Embed Size (px)
Citation preview

A R T I C L E S
Inherited human genetic disorders are usually caused by mutationsthat disrupt the coding sequences of the disease gene or its conserved,local cis-acting sequences, including promoters or mRNA processingsignals (for example, donor or acceptor splice sites and poly(A)+
addition signals; ref. 1). Much less frequently, gene expression isdownregulated by inherited deletions of remote regulatory elementswhich can be located far (∼ 5 to more than 100 kb) upstream or down-stream of the genes that they control2,3.
Although the first examples of most mutations in each of theseclasses were described in the human globin genes, giving rise toα- and β-thalassemia4, the same categories of mutation are foundunderlying most genetic disorders that have been studied in detail.For many diseases, however, there are subgroups of affected individu-als in whom the mechanism of abnormal gene expression isunknown. Undoubtedly, some of these will be shown to result fromdeletions of currently unrecognized regulatory elements. It has beensuggested that others may result from chromosomal position effectsin which gene expression is perturbed by moving a structurally nor-mal gene to an ‘unfavorable’ chromatin environment as a result of adeletion, inversion or chromosomal translocation2,3. For most dis-eases, however, we do not have enough information about the normalcis-acting sequences that regulate expression of the relevant gene orabout the epigenetic modifications (for example, nuclear localization,pattern of replication, chromatin structure and pattern of DNAmethylation) associated with its normal chromosomal environmentto determine the mechanism of abnormal gene silencing.
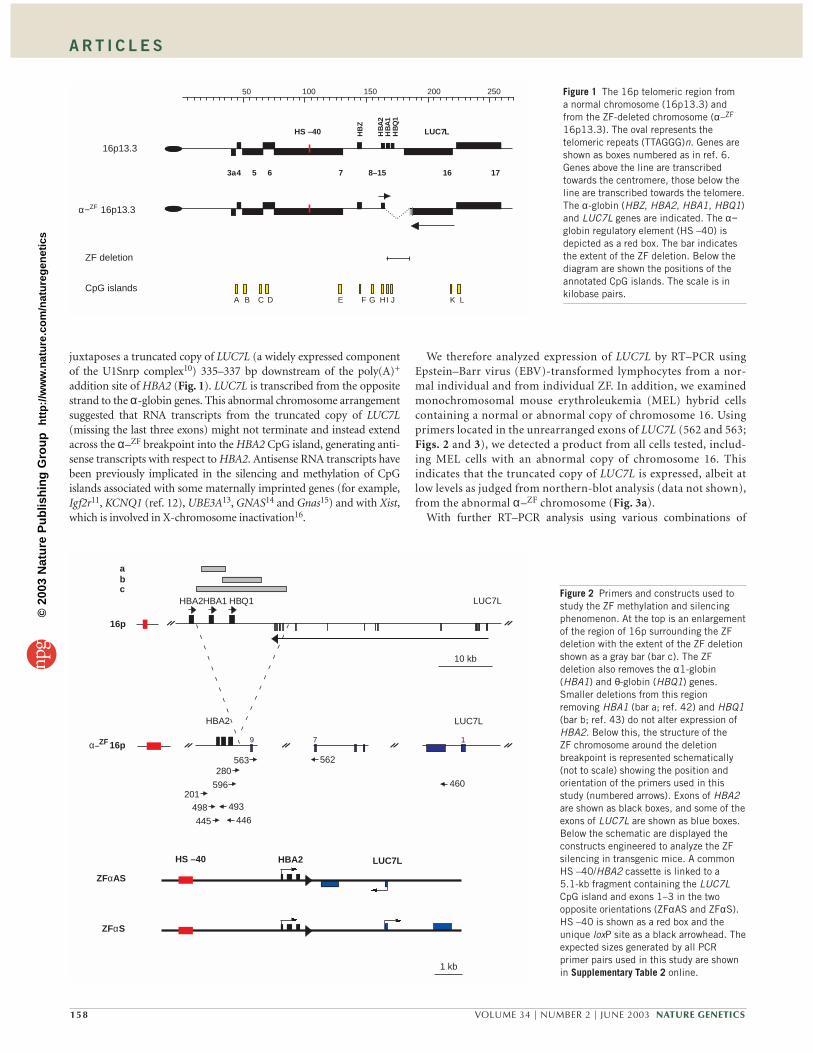
We recently reported an individual (called ZF) with α-thalassemiadue to a unique deletion (called α–ZF) that removes the α1-globin
(HBA1) and θ-globin (HBQ1) genes and juxtaposes a region that nor-mally lies ∼ 18 kb downstream of the human α-globin cluster next to thestructurally normal adult α2-globin (HBA2) gene5 (Fig. 1). The humanα-globin genes, which are exclusively expressed in erythroid cells in adevelopmental stage–specific manner, lie in a fully characterized, GC-rich, gene-dense region near the telomere of the short arm of chromo-some 16 (ref. 6; Fig. 1). The promoters of all the genes in this area,including the α-globin genes, are associated with unmethylated CpGislands6,7 (A–L; Fig. 1). The α–ZF deletion does not remove any positivecis-acting sequences5, but expression of the structurally intact α-globingene is stably silenced and, during development, its CpG island becomesdensely methylated and insensitive to endonucleases over a region ofapproximately 2 kb5. These epigenetic changes are not associated withany alterations in the timing of replication8 or nuclear compartmental-ization9 of the abnormal allele. Other CpG islands associated with genesflanking the α-globin cluster are unaffected by the deletion5. Therefore,by comparison with the normal allele, in which the α-globin gene andits regulatory elements have an identical structure and share the samenuclear milieu, the α-globin gene on the rearranged chromosome seemsto be silenced epigenetically. In this study we sought to understand themechanism by which this non-imprinted, autosomal CpG islandbecomes densely methylated and expression of its associated α-globingene becomes silenced during development to cause α-thalassemia.
RESULTSAntisense RNA transcripts extend into HBA2The previously described α–ZF deletion removes sequences betweencoordinates 164,044–5 and 182,395–6 of the α-globin cluster and
MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Headington, Oxford OX3 9DS, UK.Correspondence should be addressed to D.R.H. ([email protected]).
Transcription of antisense RNA leading to genesilencing and methylation as a novel cause of humangenetic diseaseCristina Tufarelli, Jackie A Sloane Stanley, David Garrick, Jackie A Sharpe, Helena Ayyub, William G Wood& Douglas R Higgs
Nearly all human genetic disorders result from a limited repertoire of mutations in an associated gene or its regulatory elements.We recently described an individual with an inherited form of anemia (α-thalassemia) who has a deletion that results in atruncated, widely expressed gene (LUC7L) becoming juxtaposed to a structurally normal α-globin gene (HBA2). Although itretains all of its local and remote cis-regulatory elements, expression of HBA2 is silenced and its CpG island becomes completelymethylated early during development. Here we show that in the affected individual, in a transgenic model and in differentiatingembryonic stem cells, transcription of antisense RNA mediates silencing and methylation of the associated CpG island. Thesefindings identify a new mechanism underlying human genetic disease.
NATURE GENETICS VOLUME 34 | NUMBER 2 | JUNE 2003 157NATURE GENETICS VOLUME 34 | NUMBER 2 | JUNE 2003 157
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics
A R T I C L E S
juxtaposes a truncated copy of LUC7L (a widely expressed componentof the U1Snrp complex10) 335–337 bp downstream of the poly(A)+
addition site of HBA2 (Fig. 1). LUC7L is transcribed from the oppositestrand to the α-globin genes. This abnormal chromosome arrangementsuggested that RNA transcripts from the truncated copy of LUC7L(missing the last three exons) might not terminate and instead extendacross the α–ZF breakpoint into the HBA2 CpG island, generating anti-sense transcripts with respect to HBA2. Antisense RNA transcripts havebeen previously implicated in the silencing and methylation of CpGislands associated with some maternally imprinted genes (for example,Igf2r11, KCNQ1 (ref. 12), UBE3A13, GNAS14 and Gnas15) and with Xist,which is involved in X-chromosome inactivation16.
We therefore analyzed expression of LUC7L by RT–PCR usingEpstein–Barr virus (EBV)-transformed lymphocytes from a nor-mal individual and from individual ZF. In addition, we examinedmonochromosomal mouse erythroleukemia (MEL) hybrid cellscontaining a normal or abnormal copy of chromosome 16. Usingprimers located in the unrearranged exons of LUC7L (562 and 563;Figs. 2 and 3), we detected a product from all cells tested, includ-ing MEL cells with an abnormal copy of chromosome 16. Thisindicates that the truncated copy of LUC7L is expressed, albeit atlow levels as judged from northern-blot analysis (data not shown),from the abnormal α–ZF chromosome (Fig. 3a).
With further RT–PCR analysis using various combinations of
158 VOLUME 34 | NUMBER 2 | JUNE 2003 NATURE GENETICS
CpG islandsA B C D E F G HI J K L
HS –40 HB
Z
HB
A2
HB
A1
LUC7LHB
Q1
50
4 5 6 7 168–15 173a
ZF deletion
16p13.3
α−ZF 16p13.3
100 150 200 250 Figure 1 The 16p telomeric region froma normal chromosome (16p13.3) andfrom the ZF-deleted chromosome (α–ZF
16p13.3). The oval represents thetelomeric repeats (TTAGGG)n. Genes areshown as boxes numbered as in ref. 6.Genes above the line are transcribedtowards the centromere, those below theline are transcribed towards the telomere.The α-globin (HBZ, HBA2, HBA1, HBQ1)and LUC7L genes are indicated. The α−globin regulatory element (HS –40) isdepicted as a red box. The bar indicatesthe extent of the ZF deletion. Below thediagram are shown the positions of theannotated CpG islands. The scale is inkilobase pairs.
LUC7LHBA1 HBQ1HBA2
ZFαS
ZFαAS
HBA2 LUC7LHS –40
1 kb
16p
562280
596
563
201498 493
460
445 446
α–ZF 16p
LUC7L
9 17
10 kb
cba
HBA2
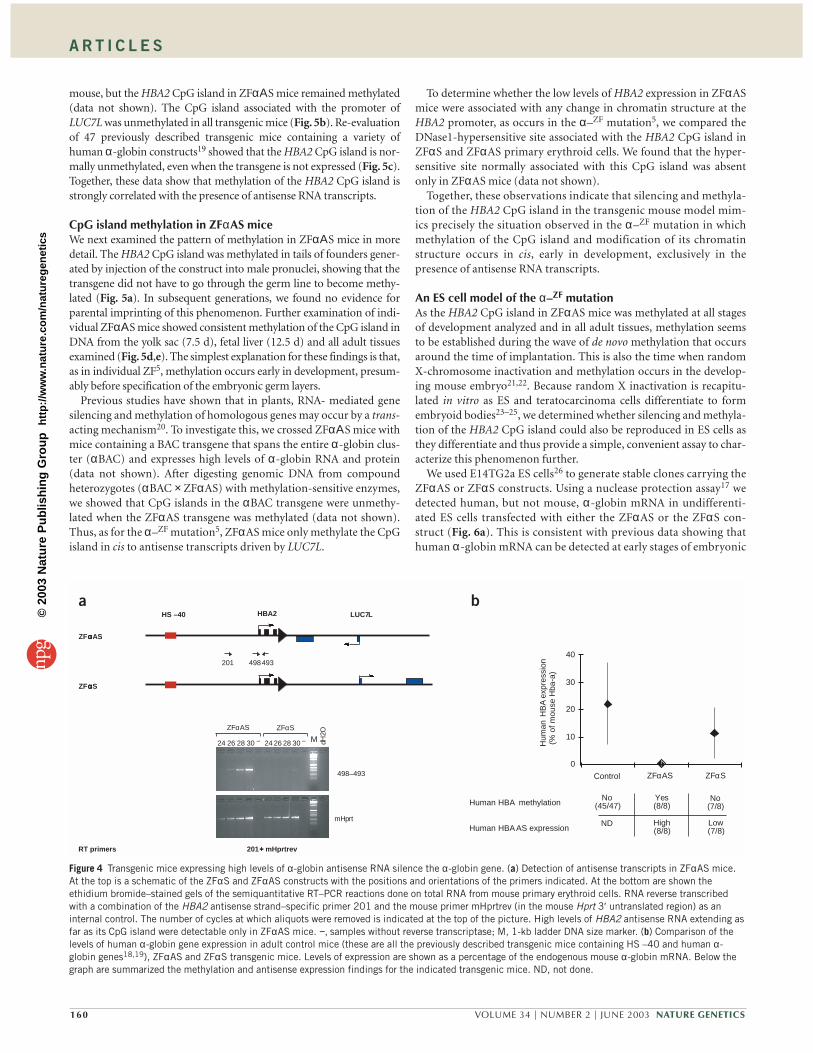
Figure 2 Primers and constructs used tostudy the ZF methylation and silencingphenomenon. At the top is an enlargementof the region of 16p surrounding the ZFdeletion with the extent of the ZF deletionshown as a gray bar (bar c). The ZFdeletion also removes the α1-globin(HBA1) and θ-globin (HBQ1) genes.Smaller deletions from this regionremoving HBA1 (bar a; ref. 42) and HBQ1(bar b; ref. 43) do not alter expression ofHBA2. Below this, the structure of theZF chromosome around the deletionbreakpoint is represented schematically(not to scale) showing the position andorientation of the primers used in thisstudy (numbered arrows). Exons of HBA2are shown as black boxes, and some of theexons of LUC7L are shown as blue boxes.Below the schematic are displayed theconstructs engineered to analyze the ZFsilencing in transgenic mice. A commonHS –40/HBA2 cassette is linked to a5.1-kb fragment containing the LUC7LCpG island and exons 1–3 in the twoopposite orientations (ZFαAS and ZFαS).HS –40 is shown as a red box and theunique loxP site as a black arrowhead. Theexpected sizes generated by all PCRprimer pairs used in this study are shownin Supplementary Table 2 online.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics

A R T I C L E S
primers located in the exons of LUC7L and others on the α-globinside of the α–ZF breakpoint (280 and 562; Figs. 2 and 3 andSupplementary Table 1 online), we identified abnormal transcriptsrunning into HBA2 only in cell lines containing the α–ZF chromo-some (Fig. 3a). Sequence analysis confirmed that the abnormal tran-scripts corresponded to correctly spliced mRNA driven from theLUC7L promoter and showed that the α–ZF deletion removed thenormal transcription termination site of LUC7L.
To determine how far the antisense transcripts extend into HBA2,we carried out reverse transcription using primers in HBA2 thatspecifically prime antisense RNA so that cDNAs would be synthesizedonly from RNA transcribed in the opposite orientation to HBA2.Antisense transcripts were readily detected using cDNA synthesizedwith such primers (Fig. 3b and data not shown), the most telomericof which (201; Fig. 2) lies in the HBA2 CpG island.
Together, these data show that in both erythroid and nonerythroidcells, low levels of alternatively spliced antisense transcripts originatingat the LUC7L promoter run across the α–ZF breakpoint in the abnormalchromosome and extend at least as far as the HBA2 CpG island.
A transgenic model recapitulates the α–ZF mutationTo investigate the potential role of antisense transcription in silencingand methylating HBA2, we made a mouse model to mimic the effectsof the α–ZF deletion. We first made a construct in which the primaryremote α-globin regulatory element (HS –40; Figs. 1 and 2), anessential cis-acting enhancer of α-globin expression in erythroidcells17, was linked to HBA2. This fragment was joined to a 5.1-kb BclIfragment encompassing the CpG island, the promoter and exons 1–3of LUC7L (Fig. 2). We linked the fragments so that the sense strand ofLUC7L was in the opposite direction to that of HBA2, as found in thenatural α–ZF deletion. As a control, we made a construct in which theLUC7L fragment was reversed so that it was in the same sense orienta-tion as HBA2. We used these constructs, and modified versions con-taining a unique loxP site to allow further manipulation, to generateseveral lines of transgenic mice.
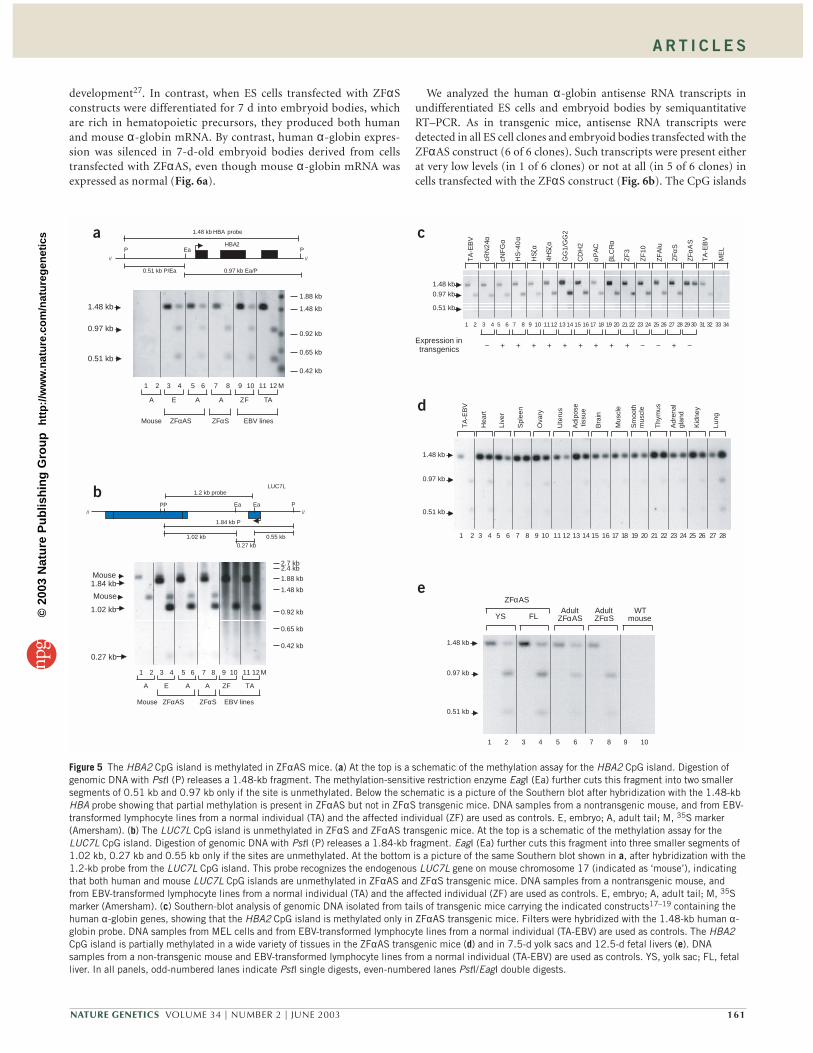
If the LUC7L promoter in these constructs could drive expressionof the truncated LUC7L gene in transgenic mice, antisense transcriptsrunning into the linked HBA2 gene should be present in transgenicmice with the antisense construct (ZFαAS mice) but not in transgenicmice with the sense construct (ZFαS mice). To examine this, we car-ried out RT–PCR on total RNA purified from primary erythroid cellsisolated from the spleens of transgenic mice. We found that the trun-cated LUC7L gene was expressed in both ZFαS and ZFαΑS mice(Supplementary Fig. 1 online). To look specifically for antisensetranscripts, we synthesized cDNA with an HBA2 antisensestrand–specific primer (Fig. 2), which lies in the HBA2 CpG island.Semiquantitative PCR analysis showed that readily detectable levels ofantisense transcripts (with respect to α-globin) were consistently pro-duced in ZFαAS mice (Fig. 4a). Further characterization of the anti-sense transcripts (data not shown) showed that they originated fromthe LUC7L promoter and were alternatively spliced in a manner thatis consistent with known alternative splice forms of the intact LUC7Lgene in vivo10.
Having faithfully recapitulated antisense RNA transcription, we nextexamined α-globin gene expression. Whereas the ZFαS adult mice hadlevels of α-globin expression similar to those of previously analyzedtransgenic mice containing HS –40 linked to HBA alone18,19, expressionfrom ZFαAS mice was low in embryos and virtually absent (<1% of themouse α-globin RNA) in adults (Fig. 4b) as judged by a nuclease protec-tion assay. Therefore, as in individual ZF, α-globin expression is virtuallyabolished in the presence of antisense RNA transcripts.
We have previously shown that the HBA2 CpG island in cis to theα–ZF deletion is densely methylated in the peripheral blood but not thegerm line of individual ZF5. Therefore, we examined the methylationstatus of this region in ZFαS and ZFαΑS mice using the methylation-sensitive enzyme EagI. Complete or partial methylation (>50%) wasobserved in whole embryos and adult tail samples in 8 of 8 ZFαΑS linesbut in only 1 of 8 ZFαS lines (Figs. 5a and 4b). When transgenes werereduced to a single copy using the Cre–loxP system, a normal pattern ofmethylation was restored to the single, partially methylated ZFαS
NATURE GENETICS VOLUME 34 | NUMBER 2 | JUNE 2003 159
ME
L ×
16
ME
L
TA
-EB
V
ZF
-EB
V
K56
2
M+ + + + + +
562–563
562–280
dH2OM
EL
× Z
F
Probe oligo 563
596 RT
PCR 280–460
1.47 kb
0.85 kb
+ − + − + −
ME
L
ME
L ×
16
ME
L ×
ZF
9–432
29
562563
LUC7L 9 78
LUC7L
562280
HBA27
– – – – – –
a
b
Figure 3 Detection of antisense transcripts in ZF cell lines. (a) Ethidiumbromide–stained gels of RT–PCRs done with (+) or without (−) reversetranscriptase on MEL cells, on MEL interspecific hybrids bearing theabnormal α–ZF chromosome (MEL × ZF) or a normal copy of humanchromosome 16 (MEL × 16), on EBV-transformed lymphocytes from anormal individual (TA-EBV) and individual ZF (ZF-EBV) and on a K562human erythroleukemia cell line using random primers for the reversetranscription and the indicated primer pairs for PCR. Transcripts extendingacross the breakpoint can be amplified from cell lines containing theabnormal α–ZF chromosome. M, 1-kb ladder DNA size marker. No antisensetranscripts originating from LUC7L or elsewhere were detected across theα-globin gene in a hybrid derived from a normal individual (SupplementaryFig. 2 online). (b) Autoradiograph of the RT–PCRs done with (+) or without(–) reverse transcriptase on MEL cells and on MEL × 16 and MEL × ZFinterspecific hybrids. Strand-specific reverse transcriptions (RT) were donewith primer 596 in HBA2 exon 3. The PCR reactions were done with theprimer pair 280 (upstream of the ZF breakpoint) and 460 (in LUC7L exon2). The Southern blot was hybridized using the 563 oligo probe located inLUC7L exon 9. The expected 1.47-kb band is visible in samples carrying theα–ZF chromosome. The additional 0.85-kb band seen in MEL × ZF is theexpected size for the alternative splicing between exons 2 and 9 of LUC7L.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics
A R T I C L E S
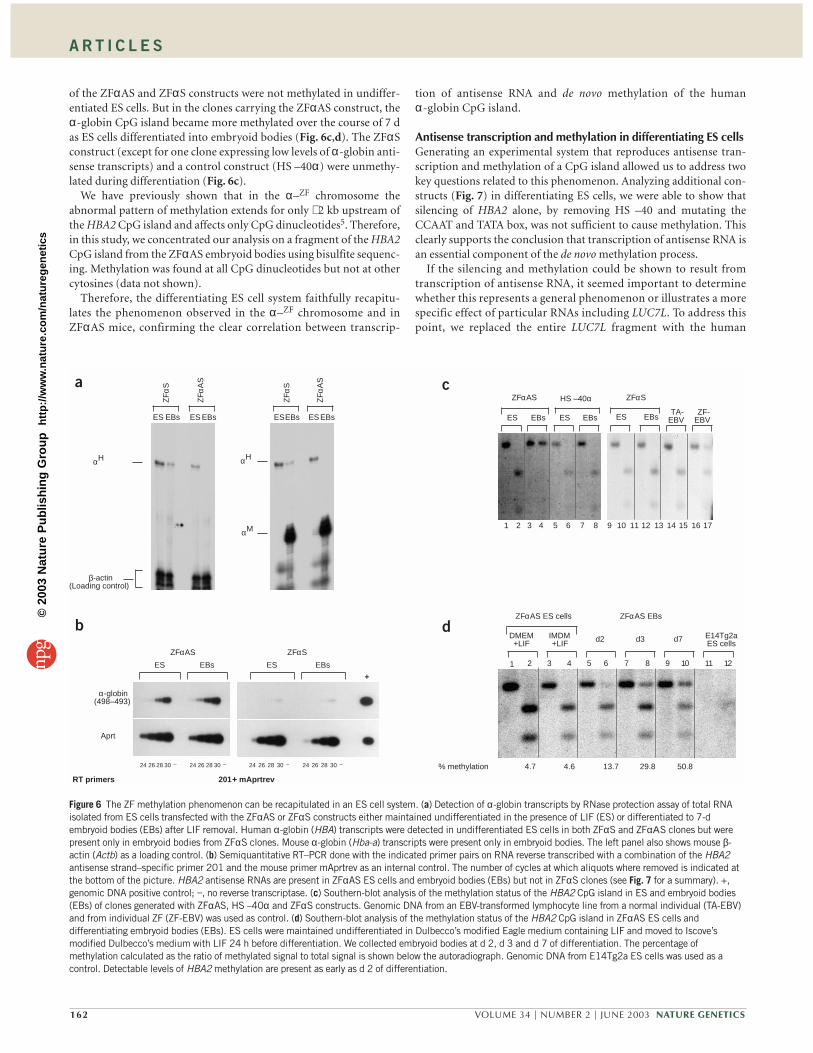
mouse, but the HBA2 CpG island in ZFαΑS mice remained methylated(data not shown). The CpG island associated with the promoter ofLUC7L was unmethylated in all transgenic mice (Fig. 5b). Re-evaluationof 47 previously described transgenic mice containing a variety ofhuman α-globin constructs19 showed that the HBA2 CpG island is nor-mally unmethylated, even when the transgene is not expressed (Fig. 5c).Together, these data show that methylation of the HBA2 CpG island isstrongly correlated with the presence of antisense RNA transcripts.
CpG island methylation in ZFαAS miceWe next examined the pattern of methylation in ZFαΑS mice in moredetail. The HBA2 CpG island was methylated in tails of founders gener-ated by injection of the construct into male pronuclei, showing that thetransgene did not have to go through the germ line to become methy-lated (Fig. 5a). In subsequent generations, we found no evidence forparental imprinting of this phenomenon. Further examination of indi-vidual ZFαΑS mice showed consistent methylation of the CpG island inDNA from the yolk sac (7.5 d), fetal liver (12.5 d) and all adult tissuesexamined (Fig. 5d,e). The simplest explanation for these findings is that,as in individual ZF5, methylation occurs early in development, presum-ably before specification of the embryonic germ layers.
Previous studies have shown that in plants, RNA- mediated genesilencing and methylation of homologous genes may occur by a trans-acting mechanism20. To investigate this, we crossed ZFαΑS mice withmice containing a BAC transgene that spans the entire α-globin clus-ter (αBAC) and expresses high levels of α-globin RNA and protein(data not shown). After digesting genomic DNA from compoundheterozygotes (αBAC × ZFαAS) with methylation-sensitive enzymes,we showed that CpG islands in the αBAC transgene were unmethy-lated when the ZFαAS transgene was methylated (data not shown).Thus, as for the α–ZF mutation5, ZFαAS mice only methylate the CpGisland in cis to antisense transcripts driven by LUC7L.
To determine whether the low levels of HBA2 expression in ZFαASmice were associated with any change in chromatin structure at theHBA2 promoter, as occurs in the α–ZF mutation5, we compared theDNase1-hypersensitive site associated with the HBA2 CpG island inZFαS and ZFαAS primary erythroid cells. We found that the hyper-sensitive site normally associated with this CpG island was absentonly in ZFαAS mice (data not shown).
Together, these observations indicate that silencing and methyla-tion of the HBA2 CpG island in the transgenic mouse model mim-ics precisely the situation observed in the α–ZF mutation in whichmethylation of the CpG island and modification of its chromatinstructure occurs in cis, early in development, exclusively in thepresence of antisense RNA transcripts.
An ES cell model of the α–ZF mutationAs the HBA2 CpG island in ZFαAS mice was methylated at all stagesof development analyzed and in all adult tissues, methylation seemsto be established during the wave of de novo methylation that occursaround the time of implantation. This is also the time when randomX-chromosome inactivation and methylation occurs in the develop-ing mouse embryo21,22. Because random X inactivation is recapitu-lated in vitro as ES and teratocarcinoma cells differentiate to formembryoid bodies23–25, we determined whether silencing and methyla-tion of the HBA2 CpG island could also be reproduced in ES cells asthey differentiate and thus provide a simple, convenient assay to char-acterize this phenomenon further.
We used E14TG2a ES cells26 to generate stable clones carrying theZFαAS or ZFαS constructs. Using a nuclease protection assay17 wedetected human, but not mouse, α-globin mRNA in undifferenti-ated ES cells transfected with either the ZFαAS or the ZFαS con-struct (Fig. 6a). This is consistent with previous data showing thathuman α-globin mRNA can be detected at early stages of embryonic
160 VOLUME 34 | NUMBER 2 | JUNE 2003 NATURE GENETICS
0
10
20
30
40
Control ZFαAS ZFαS
Human HBA methylation
Human HBA AS expression
No(45/47)
High(8/8)
Low(7/8)
Yes(8/8)
No(7/8)
Hum
an H
BA
expr
essi
on(%
of m
ouse
Hba
-a)
RT primers 201 ++++ mHprtrev
498–493
mHprt
ZFαAS ZFαS
24 26 28 −30 24 26 28 −30 dH2O
ZFααααS
493498201
HS –40 LUC7LHBA2
ZFααααAS
M
ND
a b
Figure 4 Transgenic mice expressing high levels of α-globin antisense RNA silence the α-globin gene. (a) Detection of antisense transcripts in ZFαAS mice.At the top is a schematic of the ZFαS and ZFαAS constructs with the positions and orientations of the primers indicated. At the bottom are shown theethidium bromide–stained gels of the semiquantitative RT–PCR reactions done on total RNA from mouse primary erythroid cells. RNA reverse transcribedwith a combination of the HBA2 antisense strand–specific primer 201 and the mouse primer mHprtrev (in the mouse Hprt 3′ untranslated region) as aninternal control. The number of cycles at which aliquots were removed is indicated at the top of the picture. High levels of HBA2 antisense RNA extending asfar as its CpG island were detectable only in ZFαAS mice. −, samples without reverse transcriptase; M, 1-kb ladder DNA size marker. (b) Comparison of thelevels of human α-globin gene expression in adult control mice (these are all the previously described transgenic mice containing HS –40 and human α-globin genes18,19), ZFαAS and ZFαS transgenic mice. Levels of expression are shown as a percentage of the endogenous mouse α-globin mRNA. Below thegraph are summarized the methylation and antisense expression findings for the indicated transgenic mice. ND, not done.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics
A R T I C L E S
development27. In contrast, when ES cells transfected with ZFαSconstructs were differentiated for 7 d into embryoid bodies, whichare rich in hematopoietic precursors, they produced both humanand mouse α-globin mRNA. By contrast, human α-globin expres-sion was silenced in 7-d-old embryoid bodies derived from cellstransfected with ZFαAS, even though mouse α-globin mRNA wasexpressed as normal (Fig. 6a).
We analyzed the human α-globin antisense RNA transcripts inundifferentiated ES cells and embryoid bodies by semiquantitativeRT–PCR. As in transgenic mice, antisense RNA transcripts weredetected in all ES cell clones and embryoid bodies transfected with theZFαAS construct (6 of 6 clones). Such transcripts were present eitherat very low levels (in 1 of 6 clones) or not at all (in 5 of 6 clones) incells transfected with the ZFαS construct (Fig. 6b). The CpG islands
NATURE GENETICS VOLUME 34 | NUMBER 2 | JUNE 2003 161
1.48 kb HBA probe
0.51 kb P/Ea 0.97 kb Ea/P
PEaPHBA2
// //
1.48 kb
0.97 kb
0.51 kb
ZFαAS ZFαS EBV linesMouse
E A A ZF TAA
1 2 3 4 5 6 7 8 10 11 12 M9
1.88 kb
1.48 kb
0.92 kb
0.65 kb
0.42 kb
1.2 kb probe
PEaEaP
1.84 kb P
1.02 kb
0.27 kb
0.55 kb
////
P
LUC7L
1.88 kb
1.48 kb
0.92 kb
0.65 kb
0.42 kb
2.4 kb2.7 kb
1 2 3 4 5 6 7 8 10 11 12M9
ZFαAS ZFαS
E A A
EBV lines
ZF TA
Mouse
A
0.27 kb
1.02 kb
1.84 kbMouse
Mouse
1.48 kb
0.97 kb
0.51 kb
1 2 3 4 5 6 7 8 10 11 12 13 14 15 169 7 18 19 20 21 22 23 24 25 26 27 28
Live
r
Spl
een
Ova
ry
Ute
rus
Adi
pose
tiss
ue
Bra
in
Mus
cle
TA
-EB
V
Hea
rt
Sm
ooth
mus
cle
Thy
mus
Adr
enal
glan
d
Kid
ney
Lung
Expression intransgenics − + − − −+ + + + + + + + +
0.97 kb
0.51 kb
1.48 kb
1 2 3 4 5 6 7 8 10 1112 13 14 15 169 17 18 19 20 21 22 23 24 25 26 27 28 2930 31 32 33 34
cNF
Gα
4HSζ
α
HS−
40α
GG
1/G
G2
CD
H2
αPA
C
βLC
Rα
TA
-EB
V
cRN
24α
ZF
3
ZF
10
ZF
Alu
ZFα
S
TA
-EB
V
ZFα
ΑS
ME
L
HSζ
α
YS FLAdult
ZFαASAdultZFαS
WTmouse
ZFαAS
0.97 kb
0.51 kb
1.48 kb
1 2 3 4 5 6 7 8 109
1
a
b
c
d
e
Figure 5 The HBA2 CpG island is methylated in ZFαAS mice. (a) At the top is a schematic of the methylation assay for the HBA2 CpG island. Digestion ofgenomic DNA with PstI (P) releases a 1.48-kb fragment. The methylation-sensitive restriction enzyme EagI (Ea) further cuts this fragment into two smallersegments of 0.51 kb and 0.97 kb only if the site is unmethylated. Below the schematic is a picture of the Southern blot after hybridization with the 1.48-kbHBA probe showing that partial methylation is present in ZFαAS but not in ZFαS transgenic mice. DNA samples from a nontransgenic mouse, and from EBV-transformed lymphocyte lines from a normal individual (TA) and the affected individual (ZF) are used as controls. E, embryo; A, adult tail; M, 35S marker(Amersham). (b) The LUC7L CpG island is unmethylated in ZFαS and ZFαAS transgenic mice. At the top is a schematic of the methylation assay for theLUC7L CpG island. Digestion of genomic DNA with PstI (P) releases a 1.84-kb fragment. EagI (Ea) further cuts this fragment into three smaller segments of1.02 kb, 0.27 kb and 0.55 kb only if the sites are unmethylated. At the bottom is a picture of the same Southern blot shown in a, after hybridization with the1.2-kb probe from the LUC7L CpG island. This probe recognizes the endogenous LUC7L gene on mouse chromosome 17 (indicated as ‘mouse’), indicatingthat both human and mouse LUC7L CpG islands are unmethylated in ZFαAS and ZFαS transgenic mice. DNA samples from a nontransgenic mouse, andfrom EBV-transformed lymphocyte lines from a normal individual (TA) and the affected individual (ZF) are used as controls. E, embryo; A, adult tail; M, 35Smarker (Amersham). (c) Southern-blot analysis of genomic DNA isolated from tails of transgenic mice carrying the indicated constructs17–19 containing thehuman α-globin genes, showing that the HBA2 CpG island is methylated only in ZFαAS transgenic mice. Filters were hybridized with the 1.48-kb human α-globin probe. DNA samples from MEL cells and from EBV-transformed lymphocyte lines from a normal individual (TA-EBV) are used as controls. The HBA2CpG island is partially methylated in a wide variety of tissues in the ZFαAS transgenic mice (d) and in 7.5-d yolk sacs and 12.5-d fetal livers (e). DNAsamples from a non-transgenic mouse and EBV-transformed lymphocyte lines from a normal individual (TA-EBV) are used as controls. YS, yolk sac; FL, fetalliver. In all panels, odd-numbered lanes indicate PstI single digests, even-numbered lanes PstI/EagI double digests.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics
A R T I C L E S
of the ZFαAS and ZFαS constructs were not methylated in undiffer-entiated ES cells. But in the clones carrying the ZFαAS construct, theα-globin CpG island became more methylated over the course of 7 das ES cells differentiated into embryoid bodies (Fig. 6c,d). The ZFαSconstruct (except for one clone expressing low levels of α-globin anti-sense transcripts) and a control construct (HS –40α) were unmethy-lated during differentiation (Fig. 6c).
We have previously shown that in the α–ZF chromosome theabnormal pattern of methylation extends for only ∼ 2 kb upstream ofthe HBA2 CpG island and affects only CpG dinucleotides5. Therefore,in this study, we concentrated our analysis on a fragment of the HBA2CpG island from the ZFαAS embryoid bodies using bisulfite sequenc-ing. Methylation was found at all CpG dinucleotides but not at othercytosines (data not shown).
Therefore, the differentiating ES cell system faithfully recapitu-lates the phenomenon observed in the α–ZF chromosome and inZFαAS mice, confirming the clear correlation between transcrip-
tion of antisense RNA and de novo methylation of the humanα-globin CpG island.
Antisense transcription and methylation in differentiating ES cellsGenerating an experimental system that reproduces antisense tran-scription and methylation of a CpG island allowed us to address twokey questions related to this phenomenon. Analyzing additional con-structs (Fig. 7) in differentiating ES cells, we were able to show thatsilencing of HBA2 alone, by removing HS –40 and mutating theCCAAT and TATA box, was not sufficient to cause methylation. Thisclearly supports the conclusion that transcription of antisense RNA isan essential component of the de novo methylation process.
If the silencing and methylation could be shown to result fromtranscription of antisense RNA, it seemed important to determinewhether this represents a general phenomenon or illustrates a morespecific effect of particular RNAs including LUC7L. To address thispoint, we replaced the entire LUC7L fragment with the human
162 VOLUME 34 | NUMBER 2 | JUNE 2003 NATURE GENETICS
% methylation 4.6 13.7 29.8 50.84.7
d2IMDM+LIF d3 d7 E14Tg2a
ES cellsDMEM+LIF
ZFαAS ES cells ZFαAS EBs
1 2 3 4 5 6 7 8 10 11 129
ZF-EBV
TA-EBVES EBs
ZFαS
10 11 12 13 14 15 169 17
ES EBs ES EBs
ZFαAS HS –40α
1 2 3 4 5 6 7 8
201 ++++ mAprtrevRT primers
Aprt
α-globin(498–493)
24 26 28 30 − 24 26 28 30 −
ZFαAS
ES EBs
+
ZFαS
24 26 28 30 −24 26 28 30 −
ES EBs
αH
β-actin(Loading control)
ZFα
AS
ZFα
S
ES EBs ES EBs
ZFα
AS
ZFα
S
ESEBs ESEBs
αH
αM
a
b
c
d
Figure 6 The ZF methylation phenomenon can be recapitulated in an ES cell system. (a) Detection of α-globin transcripts by RNase protection assay of total RNAisolated from ES cells transfected with the ZFαAS or ZFαS constructs either maintained undifferentiated in the presence of LIF (ES) or differentiated to 7-dembryoid bodies (EBs) after LIF removal. Human α-globin (HBA) transcripts were detected in undifferentiated ES cells in both ZFαS and ZFαΑS clones but werepresent only in embryoid bodies from ZFαS clones. Mouse α-globin (Hba-a) transcripts were present only in embryoid bodies. The left panel also shows mouse β-actin (Actb) as a loading control. (b) Semiquantitative RT–PCR done with the indicated primer pairs on RNA reverse transcribed with a combination of the HBA2antisense strand–specific primer 201 and the mouse primer mAprtrev as an internal control. The number of cycles at which aliquots where removed is indicated atthe bottom of the picture. HBA2 antisense RNAs are present in ZFαAS ES cells and embryoid bodies (EBs) but not in ZFαS clones (see Fig. 7 for a summary). +,genomic DNA positive control; −, no reverse transcriptase. (c) Southern-blot analysis of the methylation status of the HBA2 CpG island in ES and embryoid bodies(EBs) of clones generated with ZFαAS, HS –40α and ZFαS constructs. Genomic DNA from an EBV-transformed lymphocyte line from a normal individual (TA-EBV)and from individual ZF (ZF-EBV) was used as control. (d) Southern-blot analysis of the methylation status of the HBA2 CpG island in ZFαAS ES cells anddifferentiating embryoid bodies (EBs). ES cells were maintained undifferentiated in Dulbecco’s modified Eagle medium containing LIF and moved to Iscove’smodified Dulbecco’s medium with LIF 24 h before differentiation. We collected embryoid bodies at d 2, d 3 and d 7 of differentiation. The percentage ofmethylation calculated as the ratio of methylated signal to total signal is shown below the autoradiograph. Genomic DNA from E14Tg2a ES cells was used as acontrol. Detectable levels of HBA2 methylation are present as early as d 2 of differentiation.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics

A R T I C L E S
ubiquitin C (UBC) promoter28. This construct produced antisenseRNA transcripts running through the α-globin gene and, as before,this was associated with methylation of the α-globin CpG island,suggesting that methylation mediated by antisense RNA is a generaleffect rather than an isolated phenomenon related to this particularα–ZF chromosomal rearrangement.
DISCUSSIONHere we pursued a common situation in human genetics in which thereis a well defined clinical phenotype, in this case α-thalassemia, and yetno mutation can be found in the associated gene or its known regulatoryelements. Full characterization of this particular example, including theanalysis of long-range chromosome structure and detailed analysis ofepigenetic changes throughout the previously defined regulatorydomain, showed that the gene HBA2 has become insensitive to nuclease,the associated CpG island has been methylated and the gene has beensilenced by a mechanism dependent on the transcription of antisenseRNA. This adds an entirely new class of mutation to those currently con-sidered as causes of human genetic disease.
Antisense RNA transcripts have been previously implicated in theinitiation of genomic imprinting11–15,29,30 and X inactivation16,31,32.Recent studies in mouse have shown that expression of Xist, a non-coding RNA involved in X inactivation, is regulated by expression ofits antisense RNA Tsix16,32, driven by a promoter downstream of theXist gene. Similarly, silencing and methylation of CpG islands associ-ated with the promoters of imprinted genes are often associated withantisense RNA transcripts originating from CpG islands locateddownstream in the body of the imprinted gene (for example, Igf2r).Epigenetic changes and associated gene silencing often spread tomuch larger regions (over 100 kb) of the imprinted chromosome byunknown mechanisms. By contrast, silencing and methylation on theα–ZF chromosome is restricted to HBA2 and its CpG island. The pat-tern of methylation and chromatin structure of surrounding CpGislands seem to be unaffected5, indicating that the processes of RNA-mediated silencing and spreading can be dissociated.
Notably, the observations described here clearly show that the asso-ciation of antisense RNA transcripts with CpG methylation andsilencing is not unique to imprinting and the initiation of X inactiva-tion but can also occur at nonimprinted autosomal loci. Because wewere able to reproduce this phenomenon experimentally using anentirely different promoter from that driving antisense transcripts inthe unique α–ZF mutation, this is probably a general mechanism thatcould play a part in the normal regulation of gene expression (forexample, see ref. 33). Furthermore, this mechanism will probably beshown to underlie other inherited disorders, particularly those inwhich the locus associated with the disease lies in a gene-dense regionof the genome.
If DNA methylation induced by antisense RNA transcription fallswithin the natural repertoire of cellular responses, could it have a rolein acquired genetic disorders? It seems plausible that in malignantcells, where the bulk of the genome becomes hypomethylated, theresulting de-repression of SINEs, LINEs and retroviral sequences34,35
might generate transcriptional ‘noise’ leading to aberrant RNA tran-scripts, some of which may randomly initiate methylation of the CpGislands of key oncogenes and tumor suppressors. Such epigeneticmutations could contribute to the reservoir of genetic defects onwhich selection may operate in the evolution of malignant clones.
Here we reproduced antisense RNA–mediated gene silencing andde novo methylation of a CpG island with a relatively simple constructin a well defined experimental system compared with those previ-ously used to recapitulate the same phenomena from imprintedgenes. This model will allow investigators to study the closely associ-ated changes in methylation and chromatin that occur during devel-opment. In addition, it will help to determine the order of events andhierarchy of the epigenetic changes associated with methylation ofCpG islands and silencing of gene expression.
METHODSCell lines and culture conditions. We cultured K562, EBV-transformed lym-phocytes and MEL cells in RPMI 1640 medium (Sigma) supplemented with
NATURE GENETICS VOLUME 34 | NUMBER 2 | JUNE 2003 163
ZFαAS
ZFαS
HS –40α
HBA2
HBA2m
HS –40 α2ASUBC
HS –40 LUC7LHBA2
CCAAT ∆∆∆∆TATA
UBC
hαAS expressionES cells and EBs
RT–PCR
Yes(6/6)
No(5/6)
No(6/6)
No(2/2)
No(2/2)
Yes(3/6)
hα methylationEBs
Southern blot
Yes(6/6)
No(5/6)
No(6/6)
No(2/2)
No(2/2)
Yes(3/6)
Figure 7 Correlation between methylation andexpression of antisense transcripts. The variousconstructs used in the ES cell system are shown.HS –40 is indicated as a red box, HBA2 exons asblack boxes and LUC7L exons as blue boxes. Ingray is shown the UBC promoter fragment used todrive antisense transcription in HS−40α2ASUBCclones. The two columns on the right summarizethe results of the methylation studies in embryoidbodies (EBs) of the clones generated with thevarious constructs. In brackets is indicated thenumber of clones with the indicated behavior outof the total analyzed. HBA2 antisense (hα AS)expression was analyzed by RT–PCR as describedin Figure 6b. HBA2 methylation (hα methylation)was studied by Southern-blot analysis as in Figure6c for all the clones analyzed and by bisulfitesequencing for two ZFαAS clones (data notshown). Methylation of the human HBA2 CpGisland was present only in the clones that expressHBA2 antisense RNA. Only 3 of 6 cell linestransfected with HS–40α2ASUBC expressedantisense RNA transcripts, presumably due toposition effects. In support of the role ofantisense RNA transcription, however,methylation was seen only in these three celllines expressing antisense RNA.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics

A R T I C L E S
15% v/v fetal calf serum (Sigma), 50 U ml–1 penicillin (Gibco BRL), 50 µg ml–1
streptomycin (Gibco BRL) and 2 mM L-glutamine (Gibco BRL). We appliedselective pressure for chromosome 16 interspecific somatic hybrid cell linesbearing a single copy of a normal human chromosome 16 or of the α−ZF chro-mosome 16 in a MEL background by adding 1 mM adenine (Sigma), 1 mMmethotrexate (Lederle Laboratories) and 30 µM thymidine (Sigma). We incu-bated cultures incubated at 37 oC and gassed them with 5% carbon dioxide.
Engineering of constructs to study ZF silencing in mice and ES cells. We gen-erated constructs using the HS−40HBA2 cassette cloned into the SalI andBamHI sites of pSL301 to generate pSLHS−40HBA2. To include one loxP site,we linearized the plasmid with BamHI and ligated it with a double-strandedoligonucleotide obtained by annealing the oligo-loxP3 and loxP4. We excisedthe cassette HS–40HBA2 by digestion with EcoRV and released the HBA2 con-struct by digestion with XhoI.
To generate the pZFαAS and pZFαS with or without the loxP site, we cloneda 5.1-kb BclI fragment of the phage pHR11 (ref. 6) spanning the CpG islandand exons 1–3 of LUC7L into the BamHI site of plasmid pSLHS−40HBA2. Weobtained plasmids with the fragment in either orientation (pZFαAS andpZFαS). We released the fragments used for transgenesis and electroporationfrom the vector backbone by digestion with EcoRV.
To generate the plasmid pHBA2m, we cloned a 553-bp AatII–BbrPI frag-ment including the HBA2 promoter and exons 1 and 2 in pSL301 and mutatedit by PCR using primer pair ∆TATAF and ∆TATAR to remove the TATA box,followed by mutagenesis with primer pair CCAATF and CCAATR to introducethe CCAAT box mutation previously found to severely reduce α-globinexpression in transient assays36. We excised the 4.1-kb mutated HBA2 frag-ment from the vector by digestion with XhoI.
Finally, we cloned a 1.2-kb PstI UBC promoter fragment28 in the BamHI siteof pSLHS−40HBA2loxPB to generate the HS–40HBA2ASUBC construct. Wereleased the fragment used for electroporation from the vector backbone bydigestion with EcoRV. We gel-purified the fragments by electroelution beforetransgenesis and electroporation.
Generation and analysis of transgenic mice. All mouse experiments were car-ried out under UK Home Office license. We produced transgenic mice asdescribed17. We collected adult tissues and immediately froze them in liquidnitrogen for subsequent DNA or RNA extraction. To collect blood, we injectedmice with 100 U of Heparin just before death and collected the blood from thechest cavity after severing the major vessels.
To reduce the copy number in transgenic mice carrying loxP sites, we matedthem with mice carrying Cre recombinase driven by a GATA-1 promoter thatexpresses the recombinase very early in development37. We obtained embryosfrom mating male transgenic mice to wild-type (CBA × C57B) F1 females. Themorning on which a copulatory plug was noted was considered 0.5 d postcoitus. We extracted RNA from the yolk sac and blood, and we extracted DNAfrom the carcass to identify transgenic mice. We isolated primary mouse ery-throblasts from the spleens of adult mice treated with phenylhydrazine tostimulate erythropoiesis in the spleen38. We carried out RNase protectionassays as described17. We assayed17 DNaseI-hypersensitive sites in nuclei iso-lated from primary mouse erythroblasts.
Generation of ES cell clones and their differentiation. We maintained E14Tg2aES cells undifferentiated in gelatin-treated flasks in BHK-21 Glasgow minimalessential medium supplemented with 103 U ml–1 leukemia inhibitory factor (LIF;Esgro, Gibco BRL), 10% fetal calf serum (Globepharm), 6 × 10-4 M β-mercap-toethanol (Sigma) and 1× each of glutamine, sodium pyruvate and nonessentialamino acids (Gibco BRL). We obtained stable clones by co-electroporation of1–2 × 107 ES cells with 50 µg of purified construct fragment and 2 µg of NotI-lin-earized pPNT plasmid containing the neomycin resistance gene39. We isolatedG418-resistant colonies and analyzed them by PCR and Southern blotting forconstruct integration. We obtained embryoid bodies by plating the ES cells inIscove’s modified Dulbecco’s medium in the absence of LIF in bacterial petridishes to avoid cell adherence40. In general, we collected embryoid bodies after7 d of differentiation and we extracted DNA and RNA using standard methods.
RT–PCR analysis. We extracted total RNA from cells and tissues using the TRIreagent (Sigma) according to the manufacturer’s instructions. We directly
resuspended cell pellets from about 108 cells in 5 ml of TRI reagent and lysedthem by vortexing. We homogenized embryoid bodies and up to 0.4 g of tissuesamples in 5 ml of TRI reagent using a tissue homogenizer.
We used 1–5 µg of total RNA as substrate for the reverse transcriptionreactions using RNA-directed DNA polymerase Expand ReverseTranscriptase (Roche). We used 10 pmol oligo(dT), 12 pmol random hexa-mers or 25 pmol specific oligonucleotides per 50 µl reaction to prime DNAsynthesis. We denatured total RNA mixed with the primers and water for10 min at 65 oC. After denaturation, we added to the tubes 10 µl of 5×buffer (250 mM Tris-HCl, 200 mM KCl, 25 mM MgCl2, 2.5% Tween 20(v/v), pH 8.3), 2 µl dNTPs mix at 25 mM each, 5 µl 100 mM dithiothreitol,1 µl of a 40 U µl–1 RNase inhibitor RNAsin (Promega) and 1 µl of Expand(50U µl–1). We incubated the reactions for 1 h at 42 oC or at 37 oC whenrandom primers were used. We carried out PCR reactions using 1 µl of thefirst-strand cDNA generated by reverse transcription, 25 µl of 2× dimethyl-sulfoxide buffer (32 mM (NH4)2SO4, 134 mM Tris-HCl pH 8.8,20% dimethylsulfoxide (Sigma), 20 mM β-mercaptoethanol) supple-mented with 3.5 µl of 25 mM MgCl2 to a final concentration of 1.75 mM,0.4 µl of 100mM dNTPs (25 mM each, Pharmacia), 12.5 pmol of eachprimer, 2.5 U of Taq polymerase (Roche) and distilled water to a final vol-ume of 50 µl. The only exceptions were the PCRs for the amplification ofthe mouse Hba-a, Hprt and Aprt cDNAs, which we carried out using the10× buffer supplied with the Taq polymerase (Roche). After 5 min incuba-tion at 94 oC, we cycled reactions at 94 oC for 30 s, 56–60 oC for 30 s and72 oC for 30–120 s in a PTC-225 Peltier Thermal Cycler (MJ Research). Wecarried out reactions for 30 cycles, unless otherwise indicated.
DNA methylation assays. We digested 10 µg genomic DNA with PstI alone orwith PstI and the methylation-sensitive enzyme EagI. We hybridized Southernblots with the 1.48-kb PstI HBA probe (Fig. 5a), stripped them and probedthem again with a 1.2-kb fragment from the LUC7L CpG island (Fig. 5b).
We modified genomic DNA from ZFαAS embryoid bodies with bisul-fite41. We carried out two rounds of PCR using primer pairs F1 and APS1followed by amplification with nested primer pairs F2 and APS2 to amplifya 567-bp fragment from the human α-globin promoter. We cloned thisfragment in the EcoRV site of pZErO2k and sequenced it using the BigDyeterminator chemistry on an Applied Biosystems (ABI) 3700 sequencer.Sequences of all the primers used in this study are available on request.
Note: Supplementary information is available on the Nature Genetics website.
ACKNOWLEDGMENTSWe are grateful to L. Rose for preparation of the manuscript, C. Porcher for helpsetting up the ES cell differentiation assay, E. Li and T. Chen for helpful adviceand on-going collaboration and V. Samara and S. Butler for technical assistance.D.G. was supported by the Medical Research Council and the Staines ResearchFellowship, Exeter College, Oxford. This work was supported by the MedicalResearch Council.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 27 January; accepted 4 April 2003Published online 5 May 2003; doi:10.1038/ng1157
1. Cooper, D.N. & Krawczak, M. Human Gene Mutation (BIOS Scientific, Oxford,1993).
2. Bedell, M.A., Jenkins, N.A. & Copeland, N.G. Good genes in bad neighbourhoods.Nat. Genet. 12, 229–232 (1996).
3. Kleinjan, D.-J. & van Heyningen, V. Position effect in human genetic disease. Hum.Mol. Genet. 7, 1611–1618 (1998).
4. Steinberg, M.H., Forget, B.G., Higgs, D.R. & Nagel, R.L. Disorders of Hemoglobin(Cambridge University Press, Cambridge, 2001).
5. Barbour, V.M. et al. α-thalassemia resulting from a negative chromosomal positioneffect. Blood 96, 800–807 (2000).
6. Flint, J. et al. The relationship between chromosome structure and function at ahuman telomeric region. Nat. Genet. 15, 252–257 (1997).
7. Bird, A.P., Taggart, M.H., Nicholls, R.D. & Higgs, D.R. Non-methylated CpG-richislands at the human α-globin locus: implications for evolution of the α-globinpseudogene. EMBO J. 6, 999–1004 (1987).
8. Smith, Z.E. & Higgs, D.R. The pattern of replication at a human telomeric region(16p13.3): its relationship to chromosome structure and gene expression. Hum.Mol. Genet. 8, 1373–1386 (1999).
164 VOLUME 34 | NUMBER 2 | JUNE 2003 NATURE GENETICS
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics

A R T I C L E S
9. Brown, K.E. et al. Expression of α- and β-globin genes occurs within differentnuclear domains in haemopoietic cells. Nat. Cell Biol. 3, 602–606 (2001).
10. Tufarelli, C., Frischauf, A.-M., Hardison, R., Flint, J. & Higgs, D.R. Characterisationof a widely expressed gene (LUC7-LIKE) defining the centromeric boundary of thehuman α-globin domain. Genomics 71, 307–314 (2001).
11. Wutz, A. et al. Imprinted expression of the lgf2r gene depends on an intronic CpGisland. Nature 389, 745–749 (1997).
12. Smilinich, N.J. et al. A maternally methylated CpG island in KvLQT1 is associatedwith an antisense paternal transcript and loss of imprinting in Beckwith–Wiedemannsyndrome. Proc. Natl. Acad. Sci. USA 96, 8064–8069 (1999).
13. Rougeulle, C., Cardoso, C., Fontes, M., Colleaux, L. & Lalande, M. An imprintedantisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat.Genet. 19, 15–16 (1998).
14. Hayward, B.E. & Bonthron, D.T. An imprinted antisense transcript at the humanGNAS1 locus. Hum. Mol. Genet. 9, 835–841 (2000).
15. Wroe, S.F. et al. An imprinted transcript, antisense to Nesp, adds complexity to thecluster of imprinted genes at the mouse Gnas locus. Proc. Natl. Acad. Sci. USA 97,3342–3346 (2000).
16. Lee, J.T. & Lu, N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation.Cell 99, 47–57 (1999).
17. Higgs, D.R. et al. A major positive regulatory region located far upstream of thehuman α-globin gene locus. Genes Dev. 4, 1588–1601 (1990).
18. Sharpe, J.A. et al. Analysis of the human α-globin gene cluster in transgenic mice.Proc. Natl. Acad. Sci. USA 90, 11262–11266 (1993).
19. Higgs, D.R., Sharpe, J.A. & Wood, W.G. Understanding α-globin gene expression: astep towards effective gene therapy. Semin. Hematol. 35, 93–104 (1998).
20. Matzke, M., Matzke, A.J.M. & Kooter, J.M. RNA: guiding gene silencing. Science293, 1080–1083 (2001).
21. Epstein, C.J., Smith, S., Travis, B. & Tucker, G. Both X chromosomes function beforevisible X-chromosome inactivation in female mouse embryos. Nature 274, 500–503(1978).
22. Monk, M. & Harper, M.I. Sequential X chromosome inactivation coupled with cellu-lar differentiation in early mouse embryos. Nature 281, 311–313 (1979).
23. Martin, G.R. et al. X-chromosome inactivation during differentiation of female tera-tocarcinoma stem cells in vitro. Nature 271, 829–333 (1978).
24. Panning, B., Dausman, J. & Jaenisch, R. X-chromosome inactivation is mediated byXist RNA stabilization. Cell 90, 907–916 (1997).
25. Sheardown, S.A. et al. Stabilization of Xist RNA mediates initiation of X-chromo-some inactivation. Cell 91, 99–107 (1997).
26. Hooper, M., Hardy, K., Handyside, A., Hunter, S. & Monk, M. HPRT-deficient(Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells.Nature 326, 292–295 (1987).
27. Daniels, R., Lowell, S., Bolton, V. & Monk, M. Transcription of tissue-specific genesin human preimplantation embryos. Hum. Reprod. 12, 2251–2256 (1997).
28. Schorpp, M. et al. The human ubiquitin C promoter directs high ubiquitous expres-sion of transgenes in mice. Nucleic Acids Res. 24, 1787–1788 (1996).
29. Sleutels, F., Zwart, R. & Barlow, D.P. The non-coding Air RNA is required for silenc-ing autosomal imprinted genes. Nature 41, 810–813 (2002).
30. Sleutels, F., Barlow, D.P. & Lyle, R. The uniqueness of the imprinting mechanism.Curr. Opin. Genet. Dev. 10, 229–233 (2000).
31. Lee, J.T. & Jaenisch, R. Long-range cis effects of ectopic X-inactivation centres on amouse autosome. Nature 386, 275–279 (1997).
32. Lee, J.T., Davidow, L.S. & Warshawsky, D. Tsix, a gene antisense to Xist at theX-inactivation centre. Nat. Genet. 21, 400–404 (1999).
33. Futscher, B.W. et al. Role for DNA methylation in the control of cell type-specificmaspin expression. Nat. Genet. 31, 175–179 (2002).
34. Ehrlich, M. DNA hypomethylation and cancer. in DNA Alternations in Cancer (ed.Ehrlich, M.) 273–291 (Eaton, Natick, Massachusetts, 2000).
35. Baylin, S.B. & Herman, J.G. Epigenetics and loss of gene function in cancer. in DNAAlternations in Cancer (ed. Ehrlich, M.) 293–309 (Eaton, Natick, Massachusetts,2000).
36. Rombel, I. et al. Transcriptional activation of human adult α-globin genes by hyper-sensitive site-40 enhancer: function of nuclear factor-binding motifs occupied inerythroid cells. Proc. Natl. Acad. Sci. USA 92, 6454–6458 (1995).
37. Mao, X., Fujiwara, Y., Chapdelaine, A., Yang, H. & Orkin, S.H. Activation of EGFPexpression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood97, 324–326 (2001).
38. Spivak, J.L., Toretti, D. & Dickerman, H.W. Effect of phenylhydrazine-inducedhemolytic anemia on nuclear RNA polymerase activity of the mouse spleen. Blood42, 257–266 (1973).
39. Tybulewicz, V.L., Crawford, C.E., Jackson, P.K., Bronson, R.T. & Mulligan, R.C.Neonatal lethality and lymphopenia in mice with a homozygous disruption of thec-abl proto-oncogene. Cell 65, 1153–1163 (1991).
40. Keller, G., Kennedy, M., Papayannopoulou, T. & Wiles, M.V. Hematopoietic commit-ment during embryonic stem cell differentiation in culture. Mol. Cell. Biol. 13,473–486 (1993).
41. Clark, S.J., Harrison, J., Paul, C.L. & Frommer, M. High sensitivity mapping ofmethylated cytosines. Nucleic Acids Res. 11, 2990–2997 (1994).
42. Kulozik, A.E., Kar, B.C., Serjeant, G.R., Serjeant, B.E. & Weatherall, D.J. The mole-cular basis of α-thalassemia in India. Its interaction with the sickle cell gene. Blood71, 467–472 (1988).
43. Fei, Y.J., Fujita, S. & Huisman, T.H.J. Two different theta (θ)-globin gene deletionsobserved among black newborn babies. Br. J. Haematol. 68, 249–254 (1988).
NATURE GENETICS VOLUME 34 | NUMBER 2 | JUNE 2003 165
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reg
enet
ics














![Cytosine methylation is a conserved epigenetic feature ...€¦ · and repetitive element silencing [6]. Metazoan DNA methyltransferases (DNMT1, DNMT2, DNMT3a/3b [7]) catalyse this](https://img.pdfslide.us/doc/110x75/5eab33730f2ba76ce938ef9e/cytosine-methylation-is-a-conserved-epigenetic-feature-and-repetitive-element.jpg)













