Embed Size (px)
Citation preview

Time-Dependent Long-Range-CorrectedDouble-Hybrid Density Functionals with
Spin-Component and Spin-Opposite Scaling:A Comprehensive Analysis of Singlet-Singlet
and Singlet-Triplet Excitation Energies
Marcos Casanova-Páez∗ and Lars Goerigk∗
School of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia
E-mail: [email protected]; [email protected]: +61 3 834 46784
Abstract
Following the work on spin-component and spin-opposite scaled (SCS/SOS) global doublehybrids for singlet-singlet excitations by Schwabe and Goerigk [J. Chem. Theory Comput.2017, 13, 4307-4323] and our own works on new long-range corrected (LC) double hybridsfor singlet-singlet and singlet-triplet excitations [J. Chem. Theory Comput. 2019, 15, 4735-4744; J. Chem. Phys. 2020, 153, 064106], we present new LC double hybrids with SCS/SOSthat demonstrate further improvement over previously published results and methods. Weintroduce new unscaled and scaled versions of different global and LC double hybrids basedon Becke88 or PBE exchange combined with LYP, PBE or P86 correlation. For singlet-singletexcitations, we cross-validate them on six benchmark sets that cover small to medium-sizedchromophores with different excitation types (local valence, Rydberg, and charge transfer). Forsinglet-triplet excitations, we perform the cross-validation on three different benchmark setsfollowing the same analysis as in our previous work in 2020. In total, 203 unique excitations areanalyzed. Our results confirm and extend those of Schwabe and Goerigk regarding the superiorperformance of SCS and SOS variants compared to their unscaled parents by decreasing meanabsolute deviations, root-mean-square deviations or error spans by more than half and bringingabsolute mean deviations closer to zero. Our SCS/SOS variants show to be highly efficient androbust for the computation of vertical excitation energies, which even outperform specializeddouble hybrids that also contain an LC in their perturbative part. In particular, our newSCS/SOS-ωPBEPP86 and SCS/SOS-ωB88PP86 functional are four of the most accurate androbust methods tested in this work and we fully recommend them for future applications.However, if the relevant SCS and SOS algorithms are not available to the user, we suggestωB88PP86 as the best unscaled method in this work.
1 IntroductionDensity functional theory (DFT) is nowadaysthe most widely used methodology for the com-putation of ground state properties given itsgood compromise between accuracy and favor-able scaling, making it possible to treat rela-
tively large molecules, something that some-times is infeasible for high-accurate, electroncorrelation wave function theory (WFT) meth-ods. While its foundations, based on theHohenberg-Kohn theorems1 and the Kohn-Sham (KS) equations,2 are only valid for
1

ground state properties, Runge and Grossformulated the equivalent of the Hohenberg-Kohn theorems for the description of excitedstates in 1984,3 laying the foundations oftime-dependent (TD) DFT. For weak exter-nal electromagnetic fields, linear-response TD-DFT within the adiabatic approximation4 hasbecome as important to many different fieldsworking on electronically excited-state prob-lems as DFT is the preferred method for groundstate properties.A ground state DFT calculation precedes
the computation of electronic excitation en-ergies within TD-DFT; therefore, both for-malisms share the same shortcomings deter-mined by the choice of the underlying den-sity functional approximation (DFA) to the un-known “true” exchange-correlation functional.This work focuses on double-hybrid densityfunctional approximations (DHDFAs) that fol-low Grimme’s definition from 2006,5 whose out-standing performance have been demonstratedseveral times for ground state properties, asfor example shown in Refs. 6–17. Shortlyafter the demonstrated success of the firstDHDFA B2PLYP5 for ground state properties,the DHDFA idea was extended to the compu-tation of electronic vertical excitation energiesin 2007 when Grimme and Neese18 combinedHead-Gordon’s configuration interaction singleswith perturbative doubles [CIS(D)]19 approachwith the TD-DFT formalism and the closely re-lated Tamm-Dancoff Approximation20 (TDA).Herein, we exclusively deal with Grimme andNeese’s approach, when we use the acronymsTD-DHDFAs and TDA-DHDFAs. We acknowl-edge other definitions of the DHDFA con-cept,9,21–25 but also point out that not all ofthem are compatible with the TD(A)-DHDFAscheme proposed by Grimme and Neese, whichis why they will not be considered in the presentwork.Grimme and Neese’s first TD(A)-B2PLYP
study showed how the inclusion of the per-turbative CIS(D) correction reduced the errorspan by about 0.3 eV compared to the TD(A)-DFT hybrid portion of B2PLYP.18 That workalso showed promising results for singlet-triplettransitions in small (in)organic molecules and
for doublet-doublet transitions in small radi-cals, which were limited to the TDA variant ofB2PLYP. Subsequent applications on DHDFAsfor excited states involved almost exclusivelyTD-DHDFA treatments of singlet-singlet tran-sitions in organic systems. In 2009, the TD-DHDFA idea was applied for the first time toB2GP-PLYP26 on the famous test set contain-ing small organic molecules proposed by Thieland co-workers,27 where TD-B2GP-PLYP out-performed TD-B2PLYP and a range of lower-rung DFAs.28 In 2013, the “non-empirical”functionals PBE0-DH29 and PBE0-230 werealso assessed on the same test set in their TDAversions, but it was demonstrated that theywere by on average up to 0.08 eV less accuratethan TD-B2(GP-)PLYP.31 Indeed, the label“non-empirical” should not be misunderstoodas a synonym for quality or reliability, whichwas later demonstrated for DHDFAs applied toground state properties, where semi-empiricalDHDFAs by far outperformed non-empiricalones.10,32 Several studies have been conductedsince Grimme and Neese’s initial idea, andTD(A)-DHDFAs have been shown to be themost accurate DFAs for excited-state calcu-lations and absorption spectra,18,28,33–41 evenrivaling standard WFT methods.34,39–41 Whilewe refer the reader to a recently published de-tailed review on TD(A)-DHDFAs,42 we high-light only some of the advantages of those overthe conventional TD(A)-DFT approaches: abetter description of electronic circular dichro-ism (ECD) spectra,33 more accurate ver-tical singlet-singlet18,28,34,39–43 and singlet-triplet18,41 excitations in organic molecules—where TD(A)-DHDFAs even outperformed theapproximate coupled-cluster singles doublesWFT approach44,45 [(SCS-)CC2]34— a morebalanced description of the problematic firsttwo excited states in polycyclic aromatic hydro-carbons (PAHs),35,40 and no misleading resultsfrom artificial “ghost states”.28,33,34,36 Anotherlittle-known but important finding in the con-text of ECD spectroscopy is that TD-DHDFAswere the only methods that could successfullyreproduce an exciton-coupled spectrum of amerocyanine dimer aggregate,36 for which evenlong-range corrected (LC) (or range-separated)
2

hybrid functionals failed and which initially hadonly been successfully described with WFT ap-proaches.46A successful strategy to improve DHDFAs
for ground state thermochemistry has beenthe introduction of spin-component and spin-opposite scaling47–49 (SCS/SOS) to the treat-ment of the non-local electron-correlation com-ponent.6,11–14,21,50,51 SOS-based methods havethe advantage of having a reduced formal scal-ing behavior if a Laplace transform algorithmis employed.48,52 We also mention in pass-ing that the currently best-performing DHD-FAs for molecular thermochemistry are theSCS-LC-DHDFAs ωDSD3-PBEPP86-D417 andωDSD72-PBEPP86-D416 based on studies withthe GMTKN558 database for general main-group thermochemistry, kinetics and noncova-lent interactions; however, note that some ofthese methods follow different strategies com-pared to Grimme’s original DHDFA definitionfrom 2006.In 2017, significant improvements were
achieved by Schwabe and Goerigk when theysuccessfully applied the SCS and SOS tech-niques to the CIS(D) component of globalTD(A)-DHDFAs. A fitting procedure of theSCS/SOS parameters of six global DHDFAswas conducted against a high-level referencedata training set. Successful cross-validationstudies were carried out for vertical local-valence, vertical Rydberg, and 0-0 singlet-singlet excitations using the TD- and TDA-DHDFA schemes.39 Scaled and unscaled DHD-FAs were again superior to global and LC hy-brids.Their remarkable performance notwith-
standing, global TD(A)-DHDFAs sufferfrom the same flaws as any other globalDFA,40,41,43,53–55 namely the inability to ad-equately describe charge-transfer (CT) exci-tations. It was not until recently, in mid-2019, that we closed this gap by introduc-ing the first LC-DHDFAs optimized for ver-tical singlet-singlet excitation energies, namelyTD-ωB2PLYP and TD-ωB2GP-PLYP,40 bothbased on the original BLYP-based56,57 globalDHDFAs B2PLYP and B2GP-PLYP, respec-tively. Therein, we showed how the inclusion of
a single parameter, which controls the interplaybetween the long- and short-range regimes, canimprove long-range excitations such as Ryd-berg states and CT excitations without muchloss in accuracy for local-valence transitions.We particularly note that both functionals de-livered the best TD-DFT results to date for Laand Lb transitions in PAHs,40 and that theyhave already been successfully used in applica-tions to systems of biological or technologicalrelevance.58–64Prior to mid-2020, most of the BLYP-
based global DHDFAs studies had been car-ried out using the full TD-DHDFA scheme,whereas most of the studies using PBE-basedglobal DHDFAs had used the TDA-DHDFAscheme, instead.37,38,65–75 Moreover, all pre-vious benchmark studies and applications hadbeen limited to singlet-singlet excitations, ex-cept for the original work by Grimme andNeese from 2007,18 leaving out other spin mul-tiplicities of crucial relevance, such as tripletexcitations, most likely due to the lack ofa code that could handle those transitions.For this reason, we assessed for the first timesinglet-singlet and singlet-triplet excitationswith global- and LC-DHDFAs based on bothBLYP and PBE expressions for the underly-ing exchange-correlation functional and com-pared them with hybrid functionals.41 OurωB2(GP-)PLYP and Adamo and co-workers’PBE-QIDH65 displayed the best results forsinglet-triplet excitations, with the latter nev-ertheless still being unable to properly describeCT excitations, as we had to point out againin 2021.43 A very important finding was thatDHDFAs do not fix the triplet instability prob-lem76–79 when the full TD-DHDFA scheme isapplied, for which the TDA-DHDFA schememust be employed instead.Herein, we revisit the idea of applying SCS
and SOS techniques to TD(A)-DHDFAs simi-lar to the work done by Schwabe and Goerigk,and we extend them even further by apply-ing the latest TD(A)-LC-DHDFAs not only forsinglet-singlet excitations but also to singlet-triplet excitations. For this purpose, we analyze22 different scaled global- and LC-DHDFAs.Among the 8 unscaled DHDFAs, three are
3

global and five are LC ones. 6 of those unscaledmethods had been analyzed by us before, butwe also present two new LC-DHDFAs namedωPBEPP86 and ωB88PP86. The remaining 14methods are newly developed scaled versionsof the 8 unscaled ones. Lastly, we also com-pare those 22 methods with Mester and Kállay’sthree newest “Range-Separated Double-Hybridfunctional” (RS-DH) methods.80 The novelty ofthese RS-DHs is that they have been designedto be long-range corrected not only in the ex-change component (as usual LC-methods) butalso in the perturbative non-local CIS(D) cor-rection. Note that in their original work, theyonly analyzed singlet-singlet excitations usingthe TDA-DHDFA scheme and our comparisonwill be limited to those values.In this work, we briefly review the theoretical
background required for our new method de-velopments. We then discuss the definition ofour new unscaled LC-DHDFAs ωB88PP86 andωPBEPP86, followed by the development ofSCS and SOS extensions for each tested methodin this work (except the RS-DH methods). Thenew SCS and SOS TD(A)-DHDFAs are thencross-validated in a series of benchmark setsthat cover singlet-singlet and singlet-triplet ex-citations. We finally conclude with a discussionof the general robustness of the newly proposedmethods and some final remarks for the usercommunity.
2 Theoretical Background
2.1 Linear-Response TD-DFT
The most common method for computingexcited-state properties is linear-response81–83time-dependent TD-DFT within the adia-batic approximation.4,84 The latter allowsus to replace the time-dependent exchange-correlation kernel with the time-independentone from ground state DFT. The technicalitiesof TD-DFT are well-documented in the lit-erature,18,39,40,73 which is why we keep ourexplanations as brief as possible. For realorbitals, the vertical excitation energies arecomputed as the solution of the random-phase-
approximation (RPA)-type eigenvalue prob-lem:85(
A BB A
)(X(n)
Y(n)
)= ΔE(n)
(1 00 –1
)(X(n)
Y(n)
), (1)
ΔE(n) is the vertical excitation energy for thenth excited state, with X(n) and Y(n) being thecorresponding eigenvectors for single-particleexcitations and de-excitations, respectively. Aand B are matrices that contain the informationon these excitations and de-excitations, whosecomponents are given by:83
Aiaσ,jbτ = δστδijδab(εaσ – εiτ) + (iσaσ | jτbτ)
– δστax(iσjσ | aτbτ) + (1 – ax)(iσaσ | fXC | jτbτ)
(2)
and
Biaσ,jbτ =(iσaσ | bτjτ) – δστax(iσbσ | aτjτ)
+ (1 – ax)(iσaσ | fXC | bτjτ) .(3)
As usual, σ and τ are spin variables, the in-dices i, j, k . . . in this paper refer to occupiedorbitals, a, b, c . . . to virtual orbitals, and aX isthe Fock exchange scaling parameter. The lastterm in these equations has DFT origin, wherefXC is the exchange-correlation kernel and de-fined as the second derivative of the exchange-correlation functional EXC with respect to theelectronic density ρ:
fXC(r1, r2) =∂2EXC
∂ρ(r1)∂ρ(r2)(4)
with r1/2 being spatial variables.By neglecting the B matrix from Eq.(1),
we can obtain a simplified version known asthe Tamm-Dancoff approximation (TDA),20resulting in an eigenvalue problem that reads
AX(n) = ΔE(n)X(n). (5)
The label (n) will be dropped in the following.
2.2 Vertical Excitation Energieswith DHDFAs
We follow Grimme’s original definition of DHD-FAs18 alongside the LC scheme,86 similar as in
4

our construction of ωB2(GP-)PLYP:40
ELC–DHDFAXC = aXESR–HF
X (ω) + aX,DFAESR–DFTX (ω)
+ ELR–HFX (ω) + aC,DFAEDFT
C + aCEPT2C ,
(6)
where the first term corresponds to short-range Fock exchange, followed by the short-range DFT-exchange functional. The thirdterm is the full, long-range Fock exchange toensure the correct 1/r12 decay in the potential,where r12 is the interelectronic distance. Thelast two terms are the DFT-correlation func-tional and the second-order perturbative, non-local-correlation correction given by MP287for ground state or CIS(D) for excitation en-ergy calculations. Finally, aX,DFA and aC,DFAare scaling parameters for DFT exchange andcorrelation, whereas aX and aC scale short-range Fock exchange and non-local correla-tion, respectively; usually, but not always,those parameter values depend on one an-other.5,29,31,65,88 The interplay between short-and long-range regimes is controlled by therange-separation parameter ω.In the spirit of ground state DHDFA cal-
culations, Grimme and Neese’s vertical ex-citation energies at the double-hybrid level(ΔETD(A)–DHDFA) are obtained in a two-stepprocedure;18 first, we solve either the full TD-(Eq.1) or TDA-DFT (Eq.5) eigenvalue prob-lem using only the hybrid part of the DHDFA,i.e., all terms in Eq. 6 excluding the per-turbative portion. This gives rise to a ver-tical excitation energy of hybrid-DFT quality(ΔETD(A)–hybrid), which is then perturbativelycorrected with Head-Gordon’s CIS(D) correc-tion (ΔCIS(D)):
19
ΔETD(A)–DHDFA = ΔETD(A)–hybrid + aCΔCIS(D), (7)
where the parameter aC scales the CIS(D)-type contribution. More details on TD(A)-DHDFAs are given in our recent free-access ac-count in Ref. 42.
2.3 The CIS(D) Correction
The CIS(D) formalism for TD(A)-DHDFAs hasbeen extensively explained in the literature and
we refer the reader to Refs 18 and 39 for moredetailed information. In the spirit of MP2 be-ing a second-order perturbative correlation cor-rection to a Hartree-Fock (HF) ground stateenergy, the second-order perturbative CIS(D)correction19 introduces some electron correla-tion to configuration interaction singles (CIS)excitation energies. The CIS(D) energy correc-tion to the total CIS energy of an excited stateis:
ECIS(D)C = 〈ΦCIS|V|U2Φ0〉+ 〈ΦCIS|V|T2U1Φ0〉, (8)
where the first and second terms are knownas the “direct” and “indirect” terms, respec-tively. U1 and U2 are operators that generatethe singly- and doubly-excited wave functionsfrom the HF determinant (Φ0), T2 is the op-erator that generates the double excitation oftwo CIS-inactive electrons, and V is a pertur-bation potential. ΦCIS is the CIS wave functionaccording to:
ΦCIS = U1Φ0 =∑ia
tai Φai , (9)
where tai are the CIS single-excitation ampli-tudes and Φai is the singly excited determinant.The T2 operator acting on the HF determinant,Φ0, is given by
T2Φ0 =14
∑ijab
tabij Φabij
= –14
∑ijab
(ij||ab)εa + εb – εi – εj
Φabij ,
(10)
where we have used Mulliken’s notation forthe antisymmetrized two-electron integral(ij || ab) = (ij | ab) – (ia | jb) and ε stands againfor the orbital energy. Φabij is the doubly-exciteddeterminant from the HF ground state createdby the operator U2:
U2Φ0 = –14
∑ijab
〈Φabij |V|U1Φ0〉εa + εb – εi – εj – ΔECIS
Φabij (11)
The actual CIS(D) correction, ΔCIS(D), tothe CIS excitation energy, ΔECIS, is obtainedas the difference between the second-order per-turbative correlation energies for the excited
5

and the ground states:
ΔCIS(D) = ECIS(D)C – EMP2
C
= –14
∑ijab
(uabij )2
εa + εb – εi – εj – ΔECIS+∑ia
tai vai ,
(12)
with the corresponding definitions
uabij =∑c(ab || cj)tci –(ab || ci)tcj –
∑k
(ka || ij)tbk–(kb || ij)tak (13)
and
vai =12
∑jkbc
(jk || bc)[tbi tcajk + taj t
cbik + 2tbj t
acik ] (14)
A TD(A)-DHDFA excitation energy is calcu-lated by employing Eq.12 with KS orbitals in-stead of the HF ones and with ΔETD(A)–hybridexcitation energies instead ofΔECIS. For TDA-DHDFA, it is straightforward to employ the Xvector (Eq.5) for the single-excitation ampli-tudes tai , whereas for the full TD-DHDFA the Xvector is employed but the Y vector neglected(Eq.1).
2.4 The SCS- and SOS-CIS(D)Variants
Similar to Grimme’s SCS-MP2 method,47 theelectron pair contributions to the correlationenergy of an excitation energy can be separatedinto same (SS) and opposite spin (OS) contri-butions, which are then scaled differently . Thefirst implementation of the SCS-CIS(D) ideadates back to 2004 when Grimme and Izgoro-dina applied the same scaling parameters as inSCS-MP2 ground state theory to the “indirect”term in Eq. 8.89 Later, Rhee and Head-Gordonextended this idea and applied additional scaleparameters to the “direct” term.90 Therefore,we can break down the two terms in Eq.(8) intosame- and opposite spin components accordingto:
ESCS–CIS(D)c =
⟨ΦCIS | V |
(cOSU U
OS2 + cSSU U
SS2
)Φ0
⟩+⟨ΦCIS | V |
(cOST T
OS2 + cSST T
SS2
)U1Φ0
⟩,
(15)
where cSSU and cOSU are the same- and opposite-
spin scale parameters for the direct term and
cSST and cOST the related parameters for the in-
direct term. We refer the reader to e.g. Ref.39 for the explicit form of the SS and OS termsshown in Eq.(15). By setting the SS parame-ters to zero, one reduces the SCS- to the SOS-CIS(D) approach, which can benefit from a bet-ter formal scaling behavior in the same way asSOS-MP2.48Coming back to TD(A)-DHDFs, the ex-
pression for the SCS/SOS-CIS(D) correction(ΔSCS/SOS–CIS(D)), which is added to the ex-citation energy of the hybrid portion of theDHDFA, follows the same structure as ΔCIS(D)in Eq.(7), once the four SS/OS terms aretaken into consideration, giving the TD(A)-SCS/SOS-DHDF excitation energy:
ΔETD(A)–SCS/SOS–DHDFA = ΔETD(A)–hybrid +ΔSCS/SOS–CIS(D).(16)
Note that in the work by Rhee and Head-Gordon, the definition of the USS/OS
2 opera-tor considers an extra “damping” parameter (λ)for ΔECIS and two different versions of SCS-CIS(D) were developed by setting λ=0 and λ=1.Herein, we set this parameter to unity followingthe same protocol as in Refs. 34 and 39.
3 Computational DetailsAll molecular geometries were taken from theliterature, and all calculations were performedwith the same AO basis set used in the respec-tive works that developed the reference valuesto enable a direct comparison. In this con-text, we refrain from any basis set dependencestudy as it has already been conducted.39 Ithas been established that even for full CIS(D)(aC=1), local-valence and Rydberg excitationenergies only differed by about 0.03 eV whengoing from a triple to a large quadruple-ζ ba-sis set. Unsurprisingly, this difference was evenlower for DHDFAs with smaller aC. For in-stance, there was an average difference of just0.01 eV for TD-B2PLYP (aC=0.27) and TD-PBE0-DH (aC=0.125).39 We implemented theSCS/SOS-CIS(D) code for singlet and tripletexcitations into a local version of the ORCAcode,91–93 and it will be available in the upcom-ing release of ORCA5. The SCF convergence
6

criterion was set to 10–7Eh (10–8Eh for the up-dated Gordon set) along with ORCA’s numer-ical quadrature grid “4” (grid “5” and “finalgrid6” for the updated Gordon set). The resolution-of-the-identity technique was used with appro-priate auxiliary basis sets for the perturbativesteps.94,95Hybrid density functionals are not assessed
in this work because we aim to improve ourpreviously published DHDFA results by intro-ducing two new exchange-correlation combina-tions and also the SCS/SOS technique. We re-fer the reader to our previous works in Refs.40 and 41 for more information about the per-formance of global and LC hybrid functionals,which are shown to be less accurate and robust;in this context, we would just like to mentionthat the LC hybrid ωB97X96 sometimes out-performed some DHDFAs such as B2PLYP,5PBE0-DH,29 and RSX-QIDH,97 but not otherDHDFAs tested by us. Due to a lack of high-level reference data for triplet excitations inthe PAH and CT sets, we expand our analysisby also assessing the full TD-DHDFA schemeon these two sets without any further opti-mization procedure. In addition, we also as-sessed the new RS-DH methods by Mester andKállay,80 namely, RS-B88-LYP, RS-PBE-PBE,and RS-PBE-P86 which had been optimizedwith Schwabe and Goerigk’s “updated Gor-don set” 39,98 for singlet-singlet transitions us-ing the TDA-DHDFA scheme; note that forthese methods, we took the relevant values di-rectly from the original work.It is also important to consider the definition
of “chemical accuracy” for excitation energy cal-culations. It is a somewhat arbitrarily definedvalue and some studies have suggested a valueof 0.05 eV,67,99 whereas others prefer to regardan accuracy of 0.1 eV as a success.28,100 Ourstudy can be interpreted within the context ofeither of these two definitions.
Table 1: Optimized parameters for our new LC-DHDFAs following Eq. .
Functional aX aX,DFA aC,DFA aC ω[bohrs–1]ωB88PP86 0.65 0.35 0.58 0.42 0.20ωPBEPP86 0.70 0.30 0.68 0.48 0.18
4 Development
4.1 Introducing ωB88PP86 andωPBEPP86
Our new ωB88PP86 and ωPBEPP86 function-als are based on Becke88 and PBE exchange,respectively, however with modifications pro-posed by Hirao and co-workers86 in order tomake them applicable within the LC scheme.Both methods rely on Perdew’s correlationfunctional from 1986 (P86)101 with the Vosko,Wilk, and Nusair “VWN-V” local spin-densitycomponent.102 The choice of those exchangeand correlation functionals is based on the goodperformance of the global PBE-based DHDFAfrom our previous study,41 namely PBE-QIDH,where it was able to properly describe Ryd-berg transitions, the RS-PBE-P86 method thatperformed well for the updated Gordon testset,80 and the DSD-PBEP8612 ground stateDHDFA that performed well for ground statethermochemistry8 and some excited-state prop-erties.39Both methods follow the LC-DHDFA defini-
tion from Eq.6 and were optimized for singlet-singlet excitations, using the TDA-DHDFAscheme, by minimizing the root-mean-squaredeviation (RMSD) for the well-balanced up-dated Gordon set. We performed the optimiza-tion for singlet-singlet excitations using onlythe TDA-DHDFA scheme due to its more af-fordable computational cost and to make themapplicable to singlet-triplet excitations, basedon our previous experience with TDA- andTD-DHDFAs.41 We optimized these methodsonly for singlet-singlet transitions to make themcomparable to previously published methods.The original benchmark set by Gordon andco-workers98 comprised a total of 63 verticalsinglet-singlet excitations made up of 32 valenceand 31 Rydberg states in 14 different moleculesbut was based on different types of geometriesand experimental reference values. Schwabeand Goerigk re-optimized the geometries witha consistent level of theory in 2017,39 and alsoprovided new full and estimated ApproximateCoupled-Cluster Singles Doubles Triples data(CC3103/aug-cc-pVTZ104,105).
7

For the optimization procedure of ωB88PP86and ωPBEPP86, we set the initial scaling pa-rameters for the DFT, Fock exchange, andperturbative correlation components to thoseof B2GP-PLYP and PBE-QIDH, respectively.Those starting parameters were chosen, as bothfunctionals performed reasonably well for local-valence excitations. We then optimized ω. Fi-nally, a “full” optimization of all remaining scaleparameters was carried out for that ω value,giving rise to the final values presented in Ta-ble 1.Interestingly, our resulting scale parameters
for Fock exchange and perturbative correla-tion follow Toulouse et al.’s one-parameterquadratic relation (a2X=aC),88 even though thelatter had not been derived for excitation ener-gies. Results for the new methods for the fit setwill be discussed further below after the intro-duction of the new SCS/SOS methods.
4.2 New TDA-DHDFAs with SCSand SOS Components for Ex-citation Energies
4.2.1 Fitting Procedure
We combined the TD(A)-DHDFA idea withthe SCS/SOS-CIS(D) approach as pre-sented earlier in Eq. 16. For that pur-pose, we considered eight different unscaledDHDFA methods: B2PLYP,5 B2GP-PLYP,26ωB2PLYP,40 ωB2GP-PLYP,40 PBE-QIDH,65RSX-QIDH,97 ωB88PP86, and ωPBEPP86.According to our previous work in Ref. 41,PBE0-DH and RSX-0DH106 do not performwell, particularly the latter, which is why we re-frained from including them in this work. SinceSCS and SOS variants of B2PLYP and B2GP-PLYP have already been presented in Ref. 39,we will label our new variants as SCS/SOS-B2PLYP21 and SCS/SOS-B2GP-PLYP21, re-spectively. We re-optimized these two DHDFAsgiven that this time we also include singlet-triplet excitations in the fitting procedure (seebelow). For each one of these functionals, weanalyzed two different flavors for the scalingparameters:
• A free fit of all four parameters (SCS vari-ant).
• An SOS variant with cSST = cSSU = 0 anda free fit of the remaining two OS param-eters.
On rare occasions, the free fit of the four pa-rameters gave negative, unphysical values forone or both SS parameters. In those cases,we set those parameters to zero. In two cases,namely B2PLYP and ωB2PLYP, the SCS fit ledto the SOS variant. Hence, the subsequentlyused label “SCS/SOS-B2PLYP21” refers to oneand the same method; the same is true for“SCS/SOS-ωB2PLYP.The fitting procedure was conducted with the
full updated Gordon benchmark set. This in-cludes the 63 vertical singlet-singlet excitationsmentioned in Section 4.1 and additional datafor singlet-triplet excitations developed as partof Ref. 39 and published for the first time byus in mid-2020.41 Those 38 vertical singlet-triplet excitations are made up of 27 valenceand 11 Rydberg excitations from 12 differentmolecules, all with the same full or estimatedhigh-level CC3/aug-cc-pVTZ reference data.Our parametrization procedure consisted of a
minimization of the RMSD for each method onthe entire set. The final optimized scaling pa-rameters are shown in Table 2. Therein, we alsoprovide a breakdown into which DHDFAs areglobal and which are long-range corrected. Weanalyze the singlet-singlet and singlet-tripletexcitation energies for the fit set separately inthe following two sections.
4.2.2 Analysis of Vertical Singlet-Singlet Excitations
Our results for the singlet excitations in the fitsets are broken down into the whole set andits local-valence and Rydberg components. Allrelevant statistical results are shown in Table3 for each TDA-DHDFA method. First of all,we would like to point out that many methodsshare the same RMSDs. However, to be able torank them, we decided to sort the methods ac-cording to the following criteria: RMSD, MAD,
8

Table 2: Originala parameters of unscaled DHDFAsand parameters of their new SCS and SOS variantsoptimized for the TDA-DHDFA scheme.
Method cSST cOST cSSU cOS
U
B2PLYPbOriginal 0.270 0.270 0.270 0.270SCSc 0.000 0.334 0.000 0.283SOSc 0.000 0.334 0.000 0.283
B2GP-PLYPbOriginal 0.360 0.360 0.360 0.360SCS 0.018 0.475 0.000 0.468SOS 0.000 0.489 0.000 0.484
PBE-QIDHbOriginal 0.330 0.330 0.330 0.330SCS 0.070 0.515 0.096 0.524SOS 0.000 0.547 0.000 0.573
ωB2PLYPdOriginal 0.270 0.270 0.270 0.270SCSc 0.000 0.433 0.000 0.460SOSc 0.000 0.433 0.000 0.460
ωB2GP-PLYPdOriginal 0.360 0.360 0.360 0.360SCS 0.000 0.571 0.135 0.558SOS 0.000 0.570 0.000 0.610
RSX-QIDHdOriginal 0.330 0.330 0.330 0.330SCS 0.174 0.541 0.586 0.424SOS 0.000 0.632 0.000 0.714
ωB88PP86dOriginal 0.423 0.423 0.423 0.423SCS 0.000 0.557 0.092 0.545SOS 0.000 0.559 0.000 0.599
ωPBEPP86dOriginal 0.480 0.480 0.480 0.480SCS 0.000 0.610 0.297 0.540SOS 0.000 0.613 0.000 0.669
a If all four scaling parameters are identical, theoriginal, unscaled DHDF is obtained, as all parameters
equal aC in Eq. 7. b Global DHDFA. c The SCSfitting procedure led directly to the SOS parameters
(see main text). d LC-DHDFA.
MD, and error span(Δerr). For example, PBE-QIDH’s SCS and SOS variants have identicalRMSDs, MADs, and MDs and differ only inthe error span. In this case, the SCS variantoccupies a higher place in the ranking given itslower error span (0.78 vs. 0.81 eV). Most of thetime these differences are negligible, but we de-cided to apply the same criteria throughout thepresent work to be consistent.We note that the introduction of the
SCS/SOS scheme is overall beneficial for eachtested method when considering the statisticsfor the entire set of singlet-singlet excitations.Most methods display large reductions of theirRMSD for both local-valence and Rydberg ex-citations upon introduction. For instance, theRMSD for RSX-QIDH for the complete set isreduced from 0.40 eV to 0.21/0.24 eV for itsSCS/SOS variants, respectively. This coincideswith a reduction of 0.25 eV in the RMSD forlocal-valence excitations for both the SCS andSOS methods, and a reduction of 0.10/0.04eV for SCS/SOS for the Rydberg excitations.Some other methods, on the other hand, havean overall improved RMSD despite not improv-ing for both local-valence and Rydberg excita-tions. For example, for B2PLYP the RMSDfor local-valence excitations increases slightly
from 0.24 to 0.27 eV for its SCS/SOS variant.This is accompanied by a considerable reduc-tion in the RMSD for the Rydberg excitations(from 0.53 to 0.30 eV), which leads to a reduc-tion of the overall RMSD from 0.41 to 0.29 eV.We see something similar for PBE-QIDH werean improvement in the local-valence categorycompensated for slightly increased RMSDs forRydberg states when SCS/SOS is applied.When ranking all assessed methods ac-
cording to their results for the entire set ofsinglet-singlet excitations, we find that PBE-QIDH’s SCS and SOS variants yield the lowestRMSDs with 0.16 eV in both cases, closelyfollowed by SCS-ωPBEPP86, SOS-ωB88PP86,and their related cousins SCS-ωB88PP86 andSOS-ωPBEPP86. These six methods are theonly ones with RMSDs below 0.2 eV. We alsoidentify four methods that share an RMSDof exactly 0.20 eV, namely, RS-PBE-P86,SCS-ωB2GP-PLYP, PBE-QIDH, and RS-PBE-PBE. It is noteworthy that the new RS-DHs,along with PBE-QIDH, still perform well de-spite being unscaled methods. In the same con-text, another well-performing unscaled DHDFAis ωB88PP86, which has the same RMSD of0.21 eV as SOS-ωB2GP-PLYP and SCS-RSX-QIDH. Five DHDFAs immediately follow, allwith the same RMSD of 0.22 eV, i.e., RS-B88-LYP, SCS/SOS-ωB2PLYP, ωPBEPP86, SOS-and SCS-B2GP-PLYP21. The remaining fiveDHDFAs with values below 0.3 eV are SOS-RSX-QIDH, the unscaled ωB2GP-PLYP, therelated ωB2PLYP, B2GP-PLYP , and finallySCS/SOS-B2PLYP21. Lastly, RSX-QIDH andB2PLYP are the two worst performers for thisset with RMSDs of 0.40 and 0.41 eV.The MADs follow more or less the same
trends as the RMSDs. It is remarkable thatthe SCS and SOS versions of PBE-QIDH,ωB88PP86, and ωPBEPP86 are very close tothe second definition of “chemical accuracy” forexcited states, i.e., with average absolute de-viations of up to 0.1 eV. On the other hand,the MDs show an interesting behavior oncethe SCS/SOS schemes are applied. Most ofthe tested unscaled methods, regardless of sys-tematic over or underestimation of the exci-tation energies, show a systematic improve-
9

Table 3: Statistical values (in eV) for all, local-valence, and Rydberg singlet-singlet excitations in the updatedGordon set using the TDA-DHDFA formalism.a
Overall Local-valence RydbergMethod MD MAD RMSD Δerr MD MAD RMSD Δerr MD MAD RMSD ΔerrSCS-PBE-QIDH 0.07 0.12 0.16 0.78 0.13 0.14 0.18 0.48 0.01 0.11 0.13 0.54SOS-PBE-QIDH 0.07 0.12 0.16 0.81 0.12 0.13 0.17 0.53 0.02 0.12 0.14 0.54SCS-ωPBEPP86 0.07 0.14 0.17 0.82 0.09 0.12 0.16 0.51 0.04 0.15 0.17 0.69SOS-ωB88PP86 0.05 0.14 0.18 0.94 0.11 0.13 0.17 0.54 0.00 0.16 0.19 0.79SCS-ωB88PP86 0.08 0.15 0.19 0.95 0.15 0.16 0.19 0.53 0.01 0.15 0.18 0.77SOS-ωPBEPP86 0.10 0.16 0.19 0.81 0.10 0.13 0.19 0.57 0.10 0.18 0.20 0.73RS-PBE-P86 0.04 0.14 0.20 0.88 0.13 0.18 0.24 0.86 –0.05 0.10 0.14 0.72SCS-ωB2GP-PLYP 0.04 0.14 0.20 1.10 0.13 0.15 0.20 0.67 –0.07 0.13 0.19 0.77PBE-QIDH 0.12 0.15 0.20 0.75 0.23 0.23 0.27 0.49 0.01 0.08 0.10 0.47RS-PBE-PBE 0.03 0.15 0.20 0.91 0.13 0.18 0.24 0.91 –0.07 0.11 0.15 0.73ωB88PP86 0.06 0.16 0.21 1.00 0.15 0.15 0.21 0.50 –0.04 0.16 0.21 0.85SOS-ωB2GP-PLYP 0.06 0.16 0.21 1.11 0.15 0.17 0.23 0.70 –0.04 0.14 0.19 0.79SCS-RSX-QIDH 0.12 0.17 0.21 0.83 0.14 0.15 0.21 0.57 0.10 0.19 0.21 0.73RS-B88-LYP –0.02 0.18 0.22 1.00 0.11 0.18 0.24 0.97 –0.16 0.18 0.20 0.72SCS/SOS-ωB2PLYP 0.07 0.17 0.22 1.18 0.17 0.19 0.24 0.66 –0.03 0.16 0.21 0.93ωPBEPP86 0.13 0.18 0.22 0.94 0.17 0.17 0.23 0.58 0.08 0.19 0.22 0.80SOS-B2GP-PLYP21 –0.03 0.19 0.22 1.01 0.15 0.16 0.19 0.51 –0.21 0.21 0.24 0.51SCS-B2GP-PLYP21 0.01 0.19 0.22 1.00 0.19 0.20 0.23 0.50 –0.19 0.19 0.22 0.52SOS-RSX-QIDH 0.12 0.20 0.24 0.83 0.08 0.16 0.21 0.64 0.16 0.24 0.27 0.80ωB2GP-PLYP 0.12 0.19 0.25 1.08 0.26 0.26 0.32 0.65 –0.03 0.11 0.15 0.73ωB2PLYP 0.12 0.20 0.26 1.14 0.26 0.26 0.32 0.62 –0.03 0.14 0.18 0.89B2GP-PLYP –0.13 0.24 0.28 1.10 0.06 0.14 0.19 0.70 –0.33 0.33 0.36 0.57SCS/SOS-B2PLYP21 –0.02 0.25 0.29 1.22 0.23 0.23 0.27 0.64 –0.27 0.27 0.30 0.54RSX-QIDH 0.35 0.35 0.40 0.89 0.41 0.41 0.46 0.79 0.28 0.28 0.31 0.60B2PLYP –0.29 0.34 0.41 1.33 –0.09 0.18 0.24 0.96 –0.50 0.50 0.53 0.70
a The aug-cc-pVTZ AO basis set was used in all cases. The DHDFAs are sorted by their overall RMSDs. Thevalues for the RS-DH methods were taken from Ref. 80. The statistical estimators are Mean Deviation(MD),
Mean-Absolute Deviation (MAD), Root-Mean-Square Deviation (RMSD), and Error Span (Δerr).
ment by having MDs that are closer to zero.For instance, RSX-QIDH overestimates the en-ergies by on average 0.35 eV, which is im-proved by the SCS and SOS variants withMD = 0.12 eV in both cases. B2PLYPtends to underestimate the excitation ener-gies (MD=–0.29 eV), whereas the SCS/SOS-B2PLYP21 variant has one of the best MDswith a value of only –0.02 eV. Some unscaledDHDFAs with very good MDs that we wouldlike to highlight are the three new RS-DH meth-ods (MDs range from 0.04 to –0.02 eV) andωB88PP86 (MD=0.06 eV).When focussing just on local-valence tran-
sitions, we observe similar trends as before.The best performer is SCS-ωPBEPP86 withRMSD=0.16 eV, which is closely followed bySOS-ωB88PP86 and PBE-QIDH. Other goodperformers below 0.2 eV are SOS-ωPBEPP86,B2GP-PLYP and its SOS version, and SCS-ωB88PP86. The majority of the remainingmethods display results between 0.2 eV and0.3 eV, except for ωB2PLYP, ωB2GP-PLYP(both with RMSDs of 0.32 eV), and RSX-QIDH(0.46 eV).We end the discussion of singlet excitations
by analyzing the Rydberg category. The un-scaled PBE-QIDH functional is the best per-former with an excellent value of 0.10 eV.
What is surprising is the fact that its SCSand SOS variants display slightly larger RMSDsof 0.13 eV and 0.14 eV, respectively. How-ever, as we mentioned before, this mishap isgreatly compensated for by its reduction ofabout 0.10 eV in the local-valence category. TheRS-PBE-P86 method has a value of 0.14 eVand occupies the third place in the ranking,followed by RS-PBE-PBE and ωB2GP-PLYPwith RMSDs of 0.15 eV. The next DHDFAsin our ranking are SCS-ωPBEPP86, ωB2PLYP,SCS-ωB88PP86, SCS- and SOS-ωB2GP-PLYP,and SOS-ωB88PP86. The remaining methodshave RMSDs between 0.20 eV (RS-B88-LYPand SOS-ωPBEPP86) and 0.53 eV (B2PLYP).
4.2.3 Analysis of Vertical Singlet-Triplet Excitations
Following the same idea as presented by us inRef. 41, we perform two different analyses forsinglet-triplet excitations. The first type coversall transitions between the ground state singletto the various triplet excited states, which wedenote as S0-TN, with N ≥ 1. The second typefocuses on the energy splitting between the firstsinglet and triplet excited states, denoted asS1-T1. The latter is particularly intriguing forpotential future applications of well-performingmethods in areas such as singlet-fission materi-
10

Table 4: Statistical values (in eV) for the S0-TN transitions in the updated Gordon set using the TDA-DHDFAformalism.a
Overall Local-valence RydbergMethod MD MAD RMSD Δerr MD MAD RMSD Δerr MD MAD RMSD ΔerrSCS-PBE-QIDH –0.04 0.06 0.08 0.31 –0.04 0.06 0.08 0.30 –0.04 0.05 0.06 0.15SOS-PBE-QIDH –0.04 0.07 0.08 0.33 –0.04 0.06 0.08 0.31 –0.04 0.08 0.10 0.30PBE-QIDH –0.04 0.07 0.09 0.37 0.03 0.07 0.09 0.41 –0.03 0.08 0.10 0.33SCS-ωB88PP86 –0.07 0.09 0.12 0.40 –0.03 0.07 0.10 0.35 –0.03 0.11 0.12 0.36SOS-ωPBEPP86 –0.05 0.10 0.12 0.47 –0.06 0.08 0.10 0.37 0.02 0.11 0.13 0.42ωPBEPP86 0.00 0.10 0.12 0.37 –0.04 0.08 0.10 0.37 –0.02 0.12 0.13 0.35SOS-ωB88PP86 –0.09 0.10 0.13 0.43 –0.03 0.08 0.10 0.34 –0.04 0.14 0.14 0.37SOS-ωB2GP-PLYP –0.05 0.10 0.13 0.60 0.01 0.08 0.10 0.33 –0.09 0.12 0.15 0.40ωB2GP-PLYP –0.02 0.11 0.13 0.58 –0.02 0.09 0.10 0.48 –0.13 0.13 0.15 0.28SCS-ωB2GP-PLYP –0.08 0.11 0.14 0.56 –0.08 0.09 0.11 0.35 0.04 0.14 0.16 0.49ωB88PP86 –0.07 0.11 0.14 0.45 –0.08 0.09 0.11 0.37 –0.10 0.14 0.17 0.43SCS-ωPBEPP86 –0.09 0.12 0.14 0.44 –0.05 0.09 0.11 0.45 –0.13 0.14 0.18 0.41SCS/SOS-ωB2PLYP –0.08 0.11 0.15 0.62 –0.04 0.09 0.11 0.48 –0.16 0.16 0.19 0.38SOS-B2GP-PLYP21 –0.04 0.11 0.15 0.64 0.06 0.09 0.11 0.43 0.18 0.18 0.20 0.22ωB2PLYP –0.06 0.12 0.15 0.61 –0.02 0.10 0.12 0.51 –0.16 0.16 0.21 0.40SCS-B2GP-PLYP21 –0.01 0.12 0.15 0.65 0.02 0.10 0.12 0.51 –0.19 0.19 0.21 0.28SCS-RSX-QIDH –0.10 0.13 0.15 0.49 0.09 0.11 0.13 0.42 –0.16 0.17 0.22 0.49SOS-RSX-QIDH –0.07 0.13 0.15 0.57 –0.12 0.12 0.14 0.30 –0.17 0.18 0.22 0.38RSX-QIDH 0.06 0.15 0.17 0.57 –0.12 0.13 0.15 0.36 –0.22 0.22 0.23 0.28SCS/SOS-B2PLYP21 –0.02 0.16 0.19 0.71 –0.14 0.14 0.15 0.29 –0.28 0.28 0.30 0.32B2GP-PLYP –0.13 0.16 0.22 0.62 0.01 0.13 0.16 0.57 –0.37 0.37 0.38 0.27B2PLYP –0.24 0.26 0.34 0.87 –0.11 0.14 0.17 0.45 –0.56 0.56 0.57 0.39
a The aug-cc-pVTZ AO basis set was used in all cases. The DHDFAs are sorted by their overall RMSDs. Thestatistical estimators are Mean Deviation(MD), Mean-Absolute Deviation (MAD), Root-Mean-Square Deviation
(RMSD), and Error Span (Δerr).
als.107–111 It also gives us first insights into therobustness of a method, as there is no guaran-tee that a method that performs well for singletexcitations is also suitable for triplet ones.
All Singlet-Triplet Excitations in this set(S0-TN)
In Table 4, we see that nearly every testedTDA-DHDFA seems to describe singlet-tripletexcitations with a very good or acceptableRMSD below 0.2 eV, except for B2PLYP(0.34 eV) and B2GP-PLYP (0.22 eV). PBE-QIDH and its SCS/SOS variants perform out-standingly well, with RMSDs below the sec-ond definition of “chemical accuracy” for ex-citation energies, with values of 0.08-0.09 eV.Just a few hundredths of an eV above, welocate SCS-ωB88PP86, SOS-ωPBEPP86, andthe unscaled ωPBEPP86 (RMSD = 0.12 eV),which are closely followed by SOS-ωB88PP86,SOS-ωB2GP-PLYP, and the unscaled ωB2GP-PLYP (RMSD = 0.13 eV). We then findthat nine DHDFAs, both scaled and unscaledones, present good values between 0.14-0.15 eV,and based on our other criteria outlined ear-lier we rank them from SCS-ωB2GP-PLYPto SOS-RSX-QIDH. Interestingly, RSX-QIDHand SCS/SOS-B2PLYP21 deliver acceptableresults below 0.20 eV. We also notice that,in general, most unscaled TDA-DHDFAs are
slightly outperformed by their correspondingSCS/SOS variants.The MDs display small underestimations with
absolute MDs below 0.1 eV, except for B2PLYP(–0.24 eV) and B2GP-PLYP (–0.13 eV). Wepoint out that the only DHDFA that displaysan overestimation is RSX-QIDH (0.06 eV). Ingeneral, inclusion of SCS/SOS seems to keepthe MDs nearly unchanged, such as in PBE-QIDH, or it produces a slight red shift, such asin ωPBEPP86 (see Table 4.2.3)
S1-T1 Analysis
In Table 5, we see that the statistics for the S1-T1 splitting show similar trends as for the S0-TN analysis with most RMSDs being below 0.2eV. In the updated Gordon set, most low-lyingexcitation energies belong to the local-valencecategory, and only for the water molecule wehave to analyze a Rydberg excitation.B2GP-PLYP’s SOS and SCS variants are at
the top of the ranking with RMSDs of 0.08 and0.09 eV, respectively. PBE-QIDH, its SCS vari-ant, and ωB2GP-PLYP’s SCS and SOS variantsalso display the same outstanding result withRMSD = 0.1 eV. Nine methods follow closelyranging from the unscaled B2GP-PLYP (0.12eV) to the scaled SCS-ωPBEPP86 (0.15 eV).Other acceptably well-performing DHDFAs areB2PLYP, ωB2GP-PLYP, SOS-RSX-QIDH, and
11

PBE-QIDH, and ωB2PLYP. Finally, SCS-RSX-QIDH and its unscaled variant conclude the listwith values of 0.22 and 0.33 eV, respectively.One major difference to the previous S0-TN
analysis is the fact that the MDs are mostlypositive, with only one exception (B2PLYP).The reason is simply that most DHDFAs dis-play a systematic overestimation for singlet-singlet excitations, whereas they systematicallyunderestimate singlet-triplet excitations. As aresult, it is expected that the S1-T1 energysplitting also displays an overestimation.41Our findings for the updated Gordon set are
very promising. The SCS/SOS variants showconsiderable improvements over their unscaledparents for singlet-singlet transitions, while forsinglet-triplet excitations there is a more mod-est improvement. In sections 5 and 6 wewill further discuss how our new SCS andSOS variants perform in an extensive cross-validation study that covers both singlet-singletand singlet-triplet excitations, including CT ex-citations, which have not been part of our pa-rameter training.
Table 5: Statistical values (in eV) for the S1-T1 en-ergy splitting on the updated Gordon set using theTDA-DHDFA formalism.a
Method MD MAD RMSD ΔerrSOS-B2GP-PLYP21 0.02 0.07 0.08 0.26SCS-B2GP-PLYP21 0.03 0.08 0.09 0.29SOS-PBE-QIDH 0.07 0.09 0.10 0.26SOS-ωB2GP-PLYP 0.07 0.09 0.10 0.26SCS-ωB2GP-PLYP 0.08 0.09 0.10 0.24SCS-PBE-QIDH 0.08 0.09 0.10 0.23B2GP-PLYP 0.01 0.10 0.12 0.40ωPBEPP86 0.09 0.11 0.13 0.28SCS/SOS-ωB2PLYP 0.10 0.12 0.13 0.27SOS-ωB88PP86 0.10 0.12 0.13 0.35ωB88PP86 0.11 0.12 0.14 0.33SCS-ωB88PP86 0.11 0.13 0.15 0.33SCS/SOS-B2PLYP21 0.04 0.12 0.15 0.47SOS-ωPBEPP86 0.09 0.11 0.15 0.44SCS-ωPBEPP86 0.11 0.13 0.15 0.38B2PLYP –0.03 0.13 0.16 0.50ωB2GP-PLYP 0.14 0.15 0.18 0.41SOS-RSX-QIDH 0.11 0.13 0.19 0.54PBE-QIDH 0.16 0.16 0.19 0.41ωB2PLYP 0.16 0.17 0.20 0.44SCS-RSX-QIDH 0.18 0.18 0.22 0.42RSX-QIDH 0.28 0.28 0.33 0.56
a The aug-cc-pVTZ AO basis set was used in all cases.The DHDFAs are sorted by their overall RMSDs. The
statistical estimators are Mean Deviation(MD),Mean-Absolute Deviation (MAD), Root-Mean-Square
Deviation (RMSD), and Error Span (Δerr).
5 Cross-Validation for Ver-tical Singlet-Singlet Exci-tations
5.1 The ωB2(GP-)PLYP TrainingSet
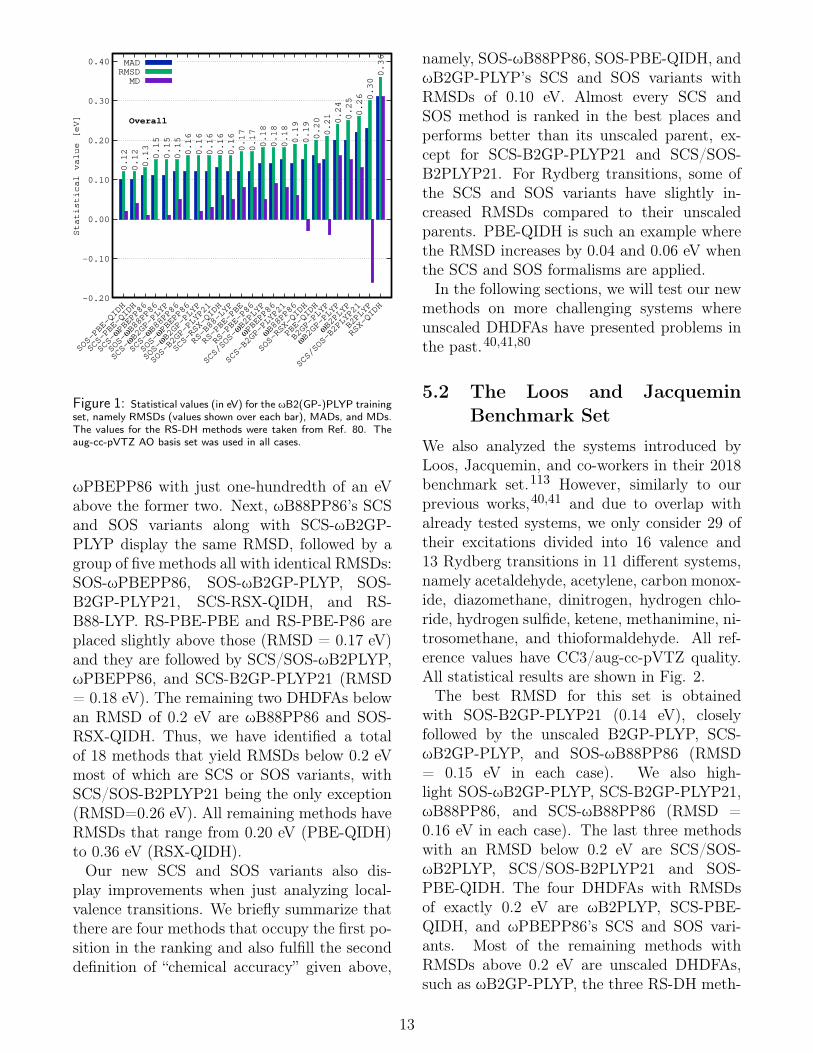
The ωB2(GP-)PLYP “training set” 40 is aslight modification of the original set used bySchwabe and Goerigk for their parametriza-tion of TD(A)-SCS/SOS-DHDFAs in 2017.39 Itcontains 34 vertical singlet-singlet excitations,made of up 28 valence and 6 Rydberg transi-tions. The reference values are based on Cou-pled Cluster Singles Doubles with PerturbativeTriples Excitation Correction112 [CCSDR(3)]with the large aug-cc-pVTZ AO basis set. Notethat herein we do not use this set for any fittingprocedure but called it “training set” to indi-cate that it was used during the developmentof our ωB2(GP-)PLYP functionals.40 Herein,we present the RMSD and MAD values for ourherein assessed TDA-DHDFAs in Fig. 1, wherethe results are, once again, broken down intolocal-valence and Rydberg transitions. Themethods are sorted according to their RMSDvalues for the overall set, which are also shownon top of the bars. All raw as well as otherstatistical data are provided in the Support-ing Information. We note in passing that the“top 10” methods are SCS/SOS variants, withthe RS-DH methods coming right after them.Indeed, the RS-DH methods have previouslybeen among the most accurate DHDFAs inRef. 80 and it was argued that it was due torange-separation in the perturbative CIS(D)treatment. We show here nevertheless thateven better results can be achieved through theSCS/SOS technique without the need of in-troducing range-separation in the perturbativetreatment. This is an encouraging outcome anda strong indicator that the SCS/SOS methodscan be transferred to systems other than theones used in the fit. We now proceed with theanalysis of the RMSDs for this set.PBE-QIDH’s SOS and SCS variants are at
the top of the ranking with equal RMSDsof 0.12 eV, but are closely followed by SCS-
12
-0.20
-0.10
0.00
0.10
0.20
0.30
0.40
SOS-PBE-QIDH
SCS-PBE-QIDH
SCS-ωPBEPP86
SOS-ωB88PP86
SCS-ωB2GP-PLYP
SCS-ωB88PP86
SOS-ωPBEPP86
SOS-ωB2GP-PLYP
SOS-B2GP-PLYP21
SCS-RSX-QIDH
RS-B88-LYP
RS-PBE-PBE
RS-PBE-P86
SCS/SOS-ωB2PLYP
ωPBEPP86
SCS-B2GP-PLYP21
ωB88PP86
SOS-RSX-QIDH
PBE-QIDH
B2GP-PLYP
ωB2GP-PLYP
ωB2PLYP
SCS/SOS-B2PLYP21
B2PLYP
RSX-QIDH
Overall
Statistical value [eV]
MAD
RMSD
MD
0.12
0.12
0.13
0.15
0.15
0.15
0.16
0.16
0.16
0.16
0.16
0.17
0.17
0.18
0.18
0.18
0.19
0.19
0.20
0.21
0.24
0.25
0.26 0.30
0.36
Figure 1: Statistical values (in eV) for the ωB2(GP-)PLYP trainingset, namely RMSDs (values shown over each bar), MADs, and MDs.The values for the RS-DH methods were taken from Ref. 80. Theaug-cc-pVTZ AO basis set was used in all cases.
ωPBEPP86 with just one-hundredth of an eVabove the former two. Next, ωB88PP86’s SCSand SOS variants along with SCS-ωB2GP-PLYP display the same RMSD, followed by agroup of five methods all with identical RMSDs:SOS-ωPBEPP86, SOS-ωB2GP-PLYP, SOS-B2GP-PLYP21, SCS-RSX-QIDH, and RS-B88-LYP. RS-PBE-PBE and RS-PBE-P86 areplaced slightly above those (RMSD = 0.17 eV)and they are followed by SCS/SOS-ωB2PLYP,ωPBEPP86, and SCS-B2GP-PLYP21 (RMSD= 0.18 eV). The remaining two DHDFAs belowan RMSD of 0.2 eV are ωB88PP86 and SOS-RSX-QIDH. Thus, we have identified a totalof 18 methods that yield RMSDs below 0.2 eVmost of which are SCS or SOS variants, withSCS/SOS-B2PLYP21 being the only exception(RMSD=0.26 eV). All remaining methods haveRMSDs that range from 0.20 eV (PBE-QIDH)to 0.36 eV (RSX-QIDH).Our new SCS and SOS variants also dis-
play improvements when just analyzing local-valence transitions. We briefly summarize thatthere are four methods that occupy the first po-sition in the ranking and also fulfill the seconddefinition of “chemical accuracy” given above,
namely, SOS-ωB88PP86, SOS-PBE-QIDH, andωB2GP-PLYP’s SCS and SOS variants withRMSDs of 0.10 eV. Almost every SCS andSOS method is ranked in the best places andperforms better than its unscaled parent, ex-cept for SCS-B2GP-PLYP21 and SCS/SOS-B2PLYP21. For Rydberg transitions, some ofthe SCS and SOS variants have slightly in-creased RMSDs compared to their unscaledparents. PBE-QIDH is such an example wherethe RMSD increases by 0.04 and 0.06 eV whenthe SCS and SOS formalisms are applied.In the following sections, we will test our new
methods on more challenging systems whereunscaled DHDFAs have presented problems inthe past.40,41,80
5.2 The Loos and JacqueminBenchmark Set
We also analyzed the systems introduced byLoos, Jacquemin, and co-workers in their 2018benchmark set.113 However, similarly to ourprevious works,40,41 and due to overlap withalready tested systems, we only consider 29 oftheir excitations divided into 16 valence and13 Rydberg transitions in 11 different systems,namely acetaldehyde, acetylene, carbon monox-ide, diazomethane, dinitrogen, hydrogen chlo-ride, hydrogen sulfide, ketene, methanimine, ni-trosomethane, and thioformaldehyde. All ref-erence values have CC3/aug-cc-pVTZ quality.All statistical results are shown in Fig. 2.The best RMSD for this set is obtained
with SOS-B2GP-PLYP21 (0.14 eV), closelyfollowed by the unscaled B2GP-PLYP, SCS-ωB2GP-PLYP, and SOS-ωB88PP86 (RMSD= 0.15 eV in each case). We also high-light SOS-ωB2GP-PLYP, SCS-B2GP-PLYP21,ωB88PP86, and SCS-ωB88PP86 (RMSD =0.16 eV in each case). The last three methodswith an RMSD below 0.2 eV are SCS/SOS-ωB2PLYP, SCS/SOS-B2PLYP21 and SOS-PBE-QIDH. The four DHDFAs with RMSDsof exactly 0.2 eV are ωB2PLYP, SCS-PBE-QIDH, and ωPBEPP86’s SCS and SOS vari-ants. Most of the remaining methods withRMSDs above 0.2 eV are unscaled DHDFAs,such as ωB2GP-PLYP, the three RS-DH meth-
13

-0.10
0.00
0.10
0.20
0.30
0.40
SOS-B2GP-PLYP21
B2GP-PLYP
SCS-ωB2GP-PLYP
SOS-ωB88PP86
SOS-ωB2GP-PLYP
SCS-B2GP-PLYP21
ωB88PP86
SCS-ωB88PP86
SCS/SOS-ωB2PLYP
SCS/SOS-B2PLYP21
SOS-PBE-QIDH
ωB2PLYP
SCS-PBE-QIDH
SOS-ωPBEPP86
SCS-ωPBEPP86
ωB2GP-PLYP
ωPBEPP86
RS-B88-LYP
B2PLYP
SOS-RSX-QIDH
PBE-QIDH
RS-PBE-PBE
SCS-RSX-QIDH
RS-PBE-P86
RSX-QIDH
Overall
Statistical value [eV]
MAD
RMSD
MD
0.14
0.15
0.15
0.15
0.16
0.16
0.16
0.16
0.17
0.19
0.19
0.20
0.20
0.20
0.20
0.21
0.22
0.23
0.24
0.24
0.24
0.26
0.26
0.27
0.38
Figure 2: Statistical values (in eV) for the Loos and Jacqueminset, namely RMSDs (values shown over each bar), MADs, and MDs.The values for the RS-DH methods were taken from Ref. 80. Theaug-cc-pVTZ AO basis set was used in all cases.
ods, or PBE-QIDH. That being said, we findthat RSX-QIDH’s SCS and SOS variants alsobelong to this range. The unscaled RSX-QIDHyields the highest RMSD of 0.38 eV, confirmingother studies that this LC-DHDFA is often lessaccurate than its global PBE-QIDH counter-part.32,41When focussing solely on local-valence ex-
citations, we notice similar trends as for thetwo previously discussed sets with most ofthe SCS and SOS variants being positionedat the top and RMSDs ranging from 0.13eV (SOS-ωPBEPP86 and SOS-PBE-QIDH) to0.16 eV (SOS-RSX-QIDH, SCS-ωB2GP-PLYP,and ωB88PP86). Note that the RS-DH meth-ods perform equally well with RMSDs of0.16-0.17 eV, values that they share withB2PLYP, B2GP-PLYP, SCS-RSX-QIDH, SOS-B2GP-PLYP21, and SOS-ωB2GP-PLYP. Thelast two methods with RMSDs below 0.2 eVare ωPBEPP86 and SCS-B2GP-PLYP21 (0.18eV). All remaining methods have RMSDs above0.20 eV.As we already pointed out in Ref. 40, we
called this set the difficult cases set becauseits Rydberg states do not seem to require
the LC scheme, contrary to the examples dis-cussed earlier. Note that this trend is alsoseen for the RS-DH methods, which show largeblue-shifts for this category (RMSD = 0.30and 0.35 eV for RS-B88-LYP and RS-PBE-P86, respectively). Hence, it is not surpris-ing that the two top places in our ranking areglobal DHDFAs methods (SOS-B2GP-PLYP21and SCS/SOS-B2PLYP21). However, our newSCS- and SOS-LC-DHDFAs improve over theirunscaled parents and yield more accurate re-sults, for instance, SCS/SOS-ωB2PLYP andωB2PLYP (0.12 vs. 0.14 eV), SCS-ωB2GP-PLYP and ωB2GP-PLYP (0.14 vs. 0.20 eV).In summary, our SCS/SOS variants of global
and LC-DHDFAs are the best-performingmethods even for the difficult cases presentedby Loos, Jacquemin, and co-workers. Noneof these systems were assessed in the previoussets, demonstrating that our new methods arerobust enough, even for the treatment of chal-lenging systems like the ones discussed in thissection.
5.3 The “Exotic-Molecules” Set
In mid-2020, Loos and Jacquemin et al. pre-sented a new benchmark set under the name“exotic molecules set”. With that name theauthors intended to imply that each of themolecules in this set includes at least one atomrepresenting elements that is uncommon inusual organic-molecule test sets, namely: F,Cl, Si, or P.114 The overall set comprises 19local-valence singlet-singlet excitations from 14different systems with high-level CC3/aug-cc-pVTZ reference values. We remind the readerthat the inclusion of these “exotic molecules”made the TD-DHDFA scheme unable to cor-rectly describe these kind of systems and betterresults were obtained with the TDA-DHDFAformalism, as we thoroughly discussed in Ref.41. Therefore, the user might take this intoconsideration when the system under studypresents any of the aforementioned species.Based on the RMSDs presented in Fig. 3, the
RS-DH methods yield the lowest RMSDs (< 0.1eV), which is an outstanding finding. B2GP-PLYP also displays an excellent value of exactly
14

-0.10
0.00
0.10
0.20
0.30
RS-B88-LYP
RS-PBE-PBE
RS-PBE-P86
B2GP-PLYP
ωPBEPP86
B2PLYP
SCS-PBE-QIDH
SCS-
ωPBEPP86
SCS-RSX-QIDH
SOS-PBE-QIDH
ωB88PP86
SOS-B2GP-PLYP21
PBE-QIDH
SCS-
ωB2GP-PLYP
SCS-B2GP-PLYP21
SCS-
ωB88PP86
SCS/SOS-
ωB2PLYP
SOS-
ωB88PP86
ωB2PLYP
SOS-
ωPBEPP86
SOS-
ωB2GP-PLYP
ωB2GP-PLYP
SCS/SOS-B2PLYP21
SOS-RSX-QIDH
RSX-QIDH
Statistical Value [eV]
MAD
RMSD
MD
0.07
0.09
0.09
0.10
0.12
0.13
0.13
0.13
0.13
0.14
0.14
0.14
0.15
0.15
0.15
0.16
0.16
0.16
0.16
0.16
0.17
0.17
0.18
0.20
0.22
Figure 3: Statistical values (in eV) for the “exotic-molecules” set,namely RMSDs (values shown over each bar), MADs, and MDs.The values for the RS-DH methods were taken from Ref. 80. Theaug-cc-pVTZ AO basis set was used in all cases.
0.10 eV for this test set, closely followed by ournew ωPBEPP86 (0.12 eV) and B2PLYP (0.13eV). Our new SCS and SOS methods feature inthe ranking from this point onward, with SCS-PBE-QIDH, SCS-ωPBEPP86, and SCS-RSX-QIDH being the first ones with good values of0.13 eV. 14 other methods, all of which devel-oped for this study, have RMSDs that rangefrom 0.14 to 0.18 eV . Lastly, SOS-RSX-QIDHand its parent RSX-QIDH are placed in last po-sition with RMSDs of 0.20 and 0.22 eV. respec-tively. We conclude that almost every assessedmethod performs fairly well compared to thepreviously sets given that their RMSDs mostlylie below 0.20 eV.In general, the MDs are well-balanced for this
set, regardless of the use of SCS or SOS. Ex-cept for SCS/SOS-B2PLYP21 and SCS-B2GP-PLYP21 (MDs=0.15 eV), and SOS-RSX-QIDH(MD=–0.10 eV), every other method has abso-lute MDs below 0.1 eV.
5.4 The Thiel Benchmark Set
To keep investigating our new SCS/SOS meth-ods’ capabilities a bit further, we also ana-lyze parts of the popular benchmark set intro-
duced by Thiel and co-workers.27 The originalset comprises 167 valence excitations, 104 ofwhich are singlet-singlet excitations with var-ious WFT reference values, out of which wechose the CC3/TZVP115 ones in agreementwith previous works in Refs. 39,41. In orderto avoid duplication with previous sets, we onlyanalyze 36 local-valence singlet excitations from8 different systems, namely: acetamide, ben-zoquinone, imidazole, norbornadiene, octate-traene, propanamide, pyridazine, and pyrimi-dine.This test set differs from previous ones given
the large RMSDs for every tested method. Af-ter a closer inspection, we found that the 21Agstate in octatetraene displays a very large over-estimation, reaching more than 1 eV in somecases. In 2011, Angeli and Pastore116 consid-ered octatetraene as a somewhat complicatedsystem due to its high double-excitation charac-ter. Previous work on TD-B2(GP-)PLYP alsoshowed this state to be problematic.28 In lightof this finding, we report the statistical resultsby considering and excluding this state, wherethe latter case is shown in parentheses (Fig. 4).When considering all excitations, PBE-
QIDH’s SCS and SOS variants are in the lead,closely followed by the unscaled B2GP-PLYPand its SOS variant, all of them with RMSDsslightly above 0.2 eV. Seven DHDFAs haveRMSDs between 0.26 and 0.29 eV; with the ex-ception of PBE-QIDH, all methods have beennewly developed for this study. Five meth-ods yield high RMSDs that range from 0.33 to0.36 eV, namely, SCS/SOS-ωB2PLYP, RSX-QIDH’s SCS and SOS variants, and ωB2GP-PLYP’s SCS and SOS variants. Finally, theRS-DH methods and the unscaled ωB2PLYPhave values of 0.40 eV, whereas ωB2GP-PLYPand RSX-QIDH come last with RMSDs of 0.43,and 0.54 eV, respectively.When excluding the 21Ag state in octate-
traene, the ranking of methods changes. Now,most of the SCS and SOS variants are placedat the top. Compared to the analysis in theprevious paragraph, those methods with thehighest RMSDs benefit from a large reduc-tion (on average 0.1 eV), whereas the oneswith the low RMSDs benefit from a smaller
15

-0.20
-0.10
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
SOS-PBE-QIDH
SCS-PBE-QIDH
B2GP-PLYP
SOS-B2GP-PLYP21
SCS-
ωPBEPP86
SCS-B2GP-PLYP21
SOS-
ωPBEPP86
SOS-
ωB88PP86
SCS-
ωB88PP86
PBE-QIDH
SCS/SOS-B2PLYP21
ωB88PP86
ωPBEPP86
B2PLYP
SCS/SOS-
ωB2PLYP
SOS-RSX-QIDH
SCS-
ωB2GP-PLYP
SCS-RSX-QIDH
SOS-
ωB2GP-PLYP
RS-B88-LYP
ωB2PLYP
RS-PBE-PBE
RS-PBE-P86
ωB2GP-PLYP
RSX-QIDH
Statistical Value [eV]
MAD
RMSD
MD
0.21 0.21 0.22 0.23
0.26 0.26 0.27 0.270.29 0.29 0.29 0.30 0.30
0.33 0.34 0.35 0.35 0.36 0.36
0.40 0.40 0.40 0.40
0.43
0.54
(0.18)(0.18)(0.21)(0.21)
(0.19)(0.24)(0.20)(0.21)(0.23)(0.26)(0.28)
(0.24)(0.24)
(0.33)(0.26)(0.23)(0.23)(0.24)(0.25)
(0.27)(0.33)(0.27)(0.28)
(0.33)
(0.45)
Figure 4: Statistical values (in eV) for the Thiel set, namely RMSDs (values shown over each bar), MADs, and MDs. Values in parenthesesare RMSDs after excluding the 21Ag state in octatetraene. The values for the RS-DH methods were taken from Ref. 80. The TZVP AO basisset was used in all cases.
reduction (on average 0.05 eV). The threebest performers this time are PBE-QIDH’sSCS and SOS variants and SCS-ωPBEPP86with RMSDs below 0.2 eV. We also see im-provements for various methods after exclud-ing the 21Ag state in octatetraene, with re-ductions in the RMSDs of up to 0.13 eV, suchas SCS-ωB2GP-PLYP, SCS/SOS-ωB2PLYP, orthe three RS-DH methods. They now all per-form similarly to unscaled PBE-QIDH. Lastly,the unscaled B2PLYP, ωB2PLYP, and ωB2GP-PLYP also have improved values (RMSD = 0.33eV in each case), whereas RSX-QIDH is still inlast place with a value of 0.45 eV. Overall, wecan conclude how most SCS/SOS variants showsuperior performance for a set that seems to bechallenging to describe.
5.5 1La and 1Lb Transitions inLinear Polycyclic AromaticHydrocarbons
Linear polycyclic aromatic hydrocarbons(PAHs) have gained particular interest dueto their possible applications in light-emittingdiodes or photovoltaics, to name two exam-ples. According to the Platt nomenclature,the two lowest singlet-singlet excitations are
usually called 1La and 1Lb.117 The formerexhibits a polarization along the short axisof the molecule, whereas the second is polar-ized along the long axis.118 It has also beensuggested that the 1La state has ionic char-acter, while the 1Lb state has covalent char-acter.119–121 Conventional TD-DFT fails todescribe both states with comparable accu-racy; as shown in detail in Refs. 35,118 and40, a good description of one state comes atthe cost of the other. In summary, LC-hybridfunctionals provide a better description of 1Laexcitations, but they tend to overestimate the1Lb ones.35,122 In 2011, it was shown for thefirst time that global TD-DHDFAs partiallysolved this problem compared to CC2 referencedata.35 In 2019, we confirmed this finding onceagain when we analyzed the same 5 linear sys-tems (from naphthalene to hexacene) with ournew TD-LC-DHDFAs40 but, in contrast to the2011 work, we relied on high-level CR-EOM-CCSD(T)123 data as well as the experimen-tal reference values taken from Ref. 118. Wefound that ωB2GP-PLYP was the best TD-DFT method at that time with results thatwere slightly better than CC2.40 Herein, were-analyze the same five systems with the samehigh-level reference values. All calculations
16

Table 6: Statistical results (in eV) for the 1La–1Lb energy gap over the five linear polyacenes from naphthaleneto hexacene using TDA (left) and full TD algorithms (right).a
TDA-DHDFA TD-DHDFAMethod MD MAD RMSD Δerr Method MD MAD RMSD ΔerrSOS-RSX-QIDHb –0.03 0.03 0.03 0.01 SCS-ωB2GP-PLYP –0.02 0.03 0.03 0.08SOS-ωB2GP-PLYP –0.08 0.08 0.08 0.05 SOS-ωPBEPP86 0.03 0.03 0.04 0.06SOS-ωPBEPP86b –0.08 0.08 0.08 0.02 SOS-ωB2GP-PLYPb 0.02 0.03 0.04 0.09SOS-ωB88PP86 –0.10 0.10 0.10 0.04 SOS-ωB88PP86 –0.04 0.04 0.04 0.06SCS-ωB2GP-PLYP –0.11 0.11 0.11 0.03 SCS-ωPBEPP86 –0.07 0.07 0.07 0.05SCS/SOS-ωB2PLYP –0.12 0.12 0.13 0.07 SOS-PBE-QIDH –0.08 0.08 0.08 0.02SOS-PBE-QIDH –0.13 0.13 0.13 0.02 SCS-ωB88PP86b –0.09 0.09 0.09 0.05SCS-ωB88PP86 –0.14 0.14 0.14 0.04 SCS/SOS-ωB2PLYPb –0.09 0.09 0.10 0.09SOS-B2GP-PLYP21 –0.17 0.17 0.17 0.04 SCS-RSX-QIDH –0.13 0.13 0.13 0.03SCS-ωPBEPP86 –0.17 0.17 0.17 0.01 SOS-RSX-QIDH 0.14 0.14 0.14 0.09SCS-PBE-QIDH –0.18 0.18 0.18 0.02 SCS-PBE-QIDH –0.14 0.14 0.14 0.02SCS-B2GP-PLYP21 –0.18 0.18 0.19 0.04 SOS-B2GP-PLYP21 –0.19 0.19 0.19 0.02RS-B88-LYP –0.23 0.23 0.24 0.19 SCS-B2GP-PLYP21 –0.21 0.21 0.21 0.01RS-PBE-PBE –0.23 0.23 0.24 0.20 ωPBEPP86 –0.23 0.23 0.23 0.05RS-PBE-P86 –0.23 0.23 0.24 0.20 ωB88PP86 –0.26 0.26 0.26 0.03SCS-RSX-QIDH –0.25 0.25 0.25 0.05 ωB2GP-PLYP –0.27 0.27 0.27 0.02ωPBEPP86 –0.26 0.26 0.26 0.05 RSX-QIDH –0.31 0.31 0.31 0.03ωB88PP86 –0.27 0.27 0.27 0.03 ωB2PLYP –0.32 0.32 0.32 0.04ωB2GP-PLYP –0.28 0.28 0.28 0.01 B2GP-PLYP –0.35 0.35 0.35 0.05ωB2PLYP –0.28 0.28 0.28 0.05 PBE-QIDH –0.38 0.38 0.38 0.04SCS/SOS-B2PLYP21 –0.29 0.29 0.29 0.07 SCS/SOS-B2PLYP21 –0.43 0.43 0.43 0.01RSX-QIDH –0.31 0.31 0.31 0.02 B2PLYP –0.44 0.44 0.44 0.03B2GP-PLYP –0.33 0.33 0.33 0.04 RS-B88-LYPc – – – –PBE-QIDH –0.36 0.36 0.36 0.03 RS-PBE-PBEc – – – –B2PLYP –0.36 0.36 0.36 0.03 RS-PBE-P86c – – – –
a All calculations were performed with the cc-pVTZ AO basis set.The values for the RS-DH methods were takenfrom Ref. 80. b These methods perform well for each one of the 1La and 1Lb states, as well as for the energetic
splitting. c TD-DHDFA data not available.
were performed with the cc-pVTZ AO basisset. Due to the lack of high-level reference datafor triplet excitations in this test set, we presentresults not only using the TDA-DHDFA schemebut also for the full TD-DHDFA scheme, as notriplet-instability problems are expected. Thisallows us also to test the transferability of ourSCS/SOS parameters to the full TD-DHDFscheme.Regarding the energy splitting between the
two states we see an improvement whenSCS/SOS is applied, however, there are twopotential reasons for this observation: (a) ei-ther both the 1La and 1Lb excitations improveand, therefore, the energy gap between themalso agrees better with the reference, or (b) theexcitations to both states are shifted similarlybut their energy difference matches with thereference. Ideally, the situation described in(a) is more desirable than (b), however, the en-ergy differences also provide information that isas important and valuable as the values for theindividual states. Those methods that belongto the situation described in (a) are highlightedin footnote b in Table 6, where we present thestatistics for the energy gap over the five sys-tems. The complete raw data and the statisticsfor each of the 1La and 1Lb states can be foundin the SI.
Similar to previous test sets, most of ourSCS/SOS variants occupy the best positionsin the ranking, with the exception of SCS-RSX-QIDH and SCS/SOS-B2PLYP21. Forthe TDA-DHDFA scheme, we identify one andthree methods that fulfill the first and seconddefinition of “chemical accuracy”, respectively,namely SOS-RSX-QIDH (RMSD = 0.03 eV),SOS-ωB2GP-PLYP (RMSD = 0.08 eV), SOS-ωPBEPP86 (RMSD = 0.08 eV), and SOS-ωB88PP86 (RMSD = 0.10 eV). Amongst thosefour methods, which all display outstanding re-sults, we highlight SOS-RSX-QIDH and SOS-ωPBEPP86 as functionals that satisfy the sit-uation described in (a), i.e., the simultane-ous improvement of both types of excitations.Eight other methods follow with RMSDs be-low 0.2 eV, the best of which are SCS-ωB2GP-PLYP, SCS/SOS-ωB2PLYP, SOS-PBE-QIDH,SCS-ωB88PP86. The three RS-DH methodsshare an RMSD of 0.24 eV, followed by SCS-RSX-QIDH with a value of 0.25 eV. Next,we find the unscaled methods ωPBEPP86,ωB88PP86 , ωB2GP-PLYP, ωB2PLYP, andSCS/SCS-B2PLYP21, all of which are the re-maining methods with RMSDs below 0.3 eV.For the TD-DHDFA scheme, the picture is
not too different, since most of the SCS/SOSmethods are at the top of the ranking, with
17

SCS/SOS-B2PLYP21 being the only exception.It is noteworthy that we now see eight methodsthat fulfill the second definition of “chemicalaccuracy”, of which the best-four methods alsofulfill the first definition (see Table 6); theseare: SCS-ωB2GP-PLYP, SOS-ωPBEPP86,SOS-ωB2GP-PLYP, and SOS-ωB88PP86,SCS-ωPBEPP86 , SOS-PBE-QIDH, SCS-ωB88PP86 , and finally SCS/SOS-ωB2PLYP.Among those methods, SOS-ωB2GP-PLYP,SCS-ωB88PP86, and SCS/SOS-ωB2PLYP be-long to category (a). Interestingly, SCS-RSX-QIDH follows this time with a good RMSDof 0.13 eV, closely followed by its SOS vari-ant and SCS-PBE-QIDH. SOS-B2GP-PLYP21is the last method with an RMSD below 0.2eV. Similarly to the TDA-DHDFA analysis,ωPBEPP86, ωB88PP86, and ωB2GP-PLYPdisplay RMSDs above 0.2 eV. The remainingunscaled methods and SCS/SOS-B2PLYP21yield RMSDs above 0.3 eV.Regarding the correct energetic order of the
states, only a few methods give the wrong or-der for anthracene with respect to the exper-imental data, where the 1La–1Lb experimen-tal gap is –0.04 eV. For the TDA-DHDFAscheme, only three methods fail, namely, SOS-ωB2GP-PLYP (gap of 0.01 eV), SOS-RSX-QIDH (0.06 eV), and SOS-ωPBEPP86 (0.01eV). For the TD-DHDFA scheme, seven meth-ods fail in this regard: SCS- and SOS-ωB2GP-PLYP (0.06 and 0.10 eV, respectively), SOS-PBE-QIDH (0.01 eV), SOS-RSX-QIDH (0.21eV), SOS-ωB88PP86 (0.04 eV), SCS- and SOS-ωPBEPP86 (0.01 and 0.11 eV, respectively).Note that the CR-EOM-CCSD(T) method alsoyields the wrong order of states for anthracene(0.10 eV) compared to experimental data.In summary, to the best of our knowledge the
results for some of the new methods developedherein constitute the best TD(A)-DFT resultspublished to date for these systems.
5.6 An Update on Charge-Transfer Excitations
The LC methods were mainly developed withthe aim to solve the charge-transfer (CT) prob-lem of TD-DFT, and we proceed with a full
assessment of our “updated” CT set presentedin Ref. 40. In 2020, we modified this set andhere we use the same systems again. Those are:2,4-dichloro-6-p-N,N-diethylaminobiphenylyl-1,3,5-triazine (DBQ),34 para-nitroaniline(pNA),124 dimethylaminobenonitrile (DMABN),124benzene-tetracyanoethylene complex (B-TCNE),124 hydrogen chloride (HCl),113 adipeptide,125 a β-dipeptide,125 and finallyN-phenylpyrrole (PP).126 Similar to the pre-vious set, we also present full TD-DHDFAresults due to the lack of data for singlet-triplet excitations. The high-level referencedata used in this set are: theoretically back-corrected from the experiment (DBQ), linear-response CCSDR(3) (pNA and peptides),EOM-CCSD(T) (DMABN and B-TCNE), andlinear-response CC3 (HCl), respectively (alsosee SI).When inspecting Table 7, it should not come
as a surprise that global TD(A)-DHDFAs arenot suitable for describing CT excitations. Wepoint out that the opposite has been claimedin 202073 but disproven by us on three occa-sions.40,41,43 This study constitutes the fourthoccasion. As already discussed by us for aryl-TCNE complexes,43 we observe again thatglobal DHDFAs artificially benefit from theTDA-DHDFA scheme thanks to a systematicblue shift in the excitation energies. In addi-tion, we see that the SCS or SOS variants ofglobal methods reduce the RMSD by half, e.g,TD(A)-B2PLYP and TD(A)-B2GP-PLYP.Inspecting the TDA-DHDFA results for
LC-DHDFAs, SOS-RSX-QIDH is the besttested method with an RMSD of 0.20 eV,closely followed by SOS-ωB88PP86 and SOS-ωPBEPP86. Three more SCS/SOS LC-DHDFAs follow before the first global DHDFAappears in the ranking, namely, SCS-B2GP-PLYP21 with an RMSD of 0.26 eV, sharing thesame value as SCS/SOS-ωB2PLYP. Next, wehave SCS-ωPBEPP86, SOS-B2GP-PLYP21,PBE-QIDH’s SCS and SOS variants, alongsideSCS-RSX-QIDH with values ranging from 0.27to 0.29 eV. Most of the remaining methodsdisplay RMSDs that range from 0.30 eV (PBE-QIDH, SCS-PBE-QIDH, and ωB88PP86) to0.66 eV (B2PLYP).
18

Table 7: Statistical results (in eV) for the full CT set using TDA (left) and full TD algorithms (right).a
TDA-DHDFA TD-DHDFAMethod MD MAD RMSD Δerr Method MD MAD RMSD ΔerrSOS-RSX-QIDH 0.05 0.17 0.20 0.70 SOS-ωB2GP-PLYP 0.03 0.12 0.15 0.47SOS-ωB88PP86 0.05 0.15 0.21 0.80 SCS-ωB2GP-PLYP 0.00 0.12 0.16 0.57SOS-ωPBEPP86 0.01 0.16 0.22 0.82 SCS/SOS-ωB2PLYP 0.07 0.12 0.16 0.48SCS-ωB2GP-PLYP 0.11 0.17 0.23 0.75 SOS-ωB88PP86 –0.04 0.14 0.18 0.68SOS-ωB2GP-PLYP 0.14 0.18 0.23 0.65 SCS-ωB88PP86 0.00 0.13 0.19 0.73SCS-ωB88PP86 0.09 0.18 0.24 0.85 SOS-RSX-QIDH –0.06 0.16 0.19 0.53SCS-B2GP-PLYP21 –0.08 0.19 0.26 0.94 SOS-ωPBEPP86 –0.08 0.16 0.20 0.69SCS/SOS-ωB2PLYP 0.18 0.21 0.26 0.66 ωB2PLYP 0.15 0.18 0.23 0.61SCS-ωPBEPP86 –0.03 0.21 0.27 1.02 ωB2GP-PLYP 0.15 0.19 0.23 0.64SOS-B2GP-PLYP21 –0.13 0.21 0.28 0.95 ωB88PP86 0.01 0.17 0.24 0.92SOS-PBE-QIDH –0.15 0.22 0.29 0.89 SCS-RSX-QIDH –0.03 0.18 0.24 0.87SCS-RSX-QIDH 0.07 0.22 0.29 1.06 SCS-B2GP-PLYP21 –0.15 0.20 0.25 0.78SCS-PBE-QIDH –0.15 0.23 0.30 0.95 ωPBEPP86 0.00 0.19 0.26 0.96PBE-QIDH –0.05 0.24 0.30 1.07 SCS-ωPBEPP86 –0.12 0.21 0.27 0.89ωB88PP86 0.09 0.24 0.30 1.04 SOS-B2GP-PLYP21 –0.20 0.23 0.29 0.79ωPBEPP86 0.08 0.25 0.31 1.09 PBE-QIDH –0.08 0.23 0.29 0.92SCS/SOS-B2PLYP21 –0.12 0.26 0.34 1.14 SOS-PBE-QIDH –0.18 0.23 0.30 0.81ωB2GP-PLYP 0.26 0.30 0.34 0.84 SCS-PBE-QIDH –0.17 0.24 0.31 0.84ωB2PLYP 0.27 0.29 0.35 0.80 RSX-QIDH 0.28 0.29 0.33 0.59B2GP-PLYP –0.28 0.34 0.44 1.17 SCS/SOS-B2PLYP21 –0.20 0.27 0.34 0.98RSX-QIDH 0.40 0.41 0.45 0.80 B2GP-PLYP –0.34 0.36 0.45 1.02B2PLYP –0.53 0.57 0.66 1.33 B2PLYP –0.58 0.59 0.69 1.18
a The “dipeptide”, “β-peptide”, and PP calculations were performed with the cc-pVTZ AO basis set. The B-TCNE,DMABN, pNA, and HCl calculations were performed with the aug-cc-pVTZ AO basis set. The DBQ calculation
was performed with the def2-TZVPP127 AO basis set.
For the TD-DHDFA results, it is noticeablethat ωB2GP-PLYP’s SCS and SOS variantshave significantly lowered their RMSDs com-pared to the TDA-DHDFA values, namely from0.23 to 0.15/0.16 eV, which position them as thebest TD-DHDFA methods. In total, there areseven new SCS/SOS-LC-DHDFA methods thatall have RMSDs below 0.2 eV and surpass thepreviously reported best results for ωB2PLYPand ωB2GP-PLYP (RMSD = 0.23 eV). The re-maining methods in this ranking are listed inTable 7; similar to the TDA-DHDFA scheme,TD-B2PLYP is the worst DHDFA with anRMSD of 0.69 eV.In Ref. 80, the RS-DH methods were as-
sessed on a slightly modified version of our CTset40 and only for the TDA-DHDFA scheme.Therein, our peptides were excluded from theanalysis. In order to compare our methods withthose, we included an additional analysis thatomits those systems in the SI. We briefly men-tion that there is a small decrease in the RMSDsfor some methods, for instance, a reduction of0.02 eV for SOS-RSX-QIDH, 0.03 eV for SOS-ωB88PP86. However, the general trend re-mains the same. The RS-DH methods are po-sitioned after the SCS and SOS variants of ourown LC-DHDFAs, with RMSDs slightly above0.30 eV.
6 Cross-Validation for Ver-tical Singlet-Triplet Exci-tations
After having presented promising results forvertical singlet-singlet excitations, we shift ourattention to the discussion of vertical singlet-triplet excitations. The first study of such exci-tations was done in 2007 for the TDA-B2PLYPfunctional,18 where Grimme and Neese exam-ined 22 excitations in seven different systemsbased on experimental data. In 2020, weassessed the TDA-DHDFAs idea for the sec-ond time and, for the first time, TD-DHDFAsfor such excitations in a more comprehensivestudy.41 Our study showed that like other func-tionals TD-DHDFAs are not useful for such ex-citations, but that TDA-DHDFAs can provideaccurate results. Herein, we follow up on ourstudy from 2020 and present the first study ofsuch excitations with SCS and SOS techniquesapplied to TDA-DHDFAs. Note that there isno data available for such excitations for thenew RS-DH methods and they will not be con-sidered in the following.
6.1 The Loos and JacqueminBenchmark Set
The original benchmark set by Loos,Jacquemin, and co-workers comprises 47
19

-0.20
-0.10
0.00
0.10
0.20
0.30
SCS-B2GP-PLYP21
SCS/SOS-B2PLYP21
SOS-B2GP-PLYP21
ωB88PP86
SCS-PBE-QIDH
ωPBEPP86
ωB2GP-PLYP
SOS-PBE-QIDH
SCS-
ωB88PP86
PBE-QIDH
SOS-
ωB2GP-PLYP
B2GP-PLYP
SCS-
ωB2GP-PLYP
SOS-
ωB88PP86
SCS-
ωPBEPP86
SOS-
ωPBEPP86
ωB2PLYP
SCS-RSX-QIDH
SCS/SOS-
ωB2PLYP
SOS-RSX-QIDH
RSX-QIDH
B2PLYP
Statistical Value [eV]
MAD
RMSD
MD
0.15
0.16
0.16
0.16
0.16
0.17
0.17
0.17
0.18
0.18
0.18
0.19
0.19
0.19
0.20
0.20
0.21
0.21
0.21
0.23
0.24 0.27
Figure 5: Statistical values (in eV) for the S0-TN transitions forthe Loos and Jacquemin set, namely RMSDs (values shown over eachbar), MADs, and MDs. The aug-cc-pVTZ AO basis set was used inall cases.
singlet-triplet transitions, of which we selected35 transitions (23 local valence, 12 Rydberg,and one CT state) in 13 different molecules,which have not been included in other hereindiscussed sets. All references are of CC3/aug-cc-pVTZ quality and taken from Ref. 113, ex-cept for HCl, which was calculated by us andreported in Ref. 41.
6.1.1 All Singlet-Triplet Excitations inthis set (S0-TN)
Fig. 5 shows the same trend as in the updatedGordon set in Section 4.2.3, thus, confirmingthe reliability of our new methods. Similar tothe updated Gordon set, our methods againperform very well, with SCS-B2GP-PLYP21being the first method in our ranking (0.15 eV),closely followed by SCS/SOS-B2PLYP21, SOS-B2GP-PLYP21, ωB88PP86, and SCS-PBE-QIDH. Furthermore, ωPBEPP86, ωB2GP-PLYP, SOS-PBE-QIDH, SCS-ωB88PP86, theunscaled PBE-QIDH, and SOS-ωB2GP-PLYPperform very well with RMSDs of about 0.18eV. The last four methods with RMSDs be-low 0.2 eV are B2GP-PLYP, SCS-ωB2GP-PLYP, SOS-ωB88PP86, and SCS-ωPBEPP86.
0.00
0.10
0.20
0.30
SOS-RSX-QIDH
SOS-
ωPBEPP86
SOS-
ωB2GP-PLYP
SOS-PBE-QIDH
SCS-PBE-QIDH
SOS-
ωB88PP86
SCS-
ωB2GP-PLYP
SOS-B2GP-PLYP21
ωPBEPP86
B2GP-PLYP
SCS-B2GP-PLYP21
SCS/SOS-
ωB2PLYP
B2PLYP
ωB88PP86
SCS-
ωB88PP86
SCS-
ωPBEPP86
SCS/SOS-B2PLYP21
SCS-RSX-QIDH
ωB2GP-PLYP
PBE-QIDH
ωB2PLYP
RSX-QIDH
Statistical Value [eV]
MAD
RMSD
MD
0.12
0.13 0.15
0.15
0.16
0.16
0.16
0.16
0.16
0.17
0.17
0.17
0.17
0.18
0.18
0.18
0.21
0.21
0.21
0.22
0.23
0.27
Figure 6: Statistical values (in eV) for the S1-T1 energy splittingon the Loos and Jacquemin set, namely RMSDs (values shown overeach bar), MADs, and MDs. The aug-cc-pVTZ AO basis set wasused in all cases.
Other functionals are shown in Fig. 5 andwe just point out that we obtained the largestRMSDs for SOS-RSX-QIDH, its unscaled vari-ant RSX-QIDH, and B2PLYP with values be-yond 0.23 eV. The MDs display the same trendas in the updated Gordon set by showing a sys-tematic underestimation for the S0-TN transi-tions.
6.1.2 S1-T1 Analysis
Despite global DHDFAs leading the S0-TNanalysis, LC-DHDFAs are the best for the S1-T1 energy splitting, as can be seen in Fig. 6,where SOS-RSX-QIDH and SOS-ωPBEPP86display very good RMSDs of about 0.12 eV,slightly above the “chemical accuracy” thresh-old. Besides those two, there are other 14methods that perform very well with RMSDsbelow 0.2 eV, six of which are SCS/SOS-LC-DHDFAs (see Fig. 6). Surprisingly, thereare two SCS/SOS methods that are positionedabove 0.2 eV, namely, SCS/SOS-B2PLYP21which was the second-best in the S0-TN analy-sis, and SCS-RSX-QIDH. The worst performeris the unscaled RSX-QIDH with an RMSD of0.27 eV.
20

-0.20
-0.10
0.00
0.10
0.20
0.30
SCS/SOS-B2PLYP21
SCS-B2GP-PLYP21
SOS-B2GP-PLYP21
SCS-PBE-QIDH
SOS-PBE-QIDH
B2GP-PLYP
ωPBEPP86
SCS-
ωB2GP-PLYP
SOS-
ωB2GP-PLYP
ωB88PP86
SCS-
ωB88PP86
ωB2GP-PLYP
SOS-
ωPBEPP86
SCS/SOS-
ωB2PLYP
PBE-QIDH
SCS-
ωPBEPP86
SOS-
ωB88PP86
B2PLYP
ωB2PLYP
SCS-RSX-QIDH
SOS-RSX-QIDH
RSX-QIDH
Statistical Value [eV]
MAD
RMSD
MD
0.07
0.09
0.10 0.15
0.15
0.15
0.15
0.17
0.17
0.18
0.18
0.18
0.19
0.20
0.20
0.20
0.20
0.21
0.22
0.23
0.24
0.25
Figure 7: Statistical values (in eV) for the S0-TN transitions forthe “Exotic-Molecules” set, namely RMSDs (values shown over eachbar), MADs, and MDs. The aug-cc-pVTZ AO basis set was used inall cases.
The MDs follow the same trend as in the up-dated Gordon set, with a systematic underesti-mation in the excitation energies for the S0-TNanalysis, whereas there is a systematic overes-timation for the S1-T1 energy splitting. In theremaining sections, we will see that this is ageneral trend and cease to comment on MDs.
6.2 The “Exotic-Molecules” Set
This set by Loos, Jacquemin, and co-workers114consists of 11 singlet-triplet transitions of local-valence character in seven different systems, allof high-level CC3/aug-cc-pVTZ quality.
6.2.1 All Singlet-Triplet Excitations inthis set (S0-TN)
Similar to Section 6.1.1, the SCS and SOS vari-ants of B2PLYP and B2GP-PLYP are also thebest methods here, with low RMSDs that rangefrom 0.07 eV for SCS/SOS-B2PLYP21 to 0.10eV for SOS-B2GP-PLYP21 (Fig. 7). Somewhathigher in energy, SCS- and SOS-PBE-QIDH,B2GP-PLYP, and ωPBEPP86 share the sameRMSD of 0.15 eV. ωB2GP-PLYP’s SCS andSOS variants, ωB88PP86, SOS-ωB88PP86, andthe unscaled ωB2GP-PLYP follow next in theranking. The last DHDFA with an RMSD be-low 0.2 eV is SOS-ωPBEPP86 with an RMSDof 0.19 eV. The remaining scaled and unscaled
0.00
0.10
0.20
0.30
SOS-RSX-QIDH
SOS-
ωB2GP-PLYP
SOS-
ωPBEPP86
SCS-
ωB2GP-PLYP
SOS-B2GP-PLYP21
SOS-PBE-QIDH
SOS-
ωB88PP86
SCS-B2GP-PLYP21
SCS/SOS-
ωB2PLYP
SCS-PBE-QIDH
SCS-
ωPBEPP86
SCS-
ωB88PP86
B2PLYP
SCS/SOS-B2PLYP21
B2GP-PLYP
ωPBEPP86
SCS-RSX-QIDH
ωB88PP86
ωB2GP-PLYP
ωB2PLYP
PBE-QIDH
RSX-QIDH
Statistical Value [eV]
MAD
RMSD
MD
0.09
0.10
0.10
0.11
0.12
0.12
0.13
0.13
0.14
0.14
0.15
0.15
0.18
0.18
0.18
0.18 0.20
0.21
0.22 0.24
0.25
0.29
Figure 8: Statistical values (in eV) for the S1-T1 energy splittingon the “Exotic-Molecules” set, namely RMSDs (values shown overeach bar), MADs, and MDs. The aug-cc-pVTZ AO basis set wasused in all cases.
methods display results that range from 0.20eV (SCS/SOS-ωB2PLYP) to 0.25 eV (RSX-QIDH).
6.2.2 S1-T1 Analysis
The S1-T1 splitting shows the same behavioras in the previous set (Fig. 8). The SOSvariants of RSX-QIDH, ωB2GP-PLYP, andωPBEPP86 are the best three performers withRMSDs within the chemical accuracy thresh-old of 0.10 eV. Other methods that also dis-play excellent results are SCS-ωB2GP-PLYP,B2GP-PLYP, PBE-QIDH, and SOS-ωB88PP86with values near 0.12 eV. SCS/SOS-ωB2PLYP,SCS-PBE-QIDH, SCS-ωPBEPP86, and SCS-ωB88PP86 perform similarly with RMSDs ofup to 0.15 eV. B2PLYP is the first unscaledmethod in the list and shares the same RMSDof 0.18 eV with SCS/SOS-B2PLYP21, B2GP-PLYP, and ωPBEPP86. The remaining meth-ods have RMSDs ranging from 0.20 eV (SCS-RSX-QIDH) to 0.29 eV (RSX-QIDH).
21

-0.20
-0.10
0.00
0.10
0.20
0.30
SCS/SOS-B2PLYP21
ωB88PP86
ωPBEPP86
SCS-
ωB88PP86
SOS-
ωB88PP86
SOS-
ωPBEPP86
SOS-B2GP-PLYP21
SCS-B2GP-PLYP21
SCS/SOS-
ωB2PLYP
B2GP-PLYP
SCS-
ωPBEPP86
SCS-PBE-QIDH
SOS-PBE-QIDH
SOS-RSX-QIDH
ωB2PLYP
PBE-QIDH
SCS-RSX-QIDH
SCS-
ωB2GP-PLYP
SOS-
ωB2GP-PLYP
ωB2GP-PLYP
RSX-QIDH
B2PLYP
Statistical Value [eV]
MAD
RMSD
MD
0.11
0.12
0.12
0.14
0.15
0.15
0.16
0.16
0.16
0.17
0.17
0.18
0.18
0.18
0.18
0.19
0.20
0.22
0.22
0.23
0.23
0.24
Figure 9: Statistical values (in eV) for the S0-TN transitions forthe Thiel set, namely RMSDs (values shown over each bar), MADs,and MDs. The TZVP AO basis set was used in all cases.
6.3 The Thiel Benchmark Set
The original set by Thiel and co-workers com-prises 63 triplet excitations with CC3/TZVP27
reference values. However, most of themolecules have already been addressed in ourpreceding analyses and therefore we decided tostudy only six molecules that have not beenassessed before, namely, acetamide, benzo-quinone, imidazole, naphthalene, norbornadi-ene, and octatetraene. For these six molecules,a total of 25 local-valence excitations need tobe analyzed.
6.3.1 All Singlet-Triplet Excitations inthis set (S0-TN)
The RMSDs are below 0.2 eV for almost ev-ery tested method (see Fig. 9). SCS/SOS-B2PLYP21 and the new unscaled DHDFAs,ωB88PP86 and ωPBEPP86, are positioned asthe best three methods with RMSDs slightlyabove the chemical accuracy threshold. Thereare 13 methods with RMSDs below 0.2 eV, sixof which are SCS/SOS-LC-DHDFAs. Surpris-ingly, we find ωB2GP-PLYP’s SCS and SOSvariants among the worst-performing methodswith an RMSD of 0.22 eV, followed by its un-
0.00
0.10
0.20
0.30
0.40
SOS-B2GP-PLYP21
SOS-PBE-QIDH
SOS-
ωPBEPP86
SCS-B2GP-PLYP21
SOS-
ωB2GP-PLYP
SOS-RSX-QIDH
SCS-PBE-QIDH
SOS-
ωB88PP86
SCS-
ωB2GP-PLYP
B2GP-PLYP
SCS/SOS-B2PLYP21
B2PLYP
ωPBEPP86
SCS/SOS-
ωB2PLYP
SCS-
ωB88PP86
SCS-
ωPBEPP86
ωB88PP86
PBE-QIDH
ωB2GP-PLYP
SCS-RSX-QIDH
ωB2PLYP
RSX-QIDH
Statistical Value [eV]
MAD
RMSD
MD
0.20
0.20
0.20
0.20
0.21
0.21
0.21
0.22
0.23
0.23
0.23
0.23
0.23
0.24
0.24
0.24
0.25 0.28
0.28
0.30
0.30
0.36
(0.13)
(0.12)
(0.12)
(0.13)
(0.13)
(0.12)
(0.13)
(0.13)
(0.14)
(0.17)
(0.16)
(0.20)
(0.16)
(0.15)
(0.15)
(0.15)
(0.17)
(0.18)
(0.19)
(0.19)
(0.20)
(0.24)
Figure 10: Statistical values (in eV) for the S1-T1 energy split-ting on the Thiel set, namely RMSDs (values shown over each bar),MADs, and MDs. The TZVP AO basis set was used in all cases.The methods are sorted according to their overall RMSDs by con-sidering the singlet excited state of π – π∗ type (21A’ symmetry) inimidazole. RMSDs based on the n – π∗ transition in imidazole areshown in parentheses.
scaled parent ωB2GP-PLYP, RSX-QIDH, andB2PLYP (more details in Fig. 9).
6.3.2 S1-T1 Analysis
Note that Thiel and co-workers argued that thefirst singlet-singlet excited state in imidazolewas of n – π∗ type and has 11A” symmetry.27However, after closer inspection of their CC3reference data provided in the same work wefound that the first singlet excited state shouldbe of π – π∗ character and have 21A’ symmetry.In light of this finding, Fig. 10 includes bothanalyses with the values based on the n – π∗transition given in parentheses.The RMSDs for the S1-T1 splitting are higher
than in previous sets when the π – π∗ type sin-glet excited state for imidazole is taken intoaccount: now the RMSDs range from 0.20eV (SOS- and SCS-B2GP-PLYP21, SOS-PBE-QIDH, and SOS-ωPBEPP86) to 0.36 eV (RSX-QIDH). Nevertheless, most of the SCS or SOSvariants still display similar behavior as in pre-vious sets, with SCS-RSX-QIDH being the onlyexception with an RMSD of 0.30 eV. Similarly,
22

the unscaled PBE-QIDH (0.28 eV), ωB2GP-PLYP (0.28 eV), ωB2PLYP (0.30 eV), andRSX-QIDH are positioned last.If we now consider the n – π∗ type singlet
excited state in imidazole, the RMSDs (val-ues in parentheses in Fig. 10) are greatly im-proved by about 0.08 eV. In this picture, thebest six methods are all SOS variants withRMSDs of 0.12-0.13 eV. Next, we identify an-other six methods all of which are SCS variantswith RMSDs of up to 0.15 eV. ωPBEPP86 isthe first unscaled DHDFA and shares the sameRMSD as SCS/SOS-B2PLYP21 of 0.16 eV. Theunscaled DHDFAs ωB88PP86, B2GP-PLYP,PBE-QIDH, ωB2GP-PLYP, and SCS-RSX-QIDH are the last five methods with RMSDsbelow 0.2 eV. Lastly, B2PLYP, ωB2PLYP, andRSX-QIDH are positioned last.
7 Overall PerformanceIn the spirit of the GMTKN55 database forground state main-group thermochemistry, ki-netics, and noncovalent interactions8 we per-form two additional analyses that help iden-tify those methods that distinguish themselvesfrom others according to their overall perfor-mance and robustness. In order to achieve suchan analysis, we opted for two different strate-gies: (a) we order the TDA-DHDFAs by theirRMSDs for each set and then rank them accord-ing to how many times a certain method yieldsthe three best or three worst RMSDs and (b)we rank them by their averaged MAD or RMSD(MAD/RMSD) in the TDA-DHDFA frameworkaccording to
MAD/RMSD =∑16
i (MAD/RMSD)i16
, (17)
where the sum runs over each of the 16 in-dividual local-valence, Rydberg, and CT ex-citation subsets and (MAD/RMSD)i is theMAD/RMSD of the respective method for theith set. For those sets for which we broke downprevious analyses into overall, local-valence,and Rydberg excitations, we have included thestatistics of the last two cases to avoid any bias
0
1
2
3
4
5
6
7
8
9
10
11
12
13
# Best 3
Best 312
9
8
7 7
6 6
5 5 5 5 5 5
4 4
2 2 2
1 1 1
0
-20-19-18-17-16-15-14-13-12-11-10-9-8-7-6-5-4-3-2-10
SCS-PBE-QIDH
SOS-PBE-QIDH
SOS-
ωPBEPP86
SOS-B2GP-PLYP21
SOS-
ωB88PP86
SOS-
ωB2GP-PLYP
SOS-RSX-QIDH
SCS-B2GP-PLYP21
SCS-
ωB88PP86
SCS-
ωPBEPP86
ωB88PP86
ωPBEPP86
PBE-QIDH
SCS-
ωB2GP-PLYP
SCS/SOS-B2PLYP21
SCS-RSX-QIDH
B2GP-PLYP
ωB2GP-PLYP
SCS/SOS-
ωB2PLYP
ωB2PLYP
B2PLYP
RSX-QIDH
# Worst 3
Worst 3
0 0 0 0 0-1
-5
0 0 0 0 0
-5
-1
-5-4
-5-6
-3
-9
-11
-18
Figure 11: Analysis of how many times a method yields the threebest and three worst RMSDs in a benchmark (sub)set.
due to possible double counting.
7.1 Top-Three Best/Worst Anal-ysis
We begin with some general comments on howthis analysis was performed. In almost everyset we observed that multiple methods yieldedthe same RMSD. For instance, in the local-valence analysis of the ωB2(GP-)PLYP train-ing set, there are four methods that share thebest RMSD of 0.10 eV (SOS-ωB88PP86, SOS-PBE-QIDH, and ωB2GP-PLYP’s SCS and SOSvariants). Therefore, we assign the first positionto each of them. We repeat the same strategyalso for the second, third, and last three posi-tions for every set. The outcome of our analysisis shown in Fig. 11.PBE-QIDH’s SCS and SOS variants are some
of the most robust functionals in this work,where SCS-PBE-QIDH is on the top twelvetimes and SOS-PBE-QIDH nine times, respec-tively. However, we have to emphasize thatneither of these methods outperform TDA-LC-DHDFAs for CT transitions, and the differenceis even larger if we consider the TD-LC-DHDFAresults (see Section 5.6 and SI). Our new SOS-ωPBEPP86 is another well-performing func-
23

tional and is one of the top-three methods eighttimes. It also comes with the advantage of per-forming well regardless of the type of excita-tion, incl. CT ones. If the nature of the transi-tion is unknown, SOS-ωPBEPP86 is thereforerobust enough to give reliable results. SOS-ωB88PP86 and SOS-B2GP-PLYP21 can alsobe recommended, keeping in mind that thefirst outperforms the latter for CT transitions.SOS-ωB2GP-PLYP and SOS-RSX-QIDH arethe first two methods that are somewhat lessrobust given that they rank among the worstthree methods once and five times, respectively.Some other methods that we recommend areSCS-ωB88PP86, SCS-ωPBEPP86, SCS-B2GP-PLYP21, ωPBEPP86, and ωB88PP86. Eventhough they only rank in the top three fivetimes, they never appear among the worst.From the unscaled PBE-QIDH onward, themethods display less robust behavior and cannot be recommended for future applications tounexplored problems if a functional of generalrobustness is required.
7.2 Averaged MAD/RMSD Anal-ysis
In Fig. 12, we present the MAD and RMSDvalues for the present work. Their trends seemto be similar to the previous analysis, whereSCS and SOS-PBE-QIDH are on top of the listwith good results of just 0.16 eV for RMSD,followed by our new methods SOS-ωB88PP86(0.17 eV), SCS-ωB88PP86 (0.18 eV), SOS-ωPBEPP86 (0.18 eV), and SCS-ωPBEPP86(0.18 eV). Other methods that we also rec-ommend are SOS-B2GP-PLYP21, SCS andSOS-ωB2GP-PLYP. Nevertheless, SOS-B2GP-PLYP21 should be used with care due to its in-ability to describe CT transitions. On the otherhand, ωPBEPP86 and ωB88PP86 still showgood results despite being unscaled methodsand are the last two with RMSD values below0.2 eV. The remaining unscaled methods dis-play values above 0.2 eV, with RSX-QIDH andB2PLYP being the ones with the poorest resultsof 0.31 and 0.33 eV, respectively. Even thoughSCS and SOS-PBE-QIDH are at the top of thelist, our new SCS/SOS-LC-DHDFA methods
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
SOS-PBE-QIDH
SCS-PBE-QIDH
SOS-
ωB88PP86
SCS-
ωB88PP86
SOS-
ωPBEPP86
SOS-B2GP-PLYP21
SCS-
ωPBEPP86
SCS-B2GP-PLYP21
SCS-
ωB2GP-PLYP
SOS-
ωB2GP-PLYP
ωPBEPP86
ωB88PP86
SCS/SOS-
ωB2PLYP
PBE-QIDH
SOS-RSX-QIDH
SCS-RSX-QIDH
SCS/SOS-B2PLYP21
ωB2GP-PLYP
B2GP-PLYP
ωB2PLYP
RSX-QIDH
B2PLYP
Averaged MAD/RMSD [eV]
Averaged MADAveraged RMSD
0.16
0.16
0.17
0.18
0.18
0.18
0.18
0.18
0.18
0.18
0.19
0.19
0.20
0.20
0.20
0.21
0.22
0.22
0.23
0.23
0.31
0.33
(0.13)
(0.14)
(0.15)
(0.15)
(0.15)
(0.15)
(0.15)
(0.15)
(0.15)
(0.15)
(0.16)
(0.16)
(0.17)
(0.17)
(0.18)
(0.18)
(0.19)
(0.18)
(0.19)
(0.19)
(0.28)
(0.29)
Figure 12: MAD/RMSD analysis (in eV). The RMSDs are shownas outlines and the MADs as solid bars. We also present their valuesabove each bar, with the MAD in parentheses. The methods aresorted according to their RMSD.
have an increased RMSD of only 0.01 eV andthey can provide more reliable results for sys-tems that might have mixed valence-CT char-acter (see Fig. 12). Indeed, our SCS/SOS-LC-DHDFAs are more accurate not only for CTtransitions but also for the PAHs.
8 ConclusionsThis has been the second time that SCS andSOS variants have been introduced and as-sessed for double hybrid density functional ap-proximations (DHDFAs) for the computationof electronic excitation energies that follow theinitial definition by Grimme and Neese (seeEq.7). The main differences to the first workof this kind in 201739 are the incorporationof the latest LC-DHDFAs40,80,97 and the anal-ysis of singlet-triplet excitations. We remindthe reader that herein we already took into ac-count the best TD(A)-DFT methods and im-proving them further poses a major challenge.We compared unscaled DHDFAs with new SCSand SOS variants and tested those on six bench-mark sets that comprised 203 excitations insmall to medium-sized molecules and different
24

excitation types. While most of this work fo-cused mostly on the Tamm-Dancoff approxima-tion (TDA-DHDFAs), we also presented resultsbased on full TD-DHDFAs for the PAH and CTbenchmark sets.We confirmed the good accuracy and robust-
ness of DHDFAs for the computation of elec-tronic excitation energies. In general, theyshowed the same accuracy as previous stud-ies. The introduction of SCS and SOS caused asystematic red-shift in the excitation energies,contrary to the previous observations made bySchwabe and Goerigk in 2017.39 Sometimes,the red-shift eliminated extreme outliers in theexcitation energies, thus reducing error spansby up to 0.3 eV. The only exceptions to thisare the BLYP-based global DHDFAs B2PLYPand B2GP-PLYP, which displayed a blue-shiftin the excitation energies. One benefit of usingSCS and SOS is a more balanced descriptioncompared to the reference values, with MDsusually being closer to zero. Our SCS and SOSvariants also showed that it is not necessary toinclude a long-range correction in the perturba-tive part of a DHDFA to obtain reasonable re-sults. In fact, the three methods following thatapproach were outperformed by many of ournewly presented ones for singlet-singlet tran-sitions (no data on those competing methodswere available for triplet transitions).One important finding is that we did not see
any particular trend of when to prefer SCSover SOS or vice-versa. In fact, SCS andSOS variants present negligible differences inthe RMSDs of up to 0.03 eV when applied toDHDFAs. Note that this is contrary to someground state wave function methods where SOScan lead to inferior results compared to SCS,compromising accuracy and efficiency.7,10 Ac-curate SOS-DHDFAs provide faster calculationof larger systems and might also open a routeto more efficient methods for the optimizationof excited-state geometries once the Laplacetransform is applied, similar to its wave func-tion counterpart.While unscaled TD(A)-DHDFAs already
constitute an improvement over conventionalTD(A)-DFT methods, as conclusively shownin the literature,18,28,33–36,40,41 we recommend
specific new SCS- and SOS-based variants pre-sented in this work for further testing andsubsequent applications. Based on our av-eraged MAD/RMSD analysis, we highly rec-ommend our SCS and SOS variants of PBE-QIDH, ωB88PP86, and ωPBEPP86. That be-ing said, SCS- and SOS-PBE-QIDH are notsuitable for the computation of CT excitations,and the SCS/SOS-ωB88PP86 and SCS/SOS-ωPBEPP86 should be employed instead. In-deed, the latter four methods can provide accu-rate excitation energies regardless of the typeof transition, making them more robust for realapplications to new systems. Finally, if onlyunscaled DHDFAs are available to the user, westrongly recommend ωB88PP86 as the best-unscaled method, which outperformed someother SCS and SOS methods presented in thiswork.Our work showed once more that TD(A)-
DHDFAs have their place in the treatment ofaccurate electronic excitation energies. All ournewly presented methods will be available inthe upcoming release of ORCA5. Finally, wewould like to encourage the implementation ofTD(A)-DHDFAs with and without SCS/SOSinto other standard software packages.
Acknowledgement M.C.-P. acknowledges a‘Melbourne Research Scholarship’ by The Uni-versity of Melbourne. L. G. acknowledgesgenerous allocation of resources by the Na-tional Computational Infrastructure (NCI) Na-tional Facility within the National Computa-tional Merit Allocation Scheme (project ID:fk5) and Research Platform Services (ResPlat)at The University of Melbourne (Project No.punim0094). This research was also sup-ported by the sustaining and strengtheningmerit-based access to the NCI LIEF Grant(LE190100021) facilitated by The University ofMelbourne.
Supporting Information Avail-ableThe Supplementary Information includes allreferences values, excitation energies and sta-
25

tistical values for all tested cases and densityfunctional approximations in two seperate xlsxfiles. Those files are available free of charge.
References(1) Hohenberg, P.; Kohn, W. Inhomogeneous
electron gas. Phys. Rev. B 1964, 136,864–871.
(2) Kohn, W.; Sham, L. J. Self-consistentequations including exchange and cor-relation effects. Phys. Rev. 1965, 140,A1133–A1138.
(3) Runge, E.; Gross, E. K. U. Density-functional theory for time-dependent sys-tems. Phys. Rev. Lett. 1984, 52, 997–1000.
(4) Bauernschmitt, R.; Ahlrichs, R. Treat-ment of electronic excitations within theadiabatic approximation of time depen-dent density functional theory. Chem.Phys. Lett 1996, 256, 454–464.
(5) Grimme, S. Semiempirical hybrid den-sity functional with perturbative second-order correlation. J. Chem. Phys. 2006,124, 034108.
(6) Goerigk, L.; Grimme, S. Efficient andaccurate double-hybrid-meta-GGA den-sity functionals-evaluation with the ex-tended GMTKN30 database for generalmain group thermochemistry, kinetics,and noncovalent interactions. J. Chem.Theory Comput. 2011, 7, 291–309.
(7) Goerigk, L.; Grimme, S. A thoroughbenchmark of density functional meth-ods for general main group thermochem-istry, kinetics, and noncovalent interac-tions. Phys. Chem. Chem. Phys. 2011,13, 6670–6688.
(8) Goerigk, L.; Hansen, A.; Bauer, C.;Ehrlich, S.; Najibi, A.; Grimme, S. Alook at the density functional theory zoowith the advanced GMTKN55 database
for general main group thermochem-istry, kinetics and noncovalent interac-tions. Phys. Chem. Chem. Phys. 2017,19, 32184–32215.
(9) Mardirossian, N.; Head-Gordon, M. Sur-vival of the most transferable at thetop of jacob’s ladder: defining and test-ing the ωB97M(2) double hybrid densityfunctional. J. Chem. Phys. 2018, 148,241736.
(10) Mehta, N.; Casanova-Páez, M.; Go-erigk, L. Semi-empirical or non-empiricaldouble-hybrid density functionals: whichare more robust? Phys. Chem. Chem.Phys. 2018, 20, 23175–23194.
(11) Kozuch, S.; Gruzman, D.; Martin, J.M. L. DSD-BLYP: A general purposedouble hybrid density functional includ-ing spin component scaling and disper-sion correction. J. Phys. Chem. C 2010,114, 20801–20808.
(12) Kozuch, S.; Martin, J. M. L. DSD-PBEP86: in search of the best double-hybrid DFT with spin-component scaledMP2 and dispersion corrections. Phys.Chem. Chem. Phys. 2011, 13, 20104–20107.
(13) Kozuch, S.; Martin, J. M. L. Spin-component-scaled double hybrids: anextensive search for the best fifth-rungfunctionals blending DFT and perturba-tion theory. J. Comput. Chem. 2013, 34,2327–2344.
(14) Santra, G.; Sylvetsky, N.; Martin, J.M. L. Minimally empirical double-hybrid functionals trained against theGMTKN55 database: revDSD-PBEP86-d4, revDOD-PBE-d4, and DOD-SCAN-d4. J. Phys. Chem. C 2019, 123, 5129–5143.
(15) Martin, J. M. L.; Santra, G. Empiricaldouble-hybrid density functional theory:A ‘third way’ in between WFT and DFT.Isr. J. Chem. 2019, 60, 787–804.
26

(16) Santra, G.; Cho, M.; Martin, J. M. L.Exploring avenues beyond revised DSDfunctionals: I. range separation, withxDSD as a special case. J. Phys.Chem. A 2021, published online, DOI:10.1021/acs.jpca.1c01294 (accessed on 30May 2021).
(17) Santra, G.; Semidalas, E.; Martin, J.M. L. Exploring avenues beyond revisedDSD functionals: II. random-phase ap-proximation and scaled MP3 corrections.J. Phys. Chem. A 2021, published on-line, DOI: 10.1021/acs.jpca.1c01294 (ac-cessed on 30 May 2021).
(18) Grimme, S.; Neese, F. Double-hybriddensity functional theory for excited elec-tronic states of molecules. J. Chem.Phys. 2007, 127, 154116.
(19) Head-Gordon, M.; Rico, R. J.; Oumi, M.;Lee, T. J. A doubles correction to elec-tronic excited states from configurationinteraction in the space of single substi-tutions. Chemical Physics Letters 1994,219, 21–29.
(20) Hirata, S.; Head-Gordon, M. Time-dependent density functional theorywithin the Tamm-Dancoff approxima-tion. Chem. Phys. Lett. 1999, 314, 291–299.
(21) Benighaus, T.; DiStasio, R. A.;Lochan, R. C.; Chai, J.-D.; Head-Gordon, M. Semiempirical double-hybriddensity functional with improved de-scription of long-range correlation. J.Phys. Chem. A 2008, 112, 2702–2712.
(22) Zhang, I. Y.; Xu, X. Reaching a uniformaccuracy for complex molecular systems:long-range-corrected XYG3 doubly hy-brid density functional. J. Phys. Chem.Lett. 2013, 4, 1669–1675.
(23) Cornaton, Y.; Fromager, E. Doublehybrid density-functional theory usingthe coulomb-attenuating method. Int. J.Quantum Chem. 2014, 114, 1199–1211.
(24) Hedegård, E. D.; Heiden, F.; Knecht, S.;Fromager, E.; Jensen, H. J. A. As-sessment of charge-transfer excitationswith time-dependent, range-separateddensity functional theory based on long-range MP2 and multiconfigurational self-consistent field wave functions. J. Chem.Phys. 2013, 139, 184308.
(25) Kalai, C.; Toulouse, J. A generalrange-separated double-hybrid density-functional theory. J. Chem. Phys. 2018,148, 164105.
(26) Karton, A.; Tarnopolsky, A.; Lamère, J.-F.; Schatz, G. C.; Martin, J. M. L. Highlyaccurate first-principles benchmark datasets for the parametrization and valida-tion of density functional and other ap-proximate methods. derivation of a ro-bust, generally applicable, double-hybridfunctional for thermochemistry and ther-mochemical kinetics. J. Phys. Chem. A2008, 112, 12868–12886.
(27) Schreiber, M.; Silva-Junior, M. R.;Sauer, S. P. A.; Thiel, W. Benchmarks forelectronically excited states: CASPT2,CC2, CCSD, and CC3. J. Chem. Phys.2008, 128, 134110.
(28) Goerigk, L.; Moellmann, J.; Grimme, S.Computation of accurate excitation en-ergies for large organic molecules withdouble-hybrid density functionals. Phys.Chem. Chem. Phys. 2009, 11, 4611–4620.
(29) Brémond, É.; Adamo, C. Seeking forparameter-free double-hybrid function-als: the PBE0-DH model. J. Chem.Phys. 2011, 135, 024106.
(30) Chai, J.-D.; Mao, S.-P. Seeking for re-liable double-hybrid density functionalswithout fitting parameters: the PBE0-2functional. Chem. Phys. Lett. 2012, 538,121–125.
(31) Goerigk, L.; Grimme, S. Double-hybriddensity functionals. Wiley Interdiscip.
27

Rev.: Comput. Mol. Sci. 2014, 4, 576–600.
(32) Najibi, A.; Casanova-Páez, M.; Go-erigk, L. Analysis of recent BLYP-and PBE-based range-separated double-hybrid density functional approximationsfor main-group thermochemistry, kinet-ics, and noncovalent interactions. J.Phys. Chem. A 2021, 125, 4026–4035.
(33) Goerigk, L.; Grimme, S. Calculationof electronic circular dichroism spectrawith time-dependent double-hybrid den-sity functional theory. J. Phys. Chem. A2009, 113, 767–776.
(34) Goerigk, L.; Grimme, S. Assessment ofTD-DFT methods and of various spinscaled CIS(D) and CC2 versions for thetreatment of low-lying valence excita-tions of large organic dyes. J. Chem.Phys. 2010, 132, 184103.
(35) Goerigk, L.; Grimme, S. Double-hybriddensity functionals provide a balanceddescription of excited 1La and 1Lb statesin polycyclic aromatic hydrocarbons. J.Chem. Theory Comput. 2011, 7, 3272–3277.
(36) Goerigk, L.; Kruse, H.; Grimme, S.Comprehensive chiroptical spectroscopy ;Wiley-Blackwell, 2012; pp 643–673.
(37) Meo, F. D.; Trouillas, P.; Adamo, C.;Sancho-García, J. C. Application of re-cent double-hybrid density functionalsto low-lying singlet-singlet excitation en-ergies of large organic compounds. J.Chem. Phys. 2013, 139, 164104.
(38) Momeni, M. R.; Brown, A. A local CC2and TDA-DFT double hybrid study onBODIPY/aza-BODIPY dimers as heavyatom free triplet photosensitizers for pho-todynamic therapy applications. J. Phys.Chem. A 2016, 120, 2550–2560.
(39) Schwabe, T.; Goerigk, L. Time-dependent double-hybrid densityfunctionals with spin-component and
spin-opposite scaling. J. Chem. TheoryComput. 2017, 13, 4307–4323.
(40) Casanova-Páez, M.; Dardis, M. B.; Go-erigk, L. ωB2PLYP and ωB2GPPLYP:the first two double-hybrid density func-tionals with long-range correction opti-mized for excitation energies. J. Chem.Theory Comput. 2019, 15, 4735–4744.
(41) Casanova-Páez, M.; Goerigk, L. Assess-ing the Tamm–Dancoff approximation,singlet–singlet, and singlet–triplet exci-tations with the latest long-range cor-rected double-hybrid density functionals.J. Chem. Phys. 2020, 153, 064106.
(42) Goerigk, L.; Casanova-Páez, M. The tripto the density functional theory zoo con-tinues: making a case for time-dependentdouble hybrids for excited-state prob-lems. Aust. J. Chem. 2021, 74, 3–15.
(43) Casanova-Páez, M.; Goerigk, L. Globaldouble hybrids do not work for chargetransfer: A comment on “double hybridsand time-dependent density functionaltheory: an implementation and bench-mark on charge transfer excited states”.J. Comput. Chem. 2021, 42, 528–533.
(44) Christiansen, O.; Koch, H.; Jørgensen, P.The second-order approximate coupledcluster singles and doubles model CC2.Chem. Phys. Lett. 1995, 243, 409–418.
(45) Hellweg, A.; Grün, S. A.; Hättig, C.Benchmarking the performance ofspin-component scaled CC2 in groundand electronically excited states. Phys.Chem. Chem. Phys. 2008, 10, 4119.
(46) Goerigk, L.; Grimme, S. Quantumchemical investigation of exciton cou-pling: super-molecular calculationsof a merocyanine dimer aggregate.ChemPhysChem 2008, 9, 2467–2470.
(47) Grimme, S. Improved second-ordermøller-plesset perturbation theoryby separate scaling of parallel- and
28

antiparallel-spin pair correlation en-ergies. J. Chem. Phys. 2003, 118,9095–9102.
(48) Jung, Y.; Lochan, R. C.; Dutoi, A. D.;Head-Gordon, M. Scaled opposite-spinsecond order Møller–Plesset correlationenergy: an economical electronic struc-ture method. J. Chem. Phys. 2004, 121,9793–9802.
(49) Grimme, S.; Goerigk, L.; Fink, R. F.Spin-component-scaled electron correla-tion methods. Wiley Interdiscip. Rev.:Comput. Mol. Sci. 2012, 2, 886–906.
(50) Chai, J.-D.; Head-Gordon, M. Long-range corrected double-hybrid densityfunctionals. J. Chem. Phys. 2009, 131,174105.
(51) Brémond, E.; Savarese, M.; Sancho-García, J. C.; Pérez-Jiménez, A. J.;Adamo, C. Quadratic integrand double-hybrid made spin-component-scaled. J.Chem. Phys. 2016, 144, 124104.
(52) Almlöf, J. Elimination of energy denomi-nators in møller-plesset perturbation the-ory by a laplace transform approach.Chem. Phys. Lett. 1991, 181, 319–320.
(53) Tozer, D. J.; Amos, R. D.; Handy, N. C.;Roos, B. O.; Serrano-Andrés, L. Doesdensity functional theory contribute tothe understanding of excited states ofunsaturated organic compounds? Mol.Phys. 1999, 97, 859–868.
(54) Tozer, D. J. Relationship betweenlong-range charge-transfer excitation en-ergy error and integer discontinuity inKohn–Sham theory. J. Chem. Phys.2003, 119, 12697–12699.
(55) Dreuw, A.; Head-Gordon, M. Failureof time-dependent density functionaltheory for long-range charge-transferexcited states:the zincbacteriochlorin-bacteriochlorin and bacteriochlorophyll-spheroidene complexes. J. Am. Chem.Soc. 2004, 126, 4007–4016.
(56) Becke, A. D. Density-functionalexchange-energy approximation withcorrect asymptotic behavior. Phys. Rev.A 1988, 38, 3098–3100.
(57) Lee, C.; Yang, W.; Parr, R. G. Devel-opment of the colle-salvetti correlation-energy formula into a functional of theelectron density. Phys. Rev. B 1988, 37,785–789.
(58) Pishchalnikov, R. Y.; Yaroshevich, I. A.;Slastnikova, T. A.; Ashikhmin, A. A.;Stepanov, A. V.; Slutskaya, E. A.;Friedrich, T.; Sluchanko, N. N.; Mak-simov, E. G. Structural peculiarities ofketo-carotenoids in water-soluble pro-teins revealed by simulation of linearabsorption. Phys. Chem. Chem. Phys.2019, 21, 25707–25719.
(59) Pollice, R.; Friederich, P.; Lavigne, C.;dos Passos Gomes, G.; Aspuru-Guzik, A.Organic molecules with inverted gaps be-tween first excited singlet and tripletstates and appreciable fluorescence rates.Matter 2021, 4, 1654–1682.
(60) Suleiman, O.; Panthi, D.; Adeyiga, O.;Odoh, S. O. Methane C-H activationby [Cu2O]2+ and [Cu3O3]2+ in copper-exchanged zeolites: computational anal-ysis of redox chemistry and X-ray absorp-tion spectroscopy. Inorg. Chem. 2021,60, 6218–6227.
(61) Gao, Z.; Chen, S.; Bai, Y.; Wang, M.;Liu, X.; Yang, W.; Li, W.; Ding, X.;Yao, J. A new perspective for evaluat-ing the photoelectric performance of or-ganic–inorganic hybrid perovskites basedon the DFT calculations of excited states.Phys. Chem. Chem. Phys. 2021, 23,11548–11556.
(62) Yaroshevich, I. A. et al. Role of hydro-gen bond alternation and charge trans-fer states in photoactivation of the Or-ange Carotenoid Protein. Commun. Biol.2021, 4, 539.
29

(63) Puget, M.; Shcherbakov, V.;Denisov, S.; Moreau, P.; Dognon, J.-P.;Mostafavi, M.; Le Caër, S. Reactionmechanisms of the degradation of flu-oroethylene carbonate, an additive oflithium-ion batteries, unraveled by ra-diation chemistry. Chem. Eur. J. 2021,n/a, n/a.
(64) Curtis, K.; Panthi, D.; Odoh, S. O.Time-dependent density functional the-ory study of copper(II) oxo active sitesfor methane-to-methanol conversion inzeolites. Inorg. Chem. 2021, 60, 1149–1159.
(65) Brémond, É.; Sancho-García, J. C.;Pérez-Jiménez, Á. J.; Adamo, C. Com-munication: double-hybrid functionalsfrom adiabatic-connection: the QIDHmodel. J. Chem. Phys. 2014, 141,031101.
(66) Vintonyak, V. V.; Warburg, K.;Kruse, H.; Grimme, S.; Hübel, K.;Rauh, D.; Waldmann, H. Identificationof thiazolidinones spiro-fused to indolin-2-ones as potent and selective inhibitorsof the mycobacterium tuberculosisprotein tyrosine phosphatase B. Angew.Chem. Int. Ed. 2010, 49, 5902–5905.
(67) Send, R.; Valsson, O.; Filippi, C. Elec-tronic excitations of simple cyanine dyes:reconciling density functional and wavefunction methods. J. Chem. TheoryComput. 2011, 7, 444–455.
(68) Markworth, P. B.; Adamson, B. D.;Coughlan, N. J. A.; Goerigk, L.;Bieske, E. J. Photoisomerization actionspectroscopy: flicking the protonatedmerocyanine-spiropyran switch in the gasphase. Phys. Chem. Chem. Phys. 2015,17, 25676–25688.
(69) Prlj, A.; Sandoval-Salinas, M. E.;Casanova, D.; Jacquemin, D.; Cormin-boeuf, C. Low-lying ππ* states of het-eroaromatic molecules: A challenge for
excited state methods. J. Chem. TheoryComput. 2016, 12, 2652–2660.
(70) Alipour, M. On the performance of time-dependent double-hybrid density func-tionals for description of absorption andemission spectra of heteroaromatic com-pounds. Theor Chem Acc 2016, 135, 1–8.
(71) Sancho-García, J. C.; Adamo, C.; Pérez-Jiménez, A. J. Describing excited statesof [n]cycloparaphenylenes by hybrid anddouble-hybrid density functionals: fromisolated to weakly interacting molecules.Theo. Chem. Acc. 2016, 135, 25.
(72) Brémond, É.; Savarese, M.; Pérez-Jiménez, Á. J.; Sancho-García, J. C.;Adamo, C. Speed-up of the excited-state benchmarking: double-hybrid den-sity functionals as test cases. J. Chem.Theory Comput. 2017, 13, 5539–5551.
(73) Ottochian, A.; Morgillo, C.; Ciofini, I.;Frisch, M. J.; Scalmani, G.; Adamo, C.Double hybrids and time-dependent den-sity functional theory: an implementa-tion and benchmark on charge transferexcited states. J. Comput. Chem. 2020,41, 1242–1251.
(74) Hernández-Martínez, L.; Brémond, E.;Pérez-Jiménez, A. J.; San-Fabián, E.;Adamo, C.; Sancho-García, J. C. Nonem-pirical (double-hybrid) density function-als applied to atomic excitation energies:A systematic basis set investigation. Int.J. Quantum Chem. 2020, 120, e26193.
(75) Brémond, É.; Ottochian, A.; Pérez-Jiménez, Á. J.; Ciofini, I.; Scalmani, G.;Frisch, M. J.; Sancho-García, J. C.;Adamo, C. Assessing challenging intra-and inter-molecular charge-transfer ex-citations energies with double-hybriddensity functionals. J. Comput. Chem.2021, 42, 970–981.
(76) Sears, J. S.; Koerzdoerfer, T.; Zhang, C.-R.; Brédas, J.-L. Communication: or-bital instabilities and triplet states from
30

time-dependent density functional the-ory and long-range corrected functionals.J. Chem. Phys. 2011, 135, 151103.
(77) Peach, M. J. G.; Tozer, D. J. Overcominglow orbital overlap and triplet instabilityproblems in TDDFT. J. Phys. Chem. A2012, 116, 9783–9789.
(78) Peach, M. J.; Warner, N.; David J. Tozer,On the triplet instability in TDDFT.Mol. Phys. 2013, 111, 1271–1274.
(79) Rangel, T.; Hamed, S. M.; Bruneval, F.;Neaton, J. B. An assessment of low-lyingexcitation energies and triplet instabil-ities of organic molecules with an abinitio bethe-salpeter equation approachand the Tamm-Dancoff approximation.J. Chem. Phys. 2017, 146, 194108.
(80) Mester, D.; Kállay, M. A simple range-separated double-hybrid density func-tional theory for excited states. J. Chem.Theory Comput. 2021, 17, 927–942.
(81) Marques, M.; Gross, E. Time-dependentdensity functional theory. Annu. Rev.Phys. Chem. 2004, 55, 427–455.
(82) Marques, M. A. L.; Ullrich, C. A.;Nogueira, F.; Rubio, A.; Burke, K.;Gross, E. K. U. Time-dependent den-sity functional theory ; Lecture notes inphysics; Springer-Verlag, 2006.
(83) Mark E. Casida, In Recent ad-vances in density functional methods ;Chong, D. P., Ed.; World ScientificSingapore, 1995; pp 155–192.
(84) Gross, E. K. U.; Kohn, W. Time-dependent density-functional theory. Ad-van. Quantum Chem. 1990, 21, 255–291.
(85) McLachlan, A. D.; Ball, M. A. Time-dependent Hartree—Fock theory formolecules. Rev. Mod. Phys. 1964, 36,844–855.
(86) Iikura, H.; Tsuneda, T.; Yanai, T.; Hi-rao, K. A long-range correction scheme
for generalized-gradient-approximationexchange functionals. J. Chem. Phys.2001, 115, 3540–3544.
(87) Møller, C.; Plesset, M. S. Note on an ap-proximation treatment for many-electronsystems. Phys. Rev. 1934, 46, 618–622.
(88) Sharkas, K.; Toulouse, J.; Savin, A.Double-hybrid density-functional theorymade rigorous. J. Chem. Phys. 2011,134, 064113.
(89) Grimme, S.; Izgorodina, E. I. Calcula-tion of 0–0 excitation energies of organicmolecules by CIS(D) quantum chemicalmethods. Chem. Phys. 2004, 305, 223–230.
(90) Rhee, Y. M.; Head-Gordon, M. Scaledsecond-order perturbation corrections toconfiguration interaction singles: effi-cient and reliable excitation energy meth-ods. J. Phys. Chem. A 2007, 111, 5314–5326.
(91) Neese, F. The ORCA program system.Wiley Interdiscip. Rev.: Comput. Mol.Sci. 2012, 2, 73–78.
(92) Neese, F. Software update: the ORCAprogram system, version 4.0. Wiley In-terdiscip. Rev.: Comput. Mol. Sci. 2018,8, e1327.
(93) Valeev, E. F. Libint: A library forthe evaluation of molecular integrals ofmany-body operators over gaussian func-tions. 2020,
(94) Vahtras, O.; Almlöf, J.; Feyereisen, M.Integral approximations for LCAO-SCFcalculations. Chem. Phys. Lett. 1993,213, 514 – 518.
(95) Weigend, F.; Köhn, A.; Hättig, C. Effi-cient use of the correlation consistent ba-sis sets in resolution of the identity MP2calculations. J. Chem. Phys 2002, 116,3175–3183.
31

(96) Chai, J.-D.; Head-Gordon, M. System-atic optimization of long-range correctedhybrid density functionals. J. Chem.Phys. 2008, 128, 084106.
(97) Brémond, É.; Savarese, M.; Pérez-Jiménez, Á. J.; Sancho-García, J. C.;Adamo, C. Range-separated double-hybrid functional from nonempirical con-straints. J. Chem. Theory Comput.2018, 14, 4052–4062.
(98) Leang, S. S.; Zahariev, F.; Gordon, M. S.Benchmarking the performance of time-dependent density functional methods. J.Chem. Phys. 2012, 136, 104101.
(99) Loos, P.-F.; Galland, N.; Jacquemin, D.Theoretical 0–0 energies with chemicalaccuracy. J. Phys. Chem. Lett. 2018, 9,4646–4651.
(100) Grimme, S. In Reviews in computationalchemistry ; Lipkowitz, K. B., Boyd, D. B.,Eds.; Wiley-VCH: New York, 2004; Vol.20, pp. 153-218.
(101) Perdew, J. P. Density-functional approx-imation for the correlation energy of theinhomogeneous electron gas. Phys. Rev.B 1986, 33, 8822–8824.
(102) Vosko, S. H.; Wilk, L.; Nusair, M. Accu-rate spin-dependent electron liquid cor-relation energies for local spin densitycalculations: a critical analysis. Can. J.Phys. 1980, 58, 1200–1211.
(103) Koch, H.; Christiansen, O.; Jør-gensen, P.; Sanchez de Merás, A. M.;Helgaker, T. The CC3 model: an itera-tive coupled cluster approach includingconnected triples. J. Chem. Phys 1997,106, 1808–1818.
(104) Dunning, T. H. Gaussian basis sets foruse in correlated molecular calculations.I. the atoms boron through neon and hy-drogen. J. Chem. Phys. 1989, 90, 1007–1023.
(105) Kendall, R. A.; Dunning, T. H.; Har-rison, R. J. Electron affinities of thefirst-row atoms revisited. systematic ba-sis sets and wave functions. J. Chem.Phys. 1992, 96, 6796–6806.
(106) Brémond, É.; Pérez-Jiménez, Á. J.;Sancho-García, J. C.; Adamo, C. Range-separated hybrid density functionalsmade simple. J. Chem. Phys. 2019, 150,201102.
(107) Paci, I.; Johnson, J. C.; Chen, X.;Rana, G.; Popović, D.; David, D. E.;Nozik, A. J.; Ratner, M. A.; Michl, J.Singlet fission for dye-sensitized solarcells: can a suitable sensitizer be found?J. Am. Chem. Soc. 2006, 128, 16546–16553.
(108) Wen, J.; Havlas, Z.; Michl, J. Captoda-tively stabilized biradicaloids as chro-mophores for singlet fission. J. Am.Chem. Soc. 2015, 137, 165–172.
(109) Grotjahn, R.; Maier, T. M.; Michl, J.;Kaupp, M. Development of a TDDFT-based protocol with local hybrid func-tionals for the screening of potential sin-glet fission chromophores. J. Chem. The-ory Comput. 2017, 13, 4984–4996.
(110) Masoomi-Godarzi, S.; Liu, M.;Tachibana, Y.; Goerigk, L.; Ghig-gino, K. P.; Smith, T. A.; Jones, D. J.Solution-processable, solid state donor-acceptor materials for singlet fission.Adv. Energy Mater. 2018, 8, 1801720.
(111) Masoomi-Godarzi, S.; Liu, M.;Tachibana, Y.; Mitchell, V. D.; Go-erigk, L.; Ghiggino, K. P.; Smith, T. A.;Jones, D. J. Liquid crystallinity as aself-assembly motif for high-efficiency,solution-processed, solid-state singletfission materials. Adv. Energy Mater.2019, 9, 1901069.
(112) Christiansen, O.; Koch, H.; Jørgensen, P.Perturbative triple excitation corrections
32

to coupled cluster singles and doubles ex-citation energies. J. Chem. Phys. 1996,105, 1451–1459.
(113) Loos, P.-F.; Scemama, A.; Blon-del, A.; Garniron, Y.; Caffarel, M.;Jacquemin, D. A mountaineering strat-egy to excited states: highly accuratereference energies and benchmarks. J.Chem. Theory Comput. 2018, 14, 4360–4379.
(114) Loos, P.-F.; Scemama, A.; Boggio-Pasqua, M.; Jacquemin, D. Mountaineer-ing strategy to excited states: highly ac-curate energies and benchmarks for ex-otic molecules and radicals. J. Chem.Theory Comput. 2020, 16, 3720–3736.
(115) Schäfer, A.; Horn, H.; Ahlrichs, R. Fullyoptimized contracted gaussian basis setsfor atoms Li to Kr. J. Chem. Phys. 1992,97, 2571–2577.
(116) Angeli, C.; Pastore, M. The lowest singletstates of octatetraene revisited. J. Chem.Phys. 2011, 134, 184302.
(117) Platt, J. R. Classification of spectra ofcata-condensed hydrocarbons. J. Chem.Phys. 1949, 17, 484–495.
(118) Lopata, K.; Reslan, R.; Kowalska, M.;Neuhauser, D.; Govind, N.; Kowal-ski, K. Excited-state studies of poly-acenes: A comparative picture usingEOMCCSD, CR-EOMCCSD(T), range-separated (LR/RT)-TDDFT, TD-PM3,and TD-ZINDO. J. Chem. Theory Com-put. 2011, 7, 3686–3693.
(119) Grimme, S.; Parac, M. Substantial er-rors from time-dependent density func-tional theory for the calculation ofexcited states of large π systems.ChemPhysChem 2003, 4, 292–295.
(120) Parac, M.; Grimme, S. A TDDFT studyof the lowest excitation energies of poly-cyclic aromatic hydrocarbons. Chem.Phys. 2003, 292, 11 – 21.
(121) Marian, C. M.; Gilka, N. Perfor-mance of the density functional The-ory/Multireference configuration interac-tion method on electronic excitation ofextended π-systems. J. Chem. TheoryComput. 2008, 4, 1501–1515.
(122) Richard, R. M.; Herbert, J. M. Time-dependent density-functional descriptionof the 1La state in polycyclic aromatichydrocarbons: charge-transfer characterin disguise? J. Chem. Theory Comput.2011, 7, 1296–1306.
(123) Kowalski, K.; Piecuch, P. New coupled-cluster methods with singles, doubles,and noniterative triples for high accuracycalculations of excited electronic states.J. Chem. Phys. 2004, 120, 1715–1738.
(124) Jacquemin, D.; Duchemin, I.; Blase, X.Is the Bethe-Salpeter formalism accuratefor excitation energies? Comparisonswith TD-DFT, CASPT2, and EOM-CCSD. J. Phys. Chem. Lett. 2017, 8,1524–1529.
(125) Serrano-Andrés, L.; Fülscher, M. P. The-oretical study of the electronic spec-troscopy of peptides. III. charge-transfertransitions in polypeptides. J. Am.Chem. Soc. 1998, 120, 10912–10920.
(126) Peach, M. J. G.; Benfield, P.; Hel-gaker, T.; Tozer, D. J. Excitation ener-gies in density functional theory: an eval-uation and a diagnostic test. J. Chem.Phys 2008, 128, 044118.
(127) Weigend, F.; Ahlrichs, R. Balanced ba-sis sets of split valence, triple zeta va-lence and quadruple zeta valence qualityfor h to rn: design and assessment of ac-curacy. Phys. Chem. Chem. Phys. 2005,7, 3297–3305.
33

Graphical TOC Entry
34





























