Embed Size (px)
Citation preview

# 2007 The Authors
Journal compilation # 2007 Blackwell Publishing Ltd
doi: 10.1111/j.1600-0854.2007.00654.xTraffic 2007; 8: 1796–1814Blackwell Munksgaard
Trafficking of the bZIP Transmembrane TranscriptionFactor CREB-H into Alternate Pathways of ERAD andStress-Regulated Intramembrane Proteolysis
Daniel Bailey, Cristina Barreca and Peter O’Hare*
Marie Curie Research Institute, The Chart,Oxted, Surrey, RH8 0TL, UK*Corresponding author: Peter O’Hare,[email protected]
CREB-H is an ATF6-related, transmembrane transcription
factor that, in response to endoplasmic reticulum (ER)-
associated stress, is cleaved by Golgi proteases and
transported to the nucleus to effect appropriate adaptive
responses. We characterize the ER processing and turn-
over of CREB-H with results which have important im-
plications for ER stress regulation and signalling. We
show that CREB-H is glycosylated and demonstrate that
both the ER and nuclear forms of CREB-H have short half-
lives. We also show that CREB-H is subject to cycles of
retrotranslocation, deglycosylation and degradation
through the ER-associated degradation (ERAD) pathway.
Proteasome inhibition resulted in accumulation of a cyto-
solic intermediate but additionally, in contrast to inhib-
ition of glycosylation, promoted specific cleavage of CREB-
H and nuclear transport of the N-terminal-truncated prod-
uct. Our data indicate that under normal conditions CREB-
H is transported back from the ER to the cytosol, where it
is subject to ERAD, but under conditions that repress
proteasome function or promote load CREB-H is diverted
from this pathway instead undergoing cleavage and
nuclear transport. Finally, we identify a cytoplasmic deter-
minant involved in CREB-H ER retention, deletion of which
results in constitutive Golgi transport and corresponding
cleavage. We present a model where cellular stresses may
be sensed at different levels by different members of the
basic and leucine zipper domain transmembrane proteins.
Key words: ATF6, CREB3L3, CREB-H, MG132, protea-
some, retrotranslocation, Site-1 protease, Site-2 protease
Received 7 March 2007, revised and accepted for publica-
tion 13 September 2007, uncorrected manuscript pub-
lished online 17 September 2007, published online 17
October 2007
The endoplasmic reticulum (ER) responds to and regulates
many key aspects of cellular metabolism and homeostasis.
Perhaps one of its most important functions is in providing
the site of translation, folding, assembly and quality control
of protein synthesis, encompassing the range of proteins
destined for membrane insertion, secretion and transport
to various locations within the network of intracellular
organelles (1–3). The ER integrates several mechanisms
to monitor protein synthetic load and the fidelity of bio-
synthetic events in protein export pathways (1,4). These
latter functions are particularly important in tissues with
a heavy secretory role such as immunoglobulin-secreting
plasma cells, the pancreas or the liver.
Misfolded or incompletely assembled proteins are re-
tained in the ER in order to facilitate proper folding, post-
translational modification and subunit interactions (5,6).
Despite the presence of numerous general and specific
chaperones which aid in these processes (2,7–9) and the
operation of ER-associatedquality controlmechanisms, fail-
ure to achieve the fully folded or assembled product can
be a frequent outcome in the biosynthetic pathway. Thus,
the ER is the site of two further key mechanisms in re-
gulating the overall levels and balance of functional pro-
teins and the cellular responses to imbalance, induced by
stresses including metabolic fluctuation, mutation or infec-
tion. These processes are termed ER-associated degrada-
tion (ERAD) and regulated intramembrane proteolysis (RIP).
ERAD is a process that usually controls the degradation of
misfolded proteins in the ER but can also be involved in the
turnover of normal ER-resident proteins (1,6). While many
mechanistic aspects are still poorly understood, it is gener-
ally accepted that ERAD involves the recognition of
unfolded or misfolded proteins by ER-resident chaperones
such asBiP, calreticulin and calnexin. This is followedby the
unfolding and retrotranslocation through the translocon to
redirect the protein out of the ER into the cytosol, a process
that is coupled with the ubiquitin–proteasome system on
the cytosolic sideof themembrane that aids in translocation
and targeting for degradation (1,10). Recent results high-
light the complexity of ERAD, wherein different pathways
may be involved in the selection and retrotranslocation of
unfolded proteins dependent upon whether the unfolded
region iswithin the cytosolic or luminal aspect of the protein
(for reviews, see 6,11,12).
RIP represents an important overlapping control process
in the homeostatic responses to the presence of unfolded
proteins and also in the modulation of fatty acid levels,
sterol synthesis and other ER-associated stresses (13–17).
The key components in these systems comprise a distinct
class of membrane-associated transcription factors, anchor-
ing partners that localize the factors to the ER and
proteases that are located in a different compartment.
The transcription factors are inserted into ER membranes,
with DNA binding and transcriptional activation domains
oriented towards the cytosolic face of the membrane
1796 www.traffic.dk

(16,18–21). The main step in controlling the activity of
these factors in specific pathways appears to reside in
their regulated release from the ER in response to specific
stimuli. They are then transported to the Golgi, where they
are cleaved in a site-specific manner by resident pro-
teases. This results in the release of the cytosolic domain,
which is then transported to the nucleus to effect tran-
scription of specific target genes (22–26).
The prototype members of this class of transcription
factors are the sterol regulatory element-binding proteins,
SREBP 1 and 2, which regulate genes involved in choles-
terol and fatty acid metabolism (14,19,27). In response to
lower cholesterol levels in ER membranes, SREBPs are
transported to the Golgi, where they are cleaved by the
subtilisin-like Golgi protease, Site-1 protease (S1P), which
cleaves at a specific motif (RSVL) in SREBP on the lumenal
side of the membrane (22,23,26,28). This cleavage is fol-
lowed by a second cleavage by the metalloprotease, Site-2
protease (S2P) (25), which cleaves at a specific residue
within the transmembrane segment, thus liberating the
SREBP N-terminal transactivation domain of the protein.
This pathway has now been shown to converge with that
of the quality control of protein folding in the ER. ATF6 is
a basic and leucine zipper domain (bZIP) transcription
factor that resides in the ER by virtue of a single trans-
membrane domain and is also activated by proteolysis. In
this case, cleavage of ATF6 is not regulated by cholesterol
but instead, in response to the accumulation of unfolded
proteins (15,16,29,30). ATF6 possesses an RxxL motif
within its lumenal domain and is subject to cleavage by the
same proteases (S1P and S2P) that cleave SREBP (17).
Recent results indicate that ATF6 is retained in the ER
through interaction with the ER chaperone BiP/GRP78
(31,32). Loss of BiP binding by ER stress appears to
unmask Golgi localization signals, allowing ATF6 to be
transported to the site of active proteases in the Golgi (31).
ATF6 then activates the transcription of chaperones such
as BiP/GRP78 and other genes involved in responding to
the accumulation of unfolded proteins and represents an
important arm of the general unfolded protein response
(UPR) pathway.
Based upon sequence homology to a central section of
ATF6 and in particular the possession of a bZIP domain
adjacent to a transmembrane domain, we and others have
proposed that certain additional factors may be subject to
RIP pathways. These factors include Luman/CREB3 (33),
OASIS/CREB3L1 (34), CREB4/AIbZIP/CREB3L4 (35,36),
BBF2H7/CREB3L2 (37) and CREB-H/CREB3L3 (38). Not-
withstanding the conservation of the bZIP domains, char-
acterization of localization, modification, processing and
activation for many of these factors remains limited.
CREB-H was originally isolated from random sequencing
of complementary DNA (cDNA) clones, derived from the
hepatoma cell line Hep2G (38). Sequence analysis indi-
cated that CREB-H contained a potential transmembrane
segment adjacent to a bZIP domain and that it had the
capacity to act as a potent transcription factor (38). The
ability of CREB-H to act as a transcription factor and to bind
DNA was confirmed in a subsequent study (39), although
it was reported that it did not respond to UPR-activating
agents such as tunicamycin or activate BiP/GRP78 expres-
sion. In contrast, it has recently been reported that CREB-
H was processed in response to tunicamycin-induced ER
stress. Moreover, CREB-H may be specifically involved in
integrating stress to different stimuli as it was activated in
response to proinflammatory cytokines such as interleukin
(IL)-6 and tumour necrosis factor-a (TNF-a), presumably
through the induction of some form of ER stress by these
ligands (40). CREB-H processing was associated with the
activation genes involved in the acute-phase inflammation
response in the liver, and a number of genes in this
pathway and in lipidogenic responses were identified
(40). This notwithstanding, there is currently limited infor-
mation on the detailed aspects of CREB-H trafficking or
the determinants involved in localization.
Here, we expand on a number of features of CREB-H with
results that are consistent with its processing by the RIP
pathway but indicate that itmay respond to different aspects
of ER-associated stress, other than that resulting from the
accumulation of unfolded proteins in the lumen. CREB-H
was localized almost exclusively to the ER and at steady
state levels was fully glycosylated with sugar modifications
sensitive to endoglycosidase H and PNGase F deglycosyla-
tion activities. However, general inhibition of glycosylation
by tunicamycin, a treatment frequently used to evoke
a UPR, did not result in detectable cleavage and nuclear
translocation of CREB-H. However, we show that CREB-H
normally undergoes retrotranslocation from the ER to the
cytosol and that proteasome inhibition actively induces the
cleavage of de novo synthesized CREB-H resulting in nuclear
accumulation of the N-terminal product. Finally, we identify
a determinant on the cytosolic side of the membrane which
is involved ER retention of CREB-H. A mutant CREB-H
containing a deletion of this determinant results in constitu-
tive Golgi-localization and corresponding proteolytic cleav-
age, suggesting that the mode of stress sensing by CREB-H
may differ from that described for ATF6. We discuss our
results in the context of general systems of balancing load,
folding and degradation pathways and propose that different
transmembrane bZIP factors recognize different molecular
signatures of ER stress akin to the growing diversity in
pathways involved in recognition and degradation of lumenal
and cytosolic misfolded proteins (41–43).
Results
A conserved multi-section domain in a subset of
bZIP transcription factors
ATF6 is a bZIP transcription factor that is anchored in the
ER and subject to regulated transport and cleavage by the
Traffic 2007; 8: 1796–1814 1797
Processing of Membrane-Bound Transcription Factor, CREB-H

Golgi enzymes S1P and S2P in response to changes in the
levels of unfolded proteins in the ER (16,17,29,30). CREB-H
is one of the five additional proteins (Figure 1A) which have
recently been reported to contain a series of conserved
domains encompassing a bZIP domain, a putative trans-
membrane domain and a consistently spaced motif con-
forming to the S1P cleavage site (44), features that define
a specific sub class of the broad group of bZIP transcription
factors. The characteristic features of this sub class of bZIP
transcription factors i.e., the presence of a conserved
hydrophobic domain immediately C-terminal to the leucine
zipper and an adjacent consensus S1P cleavage site
[Regions IV and V, respectively, using the nomenclature
defined in (44)], are expanded in the lower section of
Figure 1A. The putative transmembrane segment identi-
fied using prediction algorithms contains the motif
CØØØØØØXØØL (where Ø represents a hydrophobic
amino acid), and in ATF6, Luman and CREB4 have
been shown to be responsible for membrane localiza-
tion (16,44,45). Region V, encompassing the sequence
SRTLHN in CREB-H, represents a conserved site for
cleavage by a cellular protease, S1P. The conserved sites
within ATF6, Luman and CREB4 have been shown to be
substrates for S1P cleavage (17,44,45).
Localization and expression of CREB-H
To investigate the subcellular localization modification
and processing of CREB-H, the corresponding cDNA was
amplified from a liver cDNA library and inserted into
expression vectors under the control of the comparatively
weak herpes simplex virus thymidine kinase (TK) pro-
moter. Preliminary experiments, based on the reports for
SREBP and ATF6, confirmed that the expression of CREB-H
under the control of the strong CMV promoter could lead
to erroneous results including incomplete post-transla-
tional modification and aberrant non-specific constitutive
cleavage (Figure S3). All constructs were therefore ana-
lyzed using expression from the TK promoter. Full-length
Figure 1: Comparison of the con-
served multi-domain region in the
membrane-bound bZIP transcription
factors. Schematic and constructs used
in this study. A) Schematic of the relative
lengths and location of the conserved
domains and S1P site (5) in the bZIP
transcription factors; CREB-H, CREB4,
Luman, OASIS, BBF2H7 and ATF6. The
amino acid sequence alignment of the
predicted transmembrane domain (region
IV, long black bar) and S1P site (region V,
short black bar) are highlighted. The
blocks indicate regions with similarity or
identity in 3 out of 5 sequences. B)
Schematic of the CREB-H constructs
used in this study. The location of the
N-terminal SV5-epitope tag is marked by
black shading at position 1. C) Compara-
tive expression levels of the CREB-H
constructs following transient transfec-
tion into COS-1 cells and subsequent
analysis by SDS–PAGE/Western blot
probed for the SV5-epitope tag. TM, trans-
membrane domain.
1798 Traffic 2007; 8: 1796–1814
Bailey et al.

and mutant constructs were engineered to contain an
N-terminal or C-terminal tag for ease of analysis byWestern
blotting and immunofluorescence (Figure 1B).
Full-length CREB-H migrated as a species of apparent
molecular weight of 75 kDa, with no lower molecular
weight cleavage products observed (Figure 1C, lane 2).
The approximately 75-kDa species could be observed
to migrate as a closely spaced doublet (particularly on
lower exposures, see below), indicating potential post-
translational modification such as phosphorylation. As with
other members of this class, the apparent molecular
weight is considerably larger than the predicted mo-
lecular weight of 49 kDa, again suggesting additional
post-translational modifications, such as glycosylation
(see below). This migration is consistent with the previous
reports for CREB-H (38–40). CREB-H contains a C-terminal
sequence GDEL, which conforms to the KDEL-type ER
retrieval signal. To examine whether it was involved in
localization, we constructed a deletion mutant (CREB-
HDC1), which lacked the C-terminal 3 residues disrupting
this potential motif. This construct was expressed with
similar abundance and migration as the parental CREB-H
(Figure 1C, lane 3). We also made a variant, CREB-
HDTMC, which lacked the complete C-terminal region
including the predicted transmembrane domain, terminat-
ing at residue 323. This protein migrated at approximately
42 kDa (Figure 1C, lane 4), slightly greater than the
expected molecular weight of approximately 37 kDa, and
was also frequently observed as two or three closely
migrating species, possibly representing post-translational
modifications or short cleavage at the C-terminal end.
We next examined the location of CREB-H by immuno-
fluorescence, co-staining with antibodies to either calreti-
culin, as a marker for the ER, or trans Golgi network
(TGN)46 for the Golgi (46). CREB-H was present exclu-
sively in the cytoplasm, mostly with a reticular pattern
together with some perinuclear staining, but with little
evidence of any nuclear material (Figure 2A, panels i–iii).
CREB-H colocalized with the ER marker, calreticulin, but
not to any significant extent with the Golgi (Figure 2C).
These results are broadly consistent with those previously
reported, although a minor population was observed in the
Golgi in previous studies (38,39).
CREB-HDC1 exhibited an identical pattern of localization as
the full-length protein (Figure 2A, panels iv–vi), indicating
that the main ER localization of the steady-state population
is not because of the potential KDEL retrieval sequence,
although these results do not rule out a role for this
sequence in more detailed kinetic aspects of CREB-H
transport (see Discussion).
CREB-HDTMC (aa 1–323) terminates at the invariant
cysteine of the putative S2P site (see above, Figure 1A,
Region II, black-shaded cysteine residue) and thus repre-
sents the predicted cleavage product encompassing the
active N-terminal transcription domain. In contrast to full-
length CREB-H and CREB-HDC1, CREB-HDTMC exhibited
an almost exclusively nuclear localization, with no signifi-
cant ER staining (Figure 2A, CREB-HDTMC, panels vii–ix).
Thus the C-terminal region encompassing the transmem-
brane and lumenal domain efficiently anchored CREB-H in
the ER. Note, although that these results do not rule out
a role for the N-terminal region in retention of full-length
CREB-H in the ER (see Results below and Discussion).
Targeting of the N-terminal domain of CREB-H to the
nucleus was presumably because of a nuclear localization
signal embedded within this region, possibly within the
bZIP domain itself as has previously been observed with
several members of this class.
When originally isolated, CREB-H was reported to be
considerably more abundant in the liver than in other tis-
sues (38). We wished to examine localization of CREB-H in
a liver cell type and utilized the liver carcinoma-derived cell-
line, human hepatocarcinoma cells (HepG2) (47). Each of
the CREB-H constructs showed a qualitatively identical
pattern in HepG2 cells compared with COS-1 cells, with
the full length (and DC1 variant, data not shown) exhibiting
a cytoplasmic reticular pattern (Figure 2B) co-localizing
with calreticulin (data not shown) and CREB-HDTMC
localizing exclusively to the nucleus (Figure 2B).
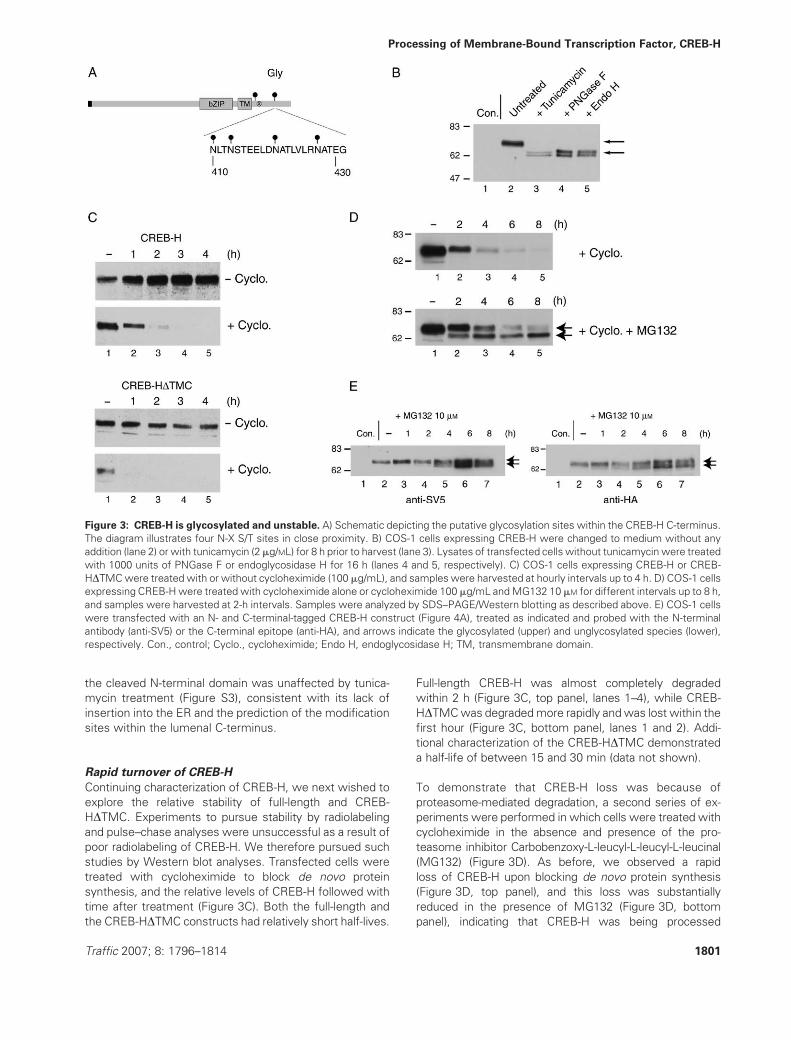
Characterization of CREB-H glycosylation
CREB-H displayed a much slower migration profile in SDS–
PAGE gels than predicted (Figure 1C), although the basis
for this has not been investigated. The CREB-H has five
potential glycosylation sites matching the motif Nx(S/T),
with four in a short 20-residue region within the predicted
lumenal domain of the protein (Figure 3A). We wished to
examine the glycosylation status of CREB-H and confirm
our prediction that the larger apparent molecular weight
was the result of glycosylation. Treatment of cells express-
ing full-length CREB-H with tunicamycin resulted in a dis-
tinct shift in mobility with all of the population being shifted
to a faster-migrating doublet (Figure 3B). Longer treatment
times or increased concentrations had no further effect. To
confirm the proposal that CREB-H was glycosylated and
that the reduction in size was because of the inhibition of
glycosylation, we also examined sensitivity of CREB-H to
treatment with deglycosylating enzymes, endoglycosidase
H and PNGase F. Incubation with either enzyme resulted in
complete conversion of CREB-H to the faster-migrating
doublet (Figure 3B, lanes 2, 4 and 5). Together, these
results provide strong evidence that CREB-H was quanti-
tatively glycosylated that tunicamycin treatment could
fully inhibit CREB-H glycosylation (see below) and that
modification was of the high mannose-type sensitive,
representing processing by enzymes of the ER. As indi-
cated, the unglycosylated (Figure 3B, lane 3) and deglyco-
sylated (Figure 3B, lanes 4 and 5) CREB-H frequently
migrated as a doublet, indicative of potential additional
post-translational modifications. As expected, migration of
Traffic 2007; 8: 1796–1814 1799
Processing of Membrane-Bound Transcription Factor, CREB-H
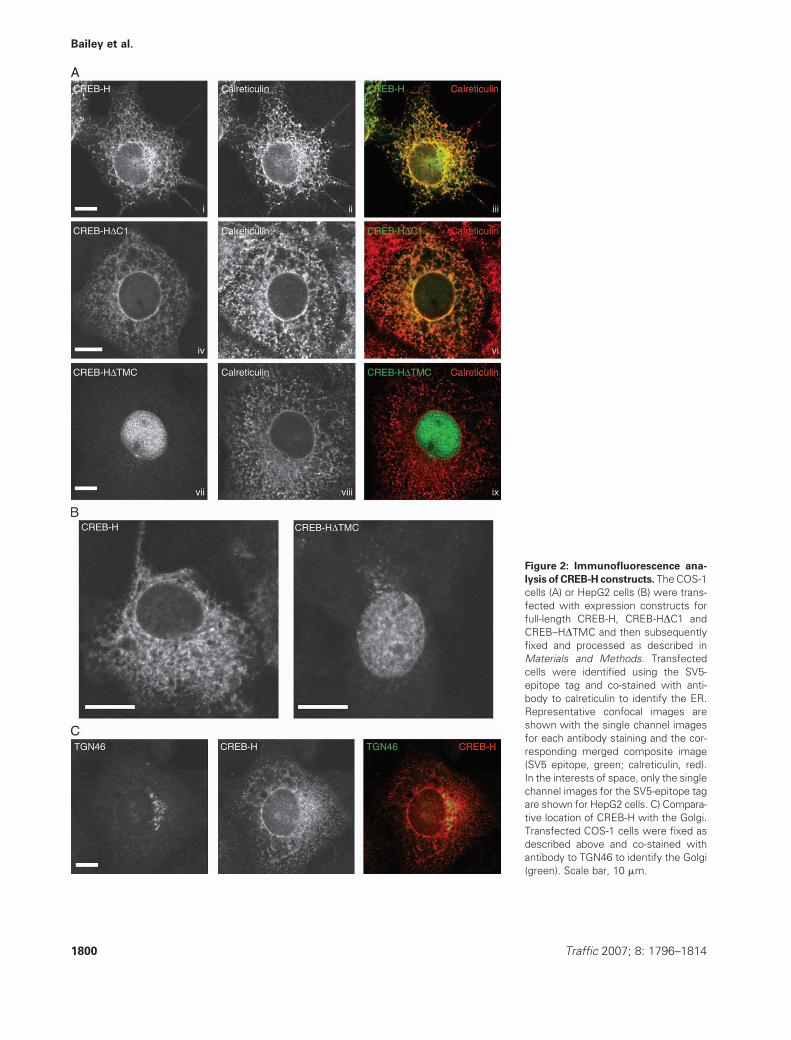
Figure 2: Immunofluorescence ana-
lysis of CREB-H constructs. The COS-1
cells (A) or HepG2 cells (B) were trans-
fected with expression constructs for
full-length CREB-H, CREB-HDC1 and
CREB–HDTMC and then subsequently
fixed and processed as described in
Materials and Methods. Transfected
cells were identified using the SV5-
epitope tag and co-stained with anti-
body to calreticulin to identify the ER.
Representative confocal images are
shown with the single channel images
for each antibody staining and the cor-
responding merged composite image
(SV5 epitope, green; calreticulin, red).
In the interests of space, only the single
channel images for the SV5-epitope tag
are shown for HepG2 cells. C) Compara-
tive location of CREB-H with the Golgi.
Transfected COS-1 cells were fixed as
described above and co-stained with
antibody to TGN46 to identify the Golgi
(green). Scale bar, 10 mm.
1800 Traffic 2007; 8: 1796–1814
Bailey et al.
the cleaved N-terminal domain was unaffected by tunica-
mycin treatment (Figure S3), consistent with its lack of
insertion into the ER and the prediction of the modification
sites within the lumenal C-terminus.
Rapid turnover of CREB-H
Continuing characterization of CREB-H, we next wished to
explore the relative stability of full-length and CREB-
HDTMC. Experiments to pursue stability by radiolabeling
and pulse–chase analyses were unsuccessful as a result of
poor radiolabeling of CREB-H. We therefore pursued such
studies by Western blot analyses. Transfected cells were
treated with cycloheximide to block de novo protein
synthesis, and the relative levels of CREB-H followed with
time after treatment (Figure 3C). Both the full-length and
the CREB-HDTMC constructs had relatively short half-lives.
Full-length CREB-H was almost completely degraded
within 2 h (Figure 3C, top panel, lanes 1–4), while CREB-
HDTMCwas degradedmore rapidly andwas lost within the
first hour (Figure 3C, bottom panel, lanes 1 and 2). Addi-
tional characterization of the CREB-HDTMC demonstrated
a half-life of between 15 and 30 min (data not shown).
To demonstrate that CREB-H loss was because of
proteasome-mediated degradation, a second series of ex-
periments were performed in which cells were treated with
cycloheximide in the absence and presence of the pro-
teasome inhibitor Carbobenzoxy-L-leucyl-L-leucyl-L-leucinal
(MG132) (Figure 3D). As before, we observed a rapid
loss of CREB-H upon blocking de novo protein synthesis
(Figure 3D, top panel), and this loss was substantially
reduced in the presence of MG132 (Figure 3D, bottom
panel), indicating that CREB-H was being processed
Figure 3: CREB-H is glycosylated and unstable. A) Schematic depicting the putative glycosylation sites within the CREB-H C-terminus.
The diagram illustrates four N-X S/T sites in close proximity. B) COS-1 cells expressing CREB-H were changed to medium without any
addition (lane 2) or with tunicamycin (2mg/ML) for 8 h prior to harvest (lane 3). Lysates of transfected cells without tunicamycin were treated
with 1000 units of PNGase F or endoglycosidase H for 16 h (lanes 4 and 5, respectively). C) COS-1 cells expressing CREB-H or CREB-
HDTMCwere treated with or without cycloheximide (100 mg/mL), and samples were harvested at hourly intervals up to 4 h. D) COS-1 cells
expressing CREB-Hwere treated with cycloheximide alone or cycloheximide 100 mg/mL andMG132 10 mM for different intervals up to 8 h,
and samples were harvested at 2-h intervals. Samples were analyzed by SDS–PAGE/Western blotting as described above. E) COS-1 cells
were transfected with an N- and C-terminal-tagged CREB-H construct (Figure 4A), treated as indicated and probed with the N-terminal
antibody (anti-SV5) or the C-terminal epitope (anti-HA), and arrows indicate the glycosylated (upper) and unglycosylated species (lower),
respectively. Con., control; Cyclo., cycloheximide; Endo H, endoglycosidase H; TM, transmembrane domain.
Traffic 2007; 8: 1796–1814 1801
Processing of Membrane-Bound Transcription Factor, CREB-H

through the ubiquitin–proteasome route. We did not detect
any retardation in CREB-H turnover in the presence of
lysosomal inhibitors (data not shown). These data are
consistent with the interpretation that CREB-H is a relatively
unstable ER protein, whose degradation is normally medi-
ated by the proteasome.
Interestingly, we observed that upon proteasome inhibi-
tion, in addition to substantial stabilization, CREB-H under-
went a shift in migration from the fully glycosylated 75-kDa
form to a lower approximately 62-kDa form, with similar
migration to the unglycosylated form of CREB-H (Figure 3D,
lower arrows). Further analysis of a CREB-H construct with
both an N-terminal tag (SV5) and a C-terminal (HA) tag
indicated that the MG132-induced product contained both
termini and thus was derived from full-length CREB-H and
not a cleaved or breakdown product (Figure 3E, SV5 and
HA; see also Figure 4). However, MG132 surprisingly did
have an effect, in that while it stabilized the full length, it
induced the appearance of a specific cleavage product of
CREB-H, as more fully described below.
CREB-H undergoes retrotranslocation through the
ERAD pathway
The appearance of the full-length but apparently under-
glycosylated form of CREB-H may have occurred either
through inhibition of glycosylation or through loss of
glycosylation. The main cellular mechanism for removing
glycosylation of ER-derived proteins occurs in the cytosol
as part of the pathway following retrotranslocation of
membrane or in the luminal ER proteins for subsequent
proteasomal degradation.
To further characterize CREB-H degradation and possible
retrotranslocation, we analyzed the full-length CREB-H con-
struct with different epitope tags at the N- and C-termini
(Figure 4A) and analyzed the disposition of the termini, in
the presence or absence of proteasome inhibitors, using
selective permeabilization with digitonin. This method has
been previously used to examine the topological distribu-
tion of transmembrane proteins and utilizes the ability of
digitonin to permeabilize the plasma membrane but not
the ER membranes, allowing access of antibody probes
to the cytosol but not the lumenal side of the ER (48).
Therefore, transfected cells were treated with or without
10 mm MG132 for 6 h, fixed in 3% paraformaldehyde,
washed with PBS and treated with either digitonin (40 mg/
mL for 3 min) to permeabilize just the plasma membrane
or with Nonidet P-40 (NP-40) (0.5% for 3 min) to per-
meabilize all membranes. Cells were processed on ice.
Fixed and permeabilized cells were then blocked and
stained using primary antibodies directed against either
the SV5-epitope tag or HA-epitope tag or calreticulin as
described above.
The results demonstrate that in the absence of protea-
some inhibitor, CREB-H was detected in the ER (Figure 4A,
upper left panels) as described above. Both the N- and
C-termini of the protein were readily detected using
NP-40 permeabilization. In marked contrast, in cells per-
meabilized with digitonin (Figure 4A, upper right panels,
�MG132), only the N-terminal epitope was detected. This
result clearly indicates that the cells were permeabilized
because the N-terminus was detected, but that the
C-terminus was not available under identical conditions,
consistent with the predicted topology of CREB-H pos-
sessing a C-terminal lumenal domain and cytoplasmic
N-terminus. (Formally, this is the first direct demonstration
confirming the likely CREB-H membrane topology). In cells
treated with MG132, the N- and C-termini were detected
using NP-40 permeabilization as expected. However, the
C-terminus of CREB-H could also now be detected using
digitonin permeabilization, in marked contrast to the result
in the absence of proteasome inhibition under identical
conditions. These results are consistent with the interpret-
ation that CREB-H is retrotranslocated out of the ER into
the cytosol (Figure 4A, lower panels, þMG132). We noted
that under conditions of proteasome inhibition when using
NP-40 permeabilization, some CREB-H could be detected
in the nucleus using the C-terminal tag, indicating that at
least a population of nuclear CREB-H may have been full
length. This was not detected under conditions of digitonin
permeabilization, consistent with the lack of nuclear per-
meabilization with this detergent. In additional control ex-
periments in which cells were stained for the ER lumenal
marker calreticulin, the results (Figure 4B) confirmed that,
as expected, digitonin permeabilization of MG132-treated
cells did not allow general detection of ER proteins in the
cytosol and thus that the observation of the carboxy
terminus of CREB-H in the cytosol after proteasome
inhibition was not some non-specific effect upon the ER.
These results strongly indicate that MG132 treatment
revealed the presence of a cytosolic population. We next
examined the colocalization between CREB-H and calreti-
culin in the presence and absence of MG132. Normally,
CREB-H completely overlapped with the ER marker
(Figure 4C, �MG132; see also Figure 2). In the presence
of MG132, while there was clear overlap with calreticulin,
a distinct more diffuse population could be readily
observed, which did not colocalize with calreticulin (Fig-
ure 4C, þMG132). This difference is highlighted by the
arrows in the inset of the colocalization patterns and is
consistent with the biochemical fractionation (see below)
likely represents the cytosolic retrotranslocated CREB-H.
Finally, to provide conclusive evidence for the proposal of
CREB-H retrotranslocation and ERAD, we performed sub-
cellular fractionation to examine the distribution of the
protein between ER membranes and cytosolic compart-
ments in the absence or presence of proteasome inhib-
ition. Transfected cells treated with or without MG132 as
above were fractionated into the ER membrane and
soluble cytosolic fractions as described in the Materials
and Methods (Figure 4D). In the absence of MG132,
CREB-H was found in the ER membrane (100 000 � g
1802 Traffic 2007; 8: 1796–1814
Bailey et al.
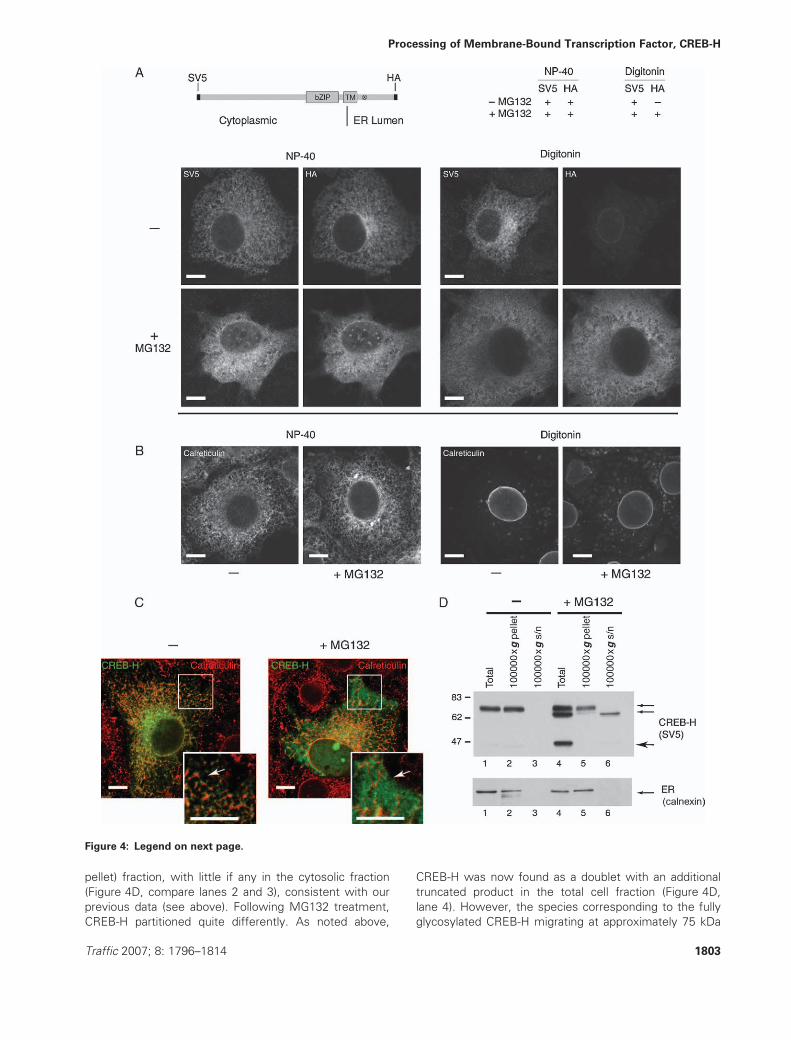
pellet) fraction, with little if any in the cytosolic fraction
(Figure 4D, compare lanes 2 and 3), consistent with our
previous data (see above). Following MG132 treatment,
CREB-H partitioned quite differently. As noted above,
CREB-H was now found as a doublet with an additional
truncated product in the total cell fraction (Figure 4D,
lane 4). However, the species corresponding to the fully
glycosylated CREB-H migrating at approximately 75 kDa
Figure 4: Legend on next page.
Traffic 2007; 8: 1796–1814 1803
Processing of Membrane-Bound Transcription Factor, CREB-H

was found almost exclusively in the ERmembrane fraction
(Figure 4D, lane 5) and none in the soluble cytosolic
fraction (lane 6). In contrast, the lower of the doublet
species (approximately 62 kDA) corresponding to the
unglycosylated form of CREB-H was detected solely in
the cytosolic fraction (Figure 4D, lane 6) with little in the ER
membrane fraction. Calnexin partitioning was used as a
control to confirm the integrity of the ER fractionation,
with none being found in the soluble fraction (Figure 4D,
lower panel). In addition, we observed in the presence of
MG132 the appearance of an approximately 45-kDa prod-
uct (Figure 4D, lane 4). The appearance of this product is
described in more detail later (Figure 5 and Discussion).
Taken together, our results firstly on the short half-life of
CREB-H, secondly on the appearance during proteasome
inhibition of a stabilized, higher mobility (but intact) isoform
co-migrating with unglycosylated CREB-H, thirdly on the
access of the C-terminus to selective probes specifically in
the presence of MG132 and finally on the differential
partitioning of the two CREB-H species to membrane
versus soluble fractions provide compelling evidence that
CREB-H is normally retrotranslocated from the ER, degly-
cosylated (presumably by peptide:N-glycanase activity)
(49) and subject to proteasome-mediated degradation.
Response of CREB-H to stress-inducing agents
Previous studies have demonstrated that chemical agents
that induce the accumulation of unfolded proteins such
as DTT, tunicamycin or thapsigargin induce forward trans-
port to the Golgi and cleavage of ATF6 (15,16) and OASIS
(50). However, contrary to these results, and despite the
ability of S1P to cleave Luman/CREB3 (45) and CREB4
(44), cleavage of these factors in response to DTT or tunica-
mycin has not been convincingly demonstrated (see
Discussion). Recently, evidence has been presented indi-
cating that CREB-H could be cleaved in response to DTT/
tunicamycin or thapsigargin (40), although a previous
report failed to observe cleavage of CREB-H in response
to tunicamycin (38).
Before examining the cleavage of CREB-H in response to
tunicamycin treatment, we wished to confirm that CREB-H
could be a substrate for S1P; we examined cleavage after
Brefeldin A (BFA) treatment as shown for SREBP (51) and
ATF6 (17). The results (Figure S1) show that within 30 min,
BFA treatment induced the appearance of a novel 45-kDa
N-terminal product, which co-migrated with CREB-HDTMC.
The BFA-induced cleavage was absolutely dependent upon
the S1P consensus site, being abolished by a single amino
acid substitution of arginine at position 361 to alanine
(R361A) (Figure S1A). The BFA treatment also induced
relocalization of CREB-H to the nucleus, consistent with
the cleavage at both the S1P and S2P sites after BFA-
induced Golgi redistribution (Figure S1B). Furthermore,
cotransfection of full-length CREB-H with the ER-localized
form of S1P (S1P.KDEL) resulted in the appearance of the
45-kDa CREB-H cleavage product and the appearance of
nuclear CREB-H. In contrast, in cells coexpressing CREB-H
and the S1P.KDAS protein, neither significant production of
the CREB-H cleavage product (Figure S2) nor any detectable
nuclear staining were observed. Taken together, these
results provide robust evidence that CREB-H is an ER
membrane-bound protein, which contains a consensus
S1P site, and is a substrate for S1P (and presumably S2P),
allowing translocation to the nucleus.
We next examined the effect of the stress-inducing
agent tunicamycin. Cells were transfected with full-length
CREB-H and treated with tunicamycin (2 mg/mL) for in-
creasing lengths of time. Under these conditions, tunica-
mycin did not induce the appearance of the approximately
45-kDaN-terminal product to anysignificant level (Figure 5A).
Tunicamycin as expected was functional, clearly resulting
in rapid loss of glycosylation of CREB-H by 2 h after
treatment such that between 4 and 8 h, only the unglyco-
sylated doublet remained (Figure 5A, lanes 5 and 6). As
tunicamycin prevents glycosylation of newly synthesized
proteins (rather than actively deglycosylating the existing
population), this result is consistent with our previous
results that the normal fully modified form is turned over
within 2–4 h through the ERAD pathway. The newly
Figure 4: CREB-H processing through the ERAD pathway. A) Schematic of the dual-tagged CREB-H construct with the N-terminal
(SV5-epitope) and C-terminal (HA-epitope) tags. Cells expressing the dual-tagged CREB-H were treated with or without MG132 (10 mM for
6 h) as indicated. After fixation with paraformaldehyde, cells were permeabilized with NP-40 to permeabilize all membranes (left-hand
panel) or digitonin to selectively permeabilize the plasma membrane (right-hand panel) as described inMaterials and Methods. Cells were
then stained with the anti-SV5 antibody to detect the N-terminus of CREB-H or the anti-HA antibody to detect the C-terminus. For clarity,
the results are summarized within the table (top right). B) Control panel showing localization of calreticulin in NP-40- or digitonin-
permeabilized cells. Cells with or without MG132 treatment were permeabilized as indicated and stained with anti-calreticulin antibody.
The ER-associated calreticulin could not be detected by digitonin permeabilization, and MG132 did not induce any altered localization.
C) MG132 induces altered cytoplasmic localization of CREB-H. Cells were treated as indicated, fixed and permeabilized with methanol,
and localization of CREB-H and calreticulin was examined. Complete colocalization was observed in the absence of MG132, while in its
presence, a distinct population (arrow) was observed without the calreticulin-associated population. This is readily seen in the inset. Scale
bar, 10 mm. D) Subcellular fractionation was performed as described inMaterials and Methods. Samples of total cell extract, ER (100 000 � g
pellet) fractions and cytosolic (100 000 � g supernatant [s/n]) fractions in the absence (lanes 1–3) or presence of MG132 (lanes 4–6) were
analyzed for CREB-H or the ER marker, calnexin. The results demonstrate the appearance of the higher mobility form in the presence of
MG132 (lane 4) and that this form was selectively present in the soluble fraction (lane 6), while the fully glycosylated form remained with
the ER membrane fraction (lane 5). The cleaved N-terminal form is also seen in the MG132-treated total lysate and partitions in the nuclear
fraction (not shown). TM, transmembrane domain.
1804 Traffic 2007; 8: 1796–1814
Bailey et al.
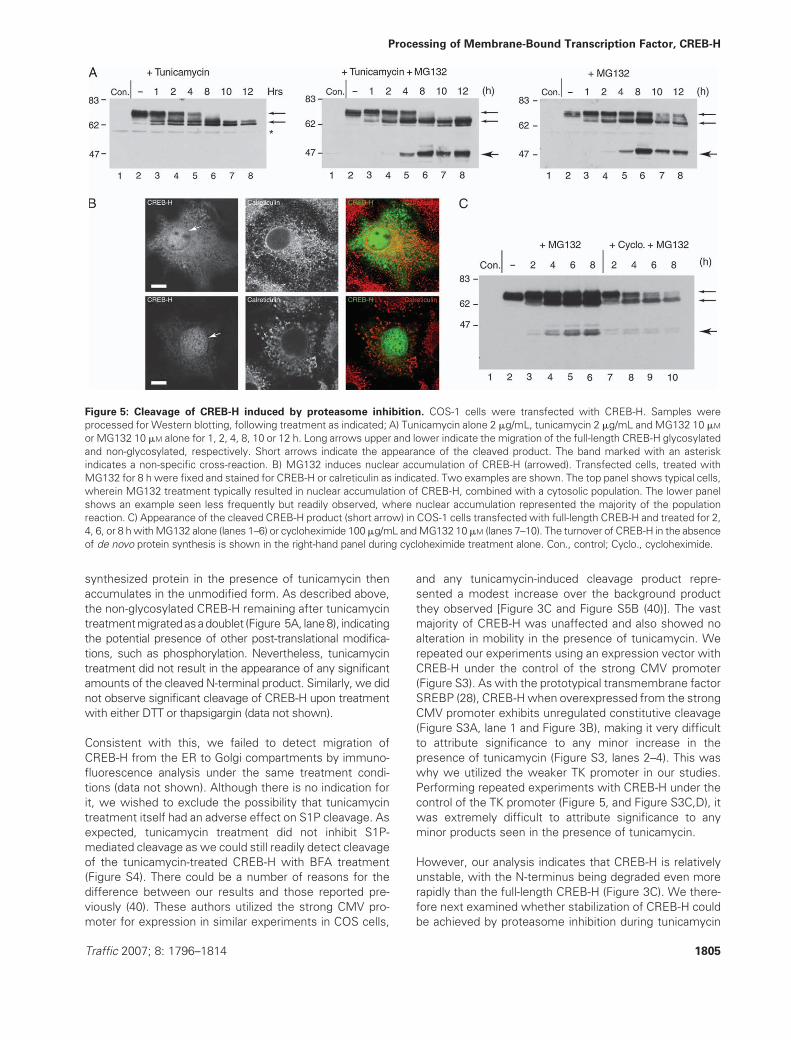
synthesized protein in the presence of tunicamycin then
accumulates in the unmodified form. As described above,
the non-glycosylated CREB-H remaining after tunicamycin
treatmentmigratedasadoublet (Figure 5A, lane8), indicating
the potential presence of other post-translational modifica-
tions, such as phosphorylation. Nevertheless, tunicamycin
treatment did not result in the appearance of any significant
amounts of the cleaved N-terminal product. Similarly, we did
not observe significant cleavage of CREB-H upon treatment
with either DTT or thapsigargin (data not shown).
Consistent with this, we failed to detect migration of
CREB-H from the ER to Golgi compartments by immuno-
fluorescence analysis under the same treatment condi-
tions (data not shown). Although there is no indication for
it, we wished to exclude the possibility that tunicamycin
treatment itself had an adverse effect on S1P cleavage. As
expected, tunicamycin treatment did not inhibit S1P-
mediated cleavage as we could still readily detect cleavage
of the tunicamycin-treated CREB-H with BFA treatment
(Figure S4). There could be a number of reasons for the
difference between our results and those reported pre-
viously (40). These authors utilized the strong CMV pro-
moter for expression in similar experiments in COS cells,
and any tunicamycin-induced cleavage product repre-
sented a modest increase over the background product
they observed [Figure 3C and Figure S5B (40)]. The vast
majority of CREB-H was unaffected and also showed no
alteration in mobility in the presence of tunicamycin. We
repeated our experiments using an expression vector with
CREB-H under the control of the strong CMV promoter
(Figure S3). As with the prototypical transmembrane factor
SREBP (28), CREB-Hwhen overexpressed from the strong
CMV promoter exhibits unregulated constitutive cleavage
(Figure S3A, lane 1 and Figure 3B), making it very difficult
to attribute significance to any minor increase in the
presence of tunicamycin (Figure S3, lanes 2–4). This was
why we utilized the weaker TK promoter in our studies.
Performing repeated experiments with CREB-H under the
control of the TK promoter (Figure 5, and Figure S3C,D), it
was extremely difficult to attribute significance to any
minor products seen in the presence of tunicamycin.
However, our analysis indicates that CREB-H is relatively
unstable, with the N-terminus being degraded even more
rapidly than the full-length CREB-H (Figure 3C). We there-
fore next examined whether stabilization of CREB-H could
be achieved by proteasome inhibition during tunicamycin
Figure 5: Cleavage of CREB-H induced by proteasome inhibition. COS-1 cells were transfected with CREB-H. Samples were
processed for Western blotting, following treatment as indicated; A) Tunicamycin alone 2 mg/mL, tunicamycin 2 mg/mL and MG132 10 mM
or MG132 10 mM alone for 1, 2, 4, 8, 10 or 12 h. Long arrows upper and lower indicate the migration of the full-length CREB-H glycosylated
and non-glycosylated, respectively. Short arrows indicate the appearance of the cleaved product. The band marked with an asterisk
indicates a non-specific cross-reaction. B) MG132 induces nuclear accumulation of CREB-H (arrowed). Transfected cells, treated with
MG132 for 8 h were fixed and stained for CREB-H or calreticulin as indicated. Two examples are shown. The top panel shows typical cells,
wherein MG132 treatment typically resulted in nuclear accumulation of CREB-H, combined with a cytosolic population. The lower panel
shows an example seen less frequently but readily observed, where nuclear accumulation represented the majority of the population
reaction. C) Appearance of the cleaved CREB-H product (short arrow) in COS-1 cells transfected with full-length CREB-H and treated for 2,
4, 6, or 8 hwithMG132 alone (lanes 1–6) or cycloheximide 100mg/mL andMG132 10mM (lanes 7–10). The turnover of CREB-H in the absence
of de novo protein synthesis is shown in the right-hand panel during cycloheximide treatment alone. Con., control; Cyclo., cycloheximide.
Traffic 2007; 8: 1796–1814 1805
Processing of Membrane-Bound Transcription Factor, CREB-H

treatment (Figure 5A, centre, lanes 4–8, short arrow). The
results demonstrated that in the presence of MG132,
tunicamycin induced the appearance of the approximately
45-kDa cleavage product beginning between 2 and 4 h
after treatment and persisting up to 12 h. Surprisingly,
however in control experiments, this product could be
detected as efficiently in the presence of MG132 alone
(Figure 5A, right, lanes 4–8, short arrow), with no signifi-
cant cumulative effect of tunicamycin and MG132 com-
bined. Consistent with the appearance of the N-terminal
cleavage product, treatment with MG132 also induced the
accumulation of nuclear CREB-H (Figure 5B, arrows).
Together, the observations of the appearance of the
45-kDa cleavage product and nuclear CREB-H indicated
that MG132 proteasome inhibition was inducing specific
cleavage and transport of the nuclear form of CREB-H.
To confirm this, we examined MG132-induced cleavage
and transport of a double mutant of CREB-H containing the
R361A S1P site substitution together with a proline to
leucine (P337L) substitution within the predicted trans-
membrane region of CREB-H, which corresponds to an
essential proline in SREBP, ATF6 and OASIS, necessary
for S2P-mediated cleavage (17,50). Upon treatment with
either MG132 or BFA, we failed to observe any cleavage or
nuclear transport of the mutant CREB-H (Figure S4).
Together, these data demonstrate specificity in the
MG132-mediated cleavage, indicating that an S1P/S2P-
specific response was involved. We note that for the
CREB-H mutant, the deglycosylated intermediate was still
generated with similar kinetics, confirming that the mutant
was still a substrate for normal processing by the retro-
translocation route (Figure S5).
These data on the generation of the 45-kDa nuclear
product are significant, and we propose two main explan-
ations. It may be that CREB-H undergoes a constitutive
low level of cleavage but at a level that cannot readily be
detected without intervention or the use of proteasome
inhibitors and that MG132 treatment stabilizes the short-
lived and otherwise undetectable product. It could how-
ever also be that inhibition of the proteasome itself is
sufficient to induce cleavage of CREB-H by inducing some
form of ER overload or stress. In either event, we find little
evidence that inhibition of glycosylation per se, and any
concomitant specifically associated stress, has any sub-
stantial effect on CREB-H cleavage. These results have
implications for the detection of ER stress events, both for
CREB-H and also within the wider context of other
membrane-bound transcription factors (see below).
Taking into account our results showing that CREB-H is
retrotranslocated through the ERAD pathway and consid-
ering the fundamental role of the proteasome in ERAD, we
therefore examined the MG132-induced cleavage in more
detail. To distinguish between constitutive but undetected
cleavage of CREB-H and cleavage actively induced by
MG132 treatment, we examined the production of the
CREB-H cleavage product in MG132-treated cells in the
presence or absence of de novo protein synthesis. To this
end, cells expressing CREB-H were treated (24 h after
transfection) with MG132 alone or MG132 in the presence
of cycloheximide for various lengths of time. MG132
treatment alone led to the progressive accumulation of
the cleaved product with similar kinetics to our earlier
observations (Figure 5C, lanes 2–6). However, treatment
with MG132 in the presence of cycloheximide led to only
a modest quantity of detectable cleavage product with no
appreciable increase over the 8-h period (Figure 5C, lanes
7–10). We interpret these results to indicate that MG132-
induced cleavage and accumulation of CREB-H require
ongoing protein synthesis and that the cleaved CREB-H
originates from de novo synthesized protein under con-
ditions of stress rather than the stabilization of a preexisting
pool of constitutively cleaved protein.
ER retention of CREB-H requires a cytosolic
determinant
In view of our observations on CREB-H cleavage induced in
response to proteasome inhibition, we wished to explore
the possibility that localization and regulation of CREB-H
might involve cytosolic determinants of the protein. Inter-
estingly, examination of the sequence alignment (Fig-
ures 1A and 6) indicates that within CREB-H and related
factors, conservation of the transmembrane segment is
more extensive than with ATF6. In particular, we noted
that while all the factors contain a conserved cysteine
leading into the conserved hydrophobic region IV, the
residues immediately N-terminal to the cysteine were
extremely well conserved in CREB-H, CREB4, Luman
and OASIS but lacking in ATF6 (Figure 6). This conserved
flanking region encompassed a motif conforming to . . .[Q]-[ST]-[SG]-T. . . immediately adjacent to the cysteine
(Figure 6A). To examine the possibility that this region
was involved in CREB-H localization or trafficking, we con-
structed a deletion mutant (CREB-HDN1) lacking 11 resi-
dues in the region from 312–323 (inclusive) and examined
expression and localization (Figure 6). The results demon-
strated that the 11-residue deletion around this region had
a marked effect on two aspects of CREB-H. Firstly, in
contrast to wildtype (w/t) CREB-H, CREB-HDN1 exhibited
significant levels of constitutive cleavage (Figure 6B, com-
pare lanes 1 and 3). The CREB-HDN1 cleavage products co-
migrated with the products induced by BFA cleavage of
w/t CREB-H, and their levels were increased moderately
in response to BFA treatment (Figure 6B). To confirm that
this constitutive cleavage was indeed because of S1P, we
examined the effect of the single substitution in the S1P
site, R361A, in the context of CREB-HDN1 (Figure 6D).
Mutation of the S1P site virtually abolished the appear-
ance of the products, confirming that constitutive cleav-
age of CREB-HDN1 was indeed because of S1P. As S1P
cleavage is thought to occur in the Golgi it may be
expected that the deletion also affected the localization.
Indeed, a second clear difference between CREB-HDN1
1806 Traffic 2007; 8: 1796–1814
Bailey et al.
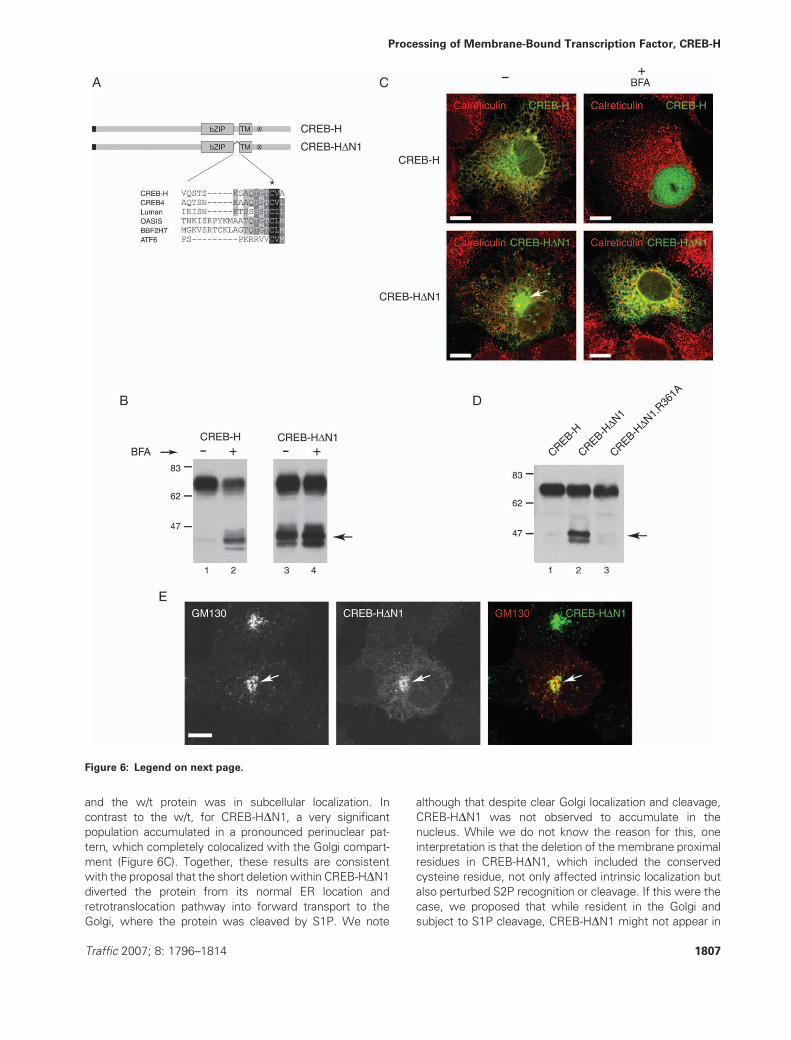
and the w/t protein was in subcellular localization. In
contrast to the w/t, for CREB-HDN1, a very significant
population accumulated in a pronounced perinuclear pat-
tern, which completely colocalized with the Golgi compart-
ment (Figure 6C). Together, these results are consistent
with the proposal that the short deletion within CREB-HDN1diverted the protein from its normal ER location and
retrotranslocation pathway into forward transport to the
Golgi, where the protein was cleaved by S1P. We note
although that despite clear Golgi localization and cleavage,
CREB-HDN1 was not observed to accumulate in the
nucleus. While we do not know the reason for this, one
interpretation is that the deletion of the membrane proximal
residues in CREB-HDN1, which included the conserved
cysteine residue, not only affected intrinsic localization but
also perturbed S2P recognition or cleavage. If this were the
case, we proposed that while resident in the Golgi and
subject to S1P cleavage, CREB-HDN1 might not appear in
Figure 6: Legend on next page.
Traffic 2007; 8: 1796–1814 1807
Processing of Membrane-Bound Transcription Factor, CREB-H

the nucleus even after treatment with BFA (which readily
induces nuclear localization of the w/t CREB-H). The results
demonstrate that while BFA treatment induced the reloc-
alization of CREB-HDN1 from the Golgi distribution pattern
to a general ER-staining pattern, consistent with general
disruption of the Golgi and with our interpretation on
constitutive Golgi localization, no nuclear accumulation
was subsequently observed (Figure 6C). This result con-
trasts with that for w/t CREB-H where nuclear accumula-
tion was readily observed after BFA treatment (Figure 6C
and Figure S1).
We cannot rule out the possibility that CREB-HDN1 is
cleaved by S2P within the Golgi and is translocated to the
nucleus but not readily observed because of its short half-
life (see above). However, this would not readily explain
the failure to detect nuclear CREB-HDN1 after BFA treat-
ment, where nuclear localization of the w/t protein is easily
observed. Also as indicated above, we did not observe
nuclear-localized CREB-HDN1 in response to MG132 treat-
ment, further suggesting that S2P site cleavage was
perturbed (data not shown). Independently of whether or
not the lack of constitutive (or BFA-induced) CREB-HDN1nuclear accumulation is because of disruption of the S2P
site, the results have significant implications for models
of the mechanism of CREB-HDN1 localization. The results
indicate that a cytoplasmic determinant in CREB-H, which
(with the exception of ATF6, see Discussion) is reasonably
conserved in other transmembrane bZIP factors, is
involved in ER localization. Disruption of this determinant
results in significant accumulation of the protein in the
Golgi and cleavage at the least by S1P in the absence of
any applied extrinsic stress.
Discussion
RIP of membrane-anchored transcription factors repre-
sents a key regulatory mechanism in responses to a range
of metabolic factors and stresses including sterol levels,
fatty acids (14,19,27), the accumulation of unfolded pro-
teins (15,16,29,30) and in yeast, to changes in oxygen
levels (52). The prototypic members of this group, SREBPs
and ATF6, are ER-resident integral membrane proteins,
which are cleaved by the Golgi-localized proteases, S1P
and S2P after forward transport out of the ER in response
to sterol levels and unfolded proteins, respectively. A
number of ATF6-related factors have now been identified
including CREB3/Luman (33,45), CREB-H (38) OASIS
(34,53), CREB4 (36) and BBF2H7 (37). Each of these
factors contains a conserved bZIP domain, together with
highly conserved defining features of an adjacent putative
transmembrane domain and consensus cleavage site for
S1P and S2P (44), indicating that they are likely to be
membrane-anchored transcription factors subjected to
RIP. Moreover, several of the factors have been shown
to be subject to stress-induced cleavage and substrates
of S1P and S2P (40,44,50). Intriguingly, CREB-H has also
been reported to integrate proinflammatory responses
with ER stress regulation. Ligands including TNF-a and
IL-6 induced CREB-H cleavage by virtue of increasing ER
stress with C-reactive protein and serum amyloid protein,
being among a number of downstream target genes (40).
However, details on characterization of CREB-H trafficking
and how it may sense stress are limited. Furthermore,
there have been inconsistent reports on the response to
stimuli, which are conventionally used to induce the
accumulation of unfolded proteins, usually tunicamycin,
DTT or thapsigargin in most studies. A number of studies
reported that these chemical treatments did not induce
detectable cleavage of CREB-H, CREB3/Luman or CREB4
(39,44,45) and that other forms of ER stress may be
involved, while recent results have reported tunicamycin-
induced cleavage of OASIS (50) and CREB-H (40).
Here, we further characterize CREB-H and examine syn-
thesis, turnover and ER stress-induced cleavage. From the
results, we propose a revised model of ER stress signalling
to CREB-H and possibly, other members of this class.
Consistent with previous reports, we show that CREB-H is
localized to the ER and quantitatively modified by glycosyl-
ation, as evidenced by its quantitative shift in mobility upon
tunicamycin or by treatment with PNGase F and Endogly-
cosidase H. Endoglycosidase H sensitivity of CREB-H
Figure 6: Constitutive Golgi localization and S1P cleavage from deletion of a cytosolic membrane proximal determinant.
A) Alignment of the residues around the beginning of the transmembrane domain in the bZIP transmembrane family members. The
transmembrane segment is aligned based on the completely conserved cysteine and following hydrophobic residues. All family members,
except ATF6, contain conserved residues upstream of the cysteine residue (*) as discussed in the text. B) Constitutive cleavage of CREB-
HDN1. Cells expressing CREB-H (lanes 1and 2) and CREB-HDN1 (lanes 3 and 4) were harvested with or without BFA treatment (1 mg/mL
for 8 h) and analyzed byWestern blotting as before. Significant cleavage of CREB-HDN1was observed evenwithout BFA treatment (lane 3)
with productsmigrating with the BFA-induced products of the w/t CREB-H (lane 2). C) Altered localization of CREB-HDN1. Cells expressingCREB-H and CREB-HDN1 as indicated were processed for immunofluorescence with or without BFA treatment (1 mg/mL for 8 h) as
indicated. Cells were stained for CREB-H and calreticulin. Pronounced altered localization of CREB-HDN1 was observed in a perinuclear
pattern (arrow). D) Constitutive cleavage of CREB-HDN1 is because of S1P cleavage. A single arginine substitution (R361A) at the
conserved S1P site abolishes BFA-induced cleavage of CREB-H (Figure S1). This same substitution was created in the background of
CREB-HDN1 and expression of CREB-H, CREB-HDN1 and CREB-HDN1.R361A analyzed. The results demonstrate that the constitutive
cleavage observed for CREB-HDN1 was virtually abolished by the single S1P site mutation. E) Additional immunofluorescence analyses
demonstrated that the CREB-HDN1 altered localization pattern corresponded to the Golgi compartment (arrowed) as determined by
colocalization with the Golgi-specific marker, GM130. Scale bar, 10 mm. TM, transmembrane domain.
1808 Traffic 2007; 8: 1796–1814
Bailey et al.

modification is consistent with the localization studies and
indicates thatCREB-H isalmostexclusively located in theER
with high mannose-type modification. We examined the
possible role of a KDEL-like ER-retrieval sequence at the
C-terminus of CREB-H (39) but found no evidence that this
sequence is involved in ER localization of CREB-H.
We show that CREB-H normally has a relatively short half-
life, and we provide robust evidence from fractionation and
selective permeabilization studies, which demonstrate
that CREB-H traffics through the ERAD route, with the
retrotranslocated, deglycosylated form accumulating
within the cytosol upon proteasome inhibition. On the
basis of these results, we propose that the short half-life of
CREB-H is because of its normal processing by the ERAD
route. While characterization of the details of CREB-H
processing by the ERAD pathway is beyond the scope of
this current work, these observations are immediately
relevant to the models for CREB-H signalling in response
to ER stress (see below).
Despite rapid and efficient BFA-induced and S1P site-
dependent cleavage of CREB-H, we were unable to dem-
onstrate significant cleavage by tunicamycin, even though
tunicamycin clearly functioned and completely blocked
CREB-H glycosylation itself. (This result applies also to
the other conventionally used chemical inducers of UPR,
DTT and thapsigargin). Zhang et al. (40) did observe
tunicamycin-induced cleavage of CREB-H, although a pre-
vious report also failed to observe the cleavage (38). We do
not know the explanation for the difference in results on
tunicamycin-induced cleavage; although consistent with
the results of Zhang et al. (39), we did observe cleavage in
the presence of cytokines IL-6 and IL-1b (Figure S6). There
is a possibility that differences were because of different
cell types or systems; although, we also repeated these
experiments in other cell types including liver cells and
found little evidence for cleavage. In our study, we em-
ployed the TK promoter to promote lower, more physio-
logical levels of gene expression compared with the CMV
promoter used in the previous study (40). In our hands,
expression of CREB-H from a CMV promoter resulted in
levels of constitutive cleavage perhaps representing gen-
eral ER stress induced through overexpression of an
exogenous factor, in a similar manner to observations for
SREBP andATF6, andmaking difficult analysis of anyminor
tunicamycin-induced population (16,28).
We considered the possibility that tunicamycin-induced
cleavage was occurring but in such a minor population of
a relatively unstable protein that the product was below
levels of detection. Consistent with this possibility in the
presence of MG132, we observed a tunicamycin-induced
cleavage product. However, in control experiments,
MG132 treatment alone, in contrast to tunicamycin treat-
ment alone, was sufficient to induce the appearance of the
CREB-H N-terminal cleavage product and nuclear trans-
port. This result suggests that CREB-H does indeed act
as a mediator of ER stress but has important implications
and two main explanations. CREB-H could be undergoing
constitutive cleavage (by virtue of some intrinsic stress) to
an unstable product degraded by the proteasome that
is essentially undetectable, with the appearance of the
product being the result of stabilizing this preexisting
population. We examined this prospect with results, which
indicate that proteasome inhibition in the absence of de
novo protein synthesis, while still allowing CREB-H retro-
translocation did not induce the accumulation of the
specific cleavage product. Although it is possible that
cycloheximide counteracted the stress imposed by
MG132 treatment, we interpret this result to indicate that
it is newly synthesized CREB-H, which is cleaved after
sensing stress because of inhibition of the proteasome.
With regard to the absence of detectable CREB-H cleav-
age by tunicamycin and like treatments, it is difficult to
attribute significance to an event which is at such a low
level to be virtually undetectable. This notwithstanding,
extremely short half-life transcription factors could have
a role in a relevant response. However, we propose an
alternative model. A priori, it is quite feasible that an ER-
anchored transcription factor, particularly one that is
undergoing retrotranslocation, may recognize the ERAD
process itself including cytosolic signals rather than the
direct lumenal signals ascribed to ATF6. We propose that
proteasome inhibition, in preventing CREB-H degradation
by the normal ERAD route, actively redirects CREB-H for
specific cleavage to the N-terminal product. The ER
location of CREB-H can be explained precisely because it
is normally subject to retrotranslocation and has a short
half-life and that its normal function may be in sensing
stress in the cytosolic aspect of this process during normal
physiological growth and metabolism.
In this regard, a very significant finding was that the
deletion of a short region on the cytosolic side of the
conserved CREB-H transmembrane domain (CREB-HDN1)had a very pronounced effect on constitutive localization
and cleavage. This deletion resulted in redirection of
CREB-H to the Golgi and constitutive cleavage by S1P.
The transmembrane domain of the CREB-HDN1 deletion
variant was obviously intact as the protein was fully
glycosylated and localised to the ER and Golgi. While there
may be other less likely explanations, in our view, the
simplest and most consistent explanation is that the
deletion disrupted a motif within CREB-H which was
normally involved in ER retention and/or directing the
protein to the ERAD pathway. Deletion of this region
resulted in constitutive forward transport, although clearly
other positive determinants could also be involved in this.
The result implies that CREB-H is localized by a retention
mechanism and that release may operate at least in part
through alteration of the interactions dictated by this cyto-
solic region of the protein.We provide convincing evidence
that CREB-H is subject to ERAD, a pathway involved in
normal cycling and turnover of ER transmembrane and
Traffic 2007; 8: 1796–1814 1809
Processing of Membrane-Bound Transcription Factor, CREB-H

lumenal proteins, as well as recognition of incorrectly
folded proteins. It will now be interesting to examine
whether any of the components currently being identified
in the ERAD pathways for cytosolic and/or lumenal recog-
nition are involved in CREB-H ERAD and more specifically
differences between the w/t CREB-H and the variant
lacking the 11-residue membrane proximal region.
If CREB-H is not regulated by lumenal ER stress, or at the
least if cytosolic regulation is the key feature of CREB-H
regulation, then the implication is that BiP, which is
considered to be the central regulator in ATF6 response,
is not the key factor in regulation of CREB-H transport and
cleavage. Previous results have demonstrated BiP binding
by ATF6, and while many ER-anchored protein with
lumenal domains will bind this abundant chaperone, the
mode of ATF6 binding was shown to be specific. The
interaction between ATF6 and BiP appears to be surpris-
ingly stable, but by virtue of specific stress recognition
motifs in its lumenal domain, ATF6 is actively released
from BiP in response to ER stress (31,54). Analysis of
binding of BiP by CREB-H will be pursued in future studies,
although BiP binding per se does not invoke relevance to
signalling as many proteins with lumenal domains are likely
to be bound. We note also that compared with the other
factors, ATF6 has a particularly long lumenal tail of approx-
imately 270 residues with specific BiP binding, ER stress-
sensing and forward Golgi transport motifs (31,32,54). By
comparison, CREB-H has a lumenal segment of about 100
residues with no obvious homology to the specific motifs
in ATF6, and indeed, the CREB-H related factor CREB4 has
a lumenal tail of just 60 residues. There is little similarity
between the interdigitated regions of ATF6 with ascribed
functions in sensing ER stress, binding BiP or forward
Golgi transport and the luminal domains of these proteins.
We find additional evidence that CREB-H and ATF6
respond to different signals. For example, DTT is also
frequently used as a strong stimulus for UPR, even though
such treatment is relatively crude and will inevitably lead to
gross changes within a cell and indeed could perturb the
very pathways in transport and enzymatic activity being
explored. In contrast to results reported for ATF6, we find
that the MG132-induced cleavage of CREB-H is actually
reduced by simultaneous treatment with DTT (data not
shown). We also point out a note of caution as although it
is not always clear from the stated methods, it is the case
that many studies utilize proteasome inhibition to detect
cleavage of ATF6, and it is also the case that in early
studies ATF6 cleavage in response to tunicamycin was not
actually readily detected (16,29). While we could find no
direct comparisons where MG132 was utilized in order to
visualize the tunicamycin (or other)-induced cleavage it
may be that proteasome inhibition was itself contributing
a different form of stress other than lumenal unglycosy-
lated or aberrantly folded proteins.
In conclusion, our work indicates that in contrast to ATF6,
the related transmembrane transcription factor CREB-H
does not efficiently respond to the traditional inducers of
UPR such as tunicamycin. However, CREB-H is cleaved in
response to stress induced by proteasome inhibition.
Although the detailed mechanism of response and cleav-
age of CREB-H in response to proteasome inhibition
remains to be explored, in essence, this is also the same
position as for tunicamycin or DTT for ATF6 as these are
relatively crude treatments and do not identify molecular
signatures. For the first time, we present evidence regard-
ing the short half-life of CREB-H and its processing through
ERAD. We also demonstrate the involvement of a cytosoli-
cally disposed membrane proximal determinant, whose
deletion causes CREB-H to be diverted from the ERAD
pathway to the Golgi, where it is constitutively cleaved by
S1P. We propose a model (Figure 7) integrating our results
on CREB-H processing by ERAD, response to proteasome
inhibition and the constitutive Golgi localization and S1P
cleavage of CREB-HDN1. We propose that CREB-H is
subject to ERAD and may monitor flux through or effi-
ciency of ERAD through a cytosolic determinant. When
this pathway is modulated or perturbed, CREB-H is redir-
ected for cleavage and activation. The cytosolic membrane
proximal region may be directly or indirectly involved in
trafficking CREB-H through the ERAD pathway. Further
work, specifically in examining the role of ERAD by
selectively disrupting components of the pathway and also
in identifying components interacting with the cytoplasmic
determinant should help to refine these proposals. Such
studies will further our general understanding of RIP and
the likely expansion of the relevant physiological environ-
mental or metabolic stimuli that affect this process.
Materials and Methods
CellsHepG2 cells [American Type Culture Collection (ATCC), HB-8065] were
grown in MEM (Eagle) containing 2 mM L-glutamine, Earle’s balanced salt
solution containing 1.5 g/L sodium bicarbonate, 0.1 mM non-essential amino
acids, 1 mM sodium pyruvate and 10% foetal bovine serum. The COS-1
cells were grown in DMEM/glutamax media, supplemented with 10%
newborn calf serum (NBCS) and penicillin and streptomycin at 100 U/mL
and 100 mg/mL, respectively. Cells were cultured at 378C in a 5% CO2
environment under standard conditions.
PlasmidsCREB-H open reading frame (ORF) was amplified from a Liver cDNA library
(Marathon Ready cDNA; Clontech) by polymerase chain reaction (PCR)
using primers CREB-HFBAM (CGCGGATCCATGAATACGGATTTAGCTGC)
and CREB-HRSAL (ACGCGTCGACCAGCTCGTCTCCCGCCGCCT) to gener-
ate an approximately 1400-bp fragment. This fragment was digested with
BamHI/SalI and inserted into pJS6 (45) and digested with BamHI/XhoI to
generate the vector pSM20, corresponding to CREB-H with an in-frame
N-terminal SV5-epitope tag and a C-terminal HA-epitope tag under the
control of the CMV promoter. Sequencing of the CREB-H ORF indicated
that our CREB-H sequence lacks a 3-bp codon for a glutamine residue at
amino acid position 52 (based on the original published sequence; GenBank
accession number: AB050902). This 3-bp deletion is also present in several
expressed sequence tags and is identical to a second reported transcript
(GenBank accession number: AB073612), suggesting that it may be the
1810 Traffic 2007; 8: 1796–1814
Bailey et al.

result of natural sequence variation. However, for clarity and consistency
with previous publications on CREB-H, we use the amino acid numbering
based on the coding sequence for transcript with GenBank accession
number AB050902. The SV5-CREB-H-HA-tag ORF was then excised as an
NheI–XbaI fragment from pSM20 and inserted into the pTK-herpes simplex
virus (HSV)-BP2 backbone and digested to generate plasmid pDJB134. All
studies reported here were performed with CREB-H under the control of
the weaker TK promoter as we and others have found aberrant processing
events when these factors are overexpressed under the control of the CMV
promoter. This feature is quite important in analyses of proteins, whose
regulation is through cleavage in responses to protein overload and stress
(see Discussion).
An SV5 epitope-tagged CREB-H construct, but lacking the C-terminal HA-
tag, was constructed by amplifying CREB-H from pSM20 with oligonucleo-
tides DB075 (ACCGGTGCTAGCCATGGCTGGAAAGCCGATCCC) and
DB073 (GAGCTCTCTAGATTACAGCTCGTCTCCCGCCGCCTC). The PCR
fragment was digested with NheI/XbaI and inserted into the pTK-HSV-
BP2 backbone and digested similarly. pTK-HSV-BP2 (ATCC) has been
described previously (28). Plasmid pDJB124, corresponding to SV5-
CREB-H with a deletion of the KDEL-like sequence at the C-terminus
(terminating at the glycine residue at aa 458), was constructed in a similar
manner to pDJB123, except that PCRwas performed with oligonucleotides
DB075 and DB074 (GAGCTCTCTAGATTATCCCGCCGCCTCCAGCCCTG).
CREB-H lacking the C-terminus transmembrane and lumenal domain
(plasmid pDJB125; CREB-HDTMC), and terminating at the predicted S2P
cleavage site (cysteine residue at aa 323), was constructed in a similar
manner to pDJB123, except that PCRwas performed with oligonucleotides
DB075 and DB076 (GAGCTCTCTAGATTAACAGGTGCCTGTCTGGGCTG).
The CREB-HDN1 construct (plasmid pDJB140) was created using over-
lapping PCR mutagenesis to delete a region in CREB-H, corresponding to
aa 312–323 inclusive. The S1P and S2P site substitution mutations were
introduced into the relevant backbone vectors using quikchange site-
directed mutagenesis (Stratagene) to introduce an arginine to alanine
substitution at aa position 361 at the putative S1P site or a proline to
leucine substitution at aa position 337 to disrupt the putative S2P cleavage.
All mutagenesis was confirmed by restriction digestion and direct sequen-
cing of the constructs.
pS1P.KDEL and pS1P.KDAS encoding the S1P containing an ER retention
signal (KDEL) or corresponding non-functional control sequence (KDAS)
have been previously described (51). These plasmids were generously
provided by Dr Joseph Goldstein.
TransfectionsTransfections were performed using the calcium phosphate precipitation
procedure modified by the use of [N, N-bis(2-hydroxyetyhl)-2-aminoetha-
nesulphonic acid]-buffered saline (pH 7.06) as previously described (55).
Routinely, 0.5–1 mg of the appropriate expression vector was transfected
with amounts of DNA normalized using pUC19 carrier DNA. HepG2
transfections were performed using Fugene 6 transfection reagent (Roche)
according to manufacturers’ instructions.
Brefeldin A, tunicamycin and proteasome inhibitor
treatmentsBrefeldin A and tunicamycin (Sigma) were prepared as 10 mg/mL stocks in
methanol or dimethyl sulphoxide (DMSO), respectively. Brefeldin A was
added to cells at a final concentration from 1 mg/mL and tunicamycin at
a final concentration of 2 mg/mL for time periods as indicated in the text.
MG132 was dissolved in DMSO and used at a final concentration of 10 mM
for various times as stated in the text.
Immunofluorescence studiesCells (1 � 105 cells/35-mm well) were plated on glass coverslips placed
in plastic tissue culture vessels. For routine immunofluorescence
analysis, cells (approximately 40 h after transfection) were washed in
PBS and fixed with ice-cold methanol and blocked in PBS/10% NBCS for
20 min. Primary antibodies were diluted in PBS/10% NBCS and applied
for 20 min. Primary antibodies used were anti-SV5 (1:1000, kindly
supplied by R. Randall) or anti-V5 (1:1000, Covance) for the SV5-tag;
anti-HA (anti-HA.11 1:100, Covance), anti-Calreticulin polyclonal (1:200,
Calbiochem) as a marker for the ER and anti-TGN46 (1:2000, Serotec) or
anti-GM130 (1:100, BD Biosciences) as markers for the Golgi apparatus.
Fluorochrome (Alexa 488 or Alexa 543)-conjugated secondary antibodies
of appropriate specificity (Molecular Probes) were diluted 1:200 in PBS/
10% NBCS and added for 20 min. Following washing, cells were
mounted in Mowiol and visualized using a Zeiss LSM 410 confocal
microscope imaging system and using a Zeiss Plan-Apochromat (�63/
1.4 numerical aperture) lens. Images for each channel were captured
sequentially with eightfold averaging at an image size of 512 � 512
pixels. Composite illustrations were prepared using Adobe software.
Example images shown are representative of numerous images gath-
ered for each test construct and condition.
To analyze CREB-H retrotranslocation during ERAD, we monitored the
appearance of the HA-tagged C-terminal end of CREB-H in the cytoplasm
under conditions of proteasome inhibition. Transfected cells were treated
Figure 7: Model for CREB-H traffick-
ing and response to stress. The
model depicts the proposal for the
normal trafficking of CREB-H through
the ERAD pathway, including retro-
translocation (1), deglycosylation (2)
and proteasome degradation (3). The
proposal is that CREB-H is normally
processed through this route and
senses perturbation, which results in
redirection (4) and transport to the Golgi
(5), followed by cleavage and nuclear
translocation (6). The protein complex
indicated by ? represents putative, as
yet, unidentified proteins that may be
involved in ER retention. Further as-
pects are discussed in the text.
Traffic 2007; 8: 1796–1814 1811
Processing of Membrane-Bound Transcription Factor, CREB-H

with or without MG132 10 mM for 6 h, fixed in 3% paraformaldehyde for
10 min, washed with PBS and treated with either digitonin (40 mg/mL for
3 min) to permeabilize just the plasma membrane or with NP-40 (0.5%
for 3 min) to permeabilize all membranes. All processing was performed on
ice. Fixed and permeabilized cells were then blocked and stained using
primary antibodies directed against either the SV5-epitope tag or HA-
epitope tag (anti-HA.11 1:100, Covance) or calreticulin as described above.
Subcellular fractionationIsolation of soluble cytosolic and membrane fractions were based upon the
procedures described (56). The COS cells (1 � 10� cells, 15-cm dish) were
transfected with CREB-H expression vector using Fugene 6 (Roche) trans-
fection reagent according to manufacturers instructions. Approximately
28-h post-transfection cells were treated with or without MG132 10 mM for
12 h and washed with PBS, followed by two washings with 0.25 M sucrose,
10 mM triethanolamine and 10 mM acetic acid, pH 7.8. Cells were then
scraped into 4 mL of ice-cold buffer containing 0.25 m sucrose, 10 mM
triethanolamine, 10 mM acetic acid, 1 mM ethylenediaminetetraacetic acid
and 0.1 mM PMSF, pH 7.8. The cell suspension was lysed by approximately
10 strokes of a Dounce homogenizer. Nuclei and unlysed cells were
removed by centrifugation at 1000 � g/10 min/48C. Supernatant was
retained and subjected to centrifugation at 100 000 � g/1 h/48C to pellet
membranes. The supernatant (cytosol fraction) was concentrated by
trichloroacetic acid precipitation (10%), acetone washed and pelleted by
centrifugation. Aliquots of the membrane and cytosol fractions were then
resuspended in sodium dodecyl sulphate-lysis buffer and subject to SDS–
PAGE and Western blotting.
Western blot analysisProteins were analyzed by separation either on 10% SDS–PAGE gels
prepared and run using the Bio-Rad Mini-Protean II apparatus or with
prepoured gel systems (Invitrogen/Novex). The proteins were transferred
to nitrocellulose membranes, which were then blocked with PBS/0.05%
Tween-20 (PBST) containing 5% non-fat dried milk. After blocking, mem-
branes were incubated with primary antibody (SV5, 1:10000 or anti-HA,
1:1000) in PBST/5% dried milk for 1 h, washed three times in PBS/1%
Triton-X-100 and incubated for a further 1 h with PBST/5% dried milk
containing the appropriate horseradish peroxidase-conjugated secondary
antibody. Following further washing in PBS/1% Triton-X-100, membranes
were processed using chemiluminescence detection reagents (Pierce).
Acknowledgments
We are grateful to Rick Randall for supplying monoclonal antibodies for
the detection of the SV5-epitope tag. We thank Dr Joseph Goldstein for
supplying the S1P constructs used in this study and Sophie Malcomber
for her assistance in the construction of the CMV promoter CREB-H ex-
pression vector. This work was supported by Marie Curie Cancer Care.
Supplementary Materials
Figure S1: The BFA-induced cleavage and relocalization of CREB-H.
A) The COS-1 cells were transfected with expression vectors for CREB-H or
the substitution mutant in the S1P site (CREB-H R361A) and mock treated or
treated with BFA 1 mg/mL for the times indicated. Samples were analyzed
with primary antibody to the SV5-epitope tag. Long arrow indicates the full-
length CREB-H, while the short arrow indicates the BFA-induced N-terminal
CREB-H cleavage product. B) The COS-1 were transfected with full-length
CREB-H. Untreated cells or cells treatedwith BFA 1 mg/mL for 2 h were fixed
and stained as for Figure 2A. Transfected cells were identified by the staining
for the SV5-epitope tag and co-stained with antibody to calreticulin to identify
the ER (SV5-epitope, green; calreticulin, red). C) Similar experiments in
HepG2 cells treated without or with BFA 1 mg/mL for 4 h and then fixed and
processed as described above. Scale bar, 10 mm.
Figure S2: Direct cleavage of CREB-H by ER-localized S1P. The COS-1
cells were cotransfected with full-length CREB-H and expression con-
structs for either S1P.KDEL or S1P.KDAS as indicated. A) Samples were
prepared and analyzed by Western blotting. CREB-H was identified with
antibody to the SV5-epitope tag. The migration of the full-length CREB-H
(long arrow) and cleaved N-terminal product (short arrow) are indicated.
B) Representative confocal images from cells prepared for indirect immuno-
fluorescence. Cells cotransfected with S1P.KDAS or S1P.KDEL and full-
length CREB-H were identified by co-staining with antibody to the Myc tag
to identify the S1P cotransfected cells (not shown) and antibody to SV5
epitope for CREB-H. Single channel images are shown (stained for the SV5
epitope). Nuclear-localized CREB-H is indicated by the white arrow. Scale
bar, 10 mm.
Figure S3: Expression of CREB-H under the control of the CMV
promoter. The COS-1 cells were transfected with CREB-H under the
control of the CMV promoter (A and B; 0.01 mg) or the TK promoter (C and
D; 1.0 mg). Cells were then changed into fresh media with (A and C) or
without (C and D) tunicamycin at 2 mg/mL for 1, 2, 4, 6 or 8 h. With
expression from the CMV promoter, the arrow indicates the appearance of
cleaved products, which were observed even in the absence of tunica-
mycin treatment (A, lane 1) and (B, lanes 1–6), making it difficult to conclude
that any minor effect of tunicamycin was significantly above background.
Use of higher amounts of the CMV promoter to increase detection simply
lead to increased constitutive cleavage. For the TK promoter, despite the
now reduced level of background cleavage (C, lane 1), it was difficult to
conclude that there was any significant tunicamycin-induced cleavage
(C, lanes 2–6) above background (D, lanes 2–6).
Figure S4: Effect of tunicamycin pretreatment on BFA-induced cleav-
age of CREB-H. The COS-1 cells were transfected with CREB-H. Cells
were either untreated (lanes 1–6) or pretreated with tunicamycin 2 mg/mL
for 8 h (lanes 7–11) prior to the addition of 1 mg/mL BFA for 0.5 (lanes 3
and 8), 1 (lanes 4 and 9), 1.5 (lanes 5 and 10) and 2 h (lanes 6 and 11).
Samples were harvested and processed for Western blotting as pre-
viously described. The long arrows indicate the position of the full-length
CREB-H (glycosylated and non-glycosylated forms, upper and lower long
arrows, respectively). Short arrow indicates the cleaved N-terminal CREB-H
product.
Figure S5: Resistance of CREB-H.P337L.R361A double mutant to
cleavage following treatment with MG132 or BFA. A) The COS-1 cells
were transfected with CREB-H (lanes 1–3 and 7) or CREB-H P337L.R361A
double mutant (lanes 4–6 and 8). Cells were either untreated (lanes 1 and 4)
or treatedMG132 for 4 and 8 h (lanes 2, 3, 5 and 6) or by addition of 1 mg/mL
BFA for 1 h (lanes 7 and 8). Samples were harvested and processed for
Western blotting as previously described. The long arrows indicate the
position of the full-length CREB-H (glycosylated and non-glycosylated
forms, upper and lower long arrows, respectively). Short arrow indicates
the cleaved N-terminal CREB-H product, which is not generated in the
double mutant. B) Immunofluorescence analysis of COS-1 cells transfected
with CREB-H P337L.R361A, and subsequently treated with BFA 1 mg/mL
for 1 h, or MG132 10 mM for 4 h. Cells were fixed and stained as described
for Figure 2. No nuclear localization was observed (compare with Figure 5B).
Scale bar, 10 mm.
Figure S6: Cleavage of CREB-H induced by cytokines IL1b and IL6
treatment. The COS-1 cells were transfected with CREB-H and treated
with a combination of IL1b and IL6 at 40 ng/mL, each for 24 or 48 h (lanes 2
and 3, respectively) or control media (lane 1) in the absence of proteasome
inhibitors. Cleavage of CREB-H was observed by 48 h after treatment.
1812 Traffic 2007; 8: 1796–1814
Bailey et al.

Supplemental materials are available as part of the online article at http://
www.blackwell-synergy.com
References
1. Brodsky JL, McCracken AA. ER protein quality control and proteasome-
mediated protein degradation. Semin Cell Dev Biol 1999;10:507–513.
2. Frydman J. Folding of newly translated proteins in vivo: the role of
molecular chaperones. Annu Rev Biochem 2001;70:603–647.
3. Ellgaard L, Molinari M, Helenius A. Setting the standards: quality
control in the secretory pathway. Science 1999;286:1882–1888.
4. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P.
Functional and genomic analyses reveal an essential coordination
between the unfolded protein response and ER-associated degrada-
tion. Cell 2000;101:249–258.
5. Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum.
Nat Rev Mol Cell Biol 2003;4:181–191.
6. Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to
destruction. Nat Cell Biol 2005;7:766–772.
7. Hampton RY. Proteolysis and sterol regulation. Annu Rev Cell Dev Biol
2002;18:345–378.
8. Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from
nascent chain to folded protein. Science 2002;295:1852–1858.
9. McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control:
chaperones culling corrupt conformations. Nat Cell Biol 2005;7:736–741.
10. McCracken AA, Brodsky JL. Recognition and delivery of ERAD sub-
strates to the proteasome and alternative paths for cell survival. Curr
Top Microbiol Immunol 2005;300:17–40.
11. Romisch K. Endoplasmic reticulum-associated degradation. Annu Rev
Cell Dev Biol 2005;21:435–456.
12. Ahner A, Brodsky JL. Checkpoints in ER-associated degradation: excuse
me, which way to the proteasome? Trends Cell Biol 2004;14:474–478.
13. Brown MS, Goldstein JL. The SREBP pathway: regulation of choles-
terol metabolism by proteolysis of a membrane-bound transcription
factor. Cell 1997;89:331–340.
14. WangX, SatoR, BrownMS,Hua X,Goldstein JL. SREBP-1, amembrane-
bound transcription factor released by sterol-regulated proteolysis. Cell
1994;77:53–62.
15. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the
cis-acting endoplasmic reticulum stress response element responsible
for transcriptional induction of mammalian glucose-regulated proteins.
Involvement of basic leucine zipper transcription factors. J Biol Chem
1998;273:33741–33749.
16. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription
factor ATF6 is synthesized as a transmembrane protein and activated
by proteolysis in response to endoplasmic reticulum stress. Mol Biol
Cell 1999;10:3787–3799.
17. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS,
Goldstein JL. ER stress induces cleavage of membrane-bound ATF6
by the same proteases that process SREBPs. Mol Cell 2000;6:
1355–1364.
18. Hua X, Sakai J, Ho YK, Goldstein JL, Brown MS. Hairpin orientation of
sterol regulatory element-binding protein-2 in cell membranes as deter-
mined by protease protection. J Biol Chem 1995;270:29422–29427.
19. Sato R, Yang J, Wang X, Evans MJ, Ho YK, Goldstein JL, Brown MS.
Assignment of the membrane attachment, DNA binding, and transcrip-
tional activation domains of sterol regulatory element-binding protein-1
(SREBP-1). J Biol Chem 1994;269:17267–17273.
20. Cox JS, Shamu CE, Walter P. Transcriptional induction of genes
encoding endoplasmic reticulum resident proteins requires a trans-
membrane protein kinase. Cell 1993;73:1197–1206.
21. Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway
from the endoplasmic reticulum to the nucleus requires a novel bifunc-
tional protein kinase/endoribonuclease (Ire1p) in mammalian cells.
Genes Dev 1998;12:1812–1824.
22. Duncan EA, Brown MS, Goldstein JL, Sakai J. Cleavage site for
sterol-regulated protease localized to a leu-Ser bond in the lumenal
loop of sterol regulatory element-binding protein-2. J Biol Chem 1997;
272:12778–12785.
23. Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL.
Sterol-regulated release of SREBP-2 from cell membranes requires
two sequential cleavages, one within a transmembrane segment. Cell
1996;85:1037–1046.
24. Sakai J, Nohturfft A, Goldstein JL, Brown MS. Cleavage of sterol
regulatory element-binding proteins (SREBPs) at site-1 requires inter-
action with SREBP cleavage-activating protein. Evidence from in vivo
competition studies. J Biol Chem 1998;273:5785–5793.
25. Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT,
Chang TY, Brown MS, Goldstein JL. Complementation cloning of S2P,
a gene encoding a putative metalloprotease required for intramem-
brane cleavage of SREBPs. Mol Cell 1997;1:47–57.
26. Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC,
Goldstein JL, Brown MS. Molecular identification of the sterol-
regulated luminal protease that cleaves SREBPs and controls lipid
composition of animal cells. Mol Cell 1998;2:505–514.
27. Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL,
Wang X. SREBP-2, a second basic-helix-loop-helix-leucine zipper pro-
tein that stimulates transcription by binding to a sterol regulatory
element. Proc Natl Acad Sci U S A 1993;90:11603–11607.
28. Hua X, Sakai J, Brown MS, Goldstein JL. Regulated cleavage of sterol
regulatory element binding proteins requires sequences on both sides
of the endoplasmic reticulum membrane. J Biol Chem 1996;271:
10379–10384.
29. Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R.
Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic
reticulum stress response. J Biol Chem 2000;275:27013–27020.
30. Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, Lee AS. ATF6 as
a transcription activator of the endoplasmic reticulum stress element:
thapsigargin stress-induced changes and synergistic interactions with
NF-Y and YY1. Mol Cell Biol 2000;20:5096–5106.
31. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6
localization by dissociation of BiP/GRP78 binding and unmasking of
Golgi localization signals. Dev Cell 2002;3:99–111.
32. Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses
endoplasmic reticulum (ER) stress and causes translocation of ATF6
from the ER to the Golgi. J Biol Chem 2002;277:13045–13052.
33. Lu R, Yang P, O’Hare P, Misra V. Luman, a new member of the CREB/
ATF family, binds to herpes simplex virus VP16-associated host cellular
factor. Mol Cell Biol 1997;17:5117–5126.
34. Honma Y, Kanazawa K, Mori T, Tanno Y, Tojo M, Kiyosawa H, Takeda J,
Nikaido T, Tsukamoto T, Yokoya S, Wanaka A. Identification of a novel
gene, OASIS, which encodes for a putative CREB/ATF family tran-
scription factor in the long-term cultured astrocytes and gliotic tissue.
Brain Res Mol Brain Res 1999;69:93–103.
35. Qi H, Fillion C, Labrie Y, Grenier J, Fournier A, Berger L, El-Alfy M,
Labrie C. AIbZIP, a novel bZIP gene located on chromosome 1q21.3
that is highly expressed in prostate tumors and of which the expression
is up-regulated by androgens in LNCaP human prostate cancer cells.
Cancer Res 2002;62:721–733.
36. Cao G, Ni X, Jiang M, Ma Y, Cheng H, Guo L, Ji C, Gu S, Xie Y, Mao Y.
Molecular cloning and characterization of a novel human cAMP response
element-binding (CREB) gene (CREB4). J Hum Genet 2002;47:373–376.
37. Storlazzi CT, Mertens F, Nascimento A, IsakssonM,Wejde J, Brosjo O,
Mandahl N, Panagopoulos I. Fusion of the FUS and BBF2H7 genes
Traffic 2007; 8: 1796–1814 1813
Processing of Membrane-Bound Transcription Factor, CREB-H

in low grade fibromyxoid sarcoma. Hum Mol Genet 2003;12:
2349–2358.
38. Omori Y, Imai J, Watanabe M, Komatsu T, Suzuki Y, Kataoka K,
Watanabe S, Tanigami A, Sugano S. CREB-H: a novel mammalian
transcription factor belonging to the CREB/ATF family and functioning
via the box-B element with a liver-specific expression. Nucleic Acids
Res 2001;29:2154–2162.
39. Chin KT, Zhou HJ, Wong CM, Lee JM, Chan CP, Qiang BQ, Yuan JG,
Ng IO, Jin DY. The liver-enriched transcription factor CREB-H is a
growth suppressor protein underexpressed in hepatocellular carcin-
oma. Nucleic Acids Res 2005;33:1859–1873.
40. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH,
Kaufman RJ. Endoplasmic reticulum stress activates cleavage of
CREBH to induce a systemic inflammatory response. Cell 2006;124:
587–599.
41. Denic V, Quan EM, Weissman JS. A luminal surveillance complex that
selects misfolded glycoproteins for ER-associated degradation. Cell
2006;126:349–359.
42. Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes
define convergent pathways for the degradation of ER proteins. Cell
2006;126:361–373.
43. Vashist S, Ng DT. Misfolded proteins are sorted by a sequential check-
point mechanism of ER quality control. J Cell Biol 2004;165:41–52.
44. Stirling J, O’Hare P. CREB4, a transmembrane bZip transcription factor
and potential new substrate for regulation and cleavage by S1P. Mol
Biol Cell 2006;17:413–426.
45. Raggo C, Rapin N, Stirling J, Gobeil P, Smith-Windsor E, O’Hare P,
Misra V. Luman, the cellular counterpart of herpes simplex virus VP16,
is processed by regulated intramembrane proteolysis. Mol Cell Biol
2002;22:5639–5649.
46. PonnambalamS,GirottiM, YaspoML,OwenCE, Perry AC, Suganuma T,
Nilsson T, Fried M, Banting G, Warren G. Primate homologues of rat
TGN38: primary structure, expression and functional implications. J Cell
Sci 1996;109:675–685.
47. Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell
lines secrete themajor plasma proteins and hepatitis B surface antigen.
Science 1980;209:497–499.
48. Swanton E, High S, Woodman P. Role of calnexin in the glycan-
independent quality control of proteolipid protein. EMBO J 2003;22:
2948–2958.
49. Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The
human cytomegalovirus US11 gene product dislocates MHC class I
heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996;
84:769–779.
50. KondoS,Murakami T, Tatsumi K, OgataM, Kanemoto S, Otori K, Iseki K,
Wanaka A, Imaizumi K. OASIS, a CREB/ATF-family member, modulates
UPR signalling in astrocytes. Nat Cell Biol 2005;7:186–194.
51. DeBose-Boyd RA, Brown MS, Li WP, Nohturfft A, Goldstein JL,
Espenshade PJ. Transport-dependent proteolysis of SREBP: relocation
of site-1 protease from Golgi to ER obviates the need for SREBP
transport to Golgi. Cell 1999;99:703–712.
52. Hughes AL, Todd BL, Espenshade PJ. SREBP pathway responds to
sterols and functions as an oxygen sensor in fission yeast. Cell 2005;
120:831–842.
53. Omori Y, Imai J, Suzuki Y, Watanabe S, Tanigami A, Sugano S.
OASIS is a transcriptional activator of CREB/ATF family with a trans-
membrane domain. Biochem Biophys Res Commun 2002;293:
470–477.
54. Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. Stable binding of
ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell
Biol 2005;25:921–932.
55. Batchelor AH, O’Hare P. Localization of cis-acting sequence require-
ments in the promoter of the latency-associated transcript of herpes
simplex virus type 1 required for cell-type-specific activity. J Virol 1992;
66:3573–3582.
56. Marsh M, Schmid S, Kern H, Harms E, Male P, Mellman I, Helenius A.
Rapid analytical and preparative isolation of functional endosomes by
free flow electrophoresis. J Cell Biol 1987;104:875–886.
1814 Traffic 2007; 8: 1796–1814
Bailey et al.





























