Embed Size (px)
Citation preview
Tutorial: Basics of electronic structure theoryIntroduction
Ralf Gehrke and Alexandre Tkatchenko
Fritz-Haber-Institut der Max-Planck-GesellschaftBerlin, 23th June 2009
Hands-on Tutorial on Ab Initio Molecular Simulations:Toward a First-Principles Understanding of Materials Properties and Functions
Goal of this tutorial� binding energy
d [Å]� bond distances
� vibrational spectra 2.85 Å
2.19 Å2.19 Å
Si3 PBE-GGA
N2
d0 = ?N2

Goal of this tutorial� structure prediction X(+)Si3 X=Sc, Ti, V, Cr
mpirun -np 2 aims.workshop.mpi.x | tee calculation.out
� two input files are required in the working directory (units: eV, Å):control.ingeometry.in
� FHI-aims is simply called and main output is written to STDOUT
� further output files are generated if requiredcharge density plotsdensity of states
� for each exercise, a separate directory should be created— mkdir tutorial1� cd tutorial1� mkdir N2
vi, emacs,...
General aspects

General aspects� directory with reference files, e.g. /usr/local/aimsfiles/tutorial1/N2
geometry.in.N2.ref control.in.pw-lda.coarse.refN.pbe.out.ref control.in.basic
� the aims-binary as well as several tools are in the binary pathmoldenvmdjmolgdis
� additional auxiliary scripts needed for this tutorial can be found in
/usr/local/aimsfiles/scripts
visualization
xmgrace gnuplot emacs vi kwrite
editor
plotting tools
� FHI-aims manual can be found on your desktop for reference
The geometry.in file
The input files
atom 0.0 0.0 0.0 Natom 1.1 0.0 0.0 N
Geometry specification� Atoms� Periodic boundary conditions� Initialization of charge/spin density
Simplest example (N2): More complex case (Cu fcc):atom 0.0 0.0 0.0 Culattice_vector 1.8 1.8 0.0lattice_vector 0.0 1.8 1.8lattice_vector 1.8 0.0 1.8

The control.in fileTwo sections:
� General section (method, spin, etc.)� Species specification (basis set, etc.)
Example:xc pw-ldaspin collinearmultiplicity 1relativistic nonecharge 0empty_states 5#occupation_type gaussian 0.001mixer pulay n_max_pulay 10 charge_mix_param 0.2
species N
The input files
The control.in fileTwo sections:
� General section (method, spin, etc.)� Species specification (basis set, etc.)
Example:
nin���1 ��n in
opt� R n inopt �
ninopt��
�
�max
�n in�� �
f � ��E F� ��1
2 �1�erf ��EF� ��
xc pw-ldaspin collinearmultiplicity 1relativistic nonecharge 0empty_states 5#occupation_type gaussian 0.001mixer pulay n_max_pulay 10 charge_mix_param 0.2
species N
see manual, chapter 2.3
The input files

The species defaults
� “light” : fast prerelaxations should always be followed by a tightly converged post-
processing calculation
� “tight” : tightly converged settings, the basis set might still need to be increased for light elements, though
/usr/local/aimsfiles/species_defaults/light
/usr/local/aimsfiles/species_defaults/tight
to be pasted into the control.in file
The input files
The main output The FHI-aims output
------------------------------------------------------------ Invoking FHI-aims ... Version 020109 Compiled on 2009/02/03 at 16:00:22
When using FHI-aims, please cite the following reference:
Volker Blum, Ralf Gehrke, Felix Hanke, Paula Havu, Ville Havu, Xinguo Ren, Karsten Reuter, and Matthias Scheffler, 'Ab Initio Molecular Simulations with Numeric Atom-Centered Orbitals: FHI-aims', Computer Physics Communications [volume], [page] (2008).
------------------------------------------------------------
Date : 20090608, Time : 145624.551 Time zero on CPU 1 : 0.199900000000000E-02 s. Internal wall clock time zero : 13704984.551 s.
Obtaining array dimensions for all initial allocations: Parsing control.in ... Parsing geometry.in ... Basic array size parameters: | Number of species : 1 | Number of atoms : 2 | Max. basis fn. angular momentum : 2 | Max. atomic/ionic basis occupied n: 2 | Max. number of basis fn. types : 2 | Max. radial fns per species/type : 3 | Max. logarithmic grid size : 1290 | Max. radial integration grid size : 71 | Max. angular integration grid size: 302 | Max. angular grid division number : 8 | Radial grid for Hartree potential : 1290 | Number of spin channels : 2
------------------------------------------------------------ Reading file control.in.------------------------------------------------------------

The main output The FHI-aims output
------------------------------------------------------------ Invoking FHI-aims ... Version 020109 Compiled on 2009/02/03 at 16:00:22
When using FHI-aims, please cite the following reference:
Volker Blum, Ralf Gehrke, Felix Hanke, Paula Havu, Ville Havu, Xinguo Ren, Karsten Reuter, and Matthias Scheffler, 'Ab Initio Molecular Simulations with Numeric Atom-Centered Orbitals: FHI-aims', Computer Physics Communications [volume], [page] (2008).
------------------------------------------------------------
Date : 20090608, Time : 145624.551 Time zero on CPU 1 : 0.199900000000000E-02 s. Internal wall clock time zero : 13704984.551 s.
Obtaining array dimensions for all initial allocations: Parsing control.in ... Parsing geometry.in ... Basic array size parameters: | Number of species : 1 | Number of atoms : 2 | Max. basis fn. angular momentum : 2 | Max. atomic/ionic basis occupied n: 2 | Max. number of basis fn. types : 2 | Max. radial fns per species/type : 3 | Max. logarithmic grid size : 1290 | Max. radial integration grid size : 71 | Max. angular integration grid size: 302 | Max. angular grid division number : 8 | Radial grid for Hartree potential : 1290 | Number of spin channels : 2
------------------------------------------------------------ Reading file control.in.------------------------------------------------------------
introduction
internal parameters
Invoking FHI-aims ...
------------------------------------------------------------ Reading file control.in.------------------------------------------------------------ XC: Using PBE gradient-corrected functionals. Spin treatment: Spin density functional theory - collinear spins. Non-relativistic treatment of kinetic energy. Occupation type: Gaussian broadening, width = 0.100000E-01 eV. Using pulay charge density mixing. Pulay mixing - number of memorized iterations: 10 Charge density mixing - mixing parameter: 0.5000 Convergence accuracy of self-consistent charge density: 0.1000E-04 Convergence accuracy of sum of eigenvalues: 0.1000E-02 Convergence accuracy of total energy: 0.1000E-05 Convergence accuracy of forces: 0.1000E-03 Maximum number of s.-c. iterations : 100 Kohn-Sham eigenvalues and eigenfunctions calculated by lapack diagonalisation. Number of empty states per atom: 5 Geometry relaxation: BFGS (simple quadratic extrapolation). Convergence accuracy for geometry relaxation: Maximum force < 0.100000E-01 eV/A. Reading configuration options for species N . | Found nuclear charge : 7
The main output The FHI-aims output
------------------------------------------------------------ Reading geometry description geometry.in.------------------------------------------------------------ Input structure read successfully. The structure contains 2 atoms, and a total of 14.000 electrons. Input geometry: | No unit cell requested. | Atomic structure: | Atom x [A] y [A] z [A] | 1: Species N 0.000000 0.000000 0.000000 | 2: Species N 0.000000 0.000000 1.100000 Initial moments and charges: | Atom Moment Charge Species | 1 (Hund's rule) 0.000000E+00 N | 2 (Hund's rule) 0.000000E+00 N Structure-dependent array size parameters: | Number of distinct atom types in initial rho : 1 | Maximum number of distinct radial functions : 6 | Maximum number of basis functions : 28 | Number of Kohn-Sham states (occupied + empty): 12------------------------------------------------------------

------------------------------------------------------------ Reading file control.in.------------------------------------------------------------ XC: Using PBE gradient-corrected functionals. Spin treatment: Spin density functional theory - collinear spins. Non-relativistic treatment of kinetic energy. Occupation type: Gaussian broadening, width = 0.100000E-01 eV. Using pulay charge density mixing. Pulay mixing - number of memorized iterations: 10 Charge density mixing - mixing parameter: 0.5000 Convergence accuracy of self-consistent charge density: 0.1000E-04 Convergence accuracy of sum of eigenvalues: 0.1000E-02 Convergence accuracy of total energy: 0.1000E-05 Convergence accuracy of forces: 0.1000E-03 Maximum number of s.-c. iterations : 100 Kohn-Sham eigenvalues and eigenfunctions calculated by lapack diagonalisation. Number of empty states per atom: 5 Geometry relaxation: BFGS (simple quadratic extrapolation). Convergence accuracy for geometry relaxation: Maximum force < 0.100000E-01 eV/A. Reading configuration options for species N . | Found nuclear charge : 7
The main output The FHI-aims output
------------------------------------------------------------ Reading geometry description geometry.in.------------------------------------------------------------ Input structure read successfully. The structure contains 2 atoms, and a total of 14.000 electrons. Input geometry: | No unit cell requested. | Atomic structure: | Atom x [A] y [A] z [A] | 1: Species N 0.000000 0.000000 0.000000 | 2: Species N 0.000000 0.000000 1.100000 Initial moments and charges: | Atom Moment Charge Species | 1 (Hund's rule) 0.000000E+00 N | 2 (Hund's rule) 0.000000E+00 N Structure-dependent array size parameters: | Number of distinct atom types in initial rho : 1 | Maximum number of distinct radial functions : 6 | Maximum number of basis functions : 28 | Number of Kohn-Sham states (occupied + empty): 12------------------------------------------------------------
summary of control.in fileReading file control.in.
------------------------------------------------------------ Reading file control.in.------------------------------------------------------------ XC: Using PBE gradient-corrected functionals. Spin treatment: Spin density functional theory - collinear spins. Non-relativistic treatment of kinetic energy. Occupation type: Gaussian broadening, width = 0.100000E-01 eV. Using pulay charge density mixing. Pulay mixing - number of memorized iterations: 10 Charge density mixing - mixing parameter: 0.5000 Convergence accuracy of self-consistent charge density: 0.1000E-04 Convergence accuracy of sum of eigenvalues: 0.1000E-02 Convergence accuracy of total energy: 0.1000E-05 Convergence accuracy of forces: 0.1000E-03 Maximum number of s.-c. iterations : 100 Kohn-Sham eigenvalues and eigenfunctions calculated by lapack diagonalisation. Number of empty states per atom: 5 Geometry relaxation: BFGS (simple quadratic extrapolation). Convergence accuracy for geometry relaxation: Maximum force < 0.100000E-01 eV/A. Reading configuration options for species N . | Found nuclear charge : 7
The main output The FHI-aims output
------------------------------------------------------------ Reading geometry description geometry.in.------------------------------------------------------------ Input structure read successfully. The structure contains 2 atoms, and a total of 14.000 electrons. Input geometry: | No unit cell requested. | Atomic structure: | Atom x [A] y [A] z [A] | 1: Species N 0.000000 0.000000 0.000000 | 2: Species N 0.000000 0.000000 1.100000 Initial moments and charges: | Atom Moment Charge Species | 1 (Hund's rule) 0.000000E+00 N | 2 (Hund's rule) 0.000000E+00 N Structure-dependent array size parameters: | Number of distinct atom types in initial rho : 1 | Maximum number of distinct radial functions : 6 | Maximum number of basis functions : 28 | Number of Kohn-Sham states (occupied + empty): 12------------------------------------------------------------
summary of control.in file
summary of geometry.in fileReading geometry description geometry.in.

The main output The FHI-aims output
------------------------------------------------------------ Begin self-consistency loop: Initialization.
Date : 20090608, Time : 145624.781------------------------------------------------------------
Initializing index lists of integration centers etc. from given atomic structure: | Number of centers in hamiltonian partition_tab : 2 | Number of centers in hartree potential : 2 | Number of centers in hartree multipole : 2 | Number of centers in electron density summation: 2 | Number of centers in basis integrals : 2 | Number of centers in integrals : 2 Partitioning the integration grid into batches with maxmin method. | Maximal weight for a single point: 1.000 | Minimal weight for a single point: 0.000 | Number of batches: 499 | Maximal batch size: 141 | Minimal batch size: 50 | Average batch size: 63.431 | Standard deviation of batch sizes: 10.671 Initializing partition tables, free-atom densities, potentials, etc. across the integration grid (initialize_grid_storage). | Net number of integration points: 31652 | of which are non-zero points : 29382 Obtaining max. number of non-zero basis functions in each batch (get_n_compute_maxes). | Maximal number of non-zero basis functions: 28 in task 0
------------------------------------------------------------ Preparing all fixed parts of the calculation.------------------------------------------------------------ Determining machine precision: 2.225073858507201E-308 Setting up grids for atomic and cluster calculations.
Creating wave function, potential, and density for free atoms.
Species: N
List of occupied orbitals and eigenvalues: n l energy [Ha] energy [eV] 1 0 -14.128445 -384.4546 2 0 -0.681395 -18.5417 2 1 -0.260121 -7.0782
Spin-polarized or charged system: Charge density initialized according to selected moments and charges.
------------------------------------------------------------ Preparing all fixed parts of the calculation.------------------------------------------------------------ Determining machine precision: 2.225073858507201E-308 Setting up grids for atomic and cluster calculations.
Creating wave function, potential, and density for free atoms.
Species: N
List of occupied orbitals and eigenvalues: n l energy [Ha] energy [eV] 1 0 -14.128445 -384.4546 2 0 -0.681395 -18.5417 2 1 -0.260121 -7.0782
Spin-polarized or charged system: Charge density initialized according to selected moments and charges.
The main output The FHI-aims output
------------------------------------------------------------ Begin self-consistency loop: Initialization.
Date : 20090608, Time : 145624.781------------------------------------------------------------
Initializing index lists of integration centers etc. from given atomic structure: | Number of centers in hamiltonian partition_tab : 2 | Number of centers in hartree potential : 2 | Number of centers in hartree multipole : 2 | Number of centers in electron density summation: 2 | Number of centers in basis integrals : 2 | Number of centers in integrals : 2 Partitioning the integration grid into batches with maxmin method. | Maximal weight for a single point: 1.000 | Minimal weight for a single point: 0.000 | Number of batches: 499 | Maximal batch size: 141 | Minimal batch size: 50 | Average batch size: 63.431 | Standard deviation of batch sizes: 10.671 Initializing partition tables, free-atom densities, potentials, etc. across the integration grid (initialize_grid_storage). | Net number of integration points: 31652 | of which are non-zero points : 29382 Obtaining max. number of non-zero basis functions in each batch (get_n_compute_maxes). | Maximal number of non-zero basis functions: 28 in task 0
Preparing all fixed parts of the calculation.
geometry-independent preparationsbasis set generation

The main output------------------------------------------------------------ Preparing all fixed parts of the calculation.------------------------------------------------------------ Determining machine precision: 2.225073858507201E-308 Setting up grids for atomic and cluster calculations.
Creating wave function, potential, and density for free atoms.
Species: N
List of occupied orbitals and eigenvalues: n l energy [Ha] energy [eV] 1 0 -14.128445 -384.4546 2 0 -0.681395 -18.5417 2 1 -0.260121 -7.0782
Spin-polarized or charged system: Charge density initialized according to selected moments and charges.
The FHI-aims output
------------------------------------------------------------ Begin self-consistency loop: Initialization.
Date : 20090608, Time : 145624.781------------------------------------------------------------
Initializing index lists of integration centers etc. from given atomic structure: | Number of centers in hamiltonian partition_tab : 2 | Number of centers in hartree potential : 2 | Number of centers in hartree multipole : 2 | Number of centers in electron density summation: 2 | Number of centers in basis integrals : 2 | Number of centers in integrals : 2 Partitioning the integration grid into batches with maxmin method. | Maximal weight for a single point: 1.000 | Minimal weight for a single point: 0.000 | Number of batches: 499 | Maximal batch size: 141 | Minimal batch size: 50 | Average batch size: 63.431 | Standard deviation of batch sizes: 10.671 Initializing partition tables, free-atom densities, potentials, etc. across the integration grid (initialize_grid_storage). | Net number of integration points: 31652 | of which are non-zero points : 29382 Obtaining max. number of non-zero basis functions in each batch (get_n_compute_maxes). | Maximal number of non-zero basis functions: 28 in task 0
Begin self-consistency loop: Initialization.
geometry-independent preparationsbasis set generation
geometry-dependent preparationsintegration gridinitialization of charge density
The main output----------------------------------------------------------- Begin self-consistency iteration # 1
Date : 20090608, Time : 145625.136------------------------------------------------------------ Evaluating new KS density. Pulay mixing of updated and previous charge densities.
The FHI-aims output
| --------------------------- | Total energy : -109.38703872 Ha -2976.57276987 eV | Total energy, T -> 0 : -109.38703872 Ha -2976.57276987 eV | Free energy : -109.38703872 Ha -2976.57276987 eV Derived energy quantities: | Kinetic energy : 109.65005028 Ha 2983.72967863 eV | Electrostatic energy : -205.37638266 Ha -5588.57571571 eV | Energy correction for multipole | error in Hartree potential : 0.00062326 Ha 0.01695968 eV | Sum of eigenvalues per atom : -839.30637513 eV | Total energy (T->0) per atom : -1488.28638494 eV | Free energy per atom : -1488.28638494 eV
Self-consistency convergence accuracy: | Change of charge/spin density : 0.2057E+00 0.9030E+00 | Change of sum of eigenvalues : 0.3707E+02 eV | Change of total energy : -.4628E+01 eV
End self-consistency iteration # 1 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.366 s 0.366 s Charge density update : 0.117 s 0.117 s Density mixing : 0.008 s 0.008 s Hartree multipole update : 0.013 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.154 s 0.154 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.000 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 2
Date : 20090608, Time : 145625.502------------------------------------------------------------

The main output----------------------------------------------------------- Begin self-consistency iteration # 1
Date : 20090608, Time : 145625.136------------------------------------------------------------ Evaluating new KS density. Pulay mixing of updated and previous charge densities.
The FHI-aims output
Begin self-consistency iteration # 1
| --------------------------- | Total energy : -109.38703872 Ha -2976.57276987 eV | Total energy, T -> 0 : -109.38703872 Ha -2976.57276987 eV | Free energy : -109.38703872 Ha -2976.57276987 eV Derived energy quantities: | Kinetic energy : 109.65005028 Ha 2983.72967863 eV | Electrostatic energy : -205.37638266 Ha -5588.57571571 eV | Energy correction for multipole | error in Hartree potential : 0.00062326 Ha 0.01695968 eV | Sum of eigenvalues per atom : -839.30637513 eV | Total energy (T->0) per atom : -1488.28638494 eV | Free energy per atom : -1488.28638494 eV
Self-consistency convergence accuracy: | Change of charge/spin density : 0.2057E+00 0.9030E+00 | Change of sum of eigenvalues : 0.3707E+02 eV | Change of total energy : -.4628E+01 eV
End self-consistency iteration # 1 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.366 s 0.366 s Charge density update : 0.117 s 0.117 s Density mixing : 0.008 s 0.008 s Hartree multipole update : 0.013 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.154 s 0.154 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.000 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 2
Date : 20090608, Time : 145625.502------------------------------------------------------------
The main output----------------------------------------------------------- Begin self-consistency iteration # 1
Date : 20090608, Time : 145625.136------------------------------------------------------------ Evaluating new KS density. Pulay mixing of updated and previous charge densities.
The FHI-aims output
| --------------------------- | Total energy : -109.38703872 Ha -2976.57276987 eV | Total energy, T -> 0 : -109.38703872 Ha -2976.57276987 eV | Free energy : -109.38703872 Ha -2976.57276987 eV Derived energy quantities: | Kinetic energy : 109.65005028 Ha 2983.72967863 eV | Electrostatic energy : -205.37638266 Ha -5588.57571571 eV | Energy correction for multipole | error in Hartree potential : 0.00062326 Ha 0.01695968 eV | Sum of eigenvalues per atom : -839.30637513 eV | Total energy (T->0) per atom : -1488.28638494 eV | Free energy per atom : -1488.28638494 eV
Self-consistency convergence accuracy: | Change of charge/spin density : 0.2057E+00 0.9030E+00 | Change of sum of eigenvalues : 0.3707E+02 eV | Change of total energy : -.4628E+01 eV
End self-consistency iteration # 1 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.366 s 0.366 s Charge density update : 0.117 s 0.117 s Density mixing : 0.008 s 0.008 s Hartree multipole update : 0.013 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.154 s 0.154 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.000 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 2
Date : 20090608, Time : 145625.502------------------------------------------------------------
Total energyTotal energy, T -> 0Free energy F�E�� S �� ,{ f n}�
E��0�
12�F ����E ����

The main output----------------------------------------------------------- Begin self-consistency iteration # 1
Date : 20090608, Time : 145625.136------------------------------------------------------------ Evaluating new KS density. Pulay mixing of updated and previous charge densities.
The FHI-aims output
| --------------------------- | Total energy : -109.38703872 Ha -2976.57276987 eV | Total energy, T -> 0 : -109.38703872 Ha -2976.57276987 eV | Free energy : -109.38703872 Ha -2976.57276987 eV Derived energy quantities: | Kinetic energy : 109.65005028 Ha 2983.72967863 eV | Electrostatic energy : -205.37638266 Ha -5588.57571571 eV | Energy correction for multipole | error in Hartree potential : 0.00062326 Ha 0.01695968 eV | Sum of eigenvalues per atom : -839.30637513 eV | Total energy (T->0) per atom : -1488.28638494 eV | Free energy per atom : -1488.28638494 eV
Self-consistency convergence accuracy: | Change of charge/spin density : 0.2057E+00 0.9030E+00 | Change of sum of eigenvalues : 0.3707E+02 eV | Change of total energy : -.4628E+01 eV
End self-consistency iteration # 1 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.366 s 0.366 s Charge density update : 0.117 s 0.117 s Density mixing : 0.008 s 0.008 s Hartree multipole update : 0.013 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.154 s 0.154 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.000 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 2
Date : 20090608, Time : 145625.502------------------------------------------------------------
Total energyTotal energy, T -> 0Free energy
Total energy
F�E�� S �� ,{ f n}�
E��0�
12�F ����E ����
clusters !
The main output----------------------------------------------------------- Begin self-consistency iteration # 1
Date : 20090608, Time : 145625.136------------------------------------------------------------ Evaluating new KS density. Pulay mixing of updated and previous charge densities.
The FHI-aims output
| --------------------------- | Total energy : -109.38703872 Ha -2976.57276987 eV | Total energy, T -> 0 : -109.38703872 Ha -2976.57276987 eV | Free energy : -109.38703872 Ha -2976.57276987 eV Derived energy quantities: | Kinetic energy : 109.65005028 Ha 2983.72967863 eV | Electrostatic energy : -205.37638266 Ha -5588.57571571 eV | Energy correction for multipole | error in Hartree potential : 0.00062326 Ha 0.01695968 eV | Sum of eigenvalues per atom : -839.30637513 eV | Total energy (T->0) per atom : -1488.28638494 eV | Free energy per atom : -1488.28638494 eV
Self-consistency convergence accuracy: | Change of charge/spin density : 0.2057E+00 0.9030E+00 | Change of sum of eigenvalues : 0.3707E+02 eV | Change of total energy : -.4628E+01 eV
End self-consistency iteration # 1 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.366 s 0.366 s Charge density update : 0.117 s 0.117 s Density mixing : 0.008 s 0.008 s Hartree multipole update : 0.013 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.154 s 0.154 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.000 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 2
Date : 20090608, Time : 145625.502------------------------------------------------------------
Change of charge/spin density: 0.2057E+00 0.9030E+00Change of sum of eigenvalues : 0.3707E+02Change of total energy : -.4628E+01
sc_accuracy_rhosc_accuracy_eevsc_accuracy_etot
scf-cycle

Self-consistency convergence accuracy: | Change of charge/spin density : 0.3928E-06 0.2390E-06 | Change of sum of eigenvalues : 0.1079E-03 eV | Change of total energy : -.1851E-08 eV
Electronic self-consistency reached - switching on the force computation.
End self-consistency iteration # 10 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.403 s 0.404 s Charge density & force component update : 0.108 s 0.108 s Density mixing : 0.057 s 0.057 s Hartree multipole update : 0.014 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.151 s 0.151 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.001 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 11
Date : 20090608, Time : 145628.908------------------------------------------------------------
atomic forces [eV/Ang]: ----------------------- atom # 1 Hellmann-Feynman + Multipole : 0.451008E-12 -.102604E-12 -.121793E+02 Pulay + GGA : 0.486819E-13 0.227680E-12 0.106895E+02 ---------------------------------------------------------------- Total forces( 1) : 0.499690E-12 0.125076E-12 -.148978E+01 atom # 2 Hellmann-Feynman + Multipole : -.296693E-12 -.817935E-13 0.121793E+02 Pulay + GGA : 0.464015E-12 0.591618E-13 -.106895E+02 ---------------------------------------------------------------- Total forces( 2) : 0.167322E-12 -.226316E-13 0.148978E+01
Self-consistency convergence accuracy: | Change of charge/spin density : 0.1034E-07 0.6491E-09 | Change of sum of eigenvalues : -.1825E-05 eV | Change of total energy : 0.1701E-10 eV | Change of forces : 0.6537E-06 eV/A
The main output The FHI-aims output
Self-consistency convergence accuracy: | Change of charge/spin density : 0.3928E-06 0.2390E-06 | Change of sum of eigenvalues : 0.1079E-03 eV | Change of total energy : -.1851E-08 eV
Electronic self-consistency reached - switching on the force computation.
End self-consistency iteration # 10 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.403 s 0.404 s Charge density & force component update : 0.108 s 0.108 s Density mixing : 0.057 s 0.057 s Hartree multipole update : 0.014 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.151 s 0.151 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.001 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 11
Date : 20090608, Time : 145628.908------------------------------------------------------------
atomic forces [eV/Ang]: ----------------------- atom # 1 Hellmann-Feynman + Multipole : 0.451008E-12 -.102604E-12 -.121793E+02 Pulay + GGA : 0.486819E-13 0.227680E-12 0.106895E+02 ---------------------------------------------------------------- Total forces( 1) : 0.499690E-12 0.125076E-12 -.148978E+01 atom # 2 Hellmann-Feynman + Multipole : -.296693E-12 -.817935E-13 0.121793E+02 Pulay + GGA : 0.464015E-12 0.591618E-13 -.106895E+02 ---------------------------------------------------------------- Total forces( 2) : 0.167322E-12 -.226316E-13 0.148978E+01
Self-consistency convergence accuracy: | Change of charge/spin density : 0.1034E-07 0.6491E-09 | Change of sum of eigenvalues : -.1825E-05 eV | Change of total energy : 0.1701E-10 eV | Change of forces : 0.6537E-06 eV/A
The main output The FHI-aims output
0.3928E-06 0.2390E-060.1079E-03 eV-.1851E-08 eV

The main output Self-consistency convergence accuracy: | Change of charge/spin density : 0.3928E-06 0.2390E-06 | Change of sum of eigenvalues : 0.1079E-03 eV | Change of total energy : -.1851E-08 eV
Electronic self-consistency reached - switching on the force computation.
End self-consistency iteration # 10 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.403 s 0.404 s Charge density & force component update : 0.108 s 0.108 s Density mixing : 0.057 s 0.057 s Hartree multipole update : 0.014 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.151 s 0.151 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.001 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 11
Date : 20090608, Time : 145628.908------------------------------------------------------------
The FHI-aims output
atomic forces [eV/Ang]: ----------------------- atom # 1 Hellmann-Feynman + Multipole : 0.451008E-12 -.102604E-12 -.121793E+02 Pulay + GGA : 0.486819E-13 0.227680E-12 0.106895E+02 ---------------------------------------------------------------- Total forces( 1) : 0.499690E-12 0.125076E-12 -.148978E+01 atom # 2 Hellmann-Feynman + Multipole : -.296693E-12 -.817935E-13 0.121793E+02 Pulay + GGA : 0.464015E-12 0.591618E-13 -.106895E+02 ---------------------------------------------------------------- Total forces( 2) : 0.167322E-12 -.226316E-13 0.148978E+01
Self-consistency convergence accuracy: | Change of charge/spin density : 0.1034E-07 0.6491E-09 | Change of sum of eigenvalues : -.1825E-05 eV | Change of total energy : 0.1701E-10 eV | Change of forces : 0.6537E-06 eV/A
0.3928E-06 0.2390E-060.1079E-03 eV-.1851E-08 eV
Electronic self-consistency reached – switching on the force computation.
force terms calculated not untilenergy is converged
save computational time
The main output Self-consistency convergence accuracy: | Change of charge/spin density : 0.3928E-06 0.2390E-06 | Change of sum of eigenvalues : 0.1079E-03 eV | Change of total energy : -.1851E-08 eV
Electronic self-consistency reached - switching on the force computation.
End self-consistency iteration # 10 : max(cpu_time) wall_clock_time(cpu1) Time for this iteration : 0.403 s 0.404 s Charge density & force component update : 0.108 s 0.108 s Density mixing : 0.057 s 0.057 s Hartree multipole update : 0.014 s 0.014 s Hartree multipole summation : 0.072 s 0.072 s Integration : 0.151 s 0.151 s Solution of K.-S. eqns. : 0.001 s 0.001 s Total energy evaluation : 0.000 s 0.001 s------------------------------------------------------------
------------------------------------------------------------ Begin self-consistency iteration # 11
Date : 20090608, Time : 145628.908------------------------------------------------------------
The FHI-aims output
atomic forces [eV/Ang]: ----------------------- atom # 1 Hellmann-Feynman + Multipole : 0.451008E-12 -.102604E-12 -.121793E+02 Pulay + GGA : 0.486819E-13 0.227680E-12 0.106895E+02 ---------------------------------------------------------------- Total forces( 1) : 0.499690E-12 0.125076E-12 -.148978E+01 atom # 2 Hellmann-Feynman + Multipole : -.296693E-12 -.817935E-13 0.121793E+02 Pulay + GGA : 0.464015E-12 0.591618E-13 -.106895E+02 ---------------------------------------------------------------- Total forces( 2) : 0.167322E-12 -.226316E-13 0.148978E+01
Self-consistency convergence accuracy: | Change of charge/spin density : 0.1034E-07 0.6491E-09 | Change of sum of eigenvalues : -.1825E-05 eV | Change of total energy : 0.1701E-10 eV | Change of forces : 0.6537E-06 eV/A
atomic forces [eV/Ang]:
Hellmann-Feynman + MultipolePulay + GGA
force terms calculated not untilenergy is converged
save computational time

The main output The FHI-aims output
Total atomic forces (unitary forces cleaned) [eV/Ang]: | 1 0.811297123282653E-28 0.000000000000000E+00 -0.148978313187762E+01 | 2 0.811297123282653E-28 0.202824280820663E-28 0.148978313187762E+01
------------------------------------------------------------ Geometry optimization: Attempting to predict improved coordinates.
Removing unitary transformations (pure translations, rotations) from forces. Before filtering: | Net force on center of mass : 0.162259E-27 0.202824E-28 0.356812E-15 eV/A | Net torque on center of mass: -.111553E-28 0.000000E+00 0.000000E+00 eV After filtering: | Net force on center of mass : 0.360288E-43 0.450360E-44 0.000000E+00 eV/A | Net torque on center of mass: 0.247698E-44 0.000000E+00 0.000000E+00 eV
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.148978E+01 eV/A. Present geometry is not yet converged.
Relaxation step number 1: Predicting new coordinates.
Advancing geometry using BFGS. Predicted maximal displacement in this step: 0.153311E-01 A Updated atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.01533115 N atom 0.00000000 0.00000000 1.11533115 N------------------------------------------------------------
The main output The FHI-aims output
Total atomic forces (unitary forces cleaned) [eV/Ang]: | 1 0.811297123282653E-28 0.000000000000000E+00 -0.148978313187762E+01 | 2 0.811297123282653E-28 0.202824280820663E-28 0.148978313187762E+01
------------------------------------------------------------ Geometry optimization: Attempting to predict improved coordinates.
Removing unitary transformations (pure translations, rotations) from forces. Before filtering: | Net force on center of mass : 0.162259E-27 0.202824E-28 0.356812E-15 eV/A | Net torque on center of mass: -.111553E-28 0.000000E+00 0.000000E+00 eV After filtering: | Net force on center of mass : 0.360288E-43 0.450360E-44 0.000000E+00 eV/A | Net torque on center of mass: 0.247698E-44 0.000000E+00 0.000000E+00 eV
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.148978E+01 eV/A. Present geometry is not yet converged.
Relaxation step number 1: Predicting new coordinates.
Advancing geometry using BFGS. Predicted maximal displacement in this step: 0.153311E-01 A Updated atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.01533115 N atom 0.00000000 0.00000000 1.11533115 N------------------------------------------------------------
Total atomic forces (unitary forces cleaned) [eV/Ang]:

The main output The FHI-aims output
Total atomic forces (unitary forces cleaned) [eV/Ang]: | 1 0.811297123282653E-28 0.000000000000000E+00 -0.148978313187762E+01 | 2 0.811297123282653E-28 0.202824280820663E-28 0.148978313187762E+01
------------------------------------------------------------ Geometry optimization: Attempting to predict improved coordinates.
Removing unitary transformations (pure translations, rotations) from forces. Before filtering: | Net force on center of mass : 0.162259E-27 0.202824E-28 0.356812E-15 eV/A | Net torque on center of mass: -.111553E-28 0.000000E+00 0.000000E+00 eV After filtering: | Net force on center of mass : 0.360288E-43 0.450360E-44 0.000000E+00 eV/A | Net torque on center of mass: 0.247698E-44 0.000000E+00 0.000000E+00 eV
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.148978E+01 eV/A. Present geometry is not yet converged.
Relaxation step number 1: Predicting new coordinates.
Advancing geometry using BFGS. Predicted maximal displacement in this step: 0.153311E-01 A Updated atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.01533115 N atom 0.00000000 0.00000000 1.11533115 N------------------------------------------------------------
Relaxation step number 1: Predicting new coodinates.
Updated atomic structure:
relaxation step
The main output The FHI-aims output
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.390021E-02 eV/A. Present geometry is converged.
------------------------------------------------------------ Final atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.00505130 N atom 0.00000000 0.00000000 1.10505130 N------------------------------------------------------------
------------------------------------------------------------ Leaving FHI-aims. Date : 20090608, Time : 145641.881
Computational steps: | Number of self-consistency cycles : 36 | Number of relaxation steps : 3
Detailed time accounting : max(cpu_time) wall_clock_time(cpu1) | Total time : 17.321 s 17.330 s | Preparation time : 0.229 s 0.229 s | Grid partitioning : 0.205 s 0.208 s | Preloading free-atom quantities on grid : 0.170 s 0.170 s | Free-atom superposition energy : 0.188 s 0.188 s | Total time for integrations : 5.780 s 5.781 s | Total time for solution of K.-S. equations : 0.036 s 0.038 s | Total time for EV reorthonormalization : 0.000 s 0.001 s | Total time for density & force components : 5.907 s 5.906 s | Total time for mixing : 0.830 s 0.834 s | Total time for Hartree multipole update : 0.493 s 0.497 s | Total time for Hartree multipole sum : 3.444 s 3.445 s | Total time for total energy evaluation : 0.002 s 0.009 s
Have a nice day.------------------------------------------------------------

The main output The FHI-aims output
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.390021E-02 eV/A. Present geometry is converged.
------------------------------------------------------------ Final atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.00505130 N atom 0.00000000 0.00000000 1.10505130 N------------------------------------------------------------
------------------------------------------------------------ Leaving FHI-aims. Date : 20090608, Time : 145641.881
Computational steps: | Number of self-consistency cycles : 36 | Number of relaxation steps : 3
Detailed time accounting : max(cpu_time) wall_clock_time(cpu1) | Total time : 17.321 s 17.330 s | Preparation time : 0.229 s 0.229 s | Grid partitioning : 0.205 s 0.208 s | Preloading free-atom quantities on grid : 0.170 s 0.170 s | Free-atom superposition energy : 0.188 s 0.188 s | Total time for integrations : 5.780 s 5.781 s | Total time for solution of K.-S. equations : 0.036 s 0.038 s | Total time for EV reorthonormalization : 0.000 s 0.001 s | Total time for density & force components : 5.907 s 5.906 s | Total time for mixing : 0.830 s 0.834 s | Total time for Hartree multipole update : 0.493 s 0.497 s | Total time for Hartree multipole sum : 3.444 s 3.445 s | Total time for total energy evaluation : 0.002 s 0.009 s
Have a nice day.------------------------------------------------------------
Maximum force component is 0.390021E-02 eV/A.Present geometry is converged.
The main output The FHI-aims output
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.390021E-02 eV/A. Present geometry is converged.
------------------------------------------------------------ Final atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.00505130 N atom 0.00000000 0.00000000 1.10505130 N------------------------------------------------------------
------------------------------------------------------------ Leaving FHI-aims. Date : 20090608, Time : 145641.881
Computational steps: | Number of self-consistency cycles : 36 | Number of relaxation steps : 3
Detailed time accounting : max(cpu_time) wall_clock_time(cpu1) | Total time : 17.321 s 17.330 s | Preparation time : 0.229 s 0.229 s | Grid partitioning : 0.205 s 0.208 s | Preloading free-atom quantities on grid : 0.170 s 0.170 s | Free-atom superposition energy : 0.188 s 0.188 s | Total time for integrations : 5.780 s 5.781 s | Total time for solution of K.-S. equations : 0.036 s 0.038 s | Total time for EV reorthonormalization : 0.000 s 0.001 s | Total time for density & force components : 5.907 s 5.906 s | Total time for mixing : 0.830 s 0.834 s | Total time for Hartree multipole update : 0.493 s 0.497 s | Total time for Hartree multipole sum : 3.444 s 3.445 s | Total time for total energy evaluation : 0.002 s 0.009 s
Have a nice day.------------------------------------------------------------
Final atomic structure: x[A] y[A] z[A] Atom atom 0.00000000 0.00000000 -0.00505130 Natom 0.00000000 0.00000000 1.10505130 N
to be used for post-processing by
copy and paste

The main output The FHI-aims output
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.390021E-02 eV/A. Present geometry is converged.
------------------------------------------------------------ Final atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.00505130 N atom 0.00000000 0.00000000 1.10505130 N------------------------------------------------------------
------------------------------------------------------------ Leaving FHI-aims. Date : 20090608, Time : 145641.881
Computational steps: | Number of self-consistency cycles : 36 | Number of relaxation steps : 3
Detailed time accounting : max(cpu_time) wall_clock_time(cpu1) | Total time : 17.321 s 17.330 s | Preparation time : 0.229 s 0.229 s | Grid partitioning : 0.205 s 0.208 s | Preloading free-atom quantities on grid : 0.170 s 0.170 s | Free-atom superposition energy : 0.188 s 0.188 s | Total time for integrations : 5.780 s 5.781 s | Total time for solution of K.-S. equations : 0.036 s 0.038 s | Total time for EV reorthonormalization : 0.000 s 0.001 s | Total time for density & force components : 5.907 s 5.906 s | Total time for mixing : 0.830 s 0.834 s | Total time for Hartree multipole update : 0.493 s 0.497 s | Total time for Hartree multipole sum : 3.444 s 3.445 s | Total time for total energy evaluation : 0.002 s 0.009 s
Have a nice day.------------------------------------------------------------
| Number of self-consistency cycles : 36| Number of relaxation steps : 3
# “ “
# “ “# “ “
The main output The FHI-aims output
Net remaining forces (excluding translations, rotations) in present geometry: Maximum force component is 0.390021E-02 eV/A. Present geometry is converged.
------------------------------------------------------------ Final atomic structure: x [A] y [A] z [A] Atom atom 0.00000000 0.00000000 -0.00505130 N atom 0.00000000 0.00000000 1.10505130 N------------------------------------------------------------
------------------------------------------------------------ Leaving FHI-aims. Date : 20090608, Time : 145641.881
Computational steps: | Number of self-consistency cycles : 36 | Number of relaxation steps : 3
Detailed time accounting : max(cpu_time) wall_clock_time(cpu1) | Total time : 17.321 s 17.330 s | Preparation time : 0.229 s 0.229 s | Grid partitioning : 0.205 s 0.208 s | Preloading free-atom quantities on grid : 0.170 s 0.170 s | Free-atom superposition energy : 0.188 s 0.188 s | Total time for integrations : 5.780 s 5.781 s | Total time for solution of K.-S. equations : 0.036 s 0.038 s | Total time for EV reorthonormalization : 0.000 s 0.001 s | Total time for density & force components : 5.907 s 5.906 s | Total time for mixing : 0.830 s 0.834 s | Total time for Hartree multipole update : 0.493 s 0.497 s | Total time for Hartree multipole sum : 3.444 s 3.445 s | Total time for total energy evaluation : 0.002 s 0.009 s
Have a nice day.------------------------------------------------------------
Have a nice day.

Overview The exercises
� the nitrogen dimerbinding curve, binding energy, spin polarizationpbe vs. pw-lda
� the silicon trimerlocal structural relaxationvibrational analysischarge density plots, HOMO plotsvisualization
� small X(+)Si3 – clusters, X=Sc,Ti,V,Cr“sampling” of cluster structuresdensity of states plots
14:30 - 15:15 (45 min)
15:15 - 16:00 (45 min)
16:00 - 18:00 (120 min)
The nitrogen dimer The exercises
/usr/local/aimsfiles/tutorial1/N2� general part of control.in file:
� species-dependent part:/usr/local/aimsfiles/species_defaults/light/07_N_default
� geometry.in file simple (just two atoms) Eb�E tot �N 2��2 E tot �N �
control.in.basic
xc xxspin xxrelativistic xx
0.8 -2962.767360.9 -2972.908881.0 -2977.15015
xmgrace, gnuplot
xmgrace, gnuplot, awk,...
d [Å]

The nitrogen dimer The exercises
the free atom� spin-polarized
� enhanced cutoff radius: 3.5 Å ��5.0 Å
spin collinear
cut_pot 5.0 1.5 1.0
The nitrogen dimer The exercises
the free atom� spin-polarized
� enhanced cutoff radius: 3.5 Å ��5.0 Å
spin collinear
cut_pot 5.0 1.5 1.0

The nitrogen dimer The exercises
the free atom� spin-polarized
� enhanced cutoff radius: 3.5 Å ��5.0 Å
spin collinear
cut_pot 5.0 1.5 1.0
################################ "First tier" hydro 2 p 1.8 hydro 3 d 6.8 hydro 3 s 5.8# "Second tier"# hydro 4 f 10.8# hydro 3 p 5.8# hydro 1 s 0.8# hydro 5 g 16# hydro 3 d 4.9# "Third tier" # hydro 3 s 16# ionic 2 p auto# hydro 3 d 6.6# hydro 4 f 11.6
The nitrogen dimer The exercises
the free atom� spin-polarized
� enhanced cutoff radius: 3.5 Å ��5.0 Å
spin collinear
cut_pot 5.0 1.5 1.0
basis set convergence
############################# "First tier" hydro 2 p 1.8 hydro 3 d 6.8 hydro 3 s 5.8# "Second tier" hydro 4 f 10.8 hydro 3 p 5.8 hydro 1 s 0.8 hydro 5 g 16 hydro 3 d 4.9# "Third tier" # hydro 3 s 16# ionic 2 p auto# hydro 3 d 6.6# hydro 4 f 11.6
tier1 tier2
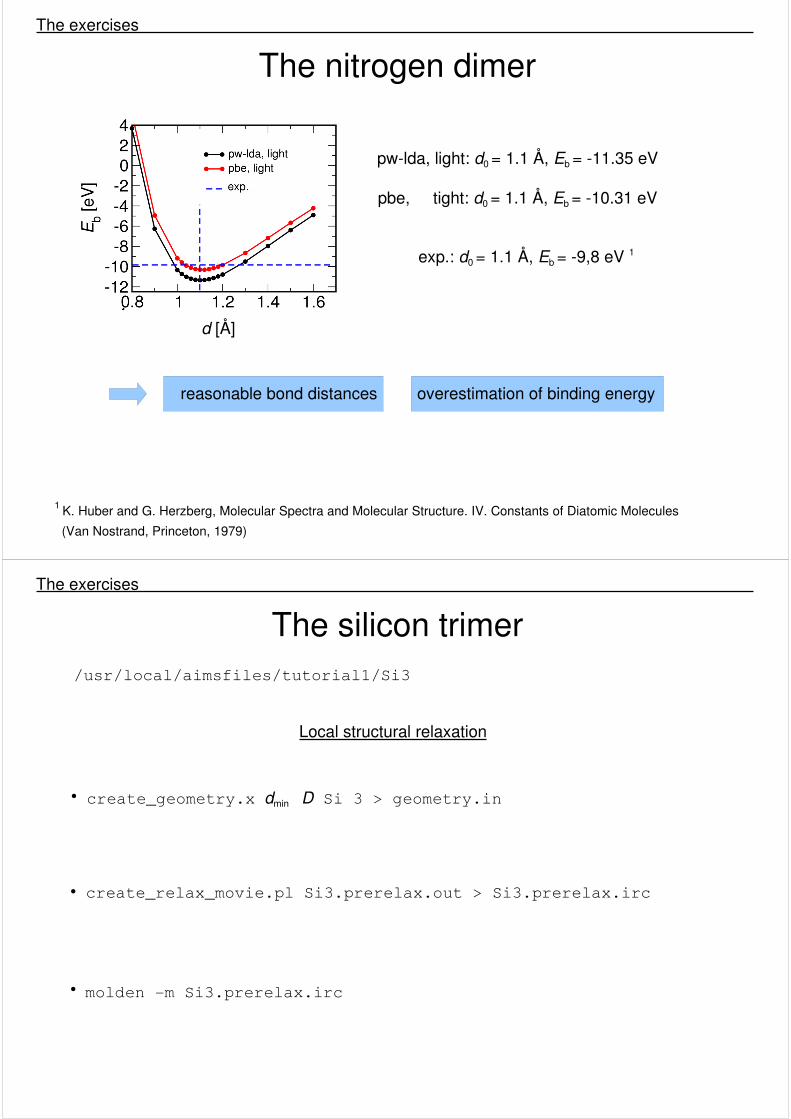
The nitrogen dimer The exercises
pw-lda, light: d0 = 1.1 Å, Eb = -11.35 eV
d [Å]
pbe, tight: d0 = 1.1 Å, Eb = -10.31 eV
exp.: d0 = 1.1 Å, Eb = -9,8 eV 1
1 K. Huber and G. Herzberg, Molecular Spectra and Molecular Structure. IV. Constants of Diatomic Molecules (Van Nostrand, Princeton, 1979)
reasonable bond distances overestimation of binding energy
The silicon trimer The exercises
� create_relax_movie.pl Si3.prerelax.out > Si3.prerelax.irc
/usr/local/aimsfiles/tutorial1/Si3
� create_geometry.x dmin D Si 3 > geometry.in
Local structural relaxation
� molden -m Si3.prerelax.irc

The silicon trimer The exercises
Visualization of local relaxation
Run through relaxation steps
Bond distancesangles
output style(balls & sticks)
The silicon trimer The exercises
Vibrational analysis:
/usr/local/aimsfiles/tutorial1/Si3/Si3_pw-lda /usr/local/aimsfiles/tutorial1/Si3/Si3_pbe
� aims.vibrations.pl Si3_pw-lda (0.005)
default value for finite displacement
Si3_pw-lda.mol
Si3_pw-lda.xyz
(molden)
(jmol)� molden Si3_pw-lda.mol
d F
d R�
�F �R� , 0����F �R� , 0���
2�d Pd R�
�P �R
�, 0����P�R� , 0���
2�
I�� d PdQ�2
IR-intensity
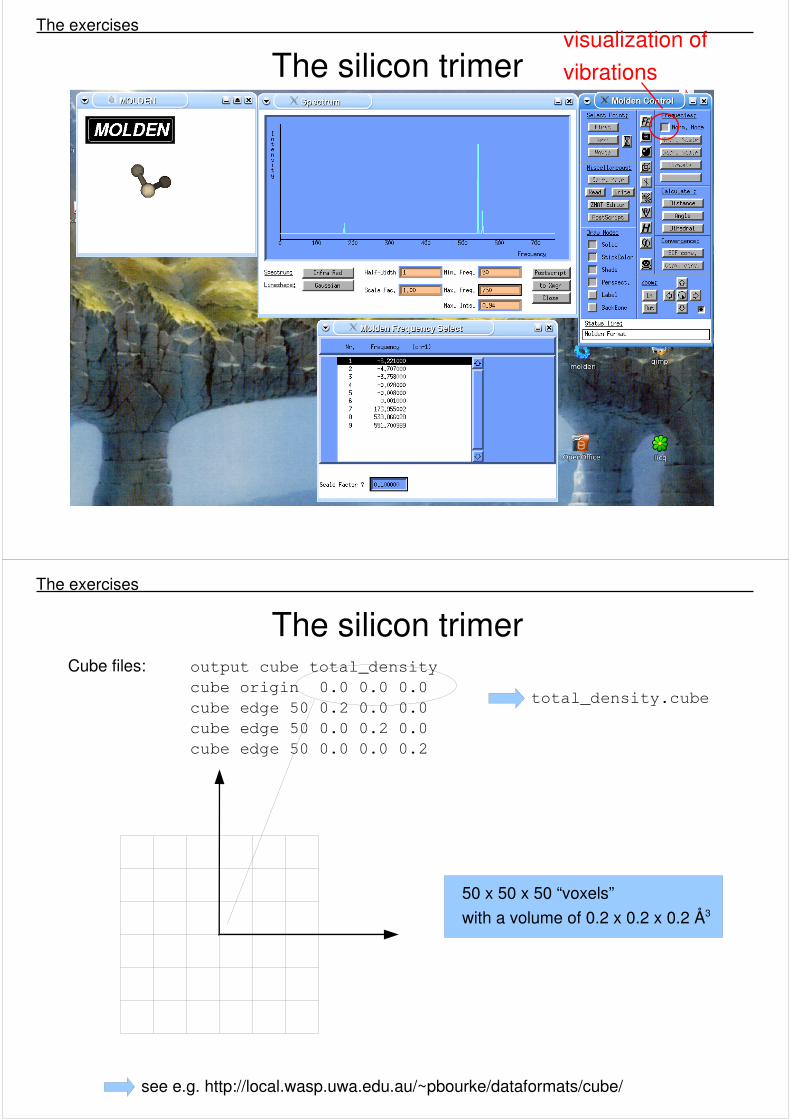
The silicon trimer The exercises
visualization of vibrations
The silicon trimer The exercises
Cube files:
total_density.cube
output cube total_densitycube origin 0.0 0.0 0.0cube edge 50 0.2 0.0 0.0cube edge 50 0.0 0.2 0.0cube edge 50 0.0 0.0 0.2
50 x 50 x 50 “voxels” with a volume of 0.2 x 0.2 x 0.2 Å3
see e.g. http://local.wasp.uwa.edu.au/~pbourke/dataformats/cube/

The silicon trimer The exercises
Enter densitymode
The silicon trimer The exercises
Read cube files
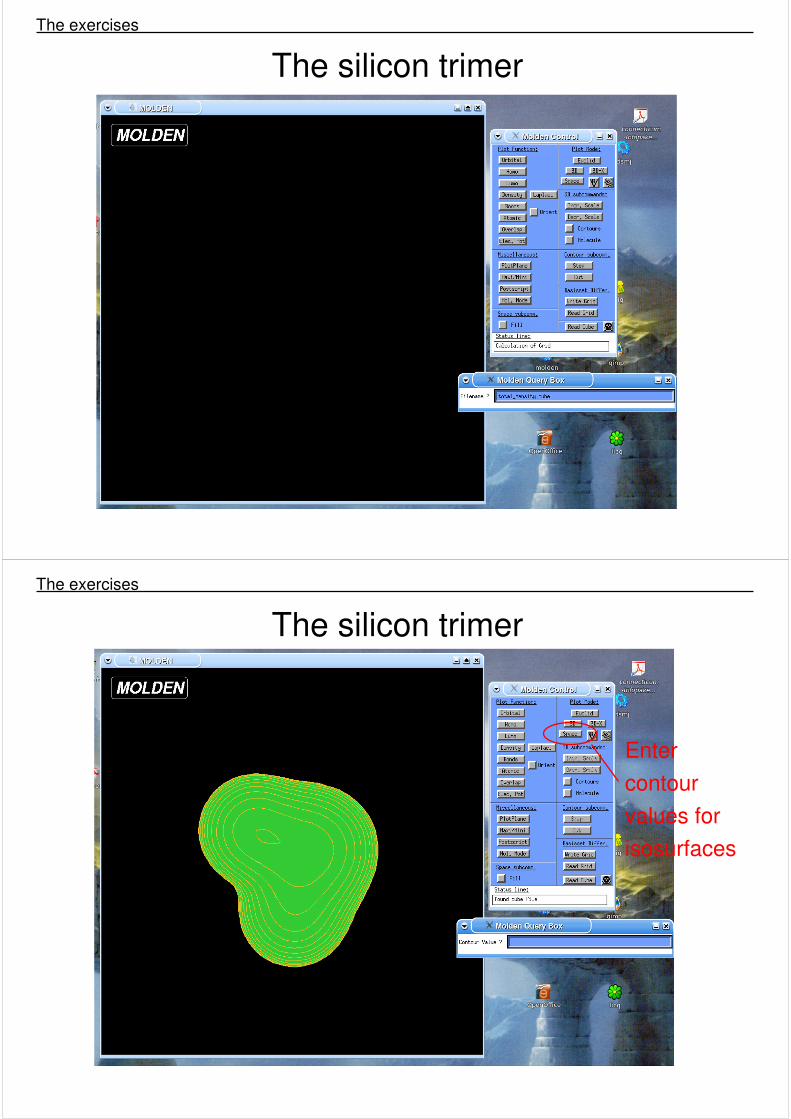
The silicon trimer The exercises
The silicon trimer The exercises
Enter contour values for isosurfaces
The silicon trimer The exercises
X(+)Si3 , X=Sc,Ti,V,Cr The exercises
� create_geometry.x dmin D Si 3 Sc 1 > geometry.in.struct1
geometry.in.struct1geometry.in.struct2geometry.in.struct3geometry.in.struct4geometry.in.struct5
control.in.prerelax
~/tutorial1/X(+)Si3/XSi3_pw-lda ~/tutorial1/X(+)Si3/XSi3_pbe~/tutorial1/X(+)Si3/X+Si3_pw-lda ~/tutorial1/X(+)Si3/X+Si3_pbe
combi_run.pl
aims.struct1.prerelax.outaims.struct2.prerelax.outaims.struct3.prerelax.outaims.struct4.prerelax.outaims.struct5.prerelax.out
� create_relax_movie.pl aims.struct1.prerelax.out > relax.1.irc
� molden -m relax.1.irc
� KS_DOS_total_raw.dat, KS_DOS_total.dat ASCII-file � xmgrace
unshifted DOS shifted w.r.t. EF
The exercises
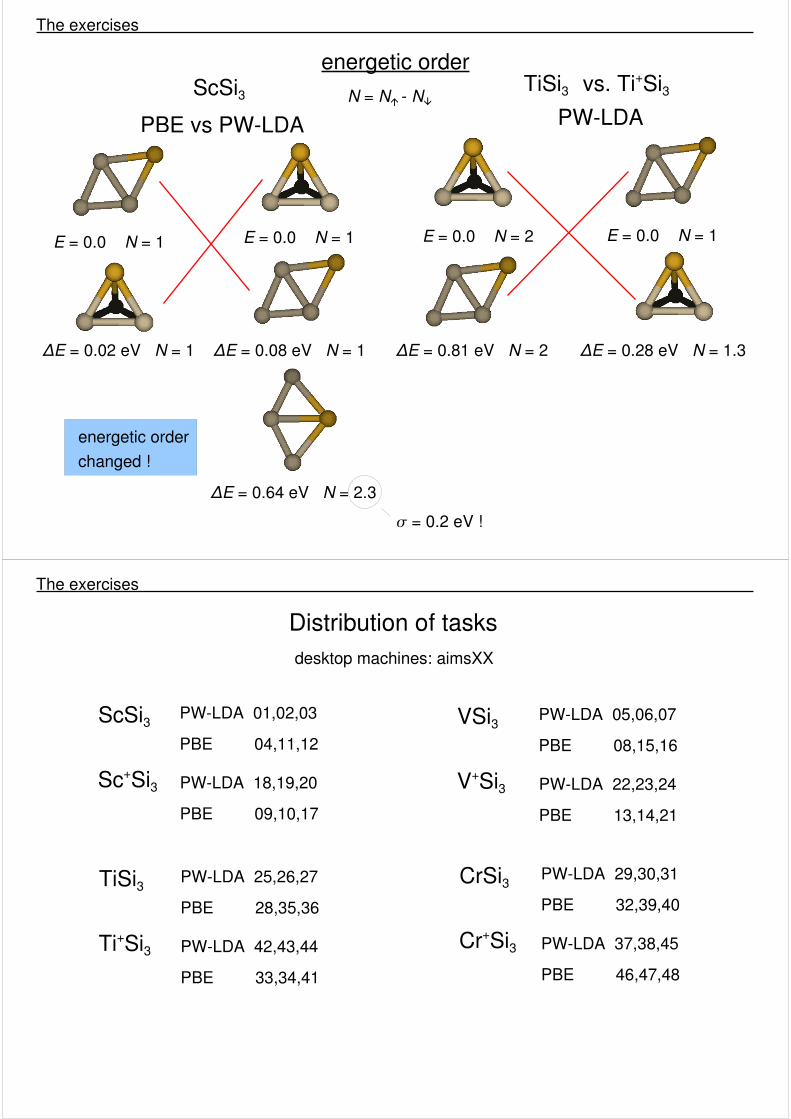
energetic order
PBE vs PW-LDAScSi3
E = 0.0 N = 1
ΔE = 0.08 eV N = 1
ΔE = 0.64 eV N = 2.3 � = 0.2 eV !
N = N� - N�
E = 0.0 N = 1
ΔE = 0.02 eV N = 1
energetic orderchanged !
TiSi3 vs. Ti+Si3PW-LDA
E = 0.0 N = 2
ΔE = 0.81 eV N = 2
E = 0.0 N = 1
ΔE = 0.28 eV N = 1.3
The exercises
Distribution of tasks
ScSi3 PW-LDA 01,02,03
PBE 04,11,12
PW-LDA 18,19,20
PBE 09,10,17
Sc+Si3
TiSi3 PW-LDA 25,26,27
PBE 28,35,36
PW-LDA 42,43,44
PBE 33,34,41
Ti+Si3
VSi3 PW-LDA 05,06,07
PBE 08,15,16
PW-LDA 22,23,24
PBE 13,14,21
V+Si3
CrSi3 PW-LDA 29,30,31
PBE 32,39,40
PW-LDA 37,38,45
PBE 46,47,48
Cr+Si3
desktop machines: aimsXX

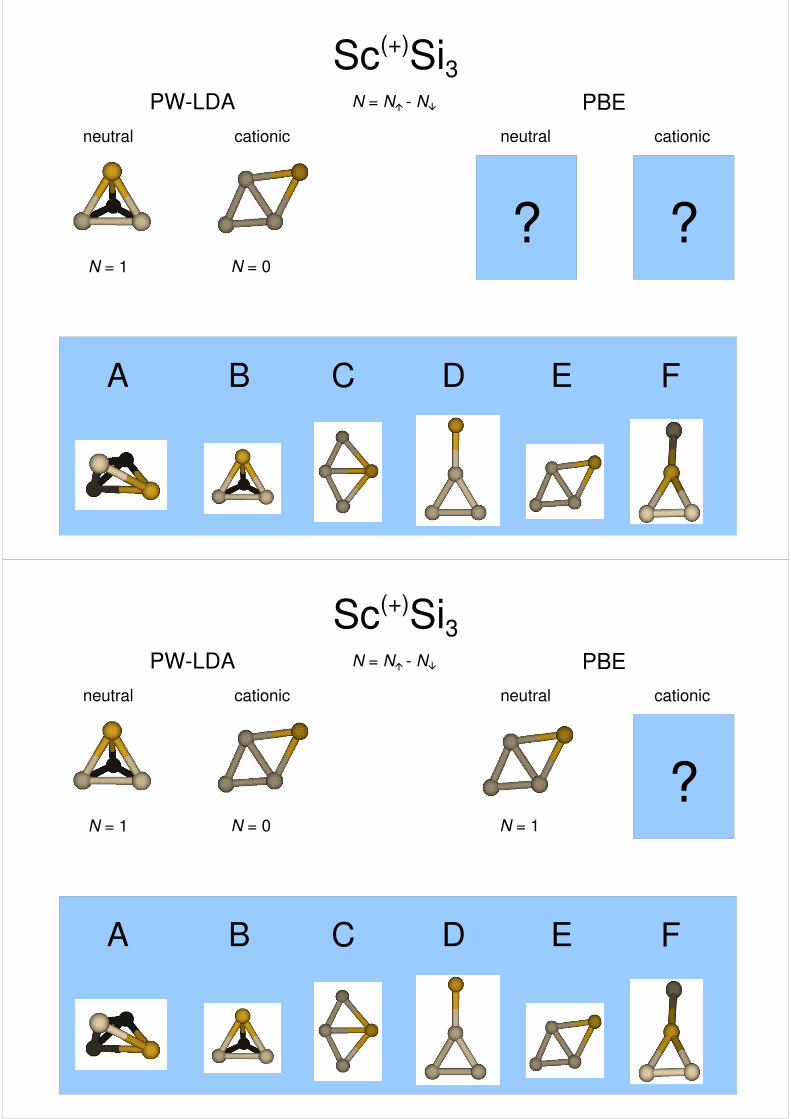
Sc(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
A B C D E F
????
Sc(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
A B C D E F
???
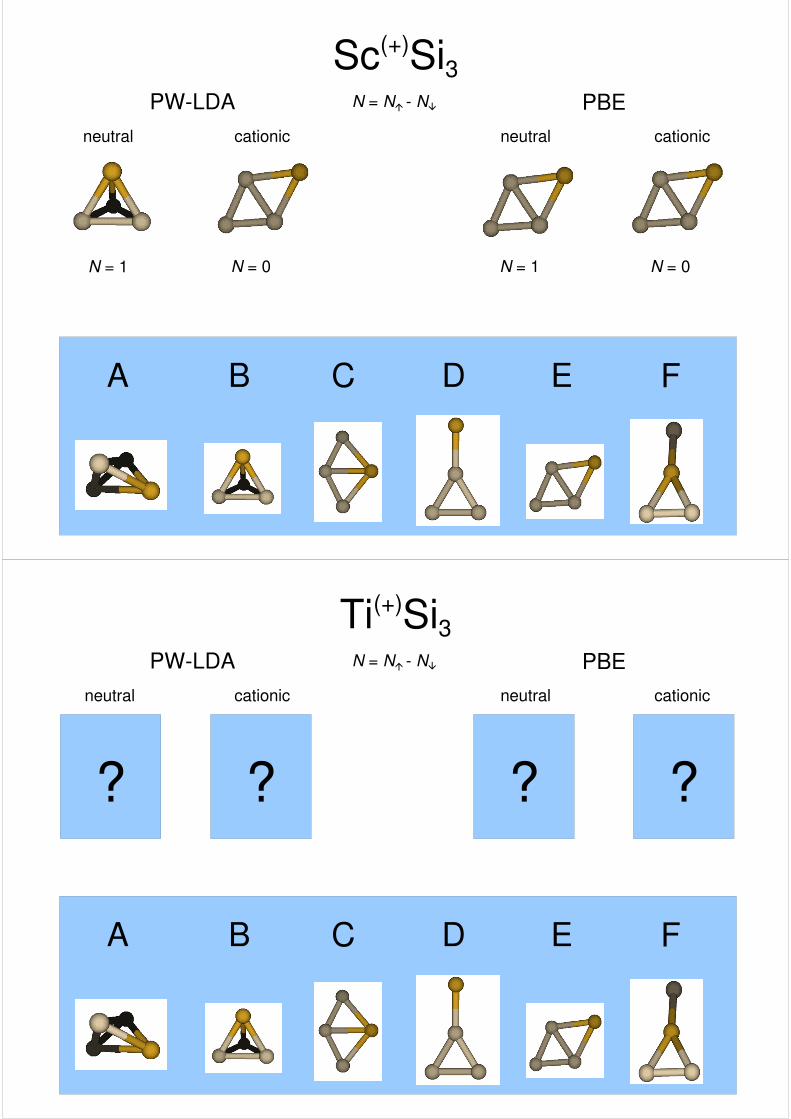
Sc(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
N = 0
A B C D E F
??
Sc(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
N = 1N = 0
A B C D E F
?
Sc(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
N = 1N = 0 N = 0
A B C D E F
Ti(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
A B C D E F
????

Ti(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 2
N = N� - N�
A B C D E F
???
Ti(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 2
N = N� - N�
N = 1
A B C D E F
??
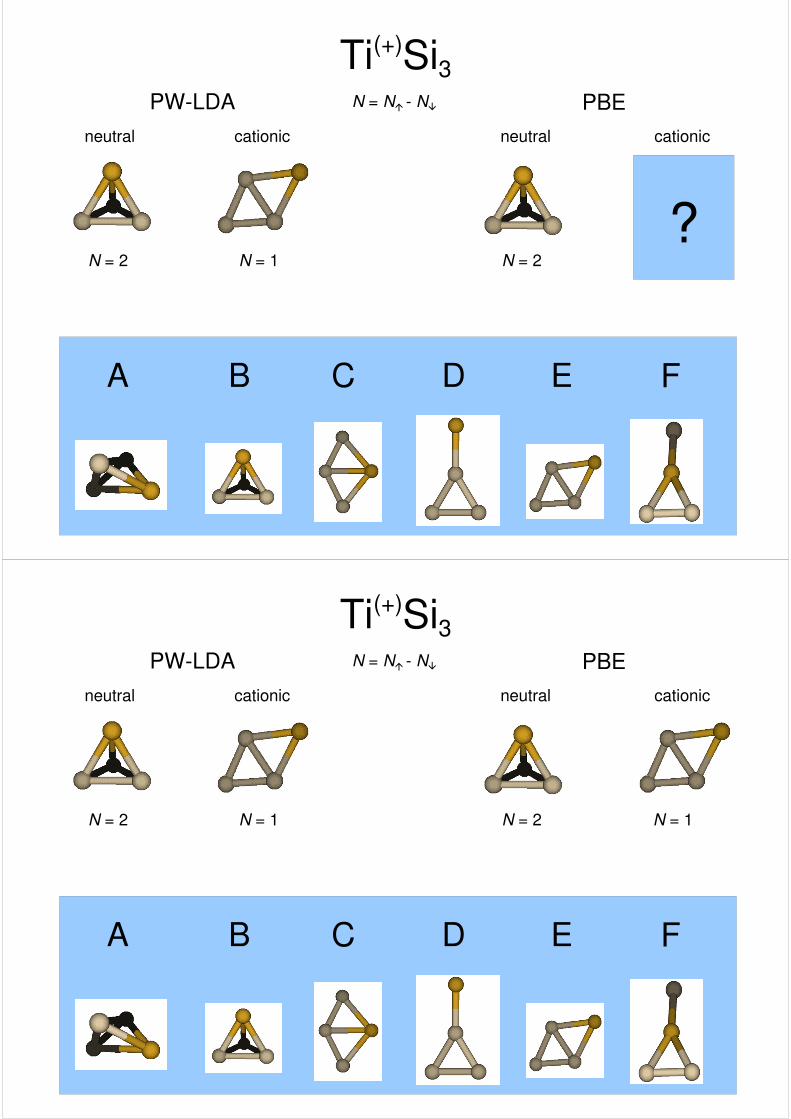
Ti(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 2
N = N� - N�
N = 2N = 1
A B C D E F
?
Ti(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 2
N = N� - N�
N = 2N = 1 N = 1
A B C D E F

V(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
A B C D E F
????
V(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
A B C D E F
???

V(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
N = 2
A B C D E F
??
V(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 1
N = N� - N�
N = 1N = 2
A B C D E F
?
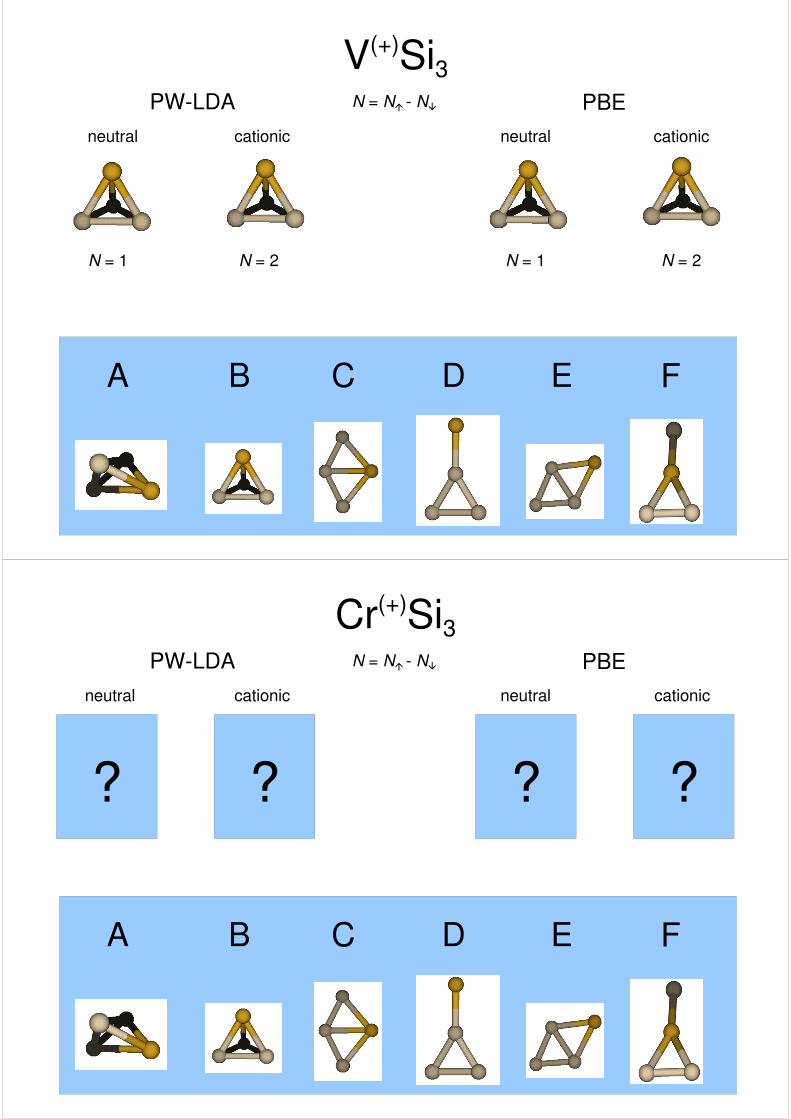
V(+)Si3PW-LDA PBE
cationicneutral neutral
N = 1
N = N� - N�
N = 1N = 2 N = 2
A B C D E F
cationic
Cr(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
A B C D E F
? ? ? ?

Cr(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
A B C D E F
? ?N = 0
?
Cr(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
A B C D E F
??N = 3N = 0
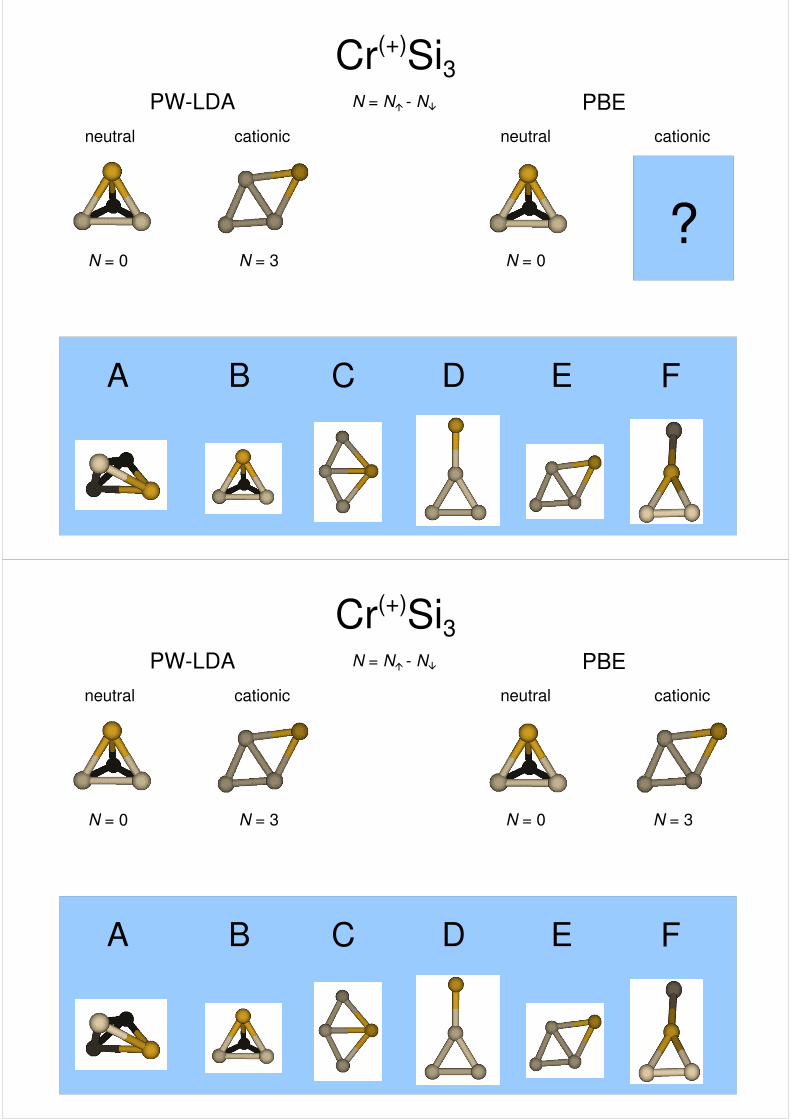
Cr(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = N� - N�
N = 0N = 3
A B C D E F
?N = 0
Cr(+)Si3PW-LDA PBE
cationic cationicneutral neutral
N = 0
N = N� - N�
N = 0N = 3 N = 3
A B C D E F
Very well done!!

![PDF] Hands-On Tutorial on Ab Initio Molecular Simulations ...Hands-On Tutorial on Ab Initio Molecular Simulations Berlin, July 12 { 21, 2011 Tutorial I: Basics of Electronic-Structure](https://img.pdfslide.us/doc/110x75/612d24fa1ecc5158694201ae/hands-on-tutorial-on-ab-initio-molecular-simulations-hands-on-tutorial-on-ab.jpg)



























