Embed Size (px)
Citation preview

THEORETISCHE PHYSIK:THERMODYNAMIK
{ VORLESUNGSSKRIPT {
von R. RedmerUniversit�at RostockFachbereich PhysikD{18051 Rostock1

2

Inhaltsverzeichnis1 Grundbegri�e der Thermodynamik 51.1 Zustandsgr�o�en im thermodynamischen Gleichgewicht . . . . . . . . . . . . . . . 51.2 Zustandsvariablen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.3 Zustandsgleichungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.4 Materialeigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92 Haupts�atze der Thermodynamik 112.1 Nullter Hauptsatz: Die Temperatur T . . . . . . . . . . . . . . . . . . . . . . . . 112.2 Erster Hauptsatz: Die innere Energie U . . . . . . . . . . . . . . . . . . . . . . . 122.3 Zweiter Hauptsatz: Die Entropie S . . . . . . . . . . . . . . . . . . . . . . . . . . 152.3.1 Irreversible Prozesse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152.3.2 Entropie und W�arme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152.3.3 Das perpetuum mobile 2. Art . . . . . . . . . . . . . . . . . . . . . . . . . 172.3.4 Gibbssche Fundamentalgleichung . . . . . . . . . . . . . . . . . . . . . . . 182.3.5 Beziehung zwischen thermischer und kalorischer Zustandsgleichung . . . . 193 Grundlegende thermodynamische Prozesse und Beziehungen 213.1 Spezi�sche W�armen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213.1.1 Erw�armung bei konstantem Volumen . . . . . . . . . . . . . . . . . . . . . 213.1.2 Erw�armung bei konstantem Druck . . . . . . . . . . . . . . . . . . . . . . 213.1.3 Beziehungen zwischen den Molw�armen . . . . . . . . . . . . . . . . . . . . 223.1.4 Abh�angigkeit von Volumen und Druck . . . . . . . . . . . . . . . . . . . . 233.1.5 Molw�armen und Zustandsgr�o�en . . . . . . . . . . . . . . . . . . . . . . . 243.2 Adiabatische und polytrope Prozesse . . . . . . . . . . . . . . . . . . . . . . . . . 263.3 Der Carnotsche Kreisprozess . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 273.3.1 Verlauf im p{V{Diagramm . . . . . . . . . . . . . . . . . . . . . . . . . . 273.3.2 Carnot{Prozess mit idealem Gas als Arbeitsmedium . . . . . . . . . . . . 283.3.3 Entropie und Wirkungsgrad f�ur den Carnot{Prozess . . . . . . . . . . . . 293.3.4 W�armepumpe und K�altemaschine . . . . . . . . . . . . . . . . . . . . . . 313.4 Thermodynamische Temperaturskala: Die absolute Temperatur T . . . . . . . . . 324 Thermodynamische Potenziale 334.1 Einkomponentensysteme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334.1.1 Entropie und innere Energie . . . . . . . . . . . . . . . . . . . . . . . . . 334.1.2 Konstruktion thermodynamischer Potenziale aus U(S; V ) . . . . . . . . . 344.1.3 Konstruktion thermodynamischer Potenziale aus S(U; V ) . . . . . . . . . 364.1.4 Die thermodynamischen Potenziale des idealen Gases . . . . . . . . . . . 374.2 Mehrkomponentensysteme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384.2.1 Entropie f�ur o�ene Systeme und chemisches Potenzial . . . . . . . . . . . 384.2.2 Gibbs{Duhemsche Gleichung . . . . . . . . . . . . . . . . . . . . . . . . . 394.2.3 Die thermodynamischen Potenziale I; J;K;L . . . . . . . . . . . . . . . . 414.3 �Ubersicht �uber thermodynamische Potenziale und Maxwell{Relationen . . . . . . 424.4 Gleichgewichts- und Stabilit�atsbedingungen . . . . . . . . . . . . . . . . . . . . . 434.4.1 Allgemeine Prinzipien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434.4.2 Auswertung der Gleichgewichts- und Stabilit�atsbedingungen . . . . . . . . 444.4.3 �Ubersicht �uber die Stabilit�atsbedingungen . . . . . . . . . . . . . . . . . . 475 Das Verhalten bei tiefen Temperaturen: Der 3. HS 495.1 Vorbemerkungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 493

5.2 Formulierung des 3. HS: Das Nernstsche W�armetheorem . . . . . . . . . . . . . . 495.3 Folgerungen aus dem 3. HS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 505.3.1 Molw�armen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 505.3.2 Thermische Koe�zienten . . . . . . . . . . . . . . . . . . . . . . . . . . . 505.3.3 Entropie und chemisches Potenzial des idealen Gases f�ur T ! 0 . . . . . . 515.4 Der Joule{Thomson{E�ekt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 535.5 Adiabatische Entmagnetisierung und negative Temperaturen . . . . . . . . . . . 556 Homogene Einkomponentensysteme 576.1 Phasen�uberg�ange . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 576.1.1 Phasendiagramm f�ur Einkomponentensysteme . . . . . . . . . . . . . . . . 576.1.2 Klassi�zierung von Phasen�uberg�angen . . . . . . . . . . . . . . . . . . . . 586.1.3 Clausius{Clapeyronsche Gleichung f�ur P�U 1. Art . . . . . . . . . . . . . . 606.1.4 P�U 2. Art und Ehrenfestsche Gleichungen . . . . . . . . . . . . . . . . . . 606.1.5 Landau{Theorie f�ur P�U 2. Art . . . . . . . . . . . . . . . . . . . . . . . . 626.2 Gase und Fl�ussigkeiten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 656.2.1 Allgemeine Beziehungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 656.2.2 van der Waalssche Zustandsgleichung . . . . . . . . . . . . . . . . . . . . 656.2.3 Virialentwicklung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 676.3 Hohlraumstrahlung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 696.3.1 Thermodynamik des Photonengases . . . . . . . . . . . . . . . . . . . . . 696.3.2 Strahlungsgesetze . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 717 Mehrkomponentensysteme 737.1 Mehrkomponentensysteme ohne chemische Reaktion . . . . . . . . . . . . . . . . 737.1.1 Gleichgewichtsbedingung f�ur heterogene Mehrkomponentensysteme . . . . 737.1.2 Gibbssche Phasenregel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 747.1.3 Ideale homogene Mischungen . . . . . . . . . . . . . . . . . . . . . . . . . 757.1.4 Mischungsentropie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 767.1.5 Reale homogene Mischungen . . . . . . . . . . . . . . . . . . . . . . . . . 777.1.6 Der osmotische Druck . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 787.1.7 Raoultsche Gesetze . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 797.2 Mehrkomponentensysteme mit chemischer Reaktion . . . . . . . . . . . . . . . . 817.2.1 Bedingung f�ur chemisches Gleichgewicht . . . . . . . . . . . . . . . . . . . 817.2.2 Das Massenwirkungsgesetz . . . . . . . . . . . . . . . . . . . . . . . . . . 827.2.3 Verschiebung des chemischen Gleichgewichts: van't Ho�sche Gleichungenund das Prinzip von Le Chatelier und Braun . . . . . . . . . . . . . . . . 837.2.4 Anwendungen zum Massenwirkungsgesetz . . . . . . . . . . . . . . . . . . 848 Literaturempfehlung 87
4

1 Grundbegri�e der Thermodynamik1.1 Zustandsgr�o�en im thermodynamischen GleichgewichtThermodynamisches System� Mikroskopische Beschreibung auf atomarem Niveau: Statistische Physik� Hier: makroskopische Beschreibung eines Systems von etwa 1023 Teilchen, z.B. Mo-lek�ule, Atome (Gase, Fl�ussigkeiten, Festk�orper), Elektronen und Protonen (Plas-men), Photonen (Schwarzk�orperstrahlung), Phononen (Festk�orpergitter) ...� Charakterisierung des Systems durch messbare Gr�o�en (Zustandsgr�o�en) wie Volu-men V , Druck p, Temperatur T , Magnetisierung ~M , Polarisation ~P : : :� Thermodynamisches System: Temperatur ist eine Zustandsgr�o�e! Thermodynamik� Wichtige Aspekte der Thermodynamik: Umwandlung verschiedener Energieformenineinander (innere Energie) und die Bewertung der Energie durch die Entropie; Pha-senumwandlungen wie Verdampfen und Kondensieren, Schmelzen und Erstarren� System: Als abgeschlossen von der Umgebung betrachtet (Vereinfachung), d.h. WWim Innern viel st�arker als mit der Umgebung. Beispiele: Sto�probe im Chemiela-bor, Thermobeh�alter (Ka�eekanne), K�uhl asche (Sticksto�), chemischer Synthese-reaktor (Ammoniaksynthese), Erdatmosph�are (Klimamodelle), Jupiter und Sonne(Astrophysik), Universum (Kosmologie)� M�ogliche Austausche mit der Umgebung:SYSTEM
δΑ
δΝ
ARBEIT
Q(mech. oder Felder) (Waermebad)
STOFF (Teilchenreservior)
WAERMEδ
� Charakterisierung des Systems:abgeschlossen: �A = 0, �Q = 0, �N = 0adiabatisch isoliert: �A 6= 0, �Q = 0, �N = 0geschlossen: �A 6= 0, �Q 6= 0, �N = 0o�en: �A 6= 0, �Q 6= 0, �N 6= 05

Thermodynamischer Gleichgewichtszustand (Erfahrungssatz)Jedes von der Umgebung isolierte thermodynamische System geht nach hinreichend lan-ger Zeit in einen Zustand �uber, den es spontan nicht wieder verl�asst. Dieser Zustandhei�t thermodynamisches Gleichgewicht. Durch ihn sind alle Eigenschaften des Systemsbestimmt.� Beispiele:{ Temperaturausgleich{ Druckausgleich{ Konzentrationsausgleich{ Spontane Magnetisierung in Ferromagnetika (z.B. Fe, Co, Ni, Gd, Dy, EuO){ Spontane Polarisation in Ferroelektrika (z.B. KDP{Kristalle wie KD2PO4, Tri-glyzinsulfat, Perovskite wie BaTiO3 oder KNbO3)� Transitivit�at des thermodynamischen Gleichgewichts: Ist A mit B im Gleichgewichtund B mit C, so ist auch A mit C im Gleichgewicht.� Nichtgleichgewichtszust�ande: Zustandsgr�o�en h�angen von Ort und Zeit ab, z.B.Anregungs- und Relaxationsprozesse (kurze intensive Laserpulse auf Ober �achen:hochangeregter Halbleiter), Injektion schneller Teilchen in ein Fusionsplasma (Hei-zung), W�armeleitung (Temperaturgradient), Di�usion (Konzentrationsgradient), elek-trischer Strom (elektrisches Feld), Hall{Widerstand (magnetisches Feld), p{n{�Uber-gang unter Last (Spannung) ...� Modell des lokalen thermodynamischen Gleichgewichts ist h�au�g n�utzlich; Problemder Wahl geeigneter Zeit- und L�angenskalen ! Nichtgleichgewichtsthermodynamikund NichtgleichgewichtsstatistikKomponenteEin System kann aus verschiedenen Komponenten (Sto�en) bestehen. Beispiel: Luft kannf�ur einfache thermodynamische Rechnungen als Gemisch aus Sticksto� (78%), Sauersto�(21%) und Argon (1%) betrachtet werden.PhaseEine Phase ist ein in physikalischer und chemischer Hinsicht homogener Bereich einesthermodynamischen Systems. Beispiel: Wasser im Gleichgewicht mit seinem Dampf istein System aus 2 Phasen.Grenz �achentrennen Phasen voneinander. Die Zustandsgr�o�en (z.B. Dichte) �andern sich sehr schnellmit dem Ort in diesen eigentlich inhomogenen, sehr schmalen �Ubergangszonen. Das Mo-dell einer mathematischen Fl�ache ist f�ur das Mehrphasensystem anwendbar.6

1.2 ZustandsvariablenZustandsvariablen sind Parameter oder Messgr�o�en, die einen thermodynamischen Gleich-gewichtszustand charakterisieren. Man unterscheidet innere (z.B. Druck p, TemperaturT , chemische Zusammensetzung) und �au�ere (Felder, Volumen V ) sowie extensive undintensive. Gleichgewichtszust�ande sind durch eine kleine Anzahl von Zustandsvariablenvollst�andig charakterisiert. Den kleinsten m�oglichen Satz von Zustandsvariablen nenntman deshalb auch vollst�andigen Satz. Die zu ihm geh�orenden Zustandsvariablen bezeich-nen wir als unabh�angig. Die Auswahl eines vollst�andigen Satzes von Zustandsvariablenist willk�urlich und erfolgt nach Zweckm�a�igkeitskriterien. Alle anderen Zustandsvariablensind Funktionen der Zustandsvariablen des vollst�andigen Satzes; man nennt sie Zustands-gr�o�en oder abh�angige Zustandsvariablen. Die Zahl der unabh�angigen Zustandsvariablengibt die Zahl der thermodynamischen Freiheitsgrade des Systems an.Extensive Zustandsgr�o�en� Proportional zur Gr�o�e des Systems, z.B. Teilchenzahl N , Sto�menge (Molzahl) n,Volumen V , Masse m, innere Energie U , Entropie S, Magnetisierung ~M , Polarisation~P� Sind in einem Mehrphasensystem aus i Phasen additiv, d.h.U = iXj=1U (j) ; S = iXj=1S(j) : : :� F�ur ihre �Anderung in einem Volumenelement �V existieren Bilanzgleichungen:dAdt = daAdt + diAdt :daA: Bilanz aus Zu uss/Ab uss von A in/aus das/dem VolumenelementdiA: Bilanz aus Vernichtung und Produktion von A im Volumenelement� Abgeschlossenes System: daA = 0. Falls weiterhin diA = 0 gilt, ist A eine Erhal-tungsgr�o�e: dAdt = 0. Beispiele: Masse m, Ladung q, Energie E, Impuls ~p, Drehimpuls~L.� Extensive Gr�o�en k�onnen von einem System auf ein anderes �ubertragen werden undsind zum Teil ineinander umwandelbar (z.B. verschiedene Energieformen, Massendurch Reaktionen).� Beachte: Die Entropie S kann in abgeschlossenen Systemen auch anwachsen (3. HS).Intensive Zustandsgr�o�en� Sind unabh�angig von der Gr�o�e des Systems, z.B. Temperatur T , Druck p� Quotienten zweier extensiver Gr�o�en sind intensiv. Bezieht man sich auf dasVolumenelement �V : �dichte, z.B. Massendichte % = �m=�VMassenelement �m: spezi�sche �, z.B. spez. W�armekapazit�at C = �Q=�mSto�menge �n: molare �, z.B. molare Entropie s = �S=�n7

Thermodynamischer Prozess� Allgemein jede �Anderung einer Zustandsgr�o�e mit der Zeit� Speziell jeder Prozess, der zum Erreichen eines Gleichgewichtszustands f�uhrt, z.B.Temperaturausgleich (W�armeleitung), Druckausgleich durch Volumen�anderung, Kon-zentrationsausgleich (Di�usion) etc.� Verlaufen von selbst oder unter dem Ein uss �au�erer Einwirkungen� Im allgemeinen irreversibel, d.h. unumkehrbar� Als Idealisierung ist der reversible (umkehrbare) Prozess anzusehen: Er l�auft nur�uber Gleichgewichtszust�ande, �andert damit die Umgebung nicht und kann wiederzum Ausgangszustand zur�uckgef�uhrt werden.� N�aherung durch sehr langsame, quasistatische Prozesse� Prozessarten:isotherm dT = 0 adiabatisch �Q = 0 polytrop c = const.isobar dp = 0 isentrop dS = 0isochor dV = 0 isenthalp dH = 01.3 ZustandsgleichungenZustandsgleichungen verkn�upfen Zustandsgr�o�en miteinander. Aus ihnen sind alle ther-modynamischen Eigenschaften des Systems ableitbar. Zustandsgleichungen kann man alsFl�achen im Zustandsraum darstellen. Der Zustandsraum wird durch eine geeignete Zahlvon Zustandsvariablen aufgespannt. F�ur ein Gas folgt z.B. aus den unabh�angigen Varia-blen Temperatur T und Volumen V die Zustands �ache f�ur den Druck p = p(T; V ). JederGleichgewichtszustand des thermodynamischen Systems entspricht einem Punkt auf derZustands �ache.Beispiel: Modell des idealen Gases� Punktf�ormige Teilchen: kein Eigenvolumen� Keine Wechselwirkung zwischen den Teilchen: H = K + V , V = 0� Gute N�aherung f�ur T !1 oder % = mV ! 0� Zustandsgleichungen f�ur das ideale Gas:p(T; V; n) = nRT=V thermische ZustandsgleichungU(T; V; n) = 32nRT kalorische Zustandsgleichung�(T; V;N) = RT ln (N�3=V ) chemisches Potenzial (1)� = q2��h2=(MkBT ) : thermische Wellenl�ange von Teilchen der Masse M�h = h=2� = 1:0546� 10�34 Ws2 : Plancksches WirkungsquantumR = 8:3145 J/(mol K) : universelle Gas{Konstante, R = LkBL = 6:0221� 1023 mol�1 : Lochschmidtsche ZahlkB = 1:38066� 10�23 Ws/K : Boltzmann{Konstante8

Einheiten:Sto�menge [n]=molAbsolute Temperatur [T]=K, 0K=�273:15 �CInnere Energie [U]=J, 1J=1Ws=107erg=0:23885calDruck [p]=Pa=N/m2, 105Pa=1bar=1.0197at=0.9869atmZustandsgleichung realer Gase:Teilchen haben eine Ausdehnung und wechselwirken miteinander. Damit ist die M�oglich-keit von Phasen�uberg�angen gegeben. Die van der Waalssche Zustandsgleichungp(V; T;N) = nRTV�B � AV 2 (2)oder �p+ av2� (v � b) = RTbezogen auf 1 mol mit v = V=n ber�ucksichtigt das Eigenvolumen der Atome (Molek�ule)(b) und die Anziehung zwischen ihnen (a=v2).1.4 MaterialeigenschaftenThermodynamische Gr�o�en, die den di�erenziellen Zuwachs einer Zustandsgr�o�e bei in�-nitesimaler �Anderung einer unabh�angigen Variable beschreiben:� = 1V @V@T !p;N isobarer Ausdehnungskoe�zient�T = � 1V @V@p !T;N isotherme Kompressibilit�at� = 1p @p@T !V;N isochorer Druckkoe�zientCv = @U@T !V;N ; Cp = @U@T !p;N W�armekapazit�aten�m = 1V 0@@ ~M@ ~H 1AT;V;N magnetische Suszeptibilit�atBeispiel ideales Gas: pV = nRT ergibt sofort� = 1T ; �T = 1p ; � = 1Tund damit den (allgemeinen) Zusammenhangp��T = � (3)9

10

2 Haupts�atze der ThermodynamikDie Haupts�atze der Thermodynamik geben das empirische Verst�andnis f�ur das Verhal-ten eines thermodynamischen Systems sowie der Zustandsgr�o�en Temperatur T , inne-re Energie U und Entropie S an; sie sind Erfahrungss�atze. Sie k�onnen gleichzeitig alsMessvorschrift f�ur die jeweilige Zustandsgr�o�e verstanden werden.2.1 Nullter Hauptsatz: Die Temperatur T� Temperatur ist eine nichtmechanische Gr�o�e� Temperatur als Zustandsgr�o�e ! thermodynamisches System� Unabh�angig von der Vorgeschichte (Evolution) des Systems� Eigenschaft des Zustands, skalare Zustandsgr�o�e� Transitivit�at gilt: TA = TB, TB = TC ! TA = TC ; man spricht auch vom thermischenGleichgewicht zwischen den jeweiligen SystemenNullter Hauptsatz: R.H. Fowler (1931)F�ur jedes thermodynamische System existiert eine skalare Zustandsgr�o�e { die Tempe-ratur T . Systeme im thermodynamischen Gleichgewicht besitzen die gleiche Temperatur.� Quanti�zierung: Temperaturskala, Messger�at: Thermometer� Messprozess: thermodynamisches System wird in thermisches Gleichgewicht mit demThermometer (m�oglichst klein) gebracht� Festlegung einer Temperaturskala:{ W�ahle die Temperatur � als Funktion zweier Zustandsvariablen als Standard:� = f(X; Y ){ Alle Punkte in der X-Y-Ebene mit � = f(X; Y ) = const: bilden eine Isotherme.{ Den Isothermen werden Zahlen an einem beliebigen Punkt zugeordnet, z.B. imSchnittpunkt der Isothermen mit der Geraden Y = Y0: �i = f(Xi; Y0).{ Die Temperatur ist dann nur noch eine Funktion von X, der thermometrischenEigenschaft: � = �(X). Man nimmt im einfachsten Fall eine lineare Skala an:�(X) = aX.{ Der Wert der Konstanten a (Dimension) wird durch einen Fixpunkt festgelegt.Entsprechend einer internationalen Regelung verwendet man den Tripelpunktvon Wasser (Eis, Wasser, Dampf im Gleichgewicht) und ordnet ihm willk�urlichdie Temperatur von 273.16 K (Kelvin) zu.{ Hat die thermometrische Eigenschaft am Tripelpunkt den Wert XT , dann folgt:�(X) = 273:16 K XXT11

{ M�ogliche thermometrische Eigenschaften: Fl�ussigkeitss�aule in GlaskapillareX =h, elektrisches Widerstandsthermometer X = Rel, Gasthermometer bei kon-stantem Druck X = V , Gasthermometer bei konstantem Volumen X = p,Thermoelement X = Uel : : :� Gasthermometer bei konstantem Volumen eignen sich besonders gut, insbesondereje geringer der Gasdruck am Tripelpunkt pT gew�ahlt wird, d.h. im Grenzfall starkerVerd�unnung.� De�nition der idealen Gastemperatur: Kelvin{Skala, SI{BasiseinheitT � � = 273:16 K limpT!0 � ppT � ; V = const: (4)� Umrechnung in gebr�auchliche Celsius{Skala: Anderer NullpunktTCelsius�C = TKelvinK � 273:15� Zusammenhang mit der Fahrenheit{Skala:TCelsius�C = 59 �TFahrenheit�F � 32�2.2 Erster Hauptsatz: Die innere Energie UThermisches Gleichgewicht wird zwischen zwei Systemen mit anf�anglich unterschiedli-chen Temperaturen TA > TB durch Austausch von W�arme hergestellt: System A wirdk�alter und B wird w�armer bis beide die gleiche Temperatur T mit TA > T > TB haben.Die W�arme Q ist eine Energieform, die zwischen Systemen �ubertragen werden kann. Sieist keine Zustandsgr�o�e und besitzt kein vollst�andiges Di�erenzial: �Q. Die �ubertrageneW�armemenge h�angt davon ab, auf welchem Weg die W�arme zu- oder abgef�uhrt wird, z.B.bei konstantem Druck oder konstantem Volumen. Dem System zugef�uhrte (abgef�uhrte)W�armemengen z�ahlen immer positiv (negativ).Der Energiebegri� hat in der Physik eine zentrale Rolle. Aus der Mechanik ist der Energie-erhaltungssatz bekannt: Ekin+Epot = const: Durch Arbeiten von Thomson, Mayer, Jouleund Helmholtz Mitte des 19. Jahrhunderts wurde dieser verallgemeinert und auf thermo-dynamische Systeme angewendet. Es hat sich durch alle Untersuchungen best�atigt, dassdie Energie eines abgeschlossenen Systems bei Ber�ucksichtigung aller Energieformen eineErhaltungsgr�o�e ist.Erster Hauptsatz: H. von Helmholtz 1857F�ur jedes thermodynamische System existiert eine extensive Zustandsgr�o�e U , die innereEnergie. Sie kann im System durch Zufuhr von W�arme �Q und Arbeit �A anwachsen:dU = �Q+ �A :F�ur abgeschlossene Systeme gilt der Energieerhaltungssatz:dU = 0 bzw: U = const:12

� Messvorschrift f�ur U : Absolutwert kann durch Wahl eines Nullpunkts �ahnlich wiebei der potenziellen Energie festgelegt werden, z.B. U = 0 f�ur T = 0 und %! 0.� Zugef�uhrte W�arme: W�arme�aquivalent (Mayer, Joule) 1 cal= 4.187 J� Am System geleistete Arbeit �A: z.B. mechanische Arbeit bei Kompression einesGases im ZylinderKraft F
xFlaeche A
V,pKolben
� Druck: p = F=A� In�nitesimale Kompression: dV = Adx < 0� Am System geleistete Arbeit:�A = �Fdx = �pdV > 0� Arbeitsdi�erenzial: �A = �pdV�Ubersicht �uber einige Arbeitsdi�erenziale:Phys. Erscheinung Zustandsvariable Arbeitsdi�erenzial �AKompression/Expansion Volumen V �pdVvon Gasen, Fl�ussigkeiten Druck pVer�anderung der Ober �ache F �dFOber �ache Ober �achenspannung �L�angen�anderung L�ange l Zdleines Drahtes Zugkraft ZMagnetisierung eines Magnetisierung ~M ~H � d ~MMediums Magnetfeldst�arke ~Helektrische Polarisation Polarisation ~P ~E � d~Peines Mediums elektrische Feldst�arke ~EGalvanisches elektrische Ladung Qel UeldQelElement elektrische Spannung Uel�Anderung der Teilchen- Molzahl ni �idnizahl einer Sorte i chemisches Potenzial �iVollst�andiges Di�erenzial einer Zustandsvariablen W (x; y; z):dW = Xdx+ Y dy + Zdz ; X = @W@x ; Y = @W@y ; Z = @W@zDie Beziehungen @X@y = @Y@x ; @X@z = @Z@x ; @Y@z = @Z@ysind die notwendige und hinreichende Bedingung daf�ur, dass dW = Xdx+Y dy+Zdz einvollst�andiges Di�erenzial ist und W damit eine Zustandsgr�o�e. Diese Bedingung lautet inintegraler Form I dW = 0 :13

Innere Energie U(T; V;N): ist ein vollst�andiges Di�erenzialdU = @U@T !V;N dT + @U@V !T;N dV + @U@N !T;V dN@2U@V @T = @2U@T@V ; @2U@V @N = @2U@N@V ; @2U@T@N = @2U@N@TH dU = 0 () U(T; V;N) ist eine Zustandsgr�o�e����
��������
V
a
b
c
2
1
T
Die �Anderung von U ist unabh�angig vomWeg (a, b, c) und allein durch Anfangs-und Endzustand gegeben. F�ur ein ge-schlossenes System mit N=const. gilt:�U = Z 21 dU = U(T2; V2)� U(T1; V1)
������
������
������
������
V
2
1
p
Die �Anderung der Arbeit A (und derW�arme Q) ist wegabh�angig. Das Um-lau�ntegral pdV verschwindet nicht undliefert die geleistete Arbeit (schra�erteFl�ache). Es gilt:I �A 6= 0 ; I �Q 6= 0Umlau�ntegrale im Zustandsraum beschreiben Kreisprozesse, die immer wieder in einenwohlde�nierten Anfangszustand zur�uckf�uhren. Solche Prozesse sind f�ur Anwendungen derThermodynamik in der Technik grundlegend (W�armekraftmaschinen). Aus H dU = 0 undH �A 6= 0, H �Q 6= 0 folgt, dass bei Kreisprozessen Arbeit und W�arme abgegeben bzw.aufgenommen werden k�onnen und die folgende Beziehung laut 1. HS erf�ullt sein muss:I dU = I �A+ I �Q = �A +�Q = 0 :Satz von der Unm�oglichkeit eines perpetuum mobile 1. Art:Es ist unm�oglich, ein perpetuum mobile 1. Art zu konstruieren, d.h. eine periodisch ar-beitende Maschine, die Arbeit abgibt, ohne Energie in irgendeiner Form (z.B. W�arme)aufzunehmen.14

2.3 Zweiter Hauptsatz: Die Entropie S2.3.1 Irreversible ProzesseDer 1. HS sagt aus, da� alle thermodynamischen Prozesse dem Energieerhaltungssatzgen�ugen m�ussen. Andererseits sind die in der Natur ablaufenden Vorg�ange irreversibel,d.h. nicht umkehrbar. Damit ist die Zeitrichtung f�ur den Ablauf von Naturvorg�angen (hierthermodynamischen Prozessen) ausgezeichnet. Man unterscheidet dabei dissipative undAusgleichsprozesse.Erfahrungss�atze:Nicht alle mit dem 1. HS vertr�aglichen Prozesse werden beobachtet. Es ist o�enbar nichtm�oglich,� dissipative Prozesse vollst�andig r�uckg�angig zu machen, d.h. solche, bei denen W�armedurch Reibung entsteht (z.B. reibungsbehaftete Str�omung, plastische Verformung,Verbrennung etc.),� Ausgleichsprozesse wieder umzukehren (z.B. Temperatur-, Druck- oder Konzentra-tionsausgleich).Es wurde z.B. niemals beobachtet, dass� sich ein Wasserbad spontan abk�uhlt und einen Stein herausschleudert,� sich eine plastische Verformung unter Abk�uhlung von selbst wieder ausbeult,� sich ein Gas spontan in einem bestimmten Bereich seines Beh�alters konzentriert,� imW�armekontakt stehende K�orper spontan eine Temperaturdi�erenz aufbauen usw.Als geeignetes Ma� f�ur die Irreversibilit�at von thermodynamischen Prozessen f�uhren wirdie Zustandsgr�o�e Entropie S ein, die noch in geeigneter Weise quanti�ziert werden mu�(Clausius, Thomson, Planck, Sommerfeld).Empirischer Befund: Bei irreversiblen Prozessen geschieht im Innern des Systems et-was, was nicht wieder r�uckg�angig gemacht werden kann.Mathematische Formulierung: Im thermodynamischen System wird bei irreversiblenProzessen eine Gr�o�e produziert, die nicht wieder vernichtet werden kann.2.3.2 Entropie und W�armeDie Entropie ist eine skalare extensive Gr�o�e, die bilanziert werden kann. Die �Anderungder Entropie in einem Volumenelement �V ist durch Erzeugung (Vernichtung) im Innernbzw. durch Zu uss (Ab uss) aus (in) die Umgebung gegeben: dS = diS + daS. Es giltdann die Bilanzgleichung: %dsdt + div ~Js = �s :15

S
∆
J
V
ds/dt �Anderung der Entropie in einem Volumen-element �V .s: Spezi�sche Entropiedichte S = R %sdV~Js: Entropiestromdichte�s: Entropieproduktionsdichte� Bei irreversiblen Prozessen in abgeschlossenen Systemen wird im Innern des SystemsEntropie erzeugt und niemals vernichtet, d.h. diS � 0 bzw. �s � 0; das Gleichheits-zeichen gilt f�ur die Idealisierung des reversiblen Prozesses.� Zusammenhang zwischen Entropie und energetischen Gr�o�en: Betrachte z.B. eindurch Reibung von T1 auf T2 erw�armtes thermodynamisches System.� Stellt man thermisches Gleichgewicht mit einem W�armebad der Temperatur T1 her,wird der urspr�unglicher Zustand T1 wieder erreicht: Das System hat Entropie durchW�arme�ubertragung auf das Bad verloren und so die Spuren des irreversiblen Pro-zesses gel�oscht.� Ansatz f�ur den Zusammenhang zwischen Entropie- und W�armestromdichte mit derabsoluten Temperatur T : ~Js = ~JQT :� Betrachte reversiblen W�armeaustausch, d.h. eine sehr kleine Temperaturdi�erenz�T ! 0, so dass der Prozess quasistatisch verl�auft, diS = 0 bzw. �s = 0 gilt unddS = daS folgt:daSdt = ZV %dasdt dV = � ZV div ~JsdV = � ZF ~Js � d~F � � ZF 1T ~JQ � d~F = 1T �Qdt{ Fall ~JQ "" d~F , d.h. W�arme wird dem System entzogen: Q z�ahlt negativ{ Fall ~JQ "# d~F , d.h. W�arme wird in das System gebracht: Q z�ahlt positiv{ d.h. oben rechts folgt immer ein positives VorzeichenDe�nition der Entropie S : daS = �QT ; diS � 0 : (5)Sommerfeldsche Formulierung des 2. HS:Jedes thermodynamische System besitzt eine extensive Zustandsgr�o�e, die Entropie S. Ih-re Zunahme bei reversiblen Prozessen errechnet sich als Quotient aus zugef�uhrter W�arme-menge �Q und der bei dieser Gelegenheit zu de�nierenden absoluten Temperatur T . Beiallen irreversiblen Prozessen wird im Innern des Systems Entropie produziert.16

� De�nition der absoluten Temperatur T : Der integrierende Nenner 1=T �uberf�uhrtdas unvollst�andige Di�erenzial der W�arme �Q in das vollst�andige Di�erenzial dSder Zustandsgr�o�e Entropie.� Es gilt allgemein: dS � �QT .� Abgeschlossene Systeme: dS � 0, d.h. die Entropie kann nur zunehmen. Solangeim System noch Prozesse von allein ablaufen, w�achst die Entropie an. Im Gleichge-wichtszustand h�ort die Entropieproduktion auf und die Entropie ist maximal.� Historische Hypothese (Clausius): Betrachte die Welt als abgeschlossenes System,in der eine Vielzahl komplizierter Prozesse ablaufen. Die Entropie kann nur An-wachsen und strebt einem Maximalwert zu. Dadurch werden tendenziell alle Tem-peraturunterschiede ausgeglichen und die M�oglichkeiten zur Verrichtung von Arbeitersch�opfen sich: W�armetod. Problem: Struktur des Universums geschlossen/o�en,statisch/expandierend/pulsierend ... ?� Natur entwickelt im Laufe der Evolution komplizierte (biologische) Strukturen: Ther-modynamik irreversibler Prozesse in o�enen Systemen (Prigogine, Glansdor�); zen-trales Problem ist die Beschreibung der Entropieproduktion.� Entropie und statistische Beschreibung (Boltzmann, Planck, Einstein, Shannon):S = kB lnWmax, W : Wahrscheinlichkeit zur Realisierung eines Systemzustands; dieEntropie ist mit dem wahrscheinlichsten Zustand verkn�upft.2.3.3 Das perpetuum mobile 2. ArtDer 1. HS regelt die Energieerhaltung bei thermodynamischen Prozessen, d.h. Energiekann nicht erzeugt werden. Es ist aber m�oglich, die verschiedenen Energieformen ineinan-der umzuwandeln, der laut 1. HS keinerlei Einschr�ankungen unterliegen. Z.B. k�onnte mandie riesigen Vorr�ate an innerer Energie in den Weltmeeren und in der Atmosph�are durchAbk�uhlung zum Antrieb von Schi�en oder Flugzeugen �uber einen geeigneten Kreisprozessnutzen. Alle Versuche zum Bau einer solchen Maschine sind gescheitert! Erfahrungssatz:Satz von der Unm�oglichkeit eines perpetuum mobile 2. Art (Planck):Es ist unm�oglich, ein perpetuum mobile 2. Art zu konstruieren, d.h. eine periodisch ar-beitende Maschine, die weiter nichts bewirkt als das Heben einer Last (Arbeitsleistung)und Abk�uhlung eines W�armereservoirs.� Die �ktive Maschine nimmt nach endlicher Zeit t die Temperatur T0 des W�armebadesan. Damit k�onnen keine W�armemengen mehr ausgetauscht werden: �Q = 0.� F�ur eine periodisch arbeitende Maschine (Kreisprozess) gilt dann: H dU = 0 !H �A = � H �Q = 0 und damit ist keine Arbeitsleistung mehr m�oglich.Alternative Formulierung (Clausius):Es existiert keine periodisch arbeitende Maschine, die keine andere dauernde Ver�ande-rung bewirkt, als dass bei einer festen Temperatur einem W�armebad W�arme entnommenwird und die gleiche W�armemenge einem anderen W�armebad bei h�oherer Temperaturzugef�uhrt wird. 17

Rolle von Energie und Entropie (Sommerfeld):In der riesigen Fabrik der Naturprozesse nimmt die Entropie die Stelle des Direktors ein,denn sie schreibt die Art und den Ablauf der Prozesse vor. Die Energie hat die Rolle desBuchhalters, der Soll und Haben ins Gleichgeicht bringt.2.3.4 Gibbssche FundamentalgleichungMan kann den 1. und 2. HS f�ur reversible Prozesse zusammenfassen:dS = 1T dU � 1T �A :Die Arbeitsdi�erenziale (siehe Tabelle) sind darstellbar als:�A = nXi=1 aidAi :Damit gilt die Gibbssche Fundamentalgleichung:dS = 1T dU � 1T nPi=1 aidAi : (6)Sie bildet die Grundlage f�ur die Gleichgewichtsthermodynamik. Speziell f�ur Gase undFl�ussigkeiten mit �A = �pdV gilt:dS = 1T dU + pT dV :� Beziehung zwischen vollst�andigen Di�erenzialen: S = S(U;Ai)� Bilde das vollst�andiges Di�erenzial und vergleiche:dS = @S(U;Al)@U dU + nXi=1 @S(U;Al)@Ai dAi ;1T = @S(U;Al)@U ; ai = �T @S(U;Al)@Ai :� Links folgt die kalorische Zustandsgleichung: T = T (U;Ai)! U = U(T;Ai)� Rechts folgt die thermische Zustandsgleichung durch Ersetzen von U : ai = ai(T;Ai)� Wichtige Eigenschaften wie die Temperatur T und die Gr�o�en ai h�angen wie dieEntropie nur von U und den Gr�o�en Ai ab; diese charakterisieren den Zustand desSystems o�enbar vollst�andig.� Die Gibbssche Fundamentalgleichung gibt also einen vollst�andigen Satz von Zu-standsvariablen an.� Thermodynamische Gr�o�en lassen sich bei Kenntnis der Entropie S(U;Ai) durcheinfaches Di�erenzieren nach den Variablen des vollst�andigen Satzes berechnen. Mannennt S(U;Ai) deshalb auch thermodynamisches Potenzial.18

2.3.5 Beziehung zwischen thermischer und kalorischer ZustandsgleichungBeide Zustandsgleichungen sind aus dem thermodynamischen Potenzial S abgeleitet wor-den und somit nicht unabh�angig. Wie lautet der explizite Zusammenhang? Betrachtenwir das Beispiel von Gasen und Fl�ussigkeiten mit den unabh�angigen Variablen T undA1 = V und gehen zu molaren Gr�o�en s = S=n, u = U=n, v = V=n in der GibbsschenFundamentalgleichung (6) �uber: Tds = du+ pdv :Das vollst�andige Di�erenzial der molaren inneren Energie ist laut kalorischer Zustands-gleichung u = u(T; v): du = @u@T !v dT + @u@v!T dv :Man erh�alt f�ur das vollst�andige Di�erenzial der molaren Entropie s = s(T; v):ds = 1T @u@T !v dT + 1T " @u@v!T + p# dv � @s@T !v dT + @s@v!T dv :Vergleich liefert: @s@T !v = 1T @u@T !v ; @s@v!T = 1T " @u@v!T + p# : (7)Die gemischten zweiten Ableitungen der molaren Entropie s(T; v) m�ussen laut Integrabi-lit�atsbedingung gleich sein,@@v ( 1T @u@T !v)T = @@T ( 1T " @u@v!T + p#)v ;so dass nach Di�erenzieren folgt:1T @2u@v@T = � 1T 2 " @u@v!T + p#+ 1T @2u@T@v + 1T @p@T !v :Die gemischten zweiten Ableitungen von u(T; v) sind wieder gleich, so dass die gew�unschteBeziehung folgt: �@u@v �T = T � @p@T �v � p : (8)Die thermische Zustandsgleichung p = p(T; v) legt die Volumenabh�angigkeit der in-neren Energie fest. Die Temperaturabh�angigkeit der inneren Energie ist dagegen nichtvollst�andig festgelegt; hier ist eine additive Temperaturfunktion frei w�ahlbar. Wird (8) indie rechte Seite von (7) eingesetzt, �ndet man eine sogenannte Maxwell{Beziehung: @s@v!T = @p@T !v : (9)Beispiel: Ideales Gas bezogen auf ein Mol pv = RT . Es folgt sofort: @u@v!T = 0 bzw: u = u(T ) ;d.h. die innere Energie des idealen Gases h�angt nicht vom Volumen ab. Experiment:Gay{Lussac{Versuch. 19

VV1 2
p,T
Irreversible Gasexpansion (Gay{Lussac{Versuch):Das System ist adiabatisch isoliert, d.h. �Q = 0. BeimEntspannen des Gases von V1 auf V1 + V2 wird kei-ne Arbeit geleistet, d.h. �A = 0. Damit ist laut 1.HS auch dU = 0; die innere Energie des idealen Ga-ses h�angt nicht vom Volumen ab. Experimentell �uberTemperaturmessung best�atigt: T = const.
20

3 Grundlegende thermodynamische Prozesse und Beziehungen3.1 Spezi�sche W�armenIm folgenden betrachten wir homogene Einkomponentensysteme mit n = konstant. Willman die Temperatur einer Substanz erh�ohen, muss man ihr W�arme zuf�uhren. Die W�arme-menge Q, die 1 Gramm Substanz um 1 Grad erw�armt, nennt man spezi�sche W�arme.Bezieht man sich auf 1 Mol mit q = Q=n, dann nennt man sie die Molw�arme cc = �qdTmit der Einheit 1 J/(mol K). Die Molw�arme ist keine Zustandsgr�o�e, da �q kein vollst�andi-ges Di�erenzial ist. Sie h�angt von der Art der Prozessf�uhrung ab.3.1.1 Erw�armung bei konstantem VolumenAus dem vollst�andigen Di�erenzial der inneren Energie u = u(T; v) und dem 1. HSdu = @u@T !v dT + @u@v!T dv ; du = �q � pdvfolgt: �q = @u@T !v dT + " @u@v!T + p# dv :Die Molw�arme ist also �uber die innere Energie allgemein durchc = �qdT = � @u@T �v + h�@u@v�T + pi dvdT (10)gegeben. F�ur konstantes Volumen folgt dv = 0 und aus (10) f�ur cv:cv � �qvdT = @u@T !v ;d.h. cv kann bei Kenntnis der kalorischen Zustandsgleichung u(T; v) sofort berechnet wer-den. Beispiel ideales Gas: u = 32RT und somit cv = 32R.3.1.2 Erw�armung bei konstantem DruckF�ur Prozesse unter konstantem Druck ist es g�unstig, anstelle der inneren Energie u(T; v)eine neue Zustandsgr�o�e einzuf�uhren, die von Temperatur T und Druck p abh�angt { diemolare Enthalpie h(T; p): h = u+ pv :Man erh�alt f�ur ihr vollst�andiges Di�erenzial mit dem 1. HS du = �q � pdv:dh = du+ pdv + vdp = �q + vdp :Die �Anderung der molaren Enthalpie dh ist bei konstantem Druck dp = 0 gleich der demSystem von au�en zugef�uhrten molaren W�arme �q. F�ur die Molw�arme c folgt damit ausdem vollst�andigen Di�erenzialdh = @h@T !p dT + @h@p!T dp21

ganz allgemein c = �qdT = � @h@T �p + h�@h@p�T � vi dpdT : (11)F�ur Prozesse bei konstantem Druck mit dp = 0 ergibt sich aus (11) speziellcp � �qpdT = @h@T !p ;d.h. cp kann bei Kenntnis der Enthalpie h(T; p) sofort berechnet werden. Beispiel idealesGas: h = u+ pv = 32RT +RT = 52RT und somit ist cp = 52R.3.1.3 Beziehungen zwischen den Molw�armenMit der Beziehung (8) zwischen kalorischer und thermischer Zustandsgleichung erh�alt manaus (10) f�ur cp sofort cp = cv + T � @p@T �v � dvdT �p : (12)Mit den De�nitionen der Materialeigenschaften und (3) ergibt sich aus (12) mit �T > 0:cp � cv = T � �p � v� = Tv �2�T > 0 :Die Beziehung (12) ist auch �uber die molare Enthalpie h(T; p) ableitbar. Ausgangspunktist der 2. HS: �q = Tds = dh � vdp. Damit folgt f�ur das vollst�andige Di�erenzial dermolaren Entropie s(T; p):ds = 1T @h@T !p dT + 1T " @h@p!T � v# dp � @s@T !p dT + @s@p!T dp :Vergleich liefert: @s@T !p = 1T @h@T !p ; @s@p!T = 1T " @h@p!T � v# : (13)Die gemischten zweiten Ableitungen der molaren Entropie s(T; p) m�ussen laut Integrabi-lit�atsbedingung gleich sein,@@p 8<: 1T @h@T !p9=;T = @@T ( 1T " @h@p!T � v#)p ;so dass nach Di�erenzieren folgt:1T @2h@p@T = � 1T 2 " @h@p!T � v#+ 1T @2h@T@p � 1T @v@T !p :Die gemischten zweiten Ableitungen von h(T; p) sind gleich und es folgt:�@h@p�T = �T � @v@T �p + v : (14)22

Die Druckabh�angigkeit der Enthalpie ist allein durch die thermische Zustandsgleichungbestimmt. Mit (14) erh�alt man aus (11) wieder die Relation (12) zwischen den Molw�armen.Setzt man (14) wieder in die rechte Seite von (13) ein, �ndet man eine weitere Maxwell{Relation: @s@p!T = � @v@T !p : (15)Beispiel ideales Gas pv = RT : Einsetzen der Ableitungen (@p=@T )v = R=v und (@v=@T )p =R=p in (12) liefert die allgemeine Beziehungcp � cv = R : (16)Die Tabelle gibt Beispiele f�ur die Molw�armen verschiedener realer Substanzen an und pr�uftdie Erf�ullung der Relation (16) f�ur ideale Gase. Die �Ubereinstimmung ist relativ gut. Ausdem �Aquipartitionstheorem der Statistischen Physik f�ur die innere Energie U = f2NkBT ,wobei f die Anzahl der Freiheitsgrade f�ur die Atome/Molek�ule ist, erh�alt man wegen u =f2RT f�ur die Molw�arme sofort cv = f2R. Damit ist cp = f+22 R und der Adiabatenexponentdurch = cpcv = cv+Rcv = 1 + Rcv = 1 + 2f gegeben.� Translationsfreiheitsgrade f trans = 3 (f�ur einatomige Gase wie z.B. He alleinigerBeitrag).� Rotationsfreiheitsgrade von Molek�ulen werden f�ur Temperaturen oberhalb �h!rot �0:01 eV � 102 K angeregt, sind abh�angig von der Molek�ulsymmetrie: f rot = 2 f�urzweiatomige Gase wie O2 oder lineare Molek�ule wie CO2, ansonsten gilt f rot = 3(drei Rotationsachsen) f�ur mehratomige Molek�ule.� Schwingungsfreiheitsgrade von Molek�ulen werden zus�atzlich bei Temperaturen ober-halb �h!vib � 0:1 eV � 103 K angeregt, ihre Abz�ahlung und Temperaturabh�angigkeitist komplizierter. N�aherungsweise ergibt sich fvib = 2.Substanz f cp [R] cv [R] cp � cv [R] He 3 2.52 1.52 1.00 1.66O2 5 3.51 2.50 1.01 1.40CO2 7 4.40 3.38 1.02 1.30C2H6 9 5.75 4.71 1.04 1.223.1.4 Abh�angigkeit von Volumen und DruckWie h�angt die Molw�arme cv vom Volumen und cp vom Druck ab? Diese Frage ist f�ur dieexperimentelle Bestimmung thermodynamischer Gr�o�en wie U , H und S wichtig, da dieMolw�armen relativ einfach zu messen sind (Kalorimetrie). Wir benutzen den Zusammen-hang mit der molaren Entropie s �uber den 2. HSc = �qdT = T dsdT ;so dass gilt: cv = T @s@T !v ; cp = T @s@T !p :23

Wir untersuchen zuerst die Abh�angigkeit von cv vom Volumen und �nden unter Aus-nutzung der Vertauschbarkeit der zweiten gemischten Ableitungen von s und mit derMaxwell{Relation (9): @cv@v !T = ( @@v "T @s@T !v#)T = T " @@T @s@v!T#v � T @2p@T 2!v :Analog erh�alt man f�ur die Druckabh�angigkeit von cp mit der Maxwell{Relation (15): @cp@p !T = 8<: @@p 24T @s@T !p359=;T = T " @@T @s@p!T#p � �T @2v@T 2!p :Die Volumenabh�angigkeit von cv und die Druckabh�angigkeit von cp werden allein von derthermischen Zustandsgleichung p = p(T; v) bestimmt. Beispiel ideales Gas pv = RT : Diezweiten Ableitungen verschwinden und damit sind cv/cp unabh�angig vom Volumen/Druck.Man �ndet dann f�ur die Molw�armen durch Integration:cv(T; v) = cv(T; v0) + vZv0 T @2p@T 2!v0 dv0 ;cp(T; p) = cp(T; p0)� pZp0 T @2v@T 2!p0 dp0 :Bei Kenntnis der thermischen Zustandsgleichung p = p(T; v) k�onnen die Funktionencv(T; v) und cp(T; p) unter Ausnutzung des allgemeinen Zusammenhangs (12) allein ausder Messung von cv in Abh�angigkeit von der Temperatur bei konstantem Volumen v0bestimmt werden.3.1.5 Molw�armen und Zustandsgr�o�enDie innere Energie u(T; v), Enthalpie h(T; p) und Entropie s(T; v) bzw. s(T; p) k�onnen ausden Molw�armen und der thermischen Zustandsgleichung p = p(T; v) berechnet werden.(1) Innere Energie: Aus dem vollst�andigen Di�erenzialdu = @u@T !v dT + @u@v!T dvfolgt mit der De�nition von cv und (8):du = cvdT + "T @p@T !v � p# dv :Integration l�angs eines Weges von (T0; v0) nach (T; v) in der T{v{Ebene ergibt die innereEnergie bis auf eine additive Konstante u0:u(T; v) = (T;v)Z(T0;v0) (cv0dT 0 + "T 0 @p@T 0!v0 � p(T 0; v0)# dv0)+ u0 :24

F�ur das ideale Gas pv = RT vereinfacht sich dieses Ergebnis und die innere Energie istallein aus der Molw�arme cv berechenbar:u(T ) = TRT0 cv(T 0)dT 0 + u0 = cv(T � T0) + u0 : (17)F�ur nicht zu gro�e Temperaturintervalle kann die Molw�arme cv als konstant angesehenwerden.(2) Enthalpie: Aus dem vollst�andigen Di�erenzialdh = @h@T !p dT + @h@p!T dpfolgt mit der De�nition von cp und (14):dh = cpdT + 24v � T @v@T !p35 dp :Integration l�angs eines Weges von (T0; p0) nach (T; p) in der T{p{Ebene ergibt die Ent-halpie bis auf eine additive Konstante h0:h(T; p) = (T;p)Z(T0;p0) 8<:cp0dT 0 + 24v(T 0; p0)� T 0 @v@T 0!p035 dp09=;+ h0 :F�ur das ideale Gas pv = RT vereinfacht sich dieses Ergebnis und die Enthalpie ist alleinaus der Molw�arme cp berechenbar:h(T ) = TRT0 cp(T 0)dT 0 + h0 = cp(T � T0) + h0 : (18)F�ur nicht zu gro�e Temperaturintervalle kann die Molw�arme cp ebenfalls als konstantangesehen werden.(3) Entropie: Aus dem 2. HS ds = �qT = 1T du + pT dv bzw. ds = �qT = 1T dh � vT dp erh�altman mit den Beziehungen (8) und (14) nach Integration soforts(T; v) = (T;v)Z(T0;v0) (cv0T 0 dT 0 + @p@T 0!v0 dv0)+ s0v ;s(T; p) = (T;p)Z(T0;p0) 8<:cp0T 0 dT 0 � @v@T 0!p0 dp09=;+ s0p :F�ur ideale Gase pv = RT und konstante Molw�armen vereinfachen sich diese Beziehungen:s(T; v) = cv ln TT0 +R ln vv0 + s0v ; s(T; p) = cp ln TT0 � R ln pp0 + s0p : (19)Ersetzt man weiterhin R = cp � cv laut (16), so ergeben sich die Gleichungen:s(T; v) = cv ln Tv �1T0v �10 !+ s0v ; s(T; p) = cp ln0B@ Tp 1� T0p 1� 0 1CA+ s0p : (20)25

3.2 Adiabatische und polytrope ProzesseUnterbindet man jeglichen W�armeaustausch des Systems mit seiner Umgebung, nenntman es adiabatisch isoliert. Alle dann noch m�oglichen Prozesse nennt man adiabatischeProzesse, die durch �q = 0 gekennzeichnet sind. Laufen die Prozesse reversibel ab, istwegen des 2. HS ds = �q=T auch ds = 0 und sie sind gleichzeitig isentrop.Polytrope Prozesse sind Prozesse, bei denen die Molw�arme konstant bleibt: c = const.Adiabatische Prozesse k�onnen wegen �q = cdT als spezielle polytrope Prozesse mit c = 0angesehen werden. Wir leiten aus�q = cdT = du+ pdv = @u@T !v dT + " @u@v!T + p# dvmit (8) die Gleichung (c� cv)dT = T @p@T !v dvab. Weiter ergibt sich mit dem Zusammenhang (12) zwischen cp und cv die di�erenziellePolytropengleichung in den Variablen T und v:dT = cp�cvc�cv �@T@v �p dv : (21)Man kann sie mit T = T (v; p) und dT = �@T@v �p dv + �@T@p �v dp auch f�ur die Variablen Tund p angeben: �@T@p �v dp = cp�cc�cv �@T@v �p dv : (22)Die entsprechenden Adiabatengleichungen erh�alt man mit c = 0:dT = � cp�cvcv �@T@v �p dv ; (23)�@T@p �v dp = � cpcv �@T@v �p dv : (24)Beispiel ideales Gas pv = RT : Mit �@T@p �v = vR , �@T@v �p = pR , cv = const., cp = const. folgtaus (22) die di�erenzielle Polytropengleichung,dpp = cp � cc� cv dvv ;die integriert mit der De�nition des Polytropenexponenten n = cp�ccv�c lautet:pvn = const: (25)Die entsprechende ideale Adiabatengleichung (Poisson{Gleichung) lautet mit c = 0 unddem Adiabatenexponenten = n(c = 0) = cpcv :pv = const: (26)26

Mit Hilfe der idealen Gasgleichung pv = RT folgen die gleichwertigen Beziehungen:Tv �1 = const:0 ; T p 1� = const:00 :Damit �ndet man aus (20) sofort, dass adiabatische Prozesse mit einem idealen Gasgleichzeitig isentrop sind, d.h. �s = 0.p
V
Adiabate c=0
Isochore c=cc<0
Isobare c=cp
v
Isotherme c->oo
c>0
c>0 c<0
n
v
1
c(n)
γ
c
Der Verlauf von Adiabaten, Isothermen, Isobaren und Isochoren im p{V{Diagramm sowiedas entsprechende Verhalten der Molw�arme c = c(n) = cv �n1�n als Funktion des Polytro-penexponenten n ist in den Abbildungen skizziert. Adiabaten verlaufen steiler als Iso-thermen. Das Auftreten von negativen Molw�armen im Bereich zwischen Adiabaten undIsothermen ist darauf zur�uckzuf�uhren, dass in diesem Bereich die bei einer Expansionnach au�en abgegebene Arbeit gr�o�er als die gleichzeitig zugef�uhrte W�arme ist. Dadurchergibt sich laut 1. HS trotz zugef�uhrter W�arme eine Abnahme der Temperatur und damitc < 0.3.3 Der Carnotsche Kreisprozess3.3.1 Verlauf im p{V{DiagrammBei Kreisprozessen wird der Anfangszustand �uber einen geschlossenen Weg, z.B. im p{V{Diagramm, wieder erreicht. W�ahrend f�ur die Zustandsgr�o�e innere Energie H dU = 0gilt, �ndet man f�ur die W�arme und Arbeit H �Q 6= 0 und H �A 6= 0. ThermodynamischeKreisprozesse bilden die Grundlage f�ur den Betrieb von W�armekraftanlagen,{ wandeln W�arme in Arbeit um,{ die W�arme wird dem in der Anlage im Kreisprozess str�omendem Fluid bei m�oglichsthoher Temperatur zugef�uhrt,{ geschlossenes System, Fluid wird zum Ausgangszustand zur�uckgef�uhrt,und Verbrennungskraftanlagen,{ chemisch gebundene Brennsto�energie wird durch Reaktion mit Sauersto� innerhalbder Maschine freigesetzt und der Prozess mit dem Verbrennungsgas fortgesetzt,{ o�enes System: Brennsto� und Luft werden zugef�uhrt, Abgase werden abgegeben.27

Die realen irreversiblen Prozesse in solchen Maschinen werden durch reversible Ersatz-prozesse beschrieben, die das thermodynamische Arbeitsprinzip m�oglichst gut wiederge-ben. Dabei ist das Prinzip des Kreisprozesses wichtig, da in der Regel eine sehr gro�eZahl von Zyklen durchlaufen werden soll. Das Ziel jedes einzelnen thermodynamischenKreisprozesses besteht in der Abgabe von Arbeit durch Zufuhr von W�arme, wobei einhoher Wirkungsgrad erreicht werden soll, d.h. das Verh�altnis aus abgegebener Arbeitund zugef�uhrter W�arme soll m�oglichst gro� sein. F�ur das prinzipielle Verst�andnis vonKreisprozessen hat der Carnotsche Prozess (CP) eine gro�e Bedeutung.p
V
3
Isothermen
Adiabaten
1
2
4T
To
u
Waermebad T
CP
Waermebad T
Arbeit A
Waerme Q
Waerme Q
o
u
u
o
Der CP besteht aus vier Schritten:1 ! 2 adiabatische Kompression: Q12 = 02 ! 3 isotherme Expansion: Q23 = Qo = �A23, Qo wird zugef�uhrt3 ! 4 adiabatische Expansion: Q34 = 04 ! 1 isotherme Kompression: Q41 = Qu = �A41, Qu wird abgef�uhrtAuf den Isothermen ist dU = 0 und damit �Q = ��A. Auf den Adiabaten ist �Q = 0 unddamit dU = �A. Die beim CP geleistete Arbeit ist schra�ert.3.3.2 Carnot{Prozess mit idealem Gas als ArbeitsmediumA = � Z V2;ToV1;Tu p(T; V )dV � Z V3;ToV2;To p(T; V )dV � Z V4;TuV3;To p(T; V )dV � Z V1;TuV4;Tu p(T; V )dV :Das Modell des idealen Gases wird f�ur das Arbeitsmedium verwendet und (17) beachtet.Entlang der Adiabaten (�Q = 0) ist �A = dU , d.h. �pdV = Cv(T )dT mit Cv = ncv.Entlang der Isothermen gilt p(T; V ) = nRT=V . Man erh�alt:A = Z ToTu Cv(T )dT � nRTo Z V3V2 dVV + Z TuTo Cv(T )dT � nRTu Z V1V4 dVV :Der erste und dritte Beitrag heben sich auf, so dass die beim CP geleistete ArbeitA = �nR �To ln V3V2 + Tu ln V1V4 �ist. Nutzt man die Adiabatengleichung TV �1 = const. aus, folgtTuV �11 = ToV �12 ; ToV �13 = TuV �14 ;28

ToTu = �V1V2� �1 = �V4V3� �1 ;so dass sich die Relation V1V2 = V4V3 ! V1V4 = V2V3ergibt und man das folgende Ergebnis erh�alt:A = �nR(To � Tu) ln V3V2 : (27)� V3 > V2, d.h. die Arbeit ist negativ und wird abgegeben� Qo = �A23 = nRTo ln V3V2 > 0 ist die aufgenommene W�arme� Qu = �A41 = nRTu ln V1V4 < 0 ist die abgegebene W�arme� Wirkungsgrad des CP: �C = abgegebene Arbeit/aufgenommene W�arme,�C = �revC = �AQo = To�TuTo = 1� TuTo < 1 : (28)� Der Wirkungsgrad des CP ist bei reversibler Prozessf�uhrung nur von der Tempe-raturdi�erenz �T = To � Tu der W�armeb�ader abh�angig und unabh�angig von derSubstanz (Arbeitsmedium).� Um einen hohen Wirkungsgrad zu erreichen, ist �T bzw. To m�oglichst gro� zuw�ahlen. Dabei ist aus wirtschaftlichen Gr�unden stets ein Kompromiss zwischen Ef-fektivit�at (�C) und technischem Aufwand (To) n�otig.� Es ist immer �C < 1, da f�ur reale CP stets Tu > 0 ist und T = 0 auch prinzipiellnicht erreicht werden kann (3. HS).3.3.3 Entropie und Wirkungsgrad f�ur den Carnot{Prozess1. HS f�ur CP: I dU = I �Q + I �A = 0 ! Qo +Qu + A = 0 :2. HS f�ur CP: I dS = I �QT = Z 32 �QTo + Z 14 �QTu = QoTo + QuTu = 0 :Clausiusscher W�armesummensatz:Die Summe der reduzierten W�armemengen Q=T verschwindet beim reversiblen Carnot{Prozess. QoTo + QuTu = 0 () QuQo = �TuTo : (29)Der Wirkungsgrad des CP f�ur reversible Prozessf�uhrung (28) ergibt sich sofort aus (29):�revC = �AQo = Qo +QuQo = 1 + QuQo � 1� TuTo :29

Irreversible Prozessf�uhrung: dS > �QT wegen diS > 0, so dass0 > QoTo + QuTu () QuQo < �TuTo :Damit gilt f�ur den Wirkungsgrad bei irreversibler Prozessf�uhrung:�irrevC = 1 + QuQo < 1� TuTo = �revC ;d.h. der Wirkungsgrad des CP ist bei irreversibler Prozessf�uhrung immer kleiner als beireversibler, �irrevC < �revC .Carnotscher Satz (1824):Von allen reversiblen Kreisprozessen, die zwischen zwei fest vorgegebenen Temperaturenverlaufen, hat der CP den gr�o�ten Wirkungsgrad.1 4
32
T
u
T
I
II
S S1 3
M
M
CP
KI
II
S
oT
T
T
CP (Linie 1{2{3{4{1) und beliebiger re-versibler Kreisprozess K (Strichpunktli-nie I{II) zwischen Tu und To im T{S{Diagramm; S1 = S2 und S3 = S4 sindfrei w�ahlbar.Zugef�uhrte W�arme auf Teilweg I: QIAbgegebene W�arme auf Teilweg II: QIIBeweis: Wir �nden f�ur die W�armemengenQI = Z S3S1 TI(S)dS = TMI (S3 � S1) ; QII = Z S1S3 TII(S)dS = TMII (S1 � S3) :Es gilt laut Mittelwertssatz der IntegralrechnungTMI < To ; TMII > Tuund damit f�ur den Wirkungsgrad jedes beliebigen reversiblen Kreisprozesses K zwischenTu und To: �revK = �AQI = QI +QIIQI = 1 + QIIQI = 1� TMIITMI < 1� TuTo = �revC ;d.h. die Beziehung �revK < �revC gilt allgemein.Der Clausiussche W�armesummensatz gilt f�ur alle reversiblen Kreisprozesse.Es ist leicht einzusehen, dass im Grenzfall n ! 1 der urspr�ungliche Kreisprozess durchdie in�nitesimal schmalen CPe immer besser approximiert wird und sich die Beitr�age dereinzelnen CPe wegheben:I dS = I �QT = limn!1 nXj=1 �Q(j)oT (j)o + �Q(j)uT (j)u ! = 0 :30

u
Adiabaten Sp
V
(i)
Isother-men T(i)
T
T (j)
(j)
o
Beweis:Ein beliebiger reversibler Kreisprozess Kwird im p{V{Diagramm durch ein Netz vonAdiabaten und Isothermen in viele schma-le CPe (Beispiel schra�ert) aufgeteilt: j =1 : : : n. F�ur jeden einzelnen gilt der Clau-siussche W�armesummensatz. Auf den Iso-thermenabschnitten T (j)o und T (j)u werden dieW�armemengen �Q(j)o und �Q(j)u �ubertragen.Damit ist gleichzeitig bewiesen, dass �Q=T das vollst�andige Di�erenzial einer Zustands-funktion, der Entropie S, ist.Weitere Kreisprozesse im p{V{Diagramm (�Ubungsaufgaben):� Joule{Prozess (Gasturbine): je zwei Isobaren und Adiabaten� Ericsson{Prozess (Gasturbine): je zwei Isobaren und Isothermen� Stirling{Prozess (Hei�gasmotor): je zwei Isochoren und Isothermen� Otto{Motor: Ersatzprozess aus je zwei Isochoren und Adiabaten� Diesel{Motor: Ersatzprozess aus Isochore, Isobare und zwei Adiabaten3.3.4 W�armepumpe und K�altemaschineWaermebad T
CP
Waermebad T
Arbeit A
Waerme Q
Waerme Q
o
u
u
o Wenn der CP umgekehrt durchlaufen wird (1{4{3{2{1), spricht man von einer W�armepumpe. Mitder aufgenommenen Arbeit A wird dem k�alterenW�armebad (Tu) die W�arme Qu entnommen unddem hei�en W�armebad (To) die W�arme jQoj = A+Qu zugef�uhrt. Es gilt immer: Qo +Qu + A = 0.W�armepumpe: Der E�ekt besteht in der Erw�armung des oberen W�armebades=) Heizung, �WP = jQojA = jQojjQoj �Qu = ToTo � Tu = 1�C > 1 :K�altemaschine: Der E�ekt besteht in der Abk�uhlung des unteren W�armebades=) K�uhlschrank, �KM = QuA = QujQoj �Qu = TuTo � Tu = 1�C � 1 :31

Technisches Problem: Wie werden die W�armemengen transferiert? Daf�ur verwendet manArbeitsmedien, die in den entsprechenden Temperaturbereichen zwischen Tu und To kon-densieren, d.h. W�arme abgeben, und Verdampfen, d.h. W�arme aufnehmen. Man verwen-det Kompressoren zur Druckerh�ohung und Drosselventile zur Entspannung, um �uber eine�Anderung des Druckes die gew�unschten Prozesse mit dem Arbeitsmedium ablaufen zulassen.3.4 Thermodynamische Temperaturskala: Die absolute Temperatur TWelche Beziehung gilt zwischen der im 2. HS de�nierten absoluten Temperatur T undder im 0. HS eingef�uhrten empirischen Temperatur �? Bisher wurde vorausgesetzt, dassT die Eigenschaften von � hat, d.h. die Funktion T = T (�) sei eineindeutig. Aus (8) folgtT @p@T !v = T @p@� !v d�dT = p+ @u@v!� :Trennung der Variablen und Integration liefert mit der Festlegung eines BezugspunktesT0 = T (�0), z.B. dem Tripelpunkt von Wasser T0 = 273:16 K:TRT0 dT 0T 0 = ln TT0 = �R�0 ( @p@� 0 )vd� 0p(� 0;v)+� @u(� 0;v)@v �� 0 : (30)Man kann also p(�; v) und u(�; v) in beliebigen empirischen Temperaturskalen � messenund dann mit (30) die absolute Temperatur T berechnen und in den Zustandsgleichungenverwenden. Alternativ l�asst sich mit dem 2. HS �q = du + pdv im Nenner von (30) dasVerh�altnis (�q=dv)� einf�uhren, wobei sich die Volumenabh�angigkeit im Quotienten unterdem Integral wegheben muss: ln TT0 = �Z�0 � @p@� 0 �v�@qdv�� 0 d� 0 :Besonders einfach wird (30) f�ur ideale Gase, da dann �@u@v �� = 0 gilt:ln TT0 = �Z�0 @p@� 0!v d� 0p = �Z�0 dpp = ln p(�; v)p(�0; v) =) T = T0 p(�; v)p(�0; v) :Der Druck in Abh�angigkeit von der empirischen idealen Gastemperatur (Celsius{Skala)bei konstantem Volumen ist durch p = p0(1 + ��) gegeben, wobei 1=� = 273:15 �C ist.Legt man den Tripelpunkt von Wasser in dieser Skala mit 0.01 �C fest, so folgtT = 273:15 K�1 + �273:15 �C� = �273:15 + ��C� K ;d.h. die empirische ideale Gastemperatur stimmt bis auf den willk�urlich w�ahlbaren Null-punkt mit der absoluten Temperatur �uberein.32

4 Thermodynamische Potenziale4.1 Einkomponentensysteme4.1.1 Entropie und innere EnergieDie Gibbssche Fundamentalgleichung (6) mit dem Arbeitsdi�erenzial �A = �pdV legt dieEntropie als vollst�andiges Di�erenzial bzgl. der Zustandsgr�o�en U; V fest:dS = 1T dU + pT dV :Ist die Entropie S = S(U; V ) bekannt, lassen sich alle anderen thermodynamischen Gr�o�enaus dem vollst�andiges Di�erenzial bestimmen:dS = @S@U !V dU + @S@V !U dV ;1T = @S@U !V ; p = T @S@V !U :Damit hat man die thermische Zustandsgleichung p(T; V ) gewonnen. Die kalorische Zu-standsgleichung U(T; V ) folgt aus (8). Die Enthalpie ist durch (14) gegeben. Die spe-zi�schen W�arme berechnet sich allgemein aus (10) bzw. (11). Man nennt die EntropieS(U; V ) auch thermodynamisches Potenzial bzgl. U und V .De�nition thermodynamischer Potenziale:Eine Zustandsgr�o�e hei�t genau dann thermodynamisches Potenzial bzgl. einesvollst�andigen Satzes von Zustandsvariablen, wenn die Kenntnis dieser Zustandsgr�o�eals Funktion des vollst�andigen Satzes gen�ugt, um alle anderen Zustandsgr�o�en zu be-stimmen.Durch Inversion ist aus der Entropie S(U; V ) die innere Energie U(S; V ) ableitbar. Ausder Gibbsschen Fundamentalgleichung �ndet man:dU = TdS � pdV :Das vollst�andige Di�erenzial der inneren Energie lautet:dU = @U@S !V dS + @U@V !S dV :Durch Vergleich �ndet man wieder:T = @U@S !V ; �p = @U@V !S :Die Gleichheit der gemischten zweiten Ableitungen liefert eine Maxwell{Beziehung: @T@V !S = � @p@S!V :33

4.1.2 Konstruktion thermodynamischer Potenziale aus U(S; V )Die thermodynamischen Potenziale S(U; V ) und U(S; V ) sind nicht besonders praktika-bel, da die Entropie direkten Messungen nicht zug�anglich ist. W�ahlt man aber andereVariablen als z.B. (S; V ), dann ist U kein thermodynamisches Potenzial mehr und manben�otigt weitere Messungen, um alle thermodynamischen Gr�o�en des Systems zu bestim-men. Daher ist die Ableitung anderer thermodynamischer Potenziale, insbesondere bzgl.der praktikablen Variablen (T; V ) und (T; p), wichtig. Dazu benutzt man die Methodeder Legendre{Transformation, die aus der klassischen Mechanik beim �Ubergang von derLagrange{Funktion L(q; _q; t) auf die Hamilton{Funktion H(q; p; t) durch Einf�uhrung ver-allgemeinerter Impulse p bekannt ist:L = L(q; _q; t) ; p � @L@ _q ;H � L(q; _q; t)� p _q = H(q; p; t) ; @H@ _q = @L@ _q � p = 0 :Damit �ndet man neue thermodynamischen Potenziale bzgl. anderer Zustandsvariablen.Wir de�nieren auf diese Weise (a) neue thermodynamischen Potenziale, (b) �nden mit derGibbsschen Fundamentalgleichung (6) TdS = dU + pdV deren vollst�andige Di�erenziale,(c) bestimmen daraus durch Vergleich weitere Zustandsgr�o�en, (d) leiten die jeweiligeMaxwell{Relation her und stellen (e) damit die innere Energie (kalorische Zustandsglei-chung) bez�uglich der neuen Variablen in Form einer Di�erenzialgleichung dar.1. Enthalpie H(S; p): H(S; p) � U � �@U@V �S V = U + pV (31)dH = dU + pdV + V dp � TdS + V dpdH = @H@S !p dS + @H@p !S dpT = @H@S !p ; V = @H@p !S =) @T@p !S = @V@S !pU(S; p) = H(S; p)� p �@H@p �S (32)� Aus (32) folgt: @U@p !S = @H@p !S � @H@p !S � p @2H@p2 !S � �p @V@p !S :� Bedeutung:W�armefunktion, misst die bei konstantem Druck zugef�uhrte W�arme,dH = TdS + V dp = TdS � �Q :� Bestimmung: kalorimetrisch �uber die Molw�armen, siehe (18).� Isenthalpe Prozesse: dH = 0. 34

2. Freie Energie F (T; V ): Helmholtzsches PotenzialF (T; V ) � U � �@U@S �V S = U � TS (33)dF = dU � TdS � SdT � �SdT � pdVdF = @F@T !V dT + @F@V !T dV�S = @F@T !V ; �p = @F@V !T =) @S@V !T = @p@T !VU(T; V ) = F (T; V )� T �@F@T �V (34)� (34) nennt man nach F (T; V ) umgeformt Helmholtzsche Di�erenzialgleichung,aus ihr folgt: @U@T !V = @F@T !V � @F@T !V � T @2F@T 2!V � T @S@T !V :� Bedeutung: gibt die bei isothermen Prozessen geleistete Arbeit andF = �SdT � pdV = �pdV � �A :3. Freie Enthalpie G(T; p): Gibbssches PotenzialG(T; p) � U � �@U@V �S V � �@U@S �V S = U + pV � TS (35)G = H � TS = F + pVdG = dF + pdV + V dp � �SdT + V dpdG = @G@T !p dT + @G@p !T dp�S = @G@T !p ; V = @G@p !T =) � @S@p!T = @V@T !pU(T; p) = G(T; p)� p �@G@p �T � T �@G@T �p (36)� Gibbssche Di�erenzialgleichung: G = H � TS,G(T; p) = H(T; p) + T �@G@T �p : (37)� Aus (37) folgt: @H@T !p = @G@T !p � @G@T !p � T @2G@T 2!p � T @S@T !p :35

� Bedeutung: G ist f�ur praktische Anwendungen besonders gut geeignet, da dieunabh�angigen Variablen Druck p und Temperatur T experimentell leicht zug�ang-lich sind und f�ur alle Bereiche eines thermodynamischen Systems im Gleichge-wichtszustand �ubereinstimmen.Die Helmholtzsche und Gibbssche Di�erenzialgleichung erlauben es, bei Kenntnis vonU(T; V ) und H(T; p) { in dieser Form sind das keine thermodynamischen Potenziale! {die freie Energie F (T; V ) und die freie Enthalpie G(T; p) zu bestimmen, allerdings nur bisauf willk�urliche Funktionen von V bzw. p. Dazu formen wir (34) und (37) um:UT 2 = FT 2 � 1T @F@T !V = � @@T �FT �V ;HT 2 = GT 2 � 1T @G@T !p = � @@T �GT �p :Man �ndet nach Integration:F (T; V )T = � Z U(T; V )T 2 dT + f(V ) ;G(T; p)T = � Z H(T; p)T 2 dT + g(p) :Damit ist gezeigt, dass eine vollst�andige Information �uber die thermodynamischen Eigen-schaften eines Systems nur aus dem thermodynamischen Potenzial bzgl. seines vollst�andi-gen Satzes an Zustandsvariablen erhalten werden kann, d.h. U(S; V ) und H(S; p) h�attenbekannt sein m�ussen.4.1.3 Konstruktion thermodynamischer Potenziale aus S(U; V )Ein weiterer Satz thermodynamischer Potenziale ergibt sich durch analoges Vorgehen ausS(U; V ). Mit Hilfe der Gibbsschen Fundamentalgleichung (6) dS = 1T dU + pT dV und demvollst�andigen Di�erenzial der Entropie kann man durch Legendre{Transformation vonden unabh�angigen Variablen (U; V ) zu den Variablen ( 1T ; pT ) �ubergehen und neue thermo-dynamische Potenziale einf�uhren, die sogenannten Planck{Massieuschen Funktionen:1. �( 1T ; V ): Massieu{Funktion (1865),�( 1T ; V ) � S � � @S@U �V U = S � UT : (38)Man �ndet � = �F=T . Das vollst�andige Di�erenzial lautet:d� = dS � 1T dU � Ud� 1T � � pT dV � Ud� 1T � :2. (U; pT ): (U; pT ) � S � � @S@V �U V = S � pT V ; (39)d = dS � pT dV � V d� pT � � 1T dU � V d� pT � :36

3. Y ( 1T ; pT ): Planck{Funktion,Y ( 1T ; pT ) � S � � @S@U �V U � � @S@V �U V = S � UT � pT V : (40)Man �ndet Y = �G=T . Das vollst�andige Di�erenzial lautet:dY = dS � 1T dU � Ud� 1T �� pT dV � V d� pT � � �Ud� 1T �� V d� pT � :4.1.4 Die thermodynamischen Potenziale des idealen GasesF�ur das ideale Gas kennen wir mit Cv = const. und dem Adiabatenexponenten = Cp=Cvdie Relation Cp � Cv = nR sowie die thermische und kalorische Zustandsgleichung:pV = nRT ; U = Cv(T � T0) + U0 ; H = Cp(T � T0) +H0 :Die Gibbssche Fundamentalgleichung (6) dS = 1T dU + pT dV lautet dann:dS = CvT dT + nRV dV :Integration in der (T; V ){Ebene und Umformung in die Variablen (T; p) liefert:S(T; V ) = Cv ln TT0 + nR ln VV0 + S0 ; S(T; p) = Cp ln TT0 + nR ln p0p + S0 :Umstellung nach der Temperatur ergibt T = T (S; V ) und T = T (S; p):T = T0 �V0V � �1 exp�S � S0Cv � ; T = T0 pp0! �1 exp S � S0Cp ! :Einsetzen in die kalorische Zustandsgleichung liefert die innere Energie U(S; V ) und nachInversion die Entropie S(U; V ). Die anderen thermodynamischen Potenziale folgen durchEinsetzen in die entsprechenden thermodynamischen Relationen (�UA):U(S; V ) = CvT0 (�V0V � �1 exp�S � S0Cv �� 1)+ U0 ;S(U; V ) = Cv ln UU0 + nR ln VV0 + S0 ;H(S; p) = CpT08<: pp0! �1 exp S � S0Cp !� 19=;+H0 ; (41)F (T; V ) = Cv(T � T0) + U0 � T �Cv ln TT0 + nR ln VV0 + S0� ;G(T; p) = Cp(T � T0) +H0 � T (Cp ln TT0 + nR ln p0p + S0) :37

4.2 Mehrkomponentensysteme4.2.1 Entropie f�ur o�ene Systeme und chemisches PotenzialWir betrachten jetzt o�ene thermodynamische Systeme, d.h. Sto�austausch mit der Um-gebung sei m�oglich. Dazu verwenden wir das Modell des homogenen Mehrkomponen-tensystems, wobei zun�achst chemische Reaktionen zwischen den einzelnen Komponentenj = 1 : : : k nicht zugelassen werden. Das vollst�andige Di�erenzial der Entropie f�ur einsolches System wird aus der Gibbsschen Fundamentalgleichung (6) gewonnen:dS = 1T dU � 1T Xi aidAi :Neben dem Arbeitsdi�erenzial f�ur Volumenarbeit am System, a1 = �p und dA1 = dV , tre-ten zus�atzliche Beitr�age auf, die mit Sto�zufuhr (Sto�ab uss) in (aus) das (dem) Systemzusammenh�angen. Die �Anderung der Molzahl einer Komponente ist mit Arbeit verbundenund wir schreiben dA2 = dnj. Damit ergibt sich:dS = 1T dU + pT dV � 1T kXj=1 ajdnj :Welche Bedeutung haben die zu den dnj konjugierten Variablen aj? Dazu stellen wir dasvollst�andige Di�erenzial der Entropie S(U; V; nj) auf:dS = @S@U !V;ni dU + @S@V !U;ni dV + kXj=1 @S@nj!U;V;ni 6=njdnj :In den partiellen Ableitungen nach den Molzahlen wird ni 6= nj vereinbart. Vergleich mitder Gibbsschen Fundamentalgleichung liefert sofort (siehe auch 4.1.1):1T = @S@U !V;ni ; pT = @S@V !U;ni ; �ajT = @S@nj!U;V;ni :Durch Inversion ist aus der Entropie S(U; V; nj) die innere Energie U(S; V; nj) ableitbar.Aus der Gibbsschen Fundamentalgleichung �ndet mandU = TdS � pdV + kXj=1 ajdnj :Das vollst�andige Di�erenzial der inneren Energie lautet:dU = @U@S !V;ni dS + @U@V !S;ni dV + kXj=1 @U@nj!S;V;ni dnj :Durch Vergleich �ndet man wieder:T = @U@S !V;ni ; �p = @U@V !S;ni ; aj = @U@nj!S;V;ni :38

Aus der Entropie bzw. der inneren Energie �ndet man damit f�ur die intensiven Gr�o�enTemperatur T , Druck p und chemisches Potenzial �j � aj der Sorte j die Relationen:T = �@U@S �V;ni ; p = � �@U@V �S;ni = ( @S@V )U;ni( @S@U )V;ni ; �j � � @U@nj �S;V;ni = �� @S@nj �U;V;ni( @S@U )V;ni : (42)Das chemische Potenzial �j gibt die �Anderung der inneren Energie mit der Molzahl an.Es hat f�ur die Beschreibung o�ener Systeme sowie des Stabilit�atsverhaltens thermodyna-mischer Systeme gro�e Bedeutung. Die Gibbssche Fundamentalgleichung lautet nun:dU = TdS � pdV + kXj=1�jdnj : (43)4.2.2 Gibbs{Duhemsche GleichungDie Entropie S(U; V; nj) ist eine extensive Gr�o�e und h�angt in dieser Form nur von anderenextensiven Gr�o�en ab. Damit ist es m�oglich, einen Skalenfaktor � einzuf�uhren:S(U 0; V 0; n0j) = S(�U; �V; �nj) = �S(U; V; nj) :Ableitung nach dem Parameter � ergibt:dSd� = @S@U 0!V 0;n0i @U 0@� + @S@V 0!U 0;n0i @V 0@� + kXj=1 @S@n0j!U 0;V 0;n0i @n0j@�= @S@U 0!V 0;n0iU + @S@V 0!U 0;n0iV + kXj=1 @S@n0j!U 0;V 0;n0inj � S(U; V; nj) :W�ahlt man speziell � = 1 und identi�ziert die partiellen Ableitungen von S mit den inten-siven thermodynamischen Gr�o�en entsprechend (42), so erh�alt man die Gibbs{DuhemscheGleichung in verschiedener Form:S(U; V; nj) = UT + pVT � kPj=1 �jnjTU � TS + pV � kPj=1�jnj = 0G � U � TS + pV = kPj=1�jnj (44)G ist die freie Enthalpie, die durch die Molzahlen und chemischen Potenziale in einemMehrkomponentensystem bestimmt ist (siehe 4.1.2). F�ur ein Einkomponentensystem er-gibt sich � = G=n � g, d.h. das chemische Potenzial ist durch die molare freie Enthalpieg gegeben.Die Gibbssche Fundamentalgleichung (43) wurde mit Hilfe der allgemeinen De�nitionder intensiven Gr�o�en T; p; �j �uber partielle Ableitungen der Entropie (bzw. der innerenEnergie) (42) integriert. Die Gibbs{Duhemsche Gleichung (44) ist zusammen mit derGibbsschen Fundamentalgleichung (43) Ausgangspunkt f�ur die Bestimmung vollst�andigerDi�erenziale thermodynamischer Gr�o�en. F�ur die freie Enthalpie G �ndet man z.B. ausdG = dU � TdS � SdT + pdV + V dp ; TdS = dU + pdV � kXj=1�jdnj ;39

sofort dG = �SdT + V dp+ kXj=1�jdnj :Damit ist G = G(T; p; nj) vollst�andiges Di�erenzial:dG = @G@T !p;nidT + @G@p !T;nidp+ kXj=1 @G@nj!T;p;nidnj :Vergleich liefert:�S = @G@T !p;ni ; V = @G@p !T;ni ; �j = @G@nj!T;p;ni :Man �ndet allgemein aus (44)dU � TdS � SdT + pdV + V dp� kXj=1�jdnj � kXj=1njd�j = 0mit Hilfe der Gibbsschen Fundamentalgleichung (43)TdS = dU + pdV � kXj=1�jdnjdie di�erenzielle Form der Gibbs{Duhemschen Gleichung:�SdT + V dp� kPj=1njd�j = 0 ; (45)d.h. die intensiven Gr�o�en T; p; �j sind nicht unabh�angig voneinander. Man kann anderer-seits den Druck p(T; �j;V ) als vollst�andiges Di�erenzial au�assen, wenn gleichzeitig dasVolumen bekannt ist: dp = SV dT + kXj=1 njV d�j :Das chemische Potenzial ist als intensive Zustandsvariable unabh�angig von der System-gr�o�e und somit unabh�angig vom Skalenfaktor �:�j(T; p;n0i) = �j(T; p;�ni) = �j(T; p;ni) :Damit folgt d�jd� = kXi=1 @�j@n0i !T;p;n0l @n0i@� = kXi=1 ni @�j@n0i !T;p;n0l = 0 :W�ahlt man wieder speziell � = 1, folgt aus der Vertauschbarkeit der zweiten Ableitungenvon G bzgl. der Molzahlen:kXi=1 ni @�j@ni !T;p;nl = kXi=1 ni @2G@ni@nj!T;p;nl = kXi=1 ni @�i@nj!T;p;nl = 0 :Diese Gleichungen nennt man Gibbs{Margulesschen Beziehungen:kPi=1ni �@�j@ni �T;p;nl = kPi=1ni � @�i@nj �T;p;nl = 0 : (46)40

4.2.3 Die thermodynamischen Potenziale I; J;K;LWir haben in 4.2.1 die Entropie S(U; V; ni) und die innere Energie U(S; V; ni) mit denMolzahlen ni als unabh�angigen Variablen eingef�uhrt. Man kann analog zum Vorgehen in4.1.2 und 4.1.3 durch Legendre{Transformation zu den chemischen Potenzialen �i �uberge-hen. Dadurch erhalten wir aus den thermodynamischen Potenzialen U;H; F;G vier weiterePotenziale und mit der Gibbs{Duhem{Beziehung (44) die Relationen:I(S; V; �i) = U(S; V; ni)� kXj=1 @U@nj!S;V;ninj = U � kXj=1�jnj = ST � pV ;K(S; p; �i) = H(S; p; ni)� kXj=1 @H@nj!S;p;ninj = H � kXj=1�jnj = ST ; (47)J(T; V; �i) = F (T; V; ni)� kXj=1 @F@nj!T;V;ninj = F � kXj=1�jnj = �pV ;L(T; p; �i) = G(T; p; ni)� kXj=1 @G@nj!T;p;ninj = G� kXj=1�jnj = 0 :Die entsprechenden Di�erenziale lauten mit Hilfe der Gibbsschen Fundamentalgleichung(43) TdS = dU + pdV � kPj=1�jdnj:dI = TdS � pdV � kXj=1njd�j ;dK = TdS + V dp� kXj=1njd�j ;dJ = �SdT � pdV � kXj=1njd�j ;dL = �SdT + V dp� kXj=1njd�j = 0 :Das Potenzial L und dessen Di�erenzial verschwinden entsprechend (44) und (45). Man�ndet z.B. f�ur das Potenzial J(T; V; �i) die Relationen�S = @J@T !V;�j ; �p = @J@V !T;�j ; �nj = @J@�j!T;V ;und aus der Vertauschbarkeit der zweiten Ableitungen die Maxwell{Beziehungen @S@V !T;�j = @p@T !V;�j ; @S@�j!T;V;�i= @nj@T !V;�i ; @p@�j!T;V;�i= @nj@V !T;�i :Das Potenzial J(T; V; �i) spielt in der statistischen Physik eine gro�e Rolle. Es ist f�uro�ene Systeme relevant, die mit einem W�armebad (dadurch wird T festgelegt) und kTeilchenreservoiren f�ur jede einzelne Komponente (damit werden die �j festgelegt) inKontakt stehen. Man nennt es auch gro�es thermodynamisches Potenzial oder Potenzialder gro�kanonischen Gesamtheit. 41
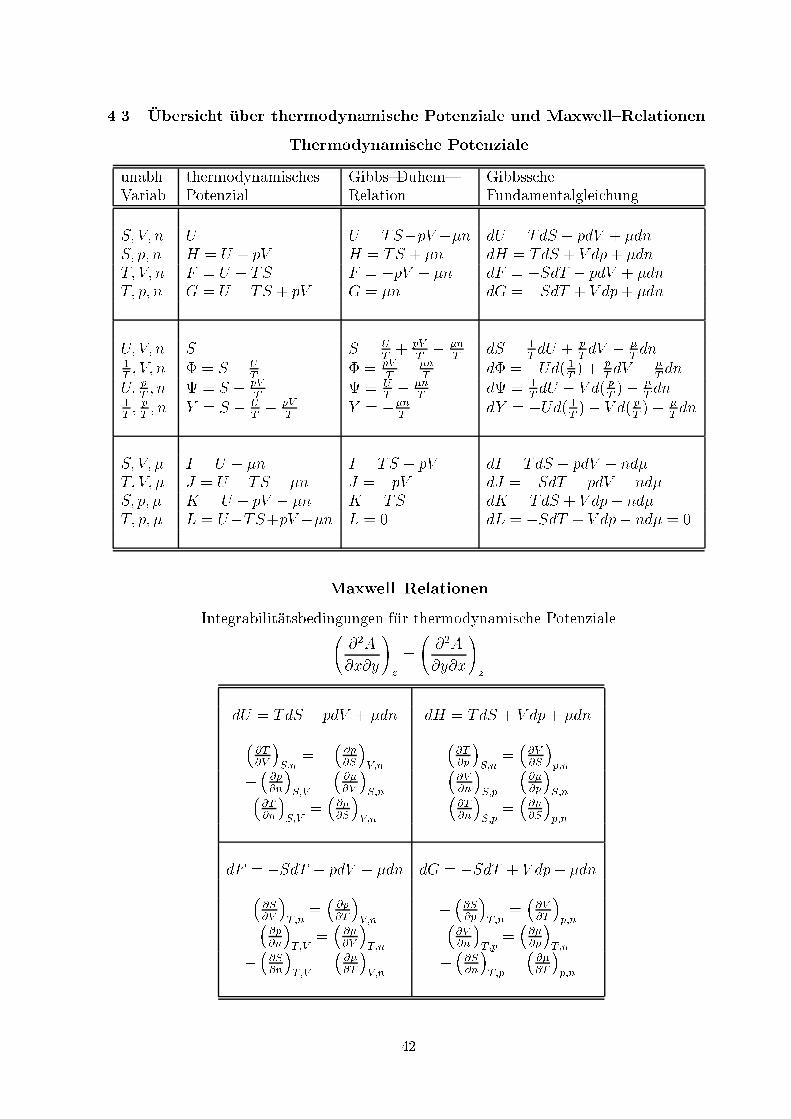
4.3 �Ubersicht �uber thermodynamische Potenziale und Maxwell{RelationenThermodynamische Potenzialeunabh. thermodynamisches Gibbs{Duhem{ GibbsscheVariab. Potenzial Relation FundamentalgleichungS; V; n U U = TS�pV +�n dU = TdS � pdV + �dnS; p; n H = U + pV H = TS + �n dH = TdS + V dp+ �dnT; V; n F = U � TS F = �pV + �n dF = �SdT � pdV + �dnT; p; n G = U � TS + pV G = �n dG = �SdT + V dp+ �dnU; V; n S S = UT + pVT � �nT dS = 1T dU + pT dV � �T dn1T ; V; n � = S � UT � = pVT � �nT d� = �Ud( 1T ) + pT dV � �T dnU; pT ; n = S � pVT = UT � �nT d = 1T dU � V d( pT )� �T dn1T ; pT ; n Y = S � UT � pVT Y = ��nT dY = �Ud( 1T )� V d( pT )� �T dnS; V; � I = U � �n I = TS � pV dI = TdS � pdV � nd�T; V; � J = U � TS � �n J = �pV dJ = �SdT � pdV � nd�S; p; � K = U + pV � �n K = TS dK = TdS + V dp� nd�T; p; � L = U�TS+pV ��n L = 0 dL = �SdT + V dp� nd� = 0Maxwell{RelationenIntegrabilit�atsbedingungen f�ur thermodynamische Potenziale @2A@x@y!z = @2A@y@x!zdU = TdS � pdV + �dn dH = TdS + V dp+ �dn� @T@V �S;n = � � @p@S�V;n �@T@p �S;n = �@V@S �p;n� � @p@n�S;V = � @�@V �S;n �@V@n �S;p = �@�@p�S;n�@T@n�S;V = �@�@S�V;n �@T@n�S;p = � @�@S�p;ndF = �SdT � pdV + �dn dG = �SdT + V dp+ �dn� @S@V �T;n = � @p@T �V;n � �@S@p�T;n = �@V@T �p;n� � @p@n�T;V = � @�@V �T;n �@V@n �T;p = �@�@p�T;n� �@S@n�T;V = � @�@T �V;n � �@S@n�T;p = � @�@T �p;n42

4.4 Gleichgewichts- und Stabilit�atsbedingungen4.4.1 Allgemeine PrinzipienAussagen �uber den thermodynamischen Gleichgewichtszustand eines Systems folgen ausdem 2. HS m.H. der Entropie. Solange noch irreversible Prozesse ablaufen, w�achst dieEntropie an: diS � 0. Hat sich der Gleichgewichtszustand eingestellt, gilt diS = 0 unddie Entropie erreicht ihren gr�o�ten Wert, S = Smax.Damit ist zur Bestimmung des Gleichgewichtszustands eines abgeschlossenen Systems eineExtremwertaufgabe mit Nebenbedingungen zu l�osen: Die Entropie S wird maximal beifestem Volumen V , innerer Energie U und Masse M (Teilchenzahlen k�onnen sich durchchemische Reaktionen �andern). Man benutzt die Methode der virtuellen Verr�uckungen,indem man virtuelle, in�nitesimal kleine Zustands�anderungen mit �V = 0, �U = 0 und�M = 0 betrachtet, die die Entropie unver�andert lassen. Die Gleichgewichtsbedingunglautet: (�S)U;V;M = 0 : (48)Neben der durch (48) garantierten Existenz eines Extremwerts f�ur die Entropie muss nochdas Maximum gefordert werden. Die Stabilit�atsbedingung lautet deshalb(�2S)U;V;M < 0 (49)und besagt, dass der Gleichgewichtszustand stabil oder mindestens metastabil gegen Zu-stands�anderungen ist. Bei metastabilen Zust�anden liegt nur ein relatives Maximum derEntropie vor. Beispiele: �uberhitzte Fl�ussigkeit, unterk�uhlte Fl�ussigkeit, Glaszustand.Gleichgewichts- und Stabilit�atsbedingungen lassen sich auch mit den anderen thermody-namischen Potenzialen f�ur nichtabgeschlossene Systeme aufstellen. Mit dem 1. und 2.HS dU = �Q� pdV ; dS � �QT ;sowie den De�nitionenH = U + pV ; F = U � TS ; G = H � TS = U + pV � TS ;�ndet man die Relationen:�Q = dU + pdV ! dU � TdS � pdV ;�Q = dH � V dp ! dH � TdS + V dp ;�Q = dF + TdS + SdT + pdV ! dF � �SdT � pdV ;�Q = dG+ TdS + SdT � V dp ! dG � �SdT + V dp :Es gibt o�enbar keine universellen Gleichgewichts- und Stabilit�atsbedingungen. F�ur diedurch die experimentellen Gegebenheiten festgelegten Nebenbedingungen muss man dasentsprechende thermodynamische Potenzial betrachten und desses Extremwert bestim-men. Man �ndet: (�U)S;V;M = 0 (�2U)S;V;M > 0(�H)S;p;M = 0 (�2H)S;p;M > 0(�F )T;V;M = 0 (�2F )T;V;M > 0(�G)T;p;M = 0 (�2G)T;p;M > 0 (50)Analoge Bedingungen lassen sich f�ur die Planck{Massieuschen Potenziale �;; Y und diePotenziale I; J;K aufstellen. 43

4.4.2 Auswertung der Gleichgewichts- und Stabilit�atsbedingungen(1) Temperaturausgleich1
1 2
U U
n n1V 2V
2Gehemmtes Gleichgewicht: Durch die w�armeisolierendeWand wird Temperaturausgleich verhindert.Gesamtsystem: U = U1 + U2; V = V1 + V2; n = n1 + n2W�armeisolierung: U1 6= U2 ! T1 6= T2Thermodynamisches Potenzial: S(U; V; n)Das gehemmte Gleichgewicht ist durch die EntropieSH = S1(U1; V1; n1) + S2(U2; V2; n2)gekennzeichnet. Beseitigt man die Hemmung, d.h. stellt man thermischen Kontakt her,wird zwischen den Untersystemen 1 und 2 W�arme �Q ausgetauscht, bis der Gleichge-wichtszustand erreicht ist. Die Wand sei nicht verr�uckbar und lasse keine Teilchen durch,d.h. �Vi = 0 und �ni = 0. Die Gleichgewichtsbedingung lautet:S(U; V; n)! Max: ; (�S)U;V;n = 0 :Durch den W�armeaustausch �andern sich die inneren Energien, so dass nach U1 = U � U2mit �U1 = ��U2 variiert werden muss:(�S)U;V;n = @S1@U1!U;V;n�U1 + @S2@U2!U;V;n�U2 = � 1T1 � 1T2� �U1 = 0 :Die Gleichgewichtsbedingung lautet: T1 = T2 � T : (51)F�ur die Auswertung der Stabilit�atsbedingung (�2S)U;V;n � 0 betrachten wir zwei gleichgro�e Teilsysteme. Da die innere Energie insgesamt konstant bleiben soll, erh�ohen wir siein 1 virtuell um �U und erniedrigen sie in 2 um den gleichen Betrag. Die �Anderung derEntropie ist bis zur 2. Ordnung in �U :�S = 12S(U + �U) + 12S(U � �U)� S(U)= 12 "S(U) + @S@U �U + 12 @2S@U2 (�U)2#+ 12 "S(U)� @S@U �U + 12 @2S@U2 (�U)2#� S(U)= 12 @2S@U2 (�U)2 � �2S :Die Stabilit�atsbedingung (�2S)U;V;n � 0 ergibt mit 1=T = (@S=@U)V;n und Cv = (@U=@T )V;ndie Forderung @2S@U2!V;n = ( @@U � 1T �)V;n = � 1T 2 @T@U !V;n = � 1T 2Cv � 0 :Der Gleichgewichtszustand ist dann stabil, wenn bei positiver Temperatur T die W�arme-kapazit�at Cv positiv ist, d.h. die innere Energie mit der Temperatur anw�achst:Cv � 0 : (52)Die analoge Bedingung Cp � 0 l�asst sich aus (�2H)S;p;n � 0 herleiten.44

(2) Druckausgleich1
1 2n
Arretierung
n1V 2V
2p p
Gehemmtes Gleichgewicht: Durch die arretierte Wandwird Druckausgleich verhindert.Gesamtsystem: U = U1 + U2; V = V1 + V2; n = n1 + n2Arretierung: p1 6= p2Thermodynamisches Potenzial: S(U; V; n)Das gehemmte Gleichgewicht ist durch die EntropieSH = S1(U1; V1; n1) + S2(U2; V2; n2)gekennzeichnet. Beseitigt man die Hemmung und macht den Kolben frei beweglich, werdensich die Teilvolumina �andern, bis der Gleichgewichtszustand erreicht ist. Die Wand lassekeine Teilchen durch (�ni = 0) und Temperaturausgleich habe bereits stattgefunden (T1 =T2 = T ). Die Gleichgewichtsbedingung lautet:S(U; V; n)! Max: ; (�S)U;V;n = 0 :Durch den beweglichen Kolben �andern sich die Teilvolumina, so dass Arbeit verrichtetwird und nach V1 = V � V2 mit �V1 = ��V2 variiert werden muss:(�S)U;V;n = @S1@V1!U;V;n�V1 + @S2@V2!U;V;n�V2 = �p1T1 � p2T2� �V1 = p1 � p2T �V1 = 0 :Die Gleichgewichtsbedingung lautet: p1 = p2 � p : (53)F�ur die Auswertung der Stabilit�atsbedingung betrachten wir wieder zwei gleich gro�eTeilsysteme. Das Volumen bleibt insgesamt konstant. Erh�ohen wir es in 1 virtuell um �Vund erniedrigen es in 2 um den gleichen Betrag, ergibt sich f�ur die �Anderung der Entropiebis zur 2. Ordnung in �V :�S = 12S(V + �V ) + 12S(V � �V )� S(V )= 12 "S(V ) + @S@V �V + 12 @2S@V 2 (�V )2#+ 12 "S(V )� @S@V �V + 12 @2S@V 2 (�V )2#� S(V )= 12 @2S@V 2 (�V )2 � �2S :Die Stabilit�atsbedingung (�2S)U;V;n � 0 ergibt mit p=T = (@S=@V )U;n die Forderung: @2S@V 2!U;n = ( @@V � pT �)U;n = 1T @p@V !T;n � 0 :Der Gleichgewichtszustand ist dann stabil, wenn sich bei Volumenabnahme der Druck imSystem erh�oht (U = const. ! T = const.):� @p@V �T;n � 0 : (54)45

(3) Chemisches Potenzial und Phasengleichgewichtp n1
Dampf
T
T np 2
Fluid
Zwei Phasen (z.B. Fluid 1 und sein Dampf 2) imGleichgewicht: Welche Molzahlen stellen sich bei ge-gebener Temperatur und Druck ein?Gesamtsystem: T1 = T2 = T; p1 = p2 = p; n1 6= n2Thermodynamisches Potenzial: freie EnthalpieG(T; p; n) = G1(T; p; n1) +G2(T; p; n2)Zwischen beiden Phasen werden Teilchen ausgetauscht, bis der Gleichgewichtszustanderreicht ist: �n1 = ��n2. Die Gleichgewichtsbedingung lautetG(T; p; n)! Min: ; (�G)T;p;n = 0 ;und ergibt bei Variation nach den Teilchenzahlen:(�G)T;p;n = @G1@n1 !T;p;n�n1 + @G2@n2 !T;p;n�n2 = (�1 � �2) �n1 = 0 :Die Gleichgewichtsbedingung lautet: �1 = �2 � � : (55)F�ur die Auswertung der Stabilit�atsbedingung betrachten wir wieder zwei gleich gro�eTeilsysteme. Die Teilchenzahl bleibt insgesamt konstant. Erh�ohen wir sie in 1 virtuell um�n und erniedrigen sie in 2 um den gleichen Betrag, ergibt sich f�ur die �Anderung der freienEnthalpie bis zur 2. Ordnung in �n:�G = 12G(n+ �n) + 12G(n� �n)�G(n)= 12 "G(n) + @G@n �n + 12 @2G@n2 (�n)2# + 12 "G(n)� @G@n �n+ 12 @2G@n2 (�n)2#�G(n)= 12 @2G@n2 (�n)2 � �2G :Die Stabilit�atsbedingung (�2G)T;p;n � 0 ergibt mit � = (@G=@n)T;p die Forderung @2G@n2 !T;p = @�@n!T;p � 0 :Der Gleichgewichtszustand ist dann stabil, wenn sich das chemische Potenzial mit derTeilchenzahl erh�oht: �@�@n�T;p � 0 : (56)Werden die Stabilit�atsbedingungen (52), (54) und (56) verletzt, k�onnen interessante phy-sikalische Vorg�ange wie Phasen�uberg�ange und Entmischungen im System ablaufen.46
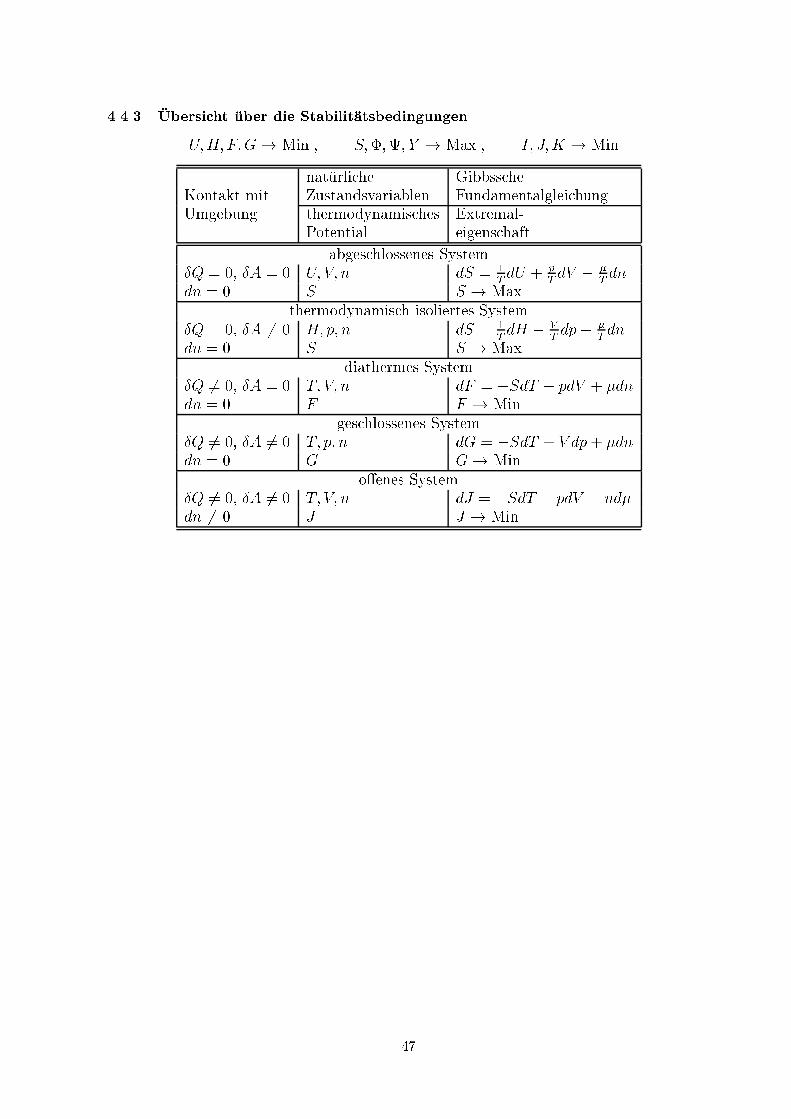
4.4.3 �Ubersicht �uber die Stabilit�atsbedingungenU;H; F;G! Min., S;�;; Y ! Max., I; J;K ! Min.nat�urliche GibbsscheKontakt mit Zustandsvariablen FundamentalgleichungUmgebung thermodynamisches Extremal-Potential eigenschaftabgeschlossenes System�Q = 0, �A = 0 U; V; n dS = 1T dU + pT dV � �T dndn = 0 S S ! Max.thermodynamisch isoliertes System�Q = 0, �A 6= 0 H; p; n dS = 1T dH � VT dp� �T dndn = 0 S S ! Max.diathermes System�Q 6= 0, �A = 0 T; V; n dF = �SdT � pdV + �dndn = 0 F F ! Min.geschlossenes System�Q 6= 0, �A 6= 0 T; p; n dG = �SdT + V dp+ �dndn = 0 G G! Min.o�enes System�Q 6= 0, �A 6= 0 T; V; n dJ = �SdT � pdV � nd�dn 6= 0 J J ! Min.
47

48

5 Das Verhalten bei tiefen Temperaturen: Der 3. HS5.1 VorbemerkungenMit dem 3. HS wird keine neue Zustandsgr�o�e eingef�uhrt, sondern das Verhalten derEntropie am absoluten Nullpunkt der Temperatur festgelegt. Die Zustandsgr�o�e Entro-pie S ist bis auf Integrationskonstanten bestimmt. Das hat f�ur Anwendungen, bei denenDi�erenzen dieser Gr�o�e zwischen Anfangs- und Endzustand relevant sind, keine Kon-sequenzen. Allerdings treten in anderen Gr�o�en wie z.B. der freien Energie F und derfreien Enthalpie G Terme TS auf, so dass deren Verwendbarkeit f�ur Prozesse mit unter-schiedlichen Temperaturen scheitert. Deshalb sind die Normierbarkeit der Entropie unddie Festlegung ihres Absolutwerts wichtige Probleme.Wir betrachten ein thermodynamisches System, in dem eine chemische Reaktion abl�auft.Die freie und innere Energie betragen vor der Reaktion F1; U1 und nach der ReaktionF2; U2. F�ur die Di�erenzen gilt die Helmholtzsche Di�erenzialgleichung (34):�F = �U + T @�F@T !V :Durch Di�erenzieren nach T erh�alt man: @�U@T !V = �T @2�F@T 2 !V :Bei konstantem Volumen gibt �U = �Q = Wv die W�armet�onung der Reaktion an. �Fnennt man A�nit�at der chemischen Reaktion bei konstantem Volumen. Untersuchungenihrer Temperaturabh�angigkeit haben f�ur T ! 0 ergeben, dass die ersten beiden Ablei-tungen (@�F=@T )V und (@2�F=@T 2)V endlich bleiben. Damit erh�alt man im Grenzfalltiefer Temperaturen die BeziehungenlimT!0�F = limT!0�U = limT!0�Q = limT!0Wvund limT!0 @�U@T !V = limT!0 @Wv@T !V = 0 :Nernst vermutete, dass dar�uberhinaus auch �F und Wv im Grenzfall T ! 0 dieselbehorizontale Tangente besitzen:limT!0 @Wv@T !V = limT!0 @�F@T !V = 0 :5.2 Formulierung des 3. HS: Das Nernstsche W�armetheoremF�ur die freie Energie gilt f�ur feste Temperaturen und Volumina laut De�nition:�F = �U � T�S = Wv � T�S :Im Grenzfall T ! 0 erh�alt man f�ur die Ableitung:limT!0 @�F@T !V = limT!0( @Wv@T !V ��S � T @�S@T !V) = 0 :49

Bleibt (@�S=@T )V endlich, folgt limT!0�S = 0.3. HS in der Formulierung von Planck:Beim absoluten Nullpunkt der Temperatur T n�ahert sich die Entropie eines Systemsim thermodynamischen Gleichgewicht einem von Parametern ai (z.B. Volumen, Druck,Aggregatzustand etc.) unabh�angigen Wert S0. Man w�ahlt in der ph�anomenologischenThermodynamik die Normierung S0 = 0.limT!0S(T; ai) = S0 � 0 ; limT!0 � @S@ai�T = 0 (57)5.3 Folgerungen aus dem 3. HS5.3.1 Molw�armenDie Molw�armen h�angen mit der Entropie zusammen (siehe 3.1.5):cv = T @s@T !v ; cp = T @s@T !p :Integriert ergeben sich die Gleichungen:s(T; v) = TZ0 cv(T 0)T 0 dT 0 + s(v) ; s(T; p) = TZ0 cp(T 0)T 0 dT 0 + s(p) :Im Grenzfall tiefer Temperaturen wird die Entropie unabh�angig von Druck und Volumen,d.h. s(v) = s(p) = 0. Die Entropie ist allein aus der Messung der Molw�armen bestimmbar.Weiter muss gefordert werden, dass cv und cp mindestens mit T gegen Null gehen, da sonstdie Integranden f�ur T ! 0 divergieren:Am absoluten Nullpunkt der Temperatur verschwinden die Molw�armen cv und cp.Es gilt z.B. c � T 3 f�ur den elastischen Festk�orper (Debye) und c � T f�ur das Elektronen-gas in Metallen (Sommerfeld).5.3.2 Thermische Koe�zientenMit Hilfe der Maxwell{Relationen (9) und (15) @S@V !T = @p@T !V ; @S@p !T = � @V@T !p ;kann man die thermischen Koe�zienten �uber die Entropie de�nieren:� = 1p @p@T !V = 1p @S@V !T ; � = 1V @V@T !p = � 1V @S@p !T :Damit �ndet man mit Hilfe des 3. HS: limT!0 � = limT!0� = 0.Im Grenzfall tiefer Temperaturen verschwinden der isochore Druckkoe�zient � und derisobare Ausdehnungskoe�zient �. 50

� Analoge Aussagen gelten auch f�ur die anderen Arbeitsdi�erenziale �A = aidAi: @S@Ai!T = � @ai@T !Ai :� Damit gilt laut 3. HS auch limT!0(@ai=@T )Ai = � limT!0(@S=@Ai)T = 0.� Die Ober �achenspannung � von uidem Helium-3 verschwindet am absoluten Null-punkt. Das Arbeitsdi�erenzial �A = �dF liefert limT!0(@�=@T )F = 0.� Die magnetische Suszeptibilit�at �m verschwindet am absoluten Nullpunkt. Das Ar-beitsdi�erenzial �A = HdM =MdH mit M = �0�mH liefert limT!0(@�m=@T )H = 0.� Allgemeine Beziehung zwischen den Molw�armen (12):limT!0 cp � cvT = limT!0 @p@T !v @v@T !p = limT!0 @S@V !T �@S@p !T = 0 :Die Di�erenz der Molw�armen cp � cv geht st�arker gegen Null als T .5.3.3 Entropie und chemisches Potenzial des idealen Gases f�ur T ! 0Die Entropie eines idealen Gases ist laut (19) durchS � S0 = Cv ln TT0 + nR ln VV0gegeben. F�ur konstantes Volumen und gegebene Anfangsbedingungen (T0; V0) divergiertdieser Ausdruck f�ur T ! 0, was dem 3. HS widerspricht. Selbst wenn man die Konstanzder W�armekapazit�at aufgibt undS � S0 = TZ0 Cv(T 0)T 0 dT 0 + nR ln VV0diskutiert, bleibt im Gegensatz zum 3. HS eine Abh�angigkeit vom Volumen bestehen.Im Grenzfall tiefer Temperaturen verliert das Modell des idealen Gases seine G�ultigkeit.Die bei tiefen Temperaturen auftretenden Abweichungen vom idealen Gasgesetz nenntman Gasentartung.� F�ur tiefe Temperaturen kondensieren die meisten Gase zu Fl�ussigkeiten und erstar-ren anschlie�end zu Festk�orpern. Damit wird das Modell des klassischen idealenGases gegenstandslos.� Ausnahmen: Helium und Wassersto� bleiben f�ur p = p0 auch bei T = 0 �ussig.� F�ur tiefe Temperaturen ist bei diesen der Ein u� von Quantene�ekten, speziell derEntartung, dominierend. Man nennt sie deshalb auch Quanten uide. Die exaktenthermodynamischen Relationen m�ussen im Rahmen der Quantenstatistik hergeleitetwerden. 51

Trotzdem ist es f�ur Anwendungen im gasf�ormigen Bereich sinnvoll, die funktionale Formder Beziehung (19) f�ur das Verhalten der Entropie zu verwenden. Dazu gehen wir zumolaren Gr�o�en �uber, s(T; v) = cv ln TT0 +R ln vv0 + s0 ;und �nden f�ur das Beispiel des einatomigen idealen Gases mit cv = 32R f�ur die molareEntropie bzgl. der Variablen (T; v) und (T; p):s(T; v) = R�32 lnT + ln v + �� ; s(T; p) = R�52 lnT � ln p+ �0� : (58)Die Entropiekonstanten lauten� = s0R � 32 lnT0 � ln v0 ; �0 = � + lnR ;und m�ussen aus der Quantenstatistik oder experimentell bestimmt werden (anstelle derwillk�urlichen Anfangsbedingungen s0; T0; v0). Verwendet man die Gibbs{Duhemsche Glei-chung (44) s = uT + pvT � �Tmit den klassischen Resultaten f�ur das ideale Gas u = 32RT und pv = RT sowie demquantenstatistischen Ausdruck f�ur das chemische Potenzial � = RT ln �NV �3�, �ndet manf�ur die Entropiekonstante nach Umformung die Sackur{Tetrode{Beziehung (�UA):�0 = ln((ekB)5=2 � m2��h2�3=2) :Sie ist allein durch Naturkonstanten und die Masse m der Teilchen bestimmt. Damit kannman auch eine Gleichung f�ur das chemische Potenzial � im gasf�ormigen Bereich mit Hilfeder Gibbs{Duhemschen Gleichung ableiten:�(T; v) = �RT �32 lnT + ln v + i� ; �(T; p) = �RT �52 lnT � ln p+ j� : (59)Dabei sind die chemischen Konstanten i; j mit den Entropiekonstanten verkn�upft:i = � � 52 ; j = �0 � 52 ! j = i+ lnR :������������������������������������������������������������
������������������������������������������������������������
T
0
gasfoermig
T TSchmelz
fest fluessig
QSchmelz VerdampfQSiedeMan kann die Entropie in der Gasphase auch empirisch �uber die bekannten W�armekapa-zit�aten in der festen und �ussigen Phase sowie die Schmelz- und Siedepunkte eichen undso die Entropiekonstante � bestimmen (�UA: Hg):s(Tboil; p0) = Z Tmelt0 cp(T )T dT + qmeltTmelt + Z TboilTmelt cp(T )T dT + qboilTboil � R ln RT 5=2boile�p0 :52

5.4 Der Joule{Thomson{E�ektZur Abk�uhlung eines Systems, um m�oglichst tiefe Temperaturen zu erreichen, sind ver-schiedene Methoden anwendbar:{ adiabatische Expansion entsprechend (26): TV �1 = const:,{ Carnot{Maschine als W�armepumpe (K�uhlschrank, siehe 3.3.4),{ adiabatische Entmagnetisierung (siehe 5.5),{ gedrosselte Entspannung durch eine por�ose Wand in einem adiabatisch isolierten Zylin-der: Joule{Thomson{Versuch, technische Anwendung im Linde{Verfahren zur Luft-ver �ussigung (Gegenstromprinzip).������������������������������������������
������������������������������������������
p p
VV
T
B2
T
1 2
1
1
2
2
Kolben 1 Kolben 2
poroese Wand
A1 B1 A2
Kolben 1 wird von Position A1 nach B1 reingedr�uckt. Gleichzeitig wird Kolben 2 vonPosition A2 nach B2 rausgezogen. Dabei seien die Dr�ucke p1; p2 konstant. Es gelte p1 > p2.Durch die adiabatische Isolierung des Zylinders wird w�ahrend des Prozesses keine W�armezu- oder abgef�uhrt: �Q = 0. Damit gilt laut 1. HS: dU = �pdV . Integration �uber dengesamten Prozess ergibt:Z BA dU = UB � UA = U2 � U1 = � Z BA pdV = � Z 0V1 p1dV � Z V20 p2dV = p1V1 � p2V2 :F�ur die Enthalpie H = U + pV erh�alt man:H2 = U2 + p2V2 = U1 + p1V1 = H1 :Der Joule{Thomson{Versuch ist ein isenthalper Prozess mitdH = @H@T !p dT + @H@p !T dp = 0 :F�ur die gesuchte Temperatur�anderung mit dem Druck ergibt sich mit (14): @T@p !H = ��@H@p �T�@H@T �p = T �@V@T �p � VCp :Verwendet man die De�nition des isobaren Ausdehnungskoe�zienten � = 1V �@V@T �p, solautet das Ergebnis f�ur den Joule{Thomson{Koe�zienten �:� � �@T@p �H = VCp (T�� 1) : (60)53

Er ist f�ur das ideale Gas pV = nRT mit � = 1=T identisch gleich Null: �id = 0. Damit kann� als Mass f�ur die Ann�aherung eines realen Gases an das Modell des idealen Gases verwen-det werden. Weiterhin ergibt sich f�ur reale Gase ein positiver (Abk�uhlung) oder negativer(Erw�armung) E�ekt entsprechend des Vorzeichens von T� � 1. Die Grenzkurve pinv(T )zwischen beiden Bereichen wird Inversionskurve genannt und aus der Gleichung T��1 = 0bestimmt. F�ur reale Gase verwendet man die van der Waalssche{Zustandsgleichung (2)in molaren Gr�o�enp(T; v) = RTv � b � av2 ! T (v) = v � bR �p+ av2� ;und erh�alt f�ur Bedingungen auf der Inversionskurve T � @v@T �p � v = 0 die Beziehung2aRT v � bv !2 � b = 0 :Umgeformt nach pinv(T ) erh�alt man (�UA):pinv(T ) = 2s2RTb ab2 � 3RT2b � ab2 : (61)Die Inversionskurve ist allein durch die Materialparameter a und b bestimmt, die einfacheRealgaskorrekturen zur idealen Zustandsgleichung angeben: Eigenvolumen der Molek�ule(b) und Anziehung zwischen ihnen (a). Man erh�alt f�ur gegebenen Druck zwei Inversions-temperaturen. Oberhalb von Ti = 2abR ist prinzipiell keine Abk�uhlung m�oglich.p
Erwaermung
Abkuehlung
a
i
3b2
2a 8a 2a9bR 9bR bR
TT =
Eine einfache mikroskopische Erkl�arung des Ef-fekts besteht darin, dass sich bei der Entspan-nung des Gases der mittlere Molek�ulabstandvergr�o�ert und damit gegen die intermolekula-ren Anziehungskr�afte \innere Arbeit" geleistetwerden muss. Das geht nur auf Kosten der ki-netischen Energie, so dass die Temperatur ab-nimmt.Inversionstemperatur f�ur einige Substanzen:Element/Verbindg. Ti in KHe-4 40H2 202N2 621Luft 603Ar 723O2 76454
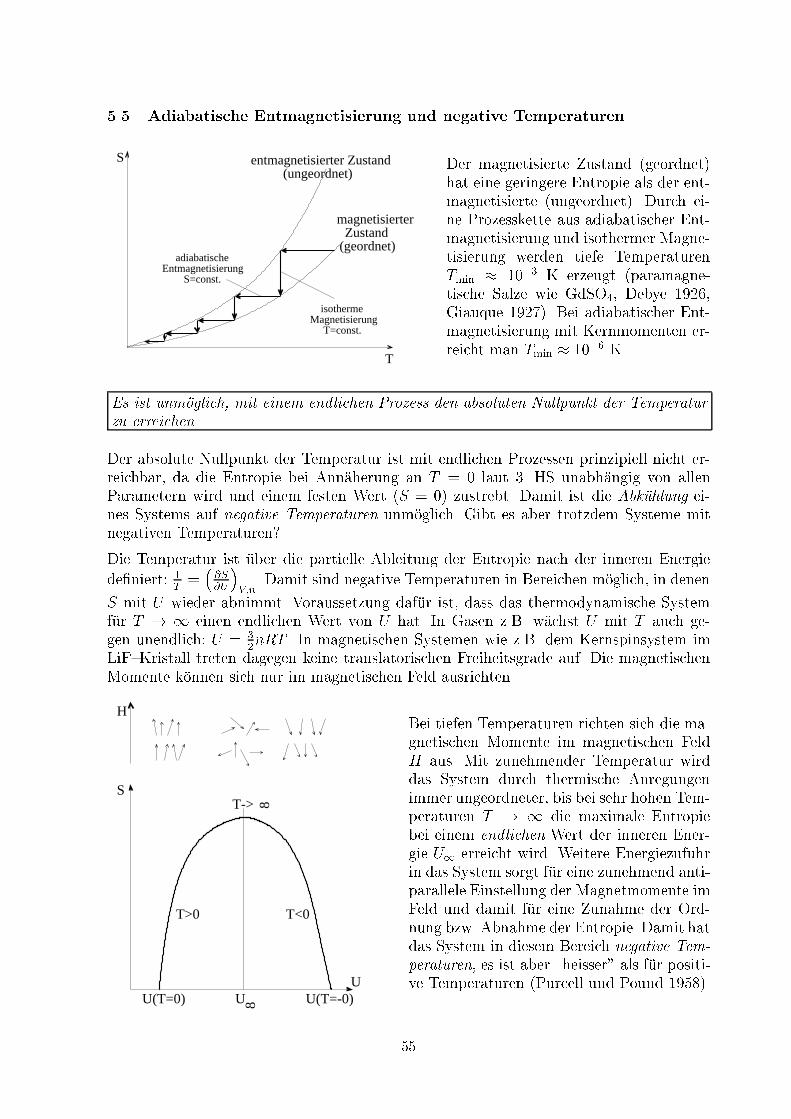
5.5 Adiabatische Entmagnetisierung und negative Temperaturen(ungeordnet)
S
T
entmagnetisierter Zustand
Magnetisierungisotherme
T=const.
Entmagnetisierungadiabatische
S=const.
Zustandmagnetisierter
(geordnet)
Der magnetisierte Zustand (geordnet)hat eine geringere Entropie als der ent-magnetisierte (ungeordnet). Durch ei-ne Prozesskette aus adiabatischer Ent-magnetisierung und isothermer Magne-tisierung werden tiefe TemperaturenTmin � 10�3 K erzeugt (paramagne-tische Salze wie GdSO4, Debye 1926,Giauque 1927). Bei adiabatischer Ent-magnetisierung mit Kernmomenten er-reicht man Tmin � 10�6 K.Es ist unm�oglich, mit einem endlichen Prozess den absoluten Nullpunkt der Temperaturzu erreichen.Der absolute Nullpunkt der Temperatur ist mit endlichen Prozessen prinzipiell nicht er-reichbar, da die Entropie bei Ann�aherung an T = 0 laut 3. HS unabh�angig von allenParametern wird und einem festen Wert (S = 0) zustrebt. Damit ist die Abk�uhlung ei-nes Systems auf negative Temperaturen unm�oglich. Gibt es aber trotzdem Systeme mitnegativen Temperaturen?Die Temperatur ist �uber die partielle Ableitung der Entropie nach der inneren Energiede�niert: 1T = � @S@U �V;n. Damit sind negative Temperaturen in Bereichen m�oglich, in denenS mit U wieder abnimmt. Voraussetzung daf�ur ist, dass das thermodynamische Systemf�ur T ! 1 einen endlichen Wert von U hat. In Gasen z.B. w�achst U mit T auch ge-gen unendlich: U = 32nRT . In magnetischen Systemen wie z.B. dem Kernspinsystem imLiF{Kristall treten dagegen keine translatorischen Freiheitsgrade auf. Die magnetischenMomente k�onnen sich nur im magnetischen Feld ausrichten.
8
S
T>0 T<0
T->
8U U(T=-0)U(T=0)U
H Bei tiefen Temperaturen richten sich die ma-gnetischen Momente im magnetischen FeldH aus. Mit zunehmender Temperatur wirddas System durch thermische Anregungenimmer ungeordneter, bis bei sehr hohen Tem-peraturen T ! 1 die maximale Entropiebei einem endlichen Wert der inneren Ener-gie U1 erreicht wird. Weitere Energiezufuhrin das System sorgt f�ur eine zunehmend anti-parallele Einstellung der Magnetmomente imFeld und damit f�ur eine Zunahme der Ord-nung bzw. Abnahme der Entropie. Damit hatdas System in diesem Bereich negative Tem-peraturen, es ist aber \heisser" als f�ur positi-ve Temperaturen (Purcell und Pound 1958).55

Praktisch erreicht man den Bereich negativer Temperaturen durch eine sehr schnelle (inetwa 10�s) Vertauschung der Feldrichtung, bei der die magnetischen Momente ihre Ori-entierung beibehalten und nun antiparallel zum magnetischen Feld ausgerichtet sind. Diecharakteristische Zeit zum Erreichen des Gleichgewichtszustands im Kernspinsystem be-tr�agt etwa 10�s. Thermisches Gleichgewicht mit dem Kristallgitter (hat immer T > 0),an das die �ubersch�ussige Energie abgegeben werden kann, wird aufgrund der schwachenWechselwirkung erst innerhalb von etwa 100s hergestellt. Damit \�uberlebt" der Zustandmit negativen Temperaturen etwa 107� die charakteristische Systemzeit.Neue Methoden zum Erreichen tiefster Temperaturen sind z.B. die Laserk�uhlung vonAtomen in magneto-optischen Fallen, bei der durch fast-resonante Absorption von Laser-licht die Atome in der Falle abgebremst werden und sich das Gas bis in den �K-Bereichabk�uhlen l�asst. Eine weitere Abk�uhlung bis in den nK-Bereich ist mit der Radiofrequenz-induzierten Verdampfungsk�uhlung m�oglich, bei der die \hei�esten" Atome durch Spin-Flip-Prozesse aus der Falle entfernt werden.Der makroskopische Quantene�ekt Bose{Einstein{Kondensation, bereits 1924 theoretischvorhergesagt, ist mit diesen Methoden erstmals in einem Gas aus Rb-Atomen bei einerTemperatur von 170 nK und einer Dichte von 3 � 1012 cm�3 beobachtet worden (Cor-nell und Wiemann 1995). Anwendungen solcher Kondensate sind z.B. das atomoptischeAnalogon zum Laser, der Atomlaser oder \Boser".
56

6 Homogene Einkomponentensysteme6.1 Phasen�uberg�ange6.1.1 Phasendiagramm f�ur EinkomponentensystemeEine Phase ist ein in physikalischer und chemischer Hinsicht homogener Bereich eines ther-modynamischen Systems. Man unterscheidet z.B. die feste, �ussige und gasf�ormige Phase,die in Abh�angigkeit von Druck, Temperatur und Volumen in einem Phasendiagramm f�urEinkomponentensysteme dargestellt werden. Phasen�uberg�ange treten auf, wenn die Sta-bilit�atsbedingungen (52), (54) oder (56) verletzt sind: @U@T !V � 0 ; @p@V !T � 0 ; @�@n!T � 0 :Zwischen den Phasen gelten die Gleichgewichtsbedingungen (51), (53) und (55):T (1) = T (2) � T ; p(1) = p(2) � p ; �(1) = �(2) � � :
T
vapor
CPsolid liquid
TL
p
VZustands �ache p(T; V ) eines einkomponentigen Systems. TL: Tripellinie, CP: kritischerPunkt, gestrichelt: kritische Isotherme, schra�ert: Koexistenzgebiete zweier Phasen.
V
solid-vapor
liquid-vapor
solid-liquid
CP
p
CP
T
TPvapor
solid liquid
p
Projektion der Zustands �ache in die p{V{ und p{T{Ebene, TP: Tripelpunkt. Koexistenz-linien p(T ) trennen zwei Phasen im p{T{Diagramm: Sublimationskurve (fest{gasf�ormig),57
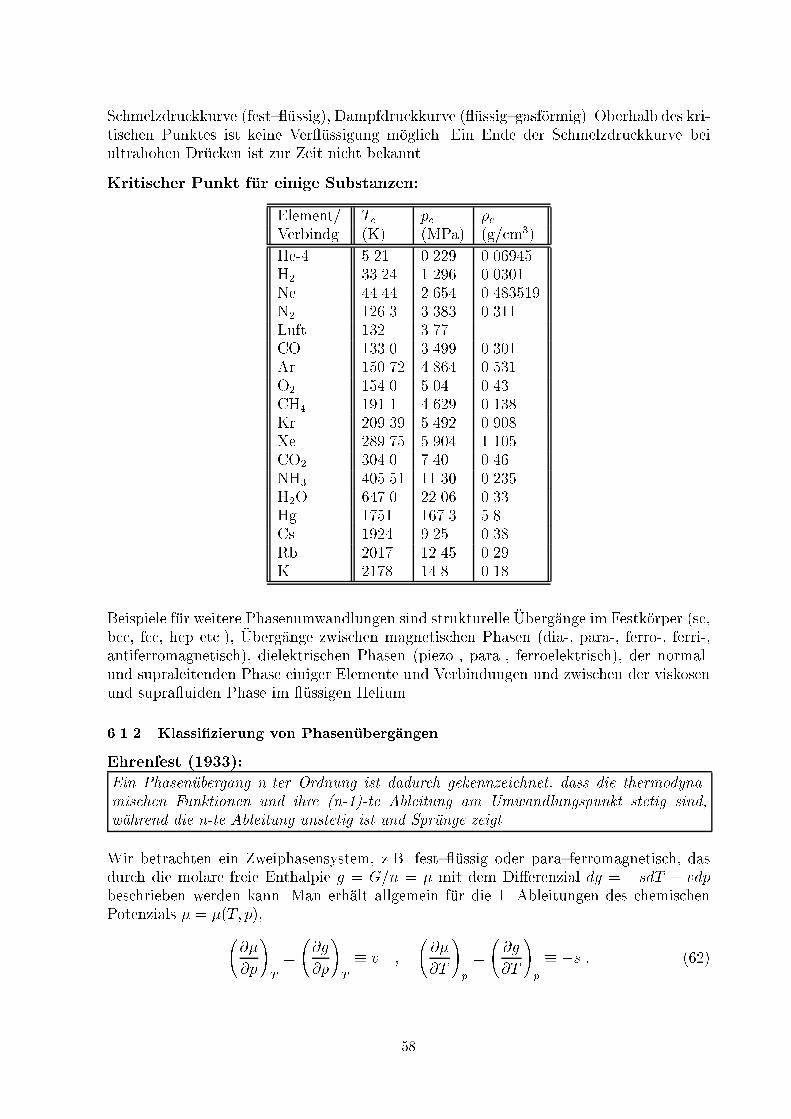
Schmelzdruckkurve (fest{ �ussig), Dampfdruckkurve ( �ussig{gasf�ormig). Oberhalb des kri-tischen Punktes ist keine Ver �ussigung m�oglich. Ein Ende der Schmelzdruckkurve beiultrahohen Dr�ucken ist zur Zeit nicht bekannt.Kritischer Punkt f�ur einige Substanzen:Element/ Tc pc �cVerbindg. (K) (MPa) (g/cm3)He-4 5.21 0.229 0.06945H2 33.24 1.296 0.0301Ne 44.44 2.654 0.483519N2 126.3 3.383 0.311Luft 132 3.77CO 133.0 3.499 0.301Ar 150.72 4.864 0.531O2 154.0 5.04 0.43CH4 191.1 4.629 0.138Kr 209.39 5.492 0.908Xe 289.75 5.904 1.105CO2 304.0 7.40 0.46NH3 405.51 11.30 0.235H2O 647.0 22.06 0.33Hg 1751 167.3 5.8Cs 1924 9.25 0.38Rb 2017 12.45 0.29K 2178 14.8 0.18Beispiele f�ur weitere Phasenumwandlungen sind strukturelle �Uberg�ange im Festk�orper (sc,bcc, fcc, hcp etc.), �Uberg�ange zwischen magnetischen Phasen (dia-, para-, ferro-, ferri-,antiferromagnetisch), dielektrischen Phasen (piezo-, para-, ferroelektrisch), der normal-und supraleitenden Phase einiger Elemente und Verbindungen und zwischen der viskosenund supra uiden Phase im �ussigen Helium.6.1.2 Klassi�zierung von Phasen�uberg�angenEhrenfest (1933):Ein Phasen�ubergang n-ter Ordnung ist dadurch gekennzeichnet, dass die thermodyna-mischen Funktionen und ihre (n-1)-te Ableitung am Umwandlungspunkt stetig sind,w�ahrend die n-te Ableitung unstetig ist und Spr�unge zeigt.Wir betrachten ein Zweiphasensystem, z.B. fest{ �ussig oder para{ferromagnetisch, dasdurch die molare freie Enthalpie g = G=n = � mit dem Di�erenzial dg = �sdT + vdpbeschrieben werden kann. Man erh�alt allgemein f�ur die 1. Ableitungen des chemischenPotenzials � = �(T; p), @�@p!T = @g@p!T � v ; @�@T !p = @g@T !p � �s ; (62)58

das molare Volumen v und die molare Entropie s. F�ur die entsprechenden 2. Ableitungen, @2�@p2 !T = @v@p!T � �v�T ; @2�@T 2!p = � @s@T !p � �cpT ; @2�@T@p = @v@T !p � v� ;(63)ergibt sich ein Zusammenhang mit der isothermen Kompressibilit�at �T , der Molw�armecp und dem isobaren Ausdehnungskoe�zienten �. Mit Hilfe der Gleichgewichtsbedingung(55) f�ur die Koexistenz zweier Phasen �1 = �2 und der Ehrenfestschen De�nition erh�altman jetzt konkrete Aussagen f�ur Phasen�uberg�ange von 1 nach 2.P�U 1. Art: Die 1. Ableitungen haben Spr�unge, d.h. es tritt ein Dichtesprung und eine�Ubergangsw�arme (latente W�arme) q12 am Umwandlungspunkt auf,�v = v2 � v1 ; �s = s2 � s1 = q12=T12 : (64)Beispiele: fest{ �ussig, �ussig{gasf�ormig, fest{gasf�ormig. Der Dichtesprung betr�agt beimP�U fest{ �ussig einige Prozent und ist sonst gro�. Die molare �Ubergangsw�arme q12 mussbei konstanter Umwandlungstemperatur T12 aufgebracht werden, um den �Ubergang von1 nach 2 zu vollziehen, in umgekehrter Richtung wird sie frei (Schmelzw�arme, Verdamp-fungsw�arme).Schmelztemperatur Tm, spezi�sche Schmelzw�arme qm, Siedetemperatur Ts undspezi�sche Verdampfungsw�arme qv f�ur einige Substanzen. Alle Angaben geltenf�ur Normaldruck. CO2 hat seinen Tripelpunkt bei 5.18 bar und 216.5 K (Trockeneis). F�urden Tripelpunkt sind unter (a) Tm und qm gegeben, w�ahrend unter (b) die Sublimations-temperatur und �w�arme f�ur Normaldruck gegeben sind.Element/ Tm qm Ts qvVerbindg. (K) (J/g) (K) (J/g)CO2 216.5a 184a 195b 574bNH3 195 339 240 1369SO2 198 117 263 402H2O 273.15 333.5 373.15 2256.5Hg 234 11.3 630 285P�U 2. Art: Die 1. Ableitungen sind stetig (kein Dichtesprung und keine latente W�arme),aber die 2. Ableitungen haben Spr�unge, d.h. die Materialeigenschaften sind am Umwand-lungspunkt unstetig,��T = �T;2 � �T;1 ; �cp = cp;2 � cp;1 ; �� = �2 � �1 : (65)Beispiele: strukturelle, magnetische, dielektrische P�U sind 2. Art, ebenso der �Ubergangvon normal- zu supraleitender Phase. Die Aufteilung in Phasen�uberg�ange h�oherer Ord-nung wird zunehmend sinnlos, da mit wachsender Ordnung die Unterschiede zwischen denPhasen immer mehr verschwinden und keine eindeutige Trennung mehr m�oglich ist.59

6.1.3 Clausius{Clapeyronsche Gleichung f�ur P�U 1. ArtT
p
vapor
liquid"1"
"2"12
T
p (T )
0
12 0
T0 +∆Τ
∆ p
Berechnung der Koexistenzlinie zwischenzwei Phasen, z.B. �ussig (1) { gasf�ormig (2).Die Gleichgewichtsbedingung (55) gilt f�ur al-le Temperaturen und legt den Verlauf derKoexistenzlinie p12(T ) fest:�1(T; p12(T )) = �2(T; p12(T )) ;�1(T0; p12(T0)) = �2(T0; p12(T0)) :Taylorentwicklung liefert f�ur beliebige Temperaturen T0 +�T :�1(T0; p12(T0))+ @�1@T !p�T+ @�1@p !T�p12 = �2(T0; p12(T0))+ @�2@T !p�T+ @�2@p !T�p12:Die Ableitungen des chemischen Potenzials ergeben(v1 � v2)�p12 = (s1 � s2)�T :Mit der �Ubergangsw�arme s2 � s1 = q12=T �ndet man im Limes �T ! 0 die Clausius{Clapeyronsche Gleichung f�ur P�U 1. Art:dp12dT = q12(T )T (v2�v1) : (66)Beispiel: P�U �ussig (1) { gasf�ormig (2) liefert mit v1 = v � v2 = vgas, dem idealenGasgesetz p12vgas = RT und q12 = const. in einem gegebenen Temperaturintervall (T0; T1)dp12dT = q12Tvgas = q12p12RT 2 ;dp12p12 = q12R dTT 2 =) ln p12 = � q12RT + const:ein Ergebnis f�ur die Dampfdruckkurve p12(T ):p12(T ) = p12(T0) exp �� q12RT � : (67)Eine Verbesserung wird durch die Ber�ucksichtigung der Temperaturabh�angigkeit der �Uber-gangsw�arme q12(T ) erhalten, die sich �uber die Molw�armen cp(T ) beschreiben l�asst.6.1.4 P�U 2. Art und Ehrenfestsche GleichungenBei P�U 2. Art sind die 1. Ableitungen der thermodynamischen Funktionen stetig, w�ahrenddie 2. Ableitungen Spr�unge zeigen. Mit Hilfe von (62) erh�alt man mit s1 = s2 und v1 = v2f�ur die totalen Di�erenziale der molaren Entropie s(T; p):ds1(T; p) = @s1@T !p dT + @s1@p !T dp = @s2@T !p dT + @s2@p !T dp = ds2(T; p) :60

Die Relationen cp = T @s@T !p ; @s@p!T = � @v@T !p = �v� ;ergeben (cp;1 � cp;2)dTT = v(�1 � �2)dp :Die 1. Ehrenfestsche Gleichung lautet:dpdT = 1Tv cp;1�cp;2�1��2 : (68)F�ur die totalen Di�erenziale des molaren Volumens v(T; p) gilt analog:dv1(T; p) = @v1@T !p dT + @v1@p !T dp = @v2@T !p dT + @v2@p !T dp = dv2(T; p) :Die Relationen �T = �1v @v@p!T ; @v@T !p = v� ;ergeben (�1 � �2)vdT = v(�T;1 � �T;2)dp :Die 2. Ehrenfestsche Gleichung lautet:dpdT = �1��2�T;1��T;2 : (69)Bezeichnet man mit �x = x1 � x2 die Di�erenz der Materialeigenschaften zwischen denbeiden Phasen, ergibt sich der allgemeine Zusammenhang:�cp��T � Tv(��)2 = 0 : (70)Bei �Ubergang zu den Variablen (T; v) anstelle von (T; p) ergibt sich aus den Gleich-gewichtsbedingungen s1 = s2 und p1 = p2 und den entsprechenden Di�erenzialen dieanaloge Gleichung: �cv�� 1�T �+ Tv (��)2 = 0 : (71)\Reine" P�U 2. Art nach der De�nition von Ehrenfest sind eher selten. Die Spr�unge inden Materialeigenschaften sind oft klein und schwer veri�zierbar. Alternativ hat Schu-bert (1968) deshalb eine Klassi�zierung von Phasen�uberg�angen �uber das Verhalten derEntropie S(T ) vorgeschlagen, die die De�nition von Ehrenfest beinhaltet und erg�anzt:1. P�U 1. Art: S(T ) verl�auft bei T0 diskontinuierlich und hat einen Sprung �S = Q12=T0mit der �Ubergangsw�arme Q12.2. P�U 2. Art: S(T ) verl�auft insgesamt stetig, die 1. Ableitung hat bei T0 einen Sprung.3. Phasenumwandlung mit ��Punkt: Wendepunkt in der T (S)�Kurve existent.4. Di�use Phasenumwandlung: Kein Wendepunkt in der T (S)�Kurve existent.61

6.1.5 Landau{Theorie f�ur P�U 2. ArtLandau hat 1937 eine ph�anomenologische Beschreibung von Phasen�uberg�angen 2. Artvorgeschlagen. Bei Phasen�uberg�angen �andert sich der Ordnungszustand im System unddamit dessen Symmetrieeigenschaften (Symmetriebrechung). Beispiele: fest{ �ussig (Nah-und Fernordnung *) Nahordnung), ferromagnetisch{paramagnetisch (spontane Magneti-sierung*) Ausrichtung der Magnetmomente im Feld) usw. Landau f�uhrte dazu den Begri�des Ordnungsparameters als neue Zustandsvariable ein. Die zentrale Idee ist dabei, dassder Ordnungsparameter � bei Ann�aherung an die Stabilit�atsgrenze des P�U verschwindet.Beispiele f�ur Ordnungsparameter in der Landau{Theorie:Phasen Phasen�ubergang, System Ordnungsparameter �gasf�ormig{ �ussig Gas{Fl�ussigkeit %Fl � %Gas �ussig{ �ussig Fl�ussigkeitsgemisch %(1)Fl � %(2)Flfest{ �ussig Festk�orper{Fl�ussigkeit %FK � %Flfest{fest Festk�orpermagnetische P�U ~Mdielektrische P�U ~Pstrukturelle P�U z.B. Phononenmodenmakroskopische Supraleitung � Gap{EnergieQuantene�ekte Supra uidit�at hi Kondensat{WFKlassi�zierung der P�U nach Landau:� P�U 1. Art:Der Ordnungsparameter �andert sich am Umwandlungspunkt unstetig. Die Phasen-umwandlung f�uhrt durch instabile Punkte auf der Stabilit�atsgrenze.� P�U h�oherer Art:Der Ordnungsparameter �andert sich am Umwandlungspunkt stetig. Die Phasenum-wandlung f�uhrt durch stabile Punkte auf der Stabilit�atsgrenze.� Kontinuierliche P�U:Der Ordnungsparameter �andert sich �uber einen gewissen Temperaturbereich stetig.Ausgangspunkt:Die freie Energie F sei ein Funktional des Ordnungsparameters � und in der N�ahe desPhasen�ubergangs in eine Potenzreihe entwickelbar. Der Ordnungsparameter wird so nor-miert, dass er auf der Stabilit�atsgrenze des Phasen�ubergangs, wo der Unterschied zwischenden Phasen verschwindet, Null ist:F � F0 + A� +B�2 + C�3 +D�4 + : : : (72)Die Koe�zienten A;B;C;D; : : : h�angen dabei von den thermodynamischen VariablenT; V; : : : ab. Aus Symmetriegr�unden [F (T; V; �) = F (T; V;��)] sollten nur gerade Poten-zen von � in der freien Energie auftreten, d.h. A = C = 0 und F = F0 +B�2 +D�4 + : : :62

Die Gleichgewichtsbedingung@F (T; V; �)@� = 2B� + 4D�3 � � = 0 (73)liefert f�ur den Ordnungsparameter auf der Phasengrenze�(2B + 4D�2) = 0mit den beiden L�osungen �1 = 0 f�ur die Phase 1 mit der niedrigeren Ordnung und �2 =q�B=2D f�ur die Phase 2 mit der h�oheren Ordnung.Die Stabilit�atsbedingung @2F (T; V; �)@�2 = 2B + 12D�2 > 0 (74)liefert die Forderung B > 0 f�ur Phase 1 mit �1 = 0 und �4B > 0 f�ur Phase 2 mit�2 = q�B=2D. Damit wechselt B auf der Phasengrenze das Vorzeichen und es gilt dortspeziell B = 0.Man macht f�ur die Temperatur- und Volumenabh�angigkeit des ersten Entwicklungspara-meters B(T; V ) in der freien Energie nahe der Phasengrenze den folgenden Ansatz mitHilfe der �Ubergangstemperatur Tc(V ) des Phasen�ubergangs:B(T; V ) = b(V )[T � Tc(V )] :Die Phasengrenze ist in der T{V{Ebene aus B = 0 durch T = Tc(V ) bestimmt. F�ur diefreie Energie (72) ergibt sich damit allgemeinF (T; V; �) = F0(T; V ) + b(V )[T � Tc(V )]�2 +D(T; V )�4und speziell f�ur die beiden Phasen:F = 8><>: F0 ; �1 = 0F0 � B24D ; �2 = r� B2D (75)Kritische Exponenten: Setzt man B(T; V ) in die L�osungen f�ur den Ordnungsparameterein, �ndet man f�ur dessen Verhalten in den beiden Phasen�1;2 = 8><>: 0 ; T > Tcrb(V )2D (Tc � T )1=2 ; T < Tc (76)Mit t � T�Tc l�asst sich das allgemein als � � (�t)� f�ur T < Tc schreiben. Den Exponenten� nennt man kritischen Exponenten.Suszeptibilit�at: Man de�niert die Suszeptibilit�at �uber� � @�@�!T = 1�@�@� �T = 1�@2F@�2 �T = 12B + 12D�2 :63

Setzt man wieder die beiden L�osungen f�ur � ein, ergibt sich�1;2 = ( 12B = 12b(T�Tc) � t� ; T > Tc� 14B = 14b(Tc�T ) � (�t)� 0 ; T < TcFreie Energie auf der Stabilit�atsgrenze � = �@F@� �T=Tc:� = 2B� + 4D�3 B=0! 4D�3 � �� :�Ubersicht �uber kritische Exponenten in der Landau{Theorie (LT): Vergleichmit Experiment, Renormalisierungsgruppentheorie nach Wilson (RT), dreidimensionalemIsing{Modell (3d{IM). rc: Korrelationsl�ange.krit. Fl�ussigkeiten magnetische Temp.- LT Experi- RT 3d-IMExp. Gase Systeme Bereich ment�0 CV � (�t)��0 CH � (�t)��0 T < Tc 0 � 0:1 0.08 0.11� CV � t�� CH � t�� T > Tc 0 � 0:1 0.08 0.11� %L � %G � (�t)� MS � (�t)� T < Tc 1/2 0.33{0.42 0.33 0.325 0 �T � (�t)� 0 �T � (�t)� 0 T < Tc 1 � 1:1 1.26 1.24 �T � t� �T � t� T > Tc 1 1.2{1.35 1.26 1.24� p� pc � j%L � %Gj� H � jM j� T = Tc 3 � 4:2 4.8 4.816�signf�%g �signfMg� 0 rc � (�t)��0 T < Tc 1/2 0.64� rc � t�� T > Tc 1/2 0.64� Abweichungen vom Experiment: Vernachl�assigung der kritischen Fluktuationen; dieKorrelationsl�ange w�achst dort stark an.� Es existieren Beziehungen zwischen den kritischen Exponenten (Rushbrooke):�+ 2� + = 2 ; � + �(� + 1) = 2 : : :� Nahe des kritischen Punktes des Phasen�ubergangs besagt die Skalenhypothese (Grif-�th), dass die physikalischen Gesetze invariant gegen L�angentransformationen derForm �r = r=u sind, falls auch alle anderen physikalischen Gr�o�en entsprechendt = T � Tc ! tuA ; � ! �B ; Hext ! HextuCersetzt werden. A;B;C sind die entsprechenden Skalenparameter.� Eine Begr�undung der Skalengesetze und die Berechnung der kritischen Exponentenist im Rahmen der Renormalisierungsgruppentheorie nach Wilson m�oglich. Dieseerlaubt auch eine Behandlung der Universalit�atshypothese, nach der das kritischeVerhalten des thermodynamischen Systems allein von der r�aumlichen Dimensionund der Symmetrie der geordneten Phase abh�angt.� Anwendung: Ginzburg{Landau{Theorie der Supraleitung als P�U 2. Art.Literaturempfehlung: Landau/Lifschitz Bd. 5 (Statistische Physik), Gebhard/Krey(Phasen�uberg�ange und kritische Ph�anomene).64

6.2 Gase und Fl�ussigkeiten6.2.1 Allgemeine BeziehungenGase und Fl�ussigkeiten geh�oren zu den einfachsten thermodynamischen Systemen. Sie ha-ben als Arbeitsmedien f�ur viele thermodynamische Prozesse und technische Anwendungeneine gro�e Bedeutung. Das Arbeitsdi�erenzial ist �A = �pdV . Ihr Zustand wird durchzwei unabh�angige Variablen, z.B. Druck p und Temperatur T beschrieben, die �uber diethermische Zustandsgleichung p = p(T; V ) mit dem Volumen zusammenh�angen, die auchin der Form f(p; T; V ) = 0 geschrieben werden kann. Die Materialeigenschaften �, �T , �und ihr allgemeiner Zusammenhang lassen sich ausdf = @f@p!T;V dp+ @f@T !p;V dT + @f@V !p;T dV = 0ableiten. Man erh�alt aus @p@V !T = �� @f@V �T;p�@f@p�T;V ; @T@p !V = ��@f@p�T;V� @f@T �p:V ; @V@T !p = �� @f@T �p;V� @f@V �p;T ;die Beziehung @V@T !p @T@p !V @p@V !T = �1 :Die Relation (3) folgt allein aus der Existenz der thermischen Zustandsgleichung und giltganz allgemein: p��T = � : (77)F�ur die Di�erenz zwischen den Molw�armen (12) erh�alt man wieder die allgemeine Bezie-hung: cp � cv = Tpv � �� : (78)6.2.2 van der Waalssche ZustandsgleichungDas Modell des idealen Gases ist nur im Grenzfall kleiner Dichten und/oder hoher Tem-peraturen anwendbar. Es versagt bei gro�en Dichten und tiefen Temperaturen, da hierKorrelationen zwischen den Teilchen zunehmend wichtig werden. Diese k�onnen durch in-teratomare/intermolekulare Potenziale beschrieben werden, die aus der Quantenmechanikberechnet werden m�ussen.Ein interessanter E�ekt ist das Auftreten von Phasen�uberg�angen, in diesem Fall �ussig{gasf�ormig. Einfache Modi�kationen der Zustandsgleichung f�ur ideale Gase erlauben es,dieses Ph�anomen zu studieren. Das Verhalten realer Gase kann qualitativ mit Hilfe dervan der Waalsschen Zustandsgleichung (2) beschrieben werden (1873):�p+ av2� (v � b) = RT :Die Materialparameter a und b beschreiben die Anziehung zwischen den Molek�ulen so-wie ihr Eigenvolumen. Man �ndet im p{V{Diagramm einen Instabilit�atsbereich, in dem65

p
V
T
fluessig-gasfoermig kr
T>T
KoexistenzkurveSpinodale
KP
B C D E
kr
kr
Fluid Gas
Isothermen
T<T
Isothermen des van der Waals{Gases mitkritischem Punkt (KP).Die Spinodale C-KP-D verbindet Punkteauf der Instabilit�atskurve, f�ur die gerade(@p=@V )T = 0 gilt. Die Koexistenzkur-ve B-KP-E entsteht nach Maxwell{Kon-struktion (siehe unten). Die kritische Iso-therme besitzt einen Wendepunkt mit ho-rizontaler Tangente.Im Instabilit�atsgebiet koexistieren �ussi-ge und gasf�ormige Phase nebeneinander.F�ur eine Isotherme mit T=const. ergibtdie Gleichgewichtsbedingung p1 = p2 =p12(T ) einen Dichtesprung von v auf vgas.(@p=@V )T � 0 verletzt ist. In diesem Bereich zerf�allt die sonst homogene Phase in einen �ussigen und einen gasf�ormigen Anteil. Der kritische Punkt KP kann aus den Bedingungen @p@v!T = 0 ; @2p@v2!T = 0 ;berechnet weren (�UA). Man �ndet:vk = 3b ; RTk = 827 ab ; pk = 127 ab2 ; pkvkRTk = 38 : (79)Man kann die van der Waalssche Zustandsgleichung auch in reduzierten Variablen ange-ben, indem man mit den kritischen Werten skaliert. Dadurch werden die Materialeigen-schaften eliminiert und alle van der Waals{Gase verhalten sich �ahnlich; man spricht vonkorrespondierenden Zust�anden �; �; � :� = ppk ; � = vvk ; � = TTk :Durch Einsetzen �ndet man sofort (�UA):�� + 3�2� (3� � 1) = 8� : (80)Maxwell{Konstruktion (1875): Mit Hilfe der Gibbs{Duhem{Relation (44) f�ur daschemische Potenzial � = u + pv � Ts �ndet man aus den Gleichgewichtsbedingungen�1 = �2 und p1 = p2 = p12(T ):s2 � s1 = u2 � u1T + p12(T )T (v2 � v1) :Wie muss der Koexistenzdruck p12(T ) gew�ahlt werden? In der Regel sind �s und �unicht bekannt. Es gilt der 1. und 2. HS:s2(T; v2)� s1(T; v1) = Z (T;v2)(T;v1) �q12(T )T = Z (T;v2)(T;v1) du+ pdvT :66

Das Integral �uber Zustandsgr�o�en kann entlang eines beliebigen Weges berechnet werden.L�angs des Zweiphasengebietes mit p12(T ) = const. ergibt sich:s2(T; v2)� s1(T; v1) = 1T [u2(T; v2)� u1(T; v1)] + p12(T )T (v2 � v1) :Entlang der (instabilen) van der Waals{Isotherme ergibt sich:s2(T; v2)� s1(T; v1) = 1T [u2(T; v2)� u1(T; v1)] + 1T Z (T;v2)(T;v1) p(T; v)dv :
fl V
p
p
V V
12
gas
Durch Vergleich �ndet man die Bedingungf�ur den Koexistenzdruck p12 im Instabilit�ats-bereich: Die Gerade p12(T ) muss so gew�ahltwerden, dass sich die Fl�achen �uber und un-ter der van der Waals{Isotherme kompen-sieren. Diese Prozedur nennt man Maxwell{Konstruktion:p12(T )(v2 � v1) = (T;v2)R(T;v1) p(T; v)dv : (81)6.2.3 VirialentwicklungDie Zustandsgleichung realer Gase l�a�t sich in Form einer Virialentwicklung darstellen:pvRT = 1 + B(T )v + C(T )v2 + : : :F�ur die van der Waalssche Zustandsgleichung erh�alt man z.B.pv = RT "1 + �b� aRT � 1v + b2v2 + b3v3 + : : :#Allgemeine Ausdr�ucke f�ur die Virialkoe�zienten B(T ); C(T ); : : : sind mit Methoden derStatistischen Physik herleitbar. In einfachster N�aherung, der bin�aren Sto�approximation,�ndet man f�ur sph�arisch symmetrische Wechselwirkungspotenziale V (r) zwischen denTeilchen f�ur den 2. Virialkoe�zienten B(T ) den AusdruckB(T ) = �2� Z 10 drr2(exp "�V (r)kBT #� 1) :Einfache Beispiele f�ur interatomare Wechselwirkungspotenziale sind das Lennard-Jones{Potenzial, das Sutherland{Potenzial und das KastenpotenzialVLJ(r) = " "��r �12 � 2��r �6# ;VSu(r) = 8<: 1 ; r < ��"0 ��r �6 ; r > � ;VKa(r) = 8><>: 1 ; 0 < r < ��" ; � < r < R�0 ; r > R� ;67
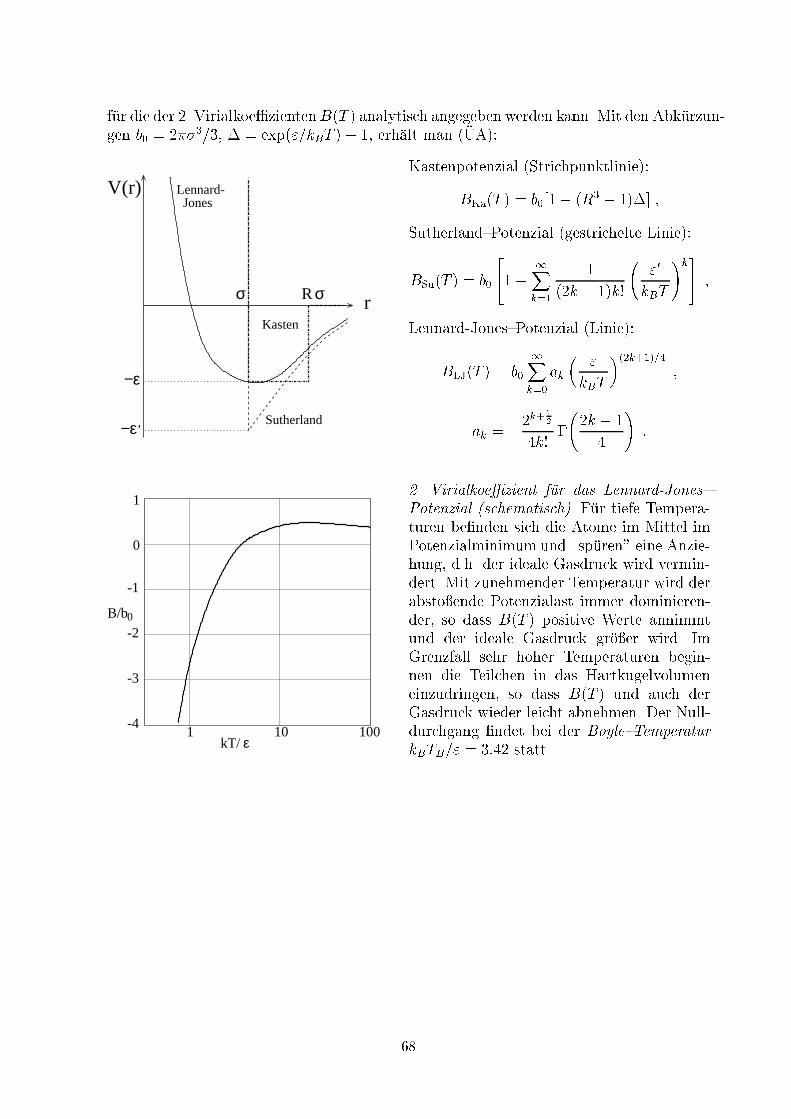
f�ur die der 2. Virialkoe�zientenB(T ) analytisch angegeben werden kann. Mit den Abk�urzun-gen b0 = 2��3=3, � = exp("=kBT )� 1, erh�alt man (�UA):σ
Lennard-Jones
V(r)
Sutherland’
R σ
Kasten
−ε
−ε
r
Kastenpotenzial (Strichpunktlinie):BKa(T ) = b0[1� (R3 � 1)�] ;Sutherland{Potenzial (gestrichelte Linie):BSu(T ) = b0 241� 1Xk=1 1(2k � 1)k! "0kBT !k35 ;Lennard-Jones{Potenzial (Linie):BLJ(T ) = b0 1Xk=0 ak � "kBT �(2k+1)=4 ;ak = �2k+ 124k! � 2k � 14 ! :0
1
B/b
-1
-2
-3
-41 10 100
kT/ ε
0
2. Virialkoe�zient f�ur das Lennard-Jones{Potenzial (schematisch). F�ur tiefe Tempera-turen be�nden sich die Atome im Mittel imPotenzialminimum und \sp�uren" eine Anzie-hung, d.h. der ideale Gasdruck wird vermin-dert. Mit zunehmender Temperatur wird derabsto�ende Potenzialast immer dominieren-der, so dass B(T ) positive Werte annimmtund der ideale Gasdruck gr�o�er wird. ImGrenzfall sehr hoher Temperaturen begin-nen die Teilchen in das Hartkugelvolumeneinzudringen, so dass B(T ) und auch derGasdruck wieder leicht abnehmen. Der Null-durchgang �ndet bei der Boyle{TemperaturkBTB=" = 3:42 statt.
68

6.3 Hohlraumstrahlung6.3.1 Thermodynamik des PhotonengasesJeder K�orper mit einer Temperatur T sendet elektromagnetische Strahlung aus (W�arme-strahlung). Beispiele sind ein gl�uhendes St�uck Kohle oder die Stahlschmelze im Hoch-ofen. Die W�armestrahlung des menschlichen K�orpers oder von Tieren liegt im infrarotenBereich. F�ur die thermodynamische Behandlung dieser Strahlung wird das Modell derHohlraumstrahlung verwendet:� Ein elektromagnetisches Strahlungsfeld be�ndet sich in einem von strahlungsun-durchl�assigen W�anden umschlossenen Volumen V .� An den W�anden stellt sich ein Gleichgewicht zwischen Absorption und Emission vonStrahlung (Photonen) ein.� Die Strahlung im Hohlraum hat damit die gleiche Temperatur wie die Wand, un-abh�angig von deren konkreten Eigenschaften.� Die Hohlraumstrahlung ist homogen, isotrop und unpolarisiert und hat damit ganzuniversellen Charakter.� Durch eine kleine �O�nung wird Strahlung ausgekoppelt und beobachtet, ohne dieEigenschaften der Hohlraumstrahlung merklich zu �andern.� Umgekehrt wird jede durch die �O�nung einfallende Strahlung von den W�andenabsorbiert und schnell thermalisiert.� Eine vollkommen absorbierende Ober �ache erscheint schwarz. Die im Innern be�nd-liche Strahlung nennt man auch Schwarzk�orperstrahlung.� Die Energiedichte u = U=V der Hohlraumstrahlung ist nicht vom Volumen sondernnur von der Temperatur abh�angig: u = u(T ).� Der Zusammenhang zwischen Strahlungsdruck und Energiedichte ist in der Elektro-dynamik �uber den Maxwellschen Spannungstensor gegeben: ps = 13u(T ); auch imRahmen der geometrischen Optik ableitbar:
dx
raum
θ
A
einfallendesPhoton
Photonreflektiertes
Hohl-
Photonendichte: n = dN=dV , dV = Adx =Avxdt = Ac cos�dt. Strahlungsdruck auf Wand:ps = FA = 1A ��pdt � dN= 1A 2p cos�dt ndV = 2pcn cos2� :Mittelung �uber �: Faktor 1=3. Nur die H�alfte al-ler Photonen hat Impuls in Wandrichtung: Fak-tor 1=2. Ergebnis mit der Photonenergie Eph =pc: ps = 122pcn13 = 13EphNV = 13 UV = 13u :69

Die Beziehung zwischen thermischer und kalorischer Zustandsgleichung (8) ergibt @U@V !T = "@(uV )@V #T = u(T ) = T @p@T !v � p � 3p ;so dass f�ur Hohlraumstrahlung der universelle ZusammenhangT @p@T !v = 4pgilt. Nach Trennung der Variablendpp = 4dTT ) ln p = lnT 4 + const:�ndet man f�ur die Zustandsgleichung des idealen Photonengases:p = aT 4u(T ) = 3aT 4 = bT 4 (82)� Der Wert der Konstanten (c: Lichtgeschwindigkeit) betr�agtb = 8�5k4B15c3h3 = 7:5659� 10�16 Wsm3K4 :� F�ur die Entropie ergibt sich mitdS = 1T (dU + pdV ) = 1T fd[u(T )V ] + pdV g = bT 3dV + 4bT 2V dT + b3T 3dV= 43bT 3dV + 4bT 2V dT � 43b d(T 3V )und S0 = 0 das Resultat: S = 43bT 3V :� Adiabatengleichung f�ur Hohlraumstrahlung:T 3V = const: ; pV 43 = const:� W�armekapazit�at Cv [Cp l�asst sich nicht sinnvoll de�nieren, da wegen (82) isobareProzesse auch isotherm sind]:Cv = @U@T !V = 4bT 3V :� Freie Energie: F = U � TS = bT 4V � 43bT 4V = �13bT 4V :� Das chemische Potenzial des Photonengases verschwindet, wie es f�ur Systeme ohnefeste Teilchenzahl (Emission/Absorption von Photonen �ndet laufend statt) charak-teristisch ist:G = �n = U + pV � TS = bT 4V + 13bT 4V � 43bT 4V � 0 :70

6.3.2 Strahlungsgesetze� Emission E: Die von einem K�orper pro Zeit- und Fl�acheneinheit abgestrahlte Ener-gie.� Absorption A: Der Anteil der einfallenden Strahlung, der von einem K�orper absor-biert wird. A = 1: schwarzer K�orper, A = 0: wei�er K�orper (idealer Spiegel).� Strahlungsdichte J : Strahlungsenergie pro Zeit- und Fl�acheneinheit.Bringt man einen schwarzen K�orper der Temperatur T in einen Hohlraum der gleichenTemperatur, hat seine Emission E = K dank des thermischen Gleichgewichts dessenStrahlungsdichte J und es gilt: K = J = 14cu :u(T)
Photonemittiertes
θ
(Detektor)
Hohlraum-strahlung
Strahlungsdichte (Emission) der Hohlraum-strahlung, im Detektor gemessen pro Zeit-und Fl�acheneinheit unter dem Winkel �:J = u(T ) � c cos� :Mittelung �uber �: Faktor 1=2. Nur die H�alftealler Photonen hat Impuls in Richtung der�O�nung: Faktor 1=2. Ergebnis: J = cu=4.Mit (82) �ndet man das Stefan{Boltzmann{Gesetz (Stefan 1879 empirisch, Boltzmann1884 theoretisch) K = �T 4 (83)mit der Konstanten � = 14bc:� = 2�5k4B15c2h3 = 5:670512� 10�8 Wm2K4 :Ein nicht schwarzer K�orper mit dem Absorptionsverm�ogen A absorbiert im Hohlraumdie Strahlungsenergie AJ , die im thermischen Gleichgewicht gleich seiner Emission seinmuss: E = AJ = AK. Das ist das Kirchho�sche Strahlungsgesetz (1859):Das Verh�altnis aus Emission und Absorptionsverm�ogen ist f�ur jeden K�orper gleich unddurch die Emission des schwarzen K�orpers gegeben: K = E=A.Wie verteilt sich die Energiedichte des Strahlungsfeldes auf die verschiedenen Frequenzin-tervalle d�? Wir de�nieren die spektrale Energiedichte bezogen auf die Frequenz � (bzw.die Wellenl�ange �) mit u�(T; �) [bzw. u�(T; �)]u(T ) = Z 10 u�(T; �)d� = Z 10 u�(T; �)d� ;f�ur die Planck 1900 seine ber�uhmte Strahlungsformel, zun�achst als Interpolationsformelzwischen den Grenzf�allen kleiner und gro�er Frequenzen (s.u.), angegeben hat:u�(T; �) = 8�c3 h�3exp(h�=kBT )�1 : (84)71

Durch Integration �uber alle Frequenzen erh�alt man wieder das das Stefan{Boltzmann{Gesetz (83) K = �T 4 mit der Konstanten (x = h�=kBT )� = 2�k4Bc2h3 Z 10 x3 dxexp(x)� 1 :� Im Grenzfall kleiner Frequenzen h�=kBT � 1 folgt die klassische Rayleigh{JeansscheStrahlungsformel (1900): u�(T; �) = 8��2kBTc3 :� Im Grenzfall gro�er Frequenzen h�=kBT � 1 folgt die Wiensche Strahlungsformel:u�(T; �) = 8�c3 h�3 exp(�h�=kBT ) :� Das Maximum der spektralen Energiedichte in Abh�angigkeit von der Temperaturwird aus (@u�=@�)T = 0 berechnet (�UA) und ergibt die Gleichung ex = 3=(3 � x).Die Nullstelle liegt bei x0 = 2:8215, so dass man�max = 2:8215kBTh = 5:8791� 1010TK s�1erh�alt. Die Frequenz der maximalen Energiedichte ist proportional zur absolutenTemperatur.� Die entsprechende Relation f�ur die Wellenl�ange erh�alt man mit c = �� und u�(T; �) =u�(T; �)d�=d� = u�(T; �)c=�2 aus der Bedingung (@u�=@�)T = 0. Die Rechnung(�UA) ergibt die Gleichung ex = 5=(5 � x) mit der Nullstelle x0 = 4:9651, so dassman �max = hc4:9651kBT = 2:8854� 10�3KT merh�alt. Das ist das Wiensche Verschiebungsgesetz: Die Wellenl�ange der maximalenEnergiedichte ist umgekehrt proportional zur absoluten Temperatur.
72

7 Mehrkomponentensysteme7.1 Mehrkomponentensysteme ohne chemische Reaktion7.1.1 Gleichgewichtsbedingung f�ur heterogene MehrkomponentensystemeIm System sollen keine chemischen Reaktionen statt�nden und keine �au�eren Felder an-liegen. Ober �achen- und Grenz �achene�ekte seien vernachl�assigbar. Gemische aus k ver-schiedenen Gasen oder Fl�ussigkeiten sind Beispiele f�ur homogeneMehrkomponentensyste-me. Bei �Anderung von Temperatur T und Druck p durch �Anderung des Volumens V oderAustausch von W�arme Q und Sto�menge nk mit der Umgebung k�onnen im System Pha-sen�uberg�ange ablaufen, so dass ein heterogenes Mehrkomponentensystem aus P Phasenvorliegt.Vapor
Solid
Fluid
Beispiel f�ur ein heterogenes Mehrkomponentensystemaus drei Phasen: Gasphase (Gasgemisch), �ussigePhase (�ubers�attigte L�osung), feste Phase (ausgef�allteSalze). In jeder k�onnen mehrere Komponenten auf-treten.Die extensiven thermodynamischen Funktionen sind additiv:V = PX�=1V (�) ; N = PX�=1N (�) ; U = PX�=1U (�) ; S = PX�=1S(�) : : :Die Gibbssche Fundamentalgleichung (6) gilt in jeder Phase � = 1 : : : P , in der i = 1 : : : kKomponenten auftreten k�onnen:dU (�) = T (�)dS(�) � p(�)dV (�) + kXi=1 �(�)i dn(�)i :Die Gibbs{Duhem{Gleichung (44) gilt analog:U (�) = T (�)S(�) � p(�)V (�) + kXi=1 �(�)i n(�)i :Die Gleichgewichtsbedingungen f�ur das System ergeben sich aus S ! Max. und lauten:T (1) = T (2) = : : : = T (P ) � T ;p(1) = p(2) = : : : = p(P ) � p ;�(1)1 = �(2)1 = : : : = �(P )1 � �1 ;�(1)2 = �(2)2 = : : : = �(P )2 � �2 ;... ... ...�(1)k = �(2)k = : : : = �(P )k � �k :Die chemischen Potenziale �(�)i sind intensive Gr�o�en, � = G=n, und damit nicht von denMolzahlen der einzelnen Komponenten in der jeweiligen Phase abh�angig, sondern nur von73

deren Konzentrationen. Man de�niert die Konzentration oder den Molenbruch x(�)i , f�urdie/den eine Summenregel gilt,x(�)i = n(�)ikPj=1n(�)j ; kXi=1 x(�)i = 1 ;so dass das chemische Potenzial von k + 1 Variablen abh�angt:�(�)i = �(�)i (T; p; x(�)1 ; x(�)2 ; : : : ; x(�)k�1) :7.1.2 Gibbssche PhasenregelDamit haben wir zur Beschreibung des heterogenen Mehrkomponentensystems k + 1Variablen in jeder der P Phasen (k + 2 � 1 Summenregel), also P (k + 1) Variableninsgesamt. Dagegen stehen (k + 2) � (P � 1) Gleichgewichtsbedingungen zwischen denP Phasen zur Verf�ugung. Damit ist die Zahl der thermodynamischen Freiheitsgradef = P (k + 1)� (k + 2)(P � 1) im System �uber die Gibbssche Phasenregel festgelegt:f = k � P + 2 : (85)Falls im System noch R chemische Reaktionen ablaufen, verringert sich die Zahl derFreiheitsgrade durch weitere Nebenbedingungen auff = k � P �R + 2 :Der thermodynamische Gleichgewichtszustand ist bei gegebener Komponenten- und Pha-senzahl durch f innere Variablen, z.B. Druck p und Temperatur T , bestimmt.Beispiel: F�ur ein Einkomponentensystem k = 1 ergibt sich f = 3 � P . Da f � 0 seinmuss, erh�alt man f�ur die m�ogliche Phasenzahl im System P � 3, d.h. es k�onnen h�ochstensdrei Phasen untereinander im Gleichgewicht stehen.� P = 1: f = 2, d.h. die Zust�ande einphasiger Einkomponentensysteme (z.B. Gas,Fl�ussigkeit, Festk�orper) existieren f�ur beliebige Kombinationen von zwei innerenZustandsvariablen [z.B. (T; p)] und bilden eine Fl�ache.� P = 2: f = 1, d.h. die Zust�ande zweiphasiger Einkomponentensysteme (z.B. Gas{Fl�ussigkeit) haben nur einen Freiheitsgrad und verlaufen auf Phasengrenzkurven[z.B. entlang der Dampfdruckkurve p(T )].� P = 3: f = 0, d.h. der Zustand eines dreiphasigen Einkomponentensystems (z.B.Gas{Fl�ussigkeit{Festk�orper) hat keinen Freiheitsgrad mehr und existiert in einemfesten Punkt des Zustandsdiagramms, dem Tripelpunkt.� In Einkomponentensystemen gibt es keine Vierphasenpunkte (z.B. wichtig f�ur Sy-steme mit mehreren festen Phasen).In Mehrkomponentensystemen mit k > 1 (bin�are, tern�are, : : :) ist das Phasendiagrammentsprechend komplizierter. Mit solchen Systemen besch�aftigt sich insbesondere die phy-sikalische Chemie. 74

7.1.3 Ideale homogene MischungenDas thermodynamische System bestehe aus einer Phase (z.B. gasf�ormig) und k Kompo-nenten (Sto�en) ohne chemische Reaktion. Die thermodynamischen Variablen seien (T; p),so dass die freie Enthalpie G als thermodynamisches Potenzial gew�ahlt werden kann:G(T; p; ni) = kXi=1 �ini ; �i = �i(T; p; x1 : : : xk�1) ; kXi=1 xk = 1 :Das chemische Potenzial �i ist intensiv und h�angt nur von den Konzentrationen xi ab,von denen eine �uber die Summenregel eliminiert werden kann. In idealen Gemischen wirddie Wechselwirkungen zwischen den verschiedenen Komponenten vernachl�assigt und dieextensiven thermodynamischen Gr�o�en sind additiv. Beispiele sind verd�unnte L�osungenund Mischungen idealer Gase. F�ur die freie Energie gilt:F (T; V; n1 : : : nk) = kXi=1 Fi(T; V; ni) :Andere thermodynamische Gr�o�en wie den Druck erh�alt man durch Ableitung:p = � @F@V !T;n = � kXi=1 @Fi@V !T;ni � kXi=1 pidi (T; V; ni) :Der Partialdruck pi der i-ten Komponente ist mit dem Gesamtdruck p �uber das DaltonscheGesetz verbunden: p = kPi=1 pidi ; pidi = ni RTV : (86)In der Form pV = kXi=1 niRT = p kXi=1 Vi ; Vi = niRTpnennt man es auch Amagatsches Gesetz, d.h. das Gesamtvolumen V des Gasgemischesergibt sich additiv aus den Teilvolumina Vi, die berechnet werden, als st�unde jede Kom-ponente unter dem Gesamtdruck p des Gemisches. F�ur die Entropie gilt analogS = � @F@T !V;n = � kXi=1 @Fi@T !V;ni � kXi=1 Sidi (T; V; ni)und man erh�alt f�ur die innere EnergieU = F + TS = kXi=1 �Fi + TSidi � � kXi=1 U idi (T; V; ni) :Das chemische Potenzial als intensive Gr�o�e ist durch�i = @F@ni!T;V;nj 6=ni = @Fi@ni!T;V = �idi (T; V; ni)! �idi �T; niV �gegeben. F�ur die Variablentransformation von (T; niV ) = (T; pi) auf (T; p) benutzt man(59) �idi (T; pi) = RT (ln pi � 52 lnT � j), so dass�idi (T; pi) = RT ln p� 52 lnT + ln pip � j! = �idi (T; p) + RT lnxi75

gilt. Dabei wurde das Daltonsche Gesetz benutzt, d.h. pip = nin = xi. Das chemischePotenzial der i-ten Komponente ist nur von der Konzentration abh�angig:�idi (T; p; ni) = �idi (T; p) +RT lnxi : (87)7.1.4 MischungsentropieT,p T,p
V. . . . .
V V Vk-1
T,p T,p
1 2 k
In einem Beh�alter be�nden sich k Kammernmit je einem idealen Gas. Die beweglichenW�ande sind undurchl�assig. Temperatur- undDruckausgleich hat stattgefunden: (T; p) f�uralle Kammern. Die Teilvolumina ergeben sichaus dem Amagatschen Gesetz: Vi = ni RTp .Die Entropie des Systems vor der Mischung ist durch die Summe der Entropien der Gasein den einzelnen Kammern gegeben:SV = kXi=1 nisidi (T; p) :Nach adiabatischem Entfernen der Trennw�ande di�undieren die Gase ineinander, bis einhomogenes Gemisch vorliegt. Die Entropie dieses Gemisches ist durch die Summe derEntropien aller Komponenten gegeben, die durch die jeweiligen Partialdr�ucke bestimmtsind: SM = kXi=1 nisidi (T; pi) :Wir benutzen wieder das Resultat (58) sidi (T; pi) = R(52 lnT � ln pi+�0) und pip = nin = xi,so dass manSM = kXi=1 niR 52 lnT � ln p + �0 + ln ppi! = kXi=1 ni �sidi (T; p) +R ln nni�erh�alt. Die Di�erenz aus der Entropie der Mischung und der Entropie des Systems vorder Mischung nennt man Mischungsentropie:�S = SM � SV = kPi=1niR ln nni > 0 : (88)Durch das Entfernen der W�ande �ndet ein irreversibler Prozess (Gasdurchmischung) stattund die Entropie des Systems w�achst an. Die Mischungsentropie �S ist unabh�angig vonden speziellen Eigenschaften der idealen Gase.Gibbssches Paradoxon: Betrachtet man z.B. ein Gemisch aus zwei Gasen mit n1 =n2 = n2 , dann ist die Mischungsentropie �S = nR ln 2. Werden die Eigenschaften derbeiden Gase im Sinne eines Gedankenexperiments gleich gemacht, �andert sich die Mi-schungsentropie nicht. Andererseits erhalten wir f�ur dieses Gas jetzt SV = nsid(T; p) undSM = nsid(T; p), also �S = 0. Dieser Widerspruch ist als Gibbssches Paradoxon bekannt.Er l�ost sich auf, wenn wir die Entropie als extensive Gr�o�e betrachten:SV = 2S �T; V2 ; n2� ; SM = S(T; V; n) = 2S �T; V2 ; n2� ! �S = 0 :76

7.1.5 Reale homogene MischungenIn realen homogenen Mischungen sind die Wechselwirkungen zwischen den Atomen undMolek�ulen zu ber�ucksichtigen, so dass Abweichungen vom idealen Verhalten auftreten.Diese sind z.B. mit �Anderungen des Volumens und der inneren Energie verbunden,�V = V (T; p; n1 : : : nk)� kXi=1 V (T; p; ni) ; �U = U(T; p; n1 : : : nk)� kXi=1 U(T; p; ni) ;so dass die entsprechende �Anderung der Enthalpie �H = �U + p�V � QM als Mi-schungsw�arme QM gemessen werden kann. Bei gegebenen Variablen (T; p; ni) ist die freieEnthalpie thermodynamisches Potenzial:G(T; p; n1 : : : nk) = kXi=1 ni�i(T; p; n1 : : : nk) :Der Ansatz von Lewis f�ur das chemische Potenzial der realen Mischung folgt dem bekann-ten idealen Verhalten (87):�i(T; p; n1 : : : nk) = �idi (T; p) +RT ln ai : (89)Dabei ist ai = xifi die Aktivit�at und fi(T; p; x1 : : : xk) der Aktivit�atskoe�zient. Man f�uhrtauch den Begri� des chemischen Exzesspotenzials �exi ein, das speziell die Wechselwir-kungsterme beschreibt: �exi = �i � �idi = RT ln fi :Dadurch hat man auch die h�au�g n�utzliche Exzessenthalpie Gex = Pki=1 ni�exi de�niert.Mit fi = 1 werden ideale Mischungen beschrieben, f�ur die �exi = 0 und Gex = 0 gilt.Beispiel: verd�unnte L�osungEine verd�unnte L�osung liegt vor, wenn in n0 Molen L�osungsmittel verschiedene Sto�e mitni Molen gel�ost sind und n0 � ni erf�ullt ist. Bei weiterem Zusatz von L�osungsmittel tretenkeine �Anderungen des Volumens und der inneren Energie auf, d.h. bzgl. des L�osungsmittelsverh�alt sich das System wie eine ideale Mischung,�0 = �id0 (T; p) +RT lnx0 ; f0 = 1 ;die Exzessanteile �ex0 = 0 und Gex0 = 0 verschwinden. Die Aktivit�atskoe�zienten dergel�osten Sto�e streben f�ur xi ! 0 gegen einen endlichen Wert, der von der Art desL�osungsmittels abh�angt. Man kann deshalb die Aktivit�at in eine Potenzreihe nach denKonzentrationen xi entwickeln, ai = �i;0xi + �i;0x2i + : : :Ber�ucksichtigt man nur den linearen Term, erh�alt man f�ur das chemische Potenzial dergel�osten Sto�e �i = �idi (T; p) +RT lnxi +RT ln�i;0 :Schl�agt man den L�osungsmittelterm zu den Idealanteilen,�idi;0(T; p) = �idi (T; p) +RT ln�i;0 ;erh�alt man eine zum Verhalten idealer Mischungen (87) analoge Form:�i = �idi;0(T; p) +RT lnxi : (90)77

Stabilit�at von Mischungen:� Gase sind immer in jedem Verh�altnis mischbar.� Bei Fl�ussigkeiten kann ein stetiger �Ubergang von vollst�andiger zu fehlender Misch-barkeit auftreten.� Entmischungserscheinungen sind stark temperaturabh�angig. Die kritische Entmi-schungstemperatur Tk bezeichnet den Bereich, f�ur den Entmischung vorkommt (ober-halb: T > Tk, unterhalb: T < Tk).� Das Entmischungsgebiet in der T{x{Ebene nennt man Mischungsl�ucke.7.1.6 Der osmotische Druckn1
(1)
n0(1)
T
=0
n(2)0
Tp(1) p(2)
n1(2)
Eine semipermeable Wand trennt zwei Teil-systeme, das reine L�osungsmittel (2) und dieverd�unnte L�osung (1). Sie ist nur f�ur dasL�osungsmittel n0 durchl�assig und verhindertso den Druckausgleich.Im thermodynamischen Gleichgewicht stellt sich in der L�osung ein h�oherer Druck als imL�osungsmittel ein. Der Druckunterschied zwischen (1) und (2) wird osmotischer Druck ge-nannt: posm = p(1)�p(2). Man berechnet ihn aus der Gleichgewichtsbedingung F (T; V; n)!Min., wobei Temperaturausgleich zwischen (1) und (2) bereits stattgefunden habe. Teil-chenaustausch durch die semipermeable Wand ist nur f�ur das L�osungsmittel m�oglich:�n(1)0 = ��n(2)0 . Die freie Energie ist also bzgl. n0 zu minimieren:F (T; V (1); V (2); n(1)0 ; n(2)0 ; n(1)1 )! Min: ; @F@n(1)0 !T;V;n �n(1)0 + @F@n(2)0 !T;V;n �n(2)0 = 0 :Man erh�alt mit � = (@F=@n)T;V die Gleichgewichtsbedingung:�(1)0 = �(1)0 (T; p(1); x(1)0 ) = �(2)0 = �(2)0 (T; p(2); x(2)0 ) :F�ur das chemische Potenzial �0 des L�osungsmittels in (1) und (2) verwenden wir dasErgebnis verd�unnter L�osungen:�(1;id)0 (T; p(1)) +RT lnx(1)0 = �(2;id)0 (T; p(2)) +RT lnx(2)0 :Man kann mitx(1)0 = n(1)0n(1)0 + n(1)1 ; x(2)0 = 1 ; p(2) = p(1) � posm ; posm � p(1); p(2)eine Entwicklung an der Stelle p(1) durchf�uhren:�(1;id)0 (T; p(1)) +RT lnx(1)0 = �(2;id)0 (T; p(1))� 0@@�(2;id)0@p 1AT;p=p(1)posm :78

Mit der Ableitung @�@p!T = 1n @G@p !T = 1nV = vund der Gleichgewichtsbedingung f�ur das reine L�osungsmittel �(1;id)0 (T; p) = �(2;id)0 (T; p)erh�alt man posmv(1)0 = �RT lnx(1)0 = RT ln0@1 + n(1)1n(1)0 1A � RT n(1)1n(1)0 :Der osmotische Druck entspricht dem idealen Gasdruck, den n(1)1 Mole des gel�osten Sto�sim Volumen V (1)0 der L�osung bei gegebener Temperatur aus�uben w�urden:posmV (1)0 = n(1)1 RT : (91)Beispiele:� Den h�oheren Druck in der L�osung nutzen P anzen aus, um N�ahrsto�e �uber Ka-pillaren von der Wurzel in den Bereich der Bl�atter zu pumpen (Pfe�ersche S�aule):posm = %gh.� Der osmotische Druck in Blutzellen betr�agt etwa 7 kPa (70 mbar). Infusionen werdendeshalb mit einer vertr�aglichen, physiologischen Kochsalzl�osung mit % = 8:8 g/ldurchgef�uhrt, die den Druck im Blut nicht absinken l�asst.7.1.7 Raoultsche Gesetzen0
(2)
T,p
Dampf
T,p
verduennte Loesung
n0 n1 (1)(1)
,
Wir betrachten jetzt das thermodynamische Gleich-gewicht zwischen einer verd�unnten L�osung und ihremDampf. Dabei wird angenommen, dass der gel�osteSto� n(1)1 nicht �uchtig ist und Teilchenaustausch nurbzgl. des L�osungsmittels mit �n(1)0 = ��n(2)0 m�oglichist. Temperatur- und Druckausgleich habe stattgefun-den, d.h. T (1) = T (2) = T und p(1) = p(2) = p.Wie �andert sich die Dampfdruckkurve p12(T ) der verd�unnten L�osung verglichen mit derdes reinen L�osungsmittels p012(T )? Dazu k�onnen wir wieder von der Gleichgewichtsbedin-gung �(1)0 = �(1)0 (T; p12; x(1)0 ) = �(2)0 = �(2)0 (T; p12; x(2)0 )ausgehen und das Ergebnis f�ur verd�unnte L�osungen verwenden:�(1;id)0 (T; p12) +RT lnx(1)0 = �(2;id)0 (T; p12) +RT lnx(2)0 :Man kann mit x(1)0 = n(1)0n(1)0 + n(1)1 ; x(2)0 = 1 ; p12 = p012 +�p12 ;79

eine Entwicklung an der Stelle p012 durchf�uhren, wobei �@�@p�T = v das Molvolumen in derjeweiligen Phase liefert:�(1;id)0 (T; p012) + v(1)0 �p12 �RT n(1)1n(1)0 = �(2;id)0 (T; p012) + v(2)0 �p12 :F�ur das reine L�osungsmittel ist die Gleichgewichtsbedingung �(1;id)0 (T; p012) = �(2;id)0 (T; p012)erf�ullt. Die daraus folgenden Raoultschen Gesetze sind universell f�ur alle Koexistenzkurveng�ultig: �p12 = RTv(1)0 �v(2)0 n(1)1n(1)0 : (92)Dampfdruck�anderung �p12 = �pvap: Phase (1) sei �ussig, Phase (2) der Dampf, dieTemperatur konstant. Allgemein gilt v(1)0 � v(2)0 und p012v(2)0 = RT im Dampf, so dass eineDampfdruckerniedrigung in der L�osung relativ zum reinen L�osungsmittel erhalten wird:�p12 = �pvap = �n(1)1n(1)0 p012 :Schmelzdruck�anderung �p12 = �pmelt: Phase (1) sei �ussig, Phase (2) fest, die Tempe-ratur konstant. Die Volumen�anderung beim Schmelzen �v = v(1)�v(2) legt das Vorzeichendes E�ekts fest, d.h. f�ur v(1) > v(2) folgt eine Schmelzdruckerh�ohung �pmelt > 0 und f�urv(1) < v(2) eine Schmelzdruckerniedrigung �pmelt < 0 (z.B. Wasser).�Anderung der �Ubergangstemperatur: Der Druck sei durch den festen Au�endruckpa gegeben, der beim Sieden durch den Dampfdruck des reinen L�osungsmittels bzw. dender verd�unnten L�osung erreicht wird:pa = p012(T 0) = p12(T 0 +�T; x(1)0 ) :Man erh�alt durch Entwicklung nach T mit p12(T; x) = p012(T ) + �p12(T; x):p012(T 0) = p12(T 0; x(1)0 ) + @p12@T !V;T=T0�T = p012(T 0)+�p12(T0; x(1)0 )+ @p12@T !V;T=T0�T :Mit Hilfe der Raoultschen Gesetze (92) f�ur �p12 und der Clausius{Clapeyronschen Glei-chung (66) f�ur (@p12=@T )V erh�alt manRTv(1)0 � v(2)0 n(1)1n(1)0 = q12(T )T (v(1)0 � v(2)0 )�T :Die �Anderung der �Ubergangstemperatur bei festem Druck betr�agt:�T = RT 2q12(T ) n(1)1n(1)0 :80
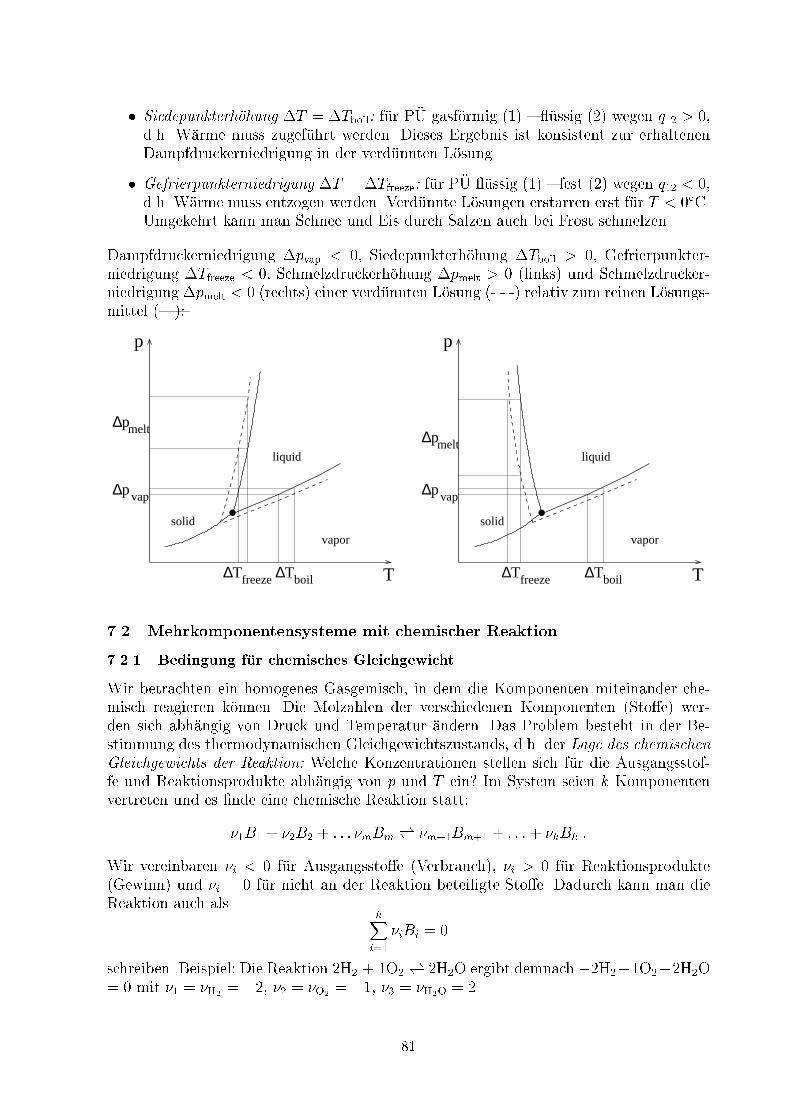
� Siedepunkterh�ohung �T = �Tboil: f�ur P�U gasf�ormig (1) { �ussig (2) wegen q12 > 0,d.h. W�arme muss zugef�uhrt werden. Dieses Ergebnis ist konsistent zur erhaltenenDampfdruckerniedrigung in der verd�unnten L�osung.� Gefrierpunkterniedrigung �T = �Tfreeze: f�ur P�U �ussig (1) { fest (2) wegen q12 < 0,d.h. W�arme muss entzogen werden. Verd�unnte L�osungen erstarren erst f�ur T < 0�C.Umgekehrt kann man Schnee und Eis durch Salzen auch bei Frost schmelzen.Dampfdruckerniedrigung �pvap < 0, Siedepunkterh�ohung �Tboil > 0, Gefrierpunkter-niedrigung �Tfreeze < 0, Schmelzdruckerh�ohung �pmelt > 0 (links) und Schmelzdrucker-niedrigung �pmelt < 0 (rechts) einer verd�unnten L�osung (- - -) relativ zum reinen L�osungs-mittel (|):liquid
T
vapor
solid
p
TT∆
∆p
∆p
melt
vap
freeze boil∆
liquid
freeze
vapor
solid
p
TT∆
∆pvap
boil
meltp∆
∆T7.2 Mehrkomponentensysteme mit chemischer Reaktion7.2.1 Bedingung f�ur chemisches GleichgewichtWir betrachten ein homogenes Gasgemisch, in dem die Komponenten miteinander che-misch reagieren k�onnen. Die Molzahlen der verschiedenen Komponenten (Sto�e) wer-den sich abh�angig von Druck und Temperatur �andern. Das Problem besteht in der Be-stimmung des thermodynamischen Gleichgewichtszustands, d.h. der Lage des chemischenGleichgewichts der Reaktion: Welche Konzentrationen stellen sich f�ur die Ausgangsstof-fe und Reaktionsprodukte abh�angig von p und T ein? Im System seien k Komponentenvertreten und es �nde eine chemische Reaktion statt:�1B1 + �2B2 + : : : �mBm *) �m+1Bm+1 + : : :+ �kBk :Wir vereinbaren �i < 0 f�ur Ausgangssto�e (Verbrauch), �i > 0 f�ur Reaktionsprodukte(Gewinn) und �i = 0 f�ur nicht an der Reaktion beteiligte Sto�e. Dadurch kann man dieReaktion auch als kXi=1 �iBi = 0schreiben. Beispiel: Die Reaktion 2H2 + 1O2 *) 2H2O ergibt demnach �2H2�1O2+2H2O= 0 mit �1 = �H2 = �2, �2 = �O2 = �1, �3 = �H2O = 2.81

Gleichgewichtsbedingung: G(T; p; n)! Min: Variiert wird bez�uglich der Molzahlen,die sich durch die chemische Reaktion �andern k�onnen:(�G)T;p;nj = kXi=1 @G@ni!T;p;nj�ni = kXi=1 �i(T; p)�ni = 0 :De�niert man eine Reaktionslaufzahl � 2 (0; 1) �uber die st�ochiometrischen Koe�zienten,�ni = �i��, �ndet man mit (�G)T;p;nj = kXi=1 �i(T; p)�i�� = 0die Bedingung f�ur das chemische Gleichgewicht der Reaktion:kPi=1 �i�i(T; p) = 0 : (93)Beispiel von oben: 2�H2 + 1�O2 = 2�H2O. Treten R Reaktionen im System auf, de�niertman entsprechend viele Reaktionslaufzahlen �R und erh�alt jeweils eine Bedingung f�ur daschemische Gleichgewicht jeder Reaktion. Die Zahl der thermodynamischen Freiheitsgra-de im System entsprechend der Gibbsschen Phasenregel (85) verringert sich durch diesezus�atzlichen Bedingungen um R: f = 2 + k � p� R.7.2.2 Das MassenwirkungsgesetzAus der Bedingung f�ur das chemische Gleichgewicht (93) m�ussen die Konzentrationen(oder Molzahlen) der beteiligten Sto�e in Abh�angigkeit von Druck und Temperatur be-rechnet werden. Dazu behandeln wir die Gasphase als ideale homogene Mischung mit�i(T; p; n1 : : : nk) = �idi (T; p) +RT lnxi ; xi = nin = pip :Dabei ist n = Pi ni und p = Pi pi.Auswertung der Gleichgewichtsbedingung (93):kXi=1 �i ��idi (T; p) +RT lnxi� = 0 ;kXi=1 �i lnxi = kXi=1 lnx�ii = � 1RT kXi=1 �i�idi (T; p) :Mit exp kXi=1 lnx�ii ! = kYi=1 exp (lnx�ii ) = kYi=1x�iifolgt das Massenwirkungsgesetz von Guldberg und Waage. Die MassenwirkungskonstanteKx(T; p) ist durch die Idealbeitr�age zum chemischen Potenzial �idi bestimmt,kQi=1 x�ii = exp � 1RT kPi=1 �i�idi (T; p)! � K idx (T; p) ; (94)82

und legt das Verh�altnis der Molenbr�uche von Ausgangssto�en und Reaktionsproduktenfest, die Ausbeute der Reaktion. F�ur das obige Beispiel 2H2 + 1O2 *) 2H2O erh�alt man:x2H2Ox2H2xO2 = K idx (T; p) :Man kann von den Molenbr�uchen xi auch zu den Partialdr�ucken pi oder den Konzentra-tionen ci �ubergehen,xi = pip ; ci = niV = pinpV = piRT ) ci = xipRT = xiv ;und erh�alt: kQi=1 p�ii = p kPi=1 �iK idx (T; p) � K idp (T; p) ; (95)kQi=1 c�ii = v� kPi=1 �iK idx (T; p) � K idc (T; p) : (96)Die Relationen (94)-(96) sind nur f�ur verd�unnte Gase und Fl�ussigkeiten anwendbar, dadie Wechselwirkungsbeitr�age zum chemischen Potenzial vernachl�assigt wurden. Formalerh�alt man mit dem Ansatz�i(T; p; n1 : : : nk) = �idi (T; p) +RT lnxi +��WWi (T; p; n1 : : : nk)dichteabh�angige Resultate, z.B.kYi=1 x�ii = exp(� 1RT kXi=1 �i ��idi (T; p) + ��WWi (T; p; n1 : : : nk)�) � KWWx (T; p) : (97)7.2.3 Verschiebung des chemischen Gleichgewichts: van't Ho�sche Gleichungenund das Prinzip von Le Chatelier und BraunWie �andert sich die Lage des chemischen Gleichgewichts bei Variation von Druck undTemperatur? Dazu geht man z.B. von Kx(T ) aus und untersucht die Ableitungen:lnKx(T; p) = � 1RT kXi=1 �i�idi (T; p) :Variation des Drucks bei fester Temperatur: 1. van't Ho�sche Gleichung,@@p lnKx(T; p) = � 1RT kXi=1 �i @�idi (T; p)@p !T = � kXi=1 �iviRT � ��vRT : (98)Die Druckabh�angigkeit der Massenwirkungskonstante wird durch die bei einmaligem Re-aktionsdurchlauf (� = 1) auftretende Volumen�anderung �v = Pki=1 �ivi bestimmt:d lnKx(T; p) = ��vRT dp = ��vv dpp = ��vv d ln p :83

� �v < 0, das Volumen der Reaktionsprodukte ist kleiner als das der Ausgangssto�e:Die Ausbeute der Reaktion wird mit zunehmendem Druck gr�o�er,Kx(T; p) � p :� �v > 0, das Volumen der Reaktionsprodukte ist gr�o�er als das der Ausgangssto�e:Die Ausbeute der Reaktion wird mit zunehmendem Druck kleiner,Kx(T; p) � 1p :Variation der Temperatur bei festem Druck: 2. van't Ho�sche Gleichung,@@T lnKx(T; p) = 1RT 2 kXi=1 �i�idi (T; p)� 1RT kXi=1 �i @�idi (T; p)@T !p :Mit (@�i=@T )p = �si und der Gibbs{Duhemschen Gleichung (44) �i = ui + pvi � Tsi =hi � Tsi erh�alt man die Relation@@T lnKx(T; p) = 1RT 2 kXi=1 �i ��idi (T; p) + Tsidi (T; p)� = 1RT 2 kXi=1 �ihi � �hRT 2 : (99)Die bei einmaligem Reaktionsdurchlauf (� = 1) auftretende Enthalpie�anderung �h =Pki=1 �ihi � qp ist bei konstantem Druck gleich der zugef�uhrten (endotherm: qp > 0) oderder freiwerdenden (exotherm: qp < 0) W�arme. Man erh�alt allgemein das Resultat:d lnKx(T; p) � qpdTRT 2 = �qpRd� 1T � ) Kx(T; p) � exp�� qpRT � :� Endotherme Reaktion mit qp > 0: Die Ausbeute der Reaktion wird mit zunehmenderTemperatur vergr�o�ert.� Exotherme Reaktion mit qp < 0: Die Ausbeute der Reaktion wird mit zunehmenderTemperatur verkleinert.Die van't Ho�schen Gleichungen bilden die Grundlage f�ur das Prinzip von Le Chatelierund Braun (1885):Ein im thermodynamischen Gleichgewichtszustand be�ndliches System weicht einem�au�eren Zwang wie der �Anderung von Temperatur und Druck aus (Prinzip vom kleinstenZwang).7.2.4 Anwendungen zum Massenwirkungsgesetz1. Ammoniaksynthese: N2 + 3H2 *) 2NH3, qp = �22 kcal (exotherm)x2NH3xN2x3H2 = K idx (T; p) :Eine systematische Erh�ohung der Ausbeute an NH3 wird mit abnehmender Tempe-ratur und zunehmendem Druck erwartet. Technisch wird die Ammoniaksynthese im84

Haber{Bosch{Verfahren bei T � 500 �C und p � 200 at mit einer Ausbeute von et-wa 18% realisiert. Bei kleineren Temperaturen ist die Reaktionsgeschwindigkeit nichtmehr �okonomisch und gr�o�ere Dr�ucke erh�ohen den technischen Aufwand (Kosten)enorm. Das NH3 wird in einem kontinuierlichen Verfahren aus dem Reaktionsgasentfernt und das nichtverbrauchte Restgas (N2, H2) plus Frischgas dem Katalysatorwieder zugef�uhrt.2. Schwache Elektrolyte:Salze, S�auren oder Basen dissoziieren in L�osungsmitteln (Auftreten von Ladungs-tr�agern { Ionen). Man �ndet z.B. f�ur die Dissoziation von Wasser H2O *) H+ +OH� unter Normalbedingungen eine Massenwirkungskonstante vonxH+xOH�xH2O � 10�14 :3. Ionisationsgleichgewicht:Das Ionisationsgleichgewicht, z.B. H+ + e� *) H in Sternatmosph�aren, ist stark vonDruck und Temperatur abh�angig. Man erh�alt das auch als Saha{Gleichung bekannteMassenwirkungsgesetz: xHxH+xe� = Kx(T; p) :Mit der Neutralit�atsforderung xH+ = xe� und der Summenregel xH+ + xe� + xH = 1folgt f�ur den Ionisationsgrad �ion �ion = xe�xe� + xHein Zusammenhang mit der Massenwirkungskonstanten:Kx(T; p) = 1� �2ion�2ion :Die Massenwirkungskonstante f�ur reale Plasmen wird unter Ber�ucksichtigung derWechselwirkungskorrekturen ��WWi (T; p) zum chemischen Potenzial entsprechend(97) berechnet.4. Dissoziationsgleichgewicht:Das Dissoziationsgleichgewicht in molekularen Fluiden unter hohem Druck, z.B. H2*) 2H im Innern der Gro�en Planeten wie Jupiter und Saturn, ist ebenfalls stark vonDruck und Temperatur abh�angig. Man erh�alt analog zum Ionisationsgleichgewichteinen Dissoziationsgrad �dis �dis = xHxH + xH2 ;der wieder unter Ber�ucksichtigung der Wechselwirkungskorrekturen entsprechend(97) zu berechnen ist.85

86

8 Literaturempfehlung- G. Adam, O. Hittmair, W�armetheorie (Vieweg, Braunschweig, 1992)- R. Becker, Theorie der W�arme (Springer, Berlin, 1966)- G. Carrington, Basic Thermodynamics (Oxford Univ. Press, Oxford, 1994)- G. Cerbe, H.-J. Ho�mann, Einf�uhrung in die Thermodynamik (Hanser, M�unchen, 1999)- S.R. De Groot, P. Mazur, Non-Equilibrium Thermodynamics (North-Holland, Amsterdam,1962)- W. Ebeling, R. Feistel, Physik der Selbstorganisation und Evolution (Akademie-Verlag, Berlin,1982)- G. Falk, Theor. Physik, Bd. II (Springer, Berlin, 1988)- E. Fermi, Thermodynamics (Dover, New York, 1956)- W. Gebhardt, U. Krey, Phasen�uberg�ange und kritische Ph�anomene (Vieweg, Braunschweig,1980)- A.M. Gueneault, Statistical Physics (Chapman & Hall, London, 1995)- W. Greiner, Lehrbuch Theor. Physik, Bd. 9 (Verlag H. Deutsch, Frankfurt/M., 1986)- T.L. Hill, Statistical Thermodynamics (Dover, New York, 1986)- R.J. Jelitto, Theoretische Physik, Bd. 6 (Aula-Verlag, Wiesbaden, 1985)- C. Kittel, H. Kr�omer, Physik der W�arme (Oldenbourg, M�unchen, 1984)- G. Kluge, G. Neugebauer, Grundlagen der Thermodynamik (Spektrum, Heidelberg, 1994)- H.J. Kreuzer, Nonequilibrium Thermodynamics and its Statistical Foundations (Oxford Uni-versity Press, Oxford, 1983)- L.D. Landau, E.M. Lifschitz, Lehrbuch Theor. Physik, Bd. 5, 9, 10 (Akademie-Verlag, Berlin)- D. Leuschner, Grundbegri�e der Thermodynamik (Akademie-Verlag, Berlin, 1979)- G. Macke, Thermodynamik und Statistik (Akademische Verlagsgesellschaft, Leipzig, 1967)- J. McLennan, Introduction to Non-Equilibrium Statistical Mechanics (Prentice Hall, Engle-wood Cli�s, 1989)- I. M�uller, Grundz�uge der Thermodynamik (Springer, Berlin, 1994)- W. Nolting, Grundkurs Theor. Physik, Bd. 4 (Zimmermann-Neufang, Ulmen, 1993)- M. Plischke, B. Bergerson, Equilibrium Statistical Physics (World Scienti�c, Singapore, 1994)- F. Reif, Statistische Physik und Theorie der W�arme (de Gruyter, Berlin, 1987)- A. Sommerfeld, Thermodynamik und Statistik (Geest & Portig, Leipzig, 1965)- H. Stumpf, A. Rieckers, Thermodynamik, Bd. 1/2 (Vieweg, Braunschweig, 1976/77)- D.N. Subarew, Statistische Thermodynamik des Nichtgleichgewichts (Akademie-Verlag, Berlin,1976)- M. Toda, R. Kubo, N. Saito, Statistical Physics I, R. Kubo, M. Toda, N. Hashitsume, StatisticalPhysics II (Springer, Berlin, 1991/1992) 87