LexA protein and determination of its specific site cleaved by the RecA protein. Cell 27: 515-522.
Huisman, 0., and R.D'Ari. 1981 An inducible DNA replication-cell division coupling mechanism in ~ coli. Nature 290:797-799.
Irbe, R.M., L.M.Morin and M.Oishi. 1981 Prophage (080) induction in Escherichia coli K-12 by specific deoxyoligonucleotides. Proc. Natl. Acad. Sci. USA 78:138-142.
Kenyon, C.J., and G.C.Walker. 1980 DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc. Natl. Acad. Sci. USA 77:2819-2823.
Kowalczykowski, S.C., R.Clow, R.Somani and A.Varghese. 1987 Effects of the Escherichia coli SSB protein on the binding of Escherichia coli RecA protein to single-stranded DNA. J. Mol. BioI. 193:81-95.
Kuhn. B., M.Abdel-Monem and J.Hoffmann-Berling. 1979 Evidence for two mechanisms for DNA unwinding catalyzed by DNA helicases. J. BioI. Chem. 254:1134311350-11350.
LeBowitz, J.H., and R.McMacken. 1986 The Escherichia coli DnaB replication protein is a DNA helicase. J. BioI. Chem. 261:4738-4748.
Lieberman, H.B., and E.M.Witkin. 1983 DNA degradation, UV sensitivity and SOS-mediated mutagenesis in strains of Escherichia coli deficient in single-strand DNA binding protein: Effects of mutations and treatments that alter levels of exonuclease V or RecA protein. Mol. Gen. Genet. 190:92-100.
Little, J.W., and P.C.Hanawalt. 1977 Induction of protein X in Escherichia £Q!i. Mol. Gen. Genet. 150:237-248.

Little, J.W., and D.W.Mount. 1982 The SOS regulatory system of Escherichia coli. Cell 29:11-22.
80
Little, J.W., S.H.Edmiston, L.Z.Pace1Ii and D.W.Mount." 1980 Cleavage of the Escherichia coli LexA protein by the RecA protease. Proc. Natl. Acad. Sci. USA 77:3225-3229.
Little, J.W. 1983 The SOS regulatory system: Control of its state by the level of RecA protease. J. Mol. BioI. 167:791-808.
Lu, A.L., S.Clark and P.Modrich. 1983 Methyl-directed repair of DNA base-pair mismatches in vitro. Proc. Natl. Acad. Sci. USA 80:4639-4643.
Lu, C., R. Scheuermann, and H. Echols. 1986 Capacity of RecA protein to bind preferentially to UV lesions and inhibit the editing (E)
subunit of DNA polymerase III: a possible mechanism for SOS-induced targeted mutagenesis. Proc. Nat1. Acad. Sci. USA 83:619-623.
Maples, V.F., and S.R.Kushner. 1982 DNA repair in Escherichia coli: Identification of the uvrD gene product. Proc. Natl. Acad. Sci. USA 79:5616-5620.
Marinus, M.G., and N.R.Morris. 1974 Biological function for 6-methyladenine residues in the DNA of Escherichia coli K-12. J. Mol. Bio. 85:309-322.
Marinus, M.G., and N.R.Morris. 1975 Pleiotropic effects of a DNA adenine" methylation mutation (dam-) in Escherichia coli K-12. Mut. Res. 28:15-26.
Marinus, M. 1984 Methylation of procaryotic DNA. In DNA Methylation. A.Razin, M.Cedar, A.Riggs, (eds.) Pp. 81-109. New York. SpringerVerlag.
Marinus, M.G. 1985 DNA methylation influences trpR promoter activity in Escherichia coli K-12. Mol. Gen. Genet. 200:185-186.
Matson, S.W. 1986 Escherichia coli he1icase II (uvrD gene product) trans locates unidirectiona1ly in a 3' to 5' direction. J. BioI. Chem. 261:10169-10175.
McEntee, K., and G.M.Weinstock. 1981 tif-1 mutation alters polynucleotide recognition by the RecA protein of Escherichia coli. Proc. Natl. Acad. Sci. USA 75:6061-6065.
McGraw. B.R., and M.G.Marinus. 1980 Isolation and characterization of dam+ revertants and suppressor mutations that modify secondary phenotypes of dam-3 strains of Escherichia coli K-12. Mol. Gen. Genet. 178:309-315.

McEntee, K., and G.M.Weinstock. 1981 tif-1 mutation alters polynucleotide recognition by the RecA protein of Escherichia coli. Proc. Nat1. Acad. Sci. USA 75:6061-6065.
81
McPartland, A., L.Green and H.Echo1s. 1980 Control of recA gene RNA in ~ coli: regulatory and signal genes. Cell 20:731-737.
Miller, J.H. 1972 Experiments in Molecular Genetics. New York: Cold Spring Harbor.
Morand./P., M.B1anco and R.Devoret. 1977 Characterization of 1exB mutations in Escherichia coli K-12. J. Bacterio1. 131:572-582.
Mount, D.W. 1979 Isolation and characterization of mutants of recA which synthesize a hyperactive RecA protein. Virology 98:484-488.
Oeda, K., T.Horiuchi and M.Sekiguchi. 1982 The uvrD gene of ~ coli encodes a DNA-dependent ATPase. Nature 298:98-100.
Ogawa, H., K.Shimada and J.Tomizawa. 1968 Studies on radiationsensitive mutants of ~ coli. I. Mutants defective in the repair synthesis. Mol. Gen. Genet. 101:227-244.
Ossanna, N., K.R.Peterson and D.W.Mount. 1986 Genetics of DNA repair in bacteria. TIGS 2:55-58.
Ossanna, N., K.R.Peterson and D.W.Mount. 1987 UV-inducib1e SOS response in Escherichia coli. Photochem Photobio1 45:905-908.
Peterson, K.R., N. Ossanna, and D.W. Mount. 1988a K-12 1exA2 gene encodes a hypoc1eavab1e repressor. (in press).
The Escherichia coli J. Bacterio1. 170:
Peterson, K.R., N.Ossanna, A.T.Th1iveris, D.G.Ennis and D.W.Mount. 1988 Derepression of specific genes promotes DNA repair and mutagenesis in Escherichia ,coli. J. Bacterio1. 170:1-4.
Peterson, K.R., K.F.Wertman, D.W.Mount and M.G.Marinus. 1985 Viability of Escherichia coli K-12 DNA adenine methylase (dam) mutants requires increased expression of specific genes in the SOS regu1on. Mol. Gen. Genet. 201:14-19.
Phizicky, E.M., and J.W.Roberts. 1981 Induction of SOS functions: Regulation of proteolytic activity of ~ coli RecA protein by interaction with DNA and nucleoside triphosphate. Cel125:259-267.
Pukki1a, P.J., J.Peterson, G.Herman, P.Modrich and M.Mese1son. 1983 Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics 104:571-582.

82
Quillardet, P., P.L.Moreau, H.Ginsburg, D.W.Mount and R.Devoret. 1982 Cell survival, UV-reactivation and induction of prophage lambda in Escherichia coli K12 overproducing RecA protein. Mol. Gen. Genet. 188:37-43.
Radding, C. 1982 Homologous pairing and strand exchange in genetic recombination. Ann. Rev. Genet. 16:405-437.
Radman, M. and R. Wagner. 1986 Mismatch repair in Escherichia coli. Ann. Rev. Genet. 20:523-538.
Resnick, J., and R.Susssman. 1982 Escherichia coli single-strand DNA binding protein from wild-type and lexCl13 mutant affects in vitro proteolytic cleavage of phage lambda repressor. Proc. Natl. Acad. Sci. USA 79:2832-2835.
Roberts, J.W., and R.Devoret. 1983 Lysogenic induction. In Lambda II. R.W. Hendrix , J.W.Roberts, F.W.Stahl, R.A.Weisberg, (eds.) Pp. 123-144. Cold Spring Harbor. Cold Spring Harbor Laboratory.
Rupp, W.D., and P.Howard-Flanders. 1968 Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Bio. 31:291-304.
Schuster, H., D.Beyersmann, M.Mikolajczyk and M.Sch1icht. 1973 Prophage induction by high temperature in thermosensitive dna mutants lysogenic for bacteriophage lambda. J. Virol. 11:879-885.
Siegel, E.C. 1973 Ultraviolet-sensitive mutator strain of Escherichia coli K-12. J. Bacteriol. 113:145-160.
Siegel, E.C. 1981 Complementation studies with the repair-deficient uvrD3, uvrE156, and recL152 mutations in Escherichia coli. Mol. Gen. Genet. 184:526-530.
Siegel, E.C. 1983 The Escherichia coli uvrD gene is inducible by DNA damage. Mol. Gen. Genet. 191:397-400.
Smirnov, G.B., and A.G.Skavronskaya. 1971 Location of uvr502 mutation on the chromosome of Escherichia coli K-12. Mol. Gen. Genet. 113:217-221.
Smith, C.L., and M.Oishi. 1978' Early events and mechanisms in the induction of bacterial SOS functions: Analysis of the phage repressor inactivation process in vivo. Proc. Natl. Acad. Sci. USA 75:1657-1661.
Smith, G., 1983 General recombination. In Lambda II. R.W.Hendrix, J.W.Roberts, F.W.Stah1, R.A.Weisberg, (eds.) Pp. 175-209. New York. Cold Spring Harbor Laboratories.

83
Taucher-Scholz, G., M.Abde1-Monem and H.Hoffmann-Ber1ing. 1983 Functions of DNA he1icases in Escherichia coli. In Mechanisms of DNA replication and recombination. N.R.Cozzare11i, (ed.) Pp. 65-76. New York. Alan R. Liss, Inc •.
Tessman, E.S., and P.Peterson. 1985 Plaque color method for rapid isolation of novel recA mutants of Escherichia coli K-12: New classes of protease-constitutive recA mutants. J. Bacterio1. 163:677-687.
Tessman, E.S., and P.K.Peterson. 1985a Isolation of Proteaseproficient, recombinase-deficient recA mutants of Escherichia coli K-12. J. Bacterio1. 163:688-695.
Walker, G.C. 1984 Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbio1. Revs. 48:60-93.
Wang, W.B., and E.S.Tessman. 1986 Location of functional regions of the Escherichia coli RecA protein by DNA sequence analysis of RecA protease-constitutive mutants. J. Bacterio1. 168:901-910.
Witkin, E.M., and T.Kogoma. 1984 Involvement of the activated form of RecA protein in SOS mutagenesis and stable DNA replication in Escherichia coli. Proc. Nat1. Acad. Sci. USA 81:7539-7543.
Witkin, E.M., J.O.McCa11, M.R.Vo1kert and I.E.Wermundsen. 1982 Constitutive expression of SOS functions and modulation of mutagenesis resulting from resolution of genetic instability at or near the recA locus of Escherichia coli. Mol. Gen. Genet. 185:43-50.
Witkin, E. 1976 Ultraviolet mutagenesis and inducb1e DNA repair in Escherichia coli. Bact. Revs. 40:869-907.
Yarranton, G.T., and M.L.Gefter. 1979 Enzyme-catalyzed DNA unwinding studies on Escherichia coli Rep protein. Proc. Nat1. Acad. Sci. USA 76:1658-16662.





























