Embed Size (px)
Citation preview

University of Connecticut University of Connecticut
OpenCommons@UConn OpenCommons@UConn
Doctoral Dissertations University of Connecticut Graduate School
5-8-2020
TRP Channels in Cardiovascular Disease TRP Channels in Cardiovascular Disease
Albert Yu University of Connecticut - Storrs, [email protected]
Follow this and additional works at: https://opencommons.uconn.edu/dissertations
Recommended Citation Recommended Citation Yu, Albert, "TRP Channels in Cardiovascular Disease" (2020). Doctoral Dissertations. 2519. https://opencommons.uconn.edu/dissertations/2519

TRP Channels in Cardiovascular Disease
Albert S. Yu, PhD
University of Connecticut, 2020
Abstract
Transient receptor potential (TRP) channels are evolutionarily conserved ion
channels that have been implicated in a wide range of physiological and
pathophysiological responses. As versatile ion channels that are permeable to calcium,
the exact nature of these ion channels have been furiously studied since its initial
discovery in Drosophila. Many TRP channels are thought to be gated by PIP2, a well-
known membrane signaling molecule. Here we show that membrane potential alters PIP2
in such a way to reduce TRPM7 activity, presumably through PIP2 depletion. This novel
mechanism of TRPM7 regulation gives us a clearer picture into a complicated protein that
has been implicated in processes ranging from embryonic development to cancer.
Furthermore, we show how TRP channels are involved in the development and
progression of various cardiovascular diseases. Our research implicates the oxidative
stress activated channel TRPM2 in the progression of atherosclerosis, with data pointing
to its role in driving inflammation by increasing circulating myeloid cell populations. We
also show that the bifunctional channel-enzyme TRPM7 plays a deleterious role in the
cardiac fibrogenesis cascade in hypertensive heart failure, and deletion of Trpm7
specifically in the cardiac fibroblast is protective against negative cardiac remodeling. My
research demonstrates not only how TRP channels are important mediators of
cardiovascular disease, but also how they are regulated at a basic molecular level.

i
TRP Channels in Cardiovascular Disease
Albert S. Yu
B.A. University of California Berkeley, 2011
A Dissertation
Submitted in Partial Fulfillment of the
Requirements for the Degree of
Doctor of Philosophy
at the
University of Connecticut
2020

ii
Copyright by
Albert S. Yu
2020

iii
APPROVAL PAGE
Doctor of Philosophy Dissertation
TRP Channels in Cardiovascular Disease
Presented by Albert S. Yu, B.A.
Major Advisor ___________________________________________________________________ Lixia Yue, PhD Associate Advisor ___________________________________________________________________ Kevin Claffey, PhD Associate Advisor ___________________________________________________________________ Kimberly Dodge-Kafka, PhD Associate Advisor ___________________________________________________________________ Linda Shapiro, PhD
University of Connecticut 2020

iv
Acknowledgments I would like to thank my friends and family both in Connecticut and California for
supporting me through this journey. I also would like to thank my committee members, my advisor Dr. Lixia Yue, past and current lab members in particular Dr. Yanlin He, Dr. Jianlin Feng, and Dr. Zhichao Yue, Dr. Hector Leonardo Aguila, the MD/PhD program especially Dr. Carol Pilbeam and Tracy Dieli, everyone in the Cell Biology and Cardiology Departments, and the Graduate School for helping me develop into the researcher and person that I am today. Finally, I would like to thank the American Heart Association for providing a gracious Predoctoral Fellowship.

v
Table of Contents Feature Page Approval Page .................................................................................................... iii. Acknowledgements ............................................................................................. iv. Table of Contents ................................................................................................. v. List of Figures ..................................................................................................... vi. Chapter and Title Page Chapter 1: Introduction ................................................................................... 1
Cardiovascular disease as a public health crisis ................... 2
A TR(i)P in ion channels ....................................................... 7 Chapter 2: Membrane potential regulation of TRPM7 .................................. 13 Chapter 3: TRPM2 modulation of inflammation and CVD ............................ 34 Chapter 4: Targeting TRPM7 to reverse cardiac fibrosis .............................. 52 Chapter 5: pH regulation of the gatekeeper, Orai ......................................... 60 Chapter 6: Conclusions, Discussion, and the future TR(i)Ps ........................ 72 Chapter 7: References ................................................................................. 75

vi
List of Figures Page Figure 1.1: A phylogenetic tree of TRP channels .......................................... 11
Figure 1.2: Membrane topology of TRPs ...................................................... 12
Figure 2.1A: Hyperpolarized membrane potential inactivates the
TRPM7 channel .......................................................................... 24
Figure 2.1B: TRPM7 inactivation shows time dependent inactivation
and recovery ............................................................................... 25
Figure 2.1C: Recovery after inactivation approaches near maximum ............. 26
Figure 2.1D: Hyperpolarized membrane potential by step protocol also
inactivates the TRPM7 channel .................................................. 27
Figure 2.1E: Preservation of intracellular contents by perforated patch technique
recapitulates whole cell results ................................................... 28
Figure 2.2: Dynamic PIP2 changes with membrane potential ........................ 29
Figure 2.3: Addition of exogenous PIP2 does not prevent TRPM7
inactivation .................................................................................. 31
Figure 2.4: Divalent ions are not likely responsible for low voltage induced
change ........................................................................................ 32
Figure 2.5: Model of TRPM7 inactivation by low voltage .............................. 33
Figure 3.1: Trpm2 deletion reduces atherosclerosis ..................................... 42
Figure 3.2: Trpm2 deletion protects against myocardial infarction ................ 43
Figure 3.3A: Flow cytometry process .............................................................. 45
Figure 3.3B: Trpm2 deletion does not alter myeloid subpopulations
in the healthy state ...................................................................... 46

vii
Figure 3.3C: Systemic changes in circulating myeloid cells
during atherosclerosis ................................................................. 47
Figure 3.3D: Myocardial infarction does not alter peripheral
myeloid populations .................................................................... 48
Figure 3.4: TRPM2 is expressed in bone marrow progenitor cells ................ 49
Figure 3.5: Model of TRPM2 induced inflammation in myeloid cells ............. 50
Figure 3.6: Myocardial specific, inducible deletion of Trpm2 to study
atherosclerosis and myocardial infarction ................................... 51
Figure 4.1: Deletion of Trpm7 attenuates fibrosis and improves
heart function .............................................................................. 57
Figure 4.2: Fibroblast specific deletion of Trpm7 recapitulates
protective effects ......................................................................... 58
Figure 4.3: Schematic of TRPM7, the chanzyme .......................................... 59
Figure 5.1: Acidic and basic external pH on Orai1/STIM1 channels ............. 66
Figure 5.2: Effects of pH on Orai2 and Orai3 ................................................ 67
Figure 5.3: pH effects on Orai1 mutants ....................................................... 68
Figure 5.4: Effects of internal pH on Orai1/STIM1 ........................................ 69
Figure 5.5: H155 as the intracellular pH sensor of Orai1/STIM1 ................... 70
Figure 5.6: Effects of external calcium on pH sensitivity of Orai1/STIM1 ...... 71

1
CHAPTER 1: INTRODUCTION TO TRP CHANNELS IN CARDIOVASCULAR DISEASE

2
Cardiovascular disease as a public health crisis
By 2030, 44% of the US population will be afflicted by cardiovascular disease
(CVD)1. Currently, CVD is the leading cause of death not only in the US but also
worldwide. The phenomenon in developed countries is driven by lifestyle factors,
particularly diet and a sedentary lifestyle. In developing countries, this trend is also taking
place due to reduced morbidity and mortality from infectious disease and a shift to a
Western lifestyle. Thus, a main priority today in healthcare and research today is finding
ways to reduce CVD morbidity and mortality.
Cardiovascular disease encompasses a wide array of diseases, including
peripheral artery disease to congenital as well as acquired heart disease 2. The most
common subtypes of CVD stem from a process known as atherosclerosis. From Greek
and Latin origins, “atherosclerosis” was first coined due to its appearance as a “porridge-
like hardening.” Biologically, an atherosclerotic plaque is composed of many different cell
types reacting to lipid deposition in the arterial walls 3. Surprisingly, studies have shown
that signs of atherosclerosis begin in adolescence, and progresses with age 4.
The initial events of atherosclerosis are thought to begin by abnormal blood flow
and stress to the arterial wall 5. The disturbed flow can be caused by increased viscosity
of the blood, for instance, by an increase in lipid content. In regions of abnormal blood
flow, endothelial cells express cell surface adhesion molecules while concurrently
allowing the entry of lipoprotein particles from the bloodstream into the arterial wall 6. The
lipoprotein particles in the arterial wall are engulfed and eliminated by resident arterial
cells, such as fibroblasts, macrophages, and endothelial precursor cells. However, during
continued deposition of lipoprotein particles into the arterial wall, the engulfing cells

3
become overloaded and insufficiently eliminate lipid particles, leading to apoptosis and
release of inflammatory cytokines. These events lead to the proliferation of resident cells,
as well as the recruitment of circulatory cells into the arterial wall. However, as lipid
deposition continues, these plaques fail to completely resolve, leading to a vicious cycle
of inflammation and plaque growth 7.
A fundamental question exists as to what dictates macrophages in atheroma
lesions to secrete inflammatory cytokines. There is a widely held belief that once
macrophages enter lesion areas and engulf oxidized lipids, the macrophages convert to
foam cells that are able to generate both a protective and inflammatory reaction 8. The
balance between inflammation and resolution must be finely maintained 9 10.
Atherosclerosis is thought to be an inflammatory process by nature. Inflammation
of the arterial wall, with concomitant recruitment of immune cells and production of
inflammatory cytokines, has long been considered as critical in the disease process.
Patients with cardiovascular disease have elevated markers of inflammation such as
circulating IL-1β and CRP. Thus, general targets of inflammation, such as IL-1β blockers,
used to treat autoimmune inflammatory disorders, have recently been considered for use
for possible treatment of atherosclerosis 11 12 13. Interestingly, data showing the benefits
of IL-1beta blockers in autoimmune disorders also shows unexpected improvements in
cardiovascular outcomes 14.
Another recent finding in atherosclerosis research has shed light into the systemic
response generated by atherosclerosis 15. In addition to the local plaque secreted factors,
there may be inflammatory cytokines that travel systemically to enhance the recruitment
of cells in an attempt to re-establish homeostasis. Furthermore, clinical data show

4
differences in the circulating monocyte populations in patients with cardiovascular
disease 16.This has been confirmed in animal models of atherosclerosis which show
increased circulating leukocytes, predominantly due to the increase in myeloid cells 17.
Furthermore, animal models have shown that these circulating myeloid cells adopt an
inflammatory state, as denoted by high expression of the inflammatory marker Ly6C,
which are also found in early atherosclerotic lesions 18. The presence of these myeloid
cells in early atheroma lesions suggests that these cells play a critical role in plaque
development and disease progression. Thus, there has been a strong interest in studying
cardiovascular disease from an immunological perspective.
As plaques grow, there are several ways blood flow can be reduced. First, the
plaque cap may become disrupted, leading to a piece of the plaque, known as an
embolus, to lodge in a smaller, downstream artery. In addition, the disrupted plaque may
cause an inflammatory reaction, or thrombus, to form at the site. Both cause a reduction
in diameter of the artery or small vessel. Furthermore, abundant plaques throughout the
circulatory system can cause reduced blood flow systemically. This leads to an increased
systolic pressure, which eventually wears down the heart and reduces blood flow to vital
organs. Thus, when blood flow is reduced, there are several main diseases that arise.
When atherosclerosis reduces blood flow to the extremities, symptoms of tingling and
pain may arise, leading to a condition known as peripheral artery disease. When blood
flow to critical arteries supplying the brain are reduced, stroke may result. Finally, when
the coronary arteries are blocked, myocardial ischemia or infarction (heart attack) may
arise.

5
Myocardial infarction is one of the major consequences of plaque buildup. Typically
affecting the main left anterior descending coronary artery that supplies blood to a major
of the left ventricle, coronary artery disease typically presents as a sharp, radiating pain
in the chest and shoulder region accompanied by shortness of breath. Depending on the
severity and extent of coronary blockage, the symptoms may resolve or may quickly
precipitate into ventricular arrhythmia and sudden cardiac death. Treatment strategies for
coronary artery disease range from treatment with drug regimens, such as aspirin and
anticoagulants to heart rhythm control agents such as beta blockers, to more invasive
procedures such as percutaneous coronary intervention, which involves catheter guided
stent placement or physical plaque disruption, or open heart surgery to revascularize
cardiac tissue from an existing blood supply using a graft, typically the left internal
mammary artery. After an initial heart attack, an acute inflammatory phase is required for
proper healing, followed by a resolution phase. However, prolongation of either of these
two phases can lead to detrimental outcomes and cardiac remodeling, which is
responsible for recurrence. Following acute intervention, long term monitoring and drug
treatment is precisely maintained in order to prevent recurrence. Unfortunately, for many
patients, an initial heart attack episode is likely not the last. In concordance with what is
seen clinically, a recent study found that myocardial infarction in mice accelerates the
inflammatory process of atherosclerosis, thus increasing susceptibility to another acute
cardiovascular episode 19.
Stroke is the second most common cause of cardiovascular disease related death.
In the case of stroke, an artery supplying a critical area of the brain is narrowed, leading
to reduced blood flow. As the brain is an organ that relies on constant blood supply, the

6
consequences of reduced or loss of blood flow and lead to irreversible, and often,
physically manifesting, outcomes. In cases of acute stroke, thrombolytics and
anticoagulants are the mainstays in terms of treatment options.
Treatment strategies for CVD can range from targeting subclinical atherosclerosis
to treating acute emergencies to managing long term recovery and repair. Primary
prevention involves modification of lifestyle factors, including diet and exercise. Early
stages of atherosclerosis can be detected by elevated blood pressure, and treatment
often involves relaxation of vessels, controlling heart rate, and reducing blood volume.
Subclinical atherosclerosis can be detected by coronary angiography and tests that
measure heart function or arterial disease in stress conditions. When atherosclerosis
presents as an acute cardiovascular event, such as peripheral ischemia, stroke, or heart
attack, acute treatment can include the use of thrombolytics, anticoagulation, or
percutaneous intervention, followed by treatment with blood thinners. Long term
treatment include anticoagulation, and for myocardial infarction, heart rate control. Both
myocardial infarction and stroke are driven by the same underlying cause-
atherosclerosis. As discussed above, many current therapies rely on symptomatic
treatment and prevention of another acute episode. However, there is a substantial need
for improvement within this field as cardiovascular disease has risen to be the top cause
of morbidity and mortality worldwide. In all these conditions, a common theme is the
presence of inflammation. Efforts to reduce or alter inflammatory signaling pathways in
human disease for the purposes of treatments have been successful to some extent.
However, there is a need to target more specific cells and pathways that cause
inflammation in cardiovascular disease. The morbidity and mortality as well as the high

7
rates of recurrence and poor prognosis are cause for concern. As many of the drugs
mentioned above mainly manage CVD, there is an urgent need to develop drugs to target
CVD before and/or during the symptom presentation, in order to halt or reverse disease.
A TR(i)P in ion channels
Ion channels are fundamental mediators of signaling. First hypothesized by
Hodgkin and Huxley and later confirmed by Katz and Miledi, ion channels serve as an
electrical conduit to mediate signaling 20. Transporters build charge, membranes store
charge, and ion channels allow a conduit for passive diffusion of ions and thus electricity.
Ions not only mediate signaling events on their own, but its movement also generates
changes in membrane potential that lead to downstream signaling events, for example in
voltage gated ion channels. One well known ion in modulating signaling events is calcium.
Calcium is a necessary cofactor/allosteric modulator in many enzymatic reactions
including the well-known regulatory regions in protein kinase C. In terms of gating, ion
channels can be activated in many additional ways. One well known example is the
activation of skeletal muscle contraction. Calcium entry through L-type calcium channels
in the cell membrane can activate the ryanodine receptors (RyR) on the sarcoplasmic
reticulum. This triggers the release of calcium from the sarcoplasmic reticulum store,
providing enough calcium for muscle contraction. Furthermore, low levels of calcium in
the endoplasmic reticulum can trigger the activation of calcium entry, known as calcium
release activation channels (CRAC) 21. Thus, calcium levels are finely maintained in a
resting cell and upon activation are meticulously increased and decreased in a space and

8
time dependent manner to allow for precise signaling. Any perturbations in this system
can ultimately lead to altered signaling or cell death.
One family of ion channels found from across species is the Transient Receptor
Potential (TRP) class of ion channels. TRP channels were originally identified in the
Drosophila melanogaster when Cosens and Manning discovered a mutant fly that
exhibited a transient response to light as compared to a sustained response in wild type
22. This led to the discovery of the first role of the TRP channel as a protein that mediates
the signal of light transduction. A couple decades later, Montell rescued the trp mutant
with its cDNA, and cloned the channel 23. The TRP channel was originally postulated to
have eight transmembrane domains based on hydrophobicity, with hydrophilic N and C
termini 24. In addition, the C termini was found to have 8-9 repeats. It is now known that
the mammalian TRP superfamily includes 28 subtypes, with related genes found in flies,
yeast, worm, and fish, suggesting conserved evolutionary roles (Figure 1.1.). The 28
mammalian Trp genes have been identified, in six subfamilies, consisting of TRPC
(canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (Ankyrin), TRPP (polycystic),
and TRPML (mucolipin), grouped mainly by their homology (Figure 1.2). Of note is that
these TRP channels lack a voltage sensor in S4. Furthermore, many channels are
permeable to calcium, a well-known signaling messenger. TRP channels are thought to
arrange in homo or heterotetramers consisting of 6 transmembrane domain subunits, and
playing roles ranging from thermosensation to hearing and sight. As such, TRP channels
are thought to be sensory channels, relying on different physical and chemical stimuli to
activate or inhibit the channel 25. Distinguishing components of each channel subtype are
the ion permeating S5-S6 transmembrane segments, as well as the N and C termini

9
domains. TRPM7, for instance, contains a C terminus domain that encodes a functional
kinase. As the understanding of the role of TRP channels progresses, a critical need
exists to identify modulators of TRP channel activity. However, the function of this kinase,
as well as its relation to ion channel activity, is being further investigated. Furthermore,
recent literature and analyses of various TRP channel structures may give additional
insight into the role of TRP channel in physiological or pathophysiological responses.
As different TRP channels were discovered, the activation mechanisms were also
brought into focus. Although the physiological activators of many TRP channels have not
been clearly characterized, the diverse responsiveness of TRP channels to sensory
stimuli indicate that TRP channels may be important in receiving different sensory
modalities and suggests their importance in regulating both physiological and
pathophysiological responses across species. For example, TRPV1-4 and TRPM3 are
activated by high temperature, and TRPM8, TRPA1, and TRPC5 are activated by low
temperature. TRP channels can also sense mechanical (for example shear and osmotic
stretch), chemical, nociceptive and lipid signals. TRPM2 can sense oxidative stress
signals like ADPR and NAD+. Furthermore, TRP channels are known to interact with other
TRPs as well as calmodulin, kinases and phospholipases 26. Some TRP channels have
ubiquitous expression throughout the human body while some channels are constitutively
active in overexpression systems. TRP channels are expressed in the heart, brain,
immune cells, kidney, bladder, digestive tract, among other organs. Thus, the study of
TRP channels will yield insight not only into the signals that activate them, but also into
how they may play a role in both human physiology and pathophysiology.

10
Recent research has elucidated the structure of many TRP channels. The super
resolution structures of these channels allows us to not only compare these channels, but
also give us a clearer picture into what activates or inhibits these channels, and how they
may potentially play a role in human disease. Though many chemical activators and
inhibitors have been identified, these chemicals lack specificity for any particular ion
channel. Structural analysis may allow us to design specific drugs to modulate channel
activity. Furthermore, one widely believed mode of gating is through phosphoinositide
signaling. Thus, structural studies will allow us to find where endogenous molecules such
as PIP2 binds to modulate channel activity 27. In addition to PIP2 binding, which signaling
pathways change PIP2 levels and the extent to which PIP2 changes or its metabolites can
be detected by TRP channels to modulate activity will be an important question to going
further. The goal of my research is to identify how TRP channels, in particular TRPM2
and TRPM7, are involved in cardiovascular disease by using animal models, and to
understand the basic mechanisms of TRP channel gating by PIP2.

11
Figure 1.1. A phylogenetic tree of human, Zebrafish, and Drosophila TRP channels. Sequence homology shows TRP channels in 6 subfamilies that comprise proteins with distinct channel properties. TRP channels are conserved from Drosophila to humans, indicating its importance in evolution and function. 28 mammalian Trp genes have been identified, in six subfamilies, consisting of TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (Ankyrin), TRPP (polycystic), and TRPML (mucolipin), grouped mainly by their homology.
12
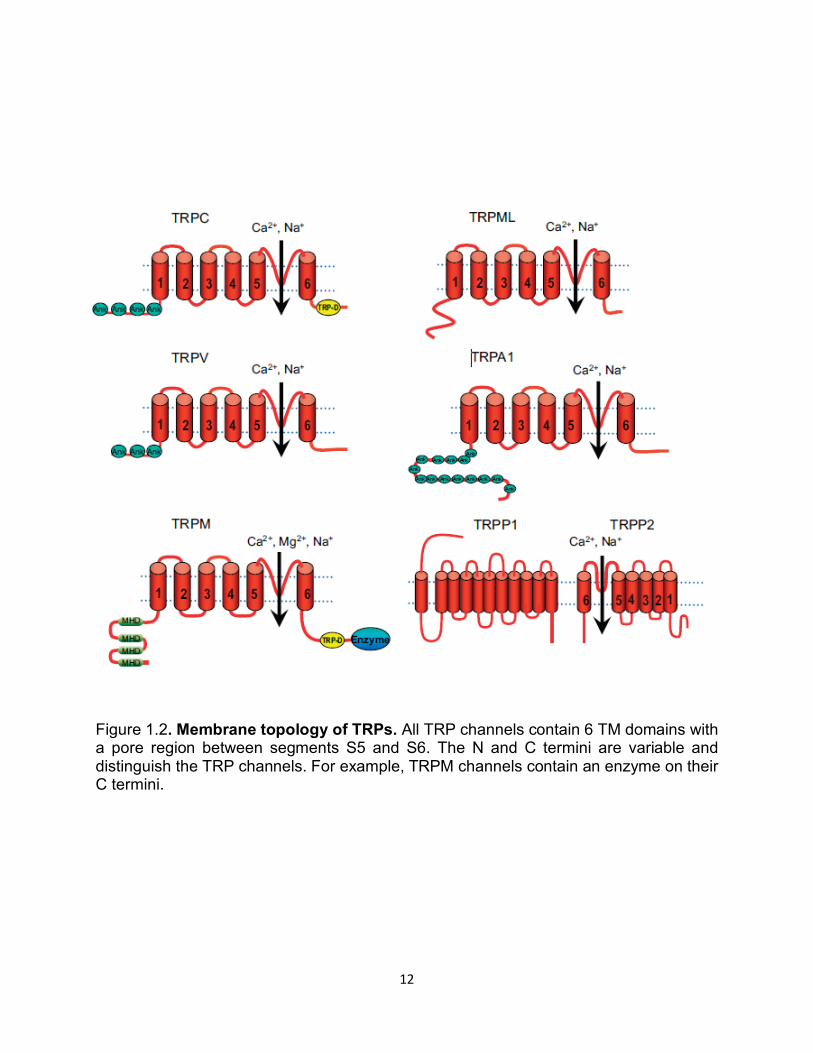
Figure 1.2. Membrane topology of TRPs. All TRP channels contain 6 TM domains with a pore region between segments S5 and S6. The N and C termini are variable and distinguish the TRP channels. For example, TRPM channels contain an enzyme on their C termini.

13
CHAPTER 2: MEMBRANE POTENTIAL REGULATION OF TRPM7

14
Introduction
Transcellular membrane potential is critical for determining ion channel activity and
cellular signaling. It is well characterized how changes in membrane potential influence
voltage gated ion channels, and how the entry of ions, such as calcium, generates a
signaling cascade. How this potential is generated and altered by transporters and ion
channels has been fundamental to our understanding of cell and human biology.
However, how this change of electrical potential across membranes influences other
cellular processes, such as enzyme activity, has not been well described. The discovery
of a voltage sensitive enzyme phosphatase in the ascidian Ciona intestinalis sperm tail,
named Ci-VSP, in 2005 has fueled speculation of the existence of similar functioning
proteins in mammals 28. Ci-VSP consists of a transmembrane S1-S4 voltage domain
similar to that of voltage gated ion channels, along with a cytoplasmic phosphatase
domain. Its phosphatase domain acts on phosphoinositides, presumably regulating
intracellular phosophoinositide levels. Ci-VSP generates the production of
phosphatidylinositol 4,5-bisphophate PtdIns(4,5)P2 from PtdIns(3,4,6)P3. Interestingly,
several years prior to the discovery of Ci-VSP, its mammalian homologues known as
transmembrane phosphatases with tensin homology (TPTE) were identified in human
testis 29 30. However, the voltage sensing property was neither examined nor described.
Thus, the question still remains as to whether a voltage sensitive enzyme functions in
mammals, and if so, how it influences human physiology.
Phosphoinositides (PI) are widely used signaling molecules that can influence the
activity of many ion channels 31. Products of PIs, such as phosphatidylinositol 4,5-
bisphophate PtdIns(4,5)P2 and PtdIns(3,4,6)P3, comprise a minority of lipids in the inner

15
cell membrane and are highly negatively charged. Through phosphorylation at the 3,4,
and 5 sites, phosphorylated PIs can form 7 distinct molecules, and thus regulate distinct
activities. Though relative few in number at the cell membrane, these phosphorylated PIs
and its breakdown products are important signal transduction molecules. PIs are thought
to be effective signaling molecules due to its transient nature upon receptor stimulation
and formation. Such receptor stimulation is thought to activate enzymes that generate
different PIs, which proceed to modulate the activity of ion channels, transporters,
enzymes, and/or other receptors. In addition, precise localization of PIs can fine tune
where and when modulation occurs. As such, PIs have a wide array of effects on ion
channels. PIs may activate certain ion channels, while the same PI may inhibit others.
Furthermore, there has been evidence to suggest that some ion channels are completely
insensitive to the changes in PIs. Thus, insight into how and which channels are regulated
by PIs will give great insight into the inner workings of the cell.
Seminal discoveries made by Hilgemann and Ball in 1996 showed that the addition
of phospholipases reduces the activity of ion transporters and channels by decreasing
PIP2 32. The authors observed that depletion of PIP2 by phospholipase C specifically
reduced sodium-calcium exchanger activity and potassium channel activity. Addition of
exogenous PIP2 was able to restore their activities. The authors also found that the metal
aluminum may interact and interfere with the PIP2 -ion channel interaction. Since then,
many channels have been identified as PIP2 sensitive, of note which are the transient
receptor potential (TRP) channels 33.
As mentioned previously, ion channels can be activated or inactivated by the level
of a particular phosphoinositide. The canonical Drosophila TRP channel is activated by

16
DAG metabolites upon PIP2 produced by rhodopsin and Gq, though the exact mechanism
is still unclear 34 35. Channel activity is maintained in wild-type channels by continuous
calcium entry and inhibition of phospholipase C, possibly through calcium-calmodulin
inhibition, thus preserving PIP2 36. Since then, PIP2 has been shown to stimulate TRPV5,
TRPM4, TRPM5, and TRPM8, while having an inhibitory effect on the heat and capsaicin
sensitive TRPV1. Inconclusive is the effect of PIP2 on TRPM7. However, it is widely
believed that receptor activation induced PLC activation leads to PIP2 hydrolysis and
TRPM7 inactivation 37. Furthermore, it was shown that TRPM8 is regulated by PIP2, and
upon calcium entry, PLC-mediated depletion of PIP2 leads to reduced channel activity 38.
TRPM7 is a member of the melastatin subfamily of TRP channels. The first TRPM
channel was characterized in 1998 where the authors found that its expression inversely
correlated with metastatic potential 39. A few years later, TRPM7 was discovered. It is a
ubiquitously expressed, divalent permeable, cation channel that has since been
implicated in many cellular processes such as adhesion, growth, and apoptosis 40. As
mentioned before, the protein contains both channel and kinase functions, however the
role of the kinase in channel function has not been clearly elucidated 41. The channel is
permeable to both calcium and magnesium, and it is thought to regulate its respective
levels. Under physiological conditions, TRPM7 is thought to mainly carry an inward
current of divalent ions, particularly Mg2+ and Ca2+, which also act as a permeation block
to monovalent ions. The channel has constitutive activity, but its activity is increased
significantly by depletion of intracellular magnesium, whereas addition of intracellular
magnesium leads to its suppression. In addition, PLC, a known binding partner of TRPM7,
activation by Galphaq linked receptors inhibits TRPM7 activity by PIP2 depletion. It was later

17
shown that the close homologue TRPM6 is also regulated by PIP242. However, what other
endogenous stimuli reduce TRPM7 activity remains a mystery.
Located on chromosome 15 in humans, Trpm7 encodes a 1,865 amino acid
protein that is involved in activities ranging from embryonic development, ischemia,
cardiovascular disease, and cancer. Global deletion of Trpm7 leads to embryonic lethality
and targeted deletion in cells is lethal 43 44. More recently it has been shown that Trpm7
deletion leads to macrothrombocytopenia in mice and humans and platelet signaling
defects 45 46. In addition, TRPM7 dysfunction is also believed to be involved in atrial
fibrillation 47. However, the exact mechanism by which TRPM7 functions and how Trpm7
deletion may influence physiology has not been elucidated. Furthermore, how TRPM7 is
modulated by its interacting partners and the relationship between the kinase and channel
functions remains to be investigated 48. As more work is currently being done using Trpm7
knockout and mutant mice, and complemented by TRPM7 cell lines, its exact function in
physiological processes will become unveiled.
Here, we show that alterations of membrane potential may serve as one method
to modulate the activity of TRPM7. We believe the modulation occurs through changes in
PIP2. This suggests that there may be an intrinsic voltage sensing mechanism in the cell
that transduces the change in membrane potential to alter PIP2 concentration, not unlike
Ci-VSP, thus regulating TRPM7 activity.

18
Results
Hyperpolarized membrane potential inactivates the TRPM7 channel.
PIP2 has been shown to regulate TRPM7 channel activity. Furthermore, it has been
shown that an invertebrate voltage sensitive phosphatase, Ci-VSP, alters PIP2 levels,
subsequently affecting ion channel activity. Thus, we set out to determine whether such
phenomenon occurs in mammalian cells. We used human embryonic kidney cells (HEK
293) and transfected TRPM7 (also known as LTRPC7). Under whole cell configuration,
TRPM7 current increased to a maximum levels about 3 minutes following rupture, after
complete dialysis of intracellular pipette solution to chelate free intracellular Mg2+. After a
steady-state current was reached, a low voltage protocol was applied. Cells were held at
either -100mV or -120mV, with interspersed ramp protocols to monitor TRPM7 activity
(Figure 2.1A-C). TRPM7 current steadily decreased in both inward and outward
directions. In addition, we used a step protocol holding at -100mV and saw a steady
decrease in inward TRPM7 activity at -100mV (Figure 2.1D). Furthermore, to verify that
perturbations of intracellular content did not affect our results, we also used perforated
patch clamp at low voltages. We saw similar decreases in current as well (Figure 2.1E).
These results suggest that a hyperpolarized membrane potential leads to reduced
TRPM7 activity.
Dynamic PIP2 changes with membrane potential
To determine what factors may be influencing TRPM7 channel activity at hyperpolarized
membrane potentials, we first looked at PIP2, a well-described molecule involved in
TRPM7 gating. In order to monitor subtle changes in PIP2 levels at the cell membrane,

19
we used Forster resonance energy transfer (FRET) using cyan fluorescent protein (CFP)
and yellow fluorescent protein (YFP) both conjugated to a Pleckstrin homology (PH)
domain, which binds to PIP2 and is natively found in proteins such as heterotrimeric G
proteins. The ratio of YFP/CFP over time has been previously shown to effectively track
the change in levels of PIP2 on the cell surface. Thus, we transfected both CFP and YFP
proteins in a LTRCP7 cell line. Using simultaneous FRET and patch clamp
electrophysiology, we were able to track both TRPM7 activity as well as PIP2 in real time.
Similar to what we observed previously, we saw TRPM7 activity decrease at negative
holding potentials. We also saw a concurrent decrease in FRET ratio, indicating that
hyperpolarized membrane potential reduces FRET at the cell membrane, suggesting that
PIP2 levels may be modified or decreased (Figure 2.2).
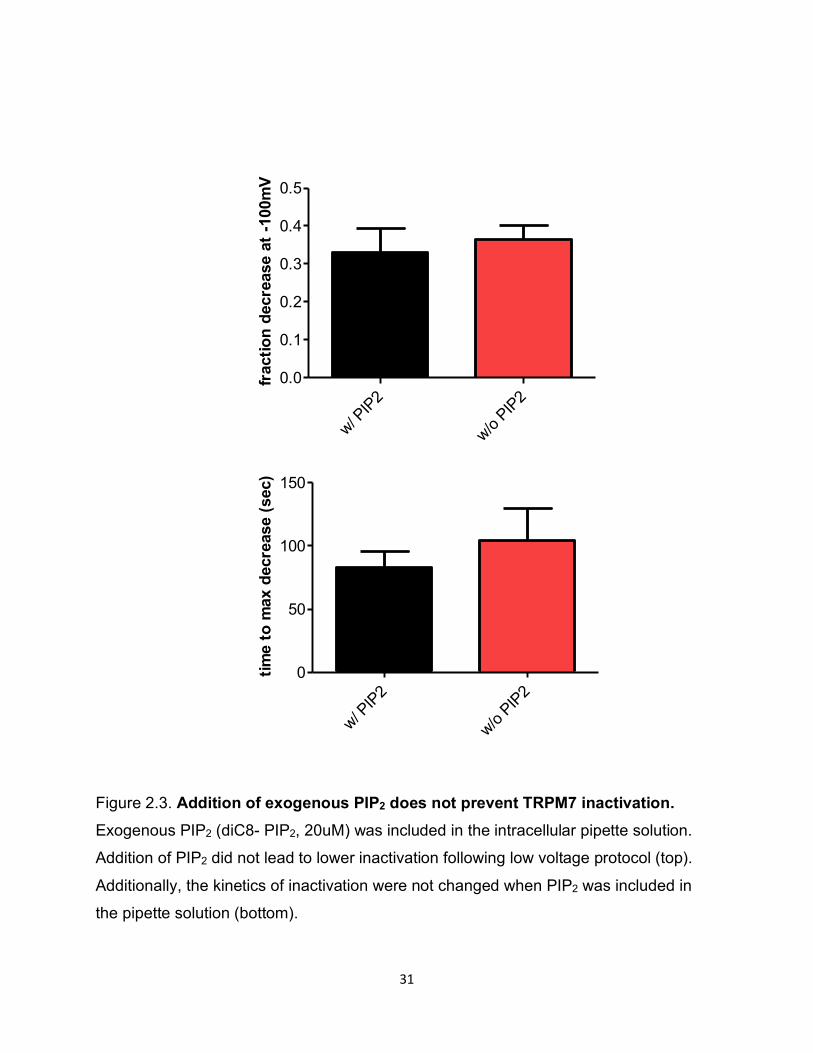
Addition of exogenous PIP2 does not prevent TRPM7 inactivation
As our FRET data indicated that PIP2 levels may be declining, we sought to test whether
exogenous PIP2 might restore TRPM7 current in the setting of hyperpolarization. Thus
we added exogenous diC8- PIP2 (20uM) in the pipette solution and ran the low voltage
protocol. We could not observe any substantial restorative effects of exogenous PIP2 on
TRPM7 current using the low voltage protocol (Figure 2.3). However, as PIP2 location
and level and its interaction with TRPM7 are tightly regulated in a time dependent manner,
the exogenous PIP2 may not be sufficient to reverse the effects of low voltage.

20
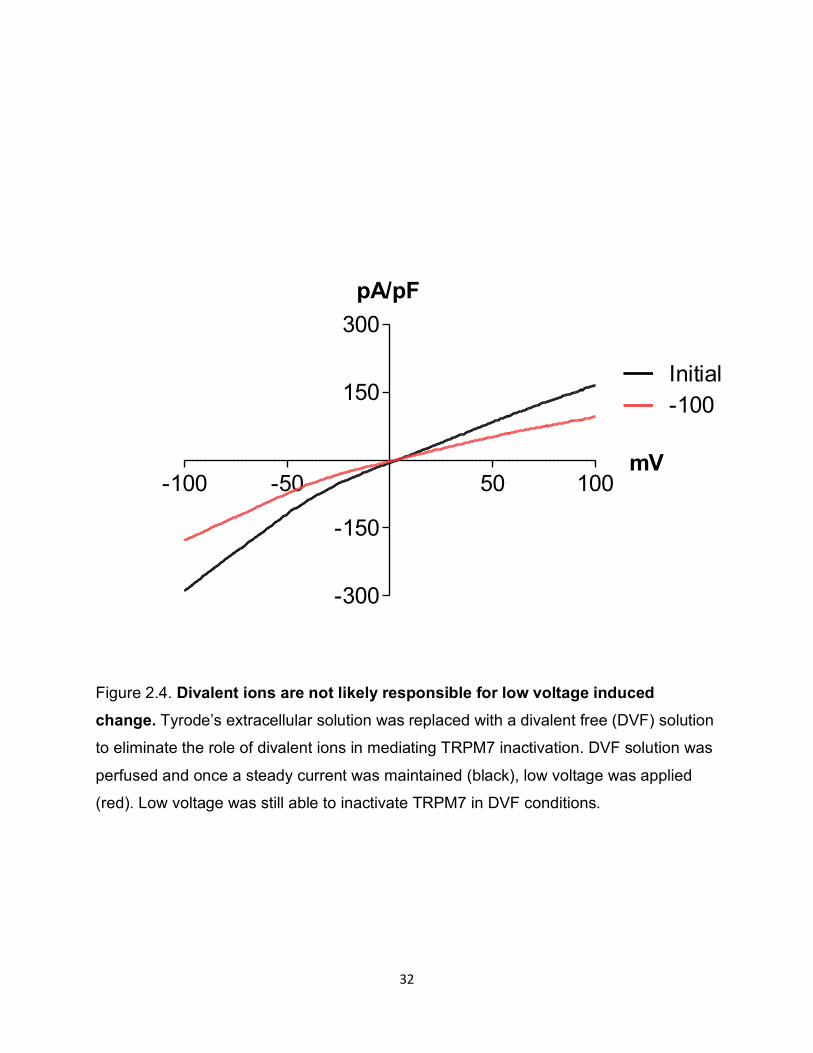
Divalent ions are not likely responsible for low voltage induced change
Although our intracellular pipette solution contained EGTA for chelating divalent cations,
we wanted to further exclude the possibility of divalent interaction with the channel and/or
PIP2 in regulating the low voltage effect that we saw. As sustained hyperpolarization
drives positive charges to enter the cell, we wanted to exclude any such divalent
interactions. It has previously been shown that multivalent cations may interfere with the
PIP2 -ion channel interaction 49 50. Thus, using our LTRPC7 cell line with the low voltage
protocol, we switched the extracellular bath from normal Tyrode’s to a solution which is
divalent free (DVF). This DVF solution altered the I-V shape of LTRPC7, as expected
(Figure 2.4). However, upon stimulation with the low voltage protocol, the decrease in
both inward and outward current persisted, suggesting that divalent cations may not play
the primary role in the low voltage response (Figure 2.4).
Discussion
We report in this paper for the first time that hyperpolarization of membrane
potential leads to decreased TRPM7 ion channel activity (Figure 2.5). We show that at
low voltage there is likely to be a change of PIP2. However, the exact mechanism of how
PIP2 levels are altered remains unclear. Our FRET data indicate that PIP2 levels may be
changing. However, there may also be a change in PIP2 availability at the cell surface at
low membrane potential, thus altering FRET and potentially ion channel interaction. Thus,
there may be a similar functioning voltage sensitive enzyme in humans as that seen in
Ciona intestinalis. However, further validation is required. One mechanism explaining our
phenomenon is the presence of functional TPTE or TPIP proteins that are homologous

21
to Ciona intestinalis Ci-VSP. Another possibility is that phosphatases or kinases may be
recruited to the cell surface by interacting proteins or lipids in order to modify PIP2 levels
at the cell surface. Thus, various kinases, phosphatases, or scaffolding proteins may be
involved in this low voltage phenomenon. We have also seen that this low voltage
response occurs in other PIP2 dependent cells. Using the same low voltage protocol, we
saw decreases in Kir2.1B channel activity, suggesting that hyperpolarization may induce
PIP2 changes that affect various ion channels and thus broadly influence cellular
physiology.
PIP2 regulates a large number of ion channels and recent studies in the crystal
structure of TRP channels may yield important clues as to how PIP2 regulation occurs. In
addition, recent work has suggested that membrane potential may influence the clustering
of PIP2 at the cell membrane to influence signaling pathways 51. Furthermore, voltage
may influence the localization of phosphoinositide regulators to influence TRP channel
activity 52. Similar to the question of how low voltage induces changes, a related question
remains as to how high voltage may influence PIP2 levels. We initially started with
hyperpolarization voltage protocols in order to keep the membrane potentials at
physiological values. However, how membrane depolarization alters PIP2 availability
remains an interesting question and to be investigated. In summary, our knowledge of
how TRP channels and other ion channels may be regulated by factors that influence
PIP2 levels is of critical importance in order to determine normal cellular physiology and
the changes that occur in pathophysiological conditions.

22
Materials and Methods
Molecular biology.
LTRPC7 cell lines were obtained from Dr. A.M. Scharenberg. PH-CFP and PH-YFP
plasmids were obtained from Dr. T. Balla.
Electrophysiology.
Whole cell and perforated patch currents were recorded using an Axopatch 200B
amplifier. Data were digitized at 5 or 10kHz, and digitally filtered off-line at 1 kHz. Patch
electrodes were pulled from borosilicate glass and heat polished to form a final resistance
around 5 MΩ when filled with internal pipette solution. Series resistance was
compensated up to 90% to reduce series resistance errors.
For whole cell current recordings, voltage stimuli lasting 200 ms were delivered at 1 s or
5 s intervals, with either voltage ramps or voltage steps ranging from -120 to +100mV or
-100mV, respectively. For low voltage holding ramp protocols, cells were held at either -
100mV or -120mV before intermittent ramp tests. A quick perfusion system was used to
exchange extracellular solutions, with complete solution exchange achieved. The internal
pipette solution consisted of (in mM) 145 Cs-methanesulfonate, 8 NaCl, 10 EGTA, and
10 HEPES, with pH adjusted to 7.2 with CsOH. In some cases, BAPTA was used as a
chelator for stronger chelating effects, but effects were similar to EGTA. For PIP2
experiments, we added 20uM diC8- PIP2 to the internal pipette solution and adjusted pH
accordingly. The standard Tyrode’s extracellular solution for whole cell recording
consisted of (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 10 HEPES, and 10 glucose, with pH
adjusted to 7.4 with NaOH. For conditions using DVF perfusion, the solution contained
145 NaCl, 10 HEPES, 5 EGTA, 2 EDTA and 10 glucose with pH adjusted to 7.4.

23
MaxChelator was used to calculate free Ca2+ and Mg2+ concentrations. For perforated
patch recordings, Nystatin (200ug/mL) was added into the pipette solution and pH
adjusted accordingly.
FRET measurements.
LTRPC7 cells were plated on 35mm cell culture dishes and transfected with PH-CFP and
PH-YFP plasmids using Lipofectamine 2000 and OptiMEM. After incubation in OptiMEM
for about 5 hours, the media was replaced with 10% BGS in DMEM F12 and 2ug/mL
tetracycline for induction. The cells were incubated overnight and split with Trypsin
(0.25%) the following day onto round glass coverslips coated with poly-L-lysine for 30
minutes. Fluorescence intensities of CFP and YFP were collected at a rate of 3 Hz using
CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon). The
YFP/CFP ratio was used for determining the percent of FRET change.
Data analysis.
Pooled data are presented as mean +/- SEM. Statistical comparisons were made using
two way analysis of variance (ANOVA) and two tailed t-test with Bonferroni correction.
p<0.05 indicate statistical significance.
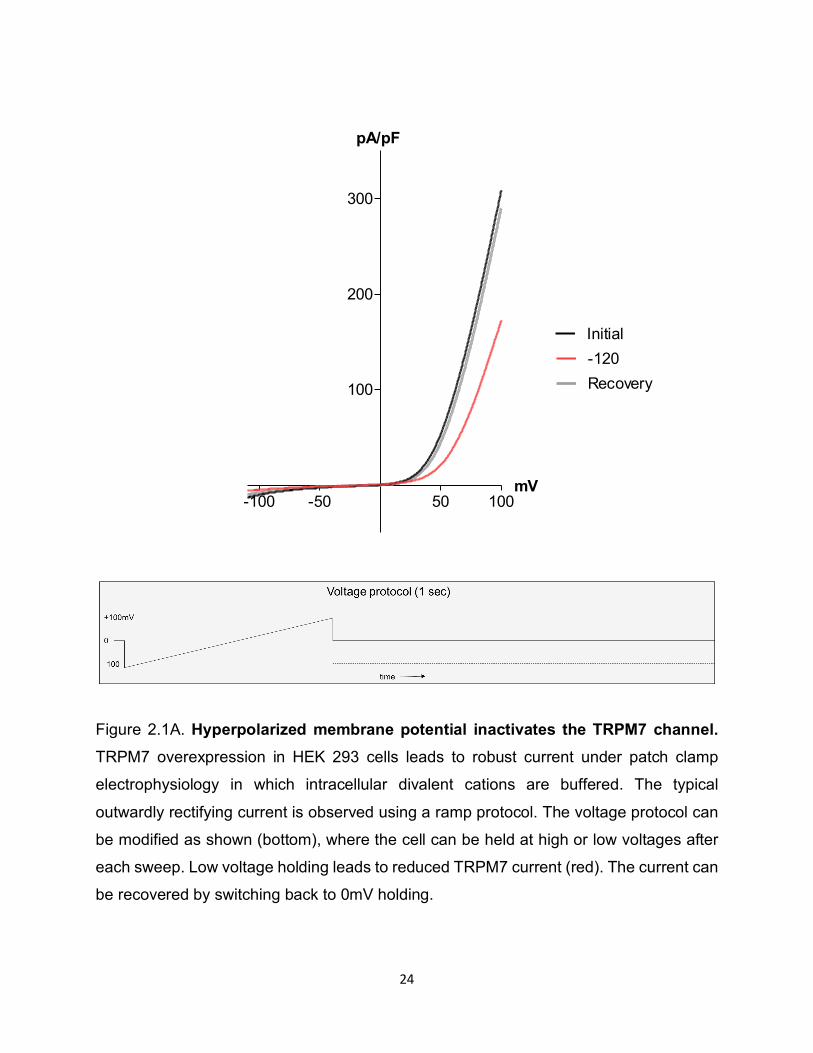
24
Figure 2.1A. Hyperpolarized membrane potential inactivates the TRPM7 channel. TRPM7 overexpression in HEK 293 cells leads to robust current under patch clamp
electrophysiology in which intracellular divalent cations are buffered. The typical
outwardly rectifying current is observed using a ramp protocol. The voltage protocol can
be modified as shown (bottom), where the cell can be held at high or low voltages after
each sweep. Low voltage holding leads to reduced TRPM7 current (red). The current can
be recovered by switching back to 0mV holding.
-100 -50 50 100
100
200
300
mV
pA/pF
Recovery
Initial-120
25
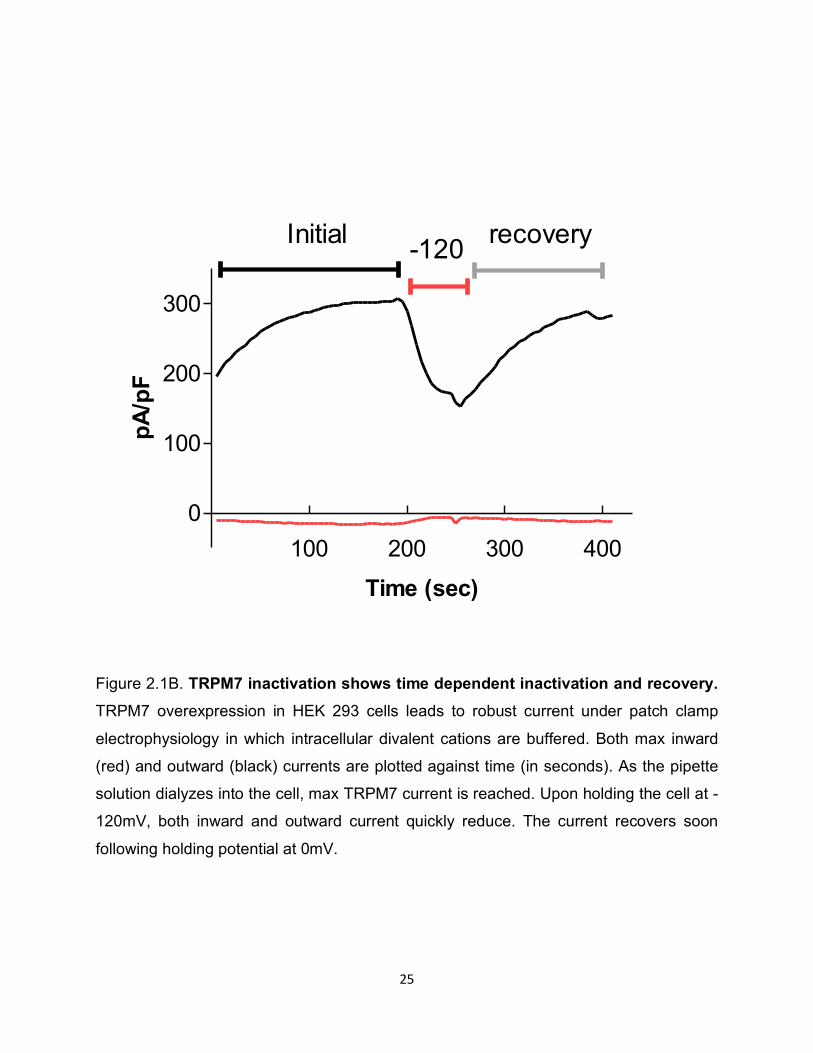
Figure 2.1B. TRPM7 inactivation shows time dependent inactivation and recovery. TRPM7 overexpression in HEK 293 cells leads to robust current under patch clamp
electrophysiology in which intracellular divalent cations are buffered. Both max inward
(red) and outward (black) currents are plotted against time (in seconds). As the pipette
solution dialyzes into the cell, max TRPM7 current is reached. Upon holding the cell at -
120mV, both inward and outward current quickly reduce. The current recovers soon
following holding potential at 0mV.
100 200 300 4000
100
200
300
Initial -120 recovery
Time (sec)
pA/p
F
26
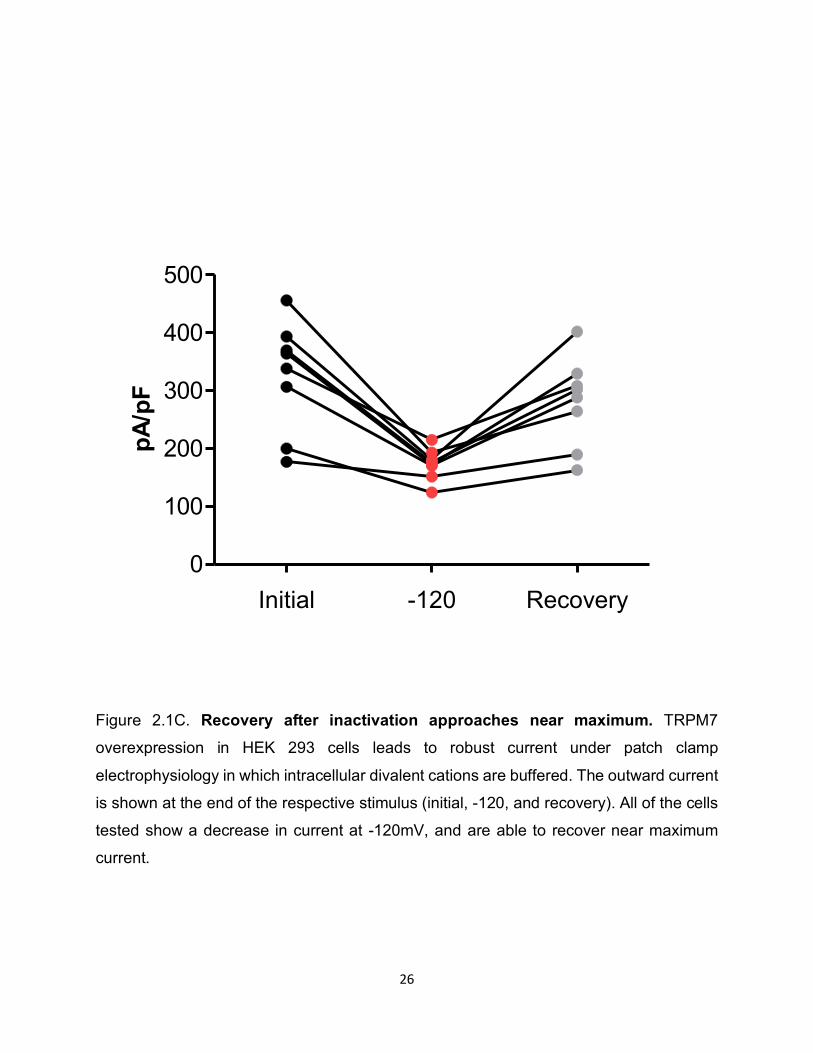
Figure 2.1C. Recovery after inactivation approaches near maximum. TRPM7
overexpression in HEK 293 cells leads to robust current under patch clamp
electrophysiology in which intracellular divalent cations are buffered. The outward current
is shown at the end of the respective stimulus (initial, -120, and recovery). All of the cells
tested show a decrease in current at -120mV, and are able to recover near maximum
current.
Initial -120 Recovery0
100
200
300
400
500
pA/pF
27
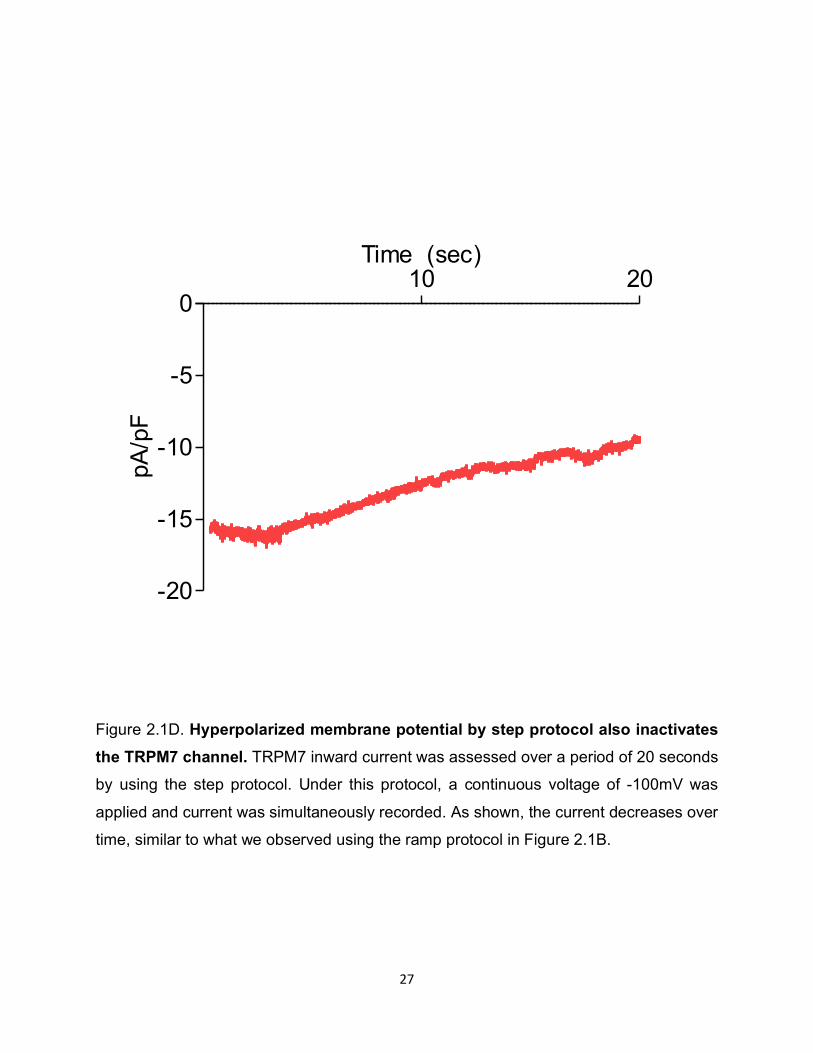
Figure 2.1D. Hyperpolarized membrane potential by step protocol also inactivates the TRPM7 channel. TRPM7 inward current was assessed over a period of 20 seconds
by using the step protocol. Under this protocol, a continuous voltage of -100mV was
applied and current was simultaneously recorded. As shown, the current decreases over
time, similar to what we observed using the ramp protocol in Figure 2.1B.
10 20
-20
-15
-10
-5
0
Time (sec)
pA/p
F
28
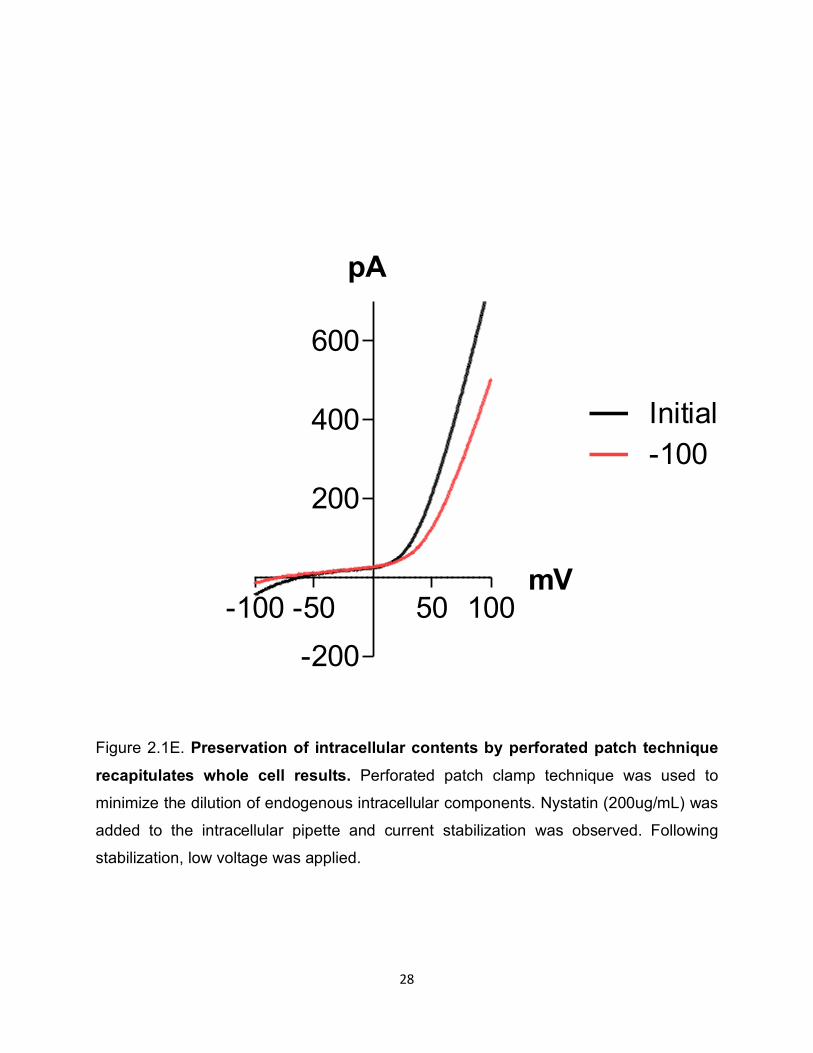
Figure 2.1E. Preservation of intracellular contents by perforated patch technique recapitulates whole cell results. Perforated patch clamp technique was used to
minimize the dilution of endogenous intracellular components. Nystatin (200ug/mL) was
added to the intracellular pipette and current stabilization was observed. Following
stabilization, low voltage was applied.
-100 -50 50 100-200
200
400
600
mV
pA
Initial-100

29
cell background0.00
0.05
0.10
0.15
0.20
Rat
io d
ecre
ase
at L
V
Initial -1200
200
400
600
pA/pF
a = 0mV b = -120mV c = 0mV

30
Figure 2.2. Dynamic PIP2 changes with membrane potential. Changes of PIP2 were
measured using FRET with PH-CFP and PH-YFP. The change in YFP/CFP ratio was
traced over time at low voltage and recovery holding potentials, while simultaneously
measuring current activity. Only the outer membrane region of the cell was included in
the ratio measurements, as this area gives the changes in membrane bound PIP2 (top).
Upon low voltage protocol, FRET decreased rapidly (middle), which also parallel a
decrease in TRPM7 current (lower left). The FRET ratio decreased about 15% at low
voltage relative to 0mV, whereas the background FRET ratio remained steady (lower
right).
31
Figure 2.3. Addition of exogenous PIP2 does not prevent TRPM7 inactivation. Exogenous PIP2 (diC8- PIP2, 20uM) was included in the intracellular pipette solution.
Addition of PIP2 did not lead to lower inactivation following low voltage protocol (top).
Additionally, the kinetics of inactivation were not changed when PIP2 was included in
the pipette solution (bottom).
w/ PIP2
w/o PIP2
0.0
0.1
0.2
0.3
0.4
0.5
frac
tion
decr
ease
at -
100m
V
w/ PIP2
w/o PIP2
0
50
100
150
time
to m
ax d
ecre
ase
(sec
)
32
Figure 2.4. Divalent ions are not likely responsible for low voltage induced change. Tyrode’s extracellular solution was replaced with a divalent free (DVF) solution
to eliminate the role of divalent ions in mediating TRPM7 inactivation. DVF solution was
perfused and once a steady current was maintained (black), low voltage was applied
(red). Low voltage was still able to inactivate TRPM7 in DVF conditions.
-100 -50 50 100
-300
-150
150
300
mV
pA/pF
Initial-100
33
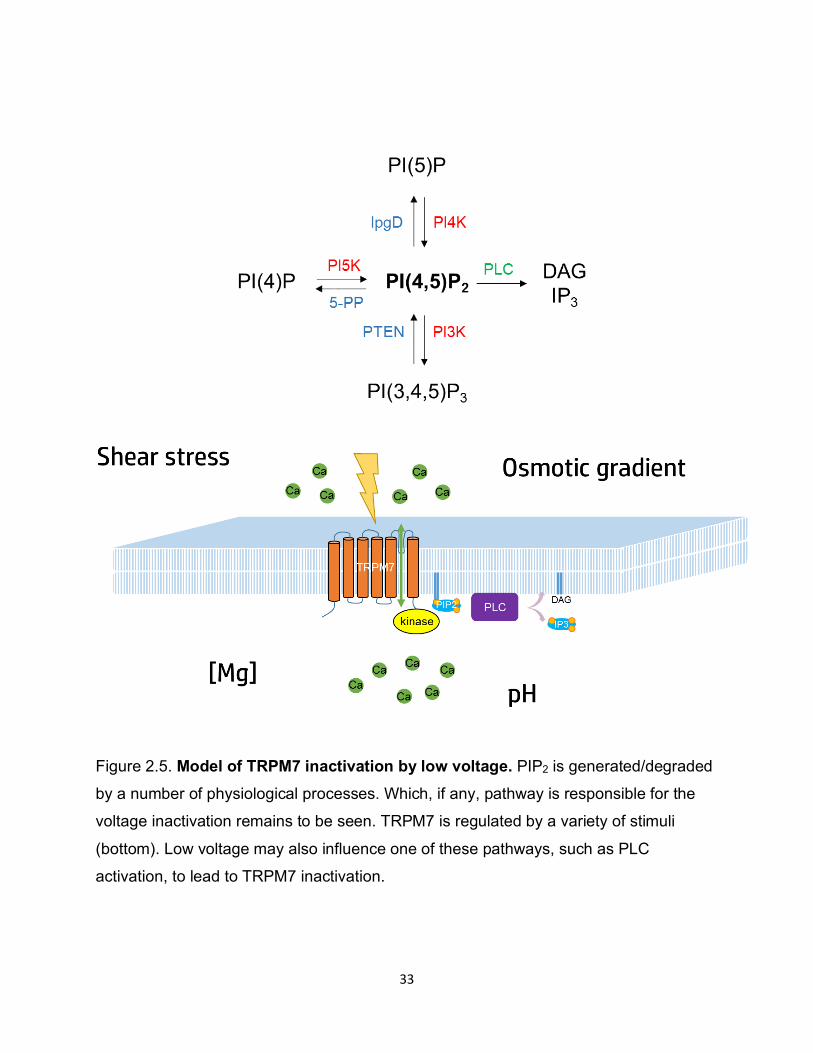
Figure 2.5. Model of TRPM7 inactivation by low voltage. PIP2 is generated/degraded
by a number of physiological processes. Which, if any, pathway is responsible for the
voltage inactivation remains to be seen. TRPM7 is regulated by a variety of stimuli
(bottom). Low voltage may also influence one of these pathways, such as PLC
activation, to lead to TRPM7 inactivation.

34
CHAPTER 3: TRPM2 MODULATION OF INFLAMMATION AND CARDIOVASCULAR DISEASE

35
Introduction
Changes that occur in heart disease are not limited to the heart, but throughout the
individual. Individuals with coronary artery disease (CAD) and subsequent myocardial
infarction (MI) are susceptible to multiple other vascular-related conditions such as stroke,
peripheral vascular disease, and ischemic kidney disease, the common underlying
connection being atherosclerosis. In patients, atherosclerosis has been associated with
high levels of the inflammatory cytokine IL-6 and the biomarker C-reactive protein along
with an increase in inflammatory peripheral monocytes 53 54. Apoe-ko mice (in combination
with a high fat diet), which lack a protein involved in hepatic uptake of circulating
cholesterol, enable atherosclerosis studies in mice 55. In mouse models of
atherosclerosis, systemic changes such as a selective expansion of circulating
inflammatory monocytes are seen. Moreover, monocytes have been shown to be involved
in MI recovery at both acute and chronic phases of infarct healing 18. Initial recruitment of
inflammatory Ly6Chi monocytes is followed by recruitment of reparative Ly6Clo monocytes
in the damaged heart, a sequence that is necessary to ensure proper healing. A recent
study showed that atherosclerosis induces a chronic elevation of inflammatory Ly6Chi
monocytes which impairs infarct healing. Furthermore, myocardial infarction has been
shown to accelerate atherosclerosis and increase the chances for a subsequent MI. Thus
a better understanding of the role of monocytes in MI using clinically relevant models of
atherosclerosis is critical. Loss of regulation of inflammatory monocytes can propagate
harmful cardiac remodeling and lead to alterations in chamber size and contractility,
ultimately leading to sudden death or recurrence, arrhythmia, heart failure, and stroke.

36
To dissect the mechanism of how inflammatory monocytes contribute to I-R injury,
we focus on TRPM2, an oxidative stress activated Ca2+-permeable ion channel 56. TRPM2
is expressed in the brain, endothelial cells, heart, and immune cells, such as
monocytes/MΦs. TRPM2 is activated through an intracellular domain that bind Ca2+ and
oxidative stress products such as hydrogen peroxide, both of which are increased in heart
disease. Moreover, TRPM2 activation by reactive oxygen species in immune cells has
been thought to play a crucial role in inflammatory diseases. Although the precise
physiological function of TRPM2 is unknown, a study has shown that TRPM2 activation
and subsequent Ca2+ entry in monocytes aggravates chemically induced colitis by
production of the paracrine signal CXCL8 which leads to an aggravation of inflammation
57. Another study has implicated neutrophil TRPM2 in exacerbating cardiac injury after
ischemia-reperfusion (I-R) injury by increased neutrophil adhesion 58. Finally, a recent
finding suggests that TRPM2-mediated Ca2+ entry is necessary in MΦs for NLRP3-
inflammasome activation and secretion of the pro-inflammatory molecule IL-1β 59.
Here we study atherosclerosis, as well as MI in a background of atherosclerosis to
closely parallel human disease, where MI is not an isolated event, but occurs in the
presence of vascular disease and inflammation. We thus overcome a barrier in
cardiovascular research which lacks such studies, where alterations in inflammatory
status, cytokine profile, and leukocyte populations abound. As survival rates after the
initial episode of MI are increasing due to improving acute interventions after MI, there is
an increasing need to develop treatments to halt the progression of heart disease. This
research takes the first steps into gaining new insight into how modulation of an ion
channel, TRPM2, after I-R injury can harness the protective aspects of inflammation,

37
while reducing its deleterious consequences. Additionally, as TRPM2 has been linked to
the inflammasome pathway, TRPM2 may serve as an effective target for other
inflammatory diseases. As there has been a push toward developing inhibitors of NLRP3
inflammasome and IL-1β for inflammatory and fibrotic diseases, the potential therapy
through TRPM2 holds promise.
Results
Trpm2 deletion protects against atherosclerosis
As TRPM2 is involved in various inflammatory conditions, we wanted to test whether
TRPM2 influences atherosclerosis disease progression. Using our mouse model of
atherosclerosis, we saw that Apoe knockout mice develop substantial aortic
atherosclerosis following treatment with a high fat diet (HFD), as measured by Oil Red
staining of the aorta. However, we were very surprised to see that the addition of global
Trpm2 knockout led to a reduction in large vessel atherosclerosis as compared to Apoe
knockout alone (Figure 3.1).
Trpm2 deletion protects against myocardial infarction
Furthermore, we wanted to see test how TRPM2 mediates other forms of cardiovascular
disease. We wanted to mimic the human scenario of MI, whether there is abundant
systemic inflammation and atherosclerosis. Thus, using the mice we used above for our
atherosclerosis studies, we induced myocardial infarction using the well-established
model of myocardial infarction. At one month following MI, we saw improved heart
function in the Trpm2 knockout mice as measured by ejection fraction. Furthermore, upon
Pressure-volume loop analysis at two months, there were increased cardiac output and

38
stroke volume, among other parameters, in the Trpm2 knockout group compared to wild-
type (Figure 3.2). This suggested that TRPM2 may be contributing to the deleterious
effects after MI, presumably by reducing inflammation.
Systemic changes in circulating myeloid cells during atherosclerosis and MI
Previous reports have shown that both atherosclerosis and MI contribute to the increased
levels of circulating inflammatory monocytes. Thus, we wanted to determine how TRPM2
influences this population by flow cytometry, which may be contributing to the
atherosclerosis and MI phenotypes we observed (Figure 3.3A). Trpm2 knockout did not
affect basal myeloid cell populations (Figure 3.3B). Flow cytometry of peripheral blood
obtained from mice treated with HFD over a long period of time showed increased levels
of circulating myeloid cells, predominantly due to the increased circulating neutrophil
population. However, Trpm2 knockout mice showed reduced neutrophil burden, more
similar to mice without atherosclerosis (Figure 3.3C). However, following myocardial
infarction, we were not able to observe a difference in circulating inflammatory Ly6Chi
cells in Trpm2 knockout mice (Figure 3.3D). Our results suggest that our method may not
have the sensitivity to detect whether TRPM2 may be influencing the circulating
peripheral blood cell population after myocardial infarction, and/or that the main
contributor effect of TRPM2 is through the change in circulating neutrophil population.
However, we do see TRPM2 expression in bone marrow progenitor cells, suggesting a
possible role in the bone marrow reserve (Figure 3.4).

39
Discussion
Our results show for that deletion of Trpm2 is protective against cardiovascular
disease (Figure 3.5). We show that global Trpm2 knockout reduces atherosclerotic
burden. Furthermore, Trpm2 knockout preserves heart function after a model of
myocardial infarction in a setting of concurrent atherosclerosis. We have also created cell
type specific knockouts of Trpm2 in the endothelium (Cdh5) and myeloid lineage (Cx3cr1
and Cd11b) in order to determine the predominant cell type involved in this process
(Figure 3.6).
Preliminary results suggest that myeloid cells are involved in the process. Flow
cytometry data indicate that circulating neutrophils are increased in atherosclerosis,
though we were not able to observe any significant differences in circulating inflammatory
monocytes after myocardial infarction. The neutrophil levels are reduced in Trpm2
knockout mice during long term atherosclerosis, suggesting that TRPM2 coordinates the
myeloid population in these disease processes. To be determined is how exactly TRPM2
mediates such deleterious effects.
One possible mechanism in which TRPM2 is involved is through changes in
cellular adhesion and/or secretion of chemokines. We believe that the pro-inflammatory
cytokine IL-1β may contribute to the inflammatory processes observed. However, other
studies have shown that TRPM2 is involved in the secretion of other chemokines such as
CXCL2, which acts to recruit other inflammatory cells. Furthermore, as circulating myeloid
levels are changed in Trpm2 knockout mice, another possible role for TRPM2 is through
its role in the release of inflammatory cells from myeloid reservoirs such as the spleen
and bone marrow. As TRPM2 has been shown to be expressed in the autonomic nervous

40
system, it may be responsible in mediating the release through actions on the sympathetic
or parasympathetic pathways 60.
Materials and Methods
Mice.
Global Trpm2-ko mice obtained from Y. Mori were used which carry a deletion in the exon
encoding for transmembrane segment 5. For atherosclerosis experiments, Apoe
knockout mice were used obtained from Jackson laboratories. All mice were in the
C57BL/6 background. Apoe-ko are referred to as “SKO” (single knockout) and Apoe-ko
with Trpm2 deletion are referred to as “DKO” (double knockout). High fat diet (HFD) food
(21% fat, 0.15% cholesterol by weight obtained from Harlan) was fed starting at 5 weeks
of age to generate atherosclerosis.
Echocardiography and PV loop.
Echocardiography heart function measurements were performed by probing the left
ventricular mid region using Vevo 770 in two-dimensional M mode. PV loop
measurements were performed using Scisense Pressure-Volume Measurement system
with catheter (Transonic) and ADVantage PV system (Scisense) and analyzed with
LabScribe (iWorx). Mice were anesthetized using 3% isoflurane.
Atherosclerosis staining.
Quantification of plaque burden was performed following HFD treatment. Aorta were
harvested from mice and stained using standard Oil Red O staining procedures.
Myocardial infarction model.

41
LAD ligation was used to produce I-R injury. This procedure followed standard techniques
of 45 minute ischemia following by reperfusion 61.
Flow cytometry.
Peripheral blood was collected at various time points using tail vein bleed. After 10-12
drops of blood were collected, leukocytes were separated and stained for flow cytometry.
Alexa Fluor 700 Rat Anti-Mouse Ly-6C (BD), FITC antimouse Ly-6G (BioLegend), Biotin
anti mouse F4/80 (BioLegend), PE CF594 streptavidin (BD), Anti-mouse CD3e PE-
cyanine 7 (eBioscience), Anti-human/mouse CD45R (B220) PE-Cyanine 7 (eBioscience),
anti-mouse NK1.1 PE-Cyanine 7 (eBioscience), Anti-mouse CD11b eFlour 450
(eBioscience), APC anti-mouse Cx3cr1 (Biolegend) were used.
Data analysis.
Pooled data are presented as mean +/- SEM. Statistical comparisons were made using
two way analysis of variance (ANOVA) and two tailed t-test with Bonferroni correction.
p<0.05 indicate statistical significance.
42
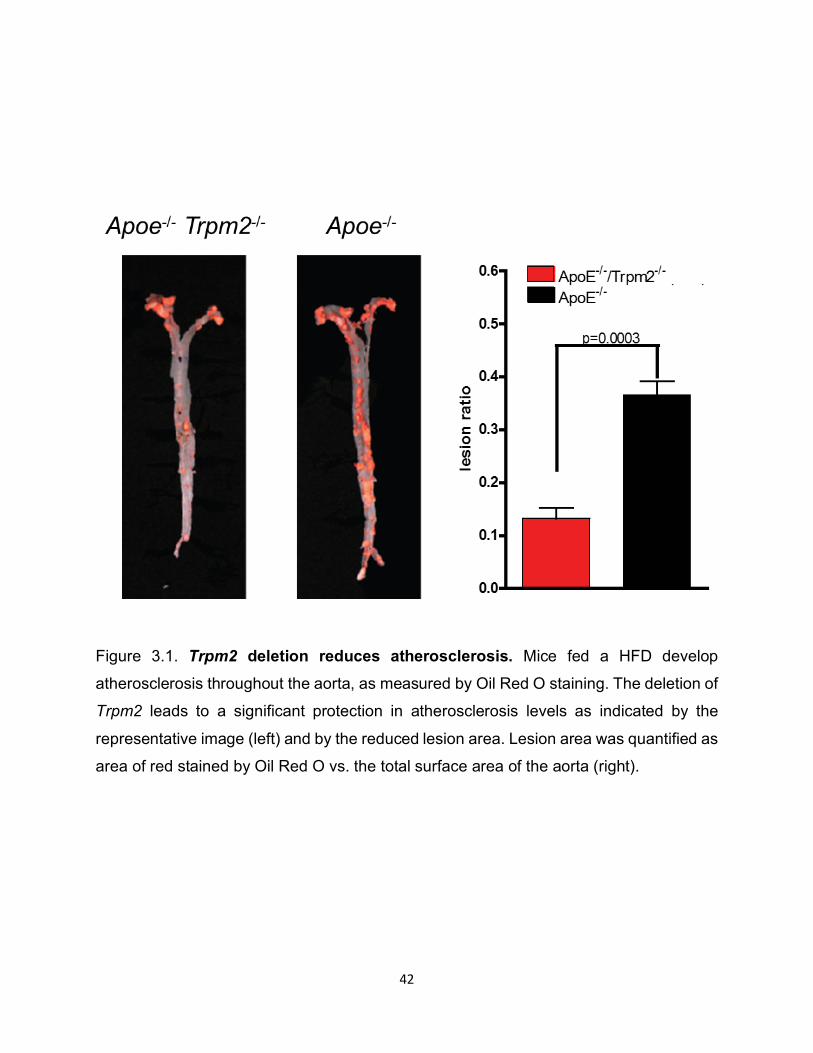
Figure 3.1. Trpm2 deletion reduces atherosclerosis. Mice fed a HFD develop
atherosclerosis throughout the aorta, as measured by Oil Red O staining. The deletion of
Trpm2 leads to a significant protection in atherosclerosis levels as indicated by the
representative image (left) and by the reduced lesion area. Lesion area was quantified as
area of red stained by Oil Red O vs. the total surface area of the aorta (right).

43

44
Figure 3.2. Trpm2 deletion protects against myocardial infarction. Deletion of Trpm2
was previously shown in our lab to protect against detrimental cardiac remodeling (top
left). Using echocardiography (top right) and PV loop analysis, we assessed how effective
Trpm2 deletion is in protecting against myocardial infarction in a model mimicking human
disease. Trpm2-ko mice in the Apoe knockout background (DKO) had improved heart
function as measured by echocardiography and PV loop analysis 3 weeks post I-R injury
(bottom).
45
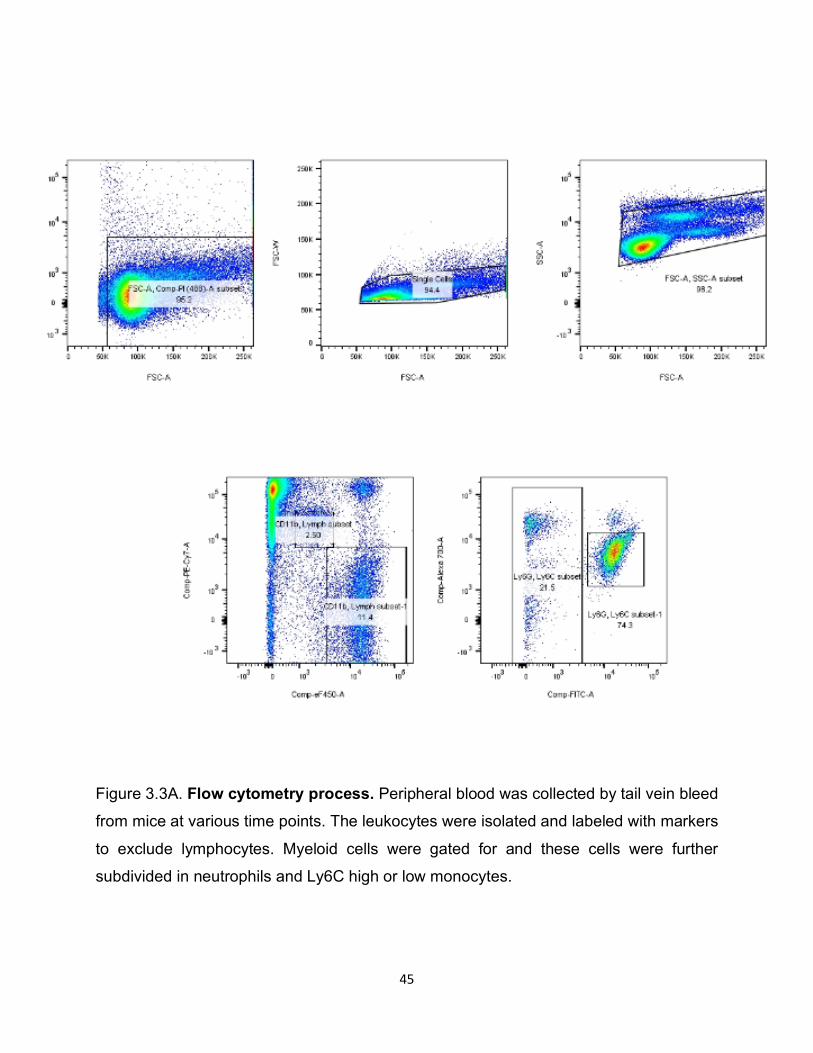
Figure 3.3A. Flow cytometry process. Peripheral blood was collected by tail vein bleed
from mice at various time points. The leukocytes were isolated and labeled with markers
to exclude lymphocytes. Myeloid cells were gated for and these cells were further
subdivided in neutrophils and Ly6C high or low monocytes.
46
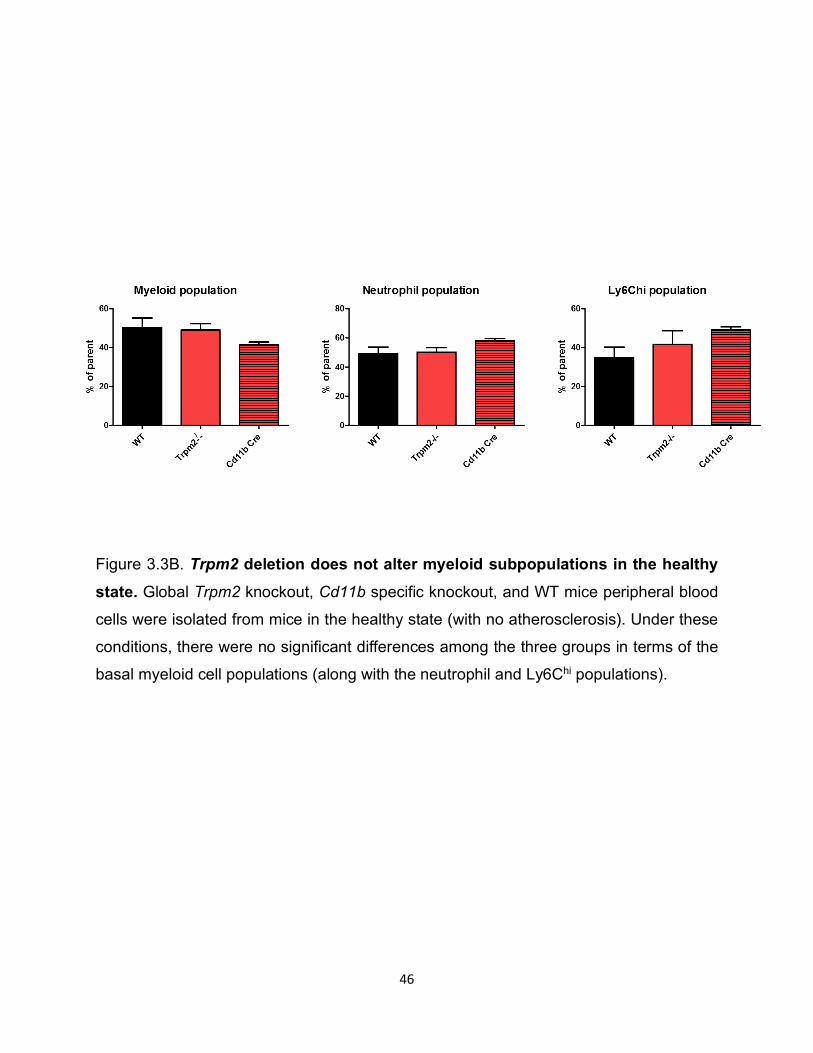
Figure 3.3B. Trpm2 deletion does not alter myeloid subpopulations in the healthy state. Global Trpm2 knockout, Cd11b specific knockout, and WT mice peripheral blood
cells were isolated from mice in the healthy state (with no atherosclerosis). Under these
conditions, there were no significant differences among the three groups in terms of the
basal myeloid cell populations (along with the neutrophil and Ly6Chi populations).
47
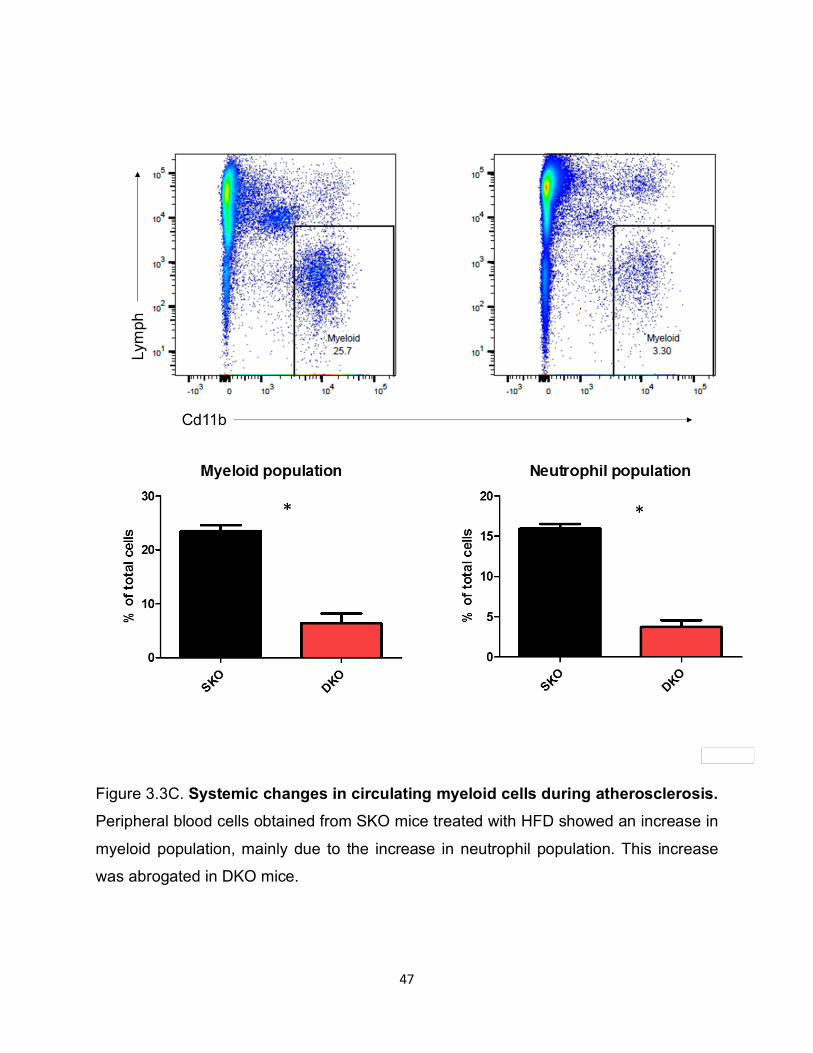
Figure 3.3C. Systemic changes in circulating myeloid cells during atherosclerosis. Peripheral blood cells obtained from SKO mice treated with HFD showed an increase in
myeloid population, mainly due to the increase in neutrophil population. This increase
was abrogated in DKO mice.
48
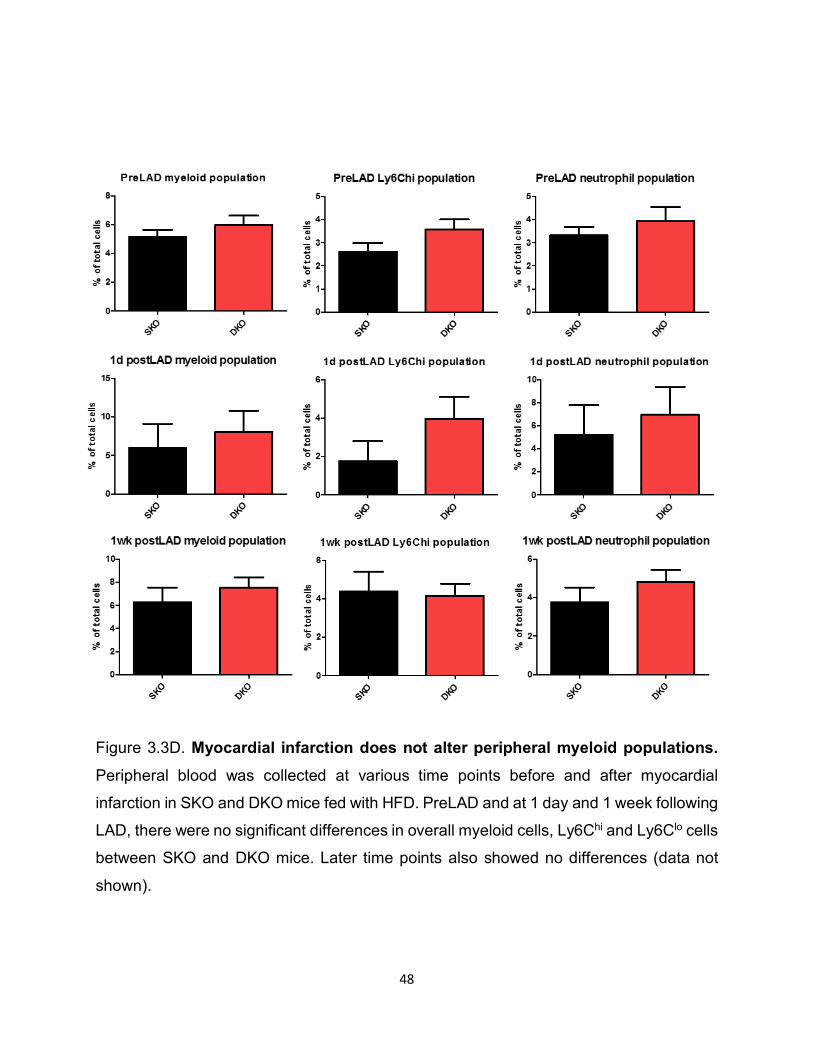
Figure 3.3D. Myocardial infarction does not alter peripheral myeloid populations. Peripheral blood was collected at various time points before and after myocardial
infarction in SKO and DKO mice fed with HFD. PreLAD and at 1 day and 1 week following
LAD, there were no significant differences in overall myeloid cells, Ly6Chi and Ly6Clo cells
between SKO and DKO mice. Later time points also showed no differences (data not
shown).
49
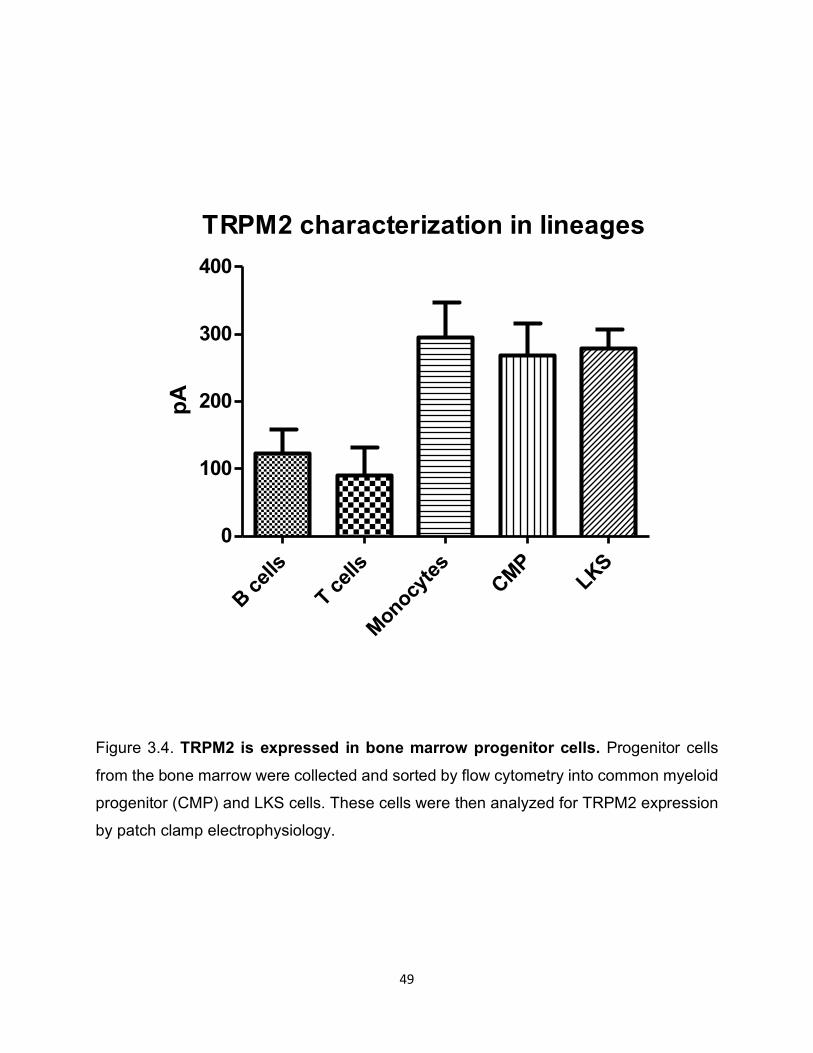
Figure 3.4. TRPM2 is expressed in bone marrow progenitor cells. Progenitor cells
from the bone marrow were collected and sorted by flow cytometry into common myeloid
progenitor (CMP) and LKS cells. These cells were then analyzed for TRPM2 expression
by patch clamp electrophysiology.
TRPM2 characterization in lineages
B cells
T cells
Monocytes CMP
LKS0
100
200
300
400
pA
50
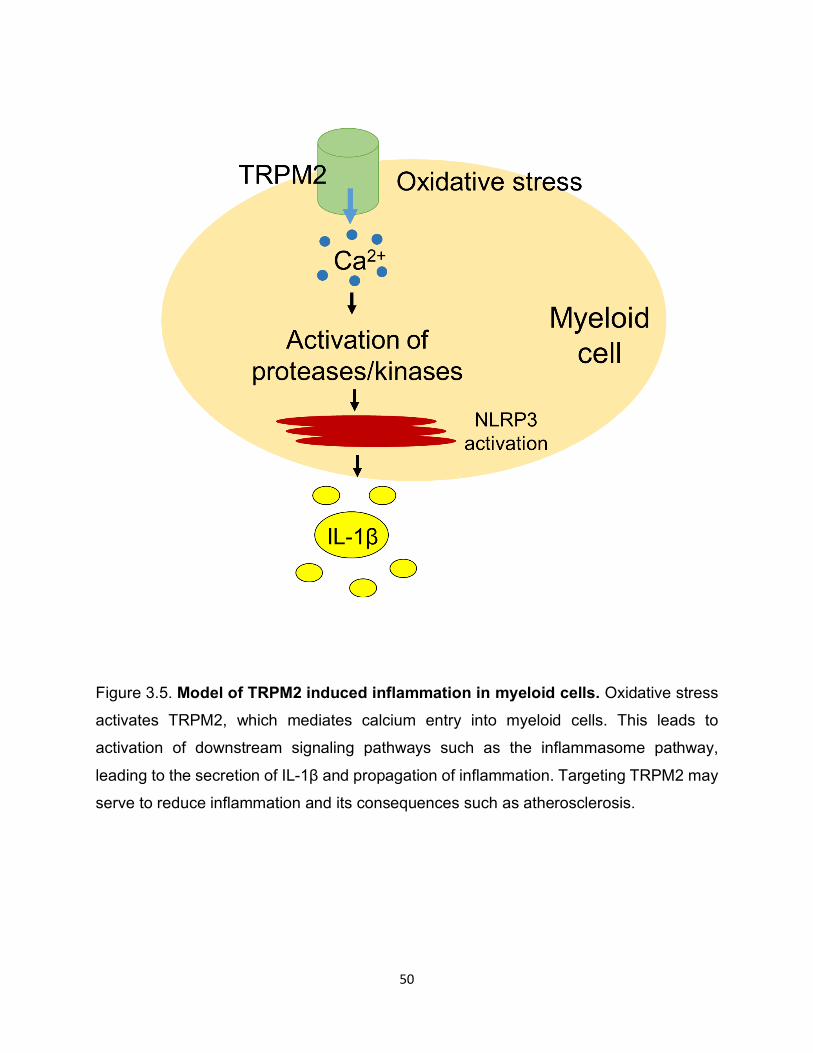
Figure 3.5. Model of TRPM2 induced inflammation in myeloid cells. Oxidative stress
activates TRPM2, which mediates calcium entry into myeloid cells. This leads to
activation of downstream signaling pathways such as the inflammasome pathway,
leading to the secretion of IL-1β and propagation of inflammation. Targeting TRPM2 may
serve to reduce inflammation and its consequences such as atherosclerosis.
51
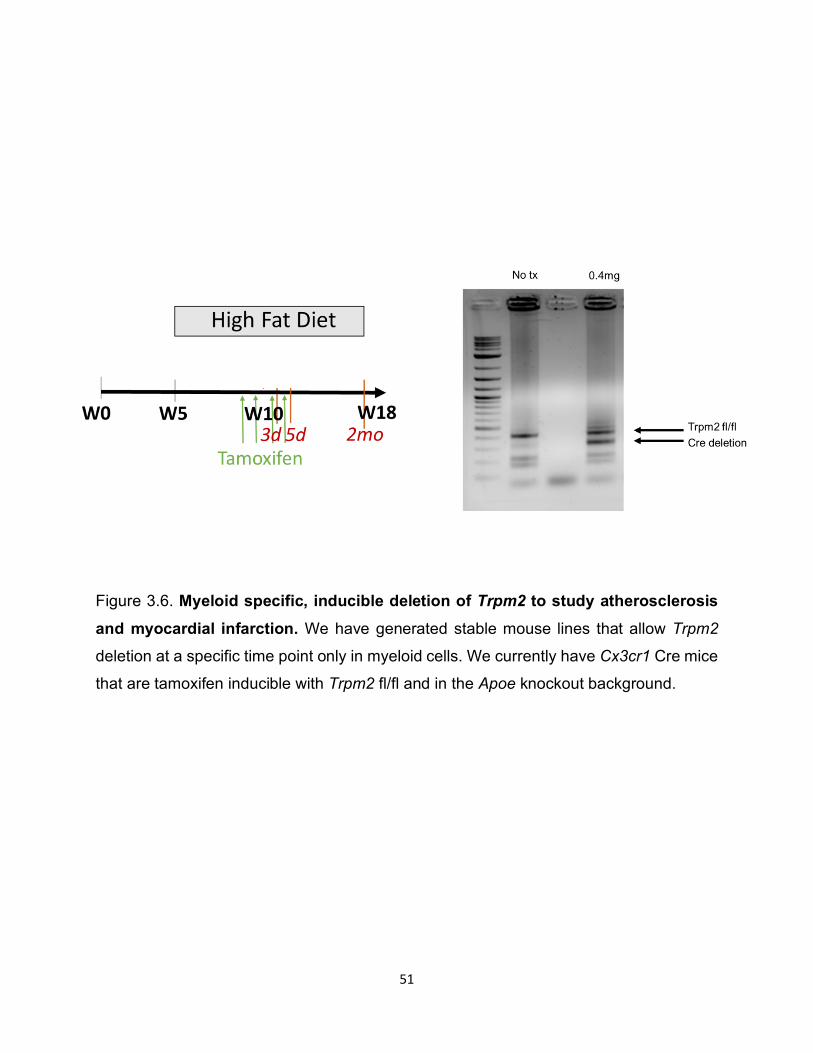
Figure 3.6. Myeloid specific, inducible deletion of Trpm2 to study atherosclerosis and myocardial infarction. We have generated stable mouse lines that allow Trpm2
deletion at a specific time point only in myeloid cells. We currently have Cx3cr1 Cre mice
that are tamoxifen inducible with Trpm2 fl/fl and in the Apoe knockout background.

52
CHAPTER 4: TARGETING TRPM7 TO REVERSE CARDIAC FIBROSIS

53
Introduction
Cardiac fibrosis is a key component in many heart diseases including cardiac
hypertrophy, heart failure, and arrhythmia. Fibrotic depositions are laid in response to
acute or chronic stressors, and are generally thought to be a protective mechanism.
However, over time, the remodeled tissue can serve as substrate for atrial fibrillation
and/or lead to reduced heart function over time due to a vicious cycle of extracellular
deposition leading to decreased contractility. Despite intensive efforts to find ways to treat
cardiac fibrosis, there has been very little progress.
The main cell type involved in the deposition of extracellular matrix in the heart is
the cardiac fibroblast (cFB) 62. Often neglected in the field of cardiac research, these
fibroblasts play a critical role in cardiac physiology and pathophysiology, with some
literature suggesting its involvement in setting the resting membrane potential and
influencing excitability. It is also believe that these fibroblasts may be both electrically and
physically coupled to cardiomyocytes. Recent studies have shown cFBs as the cell type
involved in TGFβ-induced fibrosis. Thus, targeting cFBs to reduce fibrosis has been a
topic of recent interest.
One potential target for fibrosis is through TRPM7. TRPM7, a “chanzyme” due to
its dual characteristics of being a channel while also containing a kinase domain, has
been implicated in many different types of fibrosis, ranging from liver to pulmonary to
more recently, cardiac fibrosis 63. Previous research in our lab has shown TRPM7 to be
upregulated in patients with atrial fibrillation. Furthermore, TRPM7 was shown to be
involved in the differentiation of cardiac fibroblasts into myofibroblasts following TGF-β
stimulation 64.

54
Here we show that deletion of Trpm7 in a model of hypertensive heart disease
protects against negative cardiac remodeling. In addition, using cell type specific Trpm7
knockout, we show that Trpm7 knockout specifically in cardiac fibroblasts may be critical
in providing this protective effect. Our results suggest that TRPM7 may be a novel target
for reducing cardiac fibrosis, and potentially fibrosis in other tissues as well.
Results
Deletion of Trpm7 attenuates fibrosis and improves heart function
We sought to determine if TRPM7 is involved in cardiac fibrosis. Using the
transverse aortic constriction model (TAC), we investigated the role of Trpm7 deletion.
As Trpm7 deletion in development is lethal, we used tamoxifen inducible global Trpm7
deletion. As soon as 1 week after TAC, we observed significant protection by Trpm7
deletion compared to wild type. The ejection fraction was improved, as measured by
echocardiography, up to 2 month post TAC, along with an improvement in overall survival.
Furthermore, cardiac fibroblasts isolated from TAC mice showed increased TRPM7
activity by patch clamp electrophysiology as compared to sham mice, suggesting that
TRPM7 is likely involved in negative remodeling of the heart (Figure 4.1).
Fibroblast specific deletion of Trpm7 recapitulates protective effects
To validate that TRPM7 in fibroblasts plays a key role in mediating fibrosis, we
then went ahead to use fibroblast specific Trpm7 knockout using periostin Cre and
compared the effects after TAC with myocyte specific knockout. Interestingly, we found
that Trpm7-MY-ko did not produce any protective effect in the TAC model, but Trpm7-FB-

55
ko produced similar protection as we observed using global knockout (Figure 4.2). Thus,
our results suggest that fibroblasts play a key role in mediating fibrosis through TRPM7.
Discussion
Our results show that fibroblast specific TRPM7 is involved in mediating cardiac
fibrosis. Deletion of Trpm7 produced significant protection in a pressure
overload/hypertension model. As the burden of cardiac disease and fibrosis is growing,
novel ways to reduce morbidity and mortality will need to be discovered. Here we show
that TRPM7 may serve as a potential candidate to improve heart function. However, as
TRPM7 is ubiquitously expressed, and critical for development, targeted strategies to
reduce TRPM7 activity will need to be determined.
Furthermore, the mechanism as to how TRPM7 mediates cardiac fibrosis has yet
to be determined. As TRPM7 is both a channel and kinase, one or both components may
be involved in mediating cardiac fibrosis (Figure 4.3). Future studies using channel dead
or kinase inactive forms of TRPM7 will be informative. Our lab has also shown
(unpublished data) that CaMKII may play a role in transducing the TRPM7 mediated
calcium signal during fibrogenesis, and that the TGFβ signaling pathway may also be
involved as well. Future studies to clearly elucidate the mechanism may yield great insight
into understanding fibrosis mediated by TRPM7 not only in the heart, but in other tissues
as well.

56
Methods
Mice.
Global Cre for Trpm7 knockout was induced by tamoxifen and confirmed by genotyping,
western blot and patch clamp. Control mice were the Cre- littermates. Myh6 Cre were
obtained from D. Clapham and Perisotin Cre were obtained from Dr. S.J. Conway.
Transverse aortic constriction (TAC).
A model of pressure overload was created using the established transverse aortic
constriction model 65. Briefly, a suture was place in the transverse aorta between the
brachiocephalic and left common carotid arteries. Successful constriction was measured
by Doppler before and immediately after the stricture following the procedure.
Echocardiography.
Echocardiography heart function measurements were performed by probing the left
ventricular mid region using Vevo 770 in two-dimensional M mode. Mice were
anesthetized using 3% isoflurane.
Data analysis. Pooled data are presented as mean +/- SEM. Statistical comparisons were made using
two way analysis of variance (ANOVA) and two tailed t-test with Bonferroni correction.
p<0.05 indicate statistical significance.
57
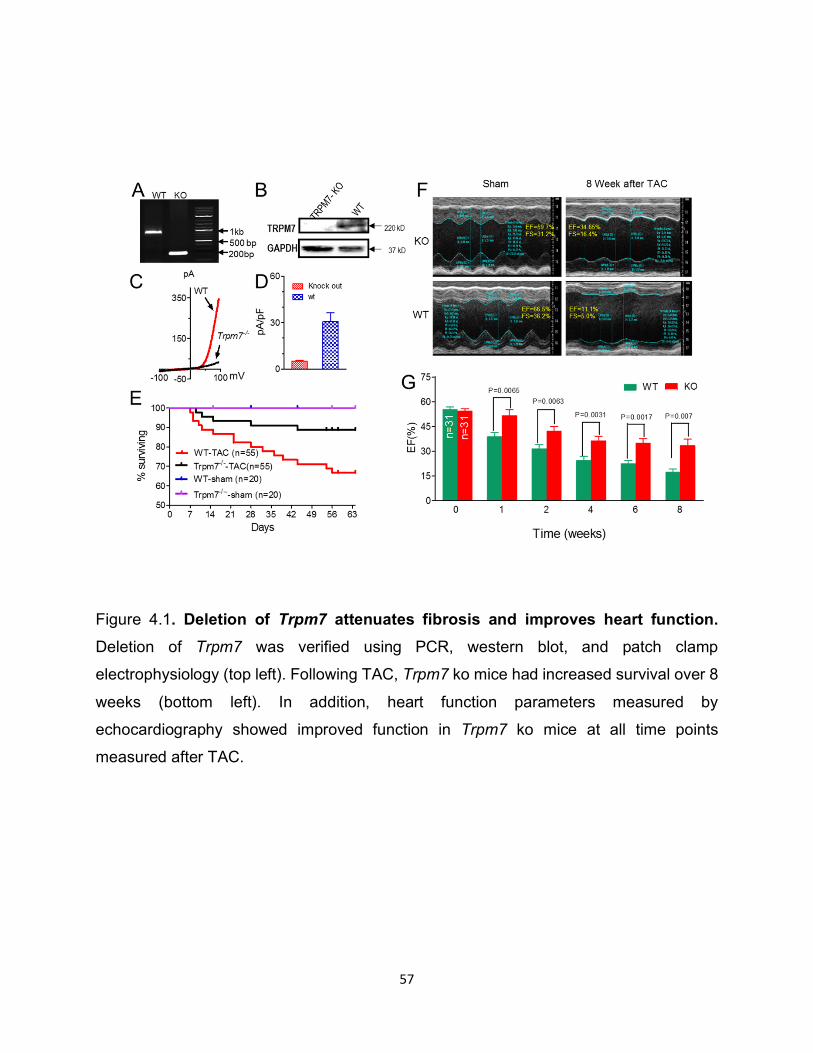
Figure 4.1. Deletion of Trpm7 attenuates fibrosis and improves heart function. Deletion of Trpm7 was verified using PCR, western blot, and patch clamp
electrophysiology (top left). Following TAC, Trpm7 ko mice had increased survival over 8
weeks (bottom left). In addition, heart function parameters measured by
echocardiography showed improved function in Trpm7 ko mice at all time points
measured after TAC.
58
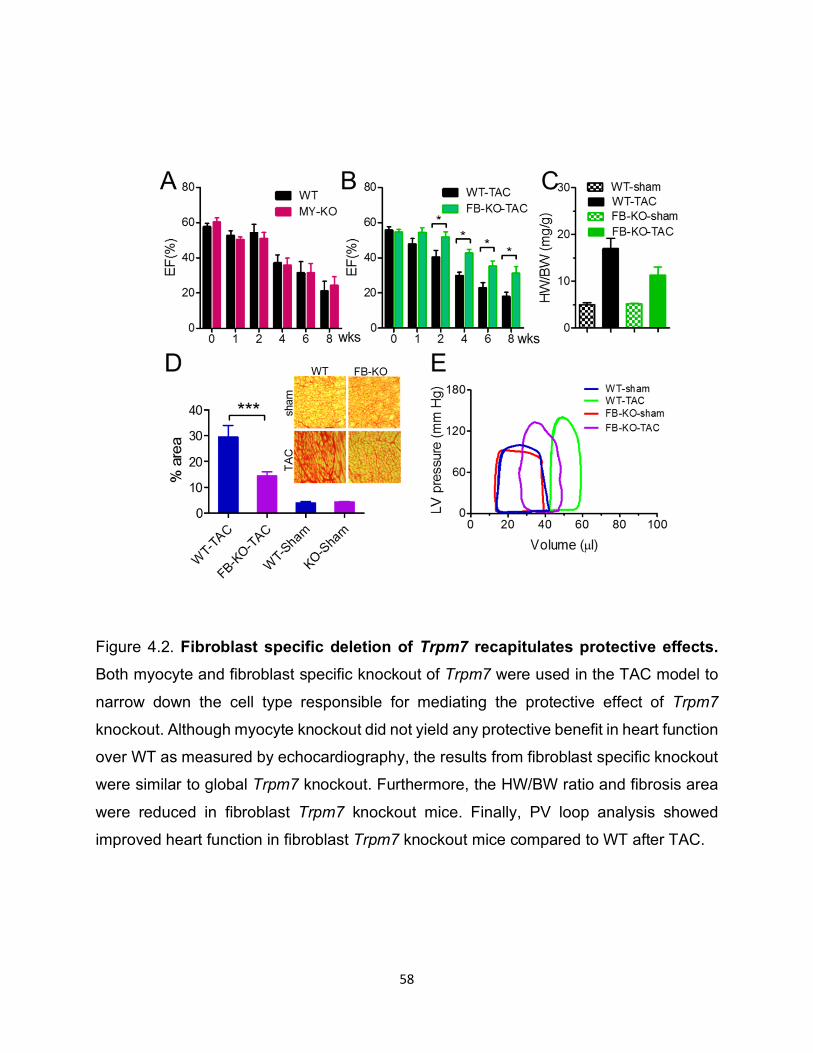
Figure 4.2. Fibroblast specific deletion of Trpm7 recapitulates protective effects. Both myocyte and fibroblast specific knockout of Trpm7 were used in the TAC model to
narrow down the cell type responsible for mediating the protective effect of Trpm7
knockout. Although myocyte knockout did not yield any protective benefit in heart function
over WT as measured by echocardiography, the results from fibroblast specific knockout
were similar to global Trpm7 knockout. Furthermore, the HW/BW ratio and fibrosis area
were reduced in fibroblast Trpm7 knockout mice. Finally, PV loop analysis showed
improved heart function in fibroblast Trpm7 knockout mice compared to WT after TAC.
59
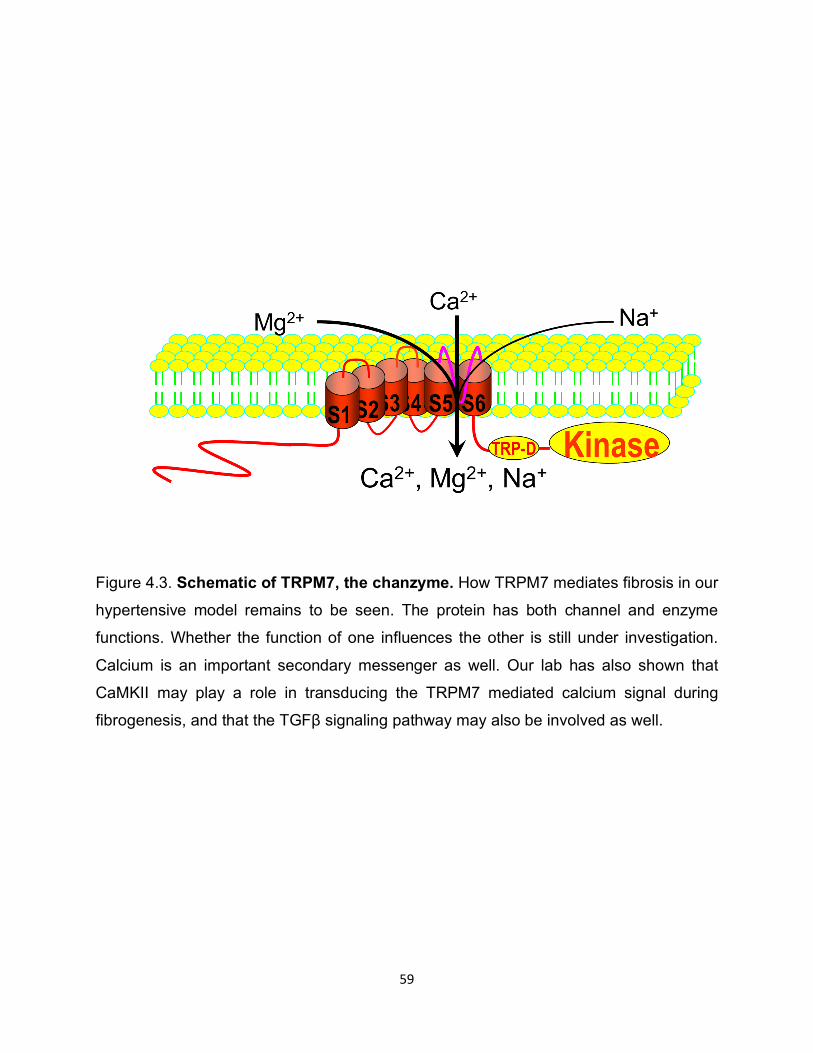
Figure 4.3. Schematic of TRPM7, the chanzyme. How TRPM7 mediates fibrosis in our
hypertensive model remains to be seen. The protein has both channel and enzyme
functions. Whether the function of one influences the other is still under investigation.
Calcium is an important secondary messenger as well. Our lab has also shown that
CaMKII may play a role in transducing the TRPM7 mediated calcium signal during
fibrogenesis, and that the TGFβ signaling pathway may also be involved as well.

60
CHAPTER 5: pH REGULATION OF THE GATEKEEPER, ORAI

61
Introduction
Calcium signaling is crucial in a variety of physiological and pathophysiological
processes, and thus its regulation is of critical importance. It is known that calcium release
activated calcium channel currents (ICRAC) are altered by conditions such as acidosis and
alkalization. Physiologically, intracellular alkalization is associated with membrane
depolarization, cell proliferation, and cellular differentiation, while intracellular acidosis
has been shown to be involved in apoptosis 66 67 68. Extracellular acidosis, which occurs
in settings such as myocardial ischemia, has been shown to inhibit ion channel activities
69 70. Thus, the question remains as to how pH is sensed by ion channels to induce
changes in the cell.
Similar to other channels, Icrac is inhibited and potentiated by acidic and basic pH,
respectively. ICRAC is composed of the Orai and STIM subunits, which act as the pore
forming and calcium sensing units respectively 71 72. Thus, pH may influence how these
two subunits bind, or may directly influence the activities of these proteins.
Here we show that acidosis and alkalosis alter Orai1/STIM channel functions. We
generated candidate mutations on transmembrane domain 3 and found that E106 is
responsible for pHo sensitivity when calcium is the permeant ion. We also identified E190
of Orai1 as the major sensor of pHo when sodium is the permeant ion. In addition, we
found H155 on the intracellular loop is responsible for sensing intracellular pH. Overall,
we show that these residues play a critical role in sensing pH to mediate acid/base
induced changes in channel activity.

62
Results
Effects of extracellular pH on Orai1/STIM1 currents
Orai1/STIM1 currents were recorded by inducing store depletion through EGTA.
The cells were perfused with extracellular DVF solutions adjusted to various pHs.
Orail1/STIM1 currents were significantly increased when the cell was exposed to high
pHo. On the other hand, alkaline pHo inhibited current amplitude (Figure 5.1). As shown
in Figure 5.1, the effects of pH were reversible. Furthermore, we tested the effects of pH
using Tyrode’s solution. Figure 5.1 shows that acidic pHo inhibited whereas basic pHo
enhanced Orai1/STIM1 current.
Effects of external pH on Orai2/STIM and Orai3/STIM
We also tested whether other Orai2 and Orai3 were similarly affected by pH. As
shown in Figure 5.2, both Orai2 and Orai3 currents were potentiated by external basic
pHo and inhibited by acidic pHo. These results indicated that all isoforms are capable of
responding to external pH cues, and that the pH sensor may reside in an Orai residue.
Mechanism of extracellular pH regulation
We first tested whether external protons directly regulated Orai1/STIM1. We
generated candidate mutations by neutralizing a series of negatively charged residues
along the external site of the channel or channel pore, in addition to E106D and E190D.
Interestingly the mutant E190D displayed smaller inhibition at acidic pHo and significantly
reduced potentiation at basic pHo compared to wild type when the cells were perfused
with DVF. Furthermore, the dose-response curve of E190D was shifted to the left,
indicating that E190 is essential for sensing extracellular pH sensitivity (Figure 5.3).
Effects of internal pH on Orai1/STIM1

63
We also tested how intracellular pH influence Orai1/STIM1 currents. We adjusted
intracellular pipette conditions to various acidic and basic pH, followed by measurement
of current amplitude. Similar to what we observed with external pH, we saw that acidic
internal pH reduced current amplitude as measured by patch clamp electrophysiology,
whereas basic pH potentiated current amplitude (Figure 5.4).
Molecular mechanisms of internal pH sensitivity of Orai1/STIM1
To identify the residues responsible for sensing internal pH, we generated a series
of mutations at candidate pH sensing residues including histidine, glutamic acid, and
cysteine. One of our candidates, H155F, showed poor response to both acidic and basic
internal pH, suggesting that this residue on Orai1 may be responsible to sensing
intracellular pH (Figure 5.5).
Discussion
Here we show that Orai/STIM1 channels involved in ICRAC are regulated by both
internal and external pH. Furthermore, we identify the residues responsible for detecting
both intracellular and extracellular pH sensitivity. In addition, the residues involved in
detecting extracellular pH vary by the type of permeant cation. We tested how
extracellular Tyrode’s solution with calcium as the permeant ion may alter pH sensitivity.
We found that the mutant E190D had similar pH sensitivity to wild type, but the mutant
E106D lost sensitivity (Figure 5.6). This suggests that E106 is the major pH sensor when
calcium is the major permeant ion. As pH fluctuates during physiological and
pathophysiological processes, how pH is sensed and how it then influences channel
activity and calcium dynamics will yield important insight into tackling diseases in which
pH changes occur 73.

64
Methods
Molecular biology.
The constructs Orai1-3 and STIM1 were purchased from Addgene. Mutations of Orai1
were performed using site directed mutagenesis (QuickChange).
Cell culture.
HEK 293 cells were grown in DMEM/F12 media supplemented with 10% FBS and
100U/ml penicillin and 100mg/ml streptomycin. The cells were incubated in a 37°C
humidity controlled incubator at 5% CO2. Plasmids were transfected using Lipofectamine
2000.
Electrophysiology.
Whole-cell currents were recorded using an Axopatch 200B amplifier. Data were digitized
at 10 or 20kHz and digitally filtered offline at 1kHz. Borosilicate glass electrodes that were
heat polished to a resistance of 5-7 MΩ were used. Series resistance was compensated
up to 90% to reduce series resistance errors. For whole cell recordings, voltage stimuli
lasting 200 ms were delivered at 1s intervals with voltage ramps ranging from -120 to
+100mV. A holding potential of 0 mV was used. The perfusion and vacuum system
allowed efficient exchange of extracellular solution.
The internal pipette solution consisted of (in mM) 145 Cs-methanesulfonate, 8 NaCl, 10
EGTA and 20 HEPES, with pH adjusted to 7.2 with CsOH. MgCl2 (3mM) was included in
the pipette solution to block any endogenous current. pH was adjusted using citrate acid
and CsOH.
Extracellular solution consisted of divalent-free solution (DVF) containing (in mM) 145 Na
SO3CH3, 20 HEPES, 5 EGTA, 2 EDTA and 10 glucose. Tyrode’s solution contained (in

65
mM) 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose. HCl was used to
adjust to acidic pH and NMDG was used to adjust to basic pH.
Data analysis.
Pooled data are presented in mean +/- SEM. Dose–response curves were fitted by an
equation of the form: E= Emax (1/[1+(EC50/C)n]), where E is the effect at concentration C,
Emax is maximal effect, EC50 is the concentration for half-maximal effect and n is the Hill
coefficient. Statistical comparisons were made using analysis of variance (ANOVA) and
two tailed t-test with Bonferroni correction. p<0.05 indicates statistical significance.
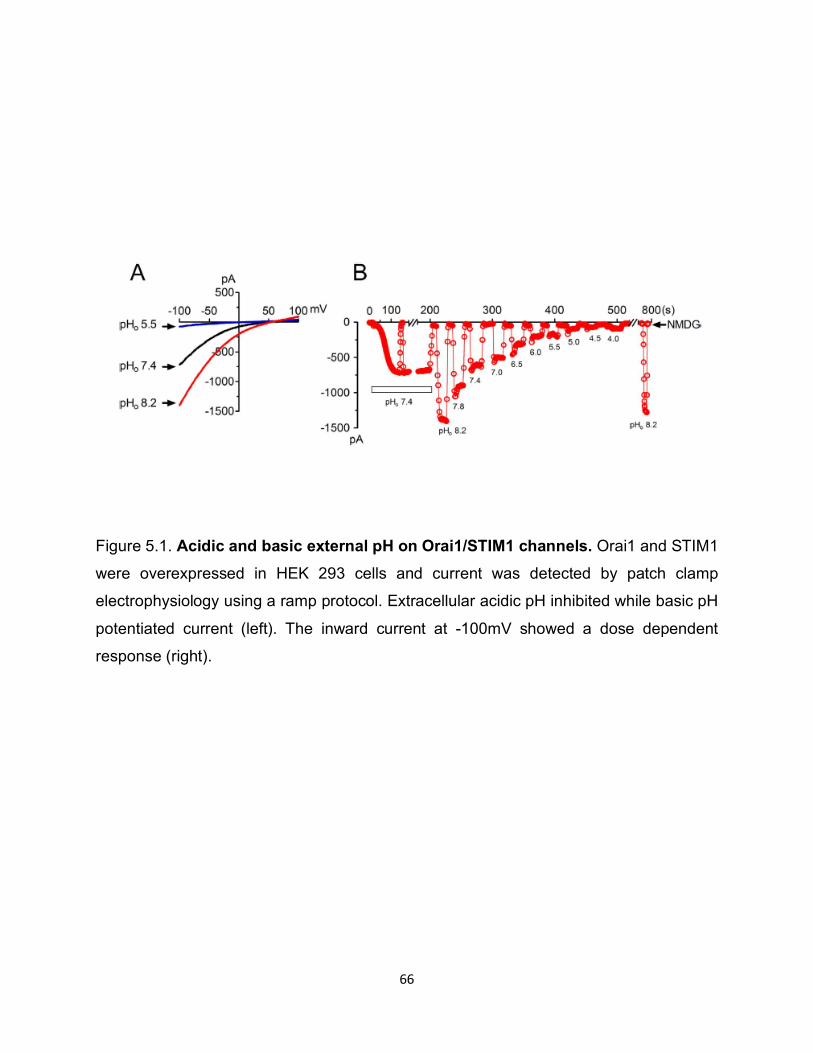
66
Figure 5.1. Acidic and basic external pH on Orai1/STIM1 channels. Orai1 and STIM1
were overexpressed in HEK 293 cells and current was detected by patch clamp
electrophysiology using a ramp protocol. Extracellular acidic pH inhibited while basic pH
potentiated current (left). The inward current at -100mV showed a dose dependent
response (right).
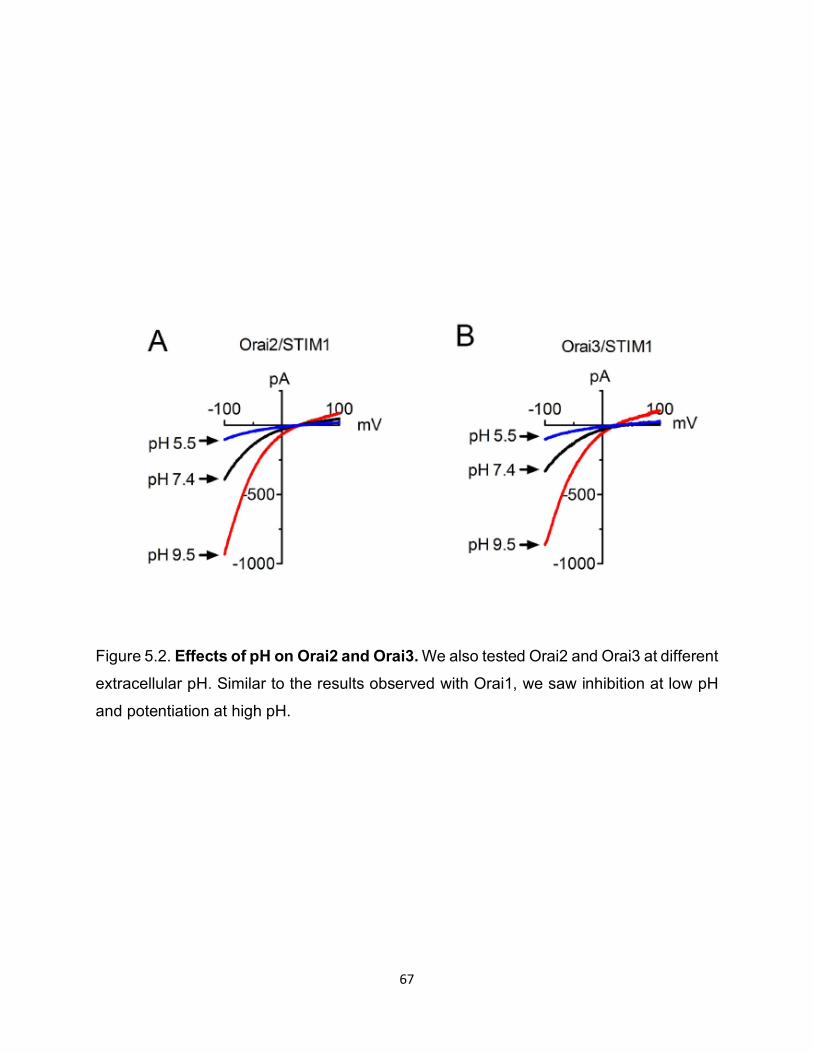
67
Figure 5.2. Effects of pH on Orai2 and Orai3. We also tested Orai2 and Orai3 at different
extracellular pH. Similar to the results observed with Orai1, we saw inhibition at low pH
and potentiation at high pH.
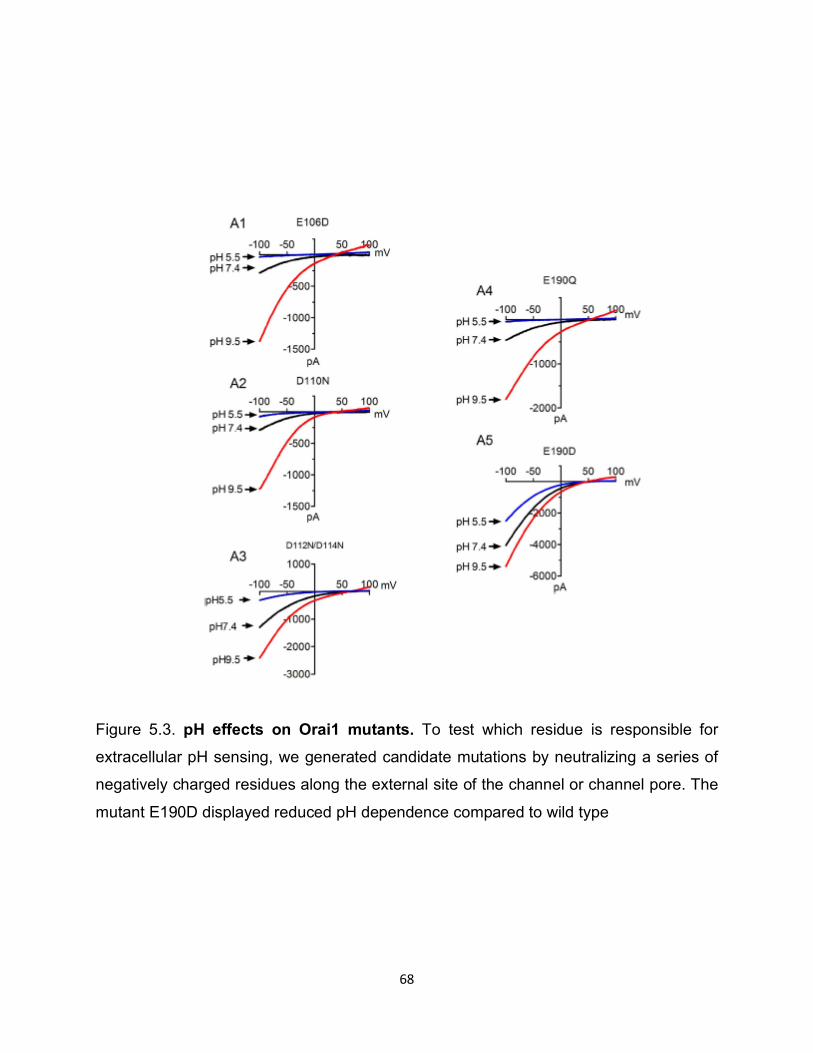
68
Figure 5.3. pH effects on Orai1 mutants. To test which residue is responsible for
extracellular pH sensing, we generated candidate mutations by neutralizing a series of
negatively charged residues along the external site of the channel or channel pore. The
mutant E190D displayed reduced pH dependence compared to wild type
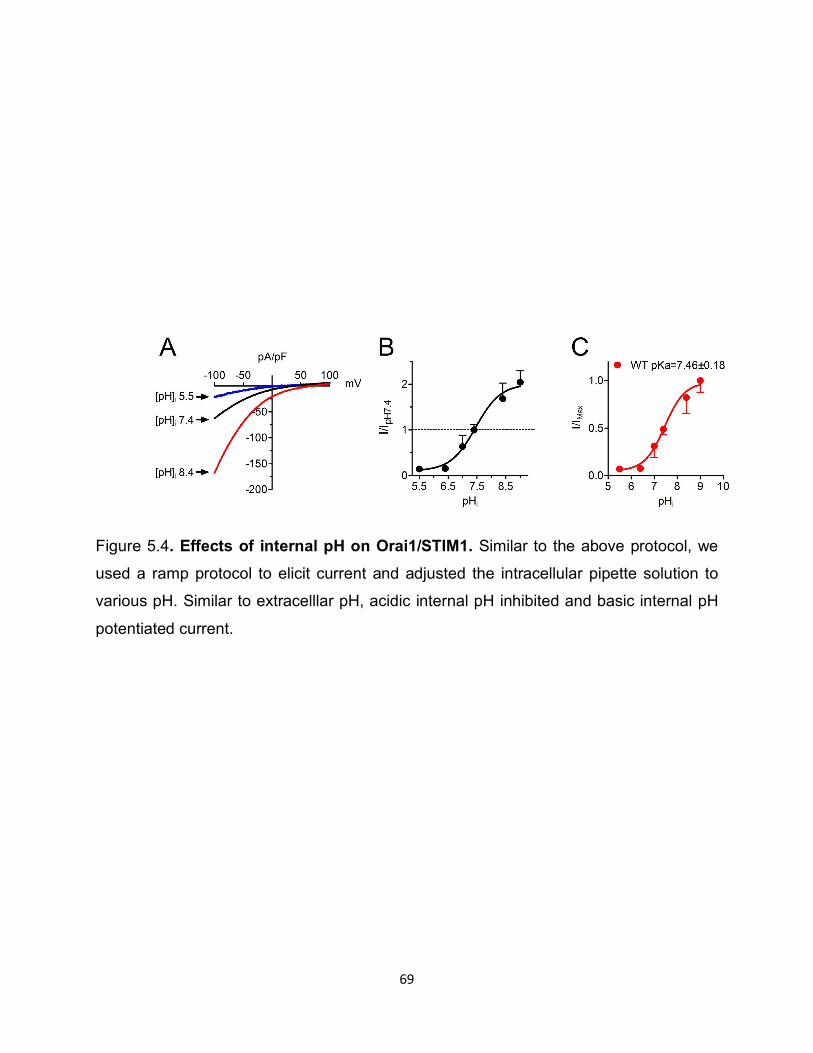
69
Figure 5.4. Effects of internal pH on Orai1/STIM1. Similar to the above protocol, we
used a ramp protocol to elicit current and adjusted the intracellular pipette solution to
various pH. Similar to extracelllar pH, acidic internal pH inhibited and basic internal pH
potentiated current.
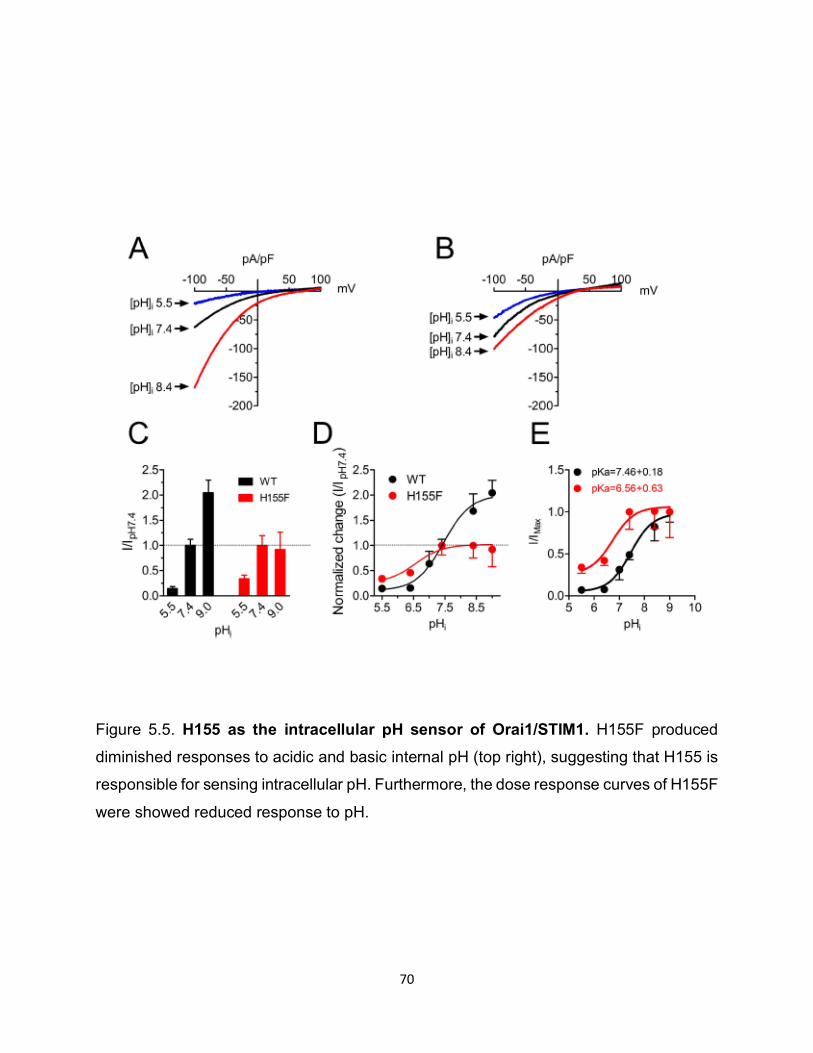
70
Figure 5.5. H155 as the intracellular pH sensor of Orai1/STIM1. H155F produced
diminished responses to acidic and basic internal pH (top right), suggesting that H155 is
responsible for sensing intracellular pH. Furthermore, the dose response curves of H155F
were showed reduced response to pH.
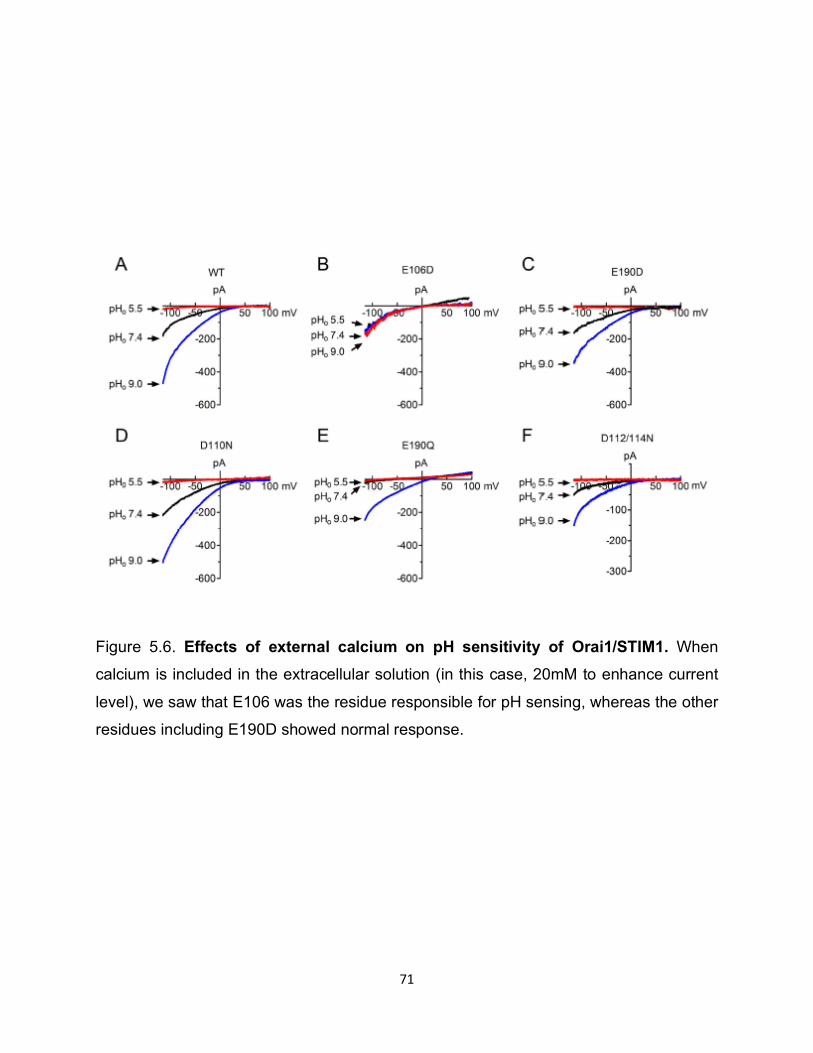
71
Figure 5.6. Effects of external calcium on pH sensitivity of Orai1/STIM1. When
calcium is included in the extracellular solution (in this case, 20mM to enhance current
level), we saw that E106 was the residue responsible for pH sensing, whereas the other
residues including E190D showed normal response.

72
CHAPTER 6:
CONCLUSIONS, DISCUSSION, AND THE FUTURE TR(I)PS

73
Since its initial discovery in Drosophila, TRP channels have become to be viewed
as versatile ion channels with diverse functions. Conserved throughout species and
present ubiquitously in almost every cell type, TRP channels mediate activities ranging
from sensation to development to cancer. These unique channels have distinct N and C
termini that confer the ability to respond to many different stimuli. None of the channels
in the family are more unique than TRPM7. As a bifunctional channel enzyme, the protein
serves as both an ion channel and an enzyme. However, the exact nature of how the ion
channel and enzyme are related in function has yet to be fully uncovered. Here we have
shown this unique channel-enzyme can be regulated by membrane potential. Specifically,
low voltage has the ability to reduce TRPM7 activity. Our results suggest that this effect
is due to a depletion in level or availability of PIP2 to TRPM7. This leads us to believe that
there may be a voltage sensitive enzyme not dissimilar to that of Ci-VSP that may be
regulating PIP2 levels at different voltages. What the specific mechanism of voltage
sensing in mammalian cells is will be exciting to see, and an avenue that our lab will
continue to pursue.
Furthermore, TRP channels have recently been identified as important players in
cardiovascular disease. TRPM2 is a well-known oxidative stress sensitive ion channel
that has been thought to mediate intercellular communication through the release of
cytokines and chemokines. We have shown that TRPM2 is involved in the atherosclerotic
disease process, as deletion of Trpm2 is protective in mouse atherosclerosis models. We
observe that mice with long term atherosclerosis have increased levels of circulating
neutrophils, and this is reversed in Trpm2 knockout mice. This highly suggests that
TRPM2 plays an essential role in mediating myeloid and neutrophil cells, though the exact

74
mechanism has not been elucidated. We have generated specific knockouts of Trpm2 in
various tissues such as the myeloid cells and endothelial cells to help us answer this
important question.
Finally, we show one of the roles of TRPM7 as a mediator of cardiac fibrosis. Our
lab has previously shown the role of TRPM7 in the differentiation of cardiac fibroblasts to
myofibroblasts. Here we show that TRPM7 in vivo can mediate fibrosis in the setting of
hypertensive heart failure. We see that deletion of Trpm7 can have a protective effect on
the heart as early as one week after induction of hypertension, which is maintained
throughout the life of the animal. Furthermore, the function of TRPM7 is primarily in the
cardiac fibroblast, as fibroblast specific knockout recapitulates the protective effects
observed in global Trpm7 knockout. The next question we will tackle is how TRPM7
mediates fibrosis and whether the chanzyme properties of the protein are utilized if at all
in this process.
Mainly considered channels important in sensory perception, the potential roles of
TRP channels in physiology and pathophysiology are now thought to be limitless. As
complex ion channels that are ubiquitous and evolutionarily conserved, we are beginning
to appreciate more and more the diverse biological roles of these channels. Similarly, as
parallel studies investigate how these channels are endogenously regulated, more insight
will be gained into their biological roles. Recent papers that have published the structure
of various TRP channels. The understanding of structural conformation will also prove to
be invaluable in the understanding of the physiological and pathophysiological roles of
TRP channels not only in cardiovascular disease but also in other diseases.

75
REFERENCES

76
1. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S,
Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage
M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman
JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB,
Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey A,
Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD,
Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW,
Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P,
American Heart Association Council on E, Prevention Statistics C, Stroke Statistics
S. Heart disease and stroke statistics-2018 update: A report from the american
heart association. Circulation. 2018;137:e67-e492
2. Schuett KA, Lehrke M, Marx N, Burgmaier M. High-risk cardiovascular patients:
Clinical features, comorbidities, and interconnecting mechanisms. Frontiers in
immunology. 2015;6:591
3. Libby P. Inflammation in atherosclerosis. Arteriosclerosis, thrombosis, and
vascular biology. 2012;32:2045-2051
4. Strong JP, Malcom GT, McMahan CA, Tracy RE, Newman WP, 3rd, Herderick EE,
Cornhill JF. Prevalence and extent of atherosclerosis in adolescents and young
adults: Implications for prevention from the pathobiological determinants of
atherosclerosis in youth study. Jama. 1999;281:727-735
5. Weber C, Noels H. Atherosclerosis: Current pathogenesis and therapeutic options.
Nature medicine. 2011;17:1410-1422

77
6. Lu H, Daugherty A. Atherosclerosis. Arteriosclerosis, thrombosis, and vascular
biology. 2015;35:485-491
7. Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis.
Circulation research. 2014;114:1757-1771
8. Leitinger N, Schulman IG. Phenotypic polarization of macrophages in
atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1120-
1126
9. Viola J, Soehnlein O. Atherosclerosis - a matter of unresolved inflammation.
Seminars in immunology. 2015;27:184-193
10. Wong BW, Meredith A, Lin D, McManus BM. The biological role of inflammation in
atherosclerosis. The Canadian journal of cardiology. 2012;28:631-641
11. Escarcega RO, Lipinski MJ, Garcia-Carrasco M, Mendoza-Pinto C, Galvez-
Romero JL, Cervera R. Inflammation and atherosclerosis: Cardiovascular
evaluation in patients with autoimmune diseases. Autoimmunity reviews. 2018
12. Libby P, Ridker PM, Hansson GK, Leducq Transatlantic Network on A.
Inflammation in atherosclerosis: From pathophysiology to practice. Journal of the
American College of Cardiology. 2009;54:2129-2138
13. Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR,
Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M.
Targeting interleukin-1beta reduces leukocyte production after acute myocardial
infarction. Circulation. 2015;132:1880-1890

78
14. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis
of cantos and beyond. Journal of the American College of Cardiology.
2017;70:2278-2289
15. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of
cytokines in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology.
2011;31:969-979
16. Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery
disease and atherosclerosis: Where are we now? Journal of the American College
of Cardiology. 2013;62:1541-1551
17. Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby
P, Pittet M, Weissleder R, Nahrendorf M. Impaired infarct healing in atherosclerotic
mice with ly-6c(hi) monocytosis. Journal of the American College of Cardiology.
2010;55:1629-1638
18. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL,
Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes
two monocyte subsets with divergent and complementary functions. The Journal
of experimental medicine. 2007;204:3037-3047
19. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y,
Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M,
Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HW, Piek JJ,
Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS,
Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R,

79
Nahrendorf M. Myocardial infarction accelerates atherosclerosis. Nature.
2012;487:325-329
20. Catterall WA, Raman IM, Robinson HP, Sejnowski TJ, Paulsen O. The hodgkin-
huxley heritage: From channels to circuits. The Journal of neuroscience : the
official journal of the Society for Neuroscience. 2012;32:14064-14073
21. Fierro L, Parekh AB. Substantial depletion of the intracellular ca2+ stores is
required for macroscopic activation of the ca2+ release-activated ca2+ current in
rat basophilic leukaemia cells. The Journal of physiology. 2000;522 Pt 2:247-257
22. Cosens DJ, Manning A. Abnormal electroretinogram from a drosophila mutant.
Nature. 1969;224:285-287
23. Montell C, Jones K, Hafen E, Rubin G. Rescue of the drosophila phototransduction
mutation trp by germline transformation. Science. 1985;230:1040-1043
24. Clapham DE, Runnels LW, Strubing C. The trp ion channel family. Nature reviews.
Neuroscience. 2001;2:387-396
25. Clapham DE. Trp channels as cellular sensors. Nature. 2003;426:517-524
26. Runnels LW, Yue L, Clapham DE. Trp-plik, a bifunctional protein with kinase and
ion channel activities. Science. 2001;291:1043-1047
27. Suh BC, Hille B. Pip2 is a necessary cofactor for ion channel function: How and
why? Annual review of biophysics. 2008;37:175-195
28. Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide
phosphatase activity coupled to an intrinsic voltage sensor. Nature.
2005;435:1239-1243

80
29. Walker SM, Downes CP, Leslie NR. Tpip: A novel phosphoinositide 3-
phosphatase. The Biochemical journal. 2001;360:277-283
30. Tapparel C, Reymond A, Girardet C, Guillou L, Lyle R, Lamon C, Hutter P,
Antonarakis SE. The tpte gene family: Cellular expression, subcellular localization
and alternative splicing. Gene. 2003;323:189-199
31. Hansen SB, Tao X, MacKinnon R. Structural basis of pip2 activation of the
classical inward rectifier k+ channel kir2.2. Nature. 2011;477:495-498
32. Hilgemann DW, Ball R. Regulation of cardiac na+,ca2+ exchange and katp
potassium channels by pip2. Science. 1996;273:956-959
33. Voets T, Nilius B. Modulation of trps by pips. The Journal of physiology.
2007;582:939-944
34. Zheng W, Cai R, Hofmann L, Nesin V, Hu Q, Long W, Fatehi M, Liu X, Hussein S,
Kong T, Li J, Light PE, Tang J, Flockerzi V, Tsiokas L, Chen XZ. Direct binding
between pre-s1 and trp-like domains in trpp channels mediates gating and
functional regulation by pip2. Cell reports. 2018;22:1560-1573
35. Leung HT, Tseng-Crank J, Kim E, Mahapatra C, Shino S, Zhou Y, An L, Doerge
RW, Pak WL. Dag lipase activity is necessary for trp channel regulation in
drosophila photoreceptors. Neuron. 2008;58:884-896
36. Hardie RC, Raghu P, Moore S, Juusola M, Baines RA, Sweeney ST. Calcium influx
via trp channels is required to maintain pip2 levels in drosophila photoreceptors.
Neuron. 2001;30:149-159
37. Runnels LW, Yue L, Clapham DE. The trpm7 channel is inactivated by pip(2)
hydrolysis. Nature cell biology. 2002;4:329-336

81
38. Rohacs T, Lopes CM, Michailidis I, Logothetis DE. Pi(4,5)p2 regulates the
activation and desensitization of trpm8 channels through the trp domain. Nature
neuroscience. 2005;8:626-634
39. Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, Tepper RI,
Shyjan AW. Down-regulation of the novel gene melastatin correlates with potential
for melanoma metastasis. Cancer research. 1998;58:1515-1520
40. Visser D, Middelbeek J, van Leeuwen FN, Jalink K. Function and regulation of the
channel-kinase trpm7 in health and disease. European journal of cell biology.
2014;93:455-465
41. Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T,
Fleig A, Scharenberg AM. Regulation of vertebrate cellular mg2+ homeostasis by
trpm7. Cell. 2003;114:191-200
42. Xie J, Sun B, Du J, Yang W, Chen HC, Overton JD, Runnels LW, Yue L.
Phosphatidylinositol 4,5-bisphosphate (pip(2)) controls magnesium gatekeeper
trpm6 activity. Scientific reports. 2011;1:146
43. Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of
trpm7 disrupts embryonic development and thymopoiesis without altering mg2+
homeostasis. Science. 2008;322:756-760
44. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T,
Kinet JP, Penner R, Scharenberg AM, Fleig A. Ltrpc7 is a mg.Atp-regulated
divalent cation channel required for cell viability. Nature. 2001;411:590-595
45. Stritt S, Nurden P, Favier R, Favier M, Ferioli S, Gotru SK, van Eeuwijk JM,
Schulze H, Nurden AT, Lambert MP, Turro E, Burger-Stritt S, Matsushita M,

82
Mittermeier L, Ballerini P, Zierler S, Laffan MA, Chubanov V, Gudermann T,
Nieswandt B, Braun A. Defects in trpm7 channel function deregulate
thrombopoiesis through altered cellular mg(2+) homeostasis and cytoskeletal
architecture. Nature communications. 2016;7:11097
46. Gotru SK, Chen W, Kraft P, Becker IC, Wolf K, Stritt S, Zierler S, Hermanns HM,
Rao D, Perraud AL, Schmitz C, Zahedi RP, Noy PJ, Tomlinson MG, Dandekar T,
Matsushita M, Chubanov V, Gudermann T, Stoll G, Nieswandt B, Braun A. Trpm7
kinase controls calcium responses in arterial thrombosis and stroke in mice.
Arteriosclerosis, thrombosis, and vascular biology. 2018;38:344-352
47. Sah R, Mesirca P, Van den Boogert M, Rosen J, Mably J, Mangoni ME, Clapham
DE. Ion channel-kinase trpm7 is required for maintaining cardiac automaticity.
Proceedings of the National Academy of Sciences of the United States of America.
2013;110:E3037-3046
48. Yu H, Zhang Z, Lis A, Penner R, Fleig A. Trpm7 is regulated by halides through its
kinase domain. Cellular and molecular life sciences : CMLS. 2013;70:2757-2771
49. Bilkova E, Pleskot R, Rissanen S, Sun S, Czogalla A, Cwiklik L, Rog T, Vattulainen
I, Cremer PS, Jungwirth P, Coskun U. Calcium directly regulates
phosphatidylinositol 4,5-bisphosphate headgroup conformation and recognition.
Journal of the American Chemical Society. 2017;139:4019-4024
50. Seo JB, Jung SR, Huang W, Zhang Q, Koh DS. Charge shielding of pip2 by cations
regulates enzyme activity of phospholipase c. PloS one. 2015;10:e0144432
51. Zhou Y, Wong CO, Cho KJ, van der Hoeven D, Liang H, Thakur DP, Luo J, Babic
M, Zinsmaier KE, Zhu MX, Hu H, Venkatachalam K, Hancock JF. Signal

83
transduction. Membrane potential modulates plasma membrane phospholipid
dynamics and k-ras signaling. Science. 2015;349:873-876
52. Hilton JK, Salehpour T, Sisco NJ, Rath P, Van Horn WD. Phosphoinositide-
interacting regulator of trp (pirt) has opposing effects on human and mouse trpm8
ion channels. The Journal of biological chemistry. 2018
53. Soeki T, Sata M. Inflammatory biomarkers and atherosclerosis. International heart
journal. 2016;57:134-139
54. Schlitt A, Heine GH, Blankenberg S, Espinola-Klein C, Dopheide JF, Bickel C,
Lackner KJ, Iz M, Meyer J, Darius H, Rupprecht HJ. Cd14+cd16+ monocytes in
coronary artery disease and their relationship to serum tnf-alpha levels.
Thrombosis and haemostasis. 2004;92:419-424
55. Vasquez EC, Peotta VA, Gava AL, Pereira TM, Meyrelles SS. Cardiac and
vascular phenotypes in the apolipoprotein e-deficient mouse. Journal of
biomedical science. 2012;19:22
56. Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y. Roles of trpm2 in oxidative
stress. Cell calcium. 2011;50:279-287
57. Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T,
Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M,
Takeshima H, Mori Y. Trpm2-mediated ca2+influx induces chemokine production
in monocytes that aggravates inflammatory neutrophil infiltration. Nature medicine.
2008;14:738-747

84
58. Hiroi T, Wajima T, Negoro T, Ishii M, Nakano Y, Kiuchi Y, Mori Y, Shimizu S.
Neutrophil trpm2 channels are implicated in the exacerbation of myocardial
ischaemia/reperfusion injury. Cardiovascular research. 2013;97:271-281
59. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L. Trpm2 links
oxidative stress to nlrp3 inflammasome activation. Nature communications.
2013;4:1611
60. Tan CH, McNaughton PA. The trpm2 ion channel is required for sensitivity to
warmth. Nature. 2016;536:460-463
61. Kolk MV, Meyberg D, Deuse T, Tang-Quan KR, Robbins RC, Reichenspurner H,
Schrepfer S. Lad-ligation: A murine model of myocardial infarction. Journal of
visualized experiments : JoVE. 2009
62. Deb A, Ubil E. Cardiac fibroblast in development and wound healing. Journal of
molecular and cellular cardiology. 2014;70:47-55
63. Xu T, Wu BM, Yao HW, Meng XM, Huang C, Ni MM, Li J. Novel insights into trpm7
function in fibrotic diseases: A potential therapeutic target. Journal of cellular
physiology. 2015;230:1163-1169
64. Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. Trpm7-
mediated ca2+ signals confer fibrogenesis in human atrial fibrillation. Circulation
research. 2010;106:992-1003
65. deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice.
Journal of visualized experiments : JoVE. 2010
66. Webb DJ, Nuccitelli R. Direct measurement of intracellular ph changes in xenopus
eggs at fertilization and cleavage. The Journal of cell biology. 1981;91:562-567

85
67. Lagadic-Gossmann D, Huc L, Lecureur V. Alterations of intracellular ph
homeostasis in apoptosis: Origins and roles. Cell death and differentiation.
2004;11:953-961
68. Fujita F, Uchida K, Moriyama T, Shima A, Shibasaki K, Inada H, Sokabe T,
Tominaga M. Intracellular alkalization causes pain sensation through activation of
trpa1 in mice. The Journal of clinical investigation. 2008;118:4049-4057
69. Lardner A. The effects of extracellular ph on immune function. Journal of leukocyte
biology. 2001;69:522-530
70. Marumo M, Suehiro A, Kakishita E, Groschner K, Wakabayashi I. Extracellular ph
affects platelet aggregation associated with modulation of store-operated ca(2+)
entry. Thrombosis research. 2001;104:353-360
71. Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential
pore subunit of the crac channel. Nature. 2006;443:230-233
72. Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz
T, Garcia KC, Dolmetsch RE, Lewis RS. Stim1 clusters and activates crac
channels via direct binding of a cytosolic domain to orai1. Cell. 2009;136:876-890
73. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG,
Lewis RS, Daly M, Rao A. A mutation in orai1 causes immune deficiency by
abrogating crac channel function. Nature. 2006;441:179-185
















![Drosophila TRP and TRPL are assembled as homomultimeric ... · Seifert, 2002; MacKinnon, 1991)], TRP channels are most likely formed by tetramers of the pore-forming subunits. Given](https://img.pdfslide.us/doc/110x75/6046e4d6b30e3452b7097508/drosophila-trp-and-trpl-are-assembled-as-homomultimeric-seifert-2002-mackinnon.jpg)











