Embed Size (px)
Citation preview

of February 13, 2018.This information is current as
Systemic Inflammatory Response SyndromeInflammation and Accelerates the Onset of TRPV1 Deletion Enhances Local
and Susan D. BrainJulie E. Keeble, Yanira Riffo-Vasquez, Kenneth D. Bruce Alam, Sarah Howat, Helen Collins, Stephen J. Thompson,Frank Kaiser, Robert Purcell, Damian W. Rivett, Saydul Elizabeth S. Fernandes, Lihuan Liang, Sarah-Jane Smillie,
ol.1102147http://www.jimmunol.org/content/early/2012/04/30/jimmun
published online 30 April 2012J Immunol
average*
4 weeks from acceptance to publicationSpeedy Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2012 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
TRPV1 Deletion Enhances Local Inflammation andAccelerates the Onset of Systemic Inflammatory ResponseSyndrome
Elizabeth S. Fernandes,* Lihuan Liang,* Sarah-Jane Smillie,* Frank Kaiser,†
Robert Purcell,* Damian W. Rivett,‡ Saydul Alam,* Sarah Howat,‡ Helen Collins,†
Stephen J. Thompson,† Julie E. Keeble,‡ Yanira Riffo-Vasquez,‡ Kenneth D. Bruce,‡ and
Susan D. Brain*
The transient receptor potential vanilloid 1 (TRPV1) is primarily localized to sensory nerve fibers and is associated with the stim-
ulation of pain and inflammation. TRPV1 knockout (TRPV1KO) mice show enhanced LPS-induced sepsis compared with wild type
(WT). This implies that TRPV1 may have a key modulatory role in increasing the beneficial and reducing the harmful components
in sepsis. We investigated immune and inflammatory mechanisms in a cecal ligation and puncture (CLP) model of sepsis over 24 h.
CLP TRPV1KO mice exhibited significant hypothermia, hypotension, and organ dysfunction compared with CLPWT mice. Anal-
ysis of the inflammatory responses at the site of initial infection (peritoneal cavity) revealed that CLP TRPV1KOmice exhibited: 1)
decreased mononuclear cell integrity associated with apoptosis, 2) decreased macrophage tachykinin NK1-dependent phagocytosis,
3) substantially decreased levels of nitrite (indicative of NO) and reactive oxygen species, 4) increased cytokine levels, and 5)
decreased bacteria clearance when compared with CLP WT mice. Therefore, TRPV1 deletion is associated with impaired
macrophage-associated defense mechanisms. Thus, TRPV1 acts to protect against the damaging impact of sepsis and may
influence the transition from local to a systemic inflammatory state. The Journal of Immunology, 2012, 188: 000–000.
Sepsis is an overwhelming and complex response initiated byinfectious microorganisms and can be lethal. It is estimatedthat .0.5 million patients worldwide are annually affected
by sepsis (1, 2). The outcome of each episode depends on theseverity of the systemic inflammation in response to the initialinfection. This systemic inflammatory response syndrome (SIRS)is characterized by activation of the immune and inflammatorysystems that, in turn, leads to multiple organ dysfunction andfailure (2, 3). Classic signs of sepsis/SIRS include hypothermia/hyperthermia, tachycardia, tachypnea, and leukocytosis/leukope-nia (2). Sepsis can be caused by a variety of microorganisms, andeffective treatments have been sought. However, as the mecha-
nisms involved in sepsis are still unclear, few drugs are successfulin treating this disease.Recently, a role for sensory nerves has been described. C and
Ad sensory fibers are distributed throughout the body and, whenstimulated, release neuropeptides such as substance P (SP) andcalcitonin gene-related peptide (CGRP) (4, 5). Transient receptorpotential vanilloid 1 (TRPV1) is a nonselective ion channel local-ized on sensory nerves (6) that acts to integrate sensory and noxiousinformation (7). TRPV1 can be activated by a range of stimuli suchas heat, protons, arachidonic acid, lipoxygenase metabolites, andcannabinoids (for review, see Ref. 8). TRPV1 has become knownas a “molecular integrator” because its activity can be enhancedby common signaling effectors such as protein kinase C (9).Evidence shows increased serum levels of CGRP and SP in
patients with sepsis (10, 11). SP is suggested to play a proin-flammatory in sepsis (12, 13), whereas a regulatory role for CGRPhas been suggested (14). Initial indications that TRPV1 couldinfluence blood pressure and temperature regulation have alsobeen reported in LPS-induced sepsis in rodents (15, 16). Protec-tion against hypotension and hypothermia has been reported inrats with endotoxemia, where capsazepine, a TRPV1 antagonist,enhances mortality (17). We have previously shown that TRPV1deletion increases TNF-a, NO, and circulating markers of organfailure during endotoxemia (18). These recent findings indicatea protective role for TRPV1 in sepsis.To date, little is known of the mechanisms via which TRPV1 is
protective and influences SIRS, a characteristic of sepsis. In thisstudy, the role of TRPV1 in sepsis was investigated using themurine cecal ligation and puncture (CLP) model used to serve asa model for human sepsis (2). We provide evidence that TRPV1protects against the immune and inflammatory responses producedin the peritoneal cavity and also the ensuing SIRS. TRPV1 dele-tion increases cytokine release, reduces reactive oxygen species
*British Heart Foundation Centre for Cardiovascular Research, King’s College Lon-don, London SE1 9NH, United Kingdom; †Immunology, Infection and InflammatoryDisease, King’s College London, London SE1 9NH, United Kingdom ‡Institute ofPharmaceutical Science, Waterloo Campus, King’s College London, London SE19NH, United Kingdom
Received for publication August 1, 2011. Accepted for publication April 2, 2012.
This work was supported by Conselho Nacional de Desenvolvimento Cientifico eTecnologico-Brazil and Arthritis Research United Kingdom (Grant 19296 to E.S.F.);the Biotechnology and Biological Sciences Research Council (to S.-J.S.); and theCapacity Building Award in Integrative Mammalian Biology supported by the Bio-technology and Biological Sciences Research Council (to J.E.K. and L.L) and theBritish Heart Foundation 4y Ph.D. programme (to S.A.).
Address correspondence and reprint requests to Dr. Susan D. Brain, Vascular BiologyGroup, Cardiovascular Division, Franklin Wilkins Building, King’s College London,Waterloo Campus, London SE1 9NH, U.K. E-mail address: [email protected]
Abbreviations used in this article: AST, aspartate aminotransferase; CGRP, calcitoningene-related peptide; CLP, cecal ligation and puncture; H2O2, hydrogen peroxide;KC, keratinocyte-derived chemokine; KO, knockout; NOX, NADPH oxidase; O2
2,superoxide; PELF, peritoneal exudate lavage fluid; ROS, reactive oxygen species;SIRS, systemic inflammatory response syndrome; SP, substance P; TREM, triggeringreceptor expressed on myeloid cell; TRPV1, transient receptor potential vanilloid 1;WT, wild type.
Copyright� 2012 by The American Association of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1102147
Published April 30, 2012, doi:10.4049/jimmunol.1102147 by guest on February 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

(ROS) and NO levels, increases apoptosis, and also reducesin vitro phagocytosis in peritoneal macrophages treated with LPS.This impaired macrophage function facilitates bacterial survival,thus promoting a worsening of the systemic inflammatory re-sponse. Deletion of TRPV1 leads to enhanced hypotension andhypothermia, indicating TRPV1 has the potential to play an im-portant role in influencing the progression of a local response toinfection and the onset of the SIRS.
Materials and MethodsAnimals
All procedures were conducted in accordance with the U.K. Animals (Sci-entific Procedures) Act of 1986. Mice were kept in a climatically controlledenvironment and given food and water ad libitum. In all studies, female andmale age- and sex-matched C57BL6/129SvJ wild type (WT) and TRPV1knockout (TRPV1KO) mice were used at 8 wk of age. TRPV1KO mice withthe exon that encodes part of the fifth and the entire sixth transmembranedomain (including the interconnecting p-loop) of the receptor replaced witha neomycin gene (6) were genotyped as previously described (18, 19). Micewere from established WT and TRPV1KO colonies held at King’s CollegeLondon and grow normally, although TRPV1KO mice show characteristicloss of thermal responsiveness (18, 19).
Sublethal sepsis induced by CLP
Experiments were designed with a premortality end point (at 24 h) accordingto the method described by Baker et al. (20), with minor modifications.Animals received a single i.m. injection containing midazolam (2 mg/kg;Roche, Welwyn, U.K.) and buprenorphine (10 mg/kg; Vetersergic, AlstoeAnimal Health, U.K.), 15 min before the surgery. During the surgicalprocedure, anesthesia was maintained with isoflurane 2% (Abbot, Kent,U.K.). Surgery was performed using aseptic techniques. In brief, an incisionof ∼2 cm was made in the ventral surface of the abdomen and the cecum wasexposed through the incision. The cecum was ligated at its base (withoutcausing bowel obstruction) with silk suture 4–0 (Ethicon; Johnson &Johnson) and perforated with a 22-gauge needle resulting in two holes.Sham-operated animals (with incision only) were used as controls. Bothsham and CLP mice were sutured with absorbable suture (Vicryl; Ethicon;Johnson & Johnson) and received 1 ml saline 0.9% (s.c.) for postsurgeryhydration. Animals were killed 24 h after surgery by cervical dislocation. Alaparotomy was performed and the peritoneal cavity was washed with 1.5ml PBS. The peritoneal exudate lavage fluid (PELF) and tissue samples(lung and liver) were collected for further analysis.
Inflammatory cell accumulation in the peritoneal cavity
Inflammatory cell migration into the peritoneal cavity was evaluatedaccording to Costa and collaborators (21) with minor modifications. Totalcell counts were performed with a Neubauer chamber and microscope(Leitz Diaplan 307-148.001; PL Fluotar310 objective; numerical aperture0.3) after diluting a sample of the collected fluid from the peritoneal cavitywith Turk solution (1:20). Cellular smears were prepared with an aliquot(50 ml) of the PELF to determine the differential leukocyte population. Fordifferential cell analysis, we considered only viable cells, that is, cells withnormal morphology. The slides were stained with Diff-Quik kit (Gamidor,Didcot, U.K.); then the analysis was carried out under an immersion ob-jective (Leitz Diaplan 307-148.001; 3100 objective; numerical aperture1.25). Images were acquired with Jenoptik ProgResC5 camera and visu-alized in Capture Pro 2.7.
In vitro phagocytosis
Phagocytosis was analyzed in the peritoneal cells obtained from WT andTRPV1KO mice. For this purpose, animals were injected i.p. with 1 ml PBScontaining 1% oyster glycogen. After 18 h, the peritoneal cavity was washedwith 10ml cold PBS, and the peritoneal cells were harvested, centrifuged (10min, 4˚C), and resuspended (final concentration of 2 3 106 cells/ml) inDMEM containing 10% FCS (v/v), glutamine (2 mM; PAA, Pasching,Austria), penicillin (13; PAA), and streptomycin (13; PAA). Cells (6 3105/well) were incubated in eight chamber culture slides (BD Falcon) at 37˚C,and after 2 h, nonadhered cells were removed and adherent cells(macrophages) were incubated in the presence and absence of 2 mmfluorescent latex beads (1:100; 5 ml/well; Sigma, Gillingham, U.K.), for 24h before analysis. Phagocytosis was evaluated in unstimulated cells andalso in cells treated with LPS (100 ng/ml, Escherichia coli 0127:B8). Therole SP (10 nM) and CGRP (10 nM) in phagocytosis was evaluated in
TRPV1KO macrophages treated with LPS. In addition, TRPV1KO mac-rophages were incubated in the presence of either the NK1 SR140333 (1mM) or the NK2 antagonist SR48968 (5 nM). In a different set of experi-ments, LPS-stimulated WT macrophages were incubated with either theTRPV1 antagonist SB366791 (20 mM) or vehicle (0.1% DMSO). After theincubation period, the cell culture medium was removed and each well waswashed three times with PBS. Wells were fixed in 2% paraformaldehyde for10 min and washed three times with PBS for the removal of excessiveparaformaldehyde. Then, 10 ml PBS (mounting medium) was placed in thetop of each well sample and slides were covered with a glass slip. Slideswere analyzed by confocal microscopy (Olympus BX51; UPlan BX2 340objective; numerical aperture 0.75; bright field). Images were acquired byan Olympus color view 3 camera and visualized in Cell P. Two lots of 100cells were counted for each sample, and the average for each sample wasconsidered as an n number. Results are expressed as percentage of cellscontaining beads and number of phagocytosed beads per 100 cells.
Caspase-3 immunocytochemistry
Caspase-3 expression was evaluated in cultured macrophages obtained fromWT and TRPV1KO mice as described earlier. After 24-h incubation withLPS (100 ng/ml), in the presence and absence of either SB366791 (20 mM)or vehicle (0.1% DMSO), the cell culture was removed and the slides werewashed three times with PBS. After the incubation period, the cell culturemedium was removed and each well was washed three times with PBS.Wells were fixed in 4% paraformaldehyde for 10 min and washed threetimes with PBS for the removal of excessive paraformaldehyde. Endoge-nous peroxidase was blocked by incubation with 3% hydrogen peroxide(H2O2) for 30 min at 4˚C in a dark chamber. Wells were washed twice withPBS (5 min) and then blocked with blocking solution (0.1% rabbit serum,Vectastain ABC kit; Vector Labs; 0.2% BSA and 2% skimmed milk). Twodrops of the rabbit polyclonal primary Ab (1:750; diluted in blockingsolution) were added to each well. Samples were incubated overnight at4˚C and then washed three times (10 min) with PBS containing 0.05%Tween 20 and 2 mM levamisole. Two drops of anti-rabbit secondary Ab(Vectastain ABC kit; Vector Labs) were added to each well and incubatedfor 1.5 h. Wells were then washed three times with PBS containing 0.05%Tween 20 and 2 mM levamisole (10 min). Two lots of 100 cells werecounted for each sample, and the average for each sample was consideredas an n number. Results are expressed as percentage of caspase-3+ cells.
Assessment of NO levels by measurement of NO22/NO3
2
The NO22/NO3
2 content was measured by the Griess assay as an indicator ofNO production in the PELF obtained from sham- and CLP-operated animals.NO3
2 was reduced to nitrite (NO22) by incubating 80 ml sample with 20 ml
of 1 U/ml nitrate reductase and 10 ml of 1 mM NADPH for 30 min at 37˚C ina 96-well plate. Next, 100 ml Griess reagent (5% v/v H3PO4 containing 1%w/v sulfanilic acid and 0.1% w/v N-1-napthylethylenediamine) was addedand incubated for 15 min at 37˚C. Absorbance at 550 nm was measuredimmediately using a spectrophotometer (Spectra Max 190; MolecularDevices, Palo Alto, CA). After subtraction of background readings, the ab-sorbance in each sample was compared with that obtained from a sodiumnitrite (0–100 mM) standard curve and expressed as NOx levels (mM).
Superoxide release from the peritoneal inflammatory cells
Superoxide (O22) release from the peritoneal inflammatory cells obtained
from sham- and CLP-operated animals was measured by chemolumines-cence using lucigenin (bis-N-methylacridinium nitrate; Sigma Chemical)as a probe (22). For this, 100 ml modified Krebs buffer (pH 7.4, compo-sition: NaCl 131 mM, KCl 5.6 mM, NaHCO3 25 mM, NaH2PO4.H2O 1mM, glucose 5 mM, HEPES 5 mM, L-arginine 100 mM, CaCl2 2.5 mM,MgCl2 1 mM, and NADPH 100 mM) was pipetted into each well of a 96-well white plate (Nunc). Then, 50,000 cells obtained from the PELF wereadded and incubated with or without O2
2 dismutase (50 U/ml; SigmaAldrich, U.K.) in 100 ml modified Krebs buffer. Immediately, the plate wasread in a chemoluminescence reader (Plate Chameleon V MicroplateReader; Noki Technologies PVT, Hidex Oy, Finland) at 37˚C, 1 s/read,each 90 s, for 1 h. Results are expressed as the difference in the cpm in thepresence and absence of O2
2 dismutase.
H2O2 production by peritoneal inflammatory cells
H2O2 production by peritoneal inflammatory cells obtained from sham-and CLP-operated animals was measured by using a H2O2/peroxidaseassay kit (Amplex Red H2O2/Peroxidase assay kit; Molecular Probes,Invitrogen, Paisley, U.K.). The assay was performed according to themanufacturer’s instructions with minor modifications. In brief, 25,000 cellsfrom the PELF were incubated with 100 ml Krebs buffer (pH 7.4, com-
2 TRPV1 AND MURINE SEPSIS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

position: NaCl 131 mM, KCl 5.6 mM, NaHCO3 25 mM, NaH2PO4.H2O1 mM, glucose 5 mM, HEPES 5 mM, CaCl2 2.5 mM, MgCl2 1 mM, andNADPH 100 mM) and 100 ml of a solution containing NaPO4 0.05 M(pH 7.4), HRP 0.2 U/ml, and Amplex Red Reagent (10-acetyl-3,7-dihydroxyphenoxazine) 25.7 mg/ml, for 2 h, at 37˚C. Samples incubatedwith Krebs buffer only were used as controls. After incubation, 100 ml ofeach sample was pipetted into 96-well plates, and reaction was read at 560nm. Absorbance readings, obtained for samples incubated in the absence orpresence of Amplex Red Reagent, were compared with a H2O2 standardcurve (0–40 mM). Results are expressed as the difference between samplesincubated in the presence of Amplex Red Reagent or Krebs (in mM).
Cytokine and chemokine measurement
PELF IL-6, IL-10, TNF-a, IL-1b, and keratinocyte-derived chemokine (KC)levels were evaluated using a proinflammatory multiplex cytokine kit (MouseProinflammatory multiplex kit; Meso-Scale Discovery, Gaithersburg, MD)according to the manufacturer’s instructions. Sample readings for each cy-tokine were compared with a standard curve (0–10,000 pg/ml). MCP-1 andMIP-1b were evaluated by using a multiplex assay (Searchlight; AushonBiosystems, Billerica, MA). Results are expressed as protein levels inpg/ml. All detection Abs exhibited,1% cross-reactivity with other analytes.
Blood pressure evaluation
The effects of sepsis on blood pressure were assessed 24 h after surgery.Blood pressure was assessed noninvasively in conscious, restrained mice bythe tail cuff technique using the CODA 6 System (Kent Scientific, Tor-rington, CT), which assesses tail blood pressure by means of volumepressure recording (18). Baseline measurements were taken before surgery.
Body temperature
Body temperature was monitored using a rectal probe (Harvard Apparatus,Holliston, MA) just before and 24 h after surgery (18).
Assessment of organ damage
Dysfunction of heart/liver, kidneys, and pancreas was assessed by mea-suring aspartate aminotransferase (AST), creatinine, and lipase levels,respectively, present in plasma obtained fromWTand TRPV1KOmice 24 hafter surgery. In a separate group, CLP WT mice were treated s.c. for6 d (twice daily) with the TRPV1 antagonists, capsazepine (50 mg/animal)or SB366791 (0.5 mg/kg), or with vehicle (120 ml; 10% DMSO in saline).Measurements were made by Nationwide Laboratories (Lancashire, U.K.).Results are expressed as UI/l (lipase and AST) and mmol/l (creatinine).
Gene expression analysis in peritoneal inflammatory cells
Quantitative mRNA expression in peritoneal inflammatory cells was de-termined by real-time PCR. In brief, peritoneal cells were collected fromWTand TRPV1KOmice 24 h after CLP and stored in RNA later until RNAextraction was performed. DNA-free total RNA was extracted from thesamples using the RNeasy Microarray kit (Qiagen, Crawley, U.K.), and 0.5mg total RNA was reverse transcribed to cDNA using the high-capacityRNA-to-cDNA kit with RNAse inhibitor (Applied Biosystems) accordingto the manufacturer’s instructions. Real-time PCR was performed ona Corbett Rotorgene (hold: 10 min at 95˚C; cycling: 45 cycles: 10 s at 95˚C,15 s at 57˚C, and 5 s at 72˚C; melt: 68–90˚C), using the SensiMixTMSYBR No-ROX Kit (Bioline). The following primers were obtained fromSigma: caspase-3 [forward: 59-GAGGCTGACTTCCTGTATGCTT-39; re-verse: 59-AACCACGACCCGTCCTTT-39], p53 [forward: 59-ATGCC-CATGCTACAGAGGAG-39; reverse: 59-AGACTGGCCCTTCTTGGTCT-39], PI3Ka [forward: 59-GACAAGAACAAGGGCGAGAT-39; reverse: 59-CAGTACCCAGCGCAGGAC-39], PI3Kb [forward: 59-GACAAGAA-CAAGGGCGAGAT-39; reverse: 59-CAGAGCGATCTCCTGTTGCT-39],TRPV1 [forward: 59-CAACAAGAAGGGGCTTACACC-39; reverse: 59-TCTGGAGAATGTAGGCCAAGAC-39], FAS [forward: 59-TGCAGA-CATGCTGTGGATCT-39; reverse: 59-CTTAACTGTGAGCCAGCAAGC-39], NADPH oxidase 2 (NOX2) [forward: 59-TGCCAACTTCCTCAGC-TACA-39; reverse: 59-GTGCACAGCAAAGTGATTGG-39], NOX4 [for-ward: 59-GAACCCAAGTTCCAAGCTCA-39; reverse: 59-AAGGC-ACAAAGGTCCAGAAA- 39], TAC-1 [forward: 59-AAGCCTCAG-CAGTTCTTTGG-39; reverse: 59-TCTGGCCATGTCCATAAAGA-39],aCGRP [forward: 59-AGCAGGAGGAAGAGCAGGA-39; reverse: 59-CAGATTCCCACACCGCTTAG-39], TLR4 [forward: 59-GGACTCT-GATCATGGCACTG-39; reverse: 59-CTGATCCATGCATTGGTAGGT-39], CD14 [forward: 59-GGACTCTGATCATGGCACTG-39; reverse: 59-CTGATCCATGCATTGGTAGGT-39], triggering receptor expressed onmyeloid cell 1 (TREM1) [forward: 59-TCTGGATTGTATCGTTGTGTGA-
39; reverse: 59-GGAGTGAACACATCTGAAGAACC-39], TREM2 [for-ward: 59-TGGGACCTCTCCACCAGTT-39; reverse: 59-GTGGTGTTGA-GGGCTTGG-39], PLA2G12A [forward: 59-TGGATATAAACCATC-TCCACCA-39; reverse: 59-GGGAAGGGATACCTATGTTCAGA-39], andB2M [forward: 59-CCTGCAGAGTTAAGCATGCC-39; reverse: 59-GATG-CTTGATCACATGTCTCG-39].
Results are expressed as copy number per microliter of pure cDNAnormalized by comparison with B2M and PLA2G12A using GeNormversion 3.4. All experiments were performed in accordance with theMinimum Information for Publication of Quantitative Real Time PCRExperiments guidelines.
Detection of bacteria in liver and lung
The presence of bacteria in liver and lung samples obtained from CLP WTand TRPV1KO mice after 24-h sepsis was assessed. For this purpose, thelevels of 16S rRNA, transcribed from a gene expressed by all bacteria (23)were determined. In brief, samples were collected and stored in RNAp-rotect Bacteria Reagent (Qiagen). Samples were processed by using en-zymatic lysis and mechanical disruption of cells present in the tissuesamples according to manufacturer’s instructions followed by DNA-freetotal RNA extraction using the RNeasy Plus Mini kit (Qiagen). A total of0.5 mg total RNAwas reverse transcribed to cDNA using the high-capacityRNA-to-cDNA kit with RNAse inhibitor (Applied Biosystems), accordingto the manufacturer’s instructions. Real-time PCR was performed on aCorbett Rotorgene (hold: 10 min at 95˚C; cycling: 45 cycles: 10 s at 95˚C,15 s at 57˚C, and 5 s at 72˚C; melt: 68–90˚C), using the SensiMixTMSYBR No-ROX Kit (Bioline). The following primers were obtained fromSigma: forward [59-CGGTGAATACGTTCYCGG-39] and reverse [59-GGWTACCTTGTTACGACT T-39] (23). A standard curve for 16S rRNAgene was derived from DNA-free total RNA extracted from an overnightculture (c. 1 3 109 CFU ml21) of Pseudomonas aeruginosa PAO1 har-vested at the end of log/start of stationary phase. Experiments were per-formed in accordance with MIQE guidelines, and results are expressed ascopy numbers per microliter.
Tissue and plasma endotoxin levels
Endotoxin levels were analyzed in plasma and liver and lung samplesobtained from CLP WT and TRPV1KO mice by using a ToxinSensorchromogenic endotoxin detection system (Genscript), which mainlymeasures endotoxin that is dissociated from bacteria (free endotoxin) (24).All samples were collected and kept in endotoxin-free tubes at280˚C untilthe endotoxin test. Just before the assay, tissue samples were homogenizedin endotoxin-free PBS. All samples were diluted 1:10 and heated at 75˚Cfor 5 min to minimize protein interference with the assay. Assay wasperformed according to manufacturer’s instructions. Results obtained fromplasma samples are expressed as endotoxin units per milliliter. Resultsobtained from tissue samples are normalized by protein content andexpressed as endotoxin units per milligram of protein. Protein levels werequantified by Bradford protein assay (Bio-Rad, Hemel Hempstead, U.K.).
Data analysis
The results are presented as the mean 6 SEM. The percentages of inhi-bition are reported as mean 6 SEM of inhibitions obtained in each indi-vidual experiment compared with control samples. Statistical comparisonsof the data were performed by ANOVA followed by Newman–Keuls andunpaired or paired t test when appropriate. The p values ,0.05 wereconsidered significant.
ResultsTRPV1 protects against organ damage and hypothermia insepsis
Systemic sepsis was evaluated 24 h after CLP by measuring plasmalevels of AST, creatinine, and lipase as indicators of heart/liver,kidney, and pancreas failure, respectively. Sepsis induction byCLP led to a significant increase in AST, but not creatinine andlipase levels, when compared with sham-operated animals, indi-cating organ dysfunction in WT mice (Fig. 1A–C). TRPV1KOexhibited further increases of AST, creatinine, and lipase pro-duction, compared with CLP-operated WT mice (Fig. 1A–C),showing that mice lacking TRPV1 suffer accelerated organdamage. No difference was observed between the sham-operatedWT and TRPV1KO mice. Similarly, 6-d treatment with the
The Journal of Immunology 3
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
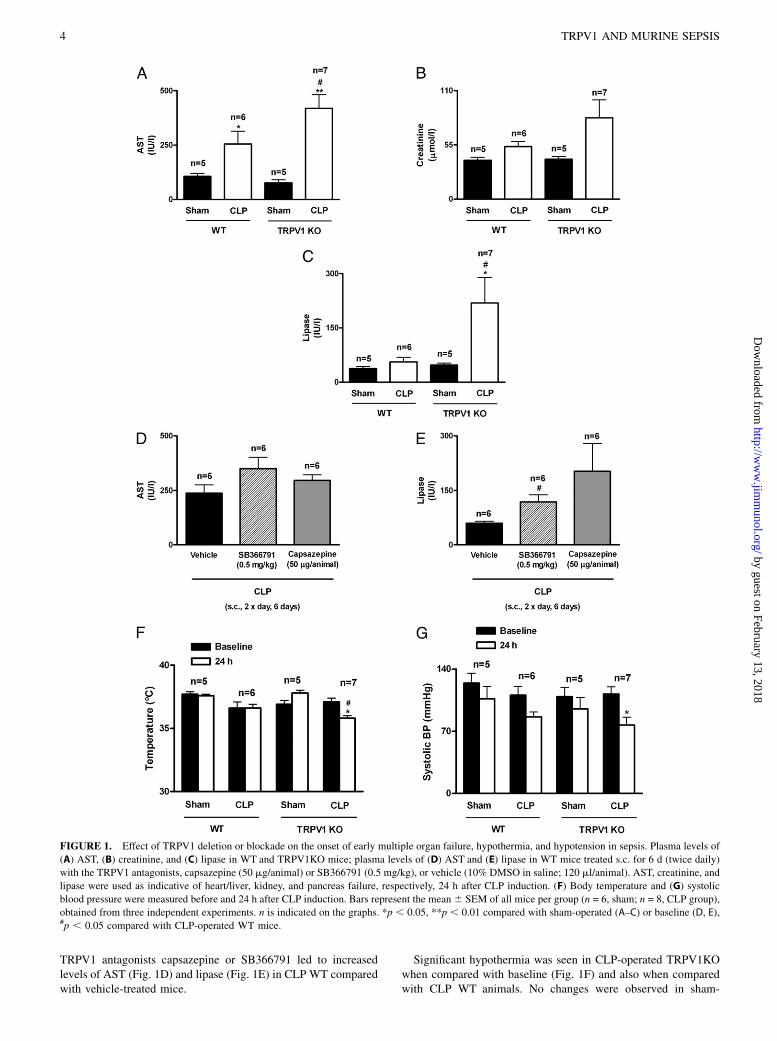
TRPV1 antagonists capsazepine or SB366791 led to increasedlevels of AST (Fig. 1D) and lipase (Fig. 1E) in CLP WT comparedwith vehicle-treated mice.
Significant hypothermia was seen in CLP-operated TRPV1KOwhen compared with baseline (Fig. 1F) and also when comparedwith CLP WT animals. No changes were observed in sham-
FIGURE 1. Effect of TRPV1 deletion or blockade on the onset of early multiple organ failure, hypothermia, and hypotension in sepsis. Plasma levels of
(A) AST, (B) creatinine, and (C) lipase in WT and TRPV1KO mice; plasma levels of (D) AST and (E) lipase in WT mice treated s.c. for 6 d (twice daily)
with the TRPV1 antagonists, capsazepine (50 mg/animal) or SB366791 (0.5 mg/kg), or vehicle (10% DMSO in saline; 120 ml/animal). AST, creatinine, and
lipase were used as indicative of heart/liver, kidney, and pancreas failure, respectively, 24 h after CLP induction. (F) Body temperature and (G) systolic
blood pressure were measured before and 24 h after CLP induction. Bars represent the mean 6 SEM of all mice per group (n = 6, sham; n = 8, CLP group),
obtained from three independent experiments. n is indicated on the graphs. *p , 0.05, **p , 0.01 compared with sham-operated (A–C) or baseline (D, E),#p , 0.05 compared with CLP-operated WT mice.
4 TRPV1 AND MURINE SEPSIS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

operated mice. A decreased systolic blood pressure was observedin both CLP WT and TRPV1KO when compared with baseline(Fig. 1G), which was significant in CLP TRPV1KO mice. Theseresults suggest TRPV1 has a role in maintaining the body ho-meostasis during the transition of local to systemic inflammatorydisease.
TRPV1 is expressed on peritoneal inflammatory cells andis critical for peritoneal mononuclear cell viability
TRPV1 is expressed in peritoneal inflammatory cells obtained fromboth sham and CLP WT mice (Fig. 2A). Thus, we hypothesizedthat TRPV1 played a role in peritoneal cell functioning. We firstevaluated the naive peritoneal cell population. TRPV1KO pre-sented lower cell counts when compared with WT mice (Fig. 2B),suggesting a possible intrinsic mechanism for lack of peritonealprotection in these animals.We analyzed the possible participation of TRPV1 in influencing
cell migration into the peritoneal cavity following CLP. Total cellcounts (Fig. 2C) were increased in both CLP WT and TRPV1KOmice when compared with shams. For differential analysis, only
viable cells were considered (Fig. 2F). CLP WT mice exhibitedincreased numbers of both polymorphonuclear and mononuclearcells (Fig. 2D, 2E). CLP TRPV1KO and WT mice presentedsimilar polymorphonuclear cell increase when compared withshams (Fig. 2D, 2E). In contrast, the number of intact mononu-clear cells was reduced (59 6 11%) in CLP-operated TRPV1KOmice compared with CLP WT mice (Fig. 2E).
Increased apoptosis and changes in cytokine and chemokinerelease in the peritoneal cavity of TRPV1KO mice
Cytokines and chemokines play a fundamental role in modulatingsepsis (25, 26). Increased levels of TNF-a, IL-6, IL-10, and IL-1bwere observed in CLP-operated WT mice when compared withshams (Fig. 3A–D). However, TNF-a, IL-6, and IL-10 levels inCLP TRPV1KO were significantly enhanced in comparison withCLP WT mice (Fig. 3A–D). MCP-1 and MIP-1b, but not KClevels, were reduced in CLP TRPV1KO when compared with WTmice (Fig. 3E–G). Analysis of the expression of FAS, a proteinassociated with apoptosis, revealed that peritoneal cells obtainedfrom CLP TRPV1KO mice present higher FAS mRNA copy
FIGURE 2. TRPV1 role in peritoneal
cells. (A) TRPV1 mRNA expression on
peritoneal cells obtained from sham and
CLP WT mice; (B) analysis of peritoneal
cell population in naive WT and
TRPV1KO mice. In vivo cell migration
was evaluated in the peritoneal cavity of
both WT and TRPV1KO, 24 h after sur-
gery for (C) total and (D) polymorphonu-
clear and (E) mononuclear cell counts. (F)
H&E staining of WT and TRPV1KO
peritoneal cells showing a viable (WT)
and a vacuolated mononuclear cell
(TRPV1KO) undergoing apoptosis. Bars
represent the mean6 SEM of all mice per
group obtained from four independent
experiments. n is indicated on the graphs.
*p , 0.05, **p , 0.01 compared with
sham-operated, #p , 0.05 compared with
CLP-operated WT mice.
The Journal of Immunology 5
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

numbers when compared with CLP WT samples (Fig. 4A). Sim-ilarly, the mRNA for the apoptosis effectors p53 and caspase-3was higher in CLP TRPV1KO than in CLP WT mice (Fig. 4B,4C). In addition, we evaluated caspase-3 protein expression inadherent macrophages cultured from CLP WT peritoneal samplestreated or not with the TRPV1 antagonist SB-366791 (20 mM).Results show that the percentage of caspase-3+ cells was greater inSB-366791–treated samples (Fig. 4D, 4E).
Reduced ROS and NO levels in the peritoneal cavity in theTRPV1KO mice
Sepsis induced by CLP in WT mice led to an increase of NO, O22,
and H2O2 release into the peritoneal cavity (Fig. 5A–C). Negli-gible amounts of these mediators were detected in sham-operatedanimals. Surprisingly, the levels of NO, H2O2, and O2
2 weresubstantially less in TRPV1KO by 85 6 7, 77 6 12, and 56 610%, respectively, compared with levels in WT mice at 24 h afterCLP. In addition, both NOX2 and NOX4 mRNA levels were re-
duced in CLP TRPV1KO in comparison with CLP WT peritonealcells (Table I).
TRPV1 deletion affects phagocytosis via the NK1 receptor forSP and by affecting PI3K expression
Unstimulated WT and TRPV1KO-derived macrophages presenteda similar ability to phagocytose beads (Fig. 6A, 6B). After LPSstimulation, the number of beads phagocytosed (Fig. 6A) by WTmacrophages, as well the number of cells able to phagocytose(Fig. 6B), remained unaltered when compared with unstimulatedcells. Surprisingly, TRPV1KO macrophages were less able tophagocytose beads after LPS with a 67 6 6% reduction whencompared with KO unstimulated cells (Fig. 6A). Similarly, thenumber of TRPV1KO macrophages containing beads was alsoreduced by 36 6 9% (Fig. 6B). SP and CGRP are released fromsensory nerves when TRPV1 is activated, but they are also presentin resident macrophages and circulating leukocytes (27–29). In-deed, SP treatment has been shown to stimulate macrophage-
FIGURE 3. Effect of TRPV1 deletion on
cytokine and chemokine generation during
sepsis. (A) TNF-a, (B) IL-6, (C) IL-10, (D) IL-
1b, (E) MCP-1, (F) MIP-b, and (G) KC were
analyzed in the peritoneal lavage collected
24 h after CLP induction. Bars represent the
mean 6 SEM of all mice per group obtained
from four independent experiments performed
in duplicate. n is indicated on the graphs.
**p , 0.01 compared with sham-operated,#p , 0.05, ##p , 0.01 compared with CLP-
operated WT mice.
6 TRPV1 AND MURINE SEPSIS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

mediated phagocytosis in vitro (30). Cotreatment of LPS-stimulated TRPV1KO macrophages with SP restored theirphagocytic ability (Fig. 6C, 6D). Preincubation of cells treatedwith LPS and SP with the selective SP NK1 receptor antagonistSR140333, but not the selective NK2 receptor antagonist SR48968, blocked this stimulatory effect of SP on TRPV1KO mac-rophages (Fig. 6C, 6D). On the other hand, CGRP coincubationhad no significant effect in the TRPV1KO macrophage response
treated with LPS (Fig. 6C). LPS-stimulated WT macrophages in-cubated with the TRPV1 antagonist SB366791 (20 mM) exhibitedless phagocytosis when compared with vehicle-treated cells (Fig.6E, 6F).PI3Ka and b are involved in phagolysosome formation and also
ROS production by macrophages (31, 32). CLP TRPV1KO peri-toneal cells express less PI3Ka and b when compared with CLPWT cells (Fig. 6G, 6H, respectively).
FIGURE 4. TRPV1KO mice exhibit
enhanced apoptosis markers. (A) FAS,
(B) p53, and (C) caspase-3 mRNA levels
in peritoneal cells obtained from WT
and TRPV1KO mice 24 h after sepsis
induction. (D and E) Caspase-3 protein
expression by immunocytochemistry in
LPS-challenged WT cells treated with
the TRPV1 antagonist SB366791 (20
mM) or vehicle (0.1% DMSO). Immu-
nocytochemistry staining was visualized
under 340 magnification. Bars repre-
sent the mean 6 SEM of all mice per
group obtained from four independent
experiments performed in duplicate.
n is indicated on the graphs. *p , 0.05
compared with vehicle-treated cells.
FIGURE 5. TRPV1 deletion decreases NO and
ROS production in the peritoneal cavity. (A)
NO22/NO3
2 levels are shown indicating NO forma-
tion (NOx); (B) H2O2 and (C) O22 release from the
peritoneal lavage cells 24 h after CLP induction. Bars
represent the mean 6 SEM of all mice per group
obtained from at least three independent experi-
ments performed in duplicate. n is indicated on the
graphs. *p , 0.05, **p , 0.01 compared with sham-
operated; ##p , 0.01 compared with CLP-operated
WT mice.
The Journal of Immunology 7
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Table I shows the mRNA copy numbers for TAC-1 (a precursorof SP) were reduced in CLP TRPV1KO when compared with CLPWT samples, whereas no difference was observed for CGRPagene.
TRPV1 does not affect pathogen recognition by TLR4
Considering that CLP is a model that involves mainly Gram-negative bacteria (2) and that we found that TRPV1KO perito-neal cells behave differently to WT peritoneal cells in the presenceof LPS, we quantified the mRNA levels of TLR4. No differencein TLR4 expression between peritoneal cell samples of CLP WTand TRPV1KO mice was observed (Table I). Also, CLP WT andTRPV1KO samples showed similar levels of mRNA for moleculesinvolved in TLR4 signaling such as CD14, TREM1, and TREM2(Table I).
TRPV1 deletion facilitates local bacterial survival
Our results show increased 16S rRNA levels in liver but not lungsamples obtained from CLP TRPV1KO when compared with WTmice (Table II). Endotoxin is mainly released from bacteria afterbeing shed or after bacterial lysis (33). CLP WT and TRPV1KOmice lung and liver samples exhibited similar levels of endotoxinper milligram of tissue (Table II). On the other hand, CLPTRPV1KO plasma samples presented reduced endotoxin unitswhen compared with CLP WT samples (Table II).
DiscussionRecent evidence shows that TRPV1 blockade or deletion in LPS-induced endotoxemia is associated with poorer survival in rats (17)and enhanced symptoms, with raised sepsis markers in mice (18).Although this protective role for TRPV1 has been demonstrated intwo species, the in vivo mechanisms are unknown. In this article,we demonstrate that TRPV1 protects against CLP-induced sepsis,considered to model human disease. This protection is linked toinflammatory cell functioning, specifically the macrophage and itsability to kill and phagocytose bacteria, pivotal to the efficiency oflocal defense mechanisms at the site of infection and the transitionfrom a local to a systemic inflammatory response as observed inSIRS.We show that TRPV1KO mice exhibited decreased body tem-
perature and systolic blood pressure after CLPwhen compared withbaseline measurements at 24 h, indicative of the onset of systemicsepsis, whereas no significant changes were observed in the CLPWT group. These results complement our findings that TRPV1deletion is associated with decreases in both temperature and meanblood pressure at 4 h after LPS treatment (18). In this study, weused the decreased temperature as a marker of impending mor-
bidity (34) in keeping with welfare considerations. AST, creati-nine, and lipase levels were measured as markers of heart/liver,kidney, and pancreas dysfunction, indicative of early signs of or-gan failure. We show that TRPV1 protects against liver, kidney,and pancreatic failure as the release of AST, creatinine, and lipaseare increased in CLP TRPV1KO mice. Similarly, increased organfailure was also observed in CLP WT animals treated with theTRPV1 antagonists capsazepine and SB366791. These data con-firm that an SIRS has been initiated by 24 h after CLP, with en-hanced hypotension, hypothermia, and markers of organ damageindicating an accelerated sepsis onset after either TRPV1 deletionor pharmacological blockade.Both human dendritic cells and T lymphocytes express func-
tional TRPV1 (35, 36). We show that WT peritoneal inflammatorycells express TRPV1 gene in both sham and septic conditions.This would enable TRPV1-mediated protection to occur at anearly stage of local gut infection after CLP. The regulation of cellmigration and inflammatory mediator release involves a fine crosstalk between several systems in the body and is critical for sepsisoutcome (1, 23, 37), but the mechanisms involved in the modu-latory effect of TRPV1 are unclear. Our results show that TRPV1can modulate sepsis at several steps of the CLP-induced inflam-matory response. SIRS and organ damage caused by 24-h CLP inmice with disrupted TRPV1 signaling was correlated with thedemonstration of a distinct enhanced inflammation in the perito-neal cavity compared with that in WT mice. The CLP-inducedperitoneal inflammation in TRPV1KO mice was associated witha reduced number of viable mononuclear cells, increased ex-pression of apoptosis markers, and increased production of keycytokines, but reduced ROS, NO, and chemokine generation inthe peritoneal cavity, facilitating an increased bacterial survivalin the peritoneal cavity. Moreover, the TRPV1KO peritoneal mac-rophages exhibited impaired phagocytosis to LPS, caused by atachykinin NK1-receptor–dependent mechanism.Mortality in sepsis is preceded by a simultaneous release of
both proinflammatory and anti-inflammatory cytokines (38). Our re-sults show that TNF-a, IL-6, and IL-10 levels are increased inthe peritoneal cavity of CLP TRPV1KO compared with CLP WTmice. This suggests that TRPV1 can influence cytokine generationduring septicemia and reinforces the concept that the inflamma-tory response observed in TRPV1KO mice is being accelerated.Indeed, there is evidence of enhanced TNF-a levels in LPS-treatedTRPV1KO, compared with WT mice (18). In addition, adminis-tration of a low dose of the TRPV1 agonist capsaicin has beenshown to reduce inflammation in septic rats, by decreasing plasmalevels of TNF-a and IL-6, but not IL-10, which was increased inthat study (39). These evidences suggest that TRPV1 activationrepresents an important step in the inflammatory response asso-ciated to sepsis.We also found that TRPV1 influences ROS and NO release from
peritoneal cells. Indeed, links between O22 production and TRPV1
upregulation, involving NOX production and subsequent O22 re-
lease, exist (40, 41). Moreover, there is evidence that TRPV1 canbe activated in the presence of H2O2 (42). We postulate that sepsistriggers O2
2 and H2O2 release from cells located in the perito-neum that, in turn, amplify TRPV1 expression to enhance ROSgeneration. We show that high levels of O2
2, H2O2, and NO arereleased from the peritoneal cells of CLP WT mice. However, oneof the most striking findings from this study is that substantiallyless ROS and NO release was detected in CLP TRPV1KO mice.This surprised us, because NO release was enhanced after 4 h inTRPV1KO in the acute response to intraperitoneal LPS (18). Theopposing effects of TRPV1 on ROS and NO release may be modeldependent. Previous studies show that capsaicin treatment reduces
Table I. Analysis of gene expression of various genes by quantitativereal-time PCR in peritoneal cell samples obtained from CLP WT andTRPV1KO mice
GeneCLP WT
(3103 Copies/ml)CLP TRPV1KO(3103 Copies/ml)
NOX2 2114.0 6 880.3 852.5 6 532.7NOX4 0.003 6 0.002 0.0006 6 0.0004TAC-1 0.23 6 0.13 0.12 6 0.06aCGRP 423 6 175.7 415.6 6 151.4TLR4 4757.3 6 2500.3 4955 6 1880.3CD14 10,900 6 3704.7 7900.2 6 3123.6TREM1 355.7 6 177.9 283.7 6 110.7TREM2 206.6 6 116.2 256.5 6 91.9
Samples were collected 24 h after surgery, and results are expressed as number ofcopies/ml (3103). Results express the mean 6 SEM of all mice per group obtainedfrom at least two independent experiments performed in duplicate; n = 5 each group.
8 TRPV1 AND MURINE SEPSIS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

plasma NO release in CLP-operated rats (39) and also reducesNO release from RAW264.7 macrophages stimulated with LPSin vitro (43), supporting the idea that TRPV1 activation can reg-
ulate NO. We found that the reduced generation of ROS and NOfrom CLP TRPV1KO peritoneal cells correlated with reducedNOX2 and NOX4 mRNA expression in these cells. Indeed, it has
FIGURE 6. TRPV1 deletion im-
pairs phagocytosis in peritoneal mac-
rophages. (A) Total beads/100 mac-
rophages obtained from TRPV1 WT
and KO mice, and (B) percentage of
macrophages containing beads with
or without LPS stimulation for 24 h
(100 ng/ml). (C) Total beads/100 mac-
rophages obtained from TRPV1 WT
and KO mice, and (D) percentage of
macrophages containing beads stim-
ulated with LPS (100 ng/ml, 24 h) in
the presence or absence of SP and
NK1 and NK2 antagonists or CGRP.
(E) Total beads/100 macrophages ob-
tained from WT mice and (F) per-
centage of macrophages containing
beads after LPS stimulation for 24 h
(100 ng/ml) in the presence of either
the TRPV1 antagonist SB366791
(20 mM) or vehicle (0.1% DMSO).
mRNA levels of (G) PI3Ka and (H)
PI3Kb levels in TRPV1 WT and KO
samples collected 24 h after CLP in-
duction. Bars represent the mean 6SEM of all mice per group obtained
from at least two independent ex-
periments performed in duplicate.
n is indicated on the graphs. *p ,0.05, **p , 0.01 compared with
WT cells, #p , 0.05 compared with
LPS-stimulated TRPV1KO cells,@p , 0.05 compared to TRPV1 KO
cells incubated with SP.
Table II. Detection of bacterial 16S rRNA by quantitative real-time PCR in tissue samples and endotoxinlevels quantification in plasma and tissue samples obtained from CLP WT and TRPV1 KO mice
16S rRNA (Copies/ml)
Tissue Endotoxin Levels(31024 EndotoxinUnits/Mg Protein)
Lung Liver Lung LiverPlasma Endotoxin Levels(Endotoxin Units/Ml)
CLP WT 129.3 6 18.2 238.5 6 22.5 2.5 6 0.6 3.0 6 0.9 8.5 6 2.2CLP TRPV1KO 129.4 6 11.4 364.3 6 42.4* 2.0 6 0.3 2.9 6 1.3 1.5 6 0.2*
Samples were collected 24 h after surgery. Results express the mean 6 SEM of all mice per group obtained from at least twoindependent experiments performed in duplicate; n = 6 CLP WT; n = 7 CLP TRPV1KO group.
*p , 0.05 compared with CLP-operated WT mice.
The Journal of Immunology 9
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

been demonstrated that NOX4 can interact with TLR4, thus reg-ulating H2O2 production (44), whereas NOX2 activation in thephagosomal membrane of macrophages leads to O2
2 generation(45). One interpretation of our data is that TRPV1 is involved inmediating the ability of phagocytes to release ROS and NOx, andkill microorganisms. This defensive mechanism is impaired inTRPV1KO mice, resulting in increased bacterial survival in theperitoneal cavity such as in the liver, and this may lead to theacceleration of the systemic phase of the disease.The decrease in ROS and NO levels in the absence of TRPV1
twinned with evidence that peritoneal cells do express TRPV1 ledus to investigate cell migration. We have shown that peritoneallavage from CLP TRPV1KO mice contained lower levels ofchemokines involved in macrophage recruitment (MCP-1 andMIP-1b) (46, 47) than that from CLP WT mice, whereas levels ofKC, a neutrophil-related chemokine (48), was similar in both WTand TRPV1KO mice. Also, in vivo analysis of cell populationsshowed that naive TRPV1KO possess fewer peritoneal residentinflammatory cells than WT mice. However, after CLP, both WTand TRPV1KO mice exhibited similar numbers of viable PMNcells in the peritoneal cavity, whereas the number of viablemononuclear cells was significantly lower in TRPV1KO com-pared with WT mice.The reduction of viable macrophages and increased TNF-a
levels observed in CLP TRPV1KO led us to investigate the ex-pression of apoptosis markers such as FAS (a death receptormember of the TNF superfamily expressed on inflammatory cells)(49), and the apoptosis effector molecules caspase-3 and p53.TNF-a itself is known to mediate apoptosis via its TNFR1 (50). Inaddition, FAS, p53, and caspase-3 mRNA levels were all increasedin peritoneal inflammatory cell samples from CLP TRPV1KOwhen compared with CLP WT mice. Similarly, WT macrophagestreated with the TRPV1 antagonist SB366791 expressed morecaspase-3 than WT cells after LPS treatment. These findings leadus to hypothesize that the increased release of TNF-a and in-creased expression of FAS and apoptosis effectors in the perito-neal cavity favors mononuclear cell apoptosis in the TRPV1KOseptic mouse. This would explain, at least in part, the reduction ofperitoneal NOx and ROS levels in the CLP TRPV1KO peritoneallavage. Furthermore, the associated increase in IL-10 and IL-6,and reduction of MCP-1 and MIP-1b generation in TRPV1KOseptic mice could all contribute to suppression of the immunesystem by deactivating macrophages in respect to their recruit-ment and their ability to phagocytose and kill microorganismsand limiting proinflammatory mediator production. This deficientimmune response could lead to a dangerous situation resulting infacilitation of bacterial replication, increasing the chances of se-vere infection and organ failure.We show that LPS-stimulated peritoneal macrophages obtained
from TRPV1KO mice exhibited a lower ability to phagocytosebeads. Similarly, treatment with the TRPV1 antagonist SB366791reduced phagocytosis when incubated with WT adherent macro-phages. Also, the gene expression of PI3Ka and b, involved inROS production, phagolysosome formation, and phagocytosis(31, 32), was considerably reduced in CLP TRPV1KO mice. Thisprovides evidence that TRPV1 can influence the response to in-fection by facilitating macrophage antibactericide function, thusacting via a protective mechanism associated with release of NOand ROS.Indeed, we present evidence of increased bacterial 16S rRNA in
liver samples obtained from CLP TRPV1KO when compared withWT mice. This is suggestive of increased bacterial colonizationin the peritoneal cavity. We also evaluated bacterial presence indistant organs such as lung, where we found similar levels of 16S
rRNA in CLP WT and TRPV1KO mice. Endotoxin is commonlyreleased from Gram-negative bacteria via shedding during growth,although a large part of its release occurs after bacterial lysis (33).Endotoxin plasma levels were increased in CLP WT but notTRPV1KO mice, whereas endotoxin tissue levels were not alteredby TRPV1 deletion. This is consistent with the suggestion thatendotoxin is being released in the circulation as bacterial cells aredestroyed by the immune system in CLP WT animals. We caninfer that at 24 h after CLP, the major area affected by TRPV1deletion is the peritoneal cavity. However, we cannot overrulea role for TRPV1 in changes in the presence of bacteria in the lungat later time points. Indeed, in support of our findings, it was re-cently demonstrated that blood samples obtained from CLPTRPV1KO exhibited a higher bacterial load 24 h after sepsis in-duction that was associated with higher mortality when comparedwith WT mice (51).We also evaluated the impact of TRPV1 disruption on pathogen
recognition. TRPV1 has been found colocalized to the TLR4 andCD14 receptor complex in sensory nerves (52). In addition, TLR4could sensitize TRPV1 via protein kinase C activation (53). Al-though these pieces of evidence suggest a sensing mechanism forbacterial infection in sensory nerves, we could not find differencesin the mRNA expression of TLR4 or in molecules involved in thepathogen recognition mediated by TLR4 and TLR4 signalingsuch as CD14, TREM1, and TREM2 (52–56). This suggests thatTRPV1 does not interfere with TLR4 functioning, but ratherreinforces the idea that TRPV1 regulates bacterial killing instead.TRPV1 is best known for its ability to release SP and CGRP (5),
neuropeptides considered as predictors of lethal sepsis outcome inpatients (11). Previously, we have shown that the enhanced LPS-induced endotoxemia observed in TRPV1KO mice is unlikely tobe due to SP (18). In this study, we show reduced levels of TAC-1mRNA in CLP TRPV1KO when compared with CLP WT peri-toneal inflammatory cells. SP is suggested to modulate immunecell function and inflammation via a range of mechanisms (28, 29,55, 57). This article shows for the first time, to our knowledge, thatSP mediates TRPV1 protection by positively regulating macro-phage phagocytosis via NK1 receptor activation. By comparison,we found no evidence for a role of CGRP in this response.In conclusion, we provide substantial new evidence that TRPV1
activation is associated with sepsis outcome. We show that TRPV1activation at the site of infection is directly linked to a series ofevents in the immune and inflammatory response, central tomacrophage functioning such as apoptosis, ROS, and NO pro-duction, and also the release of key controlling cytokines andchemokines from inflammatory cells. The sum effect of TRPV1deletion is an impairment of phagocytosis and bacterial clearanceduring septicemia. This directly influences the enhancement ofhypotension and hypothermia, and evidence of organ damage. Thisstudy demonstrates the importance of TRPV1 to the initial site ofinfection and highlights the need to learn more about fundamentalmechanisms in humans and also potential therapeutic strategies atthis early stage (e.g., when patients are undergoing operations) toattempt to combat local infections before they disseminate intoSIRS.
DisclosuresThe authors have no financial conflicts of interest.
References1. Jean-Baptiste, E. 2007. Cellular mechanisms in sepsis. J. Intensive Care Med.
22: 63–72.2. Rittirsch, D., L. M. Hoesel, and P. A. Ward. 2007. The disconnect between
animal models of sepsis and human sepsis. J. Leukoc. Biol. 81: 137–143.
10 TRPV1 AND MURINE SEPSIS
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

3. Bone, R. C. 1991. Sepsis syndrome. New insights into its pathogenesis andtreatment. Infect. Dis. Clin. North Am. 5: 793–805.
4. Holzer, P. 1991. Capsaicin as a tool for studying sensory neuron functions. Adv.Exp. Med. Biol. 298: 3–16.
5. Brain, S. D., and H. M. Cox. 2006. Neuropeptides and their receptors: innovativescience providing novel therapeutic targets. Br. J. Pharmacol. 147(Suppl. 1):S202–S211.
6. Caterina, M. J., M. A. Schumacher, M. Tominaga, T. A. Rosen, J. D. Levine, andD. Julius. 1997. The capsaicin receptor: a heat-activated ion channel in the painpathway. Nature 389: 816–824.
7. Szallasi, A., and P. M. Blumberg. 1999. Vanilloid (Capsaicin) receptors andmechanisms. Pharmacol. Rev. 51: 159–212.
8. Alawi, K., and J. Keeble. 2010. The paradoxical role of the transient receptorpotential vanilloid 1 receptor in inflammation. Pharmacol. Ther. 125: 181–195.
9. Moriyama, T., T. Higashi, K. Togashi, T. Iida, E. Segi, Y. Sugimoto,T. Tominaga, S. Narumiya, and M. Tominaga. 2005. Sensitization of TRPV1 byEP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol.Pain 1: 3.
10. Joyce, C. D., R. R. Fiscus, X. Wang, D. J. Dries, R. C. Morris, and R. A. Prinz.1990. Calcitonin gene-related peptide levels are elevated in patients with sepsis.Surgery 108: 1097–1101.
11. Beer, S., H. Weighardt, K. Emmanuilidis, M. D. Harzenetter, E. Matevossian,C. D. Heidecke, H. Bartels, J. R. Siewert, and B. Holzmann. 2002. Systemicneuropeptide levels as predictive indicators for lethal outcome in patients withpostoperative sepsis. Crit. Care Med. 30: 1794–1798.
12. Puneet, P., A. Hegde, S. W. Ng, H. Y. Lau, J. Lu, S. M. Moochhala, andM. Bhatia. 2006. Preprotachykinin-A gene products are key mediators of lunginjury in polymicrobial sepsis. J. Immunol. 176: 3813–3820.
13. Hegde, A., H. Zhang, S. M. Moochhala, and M. Bhatia. 2007. Neurokinin-1receptor antagonist treatment protects mice against lung injury in poly-microbial sepsis. J. Leukoc. Biol. 82: 678–685.
14. Orliac, M. L., R. N. Peroni, T. Abramoff, I. Neuman, E. J. Podesta, and E. Adler-Graschinsky. 2007. Increases in vanilloid TRPV1 receptor protein and CGRPcontent during endotoxemia in rats. Eur. J. Pharmacol. 566: 145–152.
15. Romanovsky, A. A. 2004. Signaling the brain in the early sickness syndrome: aresensory nerves involved? Front. Biosci. 9: 494–504.
16. Iida, T., I. Shimizu, M. L. Nealen, A. Campbell, and M. Caterina. 2005. At-tenuated fever response in mice lacking TRPV1. Neurosci. Lett. 378: 28–33.
17. Wang, Y., M. Novotny, V. Quaiserova-Mocko, G. M. Swain, and D. H. Wang.2008. TRPV1-mediated protection against endotoxin-induced hypotension andmortality in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294: R1517–R1523.
18. Clark, N., J. Keeble, E. S. Fernandes, A. Starr, L. Liang, D. Sugden, P. de Winter,and S. D. Brain. 2007. The transient receptor potential vanilloid 1 (TRPV1)receptor protects against the onset of sepsis after endotoxin. FASEB J. 21: 3747–3755.
19. Keeble, J. E., F. Russell, B. Curtis, A. Starr, E. Pinter, and S. D. Brain. 2005.Involvement of transient receptor potential vanilloid 1 in the vascular andhyperalgesic components of joint inflammation. Arthritis Rheum. 52: 3248–3256.
20. Baker, C. C., I. H. Chaudry, H. O. Gaines, and A. E. Baue. 1983. Evaluation offactors affecting mortality rate after sepsis in a murine cecal ligation andpuncture model. Surgery 94: 331–335.
21. Costa, R., E. S. Fernandes, O. Menezes-de-Lima, Jr., M. M. Campos, andJ. B. Calixto. 2006. Effect of novel selective non-peptide kinin B(1) receptorantagonists on mouse pleurisy induced by carrageenan. Peptides 27: 2967–2975.
22. Guzik, T. J., and K. M. Channon. 2005. Measurement of vascular reactive ox-ygen species production by chemiluminescence.Methods Mol. Med. 108: 73–89.
23. Smith, C. J., and A. M. Osborn. 2009. Advantages and limitations of quantitativePCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol.67: 6–20.
24. Sonesson, A., L. Larsson, A. Schutz, L. Hagmar, and T. Hallberg. 1990. Com-parison of the limulus amebocyte lysate test and gas chromatography-massspectrometry for measuring lipopolysaccharides (endotoxins) in airborne dustfrom poultry-processing industries. Appl. Environ. Microbiol. 56: 1271–1278.
25. Moine, P., and E. Abraham. 2004. Immunomodulation and sepsis: impact of thepathogen. Shock 22: 297–308.
26. Alves-Filho, J. C., A. de Freitas, F. Spiller, F. O. Souto, and F. Q. Cunha. 2008.The role of neutrophils in severe sepsis. Shock 30(Suppl. 1): 3–9.
27. Xing, L., J. Guo, and X. Wang. 2000. Induction and expression of beta-calcitoningene-related peptide in rat T lymphocytes and its significance. J. Immunol. 165:4359–4366.
28. Chavolla-Calderon, M., M. K. Bayer, and J. J. Fontan. 2003. Bone marrowtransplantation reveals an essential synergy between neuronal and hemopoieticcell neurokinin production in pulmonary inflammation. J. Clin. Invest. 111: 973–980.
29. Bracci-Laudiero, L., L. Aloe, M. C. Caroleo, P. Buanne, N. Costa, G. Starace,and T. Lundeberg. 2005. Endogenous NGF regulates CGRP expression in humanmonocytes, and affects HLA-DR and CD86 expression and IL-10 production.Blood 106: 3507–3514.
30. Rogers, D. P., C. R. Wyatt, P. H. Walz, J. S. Drouillard, and D. A. Mosier. 2006.Bovine alveolar macrophage neurokinin-1 and response to substance P. Vet.Immunol. Immunopathol. 112: 290–295.
31. Gwinn, M. R., and V. Vallyathan. 2006. Respiratory burst: role in signal trans-duction in alveolar macrophages. J. Toxicol. Environ. Health B Crit. Rev. 9: 27–39.
32. Tamura, N., K. Hazeki, N. Okazaki, Y. Kametani, H. Murakami, Y. Takaba,Y. Ishikawa, K. Nigorikawa, and O. Hazeki. 2009. Specific role of phosphoi-nositide 3-kinase p110a in the regulation of phagocytosis and pinocytosis inmacrophages. Biochem. J. 423: 99–108.
33. Mattsby-Baltzer, I., K. Lindgren, B. Lindholm, and L. Edebo. 1991. Endotoxinshedding by enterobacteria: free and cell-bound endotoxin differ in Limulusactivity. Infect. Immun. 59: 689–695.
34. Vlach, K. D., J. W. Boles, and B. G. Stiles. 2000. Telemetric evaluation of bodytemperature and physical activity as predictors of mortality in a murine model ofstaphylococcal enterotoxic shock. Comp. Med. 50: 160–166.
35. Toth, B. I., S. Benko, A. G. Szollosi, L. Kovacs, E. Rajnavolgyi, and T. Bıro.2009. Transient receptor potential vanilloid-1 signaling inhibits differentiationand activation of human dendritic cells. FEBS Lett. 583: 1619–1624.
36. Wenning, A. S., K. Neblung, B. Strauss, M. J. Wolfs, A. Sappok, M. Hoth, andE. C. Schwarz. 2011. TRP expression pattern and the functional importance ofTRPC3 in primary human T-cells. Biochim. Biophys. Acta 1813: 412–423.
37. Salvemini, D., and S. Cuzzocrea. 2002. Oxidative stress in septic shock anddisseminated intravascular coagulation. Free Radic. Biol. Med. 33: 1173–1185.
38. Osuchowski, M. F., K. Welch, J. Siddiqui, and D. G. Remick. 2006. Circulatingcytokine/inhibitor profiles reshape the understanding of the SIRS/CARS con-tinuum in sepsis and predict mortality. J. Immunol. 177: 1967–1974.
39. Demirbilek, S., M. O. Ersoy, S. Demirbilek, A. Karaman, N. Gurbuz,N. Bayraktar, and M. Bayraktar. 2004. Small-dose capsaicin reduces systemicinflammatory responses in septic rats. Anesth. Analg. 99: 1501–1507.
40. Puntambekar, P., D. Mukherjea, S. Jajoo, and V. Ramkumar. 2005. Essential roleof Rac1/NADPH oxidase in nerve growth factor induction of TRPV1 expression.J. Neurochem. 95: 1689–1703.
41. Starr, A., R. Graepel, J. Keeble, S. Schmidhuber, N. Clark, A. Grant, A. M. Shah,and S. D. Brain. 2008. A reactive oxygen species-mediated component in neu-rogenic vasodilatation. Cardiovasc. Res. 78: 139–147.
42. Keeble, J. E., J. V. Bodkin, L. Liang, R. Wodarski, M. Davies, E. S. Fernandes,Cde. F. Coelho, F. Russell, R. Graepel, M. N. Muscara, et al. 2009. Hydrogenperoxide is a novel mediator of inflammatory hyperalgesia, acting via transientreceptor potential vanilloid 1-dependent and independent mechanisms. Pain 141:135–142.
43. Chen, C. W., S. T. Lee, W. T. Wu, W. M. Fu, F. M. Ho, and W. W. Lin. 2003.Signal transduction for inhibition of inducible nitric oxide synthase andcyclooxygenase-2 induction by capsaicin and related analogs in macrophages.Br. J. Pharmacol. 140: 1077–1087.
44. Ngkelo, A., K. Meja, M. Yeadon, I. Adcock, and P. A. Kirkham. 2012. LPSinduced inflammatory responses in human peripheral blood mononuclear cells ismediated through NOX4 and Gia dependent PI-3kinase signalling. J. Inflamm.(Lond.) 9: 1.
45. Berger, S. B., X. Romero, C. Ma, G. Wang, W. A. Faubion, G. Liao, E. Compeer,M. Keszei, L. Rameh, N. Wang, et al. 2010. SLAM is a microbial sensor thatregulates bacterial phagosome functions in macrophages. Nat. Immunol. 11:920–927.
46. Serbina, N. V., T. Jia, T. M. Hohl, and E. G. Pamer. 2008. Monocyte-mediateddefense against microbial pathogens. Annu. Rev. Immunol. 26: 421–452.
47. Cheung, R., M. Malik, V. Ravyn, B. Tomkowicz, A. Ptasznik, andR. G. Collman. 2009. An arrestin-dependent multi-kinase signaling complexmediates MIP-1beta/CCL4 signaling and chemotaxis of primary human mac-rophages. J. Leukoc. Biol. 86: 833–845.
48. Mercer-Jones, M. A., M. S. Shrotri, J. C. Peyton, D. G. Remick, andW. G. Cheadle. 1999. Neutrophil sequestration in liver and lung is differentiallyregulated by C-X-C chemokines during experimental peritonitis. Inflammation23: 305–319.
49. Matiba, B., S. M. Mariani, and P. H. Krammer. 1997. The CD95 system and thedeath of a lymphocyte. Semin. Immunol. 9: 59–68.
50. Liu, Z. G. 2005. Molecular mechanism of TNF signaling and beyond. Cell Res.15: 24–27.
51. Guptill, V., X. Cui, A. Khaibullina, J. M. Keller, N. Spornick, A. Mannes,M. Iadarola, and Z. M. Quezado. 2011. Disruption of the transient receptorpotential vanilloid 1 can affect survival, bacterial clearance, and cytokine geneexpression during murine sepsis. Anesthesiology 114: 1190–1199.
52. Wadachi, R., and K. M. Hargreaves. 2006. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J. Dent. Res. 85: 49–53.
53. Aksoy, E., M. Goldman, and F. Willems. 2004. Protein kinase C epsilon: a newtarget to control inflammation and immune-mediated disorders. Int. J. Biochem.Cell Biol. 36: 183–188.
54. Turnbull, I. R., S. Gilfillan, M. Cella, T. Aoshi, M. Miller, L. Piccio,M. Hernandez, and M. Colonna. 2006. Cutting edge: TREM-2 attenuates mac-rophage activation. J. Immunol. 177: 3520–3524.
55. Peri, F., M. Piazza, V. Calabrese, G. Damore, and R. Cighetti. 2010. Exploringthe LPS/TLR4 signal pathway with small molecules. Biochem. Soc. Trans. 38:1390–1395.
56. Arts, R. J., L. A. Joosten, C. A. Dinarello, B. J. Kullberg, J. W. van der Meer, andM. G. Netea. 2011. TREM-1 interaction with the LPS/TLR4 receptor complex.Eur. Cytokine Netw. 22: 11–14.
57. Brain, S. D. 1997. Sensory neuropeptides: their role in inflammation and woundhealing. Immunopharmacology 37: 133–152.
The Journal of Immunology 11
by guest on February 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from





























