Embed Size (px)
Citation preview

Third Edition
Cutaneous T-Cell Lymphomas:
Mycosis Fungoides and Sézary Syndrome
A Guide for the Practicing Oncologist
Christiane Querfeld, H. Miles Prince, and Steven T. Rosen
City of Hope National Medical Center, Duarte, California
andPeter MacCallum Cancer Centre,
Melbourne, Victoria, Australia

Christiane Querfeld, MD, PhD, is a board-certified der-matologist and dermatopathologist who specializes in the diagnosis and treatment of cutaneous lymphoma. She is the Chief of the Division of Dermatology, Director of the Cutaneous Lymphoma Program, and Assistant Professor at City of Hope Cancer Center & Beckman Research Institute. Her clinical practice is dedicated to patients with cutane-ous lymphoma. Her research focus has been on investiga-tions of the biology of cutaneous lymphomas and novel therapies for these diseases, and she serves as co-principal
investigator on several clinical phase 1, 2, and 3 trials of cutaneous T-cell lym-phomas (CTCL). She is a member of an international collaboration of cutaneous lymphoma experts aimed toward the improved prognostication and characteriza-tion of the disease. Her laboratory-based projects are aimed to identify the con-tributing elements of the tumor microenvironment in CTCL and help elucidate the underlying mechanisms that lead to disease progression through maintenance and proliferation of malignant cells in the skin. Dr. Querfeld received her medical degree at the University of Cologne and a PhD at the University of Heidelberg. She then completed her internship at Johns Hopkins University, her dermatology training at University of Chicago and fellowships in dermatopathology and cuta-neous lymphoma at Northwestern University. A recognized authority in the field, she has published over 90 scholarly articles on cutaneous lymphoma.
Professor Miles Prince, AM, MD, is an Australian hema-tologist and Professor of Medicine at both Melbourne and Monash Universities, Director of the Centre for Blood Cell Therapies at the Peter MacCallum Cancer Centre, and Pro-fessor/Director of Cancer Immunology and Molecular On-cology at Epworth. He is a very active clinician, also over-seeing clinical and laboratory research, the latter involving stem cell research and cancer immunology. He has been in-volved in numerous clinical trials of new therapies for blood cancers and has been the principal investigator of over 80
clinical trials. Recently, his clinical research focus has been on epigenetic therapies, novel monoclonal antibodies, immunotoxins, and immunomodulatory agents for the treatment of CTCL. He has published over 400 peer-reviewed manuscripts. He leads the Cutaneous Lymphoma program in Melbourne and is Chairman of the Australian Skin Lymphoma Network. Miles is a past board member of the Interna-tional Society of Cutaneous Lymphoma.
Christiane Querfeld, MD, PhD
Miles Prince, AM, MD
About the Authors

Steven T. Rosen, MD, is the Provost, Chief Scientific Of-ficer, Director of the Comprehensive Cancer Center and the Beckman Research Institute, Irell & Manella Can-cer Center Director’s Distinguished Chair for the City of Hope. Following his graduation with distinction from Northwestern University Medical School’s 6-year honors program in 1976, Dr. Rosen completed his residency in In-ternal Medicine at Northwestern and a fellowship in Medi-cal Oncology at the National Cancer Institute. He served as the Director of the Robert H. Lurie Comprehensive Cancer
Center at Northwestern University Feinberg School of Medicine from 1989–2014. Dr. Rosen’s laboratory research focuses on experimental therapeutics and hemato-logic malignancies. Dr. Rosen has received numerous grant awards and contracts and has published more than 400 scientific papers. He is editor-in-chief of the book series Cancer Treatment And Research. Dr. Rosen is the current Chair of the Medical Science Committee of the LLS and serves on its board. He also serves on the board of American Society of Clinical Oncology’s Conquer Cancer Founda-tion. Dr. Rosen has been an advisor to more than 2 dozen NCI Comprehensive Cancer Centers. He was the recipient of Northwestern University Medical School’s Alumni Achievement Award (1994), the Martin Luther King Humanitarian Award from Northwestern Memorial Hospital (1995), the Marv Samuel Award from the Chicago Baseball Cancer Charities (1996), recognition from the Woman’s Board of Northwestern Memorial Hospital for Compassionate Care (1996), and Israel Cancer Research Fund, Man of Distinction Award (2011) and Lifetime Achieve-ment Award (2015).
Steven T. Rosen, MD

ContentsIntroduction .....................................................................................................1
Overview of CTCL .....................................................................................2Classification of cutaneous T-cell lymphomas 2Mycosis fungoides and Sézary syndrome 4Epidemiology 6Etiology and pathogenesis 6Tumor microenvironment 7Molecular and biologic characteristics 8
Screening and diagnosis ...................................................................10Clinical manifestation and differential diagnosis 10Pathology 10Specific diagnostic studies 10Evaluation of the patient 11
Staging and prognosis ..................................................................13TNMB classification for CTCL 13Prognostic markers 16
Treatment options ...................................................................................171. Topical treatments 19 Phototherapy 19 Steroids 20 Nitrogen mustard 20 Retinoids 21 Radiation 212. Systemic treatments 22 Biologic therapies 22
• Interferons 22• Retinoids 23

• Extracorporeal photopheresis 24• Alemtuzumab 25• Denileukin diftitox 26• Histone deacetylase inhibitors 26• Brentuximab vedotin 28• Mogamulizumab 28
Chemotherapy 28Single-agent chemotherapy 29• Pegylated doxorubicin 29• Nucleoside analogues 29• Antifolates 30
Multiagent chemotherapy 31 Stem cell transplant 313. Investigational treatments 32 Anti-KIR3DL2 32 Immune checkpoint inhibitors 32 Antagomirs 33 Immunomodulatory agents 33 Chimeric antigen receptor T 344. General health care 34
Outcome ............................................................................................................35
Case reports ...................................................................................................361. Approach and management of patient with early-stage MF 362. Approach and management of patient with advanced/tumor-stage MF 363. Approach and management of patient with advanced MF/SS 37 4. Approach and management of patient with Sézary syndrome 37
Summary ..........................................................................................................39
References .......................................................................................................40
Resources for additional information ...............................49


1
IntroductionCutaneous involvement by hematologic neoplasms may occur secondarily as leukemia cutis in various forms of leukemia and in non-Hodgkin lymphomas such as adult T-cell leukemia lymphoma and angioimmunoblastic T-cell lym-phoma, or as primary cutaneous lymphoma. Primary cutaneous lymphomas represent a heterogeneous group of T- and B-cell lymphomas with distinct clinical presentations, histopathologic features, treatment approaches, and outcomes. Although diagnosis and classification of primary cutaneous lymphomas was long complicated by various classification systems that had not considered the distinct clinical behavior of cutaneous versus extracutaneous lymphomas, this problem was largely overcome with the development of the World Health Organization (WHO) and European Organization for Research and Treatment of Cancer (EORTC) consensus classification for cutaneous lymphomas in 2005.1 A revision of the WHO classification of lymphoid neoplasms with clinical relevance to diag-nosis, prognosis, and treatment regimens of current entities as well as the addition of new provisional entities has since been published in 2016.2 In this guide, we focus on primary cutaneous T-cell lymphomas, with special emphasis on mycosis fungoides and Sézary syndrome.

2
Overview of cutaneous T-cell lymphomasClassification of cutaneous T-cell lymphomasThe group of cutaneous T-cell lymphomas (CTCL) account for up to 80% of all cutaneous lymphomas, and based on the WHO–EORTC classification, these can be divided into indolent and aggressive forms (Table 1).1 Mycosis fungoides (MF) with patch, plaque, and/or tumor manifestations and the aggressive variant Sézary syndrome (SS), are the most common and best studied forms of CTCL. The spectrum of CD30+ lymphoproliferative disorders is the second most common group of CTCL that includes lymphomatoid papulosis (LyP), a chronic recurrent self-healing papulonodular skin eruption, and anaplastic large-cell lymphoma (ALCL), characterized by large nodules or ulcerating tumors that may undergo spontaneous regression (Figure 1). They share overlapping features such as the histologic appearance of large atypical cells bearing a CD30+ phenotype. Both appear to have an excellent prognosis. New subtypes of LyP have been described
with similar favorable prognosis. Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a CD8+ T-cell lymphoma with an α/β T-cell receptor (TCR) phenotype that involves the subcutaneous tissue mimicking panniculitis. SPTCL is associated with a protracted clinical course with recurrent subcutaneous lesions. In addition, a heterogeneous group of peripheral T-cell lympho-mas encompass entities that do not fit into other categories and include aggressive types such as primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma and cutane-ous γ/δ T-cell lymphoma (previously known as SPTCL with a γ/δ TCR phenotype), and the rather indolent cutaneous CD4+ small-/medium-sized pleomorphic T-cell lympho-proliferative disorder (previously classified as lymphoma) and cutane-ous acral CD8+ T cell lymphoma that are nearly always localized to a single site.2 Other hematologic neoplasms include adult T-cell leukemia/lymphoma (ATLL) that is etiologi-
Figure 1(B). Patient with primary cutaneous anaplastic large-cell lymphoma presenting with a solitary erythematous nodule.
Figure 1(A). Patient with lymphomatoid papulosis presenting with clusters of erythematous papules.

3
Table 1 WHO–EORTC consensus classification for primary cutaneous lympho-mas with relative frequency and 5-year survival
WHO–EORTC Frequency (%)
5-year survival (%)
Cutaneous T-cell and NK-cell lymphomaIndolentMycosis fungoides 44 88
• Follicular MF 4 80• Pagetoid reticulosis <1 100• Granulomatous slack skin <1 100
CD30+ lymphoprolif. diseases• Anaplastic large-cell lymphoma 8 95• Lymphomatoid papulosis 12 100
Subcutaneous panniculitis-like T-cell lymphoma 1 82
CD4+ small/medium pleomorphic T-cell lymphoma 2 72
Aggressive
Sézary syndrome 3 24Cutaneous peripheral T-cell lymphoma, unspecified 2 16
• Cutaneous aggressive CD8+ T-cell lymphoma <1 18
• Cutaneous γ/δ T-cell lymphoma <1 -
Cutaneous NK-/T-cell lymphoma, nasal type <1 -
Cutaneous B-cell lymphomaIndolentFollicle center cell lymphoma 11 95
Marginal zone lymphoma 7 99
Intermediate clinical behavior
Large B-cell lymphoma of the leg 4 55
Cutaneous diffuse large B-cell lymphoma, other <1 50
Intravascular large B-cell lymphoma <1 65
Abbreviations: EORTC, European Organization for Research and Treatment of Cancer; MF, mycosis fungoides; NK, natural killer; WHO, World Health Organization. Source: Ref 1.
4
cally associated with human T-lymphotropic virus type 1 (HTLV-1). About 50% of ATLL variants are associated with skin involvement. The chronic smolder-ing form is most frequently associated with skin lesions resembling MF and may precede the onset of systemic disease. Epstein-Barr virus (EBV)-associated extranodal natural-killer (NK)-/T-cell lymphoma, nasal type, and the blastic plasmacytoid dendritic cell neoplasm (formerly classified as EBV-negative blastic NK-cell lymphoma) frequently present in the skin with or without concurrent extracutaneous manifestations.
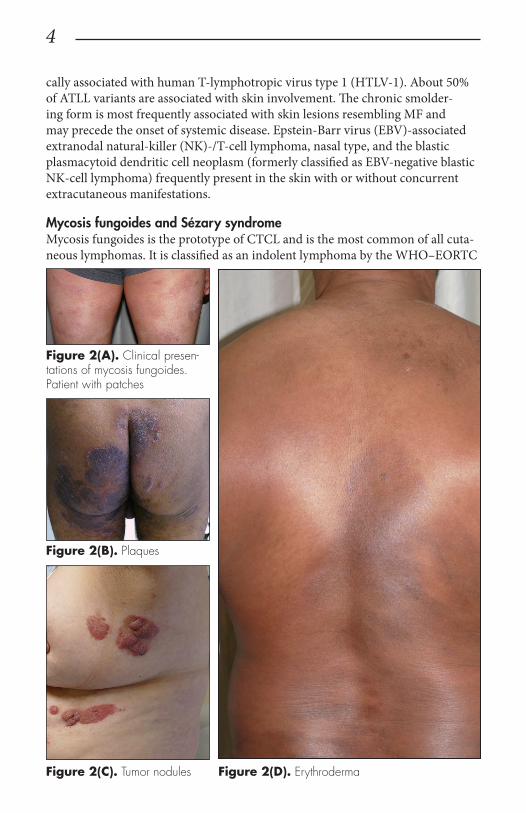
Mycosis fungoides and Sézary syndromeMycosis fungoides is the prototype of CTCL and is the most common of all cuta-neous lymphomas. It is classified as an indolent lymphoma by the WHO–EORTC
Figure 2(A). Clinical presen-tations of mycosis fungoides. Patient with patches
Figure 2(B). Plaques
Figure 2(C). Tumor nodules Figure 2(D). Erythroderma

5
Figure 3(A). Clinical presentation of a patient with Sézary syndrome presenting with generalized erythroderma.
Figure 3(B). Palmar keratoderma
and characterized by a chron-ic, slowly progressing disease with a protracted evolution. Clinically, MF is character-ized by erythematous patches, evolving into plaques, tumors, and/or erythroderma (Figure 2).3 Patients usually present with a prolonged history of a skin rash in sun-protected areas such as lower abdo-men, upper thighs, buttocks, inner arms, and breasts. The majority of patients remain in clinical stages limited to the skin; however, 20% of patients progress into more aggressive and advanced disease with either cutaneous or extracu-taneous tumor manifestations with an estimated 5-year survival rate of approximately 40%. MF has numerous clinical and histologic vari-ants. Apart from the classical Alibert-Bazin type of MF, 3 major variants have been rec-ognized in the WHO–EORTC classification including the most common variant fol-liculotropic MF, characterized by involvement of hair follicles and/or eccrine glands often leading to alopecia and associ-ated with a worse prognosis; pagetoid reticulosis (formerly named Woringer-Kolopp disease), presenting as a soli-tary psoriasiform plaque with intraepidermal (pagetoid) involvement of atypical T cells and localized to extremities with excellent control and sur-vival rates; and granulomatous

6
slack skin, which is character-ized by the development of folds of lax skin.
Sézary syndrome (SS) is part of the spectrum of erythro-dermic cutaneous T-cell lym-phomas (E-CTCL).1,4 E-CTCL can arise de novo (SS) or as a progression of preexisting MF (erythrodermic MF). Sézary
syndrome is an aggressive variant of CTCL with a leukemic component (Table 1). Approximately 3% to 5% of all newly reported cases of CTCL are patients with SS. It is characterized by circulating, atypical, malignant T lymphocytes with hy-perconvoluted (cerebriform) nuclei (Sézary cells), the presence of erythroderma, and often lymphadenopathy. Severe pruritus, ectropion, alopecia, dystrophic nails, and palmoplantar keratoderma with fissures are common associated features (Fig-ure 3). Patients with SS are immunocompromised due to defective T-cell function and therefore predisposed for opportunistic skin infections such as colonization with Staphylococcus aureus. In fact, most patients die of opportunistic infections. Patients who present with erythrodermic MF may also have coexisting patches, plaques, or tumors.
EpidemiologyCutaneous lymphomas are a group of rare diseases that represent 3.9% of all non-
Hodgkin lymphomas. MF represents the most common type of CTCL, comprising 50% of all cutaneous lymphomas. The incidence of CTCL has risen consistently since 1973, with an overall annual age-adjusted incidence of 6.4 to 9.6 cases per million according to large population-based studies.5–7 MF is typically seen at a me-dian age of 55 to 60 years. Incidence is higher among blacks and men but decreases with age. A substantial geographic variation in incidence was found based on 13 cancer registries (Surveillance, Epidemiology, and End Results Program [SEER]-13). Data suggest an interesting association with higher physician density and higher socioeconomic status. Patients with MF and SS are at significantly increased risk of developing a second primary lymphoma, especially Hodgkin lymphoma.8
Etiology and pathogenesisData on risk factors for CTCL are sparse, being mostly limited to MF. Microbio-
logic, environmental, and occupational factors have been implicated in the etiology of CTCL, but none has yet been verified. It has been suggested that the condition results from persistent antigen stimulation, with an increased risk for the develop-ment of CTCL in patients exposed to chemicals or pesticides; however, studies on the subject produced controversial results.9,10 There are studies that have shown in-creased expression of Toll-like receptors (TLRs) 2, 4, and 9 by keratinocytes in MF patients.1 Specifically, TLR2 recognizes pathogen-associated molecular patterns
Figure 3(C). Ectropion

7
(PAMPs) from S. aureus, and a number of studies provide evidence that cutaneous colonization with S. aureus influences the disease activity of MF/SS and may be the cause of the upregulation of TLRs in this disease. Significant improvement of erythroderma and extent of skin disease was seen after the eradication of S. aureus with antibiotics in patients with erythrodermic MF and SS.11 Unlike ATLL, which is etiologically associated with HTLV-1, most patients with CTCL are serologi-cally negative for HTLV-1.12 Some groups have found serologic evidence of EBV in CTCL patients, suggesting a possible role in the pathogenesis, but these suspicions have not been verified by others.13,14 Immunosuppression and/or immunosuppres-sive therapy might be a risk factor for the development of CTCL, as documented in patients following organ transplant or treatment of Hodgkin disease, in human immunodeficiency virus (HIV)-positive patients, and following pharmacologic blockade of TNF-α for immune-mediated disorders.15–19
Tumor microenvironmentSkin lesions in MF/SS are composed of malignant T cells and other nonmalig-
nant immune cells of the innate immune system (NK cells, dendritic cells [DCs], macrophages, and mast cells) and adaptive immune system (tumor-infiltrating T cells, T regulatory [Treg] cells) and stromal cells. All of these compose the tumor microenvironment in CTCL. In early-stage MF, the infiltrate is composed of pre-dominant nonmalignant T-helper cell type 1 (Th1) cells and dendritic cells inter-spersed with atypical T cells expressing a Th2 cytokine profile.20,21 A higher propor-tion of CD8+ tumor-infiltrating T cells are seen in skin biopsies of patients with patch/plaque lesions compared with those with more advanced T-stage/tumor dis-ease. In addition, a multivariate analysis confirmed the correlation between higher densities of CD8+ tumor-infiltrating T cells and improved survival rate in CTCL.22 FOXP3+ Tregs have also been correlated with superior survival rate in MF.23 Both tumor-infiltrating T cells and Tregs may contribute to an antitumor response, and their number is significantly decreased in advanced MF/SS.
The close interaction between DCs and MF cells is evident in the classic mor-phology of an epidermal Pautrier’s microabscess. One study showed that the ma-lignant CTCL cells grew in long-term cultures when they were cocultured with immature DCs.24 It is hypothesized that the growth of CTCL cells is driven by the interaction between the major histocompatibility complex (MHC) class 2 peptides on DCs and the TCR and with the interaction of CD40 with its ligand CD40L on the CTCL cell.24–26 Growth was inhibited by CD3 antibodies that bound to the TCR or interfered with the interaction of CD40 with its ligand on the CTCL cell. Dys-functional DCs are found in peripheral blood samples of patients with SS.27
Macrophages have key roles in tumor development and invasion in several hu-man cancers, but little is known about their pathogenic role in CTCL. It has been observed that the number of CD163+ macrophages in lesional skin of CTCL, atopic dermatitis, or psoriasis were significantly larger than in normal controls. Further, the numbers of CD163+ or CD68+ cells in patients with CTCL increased as more neoplastic T cells were found in the infiltrate and their number decreased after

8
treatment with topical steroid and ultraviolet light.28 Researchers have also demon-strated that the depletion of M2-like tumor-associated macrophages that express a wide array of anti-inflammatory molecules (eg, interleukin [IL]-10, TGF-β, argin-ase 1) delayed the development of this cutaneous lymphoma in xenografted human CTCL cells in immunocompromised mice.29 These studies suggest that macrophag-es may have a critical role in MF and their interaction with the neoplastic T cells through unknown mechanisms may promote progression of the skin lymphoma.
Mast cells have also been shown to play a protumorigenic role in CTCL and in cutaneous B-cell lymphomas.30 In typical CTCL tumor lesions, the number of mast cells was increased and was particularly prominent at the lymphoma margins. Higher mast cell counts were found in patients with progressing disease compared to others with nonprogressive disease. Moreover, the number of mast cells in dif-ferent stages of MF was positively correlated with advanced stages and inversely correlated with prognosis.30
Molecular and biologic characteristicsThe skin-homing malignant lymphocytes in MF/SS typically exhibit the pheno-
type of mature CD4+ memory T cells and may display differentiation markers of FOXP3 regulatory T cell (Treg), Th2, or Th17 cell phenotypes.25,31,32 Treg cells pro-duce IL10 and TGFβ, which promote an immunosuppressive microenvironment in MF. Patch/plaque lesions in early-stage MF have a Th1 cytokine profile with high expression of IL-2, IL-12, and INF-γ, whereas in advanced/tumor-stage MF the immune milieu changes from a Th1- to Th2-cytokine profile. The Th2 cytokines (IL-4, IL-5, IL-10, IL-13) may lead to peripheral eosinophilia and high serum levels of IgE, erythroderma, immunosuppression, and increased susceptibility to bacte-rial infections that are major causes of death in advanced MF/SS. The Th17 pheno-type has been observed in all stages of MF.21,33,34 The Th17 cells produce the proin-flammatory IL-17, which indirectly promotes tumor growth in CTCL through the activation of the Jak3/Stat3 pathway.32 The migration of malignant T cells to the skin is driven by the expression of various chemokine receptors and adhesion molecules involved in normal skin immunosurveillance. During the pathogenesis and progression of MF, there is a change in the chemokine receptor profile on the malignant T cells. Although all MF stages showed an increased expression of the skin homing CCR4 that facilitates migration of the neoplastic T cells into the skin, an increased expression of the lymph node homing CCR7 is also seen in tumor-stage MF and SS. CCR7 expression is correlated with loss of epidermotropism and promotes extracutaneous spread with the migration of T cells to the blood, lymph nodes, and internal organs. CTCL cells are resistant to apoptosis induced by Fas and TNF-related-apoptosis-inducing ligand (TRAIL).35 Furthermore, neoplastic T cells in MF/SS express cell surface markers that cause dysfunction of the patient’s innate and adaptive immune system.36
MF/SS is believed to develop from a background of chronic antigenic stimula-tion and is characterized by an altered immune biology and underlying cytogenetic abnormalities that lead to clonal proliferation and accumulation of malignant T

9
cells.37 CTCL cells have an aberrant immunophenotype of CD4+ T-helper/memory profile with frequent loss of CD5, CD7, and/or CD26 antigen in advanced stages.38 The malignant cells also show clonal rearrangements for the gene that encodes the TCR.39 Persistent activation of the neoplastic T cells is demonstrated by the consti-tutive phosphorylation of intracellular signaling protein STAT3.40 These cells may express the activation markers CD45RO and the IL-2 α receptor (CD25) that have provided a target for biologic therapy with denileukin diftitox.41
Recent next-generation sequencing has revolutionized the understanding of the genetic landscape of CTCL. These analyses have revealed ultraviolet radiation and recombination activating gene (RAG) endonucleases as important mutagens. Furthermore, these studies have uncovered potentially targetable oncogenic mu-tations in the T-cell signaling and differentiation (CD28, CCR4), apoptotic path-ways (FAS), NF-κB and JAK-STAT (STAT3, STA5b) signaling pathway, PI3K/AKT pathway (PTEN and RHOA), epigenetic regulators (ARID1A, DNMT3A, and KMT2C), cell cycle regulators (CDKN2A and RB), and regulators of genome integ-rity (TP53 and ATM). Collectively, these somatic mutations are believed to drive lymphomagenesis via cancer-promoting changes in proliferation, apoptosis, and T-cell effector function.42 Recent discoveries indicate that the oncogenic microRNA miR-155 is overexpressed in affected skin from CTCL patients, and its expression is mediated by the JAK/STAT 5 pathway.43

10
Screening and diagnosisClinical manifestation and differential diagnosis
Early stages of MF typically present with patches. The early patch stage may re-semble eczema, psoriasis, secondary syphilis, atopic dermatitis, or “premycotic” parapsoriasis. Diagnosis is often delayed or missed in early-stage patients pretreated with topical steroids. Most patients had multiple biopsies before being diagnosed. The interval between the onset of skin lesions to the definitive diagnosis ranges from 4 to 10 years (mean, 6 years). To further complicate diagnosis, observations about invisible MF cases with recalcitrant pruritus have been reported and have documented MF variants mimicking a plethora of benign disorders. With progres-sive invasion and infiltration of the skin by the malignant T cells, patches evolve into plaques and/or tumors. In advanced stages of MF, the patients show a com-bination of patches, plaques, and nodular/tumor lesions or diffuse erythroderma.
Patients with SS generally present with severe disabling pruritus. The skin ap-pearance may vary from mild erythema to generalized exfoliative erythroderma with keratoderma and fissures on palms and soles. Other problems include elec-trolyte imbalance, hypothermia, hair loss, and eye changes (ectropion). Lymphad-enopathy is often, but not always present. Diagnosis may be missed in the elderly whose symptoms of dry skin and pruritus are attributed to advanced age. Cutane-ous drug reactions, infectious manifestations, generalized seborrheic dermatitis, psoriasis, and chronic photosensitivity reactions may resemble E-CTCL.
Pathology Several histologic features are useful in establishing a diagnosis of MF/SS.44 In
most cases, it consists of an upper-dermal, band-like lymphocytic infiltrate with atypical lymphocytes with cerebriform nuclei, variable findings of inflammatory cells, and epidermal involvement with solitary cells or clusters of malignant lym-phocytes (Pautrier’s microabcesses). Tumor lesions express more diffuse and deep infiltrates with diminished epidermotropism. MF/SS is characterized by malignant CD4+/CCR4+ T cells that commonly lack the T-cell surface markers CD7 and/or CD26. A CD8+ phenotype has been seen in a hypopigmented variant of MF. Biopsies from erythrodermic MF or SS often lack epidermotropism or Pautrier’s microabscesses and may show a mild perivascular or superficial dermal infiltrate of atypical lymphocytes and minimal exocytosis of these cells into the epidermis. MF has numerous variants. Folliculotropic MF is the most common subtype involving adnexal structures of the skin including hair follicles and/or eccrine glands.
Specific diagnostic studies Immunophenotyping by immunohistochemistry on skin biopsies or flow cytom-
etry on skin and blood samples has an important role in the evaluation of CTCL. Studies have demonstrated an increased CD4/CD8 ratio, loss of subset markers such as CD26 and CD7, and aberrant expression or loss of pan-T-cell markers such as CD2, CD3, and CD5 in many affected patients.38
11
In its earliest stages, MF is often diagnosed as dermatitis and a definitive diagno-sis may be difficult to establish. Therefore, the presence of a dominant T-cell clone is an important diagnostic criterion. Clonal rearrangements can be detected by using the polymerase chain reaction single-strand conformational polymorphism analysis (PCR SSCP); high-throughput RNA-sequencing allows for in-depth anal-ysis of the TCR repertoire.45,46 These molecular studies can often be very useful, in association with histopathology and immunophenotyping, to support the clinical diagnosis (Table 2).
Table 2 Key diagnostic criteria for mycosis fungoides and Sézary syndrome
Criteria Early-stage MF Advanced-stage MF/SSClinical • Persistent patches/
plaques in sun protected areas
• Skin tumors• Generalized erythroderma• Lymphadenopathy
Histopathologic • Epidermotropism• Pautrier’s
microabscesses• Atypical lymphocytes• Band-like infiltrate
• Lack of epidermotropism and Pautrier’s microabscesses
• Often nondiagnostic in patients with SS
Immunophenotypic • CD4+, CD8- phenotype (skin)
• CD4+, CD8- phenotype (blood)
• Loss of T-cell subset markers: CD4, CD7, CD26
• Loss of pan T-cell markers: CD2, CD3, CD5
Molecular • Clonal T-cell receptor gene rearrangement (skin)
• Clonal T-cell receptor gene rearrangement (blood)
Laboratory • Sézary cells• Elevated LDH
Abbreviations: MF, mycosis fungoides; LDL, lactate dehydrogenase: SS, Sézary syndrome.
Evaluation of the patientAll patients with MF/SS should initially be seen and comanaged by a multidis-
ciplinary cutaneous lymphoma team consisting of members from dermatology and oncology with the support of other healthcare professionals such as radiation oncologists, pathologists, and clinical psychologists. Routine evaluation should in-clude complete physical examination, complete blood count with differential and peripheral blood smear for Sézary cell count, chemistry panel with lactate dehy-

12
drogenase (LDH), skin biopsy for histology, immunophenotyping and gene rear-rangement studies, and lymph node biopsies in cases with enlarged nodes at pres-entation to establish the diagnosis and staging (Table 3). Serologic tests for HIV and HTLV-1 should be considered in certain patients. Diagnosis in early stages of MF has been improved due to advances in TCR gene rearrangement analyses with increased sensitivity and specificity.39,47 Imaging studies should be reserved for pa-tients with clinical and laboratory findings suggestive of systemic disease or promi-nent lymphadenopathy. Bone marrow biopsy is a consideration in advanced-stage disease. Histopathologic and molecular results should be correlated with clinical findings and patients classified according to the WHO-EORTC consensus classi-fication.
Table 3 Diagnostic investigations to evaluate a patient for CTCL at initial visit
Diagnostic Procedures
CBC with differential
Chemistry panel, LDH
Skin biopsy for histopathology, immunophenotyping, and TCR gene analysis
Blood smear for Sézary cell count
Flow cytometry: T-cell subsets, CD4/CD8 ratio
TCR gene analysis (skin, blood, lymph node)
Lymph node biopsy of palpable nodes
Imaging: CT/PET scans (chest, abdomen, pelvis)*
Bone marrow biopsy*
Serologic tests: HTLV-1, HIV*
Abbreviations: CBC, complete blood count; CTCL, cutaneous T-cell lymphoma; CT/PET, computed tomography/positron emission tomography; HIV, human immunodeficiency virus; HTLV-1, human T-lymphotropic virus type 1; LDH, lactate dehydrogenase; TCR, T-cell receptor.* Selected patients based on presentation, clinical examination, and laboratory findings.

13
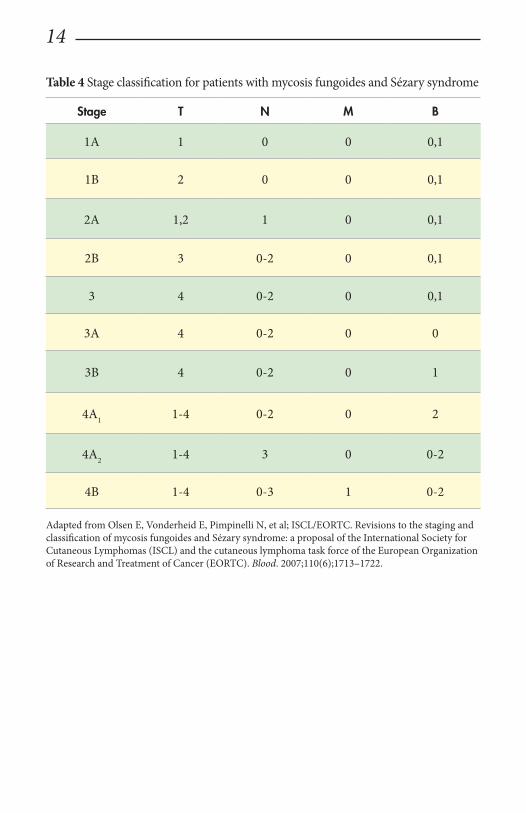
Staging and prognosisTNMB classification for cutaneous T-cell lymphoma
Accurate staging of patients with MF/SS is essential both for its prognostic value and for treatment decisions (Table 4). In contrast to the staging system that is tra-ditionally used for other lymphomas, the widely used and recommended staging system for CTCL relies on the TNMB (tumor, node, metastasis, blood) classifica-tion adopted by Bunn and Lamberg in 1979 that has been revised to include blood stage (Table 5).48
It considers the extent of skin involvement (T), presence of lymph node (N) and visceral disease (M), and detection of Sézary cells in the peripheral blood (B). Pa-tients with stage 1A disease (T1, N0, M0) have limited patch/plaque disease with less than 10% of body surface area (BSA) involvement. Patients are usually asymp-tomatic and may remain at this stage for years. As patches and plaques involve more than 10% of the BSA, patients are classified as stage 1B disease (T2, N0, M0). Stage 2A disease (T1–2, N1, M0) includes the skin findings of stage 1A or 1B dis-ease with the additional presence of peripheral lymphadenopathy. Stage 2B (T3, N0/1, M0) is associated with the development of skin tumors, which may arise de novo or in preexisting patches or plaques with or without associated lymphaden-opathy. Patients with erythroderma have stage 3 disease (3A: T4, N0-2, M0; B0; 3B: T4, N0-2, M0, B1) with or without low blood tumor burden. Erythrodermic patients with high blood tumor burden are staged as 4A1 (T1-4, N0-2, M0, B2). Involvement of nodes by tumor cells (LN3, LN4) without visceral involvement is classified as stage 4A2 (T1–4, N3, M0, B0-2), and with visceral and/or bone mar-row involvement classified as stage 4B (T1–4, N0-3, M1, B0-2). The International Society of Cutaneous Lymphoma (ISCL) has recommended definitions based on the degree of blood involvement for subsets of erythrodermic CTCL to address the differences among erythrodermic forms of CTCL (E-CTCL) (Table 6).4 B1 score would be defined as Sézary cell count of less than 1000 cells/m3 or less than 20% Sézary cells on peripheral smear. B2 score would be more than 1000 cells/m3 or greater than 20% Sézary cells on peripheral smear.
Histologic classification of lymph node (LN) involvement is also used as a prog-nostic value and for treatment decisions in patients diagnosed with MF/SS.49 It was found that the LN class correlates with disease progression and survival.49,50 Moreover, detection of a monoclonal T-cell population within nodes is associated with an inferior survival and outcome regardless of LN class.51 LN1 rating defines reactive changes, LN2 and LN3 nodes describe small or large clusters of atypical cells in paracortical T-cell regions, respectively, while LN4 nodes define frank ef-facement (Table 5).
14
Table 4 Stage classification for patients with mycosis fungoides and Sézary syndrome
Stage T N M B
1A 1 0 0 0,1
1B 2 0 0 0,1
2A 1,2 1 0 0,1
2B 3 0-2 0 0,1
3 4 0-2 0 0,1
3A 4 0-2 0 0
3B 4 0-2 0 1
4A1 1-4 0-2 0 2
4A2 1-4 3 0 0-2
4B 1-4 0-3 1 0-2
Adapted from Olsen E, Vonderheid E, Pimpinelli N, et al; ISCL/EORTC. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6);1713–1722.

15
Table 5 TNMB classification for patients with mycosis fungoides and Sézary syndrome
T (skin)T1 Limited patches, papules, and/or plaques (<10% of BSA)
T1a: Patch only; T1b: Plaque +/- patchT2 Generalized patches, papules, and/or plaques (>10% of BSA)
T2a: Patch only; T2b: Plaque +/- patchT3 One or more tumorsT4 Generalized erythroderma (>80% BSA)
N (nodes)N0 No clinically abnormal peripheral lymph nodes
N1 Clinically abnormal peripheral lymph nodes; histopathology Dutch grade 1 or NCI LN0-2
N1a: Clone negative; N1b: Clone negative
N2 Clinically abnormal peripheral lymph nodes; histopathology Dutch grade 2 or NCI LN3
N2a: Clone negative; N2b: Clone negative
N3 Clinically abnormal peripheral lymph nodes; histopathology Dutch grades 3–4 or NCI LN4; clone positive or negative
N4 Clinically abnormal peripheral lymph nodes; no histologic con-firmation
M (viscera)M0 No visceral involvement
M1 Visceral involvement (must have histopathologic confirmation and organ specified)
B (blood)B0 Absence of significant blood involvement (<5% Sézary cells)
B0a: Clone negative; B0b: Clone negativeB1 Low blood tumor burden (>5% Sézary cells; without B2 criteria)
B1a: Clone negative; B1b: Clone negativeB2 High blood tumor burden (>1000/uL Sézary cells; clone positive)
Abbreviations: BSA, body surface area; TNMB, tumor, node, metastasis, blood.
Adapted from Olsen E, Vonderheid E, Pimpinelli N, et al; ISCL/EORTC. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6);1713–1722.

16
Table 6 Proposed classification for erythrodermic CTCL and relative hematologic criteria devised by the International Society for Cutaneous Lymphoma (ISCL) in their consensus conference on erythrodermic CTCL
Erythrodermic CTCL Preexisting MF Blood findings TNMB
Sézary syndrome Rarely Leukemic T4, N0-3, M0-1, B2*
Erythrodermic MF Always Absent or minimal
T4, N0-3, M0-1, B0-1*
Erythrodermic CTCL
Absent Absent or minimal
T4, N0-3, M0-1, B0-1*
not other specified
Abbreviations: CTCL, cutaneous T-cell lymphoma; MF, mycosis fungoides; TNMB, tumor, node, metastasis, blood.*B0 <5% circulating Sézary cells; B1 Sézary cell count of <1000 cells/m3 or <20% atypical T cells on peripheral smear; B2 Sézary cell count of >1000 cells/m3 or >20% atypical T cells on peripheral smear.Source: Ref 4
Prognostic markersThe most important predictive factor for survival remains the T classification
(tumor burden), extracutaneous manifestation, and patient age.52 Inferior survival has been shown in plaque over patch disease for both limited (T1) and extensive (T2) skin disease.53 In addition, several independent adverse prognostic factors have been identified including large cell transformation, follicular mucinosis, thickness of tumor infiltrate, increased LDH, and β2 microglobulin.53–55 Patients with large circulating Sézary cells were also found to have a worse prognosis. A high Sézary cell count, loss of T-cell subset markers such as CD5 and CD7, and chromosomal abnormalities in T cells are also independently associated with a poor outcome.56 The existence of a blood clonal T-cell population, detected by PCR, was of poor predictive survival value independent of the T stage and lymph node involvement.57 Previous studies of prognostic factors have been mostly single center experiences, with a wide variation of treatment patterns. To better define the true impact of these prognostic factors, the Cutaneous Lymphoma International Consortium (CLIC) has been formed to collect well-defined prospective data to develop a Cutaneous Lymphoma International Prognostic Index (CLIPI) toward an improved prognostication and stratification for management in patients with MF/SS. Initial results from this international collaboration included data from 1275 patients with advanced-stage MF/SS and confirmed poor survival in these patients, with a median overall survival of 63 months. Univariate analyses revealed that stage 4, age older than 60 years, large cell transformation in skin, elevated WBC, and elevated LDH were significantly associated with a worse survival.58

17
Treatment optionsThe development of treatment strategies for patients with CTCL relies on the
extent of disease (TNMB stage) and its impact on a patient’s quality of life. No treat-ment is considered to be curative, and survival has not been shown to benefit from early intervention, even with multiagent chemotherapies combined with electron beam radiation.59 Early-stage (1A to 2A) MF has a favorable prognosis. The disease is mostly limited to the upper dermis and epidermis, which requires skin-directed therapies.
Current topical therapies include corticosteroids, nitrogen mustard (mechlo-rethamine), bexarotene, phototherapy with psoralens and ultraviolet light A (PUVA), ultraviolet B (UVB), or narrowband (NB)-UVB, and total skin electron beam therapy (TSEBT) (Table 7).60 Patients with refractory early-stage disease ben-efit from combination therapies such as PUVA or NB-UVB with low-dose systemic bexarotene or INF-α. Treatment goals in advanced stages of MF/SS should be to reduce tumor burden, relieve symptoms, delay disease progression, and preserve quality of life. In particular, SS is known to be refractory to most therapies and historically no treatment has been demonstrated to modify significantly the natu-ral course of this disease. Current approaches focus on biologic agents and recon-stitution of the immune function using interferons, extracorporeal photopheresis (ECP), oral bexarotene, monoclonal antibodies, recombinant fusion toxins, IL-12, and combinations of the aforementioned, for disease control. Single or localized skin tumors are ideally treated with localized radiotherapy. Mono- or polychemo-therapy is best reserved for patients with rapidly progressing disease that needs im-mediate palliation. High-dose chemotherapy followed by allogeneic bone marrow transplant is an option for younger patients with refractory advanced-stage disease.

18
Table 7 Recommended treatment approaches for patients with mycosis fungoides and Sézary syndrome
Early-stage MF
• Skin-directed SteroidsPhototherapyNitrogen mustardBexaroteneRadiation/TSEBT
Refractory early-stage MF
• Combined (skin-directed, systemic) therapy
Phototherapy & INF-αPhototherapy & bexarotene (low dose)HDAC inhibitors(vorinostat; romidepsin)
Advanced MF/SS• Biologic therapy INF-α
Retinoids/rexinoid (bexarotene)ECPHDAC inhibitors(vorinostat; romidepsin)Brentuximab vedotinAlemtuzumabLenalidomideTSEBT
• Combined therapy INF-α & phototherapyINF-α +/- bexaroteneRetinoid & phototherapyECP +/- INF-α +/- bexaroteneRomidepsin + lenalidomide
• Systemic chemotherapy• Single agent Pegylated doxorubicin
Antifolates (methotrexate, pralatrexate)Purine/pyrimidine analogues(pentostatin, gemcitabine)
Combination chemotherapy• Stem cell transplant Nonmyeloablative allogeneic • Investigational therapy Mogamulizumab
Immune checkpoint inhibitors (anti-PD1, anti-PD-L1, anti-CD47)Anti-KIR3DL2Antagomirs (miR-155)
Abbreviations: ECP, extracorporeal photopheresis; HDAC, histone deacetylase; INF-α, interferon-alpha; MF, mycosis fungoides; SS, Sézary syndrome; TSEBT, total skin electron beam therapy.

19
1. Topical treatments
PhototherapyPhototherapy is one of the mainstay skin-directed regimens in CTCL. Of the
available phototherapies, PUVA therapy has been widely used with established benefit in early-stage MF for more than 25 years. It involves treatment with orally administered 8-methoxypsoralen (8-MOP) that sensitizes the skin to ultraviolet A (UVA) irradiation. The initial UVA dosage is approximately 0.5 J/cm2 and will be increased per treatment as tolerated or up to the minimal erythema dose. Patients are required to use proper eye protection for 12 to 24 hours after a PUVA treatment to prevent the formation of cataracts. PUVA has been successfully utilized in the treatment of early-stage MF since 1976.61 Several studies confirmed high remission rates in early stages of MF, with reported complete remissions (CR) in up to 71.4% of patients.62,63 Therapy is typically given 3 times a week until CR is achieved. Ad-ditional maintenance therapy can be administered while gradually reducing PUVA to once every 4 to 6 weeks to maintain longer remission times. Common PUVA side effects include erythema, photodermatitis, pruritus, and nausea, which are generally managed with dose reduction or interruption. PUVA has also been suc-cessfully applied in the treatment of hypopigmented MF.64 Long-term remissions of more than 8 years have been reported in patients with early-stage MF.65 Disease-free survival rates for stage 1A at 5 and 10 years were 56% and 30%, respectively, and for stage 1B/2A 74% and 50%, respectively. Long-term exposure was associat-ed with an increased risk for developing chronic photodamage and non-melanoma skin cancer in about a third of patients.
Two multicenter, dose-randomized evaluations of oral low-dose bexarotene (75 mg or 150 mg daily for 24 weeks) and PUVA in patients with stage 1B-2A CTCL were performed in the US and Europe. Preliminary results have shown a response rate similar to PUVA monotherapy, but the results were achieved with lower cu-mulative Joules (Guitart J, personal communication). Although being effective in clearing superficial lesions, PUVA has its limitations in clearing deeply infiltrated lesions. PUVA is less effective in erythrodermic, folliculotropic, or tumor stage of MF; however, a combination with low-dose systemic agents (bexarotene or IFN-α) may be considered.66,67
The efficacy of broadband UVB is more limited to patch stage, while PUVA is also effective in clearing plaques. The effects of UVB phototherapy were retrospec-tively evaluated in 37 patients with MF limited to patch/plaque disease.68 Complete remission (CR) was achieved in 71%, with a median duration of 22 months. Of patients with disease limited to patches, 83% achieved remission, whereas none of the patients with plaque disease achieved remission. Narrowband UVB is consid-ered to be less carcinogenic and may offer an alternative treatment option in early-stage MF, but randomized clinical trials are needed to confirm its efficacy and long-term remission rates. In 3 small retrospective analyses, patients with clinical stage 1A/1B and parapsoriasis treated with NB-UVB showed CR rates between 54.2% and 83%.69,70 Remission times were short, however, and a maintenance schedule has

20
been difficult to establish. In patients with hypopigmented MF, NB-UVB is widely used.70 Patients with darker skin may still not respond as well to this therapy as light-skinned patients.70
SteroidsTopical corticosteroids are frequently prescribed in MF patients to induce clear-
ing of skin lesions in those with limited patches (stage 1A) and as adjunctive therapy to decrease erythema, scaling, and pruritus. In an investigational trial, 79 patients were treated daily with topical class 1 to class 3 steroids.71 Thirty-two (63%) of stage T1 patients and 7 (25%) of stage T2 patients achieved complete clearing. Thirteen patients (40%) with stage T1 and 2 patients (29%) with stage T2 relapsed; however, the median observation time was only 9 months. Reported side effects were minor skin irritation in 2 patients, reversible skin atrophy in 1 patient, and reversible de-pression of serum cortisol levels in 10 patients. A more recent clinical observation from the same treatment center reported similar efficacy in about 100 patients with T1 stage and 50 patients with T2 stage when treated with a topical class 1 steroid (clobetasol 0.05% cream or ointment) as first-line treatment.72 Treatment was ap-plied twice daily and generally well tolerated with no signs of adrenal insufficiency detected. A sustained response was not seen after steroid discontinuation.
Nitrogen mustardNitrogen mustard (mechlorethamine) belongs to a class of drugs known as
alkylating agents. The topical formulation has been widely used as a first-line treat-ment of early-stage MF since 1959. Nitrogen mustard-induced DNA damage has been the primary mechanism responsible for its systemic anticancer effects, but the mechanism of action of the topical formulation remains elusive. It does not appear to act through its alkylating properties, but rather induces cell-mediated antitumor responses via a delayed hypersensitivity reaction.73,74
Many investigators have demonstrated the efficacy of topical nitrogen mustard at a concentration of 0.1% to 0.2% in an aqueous or ointment base, with reported CR rates up to 72% of early-stage MF patients and occasional long-term remis-sions of more than 8 years.75 Updates on 203 patients with MF (clinical stage 1–3) treated with topical nitrogen mustard demonstrated its efficacy with reported CR rates of 76% to 80% for patients with stage 1A, and 35% to 68% for those with stage 1B disease.76 Skin clearance may require 6 months or longer and is followed by maintenance therapy; however, there is no evidence that prolonged maintenance is beneficial. Only 11% of patients maintain CR after 10 years. No secondary malig-nancies related to therapy were reported. A recent randomized, controlled, multi-center trial of a new nitrogen mustard 0.02% gel to evaluate its safety and efficacy by comparing it to the compounded nitrogen mustard 0.02% ointment in stage 1A–2A MF resulted in similar efficacy, leading to its approval in 2013 by the FDA for the treatment of stage 1A/2B MF patients with at least 1 previous skin-directed therapy.77 The most common adverse effects were dermatitis, pruritus, bacterial skin infection, and hyperpigmentation.

21
RetinoidsBexarotene 1% gel was approved by the FDA for early-stage MF in 2000 and is
a useful addition to skin-directed therapies in limited stage disease. It is, however, expensive and causes frequent skin irritation, which makes it unlikely that bexaro-tene gel would be used in patients with more than 10% of skin involvement.78 Bex-arotene gel is applied sparingly to patches or plaques and is most effective and best tolerated when used twice daily. Topical bexarotene has been evaluated in a dose-escalating trial with concentrations ranging from 0.1% to 1.0%.79 Median treatment duration with bexarotene gel was 10.5 months. CR rate was 21%, with a 63% overall response rate and a 75% response rate in treatment-naïve patients. Median dura-tion of remission was 24 months. A multinational phase 3 study of patients with refractory and/or persistent early-stage MF, treated with 1% bexarotene gel in a dose-frequency escalating fashion, demonstrated an overall response rate of 44% with CR in 8%.80 Median duration was less favorable at 7 months.
A pilot study with 0.1% tazarotene gel, a widely used retinoic acid receptor (RAR)-selective retinoid for psoriasis, was conducted in 20 patients with early-stage MF.81 Daily application for 6 months resulted in similar responses compared to bexarotene gel and may serve as an alternative approach to topical bexarotene.
RadiationPatients with localized tumors (stage 2B) may require systemic treatment, but
mostly respond very well to localized orthovoltage radiation. A single-fraction ra-diation of 700 to 800 cGy provides excellent palliation for CTCL lesions and is cost effective and convenient for the patient.82 Granulomatous slack skin may also benefit from radiation, although experiences are very limited.1 Total skin electron beam therapy (TSEBT) may be used in patients with more disseminated cutaneous disease. It is a treatment in which ionizing radiation is administered to the entire skin surface penetrating to the dermis. The standard total dose is 36 Gy delivered with electrons of at least 4 MeV energy and fractionated over 8 to 10 weeks. Re-ported CR rates range from 40% to 98% among patients with T1 and T2 stage, but relapse rates are high.83,84 These results were confirmed by a recent update on 57 pa-tients with T1/T2 stage MF.85 Twenty-one patients (87.5%) with T1 and 28 patients (84.8%) with T2 achieved CR. The median duration of remission was 26 months. Long-term analysis revealed that 75% of T1 patients and 25% with T2 were disease-free after 5 years. Nearly all patients developed skin-related side effects including erythema, telangiectasia, xerosis, nail dystrophy, and/or reversible alopecia.
Standard dose of TSEBT is also associated with an increased risk of non-melano-ma skin cancers. Based on skin toxicities observed with the standard dose, there was a renewed interest in defining the use of TSEBT at lower doses with the advan-tage of shorter treatment courses that also allow for repeat treatments over time, reduced treatment-related toxicities, and improved patient compliance. Results from 3 prospective trials using 12-Gy TSEBT demonstrated an ORR of 88% of skin disease in stage 1B to 3A and are comparable to the standard dose of 36 Gy.86 TSEBT may be repeated for palliative effects, although at reduced doses. Adjuvant

22
therapy including PUVA, ECP, and INF-α may improve the duration of response. Improvement in disease-free survival but not in overall survival has been reported in T1/T2 patients treated with adjuvant PUVA therapy.87
2. Systemic treatments
Biologic therapiesDecreased cell-mediated immunity with a dominant Th2 cytokine profile is ob-
served in advanced stages of MF/SS. Bexarotene, immunomodulatory cytokines such as INF-α, INF-γ, and IL-12, and ECP enhance the host antitumor response by either maintaining Th1 skewing or inhibition of Th2 cytokine production (Table 7). The increased understanding of the immunobiology of the disease has led to the development of targeted strategies. In particular, lymphoma cells express a variety of cytokine receptors such as the IL-2 receptor and surface molecules such as CD30 and CD52 that have become targets for therapeutic intervention with immunotoxin proteins or monoclonal antibodies. The mechanism of selectivity of gene expression is currently an area of intense study. Microarray data indicate that the expression of several genes within distinct apoptosis and cell cycle pathways is regulated by histone deacetylase (HDAC) inhibitors.88 Vorinostat (suberoylanilide hydroxamic acid), romidepsin (depsipeptide), and panobinostat (LBH589) are HDAC inhibitors that have demonstrated therapeutic benefit as monotherapy in CTCL.
InterferonsInterferon-alpha (INF-α) is one of the most widely used first-line treatments and
probably the most effective single agent in the treatment of CTCL. The available subtypes INF-α2a and INF-α2b do not differ in their activity and have shown a wide range of biologic effects, including antiviral, antiproliferative, and immunomodu-latory effects. Type 1 interferons (alpha/beta) work in part through a cell surface receptor activating JAK/STAT signaling and also direct cytotoxic effects.89 Resist-ance to IFN-α is associated with large cell transformation and in vitro through loss of STAT1 expression. The exact mechanism by which they exert their antitumor effects remains unknown. Th1 cytokines support cytotoxic T-cell mediated immu-nity, and it has been speculated that INF-α maintains or enhances a Th1-cell popu-lation balance for an effective cell-mediated response to malignant T lymphocytes.
INF-α was first reported in 1984 for the treatment of advanced and heavily pre-treated MF/SS with an overall response rate of 45%.48 Subsequent reported overall response rates ranged from 29% to 74% of patients with median durations from 4 to 42 months.90–92 Higher response rates were seen in early-stage MF patients.
INF-α is generally given as long-term therapy, although the optimal dose and du-ration in CTCL has not been established. Various dosages and treatment schedules have been used. The initial trials used very high doses, but significant toxicities in nearly all patients have defined the limits of its clinical application. Therapy should be initiated at low doses between 1 and 3 million units (MU) 3 times weekly with gradual escalation to 9 to 12 MU daily or as tolerated.

23
The combination therapy IFN-α and PUVA, first reported in 1995, resulted in very high response rates and showed superiority to other combinations with retin-oids or ECP.93 Thirty-nine patients with advanced disease (1B–4B) were treated with INF-α2a (12 MU 3 x weekly) and PUVA (3 x weekly) with an overall re-sponse rate of 92% and CR in 62% of patients. The median response duration was 28 months. The development of neutralizing antibodies has been associated with INF-α therapy with variable impact on response rates. The IFN-α/PUVA combina-tion has been shown to inhibit the development of neutralizing antibodies.94 Side effects are based on dosage and schedule and can be divided into acute and chronic. Initially, almost all patients develop temporary flu-like symptoms. Chronic side ef-fects can be anorexia, fatigue, depression, alopecia, cytopenia, and impaired liver function.
Pegylated interferon-alpha (PEG-IFN-α) with the advantage of once-weekly in-jection, is more convenient to administer than standard INF-α, and has demon-strated similar efficacy and tolerability in other malignancies.95 Data are limited in CTCL. A small multicenter, dose-escalation study evaluated the safety, tolerability, and efficacy of subcutaneous PEG-IFN-α-2a in patients with CTCL and showed clinical response (CR or PR) in 50% (n=2), 83% (n=5), and 66% (n=2) for the 180-, 270-, and 360-μg PEG-IFN α-2a groups, respectively.96 Four patients had stable disease; none of the patients developed disease progression. Leukopenia, elevated liver enzymes, flu-like symptoms, and thrombocytopenia were the most frequently reported adverse events with an overall increase in toxicity profile within the high-est dose group (360 μg/week).
Retinoids Retinoids are vitamin A derivatives that have important effects on cell growth,
terminal differentiation, and apoptosis and have been used to treat CTCL since the early 1980s with reported benefits in several small studies.78 Response rates ranged from 44% to 67%, with CR rates from 21% to 35% and median response duration around 8 months. Common effects consisted of skin and mucous membrane dry-ness. Bexarotene, a new retinoid X receptor (RXR)-selective retinoid, was approved in 1999 for the treatment of relapsed/refractory CTCL at early and advanced stages. In vitro studies have shown that bexarotene treatment induces apoptosis in CTCL cell lines and tumor regression in animal models, although its precise mechanisms are unknown.97 The approval of bexarotene capsules was based on 2 multicenter, open-label phase 2 and 3 clinical trials in 152 patients with early and advanced stages of CTCL patients who had failed or were refractory to 2 or more stand-ard therapies.98,99 In the early-stage trial, 58 patients randomized to doses of 6.5, 300, or 650 mg/m2 daily achieved response rates (defined as >50% improvement in skin lesions) of 20%, 54%, and 67%, respectively.98 Median time to response was 8 weeks. In the advanced-stage trial with 94 patients, response rates of 45% and 55% were observed with daily doses of 300 or 650 mg/m2, respectively, and an overall response rate of 48%. Only 6% (6/94) of patients achieved a CR, however. Median duration of response was short at 10 months.

24
A significantly higher clinical response to oral bexarotene was achieved in pa-tients by controlling dose-limiting hypertriglyceridemia with 2 concomitantly ad-ministered lipid-lowering agents compared to those taking 1 or no lipid-lowering agents. Retrospectively collected comparison data suggest that there may be little difference in efficacy between the RXR-selective retinoid bexarotene and the RAR-specific retinoid all-trans retinoic acid (ATRA). The bexarotene-treated group had an overall response rate of 21% compared to the ATRA-treated group with 12%.100 Compared to previously published data, bexarotene achieved significantly lower response rates possibly related to differences in response criteria and patient inclu-sion criteria.
Bexarotene has been combined with other therapies such as PUVA, INF-α, INF-γ, and ECP in patients with advanced disease or refractory to prior treat-ments.101 Bexarotene induces up-regulation of the high-affinity IL-2 receptor on T cells in vitro, providing the rationale for combining bexarotene with IL-2 fusion protein (denileukin diftitox) to enhance susceptibility of malignant cells.102 In a phase 1 study using this combination approach in 14 patients with MF, 8 patients (57%) achieved a response, with CR in 4 patients (28%).103
The most common significant side effects experienced in both early- and ad-vanced-stage trials were hypertriglyceridemia, hypercholesterolemia, central hy-pothyroidism, and leukopenia requiring additional treatment with lipid-lowering agents such as statins, HMG-CoA reductase inhibitors or fenofibrates, and thyroid hormone replacement.98,99 The concomitant use of gemfibrozil is contraindicated, as it increases plasma concentration of bexarotene and triglyceride levels, possi-bly related to cytochrome P450 3A4 isoenzyme inhibition. Oral bexarotene can be considered as first-line treatment for patients with stage 1B and higher as a single agent or at reduced dose in combination with PUVA, with serum lipids and thyroid parameters carefully monitored. Increased cardiac events witnessed in CTCL pa-tients may be a consequence of the lipid abnormalities associated with bexarotene treatments. The combined regimen may not increase response rates, but has the advantage of decreased dose requirements for both PUVA and bexarotene.
Extracorporeal photopheresisExtracorporeal photopheresis was approved in 1988 by the FDA for the pal-
liative treatment of patients with CTCL. It also has been effective in the treat-ment of graft-versus-host disease (GVHD) and autoimmune diseases. Circulating mononuclear cells are separated by a leukapheresis-based method, mixed with 8-MOP, exposed to UVA light (1–2 J/cm2) that activates the 8-MOP causing cross-linking of DNA, and reinfused to the patient. One suggested mechanism of action is induction of apoptosis in circulating malignant T lymphocytes with subsequent release of tumor antigens leading to a systemic antitumor response against the malignant T-cell clone. Immunomodulatory effects via the induction of antigen-specific regulatory T cells have been suggested.104 Still, the relative importance of these findings for the treatment of CTCL is not clear. Treatment is empirically given on 2 consecutive days every 14 to 28 days. It usually takes several months to

25
see an improvement and is especially of benefit for both erythrodermic MF and SS with circulating neoplastic T cells.
In 1987, investigators reported on preliminary results of the first multicenter trial for ECP in CTCL with a partial or complete response in 27 (73%) of 37 patients.105 Of the responding patients, 24 had exfoliative erythroderma. Mean time to response was 22 weeks. Subsequently performed studies on ECP have shown various degrees of overall response rates ranging from 31% to 73% of patients when used as mono-therapy.106 ECP has not been found to be as effective in patients with cutaneous patch, plaque, or tumor stages (1A to 2B).107 The United States Cutaneous Lympho-ma Consortium (USCLC) review of therapeutic options and recommendations for treatment of SS reports on various trials using ECP as monotherapy and in combi-nation, with response rates varying from 40% to 80%.108 Optimal candidates for ECP are patients with SS with less than 2 years of disease onset, modest tumor burden, and circulating neoplastic cells, and almost normal counts of circulating CD8+ T cells and NK cells. Prolonged response rates have been reported when combined with INF-α, INF-γ, bexarotene, granulocyte colony-stimulating factor (G-CSF), or TSEBT.106,109,110 Fludarabine with subsequent treatment with ECP seemed to increase the response rate, although no survival benefit was seen.111 In this nonrandomized trial, overall response rate was 63.2% versus 24% in patients with fludarabine mono-therapy. There have been few adverse events related to ECP treatment including catheter-related infection and hypotension caused by volume shifts.
Alemtuzumab Monoclonal antibodies (MoAb) augment the host antitumor response by selec-
tively targeting the malignant cells. MoAb can mediate antitumor effects through 3 major mechanisms including intrinsic cytotoxic activity, antibody-dependent cellular cytotoxicity (ADCC), and activation of complement-dependent cytolysis. Alemtuzumab is a humanized IgG1 MoAb that targets the CD52 antigen abun-dantly expressed on normal and malignant B cells and T cells, but not on hemat-opoietic stem cells. It is FDA-approved for the treatment of CLL. An analysis on results with alemtuzumab reported more than 50% response rates with 32% CR in 2 studies conducted with heavily pretreated relapsed/refractory MF/SS patients.112 These results were most promising in patients with erythrodermic MF/SS, possibly because of its depletion of central memory T cells.113,114 Median response duration was 12 months. Original studies recommended subcutaneous/intravenous doses of 30 mg 3 times weekly, but lower doses (10 mg 3 times/week) may be equally effica-cious.115 The compound, however, can be associated with significant hematologic toxicities and infectious complications consisting of reactivation of cytomegalovi-rus, herpes zoster, miliary tuberculosis, and pulmonary aspergillosis. In particular, this can be a problem when used in heavily pretreated patients. Cytopenias and prolonged immunosuppression require prophylactic antibiotic, antiviral, and anti-fungal treatment, and potential support with G-CSF. One group observed adverse cardiac events such as congestive heart failure, arrhythmia, and LV dysfunction associated with alemtuzumab therapy.

26
Denileukin diftitoxDenileukin diftitox is a recombinant fusion protein that contains the portion
of IL-2 that interacts with the IL-2R, coupled to the portion of diphtheria toxin. After binding to the IL-2R on neoplastic T cells, it is internalized and induces ap-optosis. The IL-2R consists of 3 subunits, the α-chain (CD25), β-chain (CD122), and γ-chain (CD132). Denileukin diftitox targets preferentially the intermediate (β/γ-chain) and high-affinity IL-2R (α/β/γ-chain) on malignant T lymphocytes. The degree of CD25 expression is highly variable and dependent on the tissue site. Current anti-CD25 antibodies only recognize 1 component of the receptor and therefore can be inaccurate in predicting response, although investigators have found evidence that the baseline level of CD25 expression on skin biopsies in CTCL patients correlated with their clinical response to denileukin diftitox.116 Denileukin diftitox was approved by the FDA in 1999 for the treatment of patients with CTCL refractory to standard treatment options. In general, response rates in patients with relapsed and refractory MF/SS range from 30% to 37%.41 Adverse effects include acute infusion-related events such as fever, rash, chills, dyspnea, and hypotension, and later effects such as myalgias, elevated serum transaminases, and vascular leak syndrome (VLS). The incidence of acute infusion-related events and VLS was sig-nificantly decreased with premedication of 8 mg dexamethasone, in addition to a significantly improved overall response rate of 60%. In vitro, bexarotene is known to up-regulate the expression of the high-affinity form of IL-2R in malignant T cells enhancing their susceptibility to denileukin diftitox, which prompted a phase 2 study with bexarotene combined with denileukin diftitox using escalating doses of bexarotene up to 300 mg/m2. An overall response rate of 57% was seen in mostly relapsed and refractory MF/SS patients with manageable toxicities.103 All patients, however, were premedicated with dexamethasone. There is a current ongoing in-ternational phase 2 trial of a modified formulation of denileukin diftitox (E7777).
Histone deacetylase inhibitorsThe degree of histone acetylation correlates with open chromatin, which allows
various transcription factors access to the promoter regions of target genes. In con-trast, hyperacetylation of tumor suppressor genes results in chromatin compac-tion and inactivation of genes that are frequently observed in various hematologic malignancies and solid tumors. These genes are silenced by histone deacetylases, which remove acetyl groups from histones, which form complexes with DNA (nu-cleosome). Histone deacetylase (HDAC) inhibitors may restore the expression of tumor suppressor and/or cell cycle regulatory genes by increasing the acetylation of histones, leading to inhibition of cell growth and induction of apoptosis. There also are likely nonhistone effects, as numerous nonhistone proteins are targets of HDAC inhibitors. Vorinostat, approved by the FDA for CTCL patients clinical stage 1B and higher who are refractory to at least 2 systemic therapies, is an oral HDAC class 1 and 2 inhibitor that also inactivates STAT3, which is constitutively expressed in CTCL, and enhances retinoid effects of RAR/RXR activation and gene transcrip-tion in vitro. A phase 2 trial of 400 mg vorinostat daily showed a partial response in

27
22 (29.7%) of 74 patients with only 1 CR.117 Another phase 2 trial assessed several dosing regimens. When given to 33 heavily pretreated CTCL patients, vorinostat produced an overall response rate of 24.2% (8 patients).118 Intermittent dosing was less effective than sustained dosing at 400 mg/d. The 300-mg, twice-daily regimen had higher toxicity with no additional clinical benefit over the 400-mg, once-daily regimen. No CR was observed.
Intriguingly, this study showed that nearly half of the patients with CTCL also experienced significant pruritus relief with vorinostat therapy and hence a marked improvement in their quality of life. The most common serious toxicities (grade 3 and 4) were thrombocytopenia, anemia, dehydration, nausea/vomiting, hypoten-sion, infection, sepsis, pulmonary embolism, and deep venous thrombosis. These toxicities were reversible on discontinuation of the drug.
Romidepsin (depsipeptide), approved by the FDA for advanced CTCL that is refractory to at least 1 systemic therapy, inhibits class 1 and 2 HDACs and is intra-venously administered at a weekly dose of 14 mg/m2 for 3 weeks, with 1 week off. Treatment is continued until intolerance or disease progression. Two phase 2 trials have evaluated romidepsin in advanced-stage MF.119,120 The reported ORR was 36%, with a median duration of response of 15 months. Romidepsin showed prolonged clinical responses in a subset of patients, particularly in SS patients with blood involvement with a manageable toxicity profile. Most common toxicities consisted of fatigue, gastrointestinal symptoms (nausea, vomiting, diarrhea, poor appetite), hematologic abnormalities, and infectious complications. Electrocardiographic as-sessments showed T-wave flattening in 71% of patients, less common ST depres-sion, and rare QTc prolongations (2%). Significant pruritus reduction was reported in treated patients; however, this did not correlate with clinical response. Romidep-sin may act at least in part by reducing IL-31 expression levels, thereby providing a pruritus relief in patients with CTCL.72,73 In combination with denileukin diftitox, romidepsin has shown increased expression of the IL-2 receptor and cumulative toxicity.
Panobinostat (LBH589), an oral pan-deacetylase inhibitor, has inhibited cell pro-liferation and induced apoptosis in preclinical models and exhibited efficacy in advanced-stage patients in a phase 1 study.121 Gene expression profiling on skin biopsies of treated patients showed downregulation of genes that regulate cell cycle progression and angiogenesis in response to panobinostat. A multicenter phase 2 study evaluated bexarotene-exposed and -naïve patients with advanced refrac-tory MF/SS.122 Seventy-nine bexarotene-exposed and 60 bexarotene-naïve patients received panobinostat orally (20 mg) 3 times every week. The ORR (using mod-ern multi-compartment response criteria) was 17.3% in all patients in the primary analysis (15.2% and 20.0% in the bexarotene-exposed and -naïve groups, respec-tively). The median progression-free survival was 4.2 and 3.7 months in the bexar-otene-exposed and -naïve groups, respectively. The most common adverse events were thrombocytopenia, diarrhea, fatigue, and nausea.

28
Brentuximab vedotinCutaneous infiltrates in MF/SS show variable levels of CD30 expression, with
higher expression seen in cases with large cell transformation. Brentuximab vedot-in (BV) is an antibody–drug conjugate that selectively delivers a toxic microtubule-disrupting agent into CD30-expressing cells, thereby inducing cell cycle arrest and apoptosis. A phase 2 trial in patients with refractory/advanced MF or SS showed an overall response rate of 70% (90% CI, 53%–83%).123 Of note, a wide range of CD30 expression levels (nondetectable–100%) were observed. Patients with >5% of CD30 expression within cutaneous infiltrate showed lower responses (P<.005). The most common adverse effects included peripheral neuropathy, fatigue, nausea, alopecia, and neutropenia. This drug has been granted a priority review by the FDA for patients with refractory/advanced MF/SS based on data from the phase 3 ALCANZA trial.124 Results have demonstrated a superior benefit of BV with ORR of 67% and CRR of 16% compared with ORR of 20% and CRR 2% for physician’s choice (methotrexate or BV) at a median follow-up of 17.5 months. In this trial, the rate of objective responses lasting ≥4 months was 56% in patients treated with BV, compared with 13% in patients receiving physician’s choice of standard therapies (methotrexate, bexarotene; P<.0001).
MogamulizumabMogamulizumab (KW-0761) is a humanized anti-CC-chemokine receptor 4
(CCR4) monoclonal antibody with a defucosylated Fc region leading to increased antibody-dependent cellular cytotoxicity.125 CCR4 is expressed on Tregs and T-helper memory cells and plays an important role in skin homing. Mogamulizu-mab is approved for ATLL in Japan and received breakthrough designation by the FDA. The effectiveness of mogamulizumab in CTCL has been demonstrated in separate phase 1 and 2 randomized controlled trials.125 In a phase 1/2 study, moga-mulizumab induced an overall response rate of 47.1% in SS patients and 28.6% in MF patients.126 In a multicenter Japanese phase 2 study involving 37 patients with relapsed CCR4-positive tumors, mogamulizumab treatment induced 35% of objective response, including 5 patients (14%) achieving CR.127 The most com-mon adverse effect of this treatment is lymphocytopenia (81%), and cases of severe Steven–Johnson–Lyell syndrome due to induced immune deficiency of regulatory T cells have been reported. An international phase 3 trial of mogamulizumab ver-sus vorinostat has been recently completed in previously treated CTCL patients.
ChemotherapySingle-agent and combination chemotherapies in advanced, refractory, and ag-
gressive forms of CTCL have been associated with high response rates, but short-lived durations. Importantly, a recent single-center, retrospective study compared the efficacy of systemic therapies in approximately 200 patients with MF/SS. The primary end point was time to next treatment (TTNT). The analysis revealed that chemotherapy was most efficacious when used as first-line therapy; however, it of-fered only a short median TTNT of 3.9 months. In contrast, biologic or targeted

29
therapies had a significant higher TTNT such as 8.7 months seen with IFN-α and 4.1 months seen with HDAC inhibitors.128 The use of chemotherapy agents is lim-ited to palliation of symptoms. Options include single-agent or multiagent chemo-therapy including steroids, methotrexate, chlorambucil, vincristine, doxorubicin, cyclophosphamide, etoposide, nucleoside analogues, and alkylating agents. Com-bination regimens include cyclophosphamide, doxorubicin, vincristine, and pred-nisone (CHOP) or CVP therapy.
Single-agent chemotherapySingle-agent chemotherapy with oral chlorambucil, methotrexate, or etoposide
have all been used with similar efficacy producing CR of 25% to 35% in patients with advanced stages 2B–4B, with a median response duration of 3 to 22 months. Among single-agent chemotherapies, pegylated doxorubicin, and the nucleoside analogues pentostatin and gemcitabine are reported to be particularly effective.
Pegylated doxorubicinThe pegylated liposomal encapsulated anthracycline doxorubicin was developed
to decrease the risk of cardiotoxicity experienced with conventional doxorubicin while preserving the antitumor efficacy. Anthracyclines work by deforming the DNA structure of tumor cells and terminating their biologic function. Pegylated doxorubicin was empirically tested in MF/SS patients and showed an overall re-sponse rate of 80% with CR in 6 (60%) of 10 patients.129 More published multi-center data of pegylated doxorubicin in 34 patients with recurrent or recalcitrant CTCL revealed a response rate of 88.2%.130 Twenty-seven patients (79.4%) achieved CR, with a median duration of 12 months ranging from 9.5 to 44 months. Adverse effects were generally mild compared with other chemotherapy regimens. Impor-tantly, alopecia is rarely witnessed.
Nucleoside analogues (gemcitabine, 2-CdA)The pyrimidine nucleoside analogue gemcitabine (2’,2’-diflurodeoxycytidine) is
approved for the treatment of non-small cell lung cancer and pancreatic cancer. It is metabolized intracellularly to its active metabolites gemcitabine diphosphate and triphosphate, with the latter incorporated into DNA resulting in inhibition of DNA synthesis and induction of apoptosis. Gemcitabine has shown to have clinical activity in patients with advanced CTCL with a low toxicity profile.131,132 It was investigated at 2 centers in a phase 2 trial at a dosage of 1200 mg/m2 (using traditional 3-weekly dosing every 28 days over 30 minutes) in 44 patients with clinical stage 2B-4 MF/SS.131 Twelve percent of patients achieved CR and 26% of patients achieved PR, with a median duration of 15 months and 10 months, respectively. Gemcitabine at a simi-lar dose schedule achieved higher response rates in 32 patients with untreated MF, peripheral T-cell lymphoma (unclassified), and SS.132 The overall response rate was 75%, with CR in 22% and PR in 53% of patients. The reported median duration was 10 months.
Pentostatin (deoxycoformycin) is an FDA-approved purine analogue for hairy cell

30
leukemia that inhibits adenosine deaminase. Adenosine deaminase deficiency, an in-herited disorder that has been found in patients with severe combined immunodefi-ciency characterized by a deficiency of both B cells and T cells, provided the rational to use pentostatin in clinical trials for CTCL and other non-Hodgkin lymphomas. In mostly small, nonrandomized single-center studies with only a limited number of patients (3–18 patients), the reported median response rate was 41% (range, 33%–67%), with CR in 6% of patients.133 Durations were usually short, ranging from 1.3 to 16+ months.
Fludarabine (9-β-D-arabinosyl-2-fluoroadenine) and cladribine (2-chlorodeoxy-adenosine [2-CdA]) are halogenated purine analogues that are resistant to degrada-tion by adenosine deaminase. Both are metabolized to their triphosphate forms, which lead to the inhibition of ribonucleotide reductase, DNA polymerases, and DNA repair. Experience with fludarabine as monotherapy is limited. Results of a multicenter study with 31 patients with advanced disease showed a modest response rate of 19%, with CR in 3% of patients.134 Fludarabine is associated with significant and long-lasting myelosuppression. Two studies with 2-CdA in refractory advanced-stage disease showed an overall response rate in 28% and 47% of patients, with CR in 14% and 20% of patients with short duration.135,136 The most significant toxicities encountered were prolonged myelosuppression and infectious complications.
Two nonrandomized studies have evaluated INF-α combined with fludarabine or pentostatin. Response rates have not been shown to be superior to single-agent or INF-α therapy.137 As previously mentioned, fludarabine with sequential ECP in-creased response rates but did not affect overall survival.111
Antifolates (methotrexate, pralatrexate)Methotrexate is an analog of folic acid that interferes with the synthesis of pu-
rines and pyrimidines as well as blocking the conversion of homocysteine to me-thionine. In CTCL, recent studies have shown that methotrexate enhances Fas (CD95)-dependent apoptosis in the neoplastic T cells. It has been advocated for patients with large cell transformation and advanced-stage MF.138
Methotrexate can be given orally or intravenously in a wide range of doses de-pending on the clinical indication. Low-dose weekly methotrexate (20–70 mg) administered orally once a week has an ORR of 33% and 58% for plaque (T2) MF and erythrodermic (T4) MF, respectively, with median time to treatment failure of 15 months for patients with stage T2 disease and 31 months for erythrodermic MF.139,140 Common side effects include oral mucositis/stomatitis, gastrointestinal symptoms, bone-marrow suppression, and hepatotoxicity. Folic acid supplemen-tation is recommended during methotrexate therapy to prevent common side ef-fects including oral mucositis/stomatitis, gastrointestinal symptoms, bone-mar-row suppression, and hepatotoxicity.
Pralatrexate is another folate inhibitor that has been shown to have a higher affinity for the transporter RFC-1 (reduced folate carrier-1) and increased poly-glutamylation, which results in an enhanced intracellular drug concentration as compared to methotrexate resulting in a distinct cytotoxicity profile.141 It has a

31
broader and distinct impact on gene expression alterations compared to meth-otrexate. Although it is approved for the treatment of relapsed or refractory periph-eral T-cell lymphomas (PTCLs), there is evidence of activity in CTCL as well, with an ORR of 45% with the optimized schedule of 15 mg/m2/wk IV for 3 of 4 weeks after a median of 4 cycles.142 Mucositis is the most common toxicity encountered in different studies. To lower this risk, vitamin B12 and folate supplementation and folinic acid (leucovorin rescue) are used during pralatrexate treatment. The median response duration was not reached in the study that evaluated the efficacy of prala-trexate in patients with relapsed/refractory CTCL, but the Kaplan-Meier estimates showed that 73% of responses were continuing at 6 months.
Multiagent chemotherapyThere were no randomized trials comparing combined regimens with single-agent
regimens. However, combination approaches with anthracycline-based therapies have been shown to achieve higher response rates, although a prolonged relapse-free or overall survival has not been seen. A retrospective study on 81 patients with primary cutaneous lymphomas (46 primary cutaneous B-cell lymphomas and 35 CTCL) treat-ed with COP or CHOP regimens reported an overall response rate of 40% in CTCL patients, with CR in 23% of patients. The median response duration was 5.7 months and median survival was 19 months.143 A phase 2 trial with the etoposide, vincristine, doxorubicin, bolus cyclophosphamide, and oral prednisolone (EPOCH) regimen in 15 patients with refractory CTCL resulted in an overall response rate of 80%.144 Twenty-seven percent of patients achieved a CR, and 53% of patients achieved a par-tial response (PR), with a median duration of 8 months and overall survival of 13.5 months. Significant hematologic toxicity occurred in 8 patients (61%). A combina-tion approach with ESHAP (etoposide, cisplatin, high-dose cytarabine, prednisolone) showed no advantage in patients with advanced CTCL compared to previously re-ported results. ESHAP resulted in a CR in 2 patients of 11 treated, with short duration of partial remissions (PR) in 7 patients, while severe toxicity was observed in 90% of patients, especially myelosuppression and infectious complications.145
Stem cell transplant (autologous, allogeneic) The concept of high-dose combined chemotherapy followed by autologous bone
marrow transplant or peripheral blood stem cell support has curative potential in Hodgkin lymphoma and various non-Hodgkin lymphomas, but experience in CTCL is limited. Autologous stem cell transplants (SCT) have yielded disappointing results. Despite reported effective responses with CR in most patients treated, relapses are fre-quent and may occur rapidly.146,147 Allogeneic transplantation is recognized to achieve much more durable responses, most likely due to an immune-mediated graft-versus-lymphoma (GVL) effect. Response durations as far as 6 years post transplant have been reported.148 Allogeneic transplantation is the only potential curative treatment option in patients with MF/SS; however, transplant-related mortality rates are about 25% to 30% even with the use of myeloablative and reduced-intensity conditioning (RIC) regimens including life-threatening infections and GVHD. Therefore, alloge-

32
neic transplant is usually reserved for young patients with refractory and advanced-stage MF. Patients are given bridging treatment with combination chemotherapies that are reserved for more aggressive systemic lymphomas.149 These regimens have a high response rate but a short duration of response that can be utilized to prepare the transplant. Although there is little consensus on the conditioning regimen, there are limited data from a single center that the use of TSEB before an allogenic transplan-tation provides improved skin outcomes.150 A proposed GVL effect is believed to be responsible for higher effectiveness of allogeneic transplants. Subsequently, there have been several studies further exploring the role of RIC with allogeneic transplant in MF/SS. Studies have also reiterated the role of allogeneic transplant in “down staging” CTCL in subsequent relapses with significantly lower non-relapse mortality rates as-sociated with RIC.151
3. Investigational treatmentsThere are various investigational options that have been designed to target disease-
specific pathways or immune dysfunction in CTCL. The most recent investigations are briefly discussed.
Anti-KIR3DL2 KIR3DL2 belongs to the killer Ig-like receptor (KIR) family that is expressed on
subsets of normal NK, CD4+, and CD8+ T cells, and in all stages of MF/SS includ-ing large cell transformation. IPH4102 is an anti-KIR3DL2 monoclonal antibody that depletes KIR3DL2-expressing cells through antibody-dependent cell cytotoxicity and phagocytosis. IPH4102 is currently evaluated in patients with relapsed advanced MF/SS to assess its safety profile and antitumor activity. A total of 25 patients were treated at the 10 preplanned ascending dose levels (0.0001 to 10 mg/kg). Preliminary data show excellent tolerability and promising clinical activity. IPH4102 induced drastic elimination of KIR3DL2-positive aberrant cells in skin and blood without affecting circulating normal NK cells. Accrual is ongoing (EORTC abstract presented October 2017).152
Immune checkpoint inhibitorsProgrammed death 1 (PD-1) protein, a T-cell co-inhibitory receptor, and the PD-
ligand 1 (PD-L1) play a critical role in the ability of tumor cells to evade antitumor immunity. A dysregulated immunophenotype in CTCL is a key feature in the patho-genesis of the disease and may influence patient’s response and resistance to therapy. Immunotherapy with the PD-1 inhibitor (pembrolizumab) showed encouraging re-sponses in patients with advanced-stage MF/SS.153 Results of this multicenter phase 2 trial included 24 patients with advanced, heavily pretreated MF/SS (stage 1B or higher; 16 patients = stage 4A), treated with pembrolizumab at 2 mg/kg every 3 weeks for up to 2 years and showed an ORR of 38% (1 CR, 8 PR). The median time of follow-up was 40 weeks and progression-free survival was 69% at 1 year. Of note, the PD1 expression in lesional biopsies did not correlate with response. A PD-L1 inhibitor, durvalumab, is currently being evaluated in a clinical phase 1/2 trial in patients with advanced CTCL

33
with promising responses seen. The study is ongoing.154 CD47 is an innate immune checkpoint that binds to signal regulatory protein alpha
(SIRPα) and delivers a “do not eat” signal to suppress macrophage phagocytosis. Tu-mor cells frequently overexpress CD47 to evade macrophage-mediated destruction. TTI-621 (SIRPαFc) is an immune checkpoint inhibitor consisting of the CD47 bind-ing domain of human SIRPα linked to the Fc region of human IgG1 designed to both: 1) block the CD47 “do not eat” signal, and 2) engage macrophage Fcγ receptors with IgG1 Fc to enhance phagocytosis and antitumor activity. It is hypothesized that direct intratumoral injection of TTI-621 may enhance both local and systemic antitumor activity. A phase 1, multicenter, open-label study is ongoing to characterize the safety and tolerability of intratumoral injection of TTI-621. Preliminary phase 1 data suggest that patients with MF/SS are highly responsive to a single dose of TTI-621 delivered by intratumoral injection. One patient achieved a complete response of the injected lesion, and 5 additional patients experienced decreases in tumor size and/or decreased circulating Sézary cells. Intratumoral injections were well tolerated by all patients.155
AntagomirsAntagomirs (anti-miRNAs) are chemically engineered oligonucleotides and used
to silence endogenous microRNAs. MRG-106 is an oligonucleotide inhibitor of miR-155, a microRNA that is overexpressed in skin lesions of MF. A multicenter, dose-escalating phase 1 trial is currently evaluating MRG-106 in patients with MF when administered via subcutaneous or IV injection (300–900 mg/dose). An additional co-hort received intralesional injections (75 mg/dose) to evaluate tolerability and efficacy with local delivery. Patients received 6 doses (SC/IV) in the first 26 days during cycle 1 of the study followed by weekly or biweekly doses in patients who continued beyond the first cycle. Preliminary results show that 23 (95%) of 24 enrolled patients showed improvement in either the individually treated lesion (intralesional cohort) or total skin disease (SC/IV cohorts) as measured by maximal change in skin tumor burden using the Composite Assessment of Index Lesion Severity (CAILS) or the Modified Severity Weighted Assessment Tool (mSWAT). Nine of 18 patients (stage 1B or high-er) in the SC/IV cohort reached a PR defined as 50% or greater reduction in mSWAT score. MRG-106 is well-tolerated; the most common reported side effects were fatigue, pruritus, and nausea. Treatment is ongoing.156
Immunomodulatory agentsHost immune function appears to play an integral role in mediating responses in
MF. Toll-like receptor (TLR) agonists such as imiquimod and CpG-7909 enhance im-mune responses via activation of TLR-7, 8, and/or 9. CpG-7909 (CpG oligonucleotide) is a TLR-9 agonist known to induce Th1-induced immune responses via dendritic cell activation. Preliminary results demonstrate that the cytotoxic CD8+ T-cell population increased within tumor infiltrates after systemic administration.157 Due to the small number of patients, results are inconclusive without frank evidence of efficacy.
Lenalidomide (CC-5013), an oral immunomodulatory thalidomide analogue, is currently being used in clinical trials to treat various hematologic malignancies and

34
solid tumors. The immunomodulatory properties such as T-cell costimulation with in-duction of Th1 cytokine production and cytotoxic activity along with antiangiogenic, antiproliferative, and pro-apoptotic properties provided the rationale to use this agent in patients with MF/SS. Lenalidomide was recently evaluated as a single agent for ad-vanced, refractory MF/SS in an open-label phase 2 trial and achieved an ORR of 28% with median overall survival and median progression-free survival of 43 months and 8 months, respectively.158 The most frequent adverse events included lower leg edema, anemia, fatigue, and transient flare reaction in skin, blood, and/or lymph nodes that may be associated with improvement in disease burden and was more frequently ob-served in patients with a 25-mg starting dose versus a 10-mg starting dose. Initiating lenalidomide at a dose of 25 mg /day is associated with constitutional symptoms such as fatigue and pain, and tumor flare response that mimic worsening of the patient’s disease. A slow dose escalation (eg, 5 mg in monthly increments) has been shown to improve tolerability in clinical trials and is now frequently the approach taken as the lenalidomide standard regimen for CTCL patients.
Chimeric antigen receptor T cellsChimeric antigen receptor (CAR) T cells have emerged as a novel treatment regi-
men for B-cell lymphomas. Clinical trial data have demonstrated the potent activity of anti-CD19 CAR T cells against multiple subtypes of B-cell lymphoma, including treatment-refractory diffuse large B-cell lymphoma. Anti-CD4 and anti-CD30 engi-neered CAR-T cells are being investigated in T-cell lymphomas including CTCL.
4. General health careTreatment-associated toxicities can be pronounced, particularly in elderly patients
with comorbid medical conditions, poor performance status, and/or severe skin dis-ease, or in patients whose life expectancy is short. Dose adjustments in those patients are often required to preserve quality of life, because treatment is always regarded to be palliative and greater disease elimination does not outweigh the increased risk for toxicities.
Caution should be exercised in all CTCL patients regarding skin care and avoidance of infections or sepsis. Many patients are disabled by their pruritus and skin appear-ance. Emollients should be recommended in patients with dryness and scaling. In addition, topical treatment with mid-potent steroids such as triamcinolone applied once or twice daily can minimize skin irritation and pruritus, particularly in patients with SS. A short-term course with systemic steroids often gives immediate relief of more diffuse symptoms. Oral antihistamines and/or gabapentin may be also of ben-efit for pruritus. Patients with more widespread cutaneous disease or generalized erythroderma should be screened for secondary infections such as staphylococcal or streptococcal species, and/or dermatophytes and treated with systemic antibiotics and antifungal agents. It has been shown that cutaneous colonization by S. aureus influ-ences the disease activity of CTCL, possibly by activation of Sézary cells by bacterial superantigenic exoproteins.159

35
OutcomeThe prognosis of patients with MF is excellent in early stages, with a low mortal-
ity risk. At later stages, patients have a decreased survival, with most deaths caused by infections due to the impaired immune system. Stage-dependent therapies are believed to decrease the risk of further disease progression and transformation into aggressive large-cell lymphoma. In the past, most investigational and standard treatment strategies have focused on physical measures for the primary endpoint of efficacy. CTCL is a disease with visible manifestations commonly accompanied by pruritus, burning sensations, and pain, with a significant detriment to patient’s quality of life. It may have a negative impact on the physical, emotional, and psy-chosocial wellbeing of affected patients and even their families. That patients with CTCL feel stigmatized by their condition has been established.160 The treatment of MF/SS is always palliative, and relapses are not necessarily associated with a poor outcome. Therefore, maintenance of quality of life should be one of the mainstays of therapeutic strategies.

36
Case reports1. Approach and management of patient with early-stage MF
A 31-year-old man was presented with a 5-year history of a skin rash that was limited to buttocks and lower abdomen and suddenly progressed to extensive ery-thematous, pruritic patches on trunk and extremities 6 months ago. In addition, hair loss and keratotic follicular plugs were seen within the patches. There was no lymphadenopathy noted and the patient denied constitutional symptoms. He was seen by several dermatologists in the past and intermittently treated with topical steroids without resolution. Several biopsies in the past revealed inconclusive re-sults. Histopathology displayed a band-like infiltrate with hair follicle involvement. Mucinous deposits were confirmed by PAS stain. Immunohistochemistry showed an increased CD4 to CD8 ratio and T-cell rearrangement studies of the skin dem-onstrated clonality. Laboratory investigations included a complete blood cell count with differential, chemistry panel, and LDH that were all in normal limits. Accord-ing to the clinical and histopathologic features, he was diagnosed with MF, fol-liculotropic type, clinical stage 1B (T2, N0, M0, B0). The patient received a course of PUVA therapy 3 times weekly following a baseline eye examination. Moistur-izers, applied once or twice daily, were added as barrier protection. A follow-up visit 3 months later revealed only mild improvement of his skin lesions. Treatment was switched to a combination of low-dose bexarotene with 150 mg PO daily plus PUVA therapy. The development of mild hypercholesterolemia and low TSH level after 1 month of treatment required concomitant treatment with levothyroxine and atorvastatin. Three months later, the patient achieved a complete remission. Bexarotene therapy was discontinued while PUVA therapy was maintained with a gradual decrease to once every 4 weeks at a constant dose. Two years later, PUVA maintenance was discontinued. The patient remained in remission for 5 years until he developed a few mild scaling patches on his right thigh. Localized treatment with a class 1 topical steroid cleared skin completely.
2. Approach and management of patient with advanced/tumor-stage MFA 56-year-old woman was referred with patches, plaques, and tumors on trunk
and right lower extremity. Prominent axillary and inguinal lymphadenopathy was also noted. Biopsy of the skin lesions confirmed the diagnosis of tumor stage with large cell transformation. Immunophenotypic analysis revealed that the malignant CD4+ T cells were negative for CD7 and CD26. A shotty axillary and inguinal lym-phadenopathy was confirmed by CT scans and biopsy of an axillary lymph node showed LN3 involvement. Laboratory studies revealed an elevated LDH of 324 U/L, hemoglobin of 11.1 g/dL, leukocyte count of 9700/mm3, and a Sézary cell count of less than 5%. According to the clinical and histopathologic features, the patient was diagnosed with MF, clinical stage 4A (T3, N1, M0, B0a). She was initially diagnosed with MF, clinical stage 1B 5 years ago and has failed multiple therapies including topical nitrogen mustard, topical steroids, PUVA, UVB, INF-α, bexarotene, and combinations thereof, and local radiation. Because of the progressing and refrac-

37
tory nature of her disease, a skin biopsy was performed from one of the rapidly growing tumor nodules and revealed mycosis fungoides with large cell transfor-mation. The tumor cells were positive for CD30 expression. The patient received 6 cycles of brentuximab vedotin and achieved CR that was confirmed by PET/CT scans and skin assessment. During this time, the patient was evaluated for alloge-neic transplant and received a well-matched, unrelated donor allogeneic transplant using reduced intensity regimen conditioning with melphalan and fludarabine. The patient relapsed 22 months after transplant, but reachieved CR after tapering im-munosuppression (graft-versus-lymphoma effect) and remains alive and in con-tinuous CR 8 years after transplant.
3. Approach and management of patient with advanced MF/SSA 48-year-old Asian man was presented complaining about the onset of a gradu-
ally worsening, diffuse, erythematous, pruritic rash 7 months ago. He was feeling fatigued, but denied weight loss and night sweats. He has taken oral antihistamines for symptomatic relief and the rash has responded to topical steroids. More recently, he developed lymphadenopathy for which he was referred for further evaluation. He has a history of seasonal allergy and atopic dermatitis. Physical examination re-vealed generalized exfoliative, lichenified erythroderma with axillary and inguinal lymphadenopathy. Biopsies were performed consistent with MF and positive for clonal T-cell rearrangements. Axillary lymph node biopsy revealed LN3 involve-ment (aggregates of atypical T cells with preserved nodal architecture). He was not-ed to have a Sézary cell count of 2464 cells/μL and an abnormal CD4+CD7- T-cell population in the blood. He had CT scans of chest, abdomen, and pelvis revealing a peripheral lymphadenopathy. Serologic test for HTLV-1 was negative and subse-quent performed laboratory studies were normal except for elevated LDH of 280 IU/L. Based on the clinical, histopathologic, and genetic features he was diagnosed with SS, clinical stage 4A (T4, NP1, M0, B1). The patient was initially treated with PUVA and escalating doses of INF-α 3 times weekly. Five months later, he was found to have regressed lymphadenopathy with stable skin disease and persistent pruri-tus. He has tolerated PUVA poorly and therefore treatment was just continued on INF-α. His dose went up to 6 MU 3 times weekly, tolerating it fairly well with low-grade fever and fatigue. Three months later, the patient went into clinical remission with normal laboratory studies, PET/CT scans, and negative Sézary cell count, and was subsequently discontinued from treatment. However, the patient relapsed with slowly increasing circulating Sézary cells and increasing pruritus. He was subse-quently started on romidepsin weekly for 3 weeks with 1 week off treatment.
4. Approach and management of patient with Sézary syndromeA 65-year-old woman was presented with severe pruritus, mild, diffuse eryth-
roderma, and excoriations and a 2-year history of skin rash. Blood tests revealed normal basic chemistry with elevated LDH of 257 U/L, leukocyte count of 7100/mm3, hemoglobin of 11.6 g/dL, and Sézary cell count of 4680 cells/μL. CT scans confirmed axillary and inguinal lymphadenopathy. She had an outside bone mar-

38
row biopsy demonstrating involvement of the bone marrow. Skin biopsy showed an atypical lymphocytic infiltrate with clonal T cells. Her past medical history was remarkable for diabetes and hypertension. The patient was staged as MF/SS 4B (T4, N1, M1, B2). She was initially treated with bexarotene 300 mg/m2, which she has tolerated well; however, treatment was discontinued because of severe hyper-lipidemia despite concomitant use of atorvastatin. The patient was switched to INF-α with a starting dose of 1 MU 3 times weekly. Dose escalation up to 6 MU was poorly tolerated with generalized fatigue and malaise, and signs of depression that has required intermittent dose reduction of INF-α and the additional use of an antidepressant. The intense pruritus was treated with gabapentin and antihista-mines with only mild improvement. Skin moisturizers formulated with camphor and menthol were applied daily to help relieve the discomfort of pruritic skin. Over the next 7 months, the patient did not show any response to treatment despite tem-porary coadministration of NB-UVB. She then was presented for initiation of ECP, but adequate venous access could not have been secured. The patient has received a short course of dexamethasone (4 mg daily for 4 days) to palliate her symptoms. One month later, she was enrolled in an ongoing trial with alemtuzumab that was initially given intravenously 3 times per week for 4 weeks followed by subcutane-ous administration 3 times weekly until week 12. Within 1 week, the Sézary cells have cleared completely from the blood and at the end of treatment, the patient has achieved a complete clinical and histopathologic remission with cessation of pru-ritus and ongoing to date (12 months). T-cell clones, however, remained evident.

39
SummaryTaken together, patients with early-stage (1A-2A) MF should be treated with
skin-targeted modalities to achieve complete remission. In most instances, ther-apies are not curative and recurrences of disease occur frequently; however, the long-term survival is not affected by relapse status. Patients with clinical stage 1B have a higher chance (20%) of disease progression compared to patients with clini-cal stage 1A disease (10%). Patients with advanced-stage (2B–4B) MF/SS have a significantly decreased survival. Data published in the literature show a 5-year survival of 40% for stage 2B and 25% for stage 3. Treatment goals should be dis-ease palliation, improvement of survival, and improvement of quality of life. No regimen has been proven to prolong survival in advanced stages; therefore, immu-nomodulatory regimens should be used initially to reduce the need for cytotoxic therapies with more damaging side effects. The development of new biologic thera-pies, chemotherapies, and combinations thereof, and the participation in national and international studies offers patients with CTCL various treatment concepts.

40
References 1. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lympho-
mas. Blood. 2005;105(10):3768–3785.2. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organiza-
tion classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390.3. Querfeld C, Guitart J, Kuzel TM, Rosen ST. Primary cutaneous lymphomas: a review with
current treatment options. Blood Rev. 2003;17(3):131–142.4. Vonderheid EC, Bernengo MG, Burg G, et al. Update on erythrodermic cutaneous T-cell
lymphoma: report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol. 2002;46(1):95–106.
5. Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143(7):854–859.
6. Weinstock MA, Gardstein B. Twenty-year trends in the reported incidence of mycosis fungoides and associated mortality. Am J Public Health. 1999;89(8):1240–1244.
7. Bradford PT, Devesa SS, Anderson WF, Toro JR. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113(21):5064–5073.
8. Huang K P, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sezary syndrome: evidence from pop-ulation-based and clinical cohorts. Arch Dermatol. 2007;143(1):45–50.
9. Koh HK, Charif M, Weinstock MA. Epidemiology and clinical manifestations of cutane-ous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9(5):943–960.
10. Morales-Suárez-Varela MM, Olsen J, Johansen P, et al. Occupational exposures and my-cosis fungoides. A European multicentre case-control study (Europe). Cancer Causes Control. 2005;16(10):1253–1259.
11. Talpur R, Bassett R, Duvic M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sézary syndrome. Br J Dermatol. 2008;159(1):105–112.
12. Wood GS, Schaffer JM, Boni R, et al. No evidence of HTLV-1 proviral integration in lymphoproliferative disorders associated with cutaneous T-cell lymphoma. Am J Pathol. 1997;150(2):667–673.
13. Herne KL, Talpur R, Breuer-McHam J, et al. Cytomegalovirus seropositivity is significant-ly associated with mycosis fungoides and Sézary syndrome. Blood. 2003;101(6):2132–2136.
14. Gupta RK, Ramble J, Tong CY, et al. Cytomegalovirus seroprevalence is not higher in patients with mycosis fungoides/Sezary syndrome. Blood. 2006;107(3):1241–1242.
15. Adams AE, Zwicker J, Curiel C, et al. Aggressive cutaneous T-cell lymphoma after TNF alpha blockade. J Am Acad Dermatol. 2004;51(4):660–662.
16. Beylot-Barry M, Dequidt L, Adamski H, et al. Cutaneous lymphomas in patients treat-ed by biologics: 21 cases of GFELC (French study group on cutaneous lymphomas). Abstract presented at the EORTC CLTF meeting, London, United Kingdom, October 13–15, 2017.
17. Ravat FE, Spittle MF, Russell-Jones R. Primary cutaneous T-cell lymphoma occurring after organ transplantation. J Am Acad Dermatol. 2006;54(4):668–675.
18. Kaufman D, Gordon LI, Variakojis D, et al. Successfully treated Hodgkin’s dis-ease followed by mycosis fungoides: case report and review of the literature. Cutis. 1987;39(4):291–296.
19. Wilkins K, Turner R, Dolev JC, et al. Cutaneous malignancy and human immunodefi-

41
ciency virus disease. J Am Acad Dermatol. 2006;54(2):189–206.20. Vermeer MH, van Doorn R, Dukers D, et al. CD8+ T cells in cutaneous T-cell lym-
phoma: expression of cytotoxic proteins, Fas Ligand, and killing inhibitory receptors and their relationship with clinical behavior. J Clin Oncol. 2001;19(23):4322–4329.
21. Goteri G, Filosa A, Mannello B, et al. Density of neoplastic lymphoid infiltrate, CD8+ T cells, and CD1a+ dendritic cells in mycosis fungoides. J Clin Pathol. 2003;56(6):453–458.
22. Hoppe RT, Medeiros LJ, Warnke RA, Wood GS. CD8-positive tumor-infiltrating lym-phocytes influence the long-term survival of patients with mycosis fungoides. J Am Acad Dermatol. 1995;32(3):448–453.
23. Gjerdrum LM, Woetmann A, Odum N, et al. FOXP3+ regulatory T cells in cuta-neous T-cell lymphomas: association with disease stage and survival. Leukemia. 2007;21(12):2512–2518.
24. Berger CL, Hanlon D, Kanada D, et al. The growth of cutaneous T-cell lymphoma is stimulated by immature dendritic cells. Blood. 2002;99(8):2929–2939.
25. Berger CL, Tigelaar R, Cohen J, et al. Cutaneous T-cell lymphoma: malignant prolifera-tion of T-regulatory cells. Blood. 2005:105(4):1640–1647.
26. Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part 1. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70(2):205.e1–e16.
27. Ni X, Duvic M. Dendritic cells and cutaneous T-cell lymphomas. G Ital Dermatol Vener-eol. 2011;146(2):103–113.
28. Sugaya M, Miyagaki T, Ohmatsu H, et al. Association of the numbers of CD163(+) cells in lesional skin and serum levels of soluble CD163 with disease progression of cutaneous T cell lymphoma. J Dermatol Sci. 2012;68(1):45–51.
29. Wu X, Schulte BC, Zhou Y, et al. Depletion of M2-like tumor-associated macro-phages delays cutaneous T-cell lymphoma development in vivo. J Invest Dermatol. 2014;134(11):2814–2822.
30. Rabenhorst A, Schlaak M, Heukamp LC, et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood. 2012;120(10):2042–2054.
31. Dummer R, Heald PW, Nestle FO, et al. Sézary syndrome T-cell clones display T-helper 2 cytokines and express the accessory factor-1 (interferon-gamma receptor beta-chain). Blood. 1996;88(4):1383–1389.
32. Krejsgaard T, Ralfkiaer U, Clasen-Linde E, et al. Malignant cutaneous T-cell lympho-ma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J Invest Dermatol. 2011;131:1331–1338.
33. Vowels BR, Lessin SR, Cassin M, et al. Th2 cytokine mRNA expression in skin in cutane-ous T-cell lymphoma. J Invest Dermatol. 1994;103(5):669–673.
34. Lee BN, Duvic M, Tang CK, et al. Dysregulated synthesis of intracellular type 1 and type 2 cytokines by T cells of patients with cutaneous T-cell lymphoma. Clin Diagn Lab Im-munol. 1999;6(1):79–84.
35. Kim EJ, Hess S, Richardson SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115(4):798–812.
36. Querfeld C, Wu X, Sanchez JF, et al. The miRNA profile of cutaneous T cell lym-phoma correlates with the dysfunctional immunophenotype of the disease. Blood. 2016;128:4132. (Abstract, ASH 2016)
37. Whittaker S. Biological insights into the pathogenesis of cutaneous T-cell lymphomas (CTCL). Semin Oncol. 2006;33(1 Suppl 3):S3–S6.

42
38. Sokolowska-Wojdylo M, Wenzel J, Gaffal E, et al. Absence of CD26 expression on skin-homing CLA+ CD4+ T lymphocytes in peripheral blood is a highly sensitive marker for early diagnosis and therapeutic monitoring of patients with Sézary syndrome. Clin Exp Dermatol. 2005;30(6):702–706.
39. Wood GS, Tung RM, Haeffner AC, et al. Detection of clonal T-cell receptor gamma gene rearrangements in early mycosis fungoides/Sezary syndrome by polymerase chain reaction and denaturing gradient gel electrophoresis (PCR/DGGE). J Invest Dermatol. 1994;103(1):34–41.
40. Eriksen KW, Kaltoft K, Mikkelsen G, et al. Constitutive STAT3-activation in Sezary syn-drome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001;15(5):787–793.
41. Saleh MN, LeMaistre CF, Kuzel TM, et al. Antitumor activity of DAB389IL-2 fusion toxin in mycosis fungoides. J Am Acad Dermatol. 1998;39(1):63–73.
42. Choi J, Goh G, Walradt T, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47(9):1011–1019.
43. Ralfkiaer U, Hagedorn PH, Bangsgaard N, et al. Diagnostic microRNA profiling in cuta-neous T-cell lymphoma (CTCL). Blood. 2011;118(22):5891–5900.
44. Guitart J, Kennedy J, Ronan S, et al. Histologic criteria for the diagnosis of mycosis fun-goides: proposal for a grading system to standardize pathology reporting. J Cutan Pathol. 2001;28(4):174–183.
45. Costa C, Gallardo F, Pujol RM, et al. Comparative analysis of TCR-gamma gene rear-rangements by Genescan and polyacrylamide gel-electrophoresis in cutaneous T-cell lymphoma. Acta Derm Venereol. 2004;84(1):6–11.
46. Costa C, Gallardo F, Bellosillo B, et al. Analysis of T-cell receptor gamma gene rear-rangements by PCR-Genescan and PCR-polyacrylamide gel electrophoresis in early-stage mycosis fungoides/large-plaque parapsoriasis. Dermatology. 2003;207(4):418–419.
47. Murphy M, Signoretti S, Kadin ME, Loda M. Detection of TCR-gamma gene rear-rangements in early mycosis fungoides by non-radioactive PCR-SSCP. J Cutan Pathol. 2000;27(5):228–234.
48. Bunn PA Jr, Lamberg SI. Report of the Committee on Staging and Classification of Cu-taneous T-Cell Lymphomas. Cancer Treat Rep. 1979;63(4):725–728.
49. Sausville EA, Eddy JL, Makuch RW, et al. Histopathologic staging at initial diagnosis of mycosis fungoides and the Sézary syndrome. Definition of three distinctive prognostic groups. Ann Intern Med. 1988;109(5):372–382.
50. Sausville EA, Worsham GF, Matthews MJ, et al. Histologic assessment of lymph nodes in mycosis fungoides/Sézary syndrome (cutaneous T-cell lymphoma): clini-cal correlations and prognostic import of a new classification system. Hum Pathol. 1985;16(11):1098–1109.
51. Kern DE, Kidd PG, Moe R, et al. Analysis of T-cell receptor gene rearrangement in lymph nodes of patients with mycosis fungoides. Prognostic implications. Arch Derma-tol. 1998;134(2):158–164.
52. Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139(7):857–866.
53. Diamandidou E, Colome M, Fayad L, et al. Prognostic factor analysis in mycosis fun-goides/Sézary syndrome. J Am Acad Dermatol. 1999;40(6 Pt 1):914–924.
54. Agar NS, Wedgeworth E, Crichton S. Survival outcomes and prognostic factors in my-cosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer

43
staging proposal. J Clin Oncol. 2010;28(31):4730–4739. 55. Diamandidou E, Colome-Grimmer M, Fayad L, et al. Transformation of mycosis fun-
goides/Sezary syndrome: clinical characteristics and prognosis. Blood. 1998;92(4):1150–1159.
56. Scarisbrick JJ, Whittaker S, Evans AV, et al. Prognostic significance of tumor burden in the blood of patients with erythrodermic primary cutaneous T-cell lymphoma. Blood. 2001;97(3):624–630.
57. Grange F, Hedelin G, Joly P, et al. Prognostic factors in primary cutaneous lymphomas other than mycosis fungoides and the Sézary syndrome. The French Study Group on Cutaneous Lymphomas. Blood. 1999;93(11):3637–3642.
58. Scarisbrick JJ, Prince HM, Vermeer MH, et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sezary Syndrome: effect of specific prognostic markers on survival and development of a prog-nostic model. J Clin Oncol. 2015;33(32):3766-3773.
59. Kaye FJ, Bunn PA Jr, Steinberg SM, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. N Engl J Med. 1989;321(26):1784–1790.
60. Querfeld C, Rosen ST, Kuzel TM. Cutaneous T-cell lymphoma (Mycosis fungoides and Sézary’s syndrome). In: Rakel RE, Bope ET, eds. Conn’s Current Therapy, 57th ed. Section 13, Chapter 198. Philadelphia, PA: Elsevier/Saunders; 2006;902–906.
61. Gilchrest BA, Parrish JA, Tanenbaum L, et al. Oral methoxsalen photochemotherapy of mycosis fungoides. Cancer. 1976;38(2):683–689.
62. Honigsmann H, Brenner W, Rauschmeier W, et al. Photochemotherapy for cutaneous T cell lymphoma. A follow-up study. J Am Acad Dermatol. 1984;10(2 Pt 1):238–245.
63. Herrmann JJ, Roenigk HH Jr, Hurria A, et al. Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J Am Acad Dermatol. 1995;33(2 Pt 1):234–242.
64. Akaraphanth R, Douglass MC, Lim HW. Hypopigmented mycosis fungoides: treatment and a 6(1/2)-year follow-up of 9 patients. J Am Acad Dermatol. 2000;42(1 Pt 1):33–39.
65. Querfeld C, Rosen ST, Kuzel TM, et al. Long-term follow-up of patients with early-stage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Arch Dermatol. 2005;141(3):305–311.
66. Kuzel TM, Gilyon K, Springer E, et al. Interferon alfa-2a combined with phototherapy in the treatment of cutaneous T-cell lymphoma. J Natl Cancer Inst. 1990;82(3):203–207.
67. Roenigk HH Jr, Kuzel TM, Skoutelis AP, et al. Photochemotherapy alone or combined with interferon alpha-2a in the treatment of cutaneous T-cell lymphoma. J Invest Derma-tol. 1990;95(6 suppl):198S–205S.
68. Ramsay DL, Lish KM, Yalowitz CB, Soter NA. Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol. 1992;128(7):931–933.
69. Gambichler T, Breuckmann F, Boms S, et al. Narrowband UVB phototherapy in skin conditions beyond psoriasis. J Am Acad Dermatol. 2005;52(4):660–670.
70. Gathers RC, Scherschun L, Malick F, et al. Narrowband UVB phototherapy for early-stage mycosis fungoides. J Am Acad Dermatol. 2002;47(2):191–197.
71. Zackheim HS, Kashani-Sabet M, Amin S. Topical corticosteroids for mycosis fungoides. Experience in 79 patients. Arch Dermatol. 1998;134(8):949–954.
72. Zackheim HS. Treatment of patch-stage mycosis fungoides with topical corticosteroids. Dermatol Ther. 2003;16(4):283–287.
73. Klein E, Rosner D, Holtermann OA, et al. Nonspecific antigen reactions. Natl Cancer Inst Monogr. 1976;44:87–97.

44
74. Vonderheid EC, Ekbote SK, Kerrigan K, et al. The prognostic significance of delayed hy-persensitivity to dinitrochlorobenzene and mechlorethamine hydrochloride in cutane-ous T cell lymphoma. J Invest Dermatol. 1998;110(6):946–950.
75. Vonderheid EC, Tan ET, Kantor AF, et al. Long-term efficacy, curative potential, and car-cinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. J Am Acad Dermatol. 1989;20(3):416–428.
76. Kim YH. Management with topical nitrogen mustard in mycosis fungoides. Dermatol Ther. 2003;16(4):288–298.
77. Lessin SR, Duvic M, Guitart J, et al. Topical chemotherapy in cutaneous T-cell lympho-ma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.025, gel in mycosis fungoides. JAMA Dermatol. 2013;149(1):25–32.
78. Querfeld C, Nagelli LV, Rosen ST, et al. Bexarotene in the treatment of cutaneous T-cell lymphoma. Expert Opin Pharmacother. 2006;7(7):907–915.
79. Breneman D, Duvic M, Kuzel T, et al. Phase 1 and 2 trial of bexarotene gel for skin-directed treatment of patients with cutaneous T-cell lymphoma. Arch Dermatol. 2002;138(3):325–332.
80. Heald P, Mehlmauer M, Martin AG, et al; Worldwide Bexarotene Study Group. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol. 2003;49(5):801–815.
81. Apisarnthanarax N, Talpur R, Ward S, et al. Tazarotene 0.1% gel for refractory mycosis fungoides lesions: an open-label pilot study. J Am Acad Dermatol. 2004;50(4):600–607.
82. Thomas TO, Agrawal P, Guitart J, et al. Outcome of patients treated with a single-frac-tion dose of palliative radiation for cutaneous T-cell lymphoma. Int J Radiat Oncol Biol Phys. 2013;85(3):747–753.
83. Hoppe RT, Cox RS, Fuks Z, et al. Electron-beam therapy for mycosis fungoides: the Stanford University experience. Cancer Treat Rep. 1979;63(4):691–700.
84. Jones GW, Hoppe RT, Glatstein E. Electron beam treatment for cutaneous T-cell lym-phoma. Hematol Oncol Clin North Am. 1995;9(5):1057–1076.
85. Ysebaert L, Truc G, Dalac S, et al. Ultimate results of radiation therapy for T1-T2 mycosis fungoides (including reirradiation). Int J Radiat Oncol Biol Phys. 2004;58(4):1128–1134.
86. Hoppe RT, Harrison C, Tavallaee M, et al. Low-dose total skin electron beam thera-py as an effective modality to reduce disease burden in patients with mycosis fun-goides: results of a pooled analysis from 3 phase-II clinical trials. J Am Acad Dermatol. 2015;72(2):286–292.
87. Quiros PA, Jones GW, Kacinski BM, et al. Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous T-cell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys. 1997;38(5):1027–1035.
88. van Doorn R, Zoutman WH, Dijkman R, et al. Epigenetic profiling of cutaneous T-cell lymphoma: promoter hypermethylation of multiple tumor suppressor genes including BCL7a, PTPRG, and p73. J Clin Oncol. 2005;23(17):3886–3896.
89. Sun WH, Pabon C, Alsayed Y, et al. Interferon-alpha resistance in a cutaneous T-cell lym-phoma cell line is associated with lack of STAT1 expression. Blood. 1998;91(2):570–576.
90. Vonderheid EC, Thompson R, Smiles KA, Lattanand A. Recombinant interferon alfa-2b in plaque-phase mycosis fungoides. Intralesional and low-dose intramuscular therapy. Arch Dermatol. 1987;123(6):757–763.
91. Kohn EC, Steis RG, Sausville EA, et al. Phase II trial of intermittent high-dose recom-binant interferon alfa-2a in mycosis fungoides and the Sézary syndrome. J Clin Oncol.

45
1990;8(1):155–160.92. Olsen EA, Bunn PA. Interferon in the treatment of cutaneous T-cell lymphoma. Hematol
Oncol Clin North Am. 1995;9(5):1089–1107.93. Kuzel TM, Roenigk HH Jr, Samuelson E, et al. Effectiveness of interferon alfa-2a com-
bined with phototherapy for mycosis fungoides and the Sézary syndrome. J Clin Oncol. 1995;13(1):257–263.
94. Kuzel TM, Roenigk HH Jr, Samuelson E, Rosen ST. Suppression of anti-interferon alpha-2a antibody formation in patients with mycosis fungoides by exposure to long-wave UV radiation in the A range and methoxsalen ingestion. J Natl Cancer Inst. 1992;84(2):119–121.
95. Dummer R, Mangana J. Long-term pegylated interferon-alpha and its potential in the treatment of melanoma. Biologics. 2009;3:169–182.
96. Schiller M, Tsianakas A, Sterry W, et al. Dose-escalation study evaluating pegylated in-terferon alpha-2a in patients with cutaneous T-cell lymphoma. J Eur Acad Dermatol Ve-nereol. 2017 May 30. [Epub ahead of print]
97. Zhang C, Hazarika P, Ni X, et al. Induction of apoptosis by bexarotene in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. Clin Cancer Res. 2002;8(5):1234–1240.
98. Duvic M, Martin AG, Kim Y, et al; Worldwide Bexarotene Study Group. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch Dermatol. 2001;137(5):581–593.
99. Duvic M, Hymes K, Heald P, et al; Worldwide Bexarotene Study Group. Bexarotene is ef-fective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol. 2001;19(9):2456–2471.
100. Querfeld C, Rosen ST, Guitart J, et al. Comparison of selective retinoic acid receptor- and retinoic X receptor-mediated efficacy, tolerance, and survival in cutaneous t-cell lymphoma. J Am Acad Dermatol. 2004;51(1):25–32.
101. Talpur R, Ward S, Apisarnthanarax N, et al. Optimizing bexarotene therapy for cutane-ous T-cell lymphoma. J Am Acad Dermatol. 2002;47(5):672–684.
102. Gorgun G, Foss F. Immunomodulatory effects of RXR rexinoids: modulation of high-affinity IL-2R expression enhances susceptibility to denileukin diftitox. Blood. 2002;100(4):1399–1403.
103. Foss F, Demierre MF, DiVenuti G. A phase-1 trial of bexarotene and denileukin diftitox in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2005;106(2):454–457.
104. Maeda A, Schwarz A, Kernebeck K, et al. Intravenous infusion of syngeneic apop-totic cells by photopheresis induces antigen-specific regulatory T cells. J Immunol. 2005;174(10):5968–5976.
105. Edelson R, Berger C, Gasparro F, et al. Treatment of cutaneous T-cell lymphoma by ex-tracorporeal photochemotherapy. Preliminary results. N Engl J Med. 1987;316(6):297–303.
106. Zic JA. The treatment of cutaneous T-cell lymphoma with photopheresis. Dermatol Ther. 2003;16(4):337–346.
107. Child FJ, Mitchell TJ, Whittaker SJ, et al. A randomized cross-over study to compare PUVA and extracorporeal photopheresis in the treatment of plaque stage (T2) mycosis fungoides. Clin Exp Dermatol. 2004;29(3):231–236.
108. Olsen EA, Rook AH, Zic J, et al. Sézary syndrome: immunopathogenesis, literature re-view of therapeutic options, and recommendations for therapy by the United States Cu-taneous Lymphoma Consortium (USCLC). J Am Acad Dermatol. 2011;64(2):352–404.

46
109. Wilson LD, Jones GW, Kim D, et al. Experience with total skin electron beam therapy in combination with extracorporeal photopheresis in the management of patients with erythrodermic (T4) mycosis fungoides. J Am Acad Dermatol. 2000;43(1 Pt 1):54–60.
110. Suchin KR, Cucchiara AJ, Gottlieb SL, et al. Treatment of cutaneous T-cell lymphoma with combined immunomodulatory therapy: a 14-year experience at a single institu-tion. Arch Dermatol. 2002;138(8):1054–1060.
111. Quaglino P, Fierro MT, Rossotto GL, et al. Treatment of advanced mycosis fungoides/Sézary syndrome with fludarabine and potential adjunctive benefit to subsequent ex-tracorporeal photochemotherapy. Br J Dermatol. 2004;150(2):327–336.
112. Dearden CE, Matutes E, Catovsky D. Alemtuzumab in T-cell malignancies. Med Oncol. 2002;(19 suppl):S27–S32.
113. Querfeld C, Mehta N, Rosen ST, et al. Alemtuzumab for relapsed and refractory eryth-rodermic cutaneous T-cell lymphoma: a single institution experience from the Robert H. Lurie Comprehensive Cancer Center. Leuk Lymphoma. 2009:50(12):1969–1976.
114. Clark RA, Watanabe R, Teague JE, et al. Skin effector memory T cells do not recircu-late and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4(117):117ra7.
115. Bernengo MG, Quaglino P, Comessatti A, et al. Low-dose intermittent alemtuzumab in the treatment of Sézary syndrome: clinical and immunologic findings in 14 patients. Haematologica. 2007;92(6):784–794.
116. Talpur R, Jones DM, Alencar AJ, et al. CD25 expression is correlated with histological grade and response to denileukin diftitox in cutaneous T-cell lymphoma. J Invest Der-matol. 2006;126(3):575–583.
117. Olsen E, Kim YH, Kuzel T, et al. Vorinostat (suberoylanilide hydroxamic acid, SAHA) is clinically active in advanced cutaneous T-cell lymphoma (CTCL): results of a phase IIb trial. J Clin Oncol. 2006;24(18 suppl):7500.
118. Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hy-droxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31–39.
119. Duvic M, Bates SE, Piekarz R, et al. Responses to romidepsin in patients with cutaneous T-cell lymphoma and prior treatment with systemic chemotherapy. Leuk Lymphoma. 2017;30;1–8.
120. Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, internation-al, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–4491.
121. Ellis L, Pan Y, Smyth GK, et al. Histone deacetylase inhibitor panobinostat induces clini-cal responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14(14):4500-4510.
122. Duvic M, Dummer R, Becker JC, et al. Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer. 2013;49(2):386–394.
123. Kim YH, Tavallaee M, Sundram U, et al. Phase II investigator-initiated study of brentuxi-mab vedotin in mycosis fungoides and Sézary syndrome with variable CD30 expression level: a multi-institution collaborative project. J Clin Oncol. 2015;33(32):3750–3758.
124. Prince HM, Kim YH, Horwitz SM, et al; ALCANZA Study Group. Brentuximab ve-dotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCAN-ZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet. 2017;390(10094):555–566.
125. Duvic M, Evans M, Wang C. Mogamulizumab for the treatment of cutaneous T-cell lym-

47
phoma: recent advances and clinical potential. Ther Adv Hematol. 2016;7(3):171–174.126. Duvic M, Pinter-Brown LC, Foss FM, et al. Phase 1/2 study of mogamulizumab, a de-
fucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood. 2015;125(12):1883–1889.
127. Ogura M, Ishida T, Hatake K, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol. 2014;32(11):1157–1163.
128. Hughes CF, Khot A, McCormack C, et al. Lack of durable disease control with chemo-therapy for mycosis fungoides and Sézary syndrome: a comparative study of systemic therapy. Blood. 2015;125(1):71–81.
129. Wollina U, Graefe T, Kaatz M. Pegylated doxorubicin for primary cutaneous T cell lym-phoma: a report on ten patients with follow-up. Ann N Y Acad Sci. 2001;941:214–216.
130. Wollina U, Dummer R, Brockmeyer NH, et al. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer. 2003;98(5):993–1001.
131. Zinzani PL, Baliva G, Magagnoli M, et al. Gemcitabine treatment in pretreated cutane-ous T-cell lymphoma: experience in 44 patients. J Clin Oncol. 2000;18(13):2603–2606.
132. Marchi E, Alinari L, Tani M, et al. Gemcitabine as frontline treatment for cutaneous T-cell lymphoma: phase II study of 32 patients. Cancer. 2005;104(11):2437–2441.
133. Bunn PA Jr, Hoffman SJ, Norris D, et al. Systemic therapy of cutaneous T-cell lymphomas (mycosis fungoides and the Sézary syndrome). Ann Intern Med. 1994;121(8):592–602.
134. Von Hoff DD, Dahlberg S, Hartstock RJ, Eyre HJ. Activity of fludarabine monophos-phate in patients with advanced mycosis fungoides: a Southwest Oncology Group study. J Natl Cancer Inst. 1990;82(16):1353–1355.
135. Kuzel TM, Hurria A, Samuelson E, et al. Phase II trial of 2-chlorodeoxyadenosine for the treatment of cutaneous T-cell lymphoma. Blood. 1996;87(3):906–911.
136. Saven A, Carrera CJ, Carson DA, et al. 2-Chlorodeoxyadenosine: an active agent in the treatment of cutaneous T-cell lymphoma. Blood. 1992;80(3):587–592.
137. Foss FM, Ihde DC, Breneman DL, et al. Phase II study of pentostatin and intermittent high-dose recombinant interferon alfa-2a in advanced mycosis fungoides/Sézary syn-drome. J Clin Oncol. 1992;10(12):1907–1913.
138. Wood GS, Wu J. Methotrexate and pralatrexate. Dermatol Clin. 2015;33(4):747-755.139. Zackheim HS, Kashani-Sabet M, McMillan A. Low-dose methotrexate to treat mycosis
fungoides: a retrospective study in 69 patients. J Am Acad Dermatol. 2003;49(5):873–878.140. Zackheim HS, Kashani-Sabet M, Hwang ST. Low-dose methotrexate to treat erythro-
dermic cutaneous T-cell lymphoma: results in twenty-nine patients. J Am Acad Derma-tol. 1996;34(4):626–631.
141. Izbicka E, Diaz A, Streeper R, et al. Distinct mechanistic activity profile of pralatrexate in comparison to other antifolates in in vitro and in vivo models of human cancers. Cancer Chemother Pharmacol. 2009;64(5):993–999.
142. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115–4122.
143. Fierro MT, Quaglino P, Savoia P, et al. Systemic polychemotherapy in the treatment of primary cutaneous lymphomas: a clinical follow-up study of 81 patients treated with COP or CHOP. Leuk Lymphoma. 1998;31(5-6):583–588.
144. Akpek G, Koh HK, Bogen S, et al. Chemotherapy with etoposide, vincristine, doxo-rubicin, bolus cyclophosphamide, and oral prednisone in patients with refractory cutaneous T-cell lymphoma. Cancer. 1999;86(7):1368–1376.

48
145. Mebazza A, Dupuy A, Rybojad M, et al. ESHAP for primary cutaneous T-cell lym-phomas: efficacy and tolerance in 11 patients. Hematol J. 2005;5(7):553–558.
146. Bigler RD, Crilley P, Micaily B, et al. Autologous bone marrow transplantation for advanced stage mycosis fungoides. Bone Marrow Transplant. 1991;7(2):133–137.
147. Russell-Jones R, Child F, Olavarria E, et al. Autologous peripheral blood stem cell transplantation in tumor-stage mycosis fungoides: predictors of disease-free surviv-al. Ann N Y Acad Sci. 2001;941:147–154.
148. Oyama Y, Guitart J, Kuzel TM, et al. High-dose therapy and bone marrow transplanta-tion in cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 2003;17(6):1475–1483.
149. Wu PA, Kim YH, Lavori PW, et al. A meta-analysis of patients receiving allogeneic or autologous hematopoietic stem cell transplant in mycosis fungoides and Sézary syn-drome. Biol Blood Marrow Transplant. 2009;15(8):982–990.
150. Duvic M, Donato M, Dabaja B, et al. Total skin electron beam and non-myeloablative allogeneic hematopoietic stem-cell transplantation in advanced mycosis fungoides and Sezary syndrome. J Clin Oncol. 2010;28(14):2365–2372.
151. Shiratori S, Fujimoto K, Nishimura M, et al. Allogeneic hematopoietic stem cell transplantation following reduced-intensity conditioning for mycosis fungoides and Sezary syndrome. Hematol Oncol. 2016;34(1):9–16.
152. Bagot M, Porcu P, Ram-Wolff C, et al. IPH4102, the first in class anti-KIR3DL2 MAB, is safe and clinically active in advanced cutaneous T cell lymphoma (CTCL) patients. Abstract. Presented at the EORTC CLTF meeting, London, United Kingdom, Octo-ber 13–15, 2017.
153. Khodadoust MS, Rook AH, Porcu P, et al. Pembrolizumab for treatment of relapsed/refractory mycosis fungoides and Sezary syndrome: clinical efficacy in a CITN multi-center phase 2 study. Abstract #181. Presented at the ASH 58th Annual Meeting and Exposition, San Diego, CA, December 3, 2016.
154. ClinicalTrials.gov. A phase 1/2 trial of durvalumab (medi4736) when given as a sin-gle agent or in combination with lenalidomide in patients with relapsed/ refractory peripheral t-cell lymphoma, including cutaneous t-cell lymphoma. NCT03011814.
155. Querfeld C, Thompson JA, Taylor M, et al. A single direct intratumoral injection of tti-621 (sirpαfc) induces antitumor activity in patients with relapsed/refractory my-cosis fungoides and Sézary syndrome: preliminary findings employing an immune checkpoint inhibitor blocking the cd47 “do not eat” signal. Blood. 2017;abst. 4056.
156. Querfeld C, Foss F, Pinter-Brown L, et al. Phase 1 study of the safety and effica-cy of MRG-106, a synthetic inhibitor of microRNA-155, in CTCL patients. Blood. 2017;abst. 820.
157. Wysocka M, Benoit BM, Newton S, et al. Enhancement of the host immune respons-es in cutaneous T-cell lymphoma by CpG oligodeoxynucleotides and IL-15. Blood. 2004;104(13):4142–4149.
158. Querfeld C, Rosen ST, Guitart J, et al. Results of an open-label multicenter phase 2 trial of lenalidomide monotherapy in refractory mycosis fungoides and Sézary syn-drome. Blood. 2014;123(8):1159–1166.
159. Tokura Y, Yagi H, Ohshima A, et al. Cutaneous colonization with staphylococci influ-ences the disease activity of Sézary syndrome: a potential role for bacterial superan-tigens. Br J Dermatol. 1995;133(1):6–12.
160. Demierre MF, Gan S, Jones J, Miller DR. Significant impact of cutaneous T-cell lym-phoma on patients’ quality of life: results of a 2005 National Cutaneous Lymphoma Foundation survey. Cancer. 2006;107(10):2504–2511.

49
Resources for additional informationBooks and journal articles
Gonzalez BR, Rosen ST, Querfeld C. Cutaneous T-cell lymphoma. In: Zain J, Kwak L, eds. Management of Lymphomas: A Case-Based Approach. Cham, Switzerland: Springer International Publishing AG, Adis; 2017:157–172.
Rubio-Gonzalez B, Zain J, Rosen ST, Querfeld C. Clinical manifestations and pathogenesis of cutaneous lymphomas: current status and future directions. Brit J Hematol. 2017;176(1):16–36.
Rubio-Gonzalez B, Zain J, Rosen ST, Querfeld C. Tumor microenvironment in mycosis fungoides and Sézary syndrome. Curr Opin Oncol. 2016;28(1):88–96.
Querfeld C, Rosen ST. Cutaneous T-cell lymphoma and cutaneous B-cell lymphoma. In: Abeloff MD, Armitage JO, Niederhuber JE, et al, eds. Abeloff ’s Clinical Oncology. Philadelphia, PA: Saunders Elsevier, 2013.
OrganizationsCutaneous Lymphoma FoundationAn independent, nonprofit patient advocacy organization dedicated to
supporting every person with cutaneous lymphoma by promoting awareness and education, advancing patient care, and facilitating research.
www.CLfoundation.org
Patients Against LymphomaA nonprofit foundation providing support and evidence-based information on
lymphoma and current treatments.www.lymphomation.org
International Society for Cutaneous LymphomasThe International Society for Cutaneous Lymphomas (ISCL) is a nonprofit,
worldwide association formed in 1992 during the 18th World Congress of Dermatology in New York. The aim of the society is to promote collaborative efforts between regional, national, and international groups active in clinical, histomorphologic, or research work related to cutaneous lymphoma and, consequently, to increase the knowledge of lymphoproliferative and related disorders of the skin.
www.cutaneouslymphoma.org
National Cancer InstituteA branch of the National Institutes of Health that conducts and supports research,
and provides training and health information with respect to the causes, diagnosis, and treatment of cancer. In addition, it provides a comprehensive cancer database, the Physician Data Query (PDQ), with monthly updated information on mycosis fungoides and Sézary syndrome.
www.cancer.gov

50
ClinicalTrials.govThe NCI further provides a registry of current cancer clinical cancer trials from
around the world; and directories of physicians, professionals who provide genetics services, and organizations that provide cancer care.
www.clinicaltrials.gov
Lymphoma Research FoundationThe Lymphoma Research Foundation (LRF) is a national nonprofit organization
providing funds for lymphoma research and health information for lymphoma patients. LRF offers the Lymphoma Rounds program for physicians and other healthcare professionals, meetings where they can address issues relating to the diagnosis and treatment of their patients. LRF provides in-person patient education programs, as well as teleconferences and webcasts/podcasts. It has also developed a Lymphoma Support Network to connect lymphoma patients or caregivers with volunteers who have had similar lymphoma-related experiences. LRF offers a toll-free helpline and a clinical trials information service for patients.
www.lymphoma.org
The Leukemia & Lymphoma SocietyThe Leukemia & Lymphoma Society is the world’s largest voluntary health
organization dedicated to blood cancer. Its mission is to cure leukemia, lymphoma, Hodgkin’s disease, and myeloma, to improve the quality of life of patients and their families, and to provide funding and support for research in these areas. The website provides general health information for patients and professionals, patient services, funding resources, and information about recent research and innovative treatments.
http://www.lls.org
American Academy of DermatologyThe American Academy of Dermatology (AAD) is the largest dermatology
group in the United States, representing almost all practicing dermatologists in the country and some international dermatologists.
www.aad.org
United States Cutaneous Lymphoma ConsortiumA multidisciplinary society of physicians that through collaborative efforts in
research and education improves the quality of life and prognosis of patients with cutaneous lymphoma.
www.usclc.org
American Society of HematologyWith more than 17,000 members from nearly 100 countries, the American Society
of Hematology (ASH) is the world’s largest professional society serving both clinicians and scientists around the world who are working to conquer blood diseases.
www.hematology.org

The update of this booklet was supported by a Medical Writing Grant funded by Therakos, Inc., a Mallinckrodt Pharmaceuticals company.
Clinical opinions expressed in this book are those of the authors and do not nec-essarily reflect the opinions of the sponsor, editor, and publisher, UBM Medica.
Copyright © 2017 by UBM Medica. All rights reserved.