Embed Size (px)
Citation preview

Journal of PathologyJ Pathol 2003; 201: 319–327.Published online 7 July 2003 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/path.1434
Original Paper
Tight junctional abnormality in multiple sclerosis whitematter affects all calibres of vessel and is associated withblood–brain barrier leakage and active demyelination
John Kirk,1 Jonnie Plumb,2 Meenakshi Mirakhur2 and Stephen McQuaid2*1School of Medicine Inflammation Research Centre, Queen’s University of Belfast, Northern Ireland, UK2Neuropathology Laboratory, Royal Group of Hospitals Trust, Belfast, Northern Ireland, UK
*Correspondence to:Dr Stephen McQuaid,Neuropathology and MolecularPathology Laboratories, RGHT,Institute of Pathology, GrosvenorRoad, Belfast BT12 6BL,Northern Ireland, UK.E-mail: [email protected]
Received: 25 September 2002Revised: 24 February 2003Accepted: 16 April 2003
AbstractBlood–brain barrier (BBB) hyperpermeability in multiple sclerosis (MS) is associated withlesion pathogenesis and has been linked to pathology in microvascular tight junctions (TJs).This study quantifies the uneven distribution of TJ pathology and its association with BBBleakage. Frozen sections from plaque and normal-appearing white matter (NAWM) in 14cases were studied together with white matter from six neurological and five normal controls.Using single and double immunofluorescence and confocal microscopy, the TJ-associatedprotein zonula occludens-1 (ZO-1) was examined across lesion types and tissue categories,and in relation to fibrinogen leakage. Confocal image data sets were analysed for 2198 MSand 1062 control vessels. Significant differences in the incidence of TJ abnormalities weredetected between the different lesion types in MS and between MS and control white matter.These were frequent in oil-red O (ORO)+ active plaques, affecting 42% of vessel segments,but less frequent in ORO− inactive plaques (23%), NAWM (13%), and normal (3.7%)and neurological controls (8%). A similar pattern was found irrespective of the vessel size,supporting a causal role for diffusible inflammatory mediators. In both NAWM and inactivelesions, dual labelling showed that vessels with the most TJ abnormality also showed mostfibrinogen leakage. This was even more pronounced in active lesions, where 41% of vessels inthe highest grade for TJ alteration showed severe leakage. It is concluded that disruption ofTJs in MS, affecting both paracellular and transcellular paths, contributes to BBB leakage.TJ abnormality and BBB leakage in inactive lesions suggests either failure of TJ repairor a continuing pathological process. In NAWM, it suggests either pre-lesional changeor secondary damage. Clinically inapparent TJ pathology has prognostic implications andshould be considered when planning disease-modifying therapy. Copyright 2003 JohnWiley & Sons, Ltd.
Keywords: multiple sclerosis; blood–brain barrier; contrast-enhancing lesions; tight junc-tions; ZO-1; cytokines
Introduction
The morphological correlate of the blood–brain bar-rier (BBB) in the mammalian central nervous system(CNS) is the system of belt-like ‘tight’ junctions (TJs,zonulae occludentes) found between adjacent endothe-lial cells in the lining of the vascular walls [1]. Becauseof their continuous circumferential disposition andtheir molecular architecture, they, like their ubiqui-tous epithelial counterparts [2], act both as barriers toocclude the paracellular cleft between adjacent cellsand as fences within the plane of the plasma mem-brane, to maintain apico-dorsolateral polarity [3]. Theselectively permeable BBB is rapidly responsive tophysiological and pathological stimuli and plays a keyrole in maintaining the distinctive CNS metabolic andimmunoregulatory homeostasis [1,4–6].
As knowledge of the molecular structure andcomposition of TJs improves and technology to visual-ize component molecules in their pathological context
has evolved [2], it has become apparent that TJs arepathologically affected in a wide range of diseasesincluding neoplasia, asthma, Crohn’s disease, AIDS,stroke, bacterial infection, and pre-eclampsia [1,5,6].In the CNS, for example, abnormal endothelial TJshave been detected in situations where BBB leakageoccurs, including cerebral malaria, AIDS encephalitis,and multiple sclerosis (MS) [7–10].
MS is an immune-mediated CNS disorder that isclinically and pathologically heterogeneous [11,12]. Inthe most common form, the scattered lesions are cen-tred on blood vessels, which at an early stage becomeinflamed and leaky [13]. Clinicopathological studieshave demonstrated accompanying oedema, demyelina-tion, axonal loss, and gliosis [12,14]. Overt BBB leak-age is transient, but may recur at the same or differentlocations within the neuraxis after an interval of weeks,months or years [15]. The subsequent evolution andgrowth of lesions is irregular and unpredictable, butinvolves further phases of inflammation and leakage,
Copyright 2003 John Wiley & Sons, Ltd.

320 J Kirk et al
immunologically mediated demyelination, and, to avariable degree, of axonal transection [12,16].
We recently described a spectrum of altered TJprotein (ZO-1 and occludin) expression in CNSvessels in MS. Abnormalities occurred not onlyin active demyelinating lesions, but also in thenormal-appearing white matter (NAWM) [10]. Dualimmunofluorescence revealed the coincidence in somevessels of abnormal TJs with a pre-mortem patternof perivascular serum protein leakage. We proposedthat TJ abnormality, which leads to BBB leakage, isa significant form of tissue injury in MS, alongsidedemyelination and axonopathy [10].
β-Interferon and methyl prednisolone, two therapeu-tic agents widely used for the treatment of MS, restorethe BBB, though their use has not been optimized forthis purpose [17,18]. The effective use of these andother agents to restore or stabilize the BBB in MS willdepend on a clear understanding of the extent, ampli-tude, and timescale of TJ pathology and endogenousrepair.
In this study, we have therefore assessed the rela-tionship between the nature and extent of TJ changesand the stage of development of the MS lesion. Inaddition, in order to inform the debate on pathologicalmechanisms, we studied the distribution of abnormal-ity in relation to vessel size and type. Using optimizedimmunofluorescent and confocal microscopic methods[10], we compared the incidence of TJ abnormalitiesin endothelial cells across a range of MS and controlCNS tissue categories, comprising active lesions (iecurrently or recently demyelinating), inactive (oftenchronic) lesions, NAWM, neurological control whitematter, and normal control white matter. In a relatedstudy, MS tissue blocks were dual labelled for TJ pro-teins and fibrinogen to investigate the previously notedassociation between TJ abnormality and BBB leak-age.
Materials and methods
Tissues and histology
Oil-red O (ORO) screening of snap-frozen tissue from21 clinically and neuropathologically confirmed casesof MS revealed 14 cases with sufficient blocks of opti-mum histological quality for the study. These yielded33 blocks containing active (currently or recentlydemyelinating) plaques and 18 blocks containing inac-tive plaques [19–21]. NAWM blocks (16) came fromregions of deep white matter more than 1 cm fromvisible lesions. Demographic and clinicopathologicaldetails relating to MS and control white matter sam-ples are summarized in Tables 1 and 2. Tissue sam-ples were obtained with consent for their researchuse, either from the MS Society National Brian Tis-sue Bank (registered Charity No 207495) or from theBelfast MS Donor Tissue Bank in the Department ofNeuropathology, Royal Group of Hospitals Trust.
Cryostat sections (12 µm) were cut onto amino-propyltriethoxysilane (APES)-coated glass slides andstained with haematoxylin and eosin (H&E)/ORO forlesion grading. For immunocytochemistry, all blockswere fixed in ice-cold acetone for 10 min and air-dried. Sections were labelled by indirect immunofluo-rescence for HLA-Dr, to assess microglial activation.
Immunofluorescence
Indirect immunofluorescence was carried out usingthe following antibodies and dilutions: monoclonalantibodies to HLA-Dr (Novocastra, 1 : 100) and ZO-1 (Zymed, 1 : 200) and polyclonal antibodies to ZO-1 (Zymed, 1 : 400) and glial fibrillary acidic protein(GFAP; Dako, 1 : 200). Biotinylated Ulex europaeusagglutinin (UEA; Vector, 1 : 500) was used as a markerfor endothelium. Serum protein leakage was assessedusing an antibody to fibrinogen conjugated to FITC(Dako, 1 : 100).
Table 1. Summary of the demographic data on the 14 multiple sclerosis, six other neurological disease control, and five normalcontrol cases used in this study
DiseaseNo ofcases
Mean age(range), years
Diseaseduration
Genderratio (M : F)
Death–autopsyinterval, hours
Blocksstudied
MS 14 46 (22–69) 5 months–46 years 6 : 8 1.5–29 67ONDC 6 32 (16—74) NA 5 : 1 6–10 23NC 5 57 (24–92) NA 2 : 3 4–26 26
MS = multiple sclerosis; ONDC = other neurological disease control; NC = normal control; NA = not applicable.
Table 2. Clinicopathological details of the 14 multiple sclerosis, six other neurological disease control, and five normal controlcases used in this study
MS (No of cases) ONDC (No of cases) NC (No of cases)
SPMS (12) SSPE (3) Road traffic accident (2)Acute MS (2) Head injury (1) Lung carcinoma (1)
Transverse myelitis (1) Cardiac failure (1)Metastatic bronchial carcinoma (1) Congestive heart failure (1)
MS = multiple sclerosis; SPMS = chronic secondary progressive phase of relapsing remitting MS; ONDC = other neurological disease control;SSPE = subacute sclerosing pan-encephalitis; NC = normal control.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.
TJ abnormality in multiple sclerosis and its association with BBB leakage 321
For single-label immunofluorescence, primary anti-bodies, biotinylated UEA (b-UEA), and fibrinogen-FITC were incubated and detected as described previ-ously [10]. For analysis of the relationship between TJabnormality, endothelial integrity, and fibrinogen leak-age in individual vessels [19] and of fibrinogen in rela-tion to astrocytes, sequential detection dual-labellingimmunofluorescence was carried out with polyclonalantibodies to ZO-1 or GFAP and fibrinogen-FITC orb-UEA. The polyclonal antibody was incubated for1 h at 37 ◦C, washed in PBS, and detected by incubat-ing in goat anti-rabbit Alexa 568 (Molecular Probes,1 : 500) for 1 h at 37 ◦C. Sections were incubated infibrinogen-FITC or b-UEA for 1 h at 37 ◦C and thenwashed in PBS. ZO-1/b-UEA dual-labelled sectionswere then incubated for 1 h in Z-avidin FITC (1 : 100,Zymed). All sections were mounted in citifluor forexamination under a ×40 oil-immersion objective ona Leica TCS/NT confocal microscope equipped with akrypton–argon laser. Vessel images were acquired aspreviously described [10].
Vessel measurement and semi-quantitative gradingsystems
Vessel diameters and TJ abnormalities were assessedon preparations that, in addition to the ZO-1 staining,were counterstained with propidium iodide for nuclei.This helped to outline the sometimes irregular course
of vessels and facilitated the positioning of the limitsof measuring lines. Moreover, vessels were renderedmore visible in confocal images by the tendency ofZO-1 staining to impart a faint background within theendothelial cytoplasm, in addition to the sharp junc-tional marking. Measurement to the level of accuracyrequired to assign vessels to one of four size categorieswas possible, even where abnormal ZO-1 staining wasapparent, for the latter only rarely involved the wholeof a linear segment or the entire circumference of atransversely sectioned vessel.
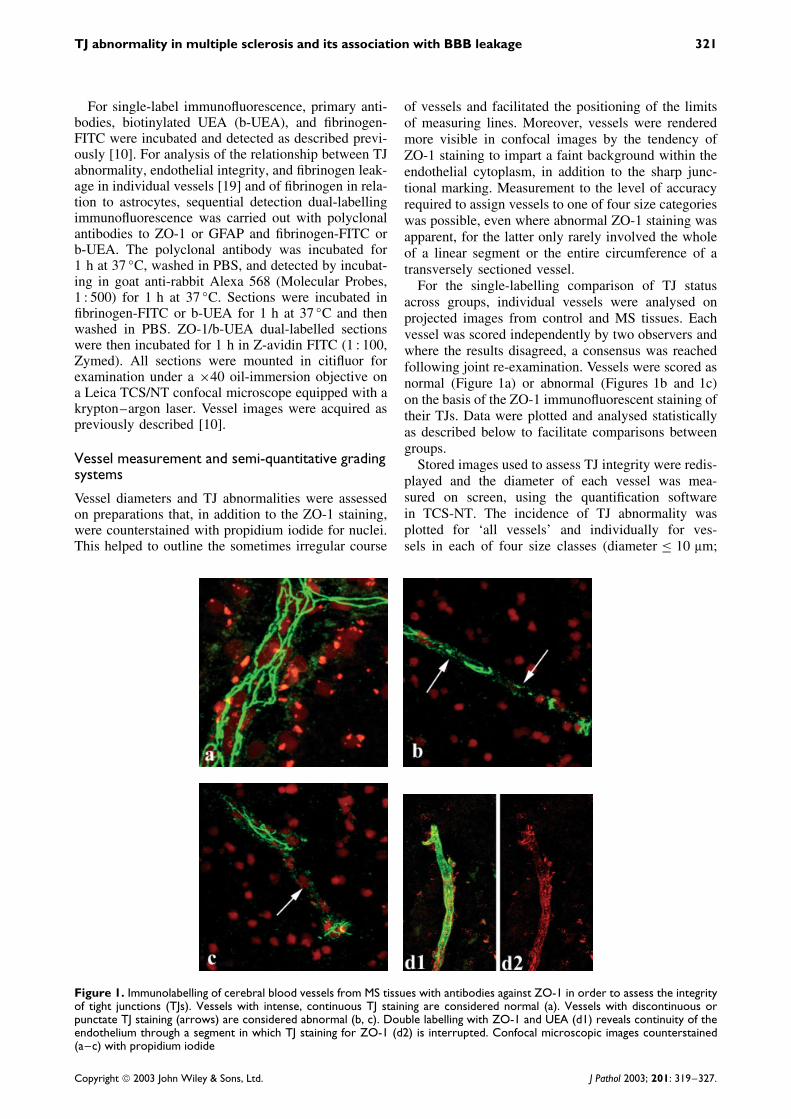
For the single-labelling comparison of TJ statusacross groups, individual vessels were analysed onprojected images from control and MS tissues. Eachvessel was scored independently by two observers andwhere the results disagreed, a consensus was reachedfollowing joint re-examination. Vessels were scored asnormal (Figure 1a) or abnormal (Figures 1b and 1c)on the basis of the ZO-1 immunofluorescent staining oftheir TJs. Data were plotted and analysed statisticallyas described below to facilitate comparisons betweengroups.
Stored images used to assess TJ integrity were redis-played and the diameter of each vessel was mea-sured on screen, using the quantification softwarein TCS-NT. The incidence of TJ abnormality wasplotted for ‘all vessels’ and individually for ves-sels in each of four size classes (diameter ≤ 10 µm;
Figure 1. Immunolabelling of cerebral blood vessels from MS tissues with antibodies against ZO-1 in order to assess the integrityof tight junctions (TJs). Vessels with intense, continuous TJ staining are considered normal (a). Vessels with discontinuous orpunctate TJ staining (arrows) are considered abnormal (b, c). Double labelling with ZO-1 and UEA (d1) reveals continuity of theendothelium through a segment in which TJ staining for ZO-1 (d2) is interrupted. Confocal microscopic images counterstained(a–c) with propidium iodide
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.
322 J Kirk et al
>10–30 µm; >30–50 µm; and >50 µm). The possi-bility of recording incomplete or undulating vessels asabnormal was minimized, because a ‘gap’ in normalZO-1 staining was only recorded as an abnormalitywhen there was additional evidence in the image forthe continuity of the vessel, as, for example, providedby the presence of PI-stained nuclei, or of punctate ordiffuse ZO-1 remnants.
In the dual-labelling study, scoring systems withthree grades were used for each variable. Thus, TJabnormalities were graded on the following scale:0 = no clear abnormality; 1 = up to 25% of theimaged vessel wall shows abnormalities in TJ proteinexpression; 2 = more than 25% of the vessel wallcontains abnormal TJs. Similarly, fibrinogen leakagewas graded as 0 = no leakage, 1 = moderate leakage,or 2 = widespread, severe leakage. A total of 292vessels was analysed from 19 active MS lesions, 243vessels from 14 areas of NAWM, and 291 vessels from17 inactive lesions. The numbers of individual vesselswith scores ranging from 0/0 (no TJ abnormality/nofibrinogen leakage), 0/1, etc to 2/2 were collated. Thepercentage of blood vessels in each leakage grade wasthen plotted against the grade of TJ abnormality foreach tissue type.
Statistical analysis
The incidence of TJ abnormality in the five tissuecategories was compared using the non-parametricKruskal–Wallis test for group differences. A pvalue below 0.05 was considered significant. WhereKruskal–Wallis was significant, Dunn’s test wassubsequently used to establish the direction andsignificance of the differences in TJ incidence betweentissue categories, using the recommended experimentalerror rate, α = 0.15 [22]. All analyses were performedusing the SPSS software package (version 11.0, SPSSInc, Chicago, IL, USA).
Results
Tissue characterization
Procedures for tissue characterization and neuropatho-logical grading have been described previously [10]. Inthe present investigation, all 18 chronic plaque blocksand all 16 NAWM blocks were ORO-negative, indicat-ing absence of active demyelination. Twelve of the 33active blocks contained scattered ORO-positive cells.A further 12 had moderate numbers of ORO-positivecells, usually aggregated in small clusters, while in theremaining nine, they were abundant, disposed in largeswathes throughout the section.
Group comparison of TJ abnormalities in MS andcontrol tissues
The data for this analysis were derived from a studyof 622 blood vessels from 33 active MS lesions, 904
vessels from 16 areas of NAWM, 672 vessels from 18chronic lesions, and 1062 vessels from 49 areas fromcontrol cases. There was no evidence of any majordamage or disruption to the endothelial lining of thesevessels, as indicated by the uniform and continuousexpression of endothelial markers (UEA, Factor VIII)(Figures 1d1 and 1d2).
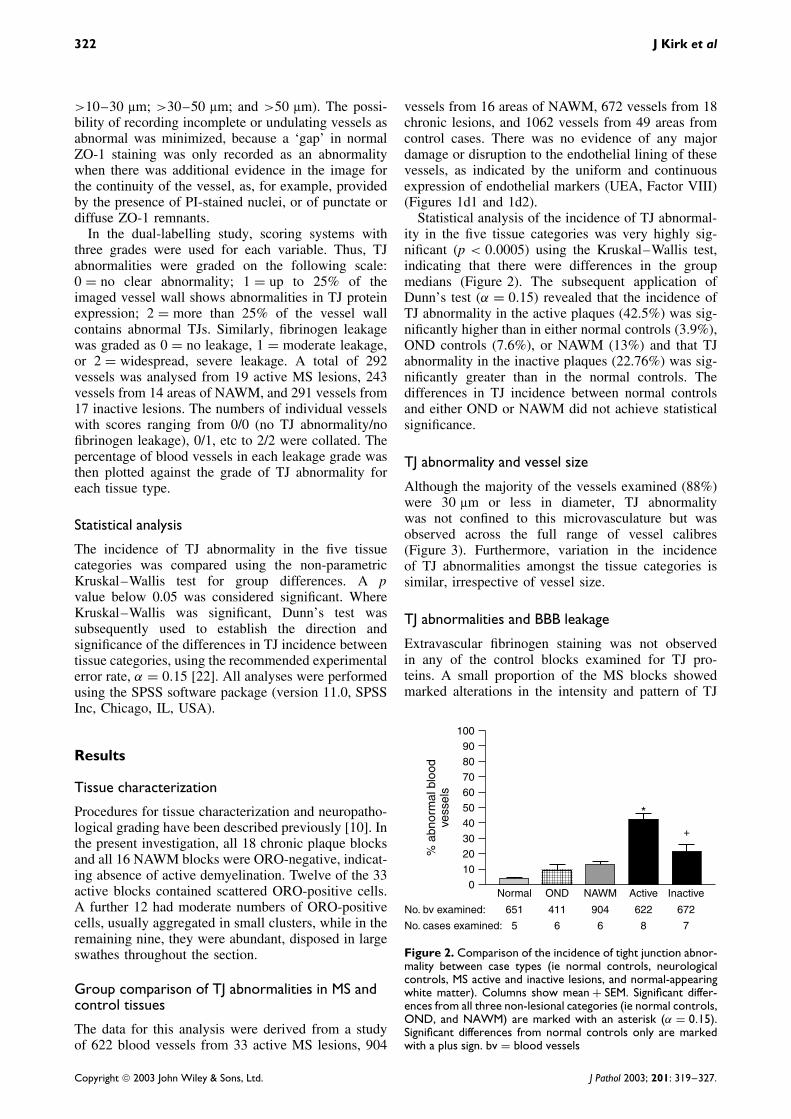
Statistical analysis of the incidence of TJ abnormal-ity in the five tissue categories was very highly sig-nificant (p < 0.0005) using the Kruskal–Wallis test,indicating that there were differences in the groupmedians (Figure 2). The subsequent application ofDunn’s test (α = 0.15) revealed that the incidence ofTJ abnormality in the active plaques (42.5%) was sig-nificantly higher than in either normal controls (3.9%),OND controls (7.6%), or NAWM (13%) and that TJabnormality in the inactive plaques (22.76%) was sig-nificantly greater than in the normal controls. Thedifferences in TJ incidence between normal controlsand either OND or NAWM did not achieve statisticalsignificance.
TJ abnormality and vessel size
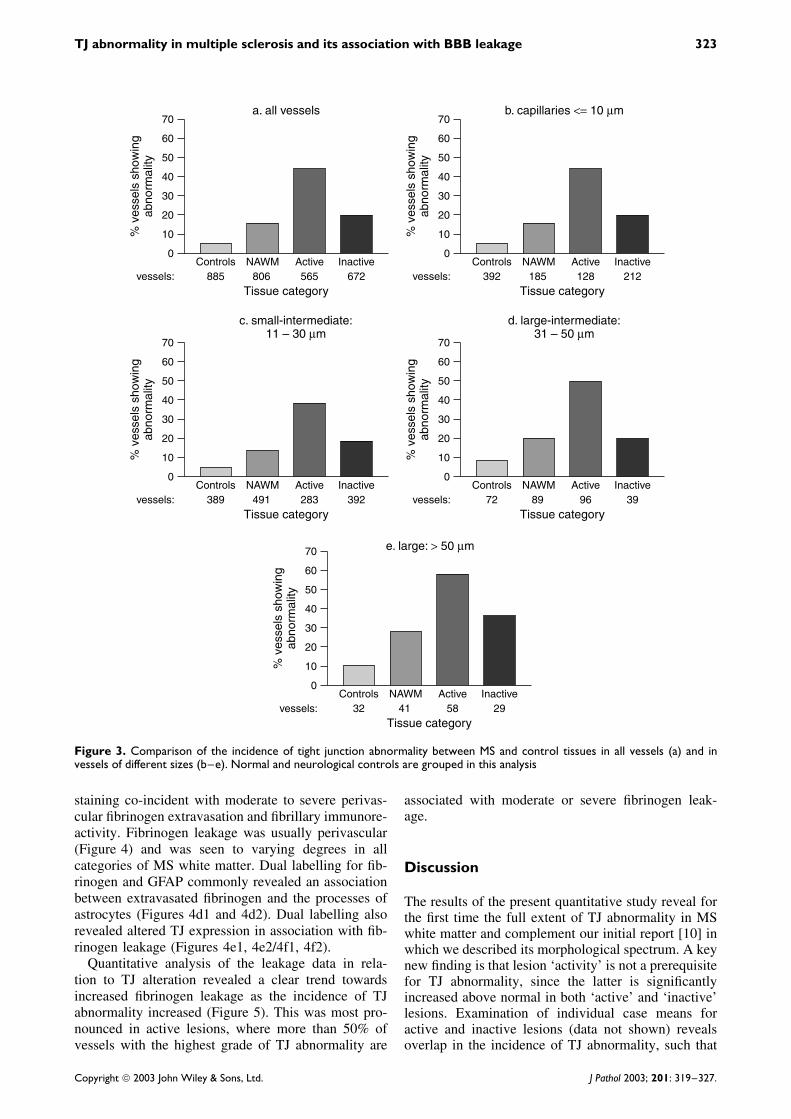
Although the majority of the vessels examined (88%)were 30 µm or less in diameter, TJ abnormalitywas not confined to this microvasculature but wasobserved across the full range of vessel calibres(Figure 3). Furthermore, variation in the incidenceof TJ abnormalities amongst the tissue categories issimilar, irrespective of vessel size.
TJ abnormalities and BBB leakage
Extravascular fibrinogen staining was not observedin any of the control blocks examined for TJ pro-teins. A small proportion of the MS blocks showedmarked alterations in the intensity and pattern of TJ
100
90
80
70
60
50
40
30
20
10
0
% a
bnor
mal
blo
odve
ssel
s
No. bv examined:
No. cases examined:
Normal
651 411 904 622 672
5 6 6
*
+
8 7
OND NAWM Active Inactive
Figure 2. Comparison of the incidence of tight junction abnor-mality between case types (ie normal controls, neurologicalcontrols, MS active and inactive lesions, and normal-appearingwhite matter). Columns show mean + SEM. Significant differ-ences from all three non-lesional categories (ie normal controls,OND, and NAWM) are marked with an asterisk (α = 0.15).Significant differences from normal controls only are markedwith a plus sign. bv = blood vessels
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.
TJ abnormality in multiple sclerosis and its association with BBB leakage 323
0
10
20
30
40
% v
esse
ls s
how
ing
abno
rmal
ity 50
60
70
Controls NAWM Active Inactive885vessels: 565
Tissue category
a. all vessels
806 672
b. capillaries <= 10 µm
0
10
20
30
40
% v
esse
ls s
how
ing
abno
rmal
ity 50
60
70
Controls NAWM Active Inactive392vessels: 128
Tissue category185 212
d. large-intermediate:31 – 50 µm
0
10
20
30
40%
ves
sels
sho
win
gab
norm
ality 50
60
70
Controls NAWM Active Inactive72vessels: 96
Tissue category89 39
c. small-intermediate:11 – 30 µm
0
10
20
30
40
% v
esse
ls s
how
ing
abno
rmal
ity 50
60
70
Controls NAWM Active Inactive389vessels: 283
Tissue category491 392
e. large: > 50 µm
0
10
20
30
40
% v
esse
ls s
how
ing
abno
rmal
ity 50
60
70
Controls NAWM Active Inactive32vessels: 58
Tissue category41 29
Figure 3. Comparison of the incidence of tight junction abnormality between MS and control tissues in all vessels (a) and invessels of different sizes (b–e). Normal and neurological controls are grouped in this analysis
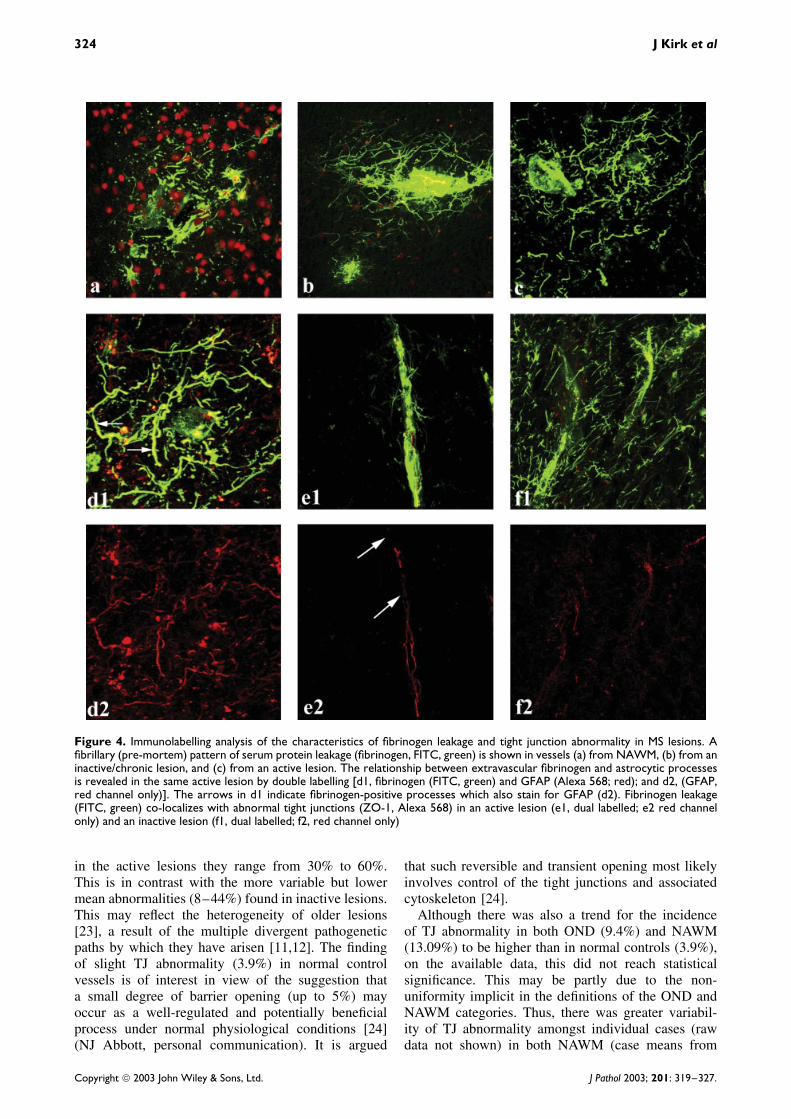
staining co-incident with moderate to severe perivas-cular fibrinogen extravasation and fibrillary immunore-activity. Fibrinogen leakage was usually perivascular(Figure 4) and was seen to varying degrees in allcategories of MS white matter. Dual labelling for fib-rinogen and GFAP commonly revealed an associationbetween extravasated fibrinogen and the processes ofastrocytes (Figures 4d1 and 4d2). Dual labelling alsorevealed altered TJ expression in association with fib-rinogen leakage (Figures 4e1, 4e2/4f1, 4f2).
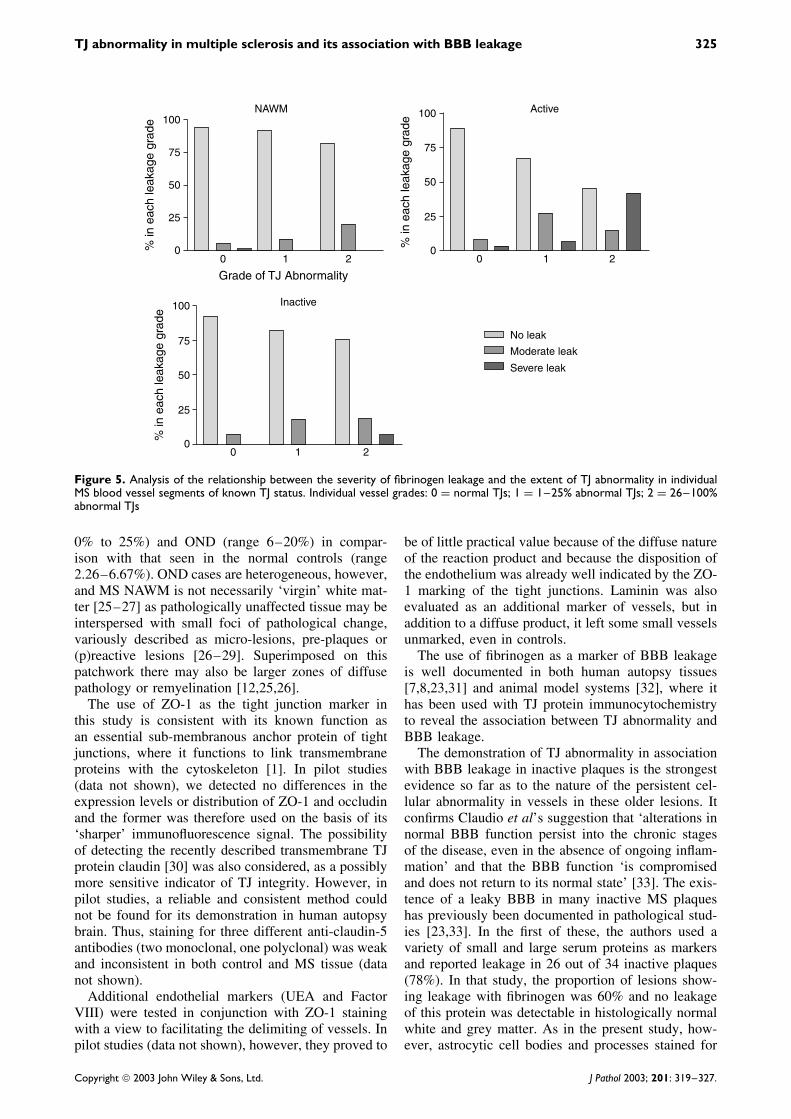
Quantitative analysis of the leakage data in rela-tion to TJ alteration revealed a clear trend towardsincreased fibrinogen leakage as the incidence of TJabnormality increased (Figure 5). This was most pro-nounced in active lesions, where more than 50% ofvessels with the highest grade of TJ abnormality are
associated with moderate or severe fibrinogen leak-age.
Discussion
The results of the present quantitative study reveal forthe first time the full extent of TJ abnormality in MSwhite matter and complement our initial report [10] inwhich we described its morphological spectrum. A keynew finding is that lesion ‘activity’ is not a prerequisitefor TJ abnormality, since the latter is significantlyincreased above normal in both ‘active’ and ‘inactive’lesions. Examination of individual case means foractive and inactive lesions (data not shown) revealsoverlap in the incidence of TJ abnormality, such that
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.
324 J Kirk et al
Figure 4. Immunolabelling analysis of the characteristics of fibrinogen leakage and tight junction abnormality in MS lesions. Afibrillary (pre-mortem) pattern of serum protein leakage (fibrinogen, FITC, green) is shown in vessels (a) from NAWM, (b) from aninactive/chronic lesion, and (c) from an active lesion. The relationship between extravascular fibrinogen and astrocytic processesis revealed in the same active lesion by double labelling [d1, fibrinogen (FITC, green) and GFAP (Alexa 568; red); and d2, (GFAP,red channel only)]. The arrows in d1 indicate fibrinogen-positive processes which also stain for GFAP (d2). Fibrinogen leakage(FITC, green) co-localizes with abnormal tight junctions (ZO-1, Alexa 568) in an active lesion (e1, dual labelled; e2 red channelonly) and an inactive lesion (f1, dual labelled; f2, red channel only)
in the active lesions they range from 30% to 60%.This is in contrast with the more variable but lowermean abnormalities (8–44%) found in inactive lesions.This may reflect the heterogeneity of older lesions[23], a result of the multiple divergent pathogeneticpaths by which they have arisen [11,12]. The findingof slight TJ abnormality (3.9%) in normal controlvessels is of interest in view of the suggestion thata small degree of barrier opening (up to 5%) mayoccur as a well-regulated and potentially beneficialprocess under normal physiological conditions [24](NJ Abbott, personal communication). It is argued
that such reversible and transient opening most likelyinvolves control of the tight junctions and associatedcytoskeleton [24].
Although there was also a trend for the incidenceof TJ abnormality in both OND (9.4%) and NAWM(13.09%) to be higher than in normal controls (3.9%),on the available data, this did not reach statisticalsignificance. This may be partly due to the non-uniformity implicit in the definitions of the OND andNAWM categories. Thus, there was greater variabil-ity of TJ abnormality amongst individual cases (rawdata not shown) in both NAWM (case means from
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.
TJ abnormality in multiple sclerosis and its association with BBB leakage 325
100
00 1
Grade of TJ Abnormality
NAWM
2
% in
eac
h le
akag
e gr
ade
25
50
75
No leak
Moderate leak
Severe leak
100
00 1
Active
2
% in
eac
h le
akag
e gr
ade
25
50
75
100
00 1
Inactive
2
% in
eac
h le
akag
e gr
ade
25
50
75
Figure 5. Analysis of the relationship between the severity of fibrinogen leakage and the extent of TJ abnormality in individualMS blood vessel segments of known TJ status. Individual vessel grades: 0 = normal TJs; 1 = 1–25% abnormal TJs; 2 = 26–100%abnormal TJs
0% to 25%) and OND (range 6–20%) in compar-ison with that seen in the normal controls (range2.26–6.67%). OND cases are heterogeneous, however,and MS NAWM is not necessarily ‘virgin’ white mat-ter [25–27] as pathologically unaffected tissue may beinterspersed with small foci of pathological change,variously described as micro-lesions, pre-plaques or(p)reactive lesions [26–29]. Superimposed on thispatchwork there may also be larger zones of diffusepathology or remyelination [12,25,26].
The use of ZO-1 as the tight junction marker inthis study is consistent with its known function asan essential sub-membranous anchor protein of tightjunctions, where it functions to link transmembraneproteins with the cytoskeleton [1]. In pilot studies(data not shown), we detected no differences in theexpression levels or distribution of ZO-1 and occludinand the former was therefore used on the basis of its‘sharper’ immunofluorescence signal. The possibilityof detecting the recently described transmembrane TJprotein claudin [30] was also considered, as a possiblymore sensitive indicator of TJ integrity. However, inpilot studies, a reliable and consistent method couldnot be found for its demonstration in human autopsybrain. Thus, staining for three different anti-claudin-5antibodies (two monoclonal, one polyclonal) was weakand inconsistent in both control and MS tissue (datanot shown).
Additional endothelial markers (UEA and FactorVIII) were tested in conjunction with ZO-1 stainingwith a view to facilitating the delimiting of vessels. Inpilot studies (data not shown), however, they proved to
be of little practical value because of the diffuse natureof the reaction product and because the disposition ofthe endothelium was already well indicated by the ZO-1 marking of the tight junctions. Laminin was alsoevaluated as an additional marker of vessels, but inaddition to a diffuse product, it left some small vesselsunmarked, even in controls.
The use of fibrinogen as a marker of BBB leakageis well documented in both human autopsy tissues[7,8,23,31] and animal model systems [32], where ithas been used with TJ protein immunocytochemistryto reveal the association between TJ abnormality andBBB leakage.
The demonstration of TJ abnormality in associationwith BBB leakage in inactive plaques is the strongestevidence so far as to the nature of the persistent cel-lular abnormality in vessels in these older lesions. Itconfirms Claudio et al’s suggestion that ‘alterations innormal BBB function persist into the chronic stagesof the disease, even in the absence of ongoing inflam-mation’ and that the BBB function ‘is compromisedand does not return to its normal state’ [33]. The exis-tence of a leaky BBB in many inactive MS plaqueshas previously been documented in pathological stud-ies [23,33]. In the first of these, the authors used avariety of small and large serum proteins as markersand reported leakage in 26 out of 34 inactive plaques(78%). In that study, the proportion of lesions show-ing leakage with fibrinogen was 60% and no leakageof this protein was detectable in histologically normalwhite and grey matter. As in the present study, how-ever, astrocytic cell bodies and processes stained for
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.

326 J Kirk et al
fibrinogen. Claudio et al’s later ultrastructural studyfound abnormalities in the endothelium and in the vas-cular basal lamina, consistent with the existence ofprofound changes in the physiology and barrier prop-erties [33]. Our findings suggest that such changesmay be a consequence of persistent TJ abnormalities,which undermine the normal endothelial polarity andfunctional integrity. Paradoxically, this may result inup-regulation of the transcellular vesicular route, aswell as the paracellular one [34].
The current confocal microscopic study confirmsthat there is a high incidence of TJ abnormality inactively demyelinating plaques, a lesion category inwhich previous studies have demonstrated importantaspects of early lesion development and expansion[12,19]. The demonstration of TJ abnormality in ves-sels of all sizes may point to the nature of causalmechanisms. If they had resulted solely from directlocal effects arising from the diapedesis of infiltrat-ing inflammatory cells [9], post-capillary venules andveins would have been expected to bear the bruntof pathological change. The involvement of all ves-sels suggests, instead, that TJ alteration is at leastpartly mediated ‘from a distance’ by cytokines orother mediators, associated with inflammatory infil-tration or immunological up-regulation [35,36]. Thisaccords with an earlier study in which vessel walldamage was reported in acute MS plaques and wasassociated with intramural deposition of complementon smooth muscle components and with an infiltra-tion of HLA-DR-positive macrophages [37]. In MSlesions, characterized by inflammation and demyeli-nation, in situ hybridization studies with digoxigenin-labelled oligonucleotide probes have shown that IL-6,IFN-γ , and TNF-α are the predominating cytokines inthe perivascular inflammatory cuffs [38]. In primarycultures of microvascular endothelial cells, both TNF-α and interferon-γ produce a striking fragmentationof ZO-1 via F-actin rearrangement [39]. Matrix met-alloproteinases (MMPs) have also been implicated inthe breakdown of the BBB in MS [40] and MMP9is thought to be an intermediate in TNF-α-inducedopening of the BBB [41].
In conclusion, our results indicate that TJ abnormal-ity is widespread in MS white matter, affecting bothlesions and NAWM, but is most common in activelesions. The involvement of vessels irrespective of sizesuggests the involvement of soluble factors in the ini-tiation of TJ damage [36,38–40]. The association ofBBB leakage with TJ alteration is most marked invessels with the highest incidence of TJ abnormality,suggesting a possible causal link, where TJ abnormal-ity is most severe. The fact that some vessels withabnormal junctions do not visibly leak suggests eithera lack of sensitivity in our detection protocol or, morelikely, that there may be a threshold of TJ injury thatmust be reached before significant BBB leakage of thelarge fibrinogen protein (molecular weight 340 kD)occurs. In vitro experiments and in vivo MRI studiessuggest that TJ pathology is responsive to current MS
therapeutic agents [17,18]. In the case of short-termor pulsed glucocorticosteroids, the effect is transient,as indicated by changes in the number of Gd-DTPA-enhancing lesions on MRI imaging [17]. IFN-β coun-teracts the inflammatory mediator-induced effects onbrain endothelial cell tight junctions in vitro [18] andhas a clear barrier-tightening effect both in vitro [18]and in vivo [42], but the contribution of these mech-anisms to the clinical benefit shown in IFN-β-treatedpatients remains to be demonstrated. A means of pre-venting the BBB damage associated with acute inflam-matory lesions is offered by a new anti-leukocyte traf-ficking drug, the α4 integrin antagonist Natalizumab(Antegren, Elan Pharmaceuticals and Biogen), whichhas recently completed phase II clinical trials for use inrelapsing MS [43]. Thus, it has been shown to reducethe formation of new Gd-DTPA-enhancing inflamma-tory brain lesions by approximately 90% over a 6-month period, though it has no detectable effect onexisting lesions. This effect fits in with the first part ofwhat Compston has identified as the therapeutic chal-lenge in MS, ie ‘to contain the disease process usingtherapy that reaches all potentially affected parts andthen to promote repair’ [44]. The widespread vascu-lar pathology described here, including a componentin inactive lesions [23,33] which may not be readilyapparent clinically or on routine Gd-enhanced imag-ing, has clear implications for prognosis and should betaken into account in the planning of a second stageof therapy, aimed at repairing established lesions andthereby preventing disease progression.
Acknowledgements
We thank Mr Gordon McGregor for his excellent technicalcontribution and Dr Gordan Cran for statistical advice. JP was arecipient of the Galen Research Fellowship from the Irish BrainResearch Trust. We thank the Neuropathology Department,Royal Hospitals Trust, Belfast and the UK MS Tissue Bankfunded by the MS Society of Great Britain and NorthernIreland, registered charity 207495, for the supply of tissuesamples. We are grateful to Dr Stanley Hawkins for his critiqueof our preliminary work.
References
1. Huber JD, Egleton RD, Davis TP. Molecular physiology andpathophysiology of tight junctions in the blood–brain barrier.Trends Neurosci 2001; 24: 719–725.
2. Fanning AS, Mitic LL, Anderson JM. Transmembrane proteins inthe tight junction barrier. J Am Soc Nephrol 1999; 10: 1337–1345.
3. Tsukita S, Furuse M, Itoh M. Structural and signaling moleculescome together at tight junctions. Curr Opin Cell Biol 1999; 11:628–633.
4. Cserr HF, Knopf PM. Cervical lymphatics, the blood–brain barrierand immunoreactivity of the brain. In Immunology of the NervousSystem, Keane RW, Hickey WF (eds). Oxford University Press:New York, 1997; 134–152.
5. Banks WA. Physiology and pathology of the blood–brain barrier:implications for microbial pathogenesis, drug delivery andneurodegenerative disorders. J Neurovirol 1999; 5: 538–555.
6. Tomkins O, Kaufer D, Korn A, et al. Frequent blood–brain barrierdisruption in the human cerebral cortex. Cell Mol Neurobiol 2001;21: 675–691.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.

TJ abnormality in multiple sclerosis and its association with BBB leakage 327
7. Dallasta LM, Pisarov LA, Esplen JE, et al. Blood–brain barriertight junction disruption in human immunodeficiency virus-1encephalitis. Am J Pathol 1999; 155: 1915–1927.
8. Brown H, Hien TT, Day N, et al. Evidence of blood–brainbarrier dysfunction in human cerebral malaria. Neuropathol ApplNeurobiol 1999; 25: 331–340.
9. Boven LA, Middel J, Verhoef J, et al. Monocyte infiltrationis highly associated with loss of the tight junction proteinzonula occludens in HIV-1-associated dementia. Neuropathol ApplNeurobiol 2000; 26: 3356–3360.
10. Plumb J, McQuaid S, Mirakhur M, Kirk J. Abnormal endothelialtight junctions in active lesions and normal-appearing white matterin multiple sclerosis. Brain Pathol 2002; 12: 154–169.
11. Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiplesclerosis: implications for diagnosis and therapy. Trends Mol Med2001; 7: 115–121.
12. Wingerchuk DM, Lucchinetti CF, Noseworthy JH. Multiple scle-rosis: current pathophysiological concepts. Lab Invest 2001; 81:263–281.
13. Kermode AG, Thompson AJ, Tofts P, et al. Breakdown of theblood–brain barrier precedes symptoms and other MRI signs ofnew lesions in multiple sclerosis. Brain 1990; 113: 1477–1489.
14. Brown WJ. The capillaries in acute and subacute multiple sclerosisplaques: a morphometric analysis. Neurology 1978; 28: 84–92.
15. Harris JO, Frank JA, Patronas N, et al. Serial gadolinium-enhanced magnetic resonance imaging scans in patients withearly relapsing-remitting multiple sclerosis: implications forclinical trials and natural history. Ann Neurol 1991; 29:548–555.
16. Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transectionin the lesions of multiple sclerosis. N Engl J Med 1998; 338:278–285.
17. Richert ND, Ostuni JL, Bash CN, et al. IFN beta 1b andmethylprednisolone promote lesion recovery in MS. Mult Scler2001; 7: 49–58.
18. Kuruganti PA, Hinojoza JR, Eaton MJ, et al. Interferon-betacounteracts inflammatory mediator-induced effects on brainendothelial cell tight junction molecules: implications for multiplesclerosis. J Neuropathol Exp Neurol 2002; 61: 710–724.
19. Gay FW, Drye TJ, Dick GWA, Esiri MM. The application ofmultifactorial cluster analysis in the staging of plaques in earlymultiple sclerosis. Identification of the primary demyelinatinglesion. Brain 1997; 120: 1461–1483.
20. Sanders V, Conrad AJ, Tourtellotte WW. On classification ofpost-mortem multiple sclerosis plaques for neuroscientists. JNeuroimmunol 1993; 46: 207–216.
21. van der Valk P, De Groot CJA. Staging of multiple sclerosis (MS)lesions: pathology of the time frame of MS. Neuropathol ApplNeurobiol 2000; 26: 2–10.
22. Daniel WW. Applied Nonparametric Statistics. Houghton–Miflin:Boston, 1978; 211–212.
23. Kwon EE, Prineas JW. Blood–brain barrier abnormalities inlongstanding multiple sclerosis lesions. An immunohistochemicalstudy. J Neuropathol Exp Neurol 1994; 53: 625–636.
24. Abbott NJ. Astrocyte–endothelial interactions and blood–brainbarrier permeability. J Anat 2002; 200: 629–638.
25. Allen IV, McKeown SR. A histological, histochemical andbiochemical study of the macroscopically normal white matter inmultiple sclerosis. J Neurol Sci 1979; 41: 81–91.
26. Goodkin DE, Rooney WD, Sloan R, et al. A serial study of new
MS lesions and the white matter from which they arise. Neurology1998; 51: 1689–1697.
27. Werring DJ, Brassat D, Droogan AG, et al. The pathogenesis oflesions and normal-appearing white matter changes in multiplesclerosis: a serial diffusion MRI study. Brain 2000; 123:1667–1676.
28. De Groot CJA, Bergers E, Kamphorst W, et al. Post-mortemMRI-guided sampling of multiple sclerosis brain lesions: increasedyield of active demyelinating and (p)reactive lesions. Brain 2001;124: 1635–1645.
29. Filippi M, Rocca MA, Martino G, et al. Magnetization transferchanges in the normal appearing white matter precede theappearance of enhancing lesions in patients with multiple sclerosis.Ann Neurol 1998; 43: 809–814.
30. Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin:claudin-5/TMVCF constitutes tight junction strands in endothelialcells. J Cell Biol 1999; 147: 185–194.
31. Akiguchi I, Tomimoto H, Suenaga T, et al. Blood–brain barrierdysfunction in Binswanger’s disease; an immunohistochemicalstudy. Acta Neuropathol (Berlin) 1998; 95: 78–84.
32. Luabeya MK, Dallasta LM, Achim CL, et al. Blood–brain barrierdisruption in simian immunodeficiency virus encephalitis.Neuropathol Appl Neurobiol 2000; 26: 454–462.
33. Claudio L, Raine CS, Brosnan CF. Evidence of persistentblood–brain barrier abnormalities in chronic progressive multiplesclerosis. Acta Neuropathol (Berlin) 1995; 90: 228–238.
34. Hawkins CP, Munro MG, Landon DN, McDonald WI. Metaboli-cally dependent blood–brain barrier breakdown in chronic relaps-ing experimental encephalomyelitis. Acta Neuropathol (Berlin)1990; 83: 630–635.
35. Edens HA, Parkos CA. Modulation of epithelial and endothelialparacellular permeability by leukocytes. Adv Drug Deliv Rev 2000;41: 315–328.
36. Walsh SV, Hopkins AM, Nusrat A. Modulation of tight junctionstructure and function by cytokines. Adv Drug Deliv Rev 2000; 41:303–313.
37. Gay D, Esiri M. Blood–brain barrier damage in acute multiplesclerosis plaques. Brain 1991; 114: 557–572.
38. Woodroofe MN, Cuzner ML. Cytokine mRNA expression ininflammatory multiple sclerosis lesions: detection by non-radioactive in situ hybridization. Cytokine 1993; 5: 583–588.
39. Blum MS, Toninelli E, Anderson JM, et al. Cytoskeletal rear-rangement mediates human microvascular endothelial tight junc-tion modulation by cytokines. Am J Physiol 1997; 273:H286–H294.
40. Cossins JA, Clements JM, Ford J, et al. Enhanced expression ofMMP-7 and MMP-9 in demyelinating multiple sclerosis lesions.Acta Neuropathol (Berlin) 1997; 94: 590–598.
41. Rosenberg GA, Dencoff JE, Correa N, et al. Effect of steroids onCSF matrix metalloproteinases in multiple sclerosis: relation toblood–brain barrier injury. Neurology 1996; 46: 1626–1632.
42. Stone LA, Frank JA, Albert PS, et al. The effect of interferon-beta on blood–brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing–remittingmultiple sclerosis. Ann Neurol 1995; 37: 611–619.
43. Miller DH, Khan OA, Sheremata WA, et al. A controlled trial ofnatalizumab for relapsing multiple sclerosis. N Engl J Med 2003;348: 15–23.
44. Compston A. Future options for therapies to limit damage andenhance recovery. Semin Neurol 1998; 18: 405–414.
Copyright 2003 John Wiley & Sons, Ltd. J Pathol 2003; 201: 319–327.





























