Embed Size (px)
Citation preview

This paper is published as part of a PCCP Themed Issue on: Metal oxide nanostructures: synthesis, properties and applications
Guest Editors: Nicola Pinna, Markus Niederberger, John Martin Gregg and Jean-Francois Hochepied
Editorial
Chemistry and physics of metal oxide nanostructures Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b905768d
Papers
Thermally stable ordered mesoporous CeO2/TiO2 visible-light photocatalysts Guisheng Li, Dieqing Zhang and Jimmy C. Yu, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b819167k
Blue nano titania made in diffusion flames Alexandra Teleki and Sotiris E. Pratsinis, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821590a
Shape control of iron oxide nanoparticles Alexey Shavel and Luis M. Liz-Marzán, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b822733k
Colloidal semiconductor/magnetic heterostructures based on iron-oxide-functionalized brookite TiO2 nanorods Raffaella Buonsanti, Etienne Snoeck, Cinzia Giannini, Fabia Gozzo, Mar Garcia-Hernandez, Miguel Angel Garcia, Roberto Cingolani and Pantaleo Davide Cozzoli, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821964h
Low-temperature ZnO atomic layer deposition on biotemplates: flexible photocatalytic ZnO structures from eggshell membranes Seung-Mo Lee, Gregor Grass, Gyeong-Man Kim, Christian Dresbach, Lianbing Zhang, Ulrich Gösele and Mato Knez, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b820436e
A LEEM/ -LEED investigation of phase transformations in TiOx/Pt(111) ultrathin films Stefano Agnoli, T. Onur Mente , Miguel A. Niño, Andrea Locatelli and Gaetano Granozzi, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821339a
Synthesis and characterization of V2O3 nanorods Alexander C. Santulli, Wenqian Xu, John B. Parise, Liusuo Wu, M.C. Aronson, Fen Zhang, Chang-Yong Nam, Charles T. Black, Amanda L. Tiano and Stanislaus S. Wong, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b822902c
Flame spray-pyrolyzed vanadium oxide nanoparticles for lithium battery cathodes See-How Ng, Timothy J. Patey, Robert Büchel, Frank Krumeich, Jia-Zhao Wang, Hua-Kun Liu, Sotiris E. Pratsinis and Petr Novák, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821389p
Mesoporous sandwiches: towards mesoporous multilayer films of crystalline metal oxides Rainer Ostermann, Sébastien Sallard and Bernd M. Smarsly, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b820651c
Surprisingly high, bulk liquid-like mobility of silica-confined ionic liquids Ronald Göbel, Peter Hesemann, Jens Weber, Eléonore Möller, Alwin Friedrich, Sabine Beuermann and Andreas Taubert, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821833a
Fabrication of highly ordered, macroporous Na2W4O13 arrays by spray pyrolysis using polystyrene colloidal crystals as templates SunHyung Lee, Katsuya Teshima, Maki Fujisawa, Syuji Fujii, Morinobu Endo and Shuji Oishi, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821209k
Nanoporous Ni–Ce0.8Gd0.2O1.9-x thin film cermet SOFC anodes prepared by pulsed laser deposition Anna Infortuna, Ashley S. Harvey, Ulrich P. Muecke and Ludwig J. Gauckler, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821473e
Surface chemistry of carbon-templated mesoporous aluminas Thomas Onfroy, Wen-Cui Li, Ferdi Schüth and Helmut Knözinger, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821505g
ZnO@Co hybrid nanotube arrays growth from electrochemical deposition: structural, optical, photocatalytic and magnetic properties Li-Yuan Fan and Shu-Hong Yu, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b823379a
Electrochemistry of LiMn2O4 nanoparticles made by flame spray pyrolysis T. J. Patey, R. Büchel, M. Nakayama and P. Novák, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821572n
Ligand dynamics on the surface of zirconium oxo clusters Philip Walther, Michael Puchberger, F. Rene Kogler, Karlheinz Schwarz and Ulrich Schubert, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b820731c
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online / Journal Homepage / Table of Contents for this issue

Thin-walled Er3+:Y2O3 nanotubes showing up-converted fluorescence Christoph Erk, Sofia Martin Caba, Holger Lange, Stefan Werner, Christian Thomsen, Martin Steinhart, Andreas Berger and Sabine Schlecht, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821304f
Wettability conversion of colloidal TiO2 nanocrystal thin films with UV-switchable hydrophilicity Gianvito Caputo, Roberto Cingolani, Pantaleo Davide Cozzoli and Athanassia Athanassiou, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b823331d
Nucleation and growth of atomic layer deposition of HfO2 gate dielectric layers on silicon oxide: a multiscale modelling investigation A. Dkhissi, G. Mazaleyrat, A. Estève and M. Djafari Rouhani, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821502b
Designing meso- and macropore architectures in hybrid organic–inorganic membranes by combining surfactant and breath figure templating (BFT) Ozlem Sel, Christel Laberty-Robert, Thierry Azais and Clément
Sanchez, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821506e
The controlled deposition of metal oxides onto carbon nanotubes by atomic layer deposition: examples and a case study on the application of V2O4 coated nanotubes in gas sensing Marc-Georg Willinger, Giovanni Neri, Anna Bonavita, Giuseppe Micali, Erwan Rauwel, Tobias Herntrich and Nicola Pinna, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821555c
In situ investigation of molecular kinetics and particle formation of water-dispersible titania nanocrystals G. Garnweitner and C. Grote, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b821973g
Chemoresistive sensing of light alkanes with SnO2 nanocrystals: a DFT-based insight Mauro Epifani, J. Daniel Prades, Elisabetta Comini, Albert Cirera, Pietro Siciliano, Guido Faglia and Joan R. Morante, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b820665a
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online

Nucleation and growth of atomic layer deposition of HfO2 gate dielectric
layers on silicon oxide: a multiscale modelling investigation
A. Dkhissi,*ab G. Mazaleyrat,ab A. Esteveab and M. Djafari Rouhaniab
Received 1st December 2008, Accepted 4th March 2009
First published as an Advance Article on the web 26th March 2009
DOI: 10.1039/b821502b
We apply our recently developed approach, combining advanced ab initio density functional
theory (DFT) methods with a probabilistic kinetic Monte Carlo (KMC) scheme, to quantify the
properties of mesoscopic size systems operating in real experimental conditions. The application
concerns the investigation of the atomic layer deposition (ALD) of HfO2 film growth on Si(100)
surface. We show that the proposed models offer guidance in the optimization of the experimental
deposition processes, in terms of OH density on the substrate, optimal growth temperature, pulse
durations, and finally growth kinetics.
I. Introduction
The reduction of the SiO2 gate oxide thickness in accordance
with Moore’s law has led to unacceptable tunnelling and
leakage current levels.1 So, the conventional SiO2 gate is
reaching its physical and electrical limitations. The search
for suitable candidates to replace SiO2 as the gate dielectric
in complementary metal-oxide-semiconductor (CMOS)
devices has recently received enormous attention. Of the many
different materials with high dielectric constant k, which have
been used as SiO2 replacements,2 hafnium oxide (HfO2) is one
of the most promising candidates. HfO2 is attractive as it
exhibits a bulk permittivity of almost 25, a wide band gap
(5.68 eV) and a good thermodynamic stability in contact with
silicon.3 However, experimentally the EOT (equivalent oxide
thickness) value of the HfO2 gate stack increases, due to the
growth of an interfacial layer between HfO2 and the silicon
substrate. Among various methods for growing high-k
dielectric films, atomic layer deposition (ALD) shows its
unique ability in depositing ultra thin films with excellent
conformity and uniformity over large areas.4 ALD, which is
a vapour deposition technique, is based on the cycling of
self-terminating surface reactions. Indeed, each precursor is
pulsed into the reaction chamber alternately, and the reaction
between the incoming precursors and surface species is self-
terminating. Thus atomic level control of film growth is
supposed to be achieved. Experimentally, ALD has been
actively investigated for depositing HfO25–21 for which HfCl4
and H2O are often used as precursors.22–24 In practice, the
density of metal atoms deposited per cycle depends on the
temperature, on the chemical nature of the precursors used
and on the density of reactive sites available on the substrate
surface. Thus, it is desired to reach full monolayer coverage
at each cycle. However, Ritala et al.22 found that only a
sub-monolayer of the HfO2 film is deposited during each cycle.
About seven deposition cycles would be needed to entirely
cover the surface25 which then shows a non-negligible
roughness. ALD growth resulting in only partial monolayer
growth per cycle is due to the steric hindrance26,27 or to the
lack of reactive sites.28 Therefore, a detailed understanding of
the basic mechanisms that take place during each cycle is
required if one wants to optimize the deposition. This is
especially true for the few first deposited layers which are of
major importance for subsequent electrical properties.
These basic atomistic mechanisms include the decomposition
of HfCl4 on the substrate and the further hydrolysis of the
resulting SiO2–O–HfCl3 surface complex, but many other reac-
tions, called ‘‘densification mechanisms’’ hereafter, are also
involved in the process. To design optimal experimental setups
in future, it is of great importance that a precise atomic level
description of the basic reaction mechanisms responsible for the
overall process of high-k film growth, and also their relation
with the thermodynamic parameters be established. Our final
aim, towards this objective, is to achieve the ambitious task of
elaborating a new generation of tools dedicated to atomic scale
simulation of technological process. Our strategy is to combine
the ab initio DFT with kinetic Monte-Carlo techniques into a
multiscale approach, which will incorporate enough reaction
mechanisms to be adequate for testing against experimental
setups. Kinetic Monte Carlo (KMC) is a suitable procedure
that can be of great help to understand mesoscopic level
processes, such as deposition of high-k materials.
This paper is dedicated to the application of our KMC tool,
enabling the treatment of HfO2 ALD from HfCl4 and H2O
precursors at the atomic scale. In particular, we present the
results concerning the following points (i) the influence of the
substrate preparation on the coverage of the surface, (ii) an
optimal growth temperature, since it is found experimentally
that HfCl4/H2O ALD of HfO2 has a broad process window
with respect to deposition temperature, between 180 and
600 1C, (iii) surface coverage saturation and (iv) growth
kinetics in different regimes.
Section II is devoted to the description of DFT and KMC
theoretical approaches. Different basic mechanisms used in
KMC simulations are also briefly listed in this section, with an
emphasis on densification mechanisms. Section III gathers
a CNRS, LAAS, 7 avenue du colonel Roche, F-31077, Toulouse,France. E-mail: [email protected]; Fax: +33 5 61 33 62 08
bUniversite de Toulouse, UPS, INSA, INP, ISAE, LAAS,F-31077, Toulouse, France
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 | 3701
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online

ab initio results, implemented as parameters in the KMC
package, and KMC simulation results, corresponding to the
first cycle deposition and to the transient and steady-state
growth regimes. Finally, conclusions are drawn in section IV.
II. Theoretical approaches
Recently we have developed an original approach within a
multiscale strategy29 to simulate the growth of HfO2 on
silicon. In particular, we have shown how this method
provides a unique and fundamental understanding of the
growth mechanism and growth kinetics. KMC is a stochastic-
based model aimed at simulating film growth at the atomic
scale, the final objective being to furnish alternative/new tools
for replacement of the macroscopic conventional TCAD
(Technology Computer Aided Design) tools used for years
by engineers in microelectronics. Such a model performs
virtually the explicit experimental-processing procedure,
atomic-scale event by atomic-scale event. The algorithm is
operated through probabilistic rules that make the overall
simulation comparable with an actual experiment.30,31 Indeed,
from the engineering point of view, the main interest is in the
microstructure that is produced under specific processing
conditions. This microstructure involves millions of atoms
processed during periods exceeding the second. Unfortunately,
the most predictive models, i.e. the quantum-based models,
are not tractable at this scale. Two options can be further
considered: molecular dynamics (MD) and kinetic Monte
Carlo (KMC). MD solves the Newton equations of motion
for the system of atoms under investigation. Trajectories are
continuous and necessitate very short integration time steps
that make the simulation cumbersome for durations that are
orders of magnitude higher than nanosecond. Another
drawback of the MD method is the general lack of adequate
inter-atomic potentials for highly disordered and hetero-
geneous systems. This is particularly true for microelectronics
semiconductor/oxide interfaces such as silicon/high-k gate
oxides. KMC appears to be a simplification compared to
MD: continuous trajectories are replaced with discrete atomic
jumps. A prior knowledge of these hoppings is needed and
must be listed from any other source: quantum-based models
or experimental characterization. Thus, KMC methods allow
the simulation of more ambitious systems in terms of size and
duration of the simulated experiment, meeting the require-
ments of the next generation of processes in microelectronics.
Basic ingredients needed to develop what we call a lattice-
based KMC model are: (i) we will define a lattice framework
able to operate the desired transition between the silicon cubic
structure and the oxide crystal, (ii) we detail the writing of
configurations that allow the association of the lattice sites
with the chemical nature of atoms occupying them, (iii) we
introduce the concept of event that must describe correctly the
chemistry of the process, and (iv) we finally indicate how
to deal with the time evolution of the KMC, namely, the
temporal dynamics of the KMC.
II.1 Coincidence lattice model
In order to define atomic positions in our KMC model, we
have to consider a two-dimensional lattice at the interface
between the two materials. Different atoms are placed within
the 2D lattice at different heights corresponding to the
successive layers. We should emphasize that, in our KMC
view, atomic positions are not defined by accurate Cartesian
coordinates, but only specified approximately around
an equilibrium position within a cell. This view allows
displacements of atoms to satisfy elastic strain usually present
in heterogeneous systems. To represent the above 2D lattice,
we superimpose the conventional cubic cell of oxide and the
silicon-based SiO2 surface in order to find a coincidence
between the two crystallographic arrangements. The silicon
(100) surface is taken as the crystal reference, an ultrathin
silicon dioxide, one atomic layer thick, being able to accom-
modate this structure perfectly. We obtain the schematic
configuration represented in Fig. 1. This resulting picture
optimizes the coincidence between the two materials. The
silicon part contains two layers in order to show the orienta-
tion of the bonds at the interface. Siloxanes bridges and
hydroxyl functions are represented as an indication. For sake
of clarity, we do not represent the dimer formation inducing
Si–O–Si species: silicon atoms are schematically located at the
nodes of the crystal lattice of the diamond structure, without
taking into account the displacements observed usually.
According to the crystallographic data,32 the distance separat-
ing two Si surface atoms is 3.84 A. In addition, if one considers
that the cubic lattice parameter of hafnia is approximately 5 A,
the distance separating two Hf atoms represents the half
diagonal of the base of the cube, approximately 3.5 A. It thus
appears possible to match the two structures, with a tension of
the cubic hafnia.
Let us imagine now that we position several HfO2 cubes in
tension on the substrate, looking for an ideal match of the two
2D lattices. We see that a tilt of the cell is required to obtain a
complete match of the atomic positions. It is then possible to
completely cover the surface without defect: we obtain three
perfectly crystalline layers of HfO2 on Si/SiO2. It is, to some
extent what we would like to obtain in real experiments. Fig. 1
is a top view of this ideal configuration where the various
species are represented by symbols for a better legibility. It
represents a (100) silicon surface, covered by single-crystal
HfO2 in its cubic phase. The square base of a conventional
cubic cell is represented in dotted line: it locates five hafnium
atoms: the four corners of the square and the central atom in
round symbols (l = 2). One atomic layer above, we find four
hafnium atoms corresponding to the centres of the four side
faces of the f.c.c. lattice, in square symbols (l = 3). Between
these two layers, the first four oxygen atoms are in dotted line
round symbols (l = 2 + 1/2). By adding two new layers, one
of oxygen atoms and one of hafnium atoms, one obtains the
totality of the conventional cube which is obviously not the
primitive cell, minimal for the construction of the lattice.
Considering the successive layers, by increasing index l, one
meets various species: silicon atoms marked as triangles, for
l = 1, Hf atoms marked as full rounds for all even layers, Hf
atoms marked as squares for all odd layers, except l = 1, and
oxygen atoms marked as dotted rounds, for all half integer
layers. For sake of clarity of Fig. 1, oxygen atoms are only
shown in the conventional cube. The cell described above
makes it possible to represent in an exhaustive way the oxide
3702 | Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 This journal is �c the Owner Societies 2009
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online
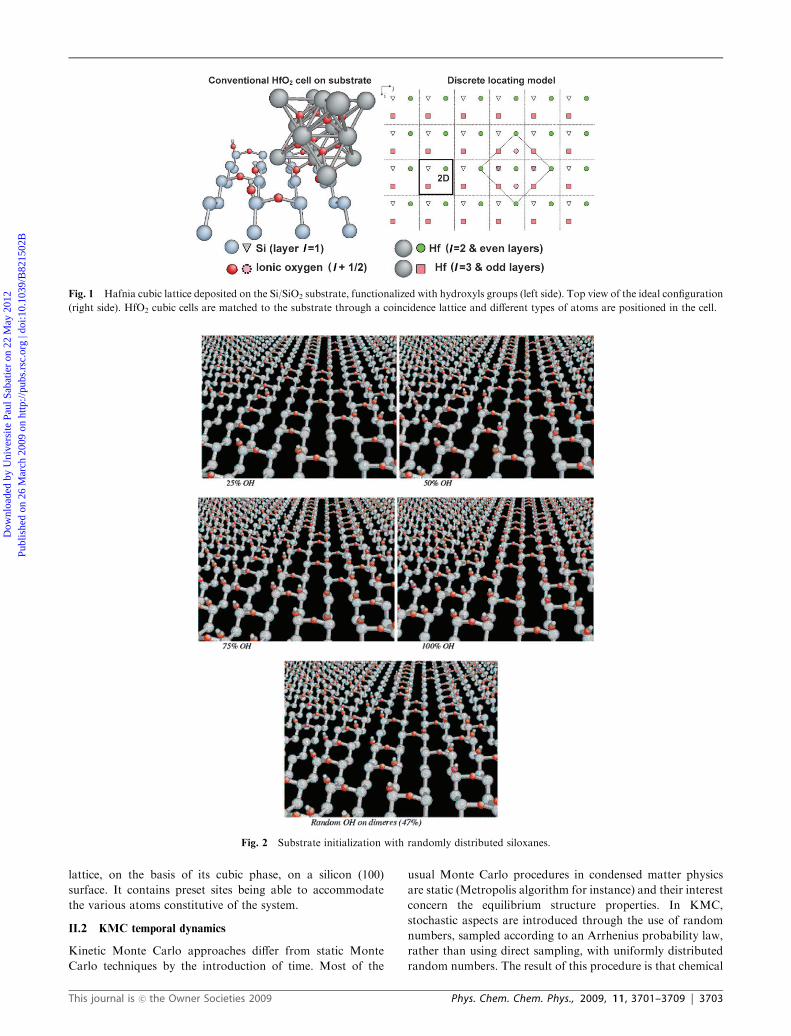
lattice, on the basis of its cubic phase, on a silicon (100)
surface. It contains preset sites being able to accommodate
the various atoms constitutive of the system.
II.2 KMC temporal dynamics
Kinetic Monte Carlo approaches differ from static Monte
Carlo techniques by the introduction of time. Most of the
usual Monte Carlo procedures in condensed matter physics
are static (Metropolis algorithm for instance) and their interest
concern the equilibrium structure properties. In KMC,
stochastic aspects are introduced through the use of random
numbers, sampled according to an Arrhenius probability law,
rather than using direct sampling, with uniformly distributed
random numbers. The result of this procedure is that chemical
Fig. 1 Hafnia cubic lattice deposited on the Si/SiO2 substrate, functionalized with hydroxyls groups (left side). Top view of the ideal configuration
(right side). HfO2 cubic cells are matched to the substrate through a coincidence lattice and different types of atoms are positioned in the cell.
Fig. 2 Substrate initialization with randomly distributed siloxanes.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 | 3703
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online

aspects are respected, but the most probable events do not
necessarily occur first. Different algorithms do exist for KMC.
In the following, we suggest a procedure that becomes effi-
cient, in comparison with the more conventional BKL (Bortz,
Kalos and Lebowitz) algorithm,33 when dealing with many
different types of events having different probabilities. This is
the case of systems where local deformation energy is taken
into account for the evaluation of the occurrence probabilities,
or where the system exhibits a complex chemistry. It should be
emphasized here that, in our KMC procedure, an event is
defined as elementary mechanism occurring at a specific site.
When the software is launched, after preliminary initializa-
tions (parameters, mechanisms, neighbourhood, configura-
tion), it makes a global scan to determine which events are
authorized and which are forbidden. Forbidden ones get a
maximum time so that they can never occur. Authorized ones
get a ‘‘specific occurrence time’’: this is the time that the
considered event typically takes to occur as soon as it becomes
possible. The ‘‘specific occurrence time’’ of the event m on site
(i,j,k) is given by
Ti;j;k;m ¼� logðZÞ
lm
with
lm ¼ n exp �DEm
kBT
� �
where Z is a random number uniformly distributed between 0
and 1, lm is the probability of occurrence of the mechanism
per unit of time, n is the attempt frequency, in the order of the
typical lattice vibration frequency, DEm the activation energy
of the mechanism, kB the Boltzmann constant and T the
temperature. The two arrival mechanisms (1-precursor arrival
and 2-water arrival) obey Maxwell–Boltzmann statistics:
l1;2 ¼cPSffiffiffiffiffiffiffiffiffiffiffiffiffiM1;2T
p
where c is a constant, P the pressure, S the elementary 2D-cell
area, M1,2 the considered species molar mass and T the
temperature. Going back to the definition of events, it becomes
obvious that different ‘‘specific occurrence times’’ are attribu-
ted to a given elementary mechanism occurring at different sites
if the deposited layer. Following this procedure, we determine a
huge list of ‘‘specific times’’, a kind of ‘‘calendar’’, just as if all
events that will occur were already foreseen. Not exactly
in fact, because this calendar will often be updated as the
configuration evolves. As soon as the calendar is updated,
the software finds the minimum ‘‘specific occurrence time’’ and
the corresponding event occurs. As this minimum time has
passed, it is withdrawn from the other times contained in the
‘‘calendar’’; a new configuration is edited; pertinent events that
are likely to become possible are added to the list and the
forbidden ones withdrawn from the list. We then go back to the
first KMC stage and search again the minimum time.
II.3 KMC application to ALD
The KMC software discussed here is built in order to simulate
the ALD experimental process. ALD consists of several cycles,
each one composed of four phases: precursor pulse, precursor
purge, hydrolysis and water purge. Each phase has its own
thermodynamic parameters and duration: some mechanisms
are always possible whereas others can only occur in a typical
phase. For instance, ‘‘Precursor arrival’’ will obviously exist
during the first phase only.
The basic chemical reactions involved in ALD process are
the incorporation of the precursors, followed by the formation
of the HCl by product, and the hydrolysis of the chlorine
terminated bonds of the incorporated precursor. Each of the
above global reactions can be decomposed into several steps,
called ‘‘elementary mechanisms’’ in our KLC approach.
The thermodynamic characteristics of these ‘‘elementary
mechanisms’’: reaction enthalpy and activation barrier, have
been determined by ab initio calculations and presented in
section III. Obviously, all implemented mechanisms are
assumed to be reversible, with probabilities defined by the
reverse activation barriers, via the Arrhenius law.
In addition to the above basic mechanisms of ALD, we have
also implemented, in our KMC model, less straightforward
mechanisms, involved in the ALD of high-k materials, which
we call ‘‘Densification mechanisms’’. Densification is defined
here as the necessary phase transition of the high-kmetal oxide
materials from their molecular structure in the gas-phase
precursors, to their solid-state structure in the deposited film.
This is particularly true in the ALD deposition of HfO2 where
the metal has a covalent bonding structure in the gas phase,
with a small coordination number in its precursor form,
whereas the metal oxide has mainly an ionic structure, with
a large coordination number, in the deposited film. This
transition is mediated by the presence of oxygen atoms:
densification could occur in the gas-phase in the presence of
pre-oxidized precursors. We have also shown that densifica-
tion can be seen as a local phenomenon where, during bridging
of two metallic centres, a local re-allocation of the oxygen
atoms occurs.
Therefore, densification mechanisms in the kinetic Monte
Carlo account for all transitions from molecular state to ionic
or crystalline states, i.e. nodes of the lattice. These include
reaction between grafted molecular ligand and the surface,
such as the bridging process of eqn (1), but also during
bridging between two grafted molecules in tree-like positions.
Si–O–HfCl3 + Si–OH - Si–O–HfCl2–O–Si + HCl (1)
The by-product is usually HCl (when bridging a Cl-terminated
tree to an OH-terminated tree) or H2O (when bridging two
OH-terminated trees).
III. Results and discussion
III.1 Ab initio results
The aim of ab initio investigations is to understand the
chemistry of possible reactions, during the first cycles of
ALD of HfO2. The related predictive method, cluster- or
periodic-based DFT, is particularly suitable for chemical
mechanisms that are difficult to reach by experiments or other
modelling strategies.
3704 | Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 This journal is �c the Owner Societies 2009
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online

The reactions between the gaseous precursors HfCl4 and
H2O with the hydroxylated SiO2 surface are predicted to
proceed via an exchange mechanism34,35 and can be separated
into two half-reactions: the first half-cycle reaction [eqn (2)]
takes place during the initial step of atomic layer deposition of
HFO2, i.e. the decomposition of HfCl4 precursor molecules:
8SiO2–OH + HfCl4 8SiO2–O–HfCl3 + HCl, (2)
This first half-cycle reaction is itself decomposed into three
elementary reversible mechanisms: (i) chemisorption of the
precursor molecule and its possible desorption, (ii) incorpora-
tion of the chemisorbed molecule and the formation of HCl by
product, and (iii) desorption of HCl which leads to the definite
incorporation of the metal atom. The decomposition of a
global reaction into several elementary mechanisms allows
the detailed examination of the incorporation process rather
than the use of global sticking coefficients.
The second half cycle reaction [eqn (3)] is water exposure
leading to the hydrolysis of Cl-terminated bonds:
8SiO2–O–Hf(OH)3�x–Clx + H2O
8SiO2–O–Hf(OH)4�x–Clx�l + HCl (3)
where x has values of 1–3.
This second half cycle reaction is also decomposed into two
elementary mechanisms: (i) chemisorption of H2O and
(ii) definite hydrolysis. We have shown that the characteristics
of both mechanisms depend on the value of x.
We have studied these reactions in detail using DFT cluster
models. Results are published in our recent papers,36 reported
here in Table 1 and implemented in our KMC package. Values
reported in Table 1 concern direct reaction characteristics:
reaction enthalpies and activation barriers. The characteristics
of reverse reactions can be easily deduced from the reported
values.
III.2 Kinetic Monte Carlo (KMC) results
A First cycle results. The starting surface is known to play
a crucial role at all stages of the device development and
operation. The first ALD cycle, i.e. precursor–substrate inter-
action, is governed by the specificity of the surface cleaning
processes and subsequent presence of nucleation sites. There-
fore, we start by investigating the dependence of the surface
coverage on the density of OH nucleation sites on the
substrate. Surfaces functionalized with hydroxyl groups can
be prepared in various ways37,38 and allow a variable concen-
tration of sites for incorporating the precursors. We thus
simulated a first ALD phase with a long duration: 100 ms
against 50 ms generally used in experiments, at 300 1C with
different starting substrates covered with 0%, 25%, 75% and
100% OH, distributed arbitrarily (Fig. 2). Such a long time is
required to overcome the poor sticking of the precursors and
to saturate efficiently the reactive sites. For a fifth case, we
used the option of initialization ‘‘no more than one OH by
dimer’’ which leads to 47% hydroxyl coverage.
The resulting coverage rates are represented in Table 2. An
increase in the total coverage is observed when one increases
the number of reactive sites of the substrate. In the case 4, it
can be noted that the total coverage amounts to approximately
55%, indicating that a majority of hafnium atoms are thus
bridged consuming each one two OH: one for the incorpora-
tion and a second for the bridging process.
Experimentally, at the end of the first ALD cycle at 300 1C,
a total coverage of 33% was measured,37 which is in good
agreement with our simulations with 50% and 47% OH
coverages. On the other hand, Zhuravlev’s results39 confirm
the 50% OH coverage of SiO2 surfaces at 300 1C (see Table 3).
The excellent agreement between the simulated total coverages
and those obtained experimentally confirms the great relia-
bility of DFT results for the first mechanisms of growth and
the success of their implementation at the mesoscopic scale.
An important issue in the ALD process is the growth
temperature. The ALD of HfO2 is often performed using
metal chloride and water successively as precursors.22–24
Experimentally, it is found that HfCl4/H2O ALD of SiO2
has a broad process window with deposition temperature
between 180 1C and 600 1C. Further the metal chloride–water
ALD process suffers from some disadvantages such as chlorine
contaminations and slow growth rates due to submonolayer
growth per cycle.40–42 As a result, high temperatures are
required to minimize the contamination. Unfortunately, the
film quality degrades at high temperatures. Our aim here is to
optimize this temperature as a compromise between these two
opposite effects, by examining KMC simulations at different
temperatures. We use the Zhuravlev results39 for the distribu-
tion of OH groups on the substrate surface as a function of
temperature (Table 3).
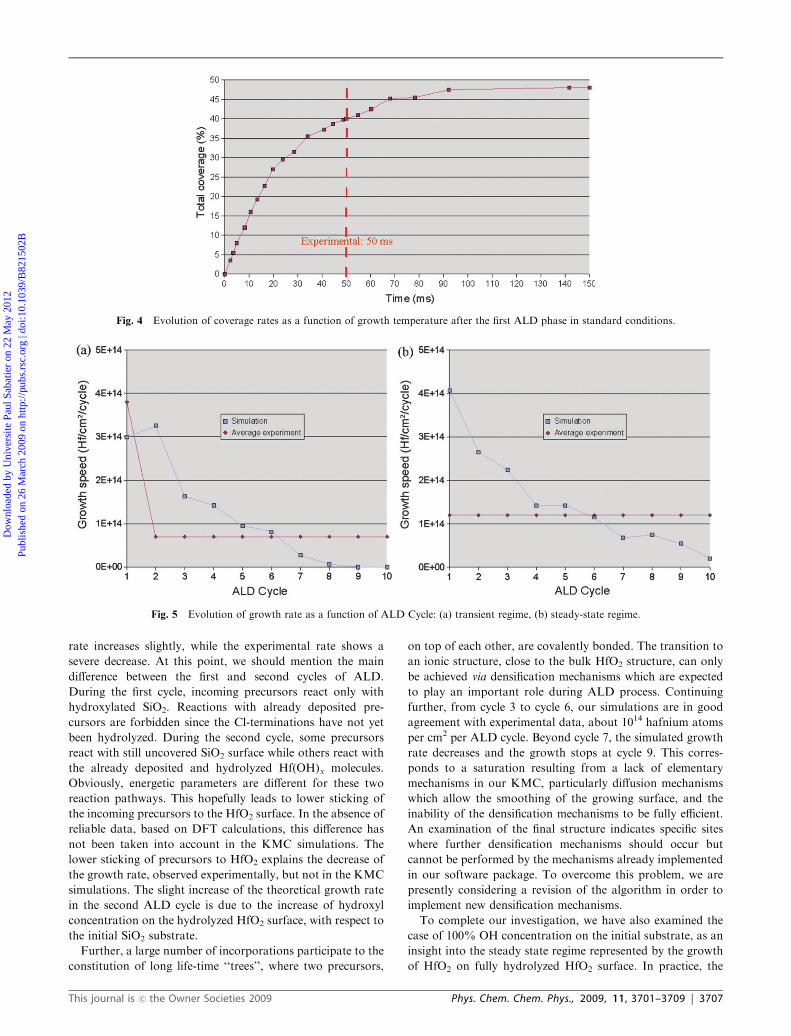
Fig. 3 shows the total coverage of the substrate with respect
to temperature, after the first ALD cycle. The total coverage
increases from 200 1C to 300 1C, then decreases until 400 1C.
This confirms the existence of an optimal growth temperature
at T = 300 1C, at least for the first ALD cycle. This predicted
temperature is in excellent agreement with the optimal experi-
mental temperature largely used for ALD of HfO2 on
SiO2/Si.43–44 This result confirms again the credibility of
our model.
Another important issue in the ALD process is the duration
of precursor pulses. Experimentally, in the case of HfO2
Table 1 Reaction enthalpies and activation barriers for precursor incorporation and hydrolysis processes
Precursor incorporation
Si–OH–HfCl4 complex TSa Si–O–HfCl3–HCl complex HCl desorptionEnergy/eV 0.48 0.88 0.22 0.12
Hydrolysis processes
M–Cl3–H2O complex TSa M–OHCl2–HCl complex HCl desorptionEnergy/eV 0.68 0.97 0.63 0.20
a TS: transition state.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 | 3705
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online
deposition, the pulse duration of HfCl4 precursor is in the
order of 50 ms. The question here is to know whether this time
is sufficient or overestimated? Fig. 4 represents the evolution of
the total coverage as a function of time. Simulations are
preformed under constant flux of HfCl4, at a pressure of
1.33 mbar, at the optimized growth temperature of 300 1C.
The results indicate a saturation of the coverage at 48%. At
the standard duration of 50 ms, the total coverage is only of
40%. The standard experimental duration cannot thus be
reduced without a strong reduction of the total coverage.
Moreover, it could be, according to our simulations, slightly
increased, at least for the very first phase of ALD. We should
mention that the standard time of 50 ms, corresponding to the
precursor pulse phase, is an experimental value, very difficult
to measure in industrial ALD equipments, since their
measurement threshold level is in this order of 50 ms. There-
fore, this experimental value must be regarded as indicative.
Examining the results of simulation of this first cycle, we can
assert the excellent validity of DFT calculations reported in
ref. 29 and 36a concerning HfCl4 decomposition and metal
incorporation, since these mechanisms are massively used in
this first cycle. A correlation is obtained between the total
coverage and the preparation of the surface: distributions of
siloxanes and hydroxyl groups. Further, using the Zhuravlev
model to initialize the starting substrate, according to
the temperature, we highlighted the existence of an optimal
temperature for the incorporation of the metal precursors
during the first phase of ALD. In our simulations, this
optimum was a consequence of a competition between two
contrary effects. However, the experimental results do not
reveal this expected behaviour at low temperatures. We can
therefore assume that some basic mechanisms are missing in
our simulator, in order to reproduce reliably the experimental
results at temperatures lower than 300 1C. This does not
constitute a serious drawback since the growth is generally
carried out at 300 1C, even higher. We finally highlighted the
saturation of the substrate by the precursors, the light over-
estimate of the pulse phase duration and coverage limit are
probably due to an overestimate of hydroxyl density obtained
by the model of Zhuravlev. This model has to be adapted to the
substrates used in the cases investigated here. In all these cases,
the tendencies were in agreement with the experimental results.
B Kinetics of growth. In experiments, several growth
modes are successively observed.37 After the first cycle
consuming a large number of metal precursors, a slow rate
transient mode allows reaching full coverage of the substrate.
Then, a faster steady-state mode, characteristics of the deposi-
tion of HfO2 on HfOx(OH)y, is established. Our aim here is to
reproduce these experimental observations. In the following,
we will investigate the kinetics of growth of HfO2 films, for
both transient and steady state regimes, up to 10 ALD cycles.
Simulation results are presented, and compared with experi-
mental data, in Fig. 5a and b, for transient and steady state
regimes, respectively. Growth rates are expressed in number of
metal atoms incorporated in the film rather than coverage, since
some precursors are incorporated on top of already deposited
metal atoms. This is a consequence of the full coverage being
only reached after a large number of ALD cycles.
As we demonstrated above, Fig. 5a shows that the first cycle
surface coverage is well reproduced by our simulations. This is
no more the case for the second cycle where simulated growth
Table 2 Coverage and crystallinity for different initial substrates
25% random OH 50% random OH 75% random OH 100% random OHRandom OHon dimers (47%) Exp.
Coverage of HfO2 18% 32% 43% 55% 30% 33%
Table 3 Zhuravlev model for substrate initialization. Fromthe Monte Carlo point of view, OH density is the percentage ofhydroxylated sites
T/1C % OH
200 68250 59300 52350 43400 34
Fig. 3 Evolution of coverage rates and crystallinity for different initial substrates.
3706 | Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 This journal is �c the Owner Societies 2009
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online
rate increases slightly, while the experimental rate shows a
severe decrease. At this point, we should mention the main
difference between the first and second cycles of ALD.
During the first cycle, incoming precursors react only with
hydroxylated SiO2. Reactions with already deposited pre-
cursors are forbidden since the Cl-terminations have not yet
been hydrolyzed. During the second cycle, some precursors
react with still uncovered SiO2 surface while others react with
the already deposited and hydrolyzed Hf(OH)x molecules.
Obviously, energetic parameters are different for these two
reaction pathways. This hopefully leads to lower sticking of
the incoming precursors to the HfO2 surface. In the absence of
reliable data, based on DFT calculations, this difference has
not been taken into account in the KMC simulations. The
lower sticking of precursors to HfO2 explains the decrease of
the growth rate, observed experimentally, but not in the KMC
simulations. The slight increase of the theoretical growth rate
in the second ALD cycle is due to the increase of hydroxyl
concentration on the hydrolyzed HfO2 surface, with respect to
the initial SiO2 substrate.
Further, a large number of incorporations participate to the
constitution of long life-time ‘‘trees’’, where two precursors,
on top of each other, are covalently bonded. The transition to
an ionic structure, close to the bulk HfO2 structure, can only
be achieved via densification mechanisms which are expected
to play an important role during ALD process. Continuing
further, from cycle 3 to cycle 6, our simulations are in good
agreement with experimental data, about 1014 hafnium atoms
per cm2 per ALD cycle. Beyond cycle 7, the simulated growth
rate decreases and the growth stops at cycle 9. This corres-
ponds to a saturation resulting from a lack of elementary
mechanisms in our KMC, particularly diffusion mechanisms
which allow the smoothing of the growing surface, and the
inability of the densification mechanisms to be fully efficient.
An examination of the final structure indicates specific sites
where further densification mechanisms should occur but
cannot be performed by the mechanisms already implemented
in our software package. To overcome this problem, we are
presently considering a revision of the algorithm in order to
implement new densification mechanisms.
To complete our investigation, we have also examined the
case of 100% OH concentration on the initial substrate, as an
insight into the steady state regime represented by the growth
of HfO2 on fully hydrolyzed HfO2 surface. In practice, the
Fig. 4 Evolution of coverage rates as a function of growth temperature after the first ALD phase in standard conditions.
Fig. 5 Evolution of growth rate as a function of ALD Cycle: (a) transient regime, (b) steady-state regime.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 | 3707
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online

simulation is performed by artificially depositing a full mono-
layer of HfO2 on the silicon substrate. We apply the Monte
Carlo procedure only after this first step. Results are very
similar to the previous case (see Fig. 5b). However, the rise of
growth rate observed previously during the second is no more
present, since the 100% coverage is artificially reached from
the beginning. In experiments, after a slow growth to cover
the substrate, a faster steady-state growth regime, around
1.2 � 1014 hafnium atoms per cm2 and ALD cycle, is observed.
In simulations, the growth rate is again too high at the
beginning, because of the inadequate parameterization model.
From cycle 4, the calculations are in good agreement with
experiments, but the growth stops later as it lacks mechanisms.
Our simulations correctly reproduce the two regimes of the
growth observed experimentally,37 at least for some ALD
cycles. Results are schematically presented in Fig. 6. In the
transient regime, after the first cycle leading to relatively high
substrate coverage, the growth rate decreases to about 0.1
layer per cycle. Our simulator suffers from a saturation effect
inherent to the method but simulations are very realistic
during approximately 5 ALD cycles. The opening of the
siloxanes bridges seems to be at the origin of the slow rate
of this mode. Siloxane openings delay the full coverage of the
surface and a more effective functionalization. The duration of
this stage is not accessible to our simulator without new
optimizations.
The steady-state regime then follows (see Fig. 6). The
growth rate is higher, because of the higher hydroxyl concen-
tration on the surface. The growth rate is in agreement with
experimental data:25,44 about 1.2 � 1014 Hf per cm2 per cycle,
i.e. 0.17 monolayer per cycle. It still remains very low
compared to what could be expected in an ‘‘atomic layer
deposition’’ technique where full monolayer coverage should
normally be reached at each cycle.
IV. Conclusions
The purpose of the present paper is to describe a new genera-
tion of tools dedicated to the modelling of high-k material
deposition on silicon substrates, providing a growth-modelling
environment for atomic layer chemical vapour deposition
(ALD) of the materials as HfO2. A multi-scale strategy has
been chosen to achieve this investigation. The corresponding
‘‘bottom-up’’ approach, from quantum level calculations to
macroscopic rate theory, is necessary to understand and
implement the most pertinent atomic scale mechanisms in
the growth models. We point out that this atomistic descrip-
tion is crucial to match the requirements of microelectronics
and emerging nanotechnology scales. Indeed, our approaches
provided interesting results concerning the substrate reactivity,
growth mechanisms and the optimization of the growth
temperature for HfO2 deposition. Moreover, some studies
are still running to make progress towards the associated
technology, in particular the calibration of activation barriers
for densification mechanisms and siloxane bridge opening.
These calibrations will allow us to draw a clear picture of
the growth mechanisms and growth kinetics, for several
deposition cycles, until a full monolayer is achieved.
Acknowledgements
This work was founded by the French National Research
Agency (ANR) through LN3M project No. ANR-05-CIGC-
0003-02.
References
1 P. A. Packan, Science., 1999, 285, 2079.2 G. D. Wilk, R. M. Wallace and J. M. Anthony, J. App. Phys.,2001, 89, 5243.
3 G. D. Wilk and D. A. Muller, App. Phys. Lett., 2003, 83, 3984.4 (a) S. M. George, A. W. OTT and J. W. Klaus, J. Phys. Chem.,1996, 100, 13121; (b) Z. Xu, M. Houssa, S. D. Gendt andM. Heyns, App. Phys. Lett., 2002, 80, 1975.
5 S. Ferrari, G. Scarel, C. Wiemer and M. Fanciulli, J. App. Phys.,2002, 92, 7675.
6 H. B. Park, M. J. Cho, J. Park, S. W. Lee, C. S. Hwang, J. P. Kim,J. H. Lee, N. I. Lee, H. K. Kang, J. C. Lee and S. J. Oh, J. App.Phys., 2003, 94, 3641.
7 T. Kawahara and K. Torii, IEICE Transactions on Electronics.,2004, E87C, 2.
8 J. J. Ganem, I. Trimaille, I. C. Vickridge, D. Blin and F. Martin,Nuclear Instruments & Methods in Physics Research SectionB-Beam Interactions with Materials and Atoms, 2004, 219, 856.
9 W. Chen, Q. Q. Sun, M. Xu, S. J. Ding, D. W. Zhang andL. K. Wang, J. Phys. Chem. C, 2007, 111, 6495.
10 D. H. Triyoso, R. I. Hegde, J. Grant, P. Fejes, R. Liu, D. Roan,M. Ramon, D. Werho, R. Rai, L. B. La, J. Baker, C. Garza,T. Guenther, B. E. White and P. J. Tobin, Journal of VacuumScience & Technology B, 2003, 22, 2121.
11 Z. M. Rittersma, F. Roozeboom, M. A. Verheijen, J. G. M. vanBerkum, T. Dao, J. H. M. Snijders, E. Vainonen-Ahlgren, E. Tois,M. Tuominen and S. Haukka, Journal of the ElectrochemicalSociety, 2004, 151, 716.
12 H. S. Chang, H. Hwang, M. H Cho and D. W. Moon, AppliedPhysics Letters, 2005, 86, 031906.
13 D. H. Triyoso, M. Ramon, R. I. Hegde, D. Roan, R. Garcia,J. Baker, X. D. Wang, P. Fejes, B. E. White and P. J. Tobin,Journal of the Electrochemical Society, 2005, 152, 203.
14 M. Cho, H. B. Park, J. Park, S. W. Lee, C. S. Hwang, J. Jeong,H. S. Kang and Y. W. Kim, Journal of the Electrochemical Society,2005, 152, 49.
15 K. Kukli, T. Aaltonen, J. Aarik, J. Lu, M. Ritala, S. Ferrari,A. Harsta and M. Leskela, Journal of the Electrochemical Society,2005, 152, F75.
16 D. H Triyoso, R. I. Hegde, B. E White and P. J. Tobin, Journal ofApplied Physics, 2005, 97, 124107.
17 D. Hellin, A. Delabie, R. L. Puurunen, P. Beaven, T. Conard,B. Brijs, S. De Gendt and C. Vinckier, Analytical Sciences, 2005,21, 845.
18 P. D. Kirsch, M. A. Quevedo-Lopez, Y. Li, H. J. Senzaki,J. J. Peterson, S. C. Song, S. A. Krishnan, N. Moumen,
Fig. 6 The three phases of growth kinetics.
3708 | Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 This journal is �c the Owner Societies 2009
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online

J. Barnett, G. Bersuker, P. Y. Hung, B. H. Lee, T. Lafford,Q. Wang, D. Gay and J. G. Ekerdt, Journal of Applied Physics,2006, 99, 023508.
19 I. S. Park, T. Lee, D. K. Choi and J. Ahn, Journal of the KoreanPhysical Society, 2006, 49, S544.
20 M. J. Cho, R. Degraeve, Pourtois, A. Delabie, L. A. Ragnarsson,T. Kauerauf, G. Groeseneken, S. De Gendt, M. Heyns andC. S. Hwang, IEEE Transactions on Electron Devices, 2007, 54, 752.
21 L. Nyns, L. Hall, T. Conard, A. Delabie, W. Deweerd, M. Heyns,S. Van Elshocht, N. Van Hoornick, C. Vinckier and S. De Gendt,Journal of the Electrochemical Society, 2006, 153, F205.
22 M. Ritala, M. Leskela, L. Niinisto, T. Prohaska, G. Friedbacherand M. Grasserbauer, Thin Solid Film, 1994, 250, 72.
23 J. Aarik, A. Aidla, A. A. Kiisler, T. Uustare and V. Sammelselg,Thin. Solid Film, 1999, 340, 110.
24 K. Kukli, J. Ihanus, M. Ritala and M. Leskela, Appl. Phys. Lett.,1996, 68, 3737.
25 M. L. Green, M. Y. Ho, B. Busch, G. D. Wilk, T. Sorsch,T. Conard, B. Brijs, W. Vandervorst, P. I. Raisanen, D. Muller,M. Bude and J. Grazul, J. App. Phys., 2002, 92, 7168.
26 M. Ylilammi, Thin Solid Films., 1996, 279, 124.27 E. L. Lakomaa, Appl. Surf. Sci., 1994, 75, 185.28 S. M. George, A. W. Ott and J. W. Klaus, J. Phys. Chem., 1996,
100, 121.29 A. Dkhissi, A. Esteve, C. Mastail, S. Olivier, G. Mazaleyrat,
L. Jeloaica and M. Djafari Rouhani, J. Chem. Theo. Comp.,2008, 4, 1915–1927.
30 M. Kotrla, Comp. Phys. Comm., 1996, 97, 82.31 P. Smilauer and D. D. Vvedensky, Phys. Rev. B, 1995, 52, 14263.32 Crystal Structure, ed. R. Wyckoff, John Wiley & Sons, New York,
1965, vol. 1.
33 A. B. Bortz, M. H. Kalos and J. L. Lebowitz, J. Comput. Phys.,1975, 17, 10.
34 M. Ritala and N. Leskela, in Handbook of Thin Film Materials,ed. H. S. Nalwa, Academic Press, New York, 2001, vol. 1.
35 A. Rahtu and M. Ritala, J. Mater. Chem., 2002, 12, 1484.36 (a) L. Jeloaica, A. Esteve, M. Djafari Rouhani and D. Esteve, App.
Phys. Lett., 2003, 83, 542; (b) A. Esteve, L. Jeloiaca,G. Mazaleyrat, A. Dkhissi, M. Djafari Rouhani, S. Ali Messaoudand N. Fazouan, MRS., 2003, 786, 35; (c) L. Jeloaica, A. Esteve,A. Dkhissi and M. Djafari Rouhani, Comp. Mater. Science., 2005,33, 59; (d) A. Dkhissi, A. Esteve, L. Jeloaica, D. Esteve andM. Djafari Rouhani, J. Am. Chem. Soc., 2005, 127, 9776;(e) A. Dkhissi, A. Esteve, L. Jeloaica, M. Djafari Rouhani andG. Landa, Chem. Phys., 2006, 323, 179.
37 D. Blin, PhD thesis, University of Montpellier, 2003.38 O. Renault, D. Samour, D. Rouchon, P. H. Holliger, A. M. Papon,
D. Blin and S. Marthon, Thin Solid Films, 2003, 428, 190.39 L. T. Zhuravlev, Colloids and Surfaces A, 2000, 173, 1.40 D. Riihela, M. Ritala, R. Matero and M. Leskela, Thin Solid
Films., 1996, 289, 250.41 J. Aarik, A. Aidla, A. Jaek, A. A. Kiisler and A. A. Tammik, Acta
Polytech. Scand., Chem. Technol. Metall. Ser., 1990, 195, 201.42 L. Hiltunen, H. Kattelus, M. Leskela, M. Makela, L. Niinisto,
E. Nykanen, P. Soininen and M. Tiitta, Mater. Chem. Phys., 1991,28, 379.
43 G. D. Wilk and D. A. Muller, App. Phys. Lett., 2003, 83, 3984.44 P. D. Kirsch, M. A. Quevedo-Lopez, H. J. Li, Y. Senzaki,
J. J. Peterson, S. C. Song, S. A. Krishnan, N. Moumen,J. Barnett, G. Bersuker, P. Y. Hung, B. H. Lee, T. Lafford,Q. Wang, D. Gay and J. G. Ekerdt, J. App. Phys., 2006, 99,023508.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 3701–3709 | 3709
Dow
nloa
ded
by U
nive
rsite
Pau
l Sab
atie
r on
22
May
201
2Pu
blis
hed
on 2
6 M
arch
200
9 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/B
8215
02B
View Online





























