Embed Size (px)
Citation preview

Towards an understanding of structure–property relationships in hole-transport materials: The influence of molecular conformation on oxidationpotential in poly(aryl)amines{
Paul J. Low,*a Michael A. J. Paterson,a Dmitry S. Yufit,a Judith A. K. Howard,a Julian C. Cherryman,b
Daniel R. Tackley,b Robert Brookc and Bev Brownc
Received 29th November 2004, Accepted 1st March 2005
First published as an Advance Article on the web 4th April 2005
DOI: 10.1039/b417962e
The influence of molecular conformation on the oxidation (ionisation) potential and electronic
structure associated with several TPD-style hole transport materials has been assessed through a
combination of single crystal X-ray diffraction, electrochemical and spectroelectrochemical
methods and DFT calculations. The introduction of methyl groups can be used to tune the
ionisation potential of these molecular species through a combination of electronic (inductive)
and thermodynamic effects, while the conformation of the biphenyl portion of the molecular
framework is found to play the greatest role in determining the Marcus-type reorganisation
energy associated with the charge transport process on the molecular level.
Introduction
Poly(aryl)amines are now widely used as hole-transport
materials in applications ranging from the Xerox process to
multi-layer organic light emitting diode based devices. In
order to maximise charge transport through a bulk sample
of these materials, it is necessary to minimise grain boundaries
caused by crystallisation in the layers of organic material.
In much work reported to date, there has therefore been a
heavy emphasis placed on the preparation and use of
amorphous polymeric compounds, and also on composite
materials derived from low molecular weight poly(aryl)amines
with an amorphous polymer support. Thus, while poly-
(aryl)amines such as TPD (1) have been the subject of
numerous experimental and computational studies, relatively
little is known about the molecular and solid-state structure
of even simple examples. Consequently, despite the intense
interest that is generated by these readily oxidised
organic materials, definitive structure–property relationships
remain elusive.
Several authors have used computational methods to
address the structure–property issues in TPD-derived mole-
cular materials, with particular emphasis on the geometric and
electronic changes which accompany oxidation/hole transport
phenomena.1–7 In the case of N,N,N9,N9-tetra(aryl)-1,19-
biphenyl-4,49-diamines, which form one of the most widely
employed classes of hole transport material the key conforma-
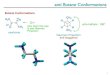
tional parameters (Fig. 1) can be considered to be: the torsion
angle between the ring systems of the biphenyl moiety, i.e. the
relative orientation of rings A and B (Fig. 1a); the relative
orientation of the NAr2 system with respect to the biphenyl
moiety i.e. the orientation of the Cx–N–Cy plane with respect
to ring A (Fig. 1b); the relative orientation of the peripheral
aromatic groups C with respect to the planar nitrogen centre
(Fig. 1c). Bond length variation should also be expected upon
oxidation, which will reflect the different localisation and
occupation of the HOMO in the neutral and hole-containing
radical cation (i.e. oxidised) form of the material.
We have recently embarked upon a study of poly(aryl)amine
based compounds with a view to obtaining experimental
evidence for how the electronic structure of TPD-related
compounds and important physical properties associated with
them, such as oxidation (or ionisation) potential and the
re-organisation energy associated with the charge transfer
process, l, are influenced by conformational changes in the
molecular structure. We considered that a firmer grasp of the
role molecular conformation plays on the molecular and bulk
properties of TPD-type systems would prove valuable in
understanding the properties of as-cast organic films of these
materials. With this goal in mind we have prepared a series of
compounds based upon the prototypical TPD framework in
which methyl groups are used to impose a degree of control
over the relative orientation of the various six-membered ring
systems. We report here the synthesis of several TPD-
derivatives and correlate the electrochemical response, the
electronic spectra of the compounds in each electrochemically
{ Electronic supplementary information (ESI) available: The opti-mised geometries of [1-H]n+–[9-H]n+ (n 5 0, 1) in chime format,together with plots of the HOMO (neutral molecules) and SOMO(oxidised species). See http://www.rsc.org/suppdata/jm/b4/b417962e/*[email protected]
PAPER www.rsc.org/materials | Journal of Materials Chemistry
2304 | J. Mater. Chem., 2005, 15, 2304–2315 This journal is � The Royal Society of Chemistry 2005
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online / Journal Homepage / Table of Contents for this issue

accessible oxidation state and electronic structures with the
molecular structures of these compounds.
Results and discussion
Syntheses
The preparation of biaryl systems through reaction of two
molecules of an aryl halide in the presence of a copper source
was first developed by Ullmann and this C–C bond forming
protocol is now known as the Ullmann coupling. Despite the
uncertainty over the precise nature of the catalytically active
species the Ullmann coupling has been widely applied to the
synthesis of many symmetrically and unsymmetrically sub-
stituted biaryl systems.8 The extension of the Ullmann
coupling to the formation of C–N and C–O bonds via the
copper-catalysed reaction of an aryl amine or phenol with an
aryl halide has been termed the Ullmann condensation. While
the reaction is notoriously capricious, with side reactions such
as halide reduction and homocoupling often encountered, the
Ullmann condensation has been utilised for over 90 years with
considerable success. Recent modifications to the reaction
conditions include the addition of hydrolytically stable co-
ligands such as phenanthrolines and 2,29-bipyridyl when
copper(I) salts are utilised as the copper source, or the use of
18-crown-6/K2CO3 with copper powder.9 Of course, while
Ullmann protocols have been widely adopted, metal catalysed
aromatic C–N coupling reactions are not limited solely to
copper, and in the last ten years or so there have been many
interesting developments in this area, particularly with regard
to palladium-catalysed processes.10–20
The compounds 2 and 3 were prepared by Ullmann
condensation of 4,49-diiodo-1,19-biphenyl with diphenyl amine
and di(p-tolyl)amine, respectively, in refluxing ortho-dichloro-
benzene in the presence of potassium carbonate, copper
powder and 18-crown-6 (Scheme 1).
Compound 4, in which methyl groups at the 2-positions of
two peripheral rings are used to increase the pitch of these
rings relative to the biphenyl rings, was prepared in a similar
manner, as described previously.24
In the case of 5a and 5b the key reagent 4,49-diiodo-
3,39-dimethyl-1,19-biphenyl was prepared by diazotisation of
the corresponding diamine, which was subsequently coupled
with the appropriate diarylamine (Scheme 2).
The acidic methylene protons of fluorene were found to
complicate the Ullmann condensation reaction with diaryl-
amines, and consequently 4,49-diiododiethylfluorene was
employed in the preparation of the fluorene derivatives
6a and 6b,21 while carbazole was found to couple readily
with 4,49-diiodo-1,19-biphenyl to give 7 under comparable
conditions.
Compounds 8 and 9 were prepared from di(p-tolyl)amine
and 2,29-dimethyl-4,49-diiodo-1,19-biphenyl or 2,29,6,69-tetra-
methyl-4,49-diiodo-1,19-biphenyl, using Ullmann chemistry
entirely analogous to that indicated in Schemes 1 and 2.22
The products 2–9 were readily purified either by column
chromatography or simple recrystallisation of the reaction
mixture, and were fully characterised by the usual spectro-
scopic methods (see Experimental).
Fig. 1 The key torsional modes available to tetra(aryl)benzidenes.
Scheme 1 The synthesis of compounds 2 and 3 via the Ullmann
condensation.
This journal is � The Royal Society of Chemistry 2005 J. Mater. Chem., 2005, 15, 2304–2315 | 2305
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

Molecular structures
Given the dearth of solid state structural data relating to the
tetra(aryl)benzidene motif, we were pleased to obtain crystals
of 5a, 6a and 7 suitable for X-ray diffraction analysis. In
keeping with the observations previously made on the solid
state structures of 1, 3, 4, 4[SbCl6] and 9a, the molecules 5a, 6a
and 7 were found to be packed into the crystal lattice without
any strong close intermolecular contacts or p-stacking motifs
in evidence.22–24 The crystals of 5a and 6a contained
disordered molecules of benzene and CH2Cl2, respectively,
filling channels formed between adjacent molecules.
Plots illustrating single molecules of 5a, 6a and 7 are given in
Fig. 2, with a comparative overlay of the molecular structures
provided in Fig. 3. Experimentally determined bond lengths
and important dihedral angles associated with 1, 3, 4, 5a, 6a, 7
and 9a are summarised in graphical format in Chart 1. The
introduction of methyl groups on the peripheral rings in
positions meta- or para- to the amine nitrogen centre have little
influence on the molecular geometry, and the geometries of 1
and 3 are similar. In each case, the twist angle between the
adjacent rings of the biphenyl moiety is approximately 35u.The DFT optimised geometries of 1 and 3 fail to reproduce the
distinct differences in orientation of the peripheral ring systems
relative to the plane defined by the N–C bonds (see below).
Since rotation of the aryl groups around the N–Caryl bonds are
relatively low energy processes, the precise geometry adopted
by the aromatic groups is probably a function of the packing
efficiency of the molecules in the solid state. However, the
geometry of 4 is remarkably well modelled by the calculation
for the gas-phase structure, suggesting that the orientation
of the 2,4-dimethylphenyl group is likely a property of the
molecule, rather than of the crystal lattice.
For 5a, the introduction of methyl groups on the biphenyl
moiety ortho- to the amine nitrogen centre (Fig. 2) results in
rotation of the diphenylamino group around the correspond-
ing N(1,2)–C(4,23) bond (Fig. 3). Obviously, this is the most
effective way to minimise unfavourable close contacts between
Scheme 2 The preparation of 5a and 5b.
Fig. 2 The molecular structure and labelling scheme of 5a (top), 6a
(middle) and 7 (bottom). Atomic displacement ellipsoids are drawn at
the 50% probability level and disordered solvent molecules of benzene
and CH2Cl2 in 5a and 6a, respectively, are omitted.
2306 | J. Mater. Chem., 2005, 15, 2304–2315 This journal is � The Royal Society of Chemistry 2005
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

the NPh2 moieties and the C(19) and C(38) methyl groups
whilst maintaining the propeller-like geometry around N(1)
and N(2). Curiously, the biphenyl group in molecule 5a is
almost planar, and the dihedral angle between the planes of the
adjacent aromatic rings is just 210.2u.The planar conformation of the central biphenyl group in
molecule 6a is enforced by the C(2)–C(19) bonds which form
the fluorene system (Fig. 2), and the molecule maintains the
usual propeller-like orientation of the aromatic rings about
the nitrogen centres. The same C(2)–C(19) bonds also restrict
the C(2)–C(1)–C(1A) bond angle in comparison with the other
molecules of this type (Chart 1, Fig. 3). The molecule 6a is
located in a special position in the crystal with a mirror plane,
which passes through the middle of the central bond, C(19)
and the ethyl groups, relating the two halves of the molecule.
Molecule 7 also occupies a special position in the crystal at
the centre of symmetry. The biphenyl fragment is remarkably
planar, the torsion angle around the central bond being 0.2u.The rigid carbazole groups formed by the C(12)–C(13) bond
are rotated around the N(1)–C(4) bond and give a torsion
angle of 46u with the plane of the bi-phenyl group, which is
similar to those found in other crystallographically charac-
terised tetraarylbenzidenes (Chart 1, Fig. 3).
Electrochemical response
The electrochemical response of 1–9 illustrate the marked
effect the substitution patterns play on the oxidation (ionisa-
tion) potential of these materials (Table 1). The parent
material 2 undergoes two sequential, one-electron oxidation
processes at E1/2 5 0.39 and 0.64 V (vs Fc/Fc+ 5 0.00 V). The
separation of the oxidation processes allows estimation of the
comproportionation constant (Kc) to be 18 000 and indicates
Fig. 3 Overlapped views of the molecules 5a, 6a and 7. Top:
view perpendicular to the biphenyl amine C–N bonds, atoms
of the symmetry related parts of the molecules and H
atoms omitted for clarity. Bottom: view perpendicular to the
mean plane of biphenyl fragment, H atoms also omitted.
Open lines correspond to molecule 5a, full lines to 6a and dashed
lines to 7.
Chart 1
This journal is � The Royal Society of Chemistry 2005 J. Mater. Chem., 2005, 15, 2304–2315 | 2307
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

Table 1 Oxidation potentials and associated parameters with 1–9 (in CH2Cl2, 0.1M NBu4BF4, 100 mV s21, all platinum electrodes, vsFc/Fc+ 5 0.0 V
E1/2(1)/V E1/2(2)/V DE/V KC
0.29 0.51 0.22 4300
0.39 0.64 0.25 18 000
0.25 0.55 0.30 133 000
0.25 0.51 0.26 20 500
0.34 0.56 0.22 5900
0.35 0.47 0.12 100
0.28 0.65 0.37 1 955 000
0.12 0.47 0.35 970 000
2308 | J. Mater. Chem., 2005, 15, 2304–2315 This journal is � The Royal Society of Chemistry 2005
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

the significant thermodynamic stability of the radical cation 2+
with respect to disproportionation.25 These parameters pro-
vide a convenient data set against which to measure the
influence of the various structural modifications made in the
other compounds.
In the case of the established hole transport material 1, and
the related materials 3 and 4, the inductively donating methyl
groups around the peripheral rings result in the expected
decrease in the first [E1/2(1)] and second [E1/2(2)] oxidation
potentials (Table 1). Compounds 3 and 4, which contain
inductively electron donating methyl groups at the 4-positions
of the peripheral ring systems, have the lowest first oxidation
potentials in this subset. Thus, while the ortho- methyl groups
in 4 can influence the conformation of the molecular system in
the solid state, the influence of this substituent on the
electrochemical behaviour appears to be inconsequential.
When compared with the observed and calculated geome-
tries of TPD and closely related materials, the dominant
structural differences in the mono-oxidised states of these
molecules include a more planar biphenyl fragment, which is
co-planar with the N–C bonds, and an increase in the torsion
angles about the N–Caryl bonds such that the peripheral ring
systems adopt orientations which enhance the propeller
geometry about the nitrogen centres.6,24 The electrochemical
response of compounds featuring the generic tetra(aryl)benzi-
dene structure are therefore likely to be sensitive to the
introduction of groups at the 2 and 3 positions of the central
biphenyl moiety (compounds 5, 6, 8, 9).
The first oxidation potentials of 5a and 5b are similar to
each other, and more positive than E1/2(1) of 1, 3 or 4. It seems
that in 5a and 5b the C(19) methyl group, which hinders
rotation about the N(1)–C(4) bond and restricts conjugation of
the nitrogen lone pairs through the biphenyl moiety, necessi-
tates a greater thermodynamic driving force for oxidation than
is the case in the unrestricted systems 1, 3 and 4. The trend in
E1/2(2) values is more keeping with the expected inductive
effects (e.g. E1/2(2) 5b , 3), further supporting the hypothesis
that upon oxidation the peripheral aromatic systems adopt an
orientation more orthogonal to the central biphenyl group and
are therefore less affected by the steric influence of the C(19)
methyl group.
Similar trends are observed when methyl groups are used to
restrict the adoption of a planar conformation in the biphenyl
moiety (8, 9).22 Thus, despite the additional inductively
electron donating methyl groups at the 2 and 29 positions of
the 1,19-biphenyl fragment in 8 the first oxidation potential is
significantly higher than that of 3. The second oxidation
potential is broadly in keeping with the trends established for
the other compounds in Table 1.
The introduction of a fluorene moiety in 6a and 6b, which
confines the biphenyl moiety to be planar, results in a
significant decrease in the first oxidation potential relative to
the model materials 1 and 3. The inductive effect of the methyl
groups on the peripheral ring systems has the expected
influence on the first and second oxidation potentials of 6b
relative to those of 6a.
The electrochemical response of the carbazole derivative 7
was complicated by the relative insolubility of the radical
cation in the presence of common anions such as BF42 and
PF62. The most reproducible electrochemical results were
obtained from THF solutions containing 0.1 M NBu4BF4 as
supporting electrolyte. The initial voltammogram revealed two
1.03 (thf) 1.50 (thf) 0.470 —
0.44 0.59 0.15 300
0.46 — — —
Table 1 Oxidation potentials and associated parameters with 1–9 (in CH2Cl2, 0.1M NBu4BF4, 100 mV s21, all platinum electrodes, vsFc/Fc+ 5 0.0 V (Continued )
E1/2(1)/V E1/2(2)/V DE/V KC
This journal is � The Royal Society of Chemistry 2005 J. Mater. Chem., 2005, 15, 2304–2315 | 2309
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

chemically irreversible oxidation processes at 1.03 and 1.50 V
(vs an internal Fc/Fc+ standard) and inspection of the working
electrode revealed an obvious film on its surface. These
chemical complications associated with 7 have been commen-
ted upon previously and we have been unable to find a
satisfactory combination of solvent and supporting electrolyte
to resolve these issues.26
Spectroelectrochemistry
Electronic spectroscopy (UV–vis–NIR) coupled with spectro-
electrochemical methods has proven to be a useful tool in the
analysis of the electronic structure of tetra(aryl)benzidenes and
closely related structures in various oxidation states27–29 and
the electronic spectra of 2n+, 3n+, 4n+ and 8n+ (n 5 0, 1, 2) have
been described in detail on previous occasions.22–24,30 The
monocations (n 5 1) derived from compounds 1–8 can be
regarded as organic ‘‘mixed-valence’’ compounds. Detailed
analyses of the NIR and/or Raman spectra of 2+, 3+, 4+ and
closely related systems which preserve the 4,49-diamino-
1,19-biphenyl substructure have concluded that these radical
cations lie at the border between Class II (valence trapped) and
Class III (delocalised) materials.24,27,30,31
The changes in the UV–vis–NIR spectral regions in the
series of compounds 1–8 that accompany oxidation to the
mono- and di-cations were followed using an OTTLE cell in
CH2Cl2 containing 0.1 M NBu4BF4 as supporting electro-
lyte.32 In general terms, oxidation of each compound to the
corresponding monocation was characterised by the shift of
the p–p* bands from 300–350 nm to 400–500 nm (Table 2) and
the appearance of a new spectral feature in the NIR region, the
energy and shape of which was sensitive to molecular structure
(Table 3). Further oxidation resulted in the collapse of the
monocation spectrum and the appearance of a new band
between 670–830 nm.
The spectroelectrochemical response of 5a was complicated
by the reactivity of the mono-oxidised form. While not
Table 2 UV–vis–NIR spectra [lmax/cm21 (e/M21dm23cm21)] of1–9n+ (in CH2Cl2, 0.1 M NBu4BF4)
Oxidation state/n
0 1 2
1 32 260 (24 280) 20 660 (28 000) 13 700 (74 800)28 300 (40 150) 71 40 (37 800) —
2 33 000 (33 100) 20 750 (26 100) 14 120 (49 300)28 500 (44 800) 7310 (27 300) —
3 33 100 (23 400) 20 660 (29 700) 12 750 (64 000)28 170 (36 600) 6820 (32 600) —
4 32 790 (24 280) 20 960 (28 000) 13 330 (56 590)28 570 (35 300) 7450 (30 680) —
5a 33 200 (40 200) — —29 950 (27 800) — —
5b 32 900 (34 450) 21 370 (11 000) 12 050 (56 600)29 400 (23 900) 14 620 (7230) —— 5260 (14 200) —
6a 31 850 (29 400) 20 200 (24 600) 14 900 (42 800)26 300 (42 200) 7520 (20 000) —
6b 31 650 (28 400) 20 700 (32 100) 12 820 (78 700)26 300 (40 200) 7520 (36 400) —
7 34 130 (38 300 — —31 550 (30 000) — —29 400 (19 600) — —
Ta
ble
3B
an
dsh
ap
ea
nd
der
ived
da
tafr
om
the
NIR
ba
nd
in1
+ –8
+(i
nC
H2C
l 2,
0.1
MN
Bu
4B
F4)
v max/c
m2
1e/
M2
1cm
21
v 1/2
(ob
s)/c
m2
1v 1
/2(H
TL
)/cm
21
v 1/2
(hig
h)
v 1/2
(lo
w)
v 1/2
(hig
h)/
v 1/2
(lo
w)
VA
B~
� nn ma
x
2/c
m2
1V
AB~
meg er
� nn ma
xV
AB~
0:0
20
5
r
�� e m
ax� nn 1=
2� nn m
ax
�� 1=2
/cm
21
17
14
03
78
00
30
80
40
50
37
20
24
40
1.5
23
57
01
90
01
89
02
73
10
27
30
03
00
54
10
03
54
02
47
01
.43
36
55
16
40
16
00
36
82
03
26
00
30
40
39
60
35
20
25
60
1.3
73
41
01
86
01
70
04
74
50
30
68
03
00
54
13
03
73
02
28
01
.63
37
25
—1
72
05
b5
26
04
72
00
32
00
34
70
37
20
26
80
1.3
92
63
01
30
01
85
06
a7
52
03
22
00
30
80
41
50
36
20
25
40
1.4
23
76
01
80
01
79
06
b7
52
03
64
00
30
85
41
60
35
30
26
40
1.3
43
76
01
78
01
90
08
53
50
57
60
03
22
03
50
03
64
02
80
01
.30
26
60
20
70
20
60
2310 | J. Mater. Chem., 2005, 15, 2304–2315 This journal is � The Royal Society of Chemistry 2005
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

apparent on the much shorter timescale of the cyclic
voltammetry experiment, [5a]+ proved to be reactive on the
longer timescales necessary for bulk electrolysis in the OTTLE
cell. Oxidative oligomerisation/polymerisation of triphenyl
amine based materials with unprotected para- positions has
been demonstrated on previous occasions.22,33–38 It is likely in
the present case that the chemical complications observed in
the spectroelectrochemical experiment are related to similar
coupling reactions.
According to the Hush model for weakly coupled (Class II)
systems, the electronic coupling parameter VAB can be deduced
from the band energy and shape of the HOMO-1ASOMO
transition. This absorption, which usually falls in the NIR
region, is often termed the ‘‘intervalence charge transfer’’, or
IVCT, band, and in the case of weakly coupled (Class II)
systems the IVCT transition corresponds to the photo-induced
intramolecular electron transfer process. In strongly coupled
(Class III) systems the optical absorption involves transitions
between delocalised molecular orbitals, and the IVCT nomen-
clature can be misleading, and the term ‘‘charge resonance
band’’ has been suggested as an alternative descriptor.39,40 In
practice, extraction of VAB from spectroscopic data using
traditional approaches requires accurate assignment of the
mixed-valence species to the correct Robin–Day class and a
reasonable estimate of the electron transfer distance, which can
be difficult when transitions which lead to the NIR absorption
band are delocalised over a significant portion of the molecular
framework.41 In this context it should be noted that Lambert
and colleagues have recently described an adaptation of the
semi-classical Marcus–Hush theory which allows calculation
of VAB in strongly coupled systems by numerical fitting of the
experimental spectrum without any assumption of the
electron-transfer distance.24,40
If we assume the radical cations 1+–8+ to lie on the Class II
side of the II–III borderline, the term VAB may be estimated
from eqn. (1), which makes no implicit assumption about the
shape of the NIR band. In eqn. (1), meg is the transition
moment (in C m) between the ground and excited states and
can be calculated (in Debye, 1 D 5 3.336 6 10230 C m) from
the integrated area under the curve from eqn. (2), vmax is the
transition energy, r is the distance between the diabatic redox
centres and e is the elementary charge.
VAB~meg
er�nnmax (1)
meg~0:09584
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiÐe(�nn)d�nn
�nnmax
s(2)
The N…N distance in the crystallographically determined
structure of the SbCl62 salt of 4+ was used to estimate r (9.89 A),
although the delocalised nature of the orbitals involved in the
transition together with very recent work by Nelson suggest that
such an approximation leads to an overestimation of the
adiabatic ET distance.41, 42 The calculated VAB terms calculated
using this approach are in good agreement with the values
calculated from the Hush relationship summarised by eqn. (3).
VAB~0:0205
r
� � ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiemax�nn1=2�nnmax
p(3)
Alternatively, if the radical cations are strongly coupled
with a vanishingly small thermal barrier to electron transfer
(i.e. the system is delocalised, Class III) then VAB is
determined from the band energy by the simple relationship
described in eqn. (4). There is an increasing body of experi-
mental support for the description of radical cations derived
from simple TPD type systems in terms of a delocalised
structure.24,30,40
VAB~�nnmax
2(4)
The shapes of the NIR bands from spectroelectrochemically
generated 1+–8+ (in CH2Cl2/0.1 M NBu4BF4) and the VAB
terms which may be calculated from eqns. (1), (3) and (4) are
summarised together with the appropriate parameters in
Table 3.
The value of VAB calculated from eqn. (4) for 1+ and 4+ is
only ca. 10% larger than that calculated using Lambert’s semi-
empirical method (1+ 3200 cm21; 4+ 3300 cm21), and therefore
eqn. (4) provides a reasonable estimate for this parameter. The
data collected in Table 3 reveal some interesting trends in
the value of VAB as a function of molecular structure. For the
mono-oxidised forms of compounds 1, 2, 3, 4 and 6a, 6b the
value of VAB calculated using eqn. (4) is essentially the same
(3650 cm21 ¡ 10%), although much lower values of the
coupling term are associated with 5b+ and 8+ (ca. 2650 cm21).
These experimental observations are consistent with con-
clusions drawn from computational work performed earlier by
others, and indicate that the electronic properties of these
redox-active tetraphenyl benzidenes are more heavily depen-
dent upon the planarity of the biphenyl core and the
conjugation of the nitrogen lone pair with the biphenyl
p-system than the orientation of the peripheral ring systems.
However, based on the much higher first oxidation potential of
the carbazole derivative 7 we are reluctant to totally neglect the
contribution of the peripheral rings to the overall stability of
the mono-oxidised forms of these materials.
Molecular modelling
Geometry optimisation (see the electronic supplementary
information (ESI) for 3D structures{) and electronic structure
calculations were undertaken for each compound 1–9 using
DFT methods (see Experimental). We designate the computa-
tional model systems as ‘‘-H’’ in order to distinguish them
from the physical samples. The calculated geometries are in
good agreement with the experimentally determined para-
meters, and the differences computed to occur following
oxidation are in broad general agreement with previous
computational4–6 and experimental studies.24
The role of the methyl substituents on the biphenyl ring
systems on the crystallographically determined structure 5a are
clearly reproduced in the calculated geometries of 5a-H and
5b-H. In the case of 8, for which no experimental structural
data are available, the calculated structure (8-H) indicates
that the 2,29-dimethyl substitution pattern imposes an almost
orthogonal orientation of the biphenyl rings, which is in full
agreement with the experimentally determined geometries of
other 2,29-dimethyl biphenyl compounds.43–45 The steric
influence of the 2,29-dimethyl groups is still evident in the
This journal is � The Royal Society of Chemistry 2005 J. Mater. Chem., 2005, 15, 2304–2315 | 2311
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

calculated geometry of [8-H]+ with the torsion angle between
the rings of the biphenyl moiety being as large as 52u.The calculated first ionisation potentials (IPs) associated
with 1-H–9-H are given in Table 4. In the case of 1-H, the IP
value of 5.38 eV obtained from our 6-31G-BPW91-DFT
results may be compared with the value of 5.73 eV from
Bredas’ earlier 6-31G**-B3LYP based calculations,6 and the
experimental value of 5.38 ¡ 0.15 eV for 1.46 Although the
computational work neglects solvation factors there is an
essentially linear correlation between the calculated IP values
and the first oxidation potential of these materials, which
lends weight to the qualitative arguments developed in the
analysis of the electrochemical response based upon structural
variation.
One of the critical parameters involved in the description of
the hole-transport process based on a Marcus-type electron
transfer reaction is the reorganisation energy, l, associated
with the change in molecular geometry which accompanies the
transfer of charge. The reorganisation energy can be estimated
from the sum of the stabilisation energy achieved by geometry
relaxation of the radical cation and neutral form of the
material (i.e. the sum of the difference in energies associated
with the material in neutral and radical cation electronic
configuration in cation and neutral geometry).5,6 Where direct
comparisons are possible, the calculated reorganisation energy
from the BPW1-DFT calculations is in good agreement with
the data reported from AM1(UHF) based calculations (2:
l 5 0.14 eV (this work) vs. 0.132 eV (AM1(UHF)7), but
considerably lower than the B3LYP estimated value (1: l 5
0.13 eV (this work) vs. 0.29 eV (B3LYP)6), reflecting the
sensitivity of the absolute value of the calculated parameters to
the computational method employed.
Within the data series for our compounds, the lowest values
of l calculated from the BPW1 determined geometries are,
unsurprisingly, associated with the rigid molecular framework
of 7, and the triphenylamine-like compound 9. The introduc-
tion of the 3,39 methyl groups on the biphenyl moiety does
not have a significant influence on the reorganisation energy
associated with the oxidation of [5a/b-H], which can be
attributed to the lowest energy conformation of [5a/b-H]+
being one which avoids steric interaction between the peri-
pheral ring system and these groups.
The reorganisation energy of 6a is curiously high compared
with that of 6b, and other compounds in the series. Given the
significantly greater experimental oxidation potentials of 6a
relative to 6b this influence seems to be of real physical
significance and not an artefact of the calculations, although
the origin is not clear at present. The largest reorganisation
parameter in the series investigated here is associated with 8,
in which the 2,29-methyl groups restrict the attainment of a
planar conformation in the biphenyl portion of the molecule.
Therefore, the present work, which has investigated a
significant number of TPD derivatives, supports the conclu-
sions reached in Malagoli and Bredas’ study of TPD, and
indicates that the conformation of the biphenyl moiety is
the dominating molecular property in determining the relative
gas-phase reorganisation energy in these tetra(aryl)benzidene-
based hole-transport materials.
Conclusion
A combination of single crystal X-ray diffraction, electro-
chemical and spectroelectrochemical measurements and DFT
based calculations have been used to interrogate the impact of
molecular conformation on oxidation (ionisation) potential,
electronic structure and reorganisation barriers to the charge
transfer process in TPD-type hole transport materials. The
oxidation (ionisation) potentials of TPD-style materials may
be tuned not only through inductive effects, but also through
the influence of Marcus-type factors, with the conformation
of the biphenyl moiety playing a significant role in determining
the reorganisation energy associated with the hole-transfer
process on the molecular level.
Experimental
Instrumentation
Mass spectra were recorded on a Micromass Autospec
instrument operating in EI mode. NMR spectra were recorded
from CDCl3 solutions on Varian XL-200, Varian Unity-300
or Varian VXR-400 spectrometers. All chemical shifts are
reported in d (ppm), referenced against solvent resonances.
UV–vis–NIR spectra were recorded on a Varian Cary-5
spectrophotometer, electrochemical measurements were made
with an AutoLab PGSTAT30 from CH2Cl2 solutions contain-
ing 0.1 M NBu4BF4 as supporting electrolyte and potentials
cited are referenced against ferrocene such that the ferrocene
couple falls at 0 V. Spectroelectrochemical experiments were
Table 4 Calculated energies (in eV), ionisation potentials and reorganisation energies for compounds 1-H–9-H
Compound E(neu)/eV E(cat-scf)/eV E(cat)/eV E(neu-scf)/eV E1/2/V IP/eV l/eV
1-H 0 5.44 5.38 0.066 0.29 5.44 0.132-H 0 5.51 5.45 0.071 0.39 5.51 0.143-H 0 5.29 5.23 0.062 0.25 5.29 0.124-H 0 5.42 5.34 0.075 0.25 5.42 0.155a-H 0 5.58 5.52 0.076 0.34 5.58 0.145b-H 0 5.36 5.30 0.063 0.35 5.36 0.126a-H 0 5.41 5.33 0.093 0.28 5.41 0.186b-H 0 5.19 5.13 0.066 0.12 5.19 0.147-H 0 6.04 6.01 0.037 1.03 6.04 0.078-H 0 5.39 5.26 0.075 0.44 5.39 0.209-H 0 5.35 5.31 0.045 0.46 5.35 0.09a E(neu) energy of the neutral molecule in the neutral geometry, chosen to be the reference point; E(cat-scf) energy of the cation in the neutralgeometry; E(cat) energy of the cation in the cation geometry; E(neu-scf) energy of the neutral molecule in the cation geometry
2312 | J. Mater. Chem., 2005, 15, 2304–2315 This journal is � The Royal Society of Chemistry 2005
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

also carried out in 0.1 M NBu4BF4/CH2Cl2 solutions using an
OTTLE cell,32 fitted with a Pt gauze mesh semi-transparent
working electrode, and Pt wire counter and pseudo-reference
electrodes, and a home-built potentiostat.
All synthetic reactions were carried out under a nitrogen
atmosphere in flame-dried glassware. Solvents were dried from
an appropriate agent and distilled. Compounds 2,29-dimethyl-
4,49-diiodobiphenyl, 2,29,6,69-tetramethyl-4,49-diiodobiphenyl,
3,39dimethyl-4,49-diiodobiphenyl,47,48 9,99-diethyl-2,7-diiodo-
fluorene,49 3, 4, 8 and 922,24 were prepared by literature
methods. TPD (1) was obtained from Avecia Ltd. All other
reagents were purchased from commercial sources and used
as received.
General procedure. In our hands, the Ullmann condensation
reactions proceeded most readily and consistently when
employing 18-crown-6 as a solubilising agent for the active
copper species and ortho-dichlorobenzene as solvent.9 In a
typical procedure, a rigorously dried two-necked 100 ml
round-bottomed flask was fitted with a magnetic stir bar,
and a condenser topped with a nitrogen gas inlet. The
apparatus was purged with dry nitrogen, and charged with
ortho-dichlorobezene (10 ml), the appropriate di(aryl)amine
(5.5 mmol) and diiodobiaryl (2.5 mmol), K2CO3 (20 mmol),
copper powder (5.0 mmol) and 18-crown-6 (0.5 mmol). The
reaction mixture was rapidly heated at the reflux point of the
solvent (ca. 180 uC) and maintained at that temperature for
12–24 h. After this time, the reaction mixture was cooled
and filtered to remove the inorganic residues. The filtrate was
reduced in volume under vacuum, and the crude product
purified by column chromatography or precipitation and
recrystallisation. Satisfactory elemental analyses were obtained
for all compounds.
N,N,N9,N9-Tetraphenyl-1,19-phenyl-4,49-diamine (2).
Diphenylamine (0.916 g, 5.43 mmol), diiodobiphenyl (1.0 g,
2.46 mmol), K2CO3 (2.72 g, 19.68 mmol), copper powder
(0.34 g, 5.3 mmol) and 18-crown-6 (0.13 g, 0.49 mmol) were
dissolved in ortho-dichlorobenzene (10 ml) and heated at reflux
for 18.5 h. The crude reaction mixture was diluted with MeOH
to precipitate the crude product which was collected, dried and
recrystallised from CH2Cl2 to afford the pure title compound
(0.68 g, 56%). 1H NMR d 7.48 (d, JHH 5 9 Hz, 4H), 7.16
(pseudo-t, JHH 5 15 Hz, 8H), 7.03 (d, JHH 5 9 Hz, 8H), 7.03
(d, JHH 5 9 Hz, 4H), 6.93 (t, JHH 5 14 Hz, 4H). 13C NMR d
147.71, 146.73, 134.74, 129.25, 127.29, 124.30, 124.08, 122.81.
EI MS m/z 488 [M]+. mp 220–222u
N,N,N9,N9-Tetraphenyl-(3,39-dimethyl)-(1,19-biphenyl)-
4,49-diamine (5a). The compounds 3,39-dimethyl-4,49-diiodo-
biphenyl (1.0 g, 2.3 mmol), diphenylamine (0.92 g, 5.75 mmol),
K2CO3 (2.54 g, 18.43 mmol), copper powder (0.31 g,
4.94 mmol), and 18-crown-6 (0.12 g, 0.46 mmol) were
dissolved in ortho-dichlorobenzene (10 ml) and heated to
reflux for 18 h. The solution was filtered and the solvent
removed in vacuo, the residue was dissolved in MeOH and
placed in the freezer, producing a pale yellow precipitate which
was purified by column chromatography (silica, hexanes–
CH2Cl2) (0.42 g, 36%). 1H NMR d 7.52 (d, JHH 5 2 Hz, 2H),
7.47 (dd, JHH 5 8 Hz, 2H), 7.26 (pseudo-t, apparent JHH 5
8 Hz, 8H), 7.21 (d, JHH 5 8 Hz, 2H), 7.05 (d, JHH 5 8 Hz, 8H),
6.97 (t, JHH 5 14 Hz, 4H), 2.12 (s, 6H, CH3). 13C NMR d
147.48, 144.65, 138.03, 136.53, 130.21, 129.75, 129.07, 125.83,
121.69, 121.51, 18.83. EI MS m/z 516 [M]+, 349 [M-NPh2]+
mp 174–176u
N,N,N9,N9-Tetra(4-methylphenyl)-(3,39-dimethyl)-1,19-
biphenyl-4,49-diamine (5b). A solution of 3,39-dimethyl-4,49-
diiodobiphenyl (1.0 g, 2.3 mmol), di(4-methylphenyldiamine
(1.0 g, 5.07 mmol), K2CO3 (2.54 g, 18.43 mmol), copper
powder (0.31 g, 4.94 mmol), and 18-crown-6 (0.12 g,
0.46 mmol) in ortho-dichlorobenzene was heated at reflux for
27 h. The solution was filtered and the solvent removed
in vacuo, the residue was recrystallised from MeOH in the
freezer. The yellow precipitate was purified by flash chroma-
tography (SiO2, CH2Cl2–hexane 1 : 9), (0.43 g, 33%). 1H NMR
d 7.38 (m, 2H), 7.19 (m, 4H), 6.88 (m, 16H), 2.22 (s, 12H,
CH3), 2.00 (s, 6H, CH3). 13C NMR d 145.35, 144.97, 137.61,
136.19, 130.72, 130.04, 129.59, 129.35, 125.62, 121.69, 20.68,
18.91. EI MS m/z 572 [M]+, 480 [M-Ar]+, 377 [M-NAr2]+.
mp 170–172u.
9,9-Diethyl-2,7-diiodofluorene. To a stirred solution of 9,9-
diethylfluorene (5.5 g, 24.7 mmol) and finely divided I2 (6.92 g,
27.3 mmol) in acetic acid (100 ml), a mixture of red fuming
nitric acid (2 ml) and concentrated sulfuric acid (10 ml) was
added dropwise. The mixture was stirred for 30 min then
poured into H2O (700 ml), neutralised with a sodium hydrogen
carbonate solution and the organics extracted with Et2O. The
solution was washed with a sodium thiosulfate solution, dried
(MgSO4) and the solvents removed in vacuo, producing an off
white solid. (3.56 g, 30%). 1H NMR d 7.49 (m, 2H, Ar), 7.26
(m, 2H, Ar), 7.11 (s, 2H, Ar), 1.82 (q, JHH 5 20 Hz, 4H, CH2),
0.16 (t, JHH 5 16Hz, 6H, CH3). 13C NMR d 150.79, 140.35,
136.12, 132.45, 121.69, 93.04, 56.30, 32.72, 8.27. EI MS m/z
474 [M]+, 347 [M-I]+, 222 [M-2I]+
N,N,N9,N9-Tetraphenyl-9,9-diethyl-2,7-diaminofluorene
(6a). A solution of 9,9-diethyl-2,7-diiodofluorene (1.5 g,
3.15 mmol), diphenylamine (1.17 g, 6.93 mmol), K2CO3
(3.48 g, 25.21 mmol), copper powder (0.43 g, 6.77 mmol),
and 18-crown-6 (0.17 g, 0.63 mmol) in ortho-dichlorobenzene
was heated at reflux for 18 h. The solution was filtered and the
solvent removed in vacuo. Purified by chromatography (silica,
hexanes–CH2Cl2) (0.531 g, 30%). 1H NMR d 7.0 (m, 26H),
1.72 (q, JHH 5 21 Hz, 4H), 0.29 (t, JHH 5 14 Hz, 6H). 13C
NMR d 151.28, 146.48, 144.32, 130.55, 129.86, 129.11, 127.72,
123.84, 122.33, 119.54, 56.25, 32.55, 8.56. EI MS m/z 556 [M]+,
404 [M-2Ph]+, 389[M-N Ph2]+. mp 191–193u
N,N,N9,N9-Tetra(4-methylphenyl)-9,9-diethyl-2,7-diamino-
fluorene (6b). A solution of 9,9-diethyl-2,7-diiodofluorene
(1.5 g, 3.15 mmol), di(4-methylphenyl)amine (1.37 g, 6.93 mmol),
K2CO3 (3.48 g, 25.21 mmol), copper powder (0.43 g,
6.77 mmol), and 18-crown-6 (0.17 g, 0.63 mmol) in ortho-
dichlorobenzene was heated at reflux for 18 h. The solution
was filtered and the solvent removed in vacuo, the residue was
recrystallised from MeOH at 220 uC (0.87 g, 45%). 1H NMR d
This journal is � The Royal Society of Chemistry 2005 J. Mater. Chem., 2005, 15, 2304–2315 | 2313
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

7.0 (m, 22H), 2.2(s, 12H), 1.71 (q, JHH 5 20 Hz, 4H), 0.29 (t,
JHH 5 20 Hz, 6H). 13C NMR d 151.28, 145.72, 142.58, 132.80,
130.56, 130.02, 127.97, 124.56, 121.69, 56.43, 32.81, 21.04,
18.31, 8.83. EI MS m/z 612 [M]+, 417 [M-NAr2]+. mp 187–189u
4,49-Dicarbazole-1,19-biphenyl (7). Carbazole (0.90 g,
5.42 mmol), diiodobiphenyl (1.0 g, 2.46 mmol), K2CO3
(2.72 g, 19.7 mmol), copper powder (0.43 g, 6.77 mmol), and
18-crown-6 (0.13 g, 0.49 mmol) were dissolved in ortho-
dichlorobenzene and heated to reflux for 19.5 h. The solution
was filtered and the solvent removed in vacuo, the residue
was recrystallised from MeOH at 220 uC (0.94 g, 79%). 1H
NMR d 8.18 (d, JHH 5 8 Hz, 4H), 7.93 (d, JHH 5 8 Hz, 4H),
7.73 (d, JHH 5 8 Hz, 4H), 7.53 (d, JHH 5 8 Hz, 4H), 7.47
(pseudo-t, apparent JHH 5 15 Hz, 4H, Hc), 7.33 (t, JHH 5
15 Hz, 4H). 13C NMR d 140.82, 139.30, 137.25, 128.52, 127.49,
126.02, 123.50, 120.38, 120.08, 109.83. EI MS m/z 484 [M]+.
mp 270–272u
Crystallographic details
The data were collected at 120(1) K on a Bruker SMART
CCD 6000 (Bruker SMART CCD 1K for 5a) diffractometer
(v-scan, 0.3u per frame) using Mo Ka radiation (l 5 0.71073 A).
No absorption correction was applied. The structures were
solved by direct method and refined by full-matrix least
squares on F2 for all data using SHELXTL software. All non-
hydrogen atoms (except disordered ones) were refined with
anisotropic displacement parameters, H atoms in structure 7
were located on the difference map and refined isotropically, in
the structures 5a and 6a H atoms were placed into ideal
calculated positions and refined in ‘‘riding-mode’’.
Crystal data for 5a. C38H32N2?C6H6, M 5 594.76, triclinic,
space group P1, a 5 10.1614(3), b 5 10.6110(4), c 5
16.4871(5) A, a 5 83.65(1), b 5 78.61(1), c 5 71.11(1)u,U 5 1646.7(1) A3, F(000) 5 632, Z 5 2, Dc 5 1.200 mg m23,
m 5 0.069 mm21. 19 694 reflections ( 2.15 ¡ h ¡ 28.5u) were
collected yielding 8276 unique data (Rmerg 5 0.048). Final
wR2(F2) 5 0.1704 for all data (412 refined parameters),
conventional R(F) 5 0.0522 for 6180 reflections with I . 2s,
GOF 5 1.064. The largest peak on the residual map (0.45 e A23)
is in the disordered benzene molecule area.{
Crystal data for 6a. C41H36N2?0.5CH3Cl, M 5 580.45,
orthorhombic, space group Pnma, a 5 8.4577(2), b 5
19.9091(5), c 5 19.2392(5) A, U 5 3239.6(1) A3, F(000) 5
1230, Z 5 4, Dc 5 1.190 mg m23, m 5 0.108 mm21. 20 716
reflections ( 2.05 ¡ h ¡ 26.0u) were collected yielding 3282
unique data (Rmerg 5 0.067). The H atom of the disordered
solvent CH3Cl molecule could not be located reliably and was
not included in the refinement. Final wR2(F2) 5 0.1600 for all
data (234 refined parameters), conventional R(F) 5 0.0522 for
2295 reflections with I . 2s, GOF 5 1.099. The largest peak
on the residual map (0.52 e A23) is located in the vicinity of the
disordered chlorine atoms.
Crystal data for 7. C36H24N2, M 5 484.57, monoclinic,
space group P21/c, a 5 8.0120(4), b 5 16.0080(7), c 5
10.2428(5) A, b 5 110.19(1)u, U 5 1232.9(1) A3, F(000) 5 508,
Z 5 2, Dc 5 1.305 mg m23, m 5 0.076 mm21. 7634 reflections
(2.47 ¡ h ¡ 29.0u) were collected yielding 3249 unique data
(Rmerg 5 0.062). Final wR2(F2) 5 0.0874 for all data
(220 refined parameters), conventional R(F) 5 0.0458 for
1739 reflections with I ¢ 2s, GOF 5 0.933. The largest peak
on the residual map is 0.20 e A23.
Computational work
All calculations were run using the Gaussian 98 or Gaussian 03
programs. For each molecule, the geometry was optimised for
both the neutral and radical cation states at a DFT level using
the BPW91 functional (Becke’s 1988 exchange functional,50
with Perdew and Wang’s 1991 gradient-corrected correlation
functional51) in conjunction with the 6-31G(d,p) basis set.52–56
Where possible, molecular symmetry constraints were used to
simplify the calculations and to aid the interpretation of the
results. Post-processing for visualisation of the molecular
orbitals generated by the DFT calculations was performed
using the Molekel programme.57
Acknowledgements
This work was supported by the University of Durham, the
EPSRC, ONE-North East and Avecia Ltd. MAJP held an
EPSRC/Avecia Ltd CASE award through the CASE for
New Academics programme (PJL).
Paul J. Low,*a Michael A. J. Paterson,a Dmitry S. Yufit,a
Judith A. K. Howard,a Julian C. Cherryman,b Daniel R. Tackley,b
Robert Brookc and Bev Brownc
aDepartment of Chemistry, University of Durham, South Road, Durham,UK DH1 3LE. E-mail: [email protected];Fax: +(44) (0)191 384 4737; Tel: +(44) (0)191 334 2114bIntertek ASG, PO Box 42, Hexagon House, Blackley,Manchester M9 8ZS, UKcAvecia Ltd., Hexagon House, Blackley, Manchester M9 8ZS, UK
References
1 M. Malagoli, M. Manoharan, B. Kippelen and J. L. Bredas, Chem.Phys. Lett., 2002, 354, 28.
2 S. Aratani, T. K. Kawanishi and A. Kakuta, Jpn. J. Appl. Phys.,1991, 30, L1656.
3 S. Aratani, T. Kawanishi and A. Kakuta, Jpn. J. Appl. Phys., 1996,35, 2184.
4 B. C. Lin, C. P. Cheng and Z. P. M. Lao, J. Phys. Chem. A, 2003,107, 5241.
5 K. Sakanoue, M. Motada, M. Sugimoto and S. Sakaki, J. Phys.Chem. A, 1999, 103, 5551.
6 M. Malagoli and J. L. Bredas, Chem. Phys. Lett., 2000, 327, 13.7 K. Sakanoue, M. Motoda, M. Sugimoto and S. Sakaki, Nonlinear
Opt., 2000, 26, 271.8 P. E. Fanta, Chem. Rev., 1964, 64, 603.9 S. Gauthier and J. M. J. Frechet, Synthesis, 1987, 383.
10 C. G. Frost and P. Mendonca, J. Chem. Soc., Perkin Trans. 1,1998, 2615.
11 J. F. Hartwig, Angew. Chem., Int. Ed. Engl., 1998, 37, 2046.12 J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609.
{ The structures of compounds 5a, 6a and 7 have been deposited withthe CCDC, reference numbers 257076–257078, respectively. See http://www.rsc.org/suppdata/jm/b4/b417962e/ for crystallographic data inCIF format.
2314 | J. Mater. Chem., 2005, 15, 2304–2315 This journal is � The Royal Society of Chemistry 2005
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online

13 B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120,7369.
14 M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118,7217.
15 A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem.,Int. Ed. Engl., 1995, 34, 1348.
16 A. S. Garam, R. A. Rennels and S. L. Buchwald, Angew. Chem.,Int. Ed. Engl., 1995, 34, 1348.
17 J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1996, 61, 1133.18 J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc.,
1996, 118, 7215.19 J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 2000, 65, 1144.20 S. M. Reid, J. T. Mague and M. J. Fink, J. Organomet. Chem.,
2000, 616, 10.21 While this work was in progress, the synthesis and molecular
structure of an unsolvated form of 6a was reported: D. Z. Cao,Z. Q. Lin, Q. Fang, G. B. Xu, G. Xue, G. Q. Liu and W. T. Yu,J. Organomet. Chem., 2004, 689, 2201.
22 P. J. Low, M. A. J. Paterson, A. E. Goeta, D. S. Yufit,J. A. K. Howard, J. C. Cherryman, D. R. Tackley and B. Brown,J. Mater. Chem., 2004, 14, 2516.
23 A. R. Kennedy, W. E. Smith, D. R. Tackley, W. I. F. David,K. Shankland, B. Brown and S. J. Teat, J. Mater. Chem., 2002, 12,168.
24 P. J. Low, M. A. J. Paterson, H. Puschmann, A. E. Goeta,J. A. K. Howard, C. Lambert, J. C. Cherryman, D. R. Tackley,S. Leeming and B. Brown, Chem.–Eur. J., 2004, 10, 83.
25 D. E. Richardson and H. Taube, Inorg. Chem., 1981, 20, 1278.26 B. E. Kone, D. E. Loy and M. E. Thompson, Chem. Mater., 1998,
10, 2235.27 C. Lambert and G. Noll, J. Am. Chem. Soc., 1999, 121, 8434.28 C. Lambert, G. Noll and J. Schelter, Nat. Mater., 2002, 1, 69.29 C. Lambert and G. Noll, J. Chem. Soc., Perkin Trans. 2, 2002,
2039.30 R. E. Littleford, M. A. J. Paterson, P. J. Low, D. R. Tackley,
L. Jayes, G. Dent, J. C. Cherryman, B. Brown and W. E. Smith,Phys. Chem. Chem. Phys., 2004, 6, 3257.
31 S. F. Nelson, Chem.–Eur. J., 2000, 6, 581.32 C. M. Duff and G. A. Heath, Inorg. Chem., 1991, 20, 2528.33 H. Sato, A. Kanega, R. Yamaguchi, K. Ogino and J. Kurjata,
Chem. Lett., 1999, 79.34 A. Petr, C. Kvarnstrom, L. Dunsch and A. Ivaska, Synth. Met.,
2000, 108, 245.35 C. Lambert and G. Noll, Synth. Met., 2003, 139, 57.
36 M. Leung, M.-Y. Chou, Y. O. Su, C. L. Chiang, H.-L. Chen,C. F. Yang, C.-C. Lin and H.-T. Chen, Org. Lett., 2003, 5, 839.
37 M.-Y. Chou, M.-K. Leung, Y. O. Su, C. L. Chiang, C.-C. Lin,J.-H. Liu, C.-K. Kuo and C.-Y. Mou, Chem. Mater., 2004, 16, 654.
38 P. J. Low, M. A. J. Paterson, A. E. Goeta, D. S. Yufit,J. A. K. Howard, J. C. Cherryman, D. R. Tackley and B. Brown,J. Mater. Chem., 2004, 14, 2516.
39 B. Badger and B. Brocklehurst, Trans. Faraday Soc., 1970, 66,2939.
40 A. V. Szeghalmi, M. Erdmann, V. Engel, M. Schmitt, S. Amthor,V. Kriegisch, G. Noll, R. Stahl, C. Lambert, D. Leusser, D. Stalke,M. Zabel and J. Popp, J. Am. Chem. Soc., 2004, 126, 7834.
41 B. S. Brunschwig, C. Cruetz and N. Sutin, Chem. Soc. Rev., 2002,31, 168.
42 Work unpublished at present cited in ref. 40.43 T. M. Barclay, A. W. Cordes, N. A. George, R. C. Haddon,
M. E. Itkis and R. T. Oakley, Chem. Commun., 1999, 2269.44 L. Jaquinod, N. Kyritsakas, J. Fischer and R. Weiss, New J.
Chem., 1995, 19, 453.45 F. Fowweather, Acta Crystallogr., 1952, 5, 820.46 J. D. Anderson, E. M. McDonald, P. A. Lee, M. L. Anderson,
E. L. Ritchie, H. K. Hall, T. Hopkins, E. A. Mash, J. Wang,A. Padias, S. Thayumanavan, S. Barlow, S. R. Marder,G. E. Jabbour, S. Shaheen, B. Kippelen, N. Peyghambarian,R. M. Wightman and N. R. Armstrong, J. Am. Chem. Soc., 1998,120, 9646.
47 S. F. Nelson, R. F. Ismagilov, K. E. Gentile and D. R. Powell,J. Am. Chem. Soc., 1999, 121, 7108.
48 A. Helms, D. Heiler and G. McLendon, J. Am. Chem. Soc., 1992,114, 6227.
49 R. C. Bansal, E. J. Eisenbraun and R. J. Ryba, Oppi Briefs, 1987,19, 258.
50 A. D. Becke, Phys. Rev. A, 1988, 38, 3098.51 J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B, 1996, 54,
16533.52 R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971,
54, 724.53 W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972,
56, 2257.54 P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209.55 M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163.56 P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213.57 S. Portmann and H. P. Luthi, Chimia, 2000, 54, 766.
This journal is � The Royal Society of Chemistry 2005 J. Mater. Chem., 2005, 15, 2304–2315 | 2315
Publ
ishe
d on
04
Apr
il 20
05. D
ownl
oade
d by
Wes
tern
Ken
tuck
y U
nive
rsity
on
29/1
0/20
14 1
8:20
:19.
View Article Online