Embed Size (px)
Citation preview

Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
Treacher Collins Syndrome: Protocol ManagementFrom Birth to Maturity
James T. Thompson, MD,* Peter J. Anderson, FRACS,Þ and David J. David, AC, FRACSÞ
Background: Management of patients with Treacher Collins syn-drome is complicated and involves multiple disciplines working inconcert to achieve a common outcome. This article reviews theexperience at the Australian Craniofacial Unit and describes theprotocol for management.Methods: Fifty patients were treated during the last 30 years. Therecords of these patients were reviewed to establish what interven-tions they required and how these fit into a protocol for management.Results: The protocol for management of Treacher Collins syn-drome can be divided into 3 epochs. In the first epoch from birthto age 2, airway and feeding problems were the main focus. Fourpatients required tracheostomy. Of these, 1 died and the others re-ceived mandibular distraction. Hearing is evaluated and addressedearly. Eleven patients (23%) required repair of a cleft palate. In thesecond epoch (aged 2Y12 y), speech therapy is critical as is a focuson integrating into the education system. During this epoch, recon-struction of the upper face was performed either with bone graftsor with vascularized bone flaps. Both required repeat bone graftslater. In the third epoch (aged 13Y18 y), orthognathic surgery wasperformed. Revision surgery and further bone grafting were per-formed again at around age 18. Patients reported being generallyhappy with their appearance and with few exceptions were able tocomplete education, gain employment, and feel socially accepted.Conclusions: Management of patients with Treacher Collins syn-drome should be through a multidisciplinary protocol to achievegood results while minimizing confusion and unnecessary surgery.
Key Words: Treacher Collins, protocol management,multidisciplinary
(J Craniofac Surg 2009;20: 2028Y2035)
Treacher Collins syndrome describes a complicated assortment ofcraniofacial malformations resulting in problems affecting the
form and function of the eyes, ears, nose, maxilla, palate, mandible,and airway. Although first described in 1889 by Berry,1 TreacherCollins’s2 later description in 1900 ultimately resulted in his epon-ymous association with the syndrome. The scientific nomenclaturemost commonly used is mandibulofacial dysostosis, although thisis not specific to Treacher Collins syndrome.
As a result of the complex assortment of deformities, it isdeemed essential that each patient receives comprehensive multi-disciplinary care for optimal outcomes. Because the incidence ofTreacher Collins is only 1 in 50,000 live births,3 much of the care ofthese patients is documented sporadically as clinical reports or de-scriptions of operative interventions by the various specialties in-volved. Therefore, the purpose of this review was to examine themanagement of patients with Treacher Collins syndrome during a30-year period from a broad perspective to determine what successesand failures occur along the way. Finally, this article will describehow best to organize the multiple disciplines involved and presenta protocol for future management and assessment of outcomes.
METHODSFifty patients were registered as patients at the Australian
Craniofacial Unit in Adelaide from 1975 to 2005. All patients had aconfirmed diagnosis of Treacher Collins syndrome by the cranio-facial team and clinical genetics. Patients were not included in thestudy if the diagnosis was uncertain. In addition, patients who wereseen but did not have adequate records were excluded. Patientrecords were examined for clinical features, interventions, andoutcomes.
RESULTSOverall, 50 patients were treated during the period reviewed.
Three of these patients had incomplete records and were notincluded in the study. Of the 47 included in the study, 23 were malesand 24 were females. Fourteen patients were treated from birth tomaturity; the remainder of the patients were evaluated and treatedeither at a later stage of childhood or as adults. One patient died ofsepticemia at the age of 1 year. This patient was a severely affectedindividual and had undergone tracheostomy but had received noother surgical treatment. Another patient died in adolescence in avehicle crash, making the syndrome-related childhood mortality 1 of47. Eight families had Treacher Collins syndrome, making up 20 ofthe patients seen, whereas 27 were new mutations.
The physical features and problems found in this patientgroup are listed in Table 1 and are generally consistent with otherreports.3 The microform of Treacher Collins is often difficult todetect. In several of the families in our series, it was not evident that1 of the parents was affected until a second affected child was born.Based on our experience, the subtle evidence of deformity in themicroform state is most evident around the zygoma with very mild
ORIGINAL ARTICLE
2028 The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009
From the *Department of Plastic and Reconstructive Surgery, Wake ForestUniversity School of Medicine, Winston-Salem, North Carolina; and†Australian Craniofacial Unit, Women’s and Children’s Hospital, in asso-ciation with the University of Adelaide, Adelaide, Australia.Received June 3, 2009.Accepted for publication June 29, 2009.Address correspondence and reprint requests to James T. Thompson, MD,
Department of Plastic and Reconstructive Surgery, Wake ForestUniversity School of Medicine, Winston-Salem, NC, USA; E-mail:[email protected]
Presented at the Biennial International Congress of the International Societyof Craniofacial Surgery, August 2007, Salvador, Bahia, Brazil.
The authors have no financial disclosures to make regarding this article.Copyright * 2009 by Mutaz B. Habal, MDISSN: 1049-2275DOI: 10.1097/SCS.0b013e3181be8788

Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
downslanting palpebral fissures and depressions in the zygomaticbody (seen in the older patient in Fig. 1).
Airway ManagementPreviously, the airway management strategy used by this unit
for retrognathism was published.4 The focus is on managing theairway with positioning, nasopharyngeal airway, or positive pressuremasks, using tracheostomy only as a last resort. No neonatal di-straction or tongue-lip adhesions were performed. In this series,4 patients required tracheostomy during infancy. One patient diedat 1 year from septicemia related to a pulmonary infection and wasthe only patient to die in this series from a disease-related problem.Three were decannulated after mandibular distraction osteogenesishad been performed (Table 2). Two other patients required emer-gency tracheostomies later in childhood. One was a result of foodaspiration; and the other, an inability to intubate for elective surgeryby a different hospital that did not routinely care for children withcraniofacial deformities.
Airway problems can extend into childhood as well. Tenpatients were found to have obstructive sleep apnea during theirchildhood years. Of these patients, 2 had no previous airwayproblems. Tonsillectomy and adenoidectomy was performed in 7patients, and 4 patients required continuous positive airway pressure(CPAP) beyond age 10. Bimaxillary advancement performed duringthe teenage years resolved the problem for 2 of the 4 patients onCPAP, whereas the other 2 are still awaiting surgery.
GeneticsOnly 4 of the patients treated at this unit had formal genetic
testing because the diagnosis is usually clinically obvious andbecause of cost constraints. Of these 4, 2 were confirmed to have amutation in the TCOF1 gene. In all, 57% of the patients were con-sidered new mutations, whereas 43% were familial.
OphthalmologyPatients with Treacher Collins syndrome experience a variety
of periorbital and ophthalmological problems. In this series,downslanting palpebral fissures were noted in all 47 patients.Other deformities in decreasing incidence included absent mediallower lid eyelashes, lower lid lacrimal deformity, epiphora, lowerlid coloboma, vision impairment requiring treatment, and ptosis.Although cataracts have been described in the literature,5 none werefound in this series.
Absence of the lower lacrimal puncta lead to documentedepiphora in 20 patients. No patient with epiphora required treatmentother than routine eye care.
Ophthalmological problems included astigmatism, hyperme-tropia, and squint. Overall, 16 of the patients treated required eithercorrective lenses or surgery to improve their vision. Three patientswere noted to have amblyopia.
Ears/HearingAlmost all of the patients in this series had a conductive
hearing loss with only 1 patient having a mixed hearing loss. Allpatients were screened for hearing in infancy and then again duringchildhood when more formalized testing could be done. The hearingloss was categorized into normal (G20 dB), mild (20Y35 dB),moderate (35Y50 dB), and severe (950 dB) by audiometry that wasusually performed at age 3 and again around age 8. Of the 46patients who were tested for hearing, the distribution was as follows:normal (5 patients), mild (5 patients), moderate (6 patients), andsevere (30 patients). All patients with hearing loss were given bone-conduction hearing aids.
Nine patients required tympanostomy tubes including 1 of the11 patients with a palatal cleft. The remainder of the patients withcleft palate did not require tubes. One patient had a middle earreconstruction with a ceramic ossicular chain. She was reported ashaving 15-dB gain but continued to require hearing aids.
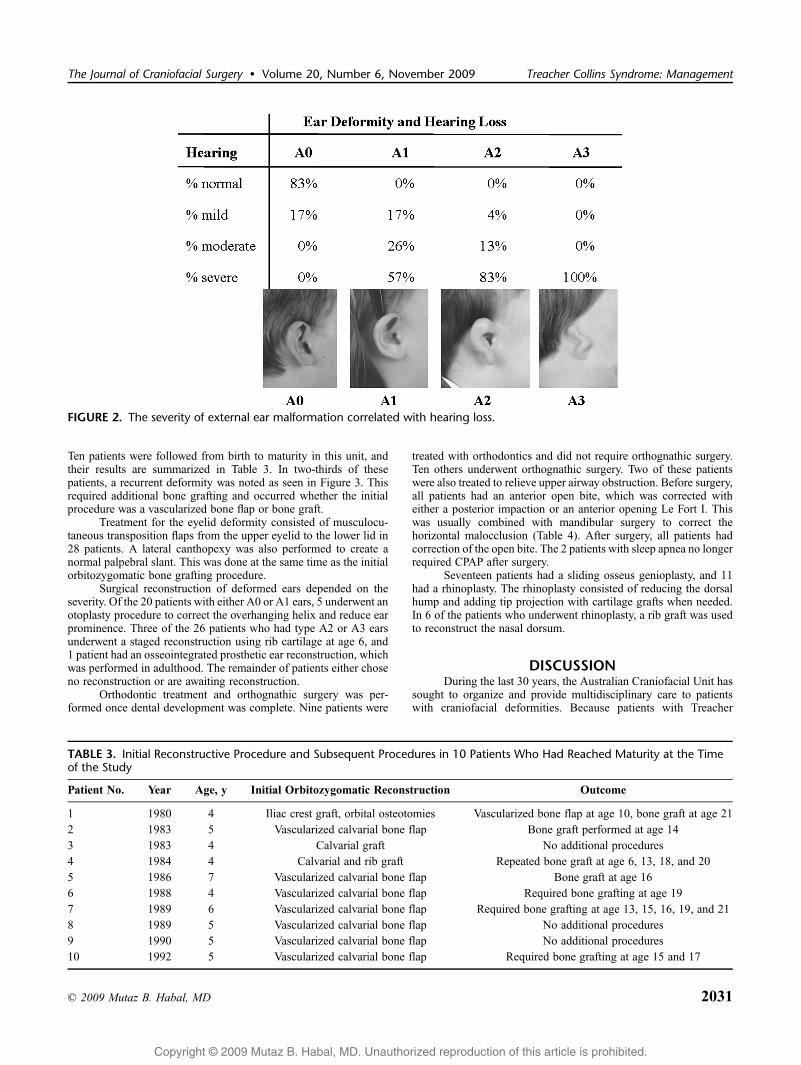
External ear appearance was classified using the sameauricular deformity classification previously published for hemi-facial microsomia.6 Under this system, a normal ear is labeled A0, asmall ear with malformation retaining the characteristic features islabeled A1, an ear with a rudimentary helix is labeled A2, and anabsent pinna with a malformed lobule is labeled A3. Auriculardeformity was present in 87% of the patients. The degree of hearingimpairment was noted to correlate with external ear deformity asnoted in Figure 2.
SpeechSpeech abnormalities affected 34 of 46 patients evaluated.
Typical speech problems included abnormal resonance dominatedby hyponasality attributed to the size restriction in the nasal passagesand oropharynx as well as hearing loss. Hypernasality was alsonoted in association with those children who had a cleft palate andvelopharyngeal insufficiency. Articulation errors were found in 17 of46 patients and were attributed to the malocclusion with anterioropen bite and retroglossa.
Treatment consisted of review by an audiologist and anotolaryngologist within the first year to evaluate and correct hearingloss where possible. After this, speech and language therapy wasinitiated as soon as possible. Particular attention was given tomaladaptive patterns related to the abnormal oral anatomy. When
TABLE 1. Features of Treacher Collins Syndrome andIncidence Noted in this Series of Patients
Feature n %
Downslanting palpebral fissures 47 100Malar hypoplasia 44 94Mandibular hypoplasia 42 89Hearing disability 42 89Malocclusion 41 87Auricular deformity 40 85Middle ear deformity 40 85Speech problems 34 74External auditory canal deformity 30 64Absent eyelashes 29 62Lacrimal deformity 27 57Airway compromise at birth 22 47Epiphora 20 43Lower eyelid coloboma 20 43Visual disability 16 34Palatopharyngeal incompetence 14 30Pierre Robin sequence 14 30Cleft palate 11 23Otitis media requiring tubes 9 19Obstructive sleep apnea beyond childhood 9 19Ptosis 7 15Choanal atresia 5 11Macrostomia 5 11Ear tags 4 9Cleft lip 1 2Inner ear deformity 0 0
The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009 Treacher Collins Syndrome: Management
* 2009 Mutaz B. Habal, MD 2029

Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
these patterns developed, the children were given exercises to correctthese patterns and ultimately prepare them for orthodontic andorthognathic correction. All patients were able to achieve excellentspeech results except one who was from an underdeveloped nationand had a severe hearing loss and was not treated until adulthood.
The incidence of cleft palate was 11 of 46 patients in thisseries. Patients who had a cleft palate were be unable to have apalatal repair at the typical time (G12 mo) because of the com-plicated airway management. The average age at palate repair inthis group was 2.1 years with only 1 patient having a cleft palaterepair before 1 year of age.
Early attention to feeding is also an important issue. Twenty-two of the patients in this series were noted to have some degree offeeding difficulty, which was attributed to respiratory problems.Nine patients required either nasogastric or gastrostomy feedingduring the first year of life of whom 8 had cleft palates. In patientswith the most severe conditions, there were also some problems withoral aversion or hypersensitivity.
PsychosocialOn psychologic testing, 2 of 46 patients evaluated were noted
to have a nonverbal intellectual disability. Most of the patients whohad severe hearing loss also had trouble on the verbal examination,but this was not considered a true intellectual disability but rather afunction of the hearing loss. It was part of the protocol to have eachchild evaluated by the psychologist before starting school and then atleast once again several years into school. Formal testing usuallyconsisted of the Wechsler Intelligence Scale for Children or theGriffith Mental Development Scale.
Of the 14 patients who were treated in this unit from birth tomaturity, 12 had successful social outcomes. They completed edu-cation to year 12 and either have pursued further education/technicaltraining or have begun employment. Of the 2 who did not achievethese goals, one had multiple medical problems and intellectualdisabilities, whereas the other has had ongoing social/legalproblems. Only 1 patient of the 14 described teasing as a significantproblem during childhood that was difficult to overcome.
Facial ReconstructionIn all, 33 of 46 patients underwent orbitozygomatic re-
construction. Before 1986, the primary method of reconstructionwas on-lay bone grafts to the deficient zygomatic area. Sevenpatients were treated during this period, 2 of whom also had orbitalosteotomies to advance the zygoma. One patient during this periodreceived a vascularized bone flap based on the temporalis muscle.Between 1986 and 2000, 15 patients received the vascularized boneflap procedure and only 5 underwent on-lay bone grafting alone, allof whom were adults at the time of their primary reconstruction.Finally, in patients treated after 2000, 5 were treated with on-laybone grafting and only 1 patient received a vascularized bone graft.
FIGURE 1. From left to right, 3 generations of Treacher Collins with increasing severity. The only feature noted in the eldestpatient was the orbitozygomatic depression.
TABLE 2. Airway Outcomes for Patients Who RequiredTracheostomy in Infancy
Patient No. Decannulation Details
1 No Died at age 1 y from sepsis2 3 y Distraction: 13-mm advancement3 1 y Distraction: 15-mm advancement4 6 y Distraction: 25-mm advancement
Thompson et al The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009
2030 * 2009 Mutaz B. Habal, MD
Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
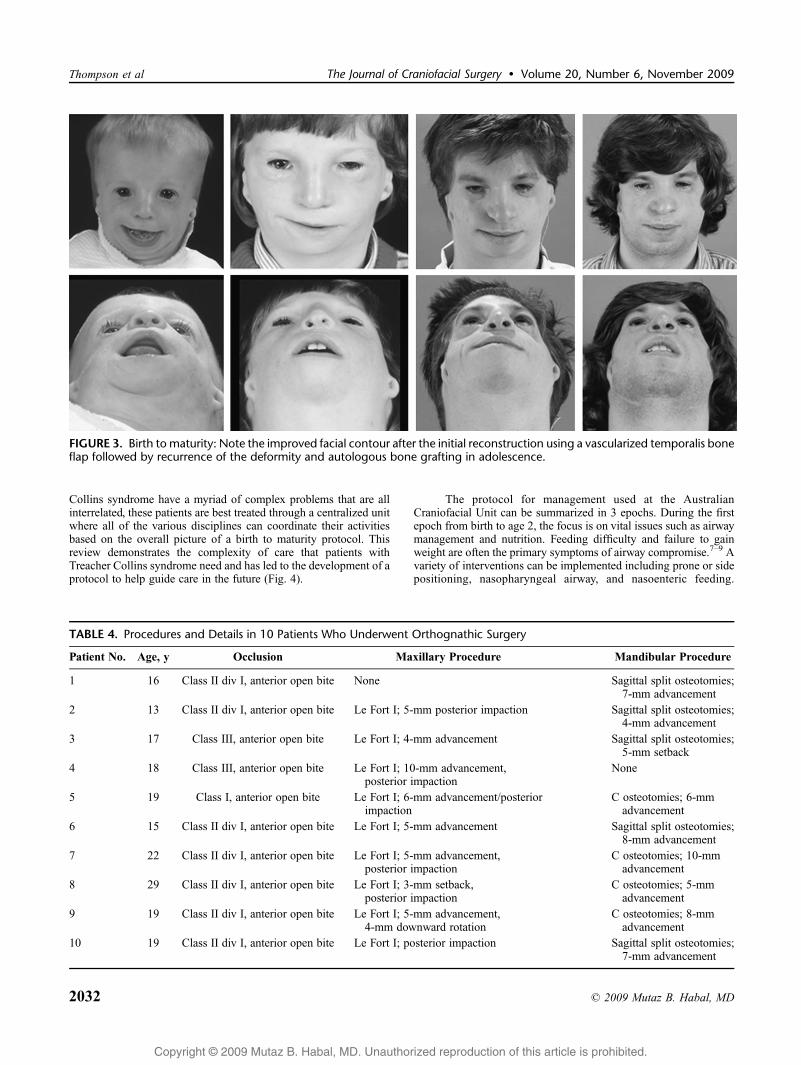
Ten patients were followed from birth to maturity in this unit, andtheir results are summarized in Table 3. In two-thirds of thesepatients, a recurrent deformity was noted as seen in Figure 3. Thisrequired additional bone grafting and occurred whether the initialprocedure was a vascularized bone flap or bone graft.
Treatment for the eyelid deformity consisted of musculocu-taneous transposition flaps from the upper eyelid to the lower lid in28 patients. A lateral canthopexy was also performed to create anormal palpebral slant. This was done at the same time as the initialorbitozygomatic bone grafting procedure.
Surgical reconstruction of deformed ears depended on theseverity. Of the 20 patients with either A0 or A1 ears, 5 underwent anotoplasty procedure to correct the overhanging helix and reduce earprominence. Three of the 26 patients who had type A2 or A3 earsunderwent a staged reconstruction using rib cartilage at age 6, and1 patient had an osseointegrated prosthetic ear reconstruction, whichwas performed in adulthood. The remainder of patients either choseno reconstruction or are awaiting reconstruction.
Orthodontic treatment and orthognathic surgery was per-formed once dental development was complete. Nine patients were
treated with orthodontics and did not require orthognathic surgery.Ten others underwent orthognathic surgery. Two of these patientswere also treated to relieve upper airway obstruction. Before surgery,all patients had an anterior open bite, which was corrected witheither a posterior impaction or an anterior opening Le Fort I. Thiswas usually combined with mandibular surgery to correct thehorizontal malocclusion (Table 4). After surgery, all patients hadcorrection of the open bite. The 2 patients with sleep apnea no longerrequired CPAP after surgery.
Seventeen patients had a sliding osseus genioplasty, and 11had a rhinoplasty. The rhinoplasty consisted of reducing the dorsalhump and adding tip projection with cartilage grafts when needed.In 6 of the patients who underwent rhinoplasty, a rib graft was usedto reconstruct the nasal dorsum.
DISCUSSIONDuring the last 30 years, the Australian Craniofacial Unit has
sought to organize and provide multidisciplinary care to patientswith craniofacial deformities. Because patients with Treacher
FIGURE 2. The severity of external ear malformation correlated with hearing loss.
TABLE 3. Initial Reconstructive Procedure and Subsequent Procedures in 10 Patients Who Had Reached Maturity at the Timeof the Study
Patient No. Year Age, y Initial Orbitozygomatic Reconstruction Outcome
1 1980 4 Iliac crest graft, orbital osteotomies Vascularized bone flap at age 10, bone graft at age 212 1983 5 Vascularized calvarial bone flap Bone graft performed at age 143 1983 4 Calvarial graft No additional procedures4 1984 4 Calvarial and rib graft Repeated bone graft at age 6, 13, 18, and 205 1986 7 Vascularized calvarial bone flap Bone graft at age 166 1988 4 Vascularized calvarial bone flap Required bone grafting at age 197 1989 6 Vascularized calvarial bone flap Required bone grafting at age 13, 15, 16, 19, and 218 1989 5 Vascularized calvarial bone flap No additional procedures9 1990 5 Vascularized calvarial bone flap No additional procedures10 1992 5 Vascularized calvarial bone flap Required bone grafting at age 15 and 17
The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009 Treacher Collins Syndrome: Management
* 2009 Mutaz B. Habal, MD 2031
Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
Collins syndrome have a myriad of complex problems that are allinterrelated, these patients are best treated through a centralized unitwhere all of the various disciplines can coordinate their activitiesbased on the overall picture of a birth to maturity protocol. Thisreview demonstrates the complexity of care that patients withTreacher Collins syndrome need and has led to the development of aprotocol to help guide care in the future (Fig. 4).
The protocol for management used at the AustralianCraniofacial Unit can be summarized in 3 epochs. During the firstepoch from birth to age 2, the focus is on vital issues such as airwaymanagement and nutrition. Feeding difficulty and failure to gainweight are often the primary symptoms of airway compromise.7Y9 Avariety of interventions can be implemented including prone or sidepositioning, nasopharyngeal airway, and nasoenteric feeding.
TABLE 4. Procedures and Details in 10 Patients Who Underwent Orthognathic Surgery
Patient No. Age, y Occlusion Maxillary Procedure Mandibular Procedure
1 16 Class II div I, anterior open bite None Sagittal split osteotomies;7-mm advancement
2 13 Class II div I, anterior open bite Le Fort I; 5-mm posterior impaction Sagittal split osteotomies;4-mm advancement
3 17 Class III, anterior open bite Le Fort I; 4-mm advancement Sagittal split osteotomies;5-mm setback
4 18 Class III, anterior open bite Le Fort I; 10-mm advancement,posterior impaction
None
5 19 Class I, anterior open bite Le Fort I; 6-mm advancement/posteriorimpaction
C osteotomies; 6-mmadvancement
6 15 Class II div I, anterior open bite Le Fort I; 5-mm advancement Sagittal split osteotomies;8-mm advancement
7 22 Class II div I, anterior open bite Le Fort I; 5-mm advancement,posterior impaction
C osteotomies; 10-mmadvancement
8 29 Class II div I, anterior open bite Le Fort I; 3-mm setback,posterior impaction
C osteotomies; 5-mmadvancement
9 19 Class II div I, anterior open bite Le Fort I; 5-mm advancement,4-mm downward rotation
C osteotomies; 8-mmadvancement
10 19 Class II div I, anterior open bite Le Fort I; posterior impaction Sagittal split osteotomies;7-mm advancement
FIGURE 3. Birth to maturity: Note the improved facial contour after the initial reconstruction using a vascularized temporalis boneflap followed by recurrence of the deformity and autologous bone grafting in adolescence.
Thompson et al The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009
2032 * 2009 Mutaz B. Habal, MD

Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
Results here indicate that most airway problems can be managedwith positioning or with a nasal airway. Once a tracheostomyhas been placed, however, it was difficult to remove and requiredchildhood distraction in all cases. Neonatal distraction was not used
here but could be considered in severe cases to avoid tracheostomy.In this series, 10 patients had breathing difficulties into childhoodincluding 2 who had no problems in infancy. This illustrates the needfor continued observation throughout development. Finally, the
FIGURE 4. The Treacher Collins protocol at the Australian Craniofacial Unit.
The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009 Treacher Collins Syndrome: Management
* 2009 Mutaz B. Habal, MD 2033

Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
difficult airway in patients with Treacher Collins syndrome shouldnot be underestimated. Experienced anesthetists play a critical rolein the multidisciplinary care.
Hearing loss is another important issue that is addressedduring the first epoch. Because 17% of patients with normal-appearing external ears still had at least a mild hearing impairment,external ear appearance should not influence audiometry screening.In fact, the more normal appearing the external ear, the more likelythe child will benefit from an intervention such as tympanostomytube placement. Middle ear reconstruction can be considered forpatients with Treacher Collins syndrome if deemed feasible whenthe child is older. However, the goal of ossicular reconstructionshould be to achieve hearing without the need for aids, which wasnot accomplished in 1 patient who had this done in this series. Anyotologic surgery planned should be performed by a surgeon familiarto the abnormal anatomy of the facial nerve in Treacher Collinssyndrome.10
Genetic counseling is offered to patients and families duringthe first epoch. Genetic testing is possible and has identifiedmutations from 48% to 93% of patients thought to have TreacherCollins depending on the how extensive the genetic screening andthe clinical accuracy of the patient population tested.11Y14 In thisstudy, 2 of 4 patients tested positive for the TCOF1 gene. Becausethis mutation is not present in similar conditions such as Goldenhar,Miller, or Nagar syndrome, the role of testing may be best suited topatients who have an equivocal clinical diagnosis.13,15,16 Based onthe cost of testing, and the relative ease of clinical diagnosis, genetictesting is not a routine part of the Treacher Collins protocol.
The second epoch, from ages 2 to 12 years, focuses onspeech, social development, and the primary surgical reconstructionof the upper face. Facial reconstruction procedures for TreacherCollins syndrome have been described in detail before.9,17Y20 In thisseries, a combination of autologous bone grafting and vascularizedbone flaps was used. Neither technique produced lasting results,leading to repeated bone grafts in two-thirds of the patients. Mostpatients treated after 2000 underwent reconstruction using autolo-gous bone grafts, and the protocol reflects the need for repeatedgrafting later in life. The lower eyelid deformity was also treated atthis time using a musculocutaneous transposition flap from theupper eyelid to the lower combined with a lateral canthopexy.Although generally achieving the desired effect of protecting thecornea from exposure, the cosmetic results remain one of the mostdifficult aspects of managing Treacher Collins.
Previously, it has been shown that the teasing craniofacialpatients receive is not necessarily specific to their deformity or itsseverity.21 Nevertheless, when faced with criticism or teasing, mostchildren have coped remarkably well.22 Therefore, the timing ofsurgical intervention in this protocol is not driven by teasing.Teasing can usually be managed through education of the parents,teachers, and peers of the patients rather than deviation from thetreatment plan. This helps them to understand what is happening ateach point in development and eliminates unnecessary surgicalinterventions.
Speech and language development must be watched closelyduring this period. Generally, resonance and articulation problemsimproved with therapy, structural growth, and orthodontic correc-tion. However, it is important to monitor articulatory proficiencyfrom an early age to detect maladaptive compensatory patterns andintervene where appropriate.
In the third epoch, from age 12 to 18, the facial reconstructionis finished. During this time, orthognathic surgery, rhinoplasty, andgenioplasty operations were performed. Ideally, these operationsshould be combined when possible, although when a Le Fort Iosteotomy was performed, the rhinoplasty was performed separately.Because repeat zygomatic bone grafting is often required, it should
be performed in conjunction with other procedures. In thisexperience, all of the patients undergoing orthognathic surgeryrequired correction of an anterior open bite along with variableamounts of horizontal malocclusion. The anterior open bite wascorrected in all cases, and no patients required repeat surgery for thisproblem.
Formulating outcome goals for patients with such a widevariety of problems can be difficult. Nonetheless, it is critical to theoverall management strategy to have a vision of what can and shouldbe achieved throughout growth and development. Clearly, withineach subspecialty, the outcomes will be dependent on the severity ofthe deformity. The goal of therapy, however, is the same. Eachpatient should have an opportunity to overcome as much of thedeformity as possible. In the future we plan to generate a survey ofthe protocols and outcome goals of other units around the globe aswe move forward in the benchmarking process through theInternational Society of Craniofacial Surgery.
REFERENCES1. Berry GA. Two case reports. Royal London Ophthal Hosp Rep
1889;12:2552. Treacher Collins E. Cases with symmetrical congenital notches in the
outer part of each lower lid and defective development of malar bones.Trans Ophthalmol Soc U K 1900;20:190
3. Gorlin RJ, Cohen MM, Levin LS. Syndromes of the Head and Neck.Oxford, England: Oxford University Press, 1990
4. Wilson AC, Moore DJ, Moore MH, et al. Late presentation of upperairway obstruction in Pierre Robin sequence. Arch Dis Child2000;83:435Y438
5. Biebesheimer JB, Fredrick DR. Delayed-onset infantile cataracts in acase of Treacher Collins syndrome. Arch Ophthalmol 2004;122:1721Y1722
6. David DJ, Mahatumarat C, Cooter RD. Hemifacial microsomia:a multisystem classification. Plast Reconstr Surg 1987;80:525Y535
7. Moore MH, Guzman-Stein G, Proudman TW, et al. Mandibularlengthening by distraction for airway obstruction in Treacher-Collinssyndrome. J Craniofac Surg 1994;5:22Y25
8. Argenta LC, Iacobucci JJ. Treacher Collins syndrome: present conceptsof the disorder and their surgical correction. World J Surg 1989;13:401Y409
9. Posnick JC. Treacher Collins syndrome: perspectives in evaluationand treatment. J Oral Maxillofac Surg 1997;55:1120Y1133
10. Takegoshi H, Kaga K, Chihara Y. Facial canal anatomy in patientswith mandibulofacial dysostosis: comparison with respect to theseverities of microtia and middle ear deformity. Otol Neurotol 2005;26:803Y808
11. Edwards SJ, Gladwin AJ, Dixon MJ. The mutational spectrum inTreacher Collins syndrome reveals a predominance of mutations thatcreate a premature-termination codon. Am J Hum Genet 1997;60:515Y524
12. Splendore A, Silva EO, Alonso LG, et al. High mutation detection ratein TCOF1 among Treacher Collins syndrome patients revealsclustering of mutations and 16 novel pathogenic changes. Hum Mutat2000;16:315Y322
13. Splendore A, Jabs EW, Passos-Bueno MR. Screening of TCOF1 inpatients from different populations: confirmation of mutational hot spotsand identification of a novel missense mutation that suggestsan important functional domain in the protein treacle. J Med Genet2002;39:493Y495
14. Teber OA, Gillessen-Kaesbach G, Fischer S, et al. Genotyping in 46patients with tentative diagnosis of Treacher Collins syndrome revealedunexpected phenotypic variation. Eur J Hum Genet 2004;12:879Y890
15. The Treacher Collins Collaborative Group. Positional cloning of a geneinvolved in the pathogenesis of Treacher Collins syndrome. The
Thompson et al The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009
2034 * 2009 Mutaz B. Habal, MD

Copyright @ 2009 Mutaz B. Habal, MD. Unauthorized reproduction of this article is prohibited.
Treacher Collins Syndrome Collaborative Group. Nat Genet1996;12:130Y136
16. Splendore A, Passos-Bueno MR, Jabs EW, et al. TCOF1 mutationsexcluded from a role in other first and second branchial arch-relateddisorders. Am J Med Genet 2002;111:324Y327
17. Posnick JC, Ruiz RL. Treacher Collins syndrome: current evaluation,treatment, and future directions. Cleft Palate Craniofac J 2000;37:434
18. van der Meulen JC, Hauben DJ, Vaandrager JM, et al. The use of atemporal osteoperiosteal flap for the reconstruction of malar hypoplasiain Treacher Collins syndrome. Plast Reconstr Surg 1984;74:687Y693
19. Tulasne JF, Tessier PL. Results of the Tessier integral procedure forcorrection of Treacher Collins syndrome. Cleft Palate J 1986;1:40Y49
20. David DJ. Treacher collins syndrome. In: Muir IFK, ed. CurrentOperative Surgery: Plastic and Reconstructive Surgery. London, UK:Bailliere Tindall, 1985
21. Carroll P. Teasing and victimization? It’s rarely simple! In: David DJ, ed.Craniofacial II: Proceedings of the Eleventh International Congress ofThe International Society of Craniofacial Surgery. Bologna, Italy:Medimond S.r.l., 2006:394Y397
22. Beaune L, Forrest CR, Keith T. Adolescents’ perspectives on livingand growing up with Treacher Collins syndrome: a qualitative study.Cleft Palate Craniofac J 2004;41:343Y350
The Journal of Craniofacial Surgery & Volume 20, Number 6, November 2009 Treacher Collins Syndrome: Management
* 2009 Mutaz B. Habal, MD 2035







![A to Z Directory – Virginia Commonwealth University · Web viewTreacher Collins Syndrome (TCS) is an autosomal disorder that mainly affects craniofacial development [1]. This syndrome](https://img.pdfslide.us/doc/110x75/61353867dfd10f4dd73c3bec/a-to-z-directory-a-virginia-commonwealth-web-view-treacher-collins-syndrome-tcs.jpg)





















