Embed Size (px)
Citation preview

www.elsevier.com/locate/apcata
Applied Catalysis A: General 330 (2007) 1–11
Thermal and catalytic oligomerisation of fatty acids
Pasi Tolvanen, Paivi Maki-Arvela, Narendra Kumar, Kari Eranen, Rainer Sjoholm,Jarl Hemming, Bjarne Holmbom, Tapio Salmi, Dmitry Yu. Murzin *
Process Chemistry Centre, Abo Akademi University, FI-20500 Turku, Finland
Received 15 January 2007; received in revised form 25 May 2007; accepted 7 June 2007
Available online 23 June 2007
Abstract
Thermal and catalytic oligomerisation of technical grade linoleic acid was investigated under argon atmosphere at 280 8C. The most efficient
catalyst was H-MCM-41 followed by H-Beta 75. Several types of products, such as cyclic and aromatic compounds were formed indicating a
complex reaction network for catalytic oligomerisation of fatty acids.
# 2007 Elsevier B.V. All rights reserved.
Keywords: Linoleic acid oligomerisation; Mesoporous materials; Size exclusion chromatography
1. Introduction
Fatty acid dimers are valuable components, which can be
used as ingredients in paints and glues [1]. Furthermore, amides
of fatty acid dimers derived from tall or soybean oil are used as
epoxy coatings, printing inks and hot-melt adhesives [1,2].
Oligomerisation of fatty acids has been intensively investigated
[3–31] and the reaction has been carried out in the presence of
homogeneous [4,31,32] and heterogeneous catalysts
[3,5,7,20,27–30,37]. Additionally, it is known that fatty acids
react during distillation in the absence of any catalyst under
high vacuum at 270 8C forming fatty acid anhydrides via
dehydration [33–36]. These anhydrides formed are unstable
and form high molecular neutral compounds.
Oligomerisation of fatty acids has been traditionally
catalyzed by homogeneous catalysts, such as alkali or alkaline
metal salts [31] and iodine [4]. Oligomerisation of a mixture
containing equimolar amounts of oleic and linoleic acid in the
presence of 0.05 wt.% iodine at 260 8C has resulted in the
formation of 22 wt.% dimers after 5 h [4]. Both Lewis acids,
such as SnCl4 [32] and Brønsted acid catalysts, e.g. resin in H+
form [37] catalyze oligomerisation of fatty acids.
Heterogeneous catalysts are environmentally more friendly
and their industrial use is more attractive than the use of
* Corresponding author.
E-mail address: [email protected] (D.Yu. Murzin).
0926-860X/$ – see front matter # 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcata.2007.06.012
homogeneous catalysts, since they can be easily separated from
the products and reused. Dimerisation of fatty acids has been
investigated intensively over clays [3,5,7,20,27–29] and
typically clay materials, such as bentonite and montmorillonite,
have been used in the oligomerisation of fatty acids [5–30].
Bentonite has been found to be a very efficient catalyst for
dimerisation of oleic acid [3,7,28]. The reaction mechanism for
fatty acid transformations was found to be complex, consisting
of dimerisation, also of hydrogenation and dehydrogenation
steps [8,9]. It has been stated, that these reactions are acid-
catalyzed and, furthermore, bicyclic dimers of linoleic acid can
be formed [15]. Dimerisation of oleic acid has additionally been
carried out in continuous mode over montmorillonite as a
catalyst giving the yields of the dimers between 40 and
60 wt.%, depending on the process parameters, such as
temperature, catalyst particle size and water content [20].
Cation-exchanged clays are active in oligomerisation of
fatty acids [5,29]. The highest yield of dimers was achieved for
tall oil at 230 8C by using 6 wt.-% clay containing montmor-
illonite and LiOH or Ca(OH)2. The conversion was 71% with
the product containing a mixture of dimers [29]. A mixture of
oleic and elaidic acid as well as tall oil derived fatty acids was
dimerized over a clay containing LiOH and as a result, 65 wt.%
of different kinds of dimers (linear, alicyclic, aromatic and
polycyclic) were formed [27]. Furthermore, it was stated in [27]
that oleic acid dimers produced over a clay exchanged with
LiOH exhibited mostly a linear structure, while linoleic acid
dimers were polycyclic. Dimerisation of oleic acid has also
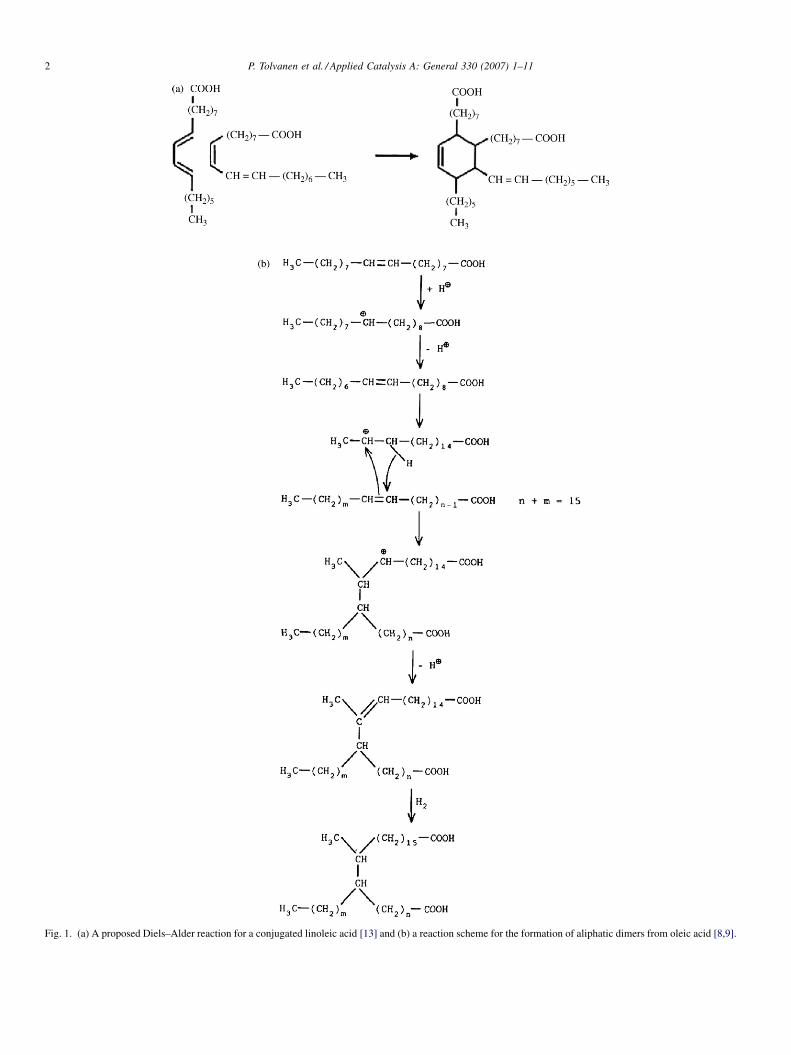
Fig. 1. (a) A proposed Diels–Alder reaction for a conjugated linoleic acid [13] and (b) a reaction scheme for the formation of aliphatic dimers from oleic acid [8,9].
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–112

P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–11 3
been studied over montmorillonite exchanged with several
different metal cations [5]. The highest yield of dimers has been
obtained over a clay containing magnesium as a cation
(42 wt.%) [5]. The cations located between the catalyst layers,
were active for isomerisation, and not the external catalyst
layer, as earlier proposed. It has thus been stated that the rate of
dimerisation was not dependent on the catalyst acidity, but
depended on the interfacial distance between the clay layers
[19].
The drawback in using clays is the large amount of by-
products formed. To avoid side reactions and to develop more
selective catalysts for oligomerisation of fatty acids, it is
necessary to know the reaction mechanism for oligomerisation
of fatty acids. Several other heterogeneous catalysts, such as
silica–alumina, SiO2, g-Al2O3 and MCM-41, were investigated
in the dimerisation of oleic acid at 255 8C [30]. The yields
of dimers were the highest with a clay montmorillonite
followed by silica–alumina > Li-montmorillonite > acid-acti-
vated montmorillonite » SiO2 > MCM-41 = g-alumina. Inter-
estingly, also an acid-activated montmorillonite exhibited a
lower activity than montmorillonite [30].
The reaction mechanism for the oligomerisation of fatty
acids over the clays is, however, not very clear, and several
different mechanisms have been proposed [6–15]. In these
experiments, mostly oleic acid or mixtures of oleic and linoleic
acids as well as conjugated linoleic acids (a mixture of 9,11-
and 10,12-linoleic acid) have been applied. The formation of
three different types of dimers has been confirmed, namely
aromatic dimers, tetra-substituted ring forms, an alicyclic
unsaturated dimer as well as alicyclic saturated dimers. It has
been proposed that conjugated linoleic acid reacts according to
a Diels–Alder mechanism, combining two molecules via
electrophilic addition in the double bond position forming a
cyclohexene ring (Fig. 1a) [13–15]. On the other hand, the
dimerisation of fatty acids was suggested to be initiated via a
cation mechanism forming cyclic dimers [13]. Clays are stated
to act as Lewis acids generating carbenium ions [38]. The
reaction proceeds thus via protonation of the double bond.
Carbenium ions were also formed in oleic acid oligomerisation
in the presence of H+ (Fig. 1b) [8,9]. It was, however, stated that
carbenium ions are not very selective in catalyzing only
oligomerisation of linoleic acid, since several side reactions can
proceed simultaneously, such as double bond isomerisation,
chain branching [39] and lactonisation.
The aim of this study was to compare the thermal
oligomerisation of technical grade linoleic acid (linoleic acid:
oleic acid ratio about 2:1) with the catalytic one. Seven different
catalysts, varying acidity and pore size were tested.
2. Experimental
2.1. Catalyst preparation and characterization
2.1.1. Catalyst preparation
The used zeolites were supplied by Zeolyst International.
Prior to their calcination at 530 8C the Na-Beta zeolites were
ion-exchanged to NH4-form with 3 M NH4Cl.
Na-MCM-41 was synthesized by using the method
described in Refs. [40,41]. As a surfactant, and as a source
for silica and aluminium tetradecyltrimethylammonium bro-
mide (Aldrich), sodium silicate solution (Merck) and sodium
aluminate (Riedel de Haen), respectively, were used. The
surfactant was removed at 540 8C. The formed Na-MCM-41
was ion-exchanged with 1 M NH4Cl solution and washed with
distilled water to remove chloride ions. H-MCM-41 was
obtained after calcination of this material at 530 8C.
Si-MCM-41 mesoporous molecular sieve was synthesized in
a 300 ml autoclave (Parr Instruments) as mentioned in the Refs.
[40,41] with some modifications. A gel mixture was prepared
by using the reagents, fumed silica (Aldrich), tetramethyl
ammonium silicate (Sachem), sodium silicate (Merck),
cetyltrimethyl ammonium bromide (Aldrich) and distilled
water. As the prepared gel, mixture was introduced into the
300 ml autoclave (Parr) and synthesis of Si-MCM-41 was
carried out in an oven at 100 8C. After the completion of
synthesis, the autoclave was quenched, and the mesoporous
material was filtered and washed with distilled water. Drying of
the sample was carried out at 110 8C for 12 h and calcination to
remove surfactant was performed in a muffle oven at 550 8C for
10 h. Alumina was supplied from UOP and Fe2O3 (FeIII oxide)
from Sigma–Aldrich (>99%).
2.1.2. Catalyst characterization
The specific surface areas of the catalysts were measured by
nitrogen adsorption using Sorptometer 1900 (Carlo Erba
Instruments). The catalysts were outgassed at 200 8C for 4 h.
Zeolites and mesoporous materials were analyzed by X-ray
powder diffractometer (Philips PW 1820) to investigate the
phase purity.
The concentration of Brønsted and Lewis acid sites was
measured by FTIR (ATI Mattson) by using pyridine (>99.5%,
a.r.) as a probe molecule [42,43]. The quantification of pyridine
was based on the molar extinction coefficient determined in
Ref. [44].
2.2. Experimental procedure
Linoleic acid oligomerisation was studied in a pressurized
reactor under argon (AGA, 99.9999%) atmosphere. The dried
catalyst, typically 0.5 g, if not otherwise stated, and 130 g
linoleic acid (Fluka, technical grade) were put into the reactor
and the reactor was closed and heated up to the desired
temperature. The monomer concentration was calculated using
the average molecular mass of technical grade linoleic acid
(65 wt.% linoleic acid, 35 wt.% oleic acid corresponding the
molecular mass of 280.7 g/mol) being 3.207 mol/l. Thus the total
molar amount of acids was initially 0.463 mol. To suppress
external and internal mass transfer resistances, small catalyst
particle (below 150 mm) and intensive stirring (1000 rpm) were
applied in the kinetic experiments. The initial sample was
withdrawn as the heating of the reactor was started. When the
desired temperature was reached, the corresponding time was
taken as zero reaction time. The time for heating up the reaction
mixture to the desired temperature varied between 20 and
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–114
35 min. Samples with a volume of 1 ml were taken out from the
reactor and stored under nitrogen atmosphere prior to analysis.
2.3. Analysis
2.3.1. SEC–HPLC analysis
The samples were analyzed by SEC–HPLC technique.
Tetrahydrofuran was used as eluent. Typically five droplets of a
sample were pipetted into a measuring flask (50 ml) and
weighed. The flasks were filled with tetrahydrofuran and the
obtained sample concentrations varied thus between 1.3 and
1.4 mg/ml. The samples were analyzed by SEC–HPLC together
with the calibration samples containing a known concentration
of soybean oil. The samples were additionally flushed with
argon prevent oxidation of unsaturated acids and kept at 4 8Cprior to their analysis.
The diluted samples were filtered with a 0.2 mm filter
(containing a membrane material PTFE, teflon) and analyzed
by SEC–HPLC equipped with three different columns (Jordi
precolumn, Jordigel DVB500A (7.8 mm � 300 mm), TSK
G3000HHR (7.8 mm � 300 mm). The two similar columns
were used in series in order to improve the separation. The
components were detected with a LT-ELS-detector (Low-
Temperature Evaporative Light-Scattering Detector, Sedex 85,
Sedere LT-ELSD). The SEC–HPLC system contained addi-
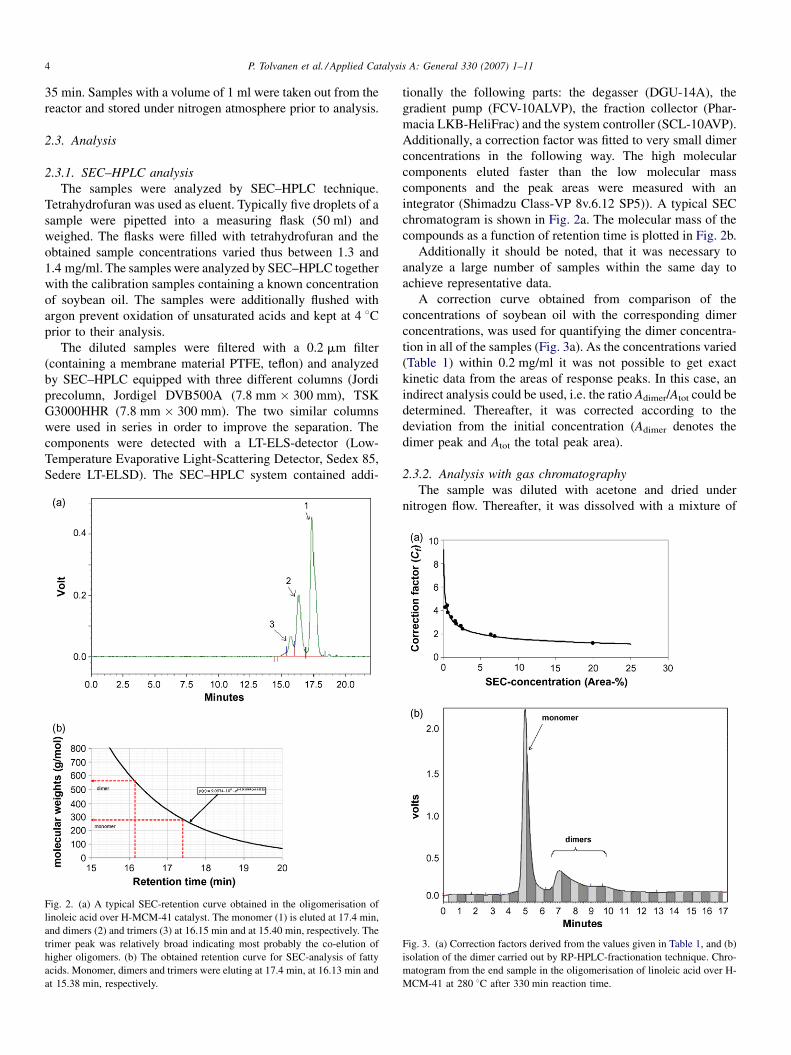
Fig. 2. (a) A typical SEC-retention curve obtained in the oligomerisation of
linoleic acid over H-MCM-41 catalyst. The monomer (1) is eluted at 17.4 min,
and dimers (2) and trimers (3) at 16.15 min and at 15.40 min, respectively. The
trimer peak was relatively broad indicating most probably the co-elution of
higher oligomers. (b) The obtained retention curve for SEC-analysis of fatty
acids. Monomer, dimers and trimers were eluting at 17.4 min, at 16.13 min and
at 15.38 min, respectively.
tionally the following parts: the degasser (DGU-14A), the
gradient pump (FCV-10ALVP), the fraction collector (Phar-
macia LKB-HeliFrac) and the system controller (SCL-10AVP).
Additionally, a correction factor was fitted to very small dimer
concentrations in the following way. The high molecular
components eluted faster than the low molecular mass
components and the peak areas were measured with an
integrator (Shimadzu Class-VP 8v.6.12 SP5)). A typical SEC
chromatogram is shown in Fig. 2a. The molecular mass of the
compounds as a function of retention time is plotted in Fig. 2b.
Additionally it should be noted, that it was necessary to
analyze a large number of samples within the same day to
achieve representative data.
A correction curve obtained from comparison of the
concentrations of soybean oil with the corresponding dimer
concentrations, was used for quantifying the dimer concentra-
tion in all of the samples (Fig. 3a). As the concentrations varied
(Table 1) within 0.2 mg/ml it was not possible to get exact
kinetic data from the areas of response peaks. In this case, an
indirect analysis could be used, i.e. the ratio Adimer/Atot could be
determined. Thereafter, it was corrected according to the
deviation from the initial concentration (Adimer denotes the
dimer peak and Atot the total peak area).
2.3.2. Analysis with gas chromatography
The sample was diluted with acetone and dried under
nitrogen flow. Thereafter, it was dissolved with a mixture of
Fig. 3. (a) Correction factors derived from the values given in Table 1, and (b)
isolation of the dimer carried out by RP-HPLC-fractionation technique. Chro-
matogram from the end sample in the oligomerisation of linoleic acid over H-
MCM-41 at 280 8C after 330 min reaction time.

Table 1
Values used in the calculations for determination of the correction factor, with a
known amount of soybean oil as standard
Concentration of
soya oil (mg/ml)
Concentration according
to peak area (mg/ml)
Value of the
correction factor (cf)
0.016 0.004 4.271
0.032 0.008 3.852
0.032a 0.009 4.419
0.048 0.014 3.416
0.063 0.020 3.088
0.063a 0.027 2.869
0.079 0.030 2.677
0.079a 0.031 2.420
0.157 0.086 1.950
0.157a 0.090 1.803
0.248 0.184 1.294
a The same amount of soya oil, but different amounts of sample.
Fig. 4. Thermal oligomerisation of linoleic acid at 280 8C under argon.
Symbols: monomer (^), dimer (&) and trimer (~).
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–11 5
BSTFA-TMCS (4:1) in pyridine and silylated at 70 8C for
45 min. The silylated monomers were analyzed both with a
short column and with a long column. The short column was a
HP-1 column (6 m, 0.53 mm, 0.15 mm film thickness), which
was used applying the following temperature programme:
100 8C (1.5 min)–12 8C/min–340 8C (18 min). The injector and
detector were SPI (septum equipped programmable injector) of
on-column type with the temperature program 80 8C (0.5 min)–
200 8C/min–340 8C (18 min) and FI-detector working at 340 8C,
respectively. The long column was a HP-1, (25 m, 0.20 mm,
0.11 mm film thickness) and FI detector at 300 8C was used. The
split ratio was 1:20. As internal standards, heneicosanoic acid
and bis(ethyl)hexylphtalate were used, respectively. Addition-
ally, the samples were analyzed by GC–MS.
2.3.3. Separation and confirmation of fatty acid dimer
A reversed-phase liquid chromatographic technique (RP-
HPLC) was used as an alternative method to separate the
products. Higher concentrations of samples, up to 100 mg/ml,
could be analyzed with RP-HPLC than with SEC-HPLC,
whereas with SEC-HPLC analysis the concentrations were
about 1–2 mg/ml. Samples could be fractioned by using RP-
HLPC technique. Both methanol and 95 wt.% ethanol in water
were tested as solvents, but only the latter solvent was able to
dissolve the samples completely at room temperature. A sample
with the concentration of 100 mg/ml was dissolved in an
ethanol–water mixture and analyzed by the RP-HPLC method.
The eluent flow (ethanol) was 20 ml/min and the total retention
time for the sample (precolumn, 20 mm � 75 mm, Phenom-
enex RP-18, 20 mm � 250 mm, detector: UV, 210 nm) was
16 min. The sample was fractioned into 30 pieces of 10 ml
sample tubes with 0.5 min time interval and a typical RP-HPLC
chromatogram is displayed in Fig. 3b, in which two peak areas
are clearly visble, the separated fractions at 5 min (fractions
8–9) and a broad peak at 7–11 min (fractions 12–19). These
peaks are the monomer and dimer peaks, which were additionally
analyzed by SEC–HPLC technique. The fractions 2–6, 8–9, 12–
14, 16–19, 20–24, and 25–30 were combined and evaporated
with nitrogen gas and dried in vacuum oven. These samples were
analysed by 1H-NMR (JEOL JNM-LA400) and GC–MS.
2.3.4. Study of aluminium leaching from alumina
The dissolution of Al from Al2O3 was measured from the
centrifuged reaction mixture by using laser ablation (New Wave
Research, UP-213) – ICP-MS (Perkin–Elmer Elan 6100 DRC
plus) technique. The LA-ICP-MS analysis of the samples was
done in a frozen state at�80 8C. The Laser Ablation Cryo-Cell
has been constructed in house.
3. Results and discussion
3.1. Catalyst characterization results
Seven different catalysts were used in the oligomerisation of
linoleic acid: g-Al2O3, Fe2O3, and three different H-Beta zeolites
exhibiting different Si/Al ratios, as well as two mesoporous
catalysts, mildly acidic H-MCM-41 and non-acidic Si-MCM-41
(Table 2). The X-ray powder diffraction pattern of Si-MCM-41
was similar to that of MCM-41. The BET surface area of Si-
MCM-41 was determined to be 1208 m2/g.
The catalysts exhibited both varying Brønsted and Lewis
acidities (Table 2). In the series of H-Beta zeolites, the most
acidic material was H-Beta-22 with the lowest Si/Al ratio. The
concentration of strong acid sites was H-Beta zeolites display
also Lewis acidity. The mesoporous H-MCM-41 contained a
higher amount of Brønsted acid sites than H-Beta-300. No
Brønsted acid sites were present in Si-MCM-41 and g-Al2O3
contained 7 mmol/gcat Brønsted acid sites, whereas it contained
the second highest amount of Lewis acid sites after H-MCM-41.
3.2. Oligomerisation results
Linoleic acid is known to oligomerize in the absence of any
catalyst via a thermal route [46]. Thus, a comparison of thermal
and catalytic oligomerisation of linoleic acid was performed in
this work.
3.2.1. Thermal oligomerisation of linoleic acid
Thermal oligomerisation of linoleic acid (Fig. 4) was carried
out at 280 8C under Ar (Table 3). The initial oligomerisation
rate, defined as a change in the molar total amount of the acids
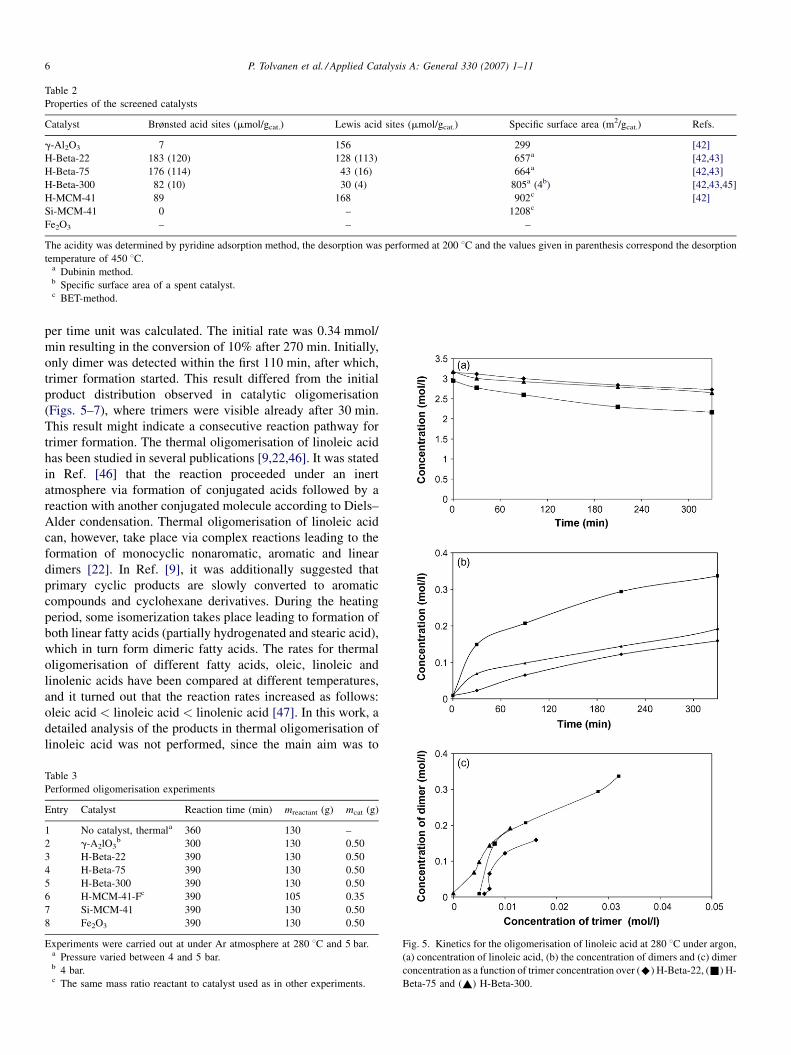
Table 2
Properties of the screened catalysts
Catalyst Brønsted acid sites (mmol/gcat.) Lewis acid sites (mmol/gcat.) Specific surface area (m2/gcat.) Refs.
g-Al2O3 7 156 299 [42]
H-Beta-22 183 (120) 128 (113) 657a [42,43]
H-Beta-75 176 (114) 43 (16) 664a [42,43]
H-Beta-300 82 (10) 30 (4) 805a (4b) [42,43,45]
H-MCM-41 89 168 902c [42]
Si-MCM-41 0 – 1208c
Fe2O3 – – –
The acidity was determined by pyridine adsorption method, the desorption was performed at 200 8C and the values given in parenthesis correspond the desorption
temperature of 450 8C.a Dubinin method.b Specific surface area of a spent catalyst.c BET-method.
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–116
per time unit was calculated. The initial rate was 0.34 mmol/
min resulting in the conversion of 10% after 270 min. Initially,
only dimer was detected within the first 110 min, after which,
trimer formation started. This result differed from the initial
product distribution observed in catalytic oligomerisation
(Figs. 5–7), where trimers were visible already after 30 min.
This result might indicate a consecutive reaction pathway for
trimer formation. The thermal oligomerisation of linoleic acid
has been studied in several publications [9,22,46]. It was stated
in Ref. [46] that the reaction proceeded under an inert
atmosphere via formation of conjugated acids followed by a
reaction with another conjugated molecule according to Diels–
Alder condensation. Thermal oligomerisation of linoleic acid
can, however, take place via complex reactions leading to the
formation of monocyclic nonaromatic, aromatic and linear
dimers [22]. In Ref. [9], it was additionally suggested that
primary cyclic products are slowly converted to aromatic
compounds and cyclohexane derivatives. During the heating
period, some isomerization takes place leading to formation of
both linear fatty acids (partially hydrogenated and stearic acid),
which in turn form dimeric fatty acids. The rates for thermal
oligomerisation of different fatty acids, oleic, linoleic and
linolenic acids have been compared at different temperatures,
and it turned out that the reaction rates increased as follows:
oleic acid < linoleic acid < linolenic acid [47]. In this work, a
detailed analysis of the products in thermal oligomerisation of
linoleic acid was not performed, since the main aim was to
Table 3
Performed oligomerisation experiments
Entry Catalyst Reaction time (min) mreactant (g) mcat (g)
1 No catalyst, thermala 360 130 –
2 g-A2lO3b 300 130 0.50
3 H-Beta-22 390 130 0.50
4 H-Beta-75 390 130 0.50
5 H-Beta-300 390 130 0.50
6 H-MCM-41-Fc 390 105 0.35
7 Si-MCM-41 390 130 0.50
8 Fe2O3 390 130 0.50
Experiments were carried out at under Ar atmosphere at 280 8C and 5 bar.a Pressure varied between 4 and 5 bar.b 4 bar.c The same mass ratio reactant to catalyst used as in other experiments.
Fig. 5. Kinetics for the oligomerisation of linoleic acid at 280 8C under argon,
(a) concentration of linoleic acid, (b) the concentration of dimers and (c) dimer
concentration as a function of trimer concentration over (^) H-Beta-22, (&) H-
Beta-75 and (~) H-Beta-300.
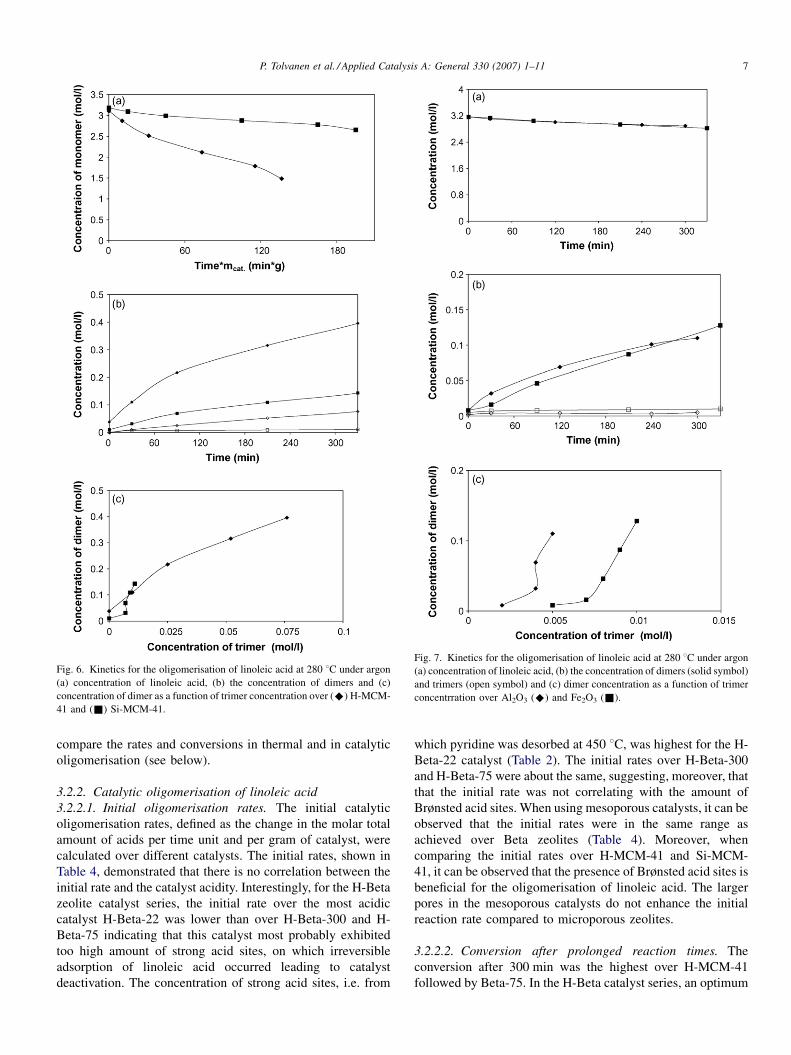
Fig. 6. Kinetics for the oligomerisation of linoleic acid at 280 8C under argon
(a) concentration of linoleic acid, (b) the concentration of dimers and (c)
concentration of dimer as a function of trimer concentration over (^) H-MCM-
41 and (&) Si-MCM-41.
Fig. 7. Kinetics for the oligomerisation of linoleic acid at 280 8C under argon
(a) concentration of linoleic acid, (b) the concentration of dimers (solid symbol)
and trimers (open symbol) and (c) dimer concentration as a function of trimer
concentrration over Al2O3 (^) and Fe2O3 (&).
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–11 7
compare the rates and conversions in thermal and in catalytic
oligomerisation (see below).
3.2.2. Catalytic oligomerisation of linoleic acid
3.2.2.1. Initial oligomerisation rates. The initial catalytic
oligomerisation rates, defined as the change in the molar total
amount of acids per time unit and per gram of catalyst, were
calculated over different catalysts. The initial rates, shown in
Table 4, demonstrated that there is no correlation between the
initial rate and the catalyst acidity. Interestingly, for the H-Beta
zeolite catalyst series, the initial rate over the most acidic
catalyst H-Beta-22 was lower than over H-Beta-300 and H-
Beta-75 indicating that this catalyst most probably exhibited
too high amount of strong acid sites, on which irreversible
adsorption of linoleic acid occurred leading to catalyst
deactivation. The concentration of strong acid sites, i.e. from
which pyridine was desorbed at 450 8C, was highest for the H-
Beta-22 catalyst (Table 2). The initial rates over H-Beta-300
and H-Beta-75 were about the same, suggesting, moreover, that
that the initial rate was not correlating with the amount of
Brønsted acid sites. When using mesoporous catalysts, it can be
observed that the initial rates were in the same range as
achieved over Beta zeolites (Table 4). Moreover, when
comparing the initial rates over H-MCM-41 and Si-MCM-
41, it can be observed that the presence of Brønsted acid sites is
beneficial for the oligomerisation of linoleic acid. The larger
pores in the mesoporous catalysts do not enhance the initial
reaction rate compared to microporous zeolites.
3.2.2.2. Conversion after prolonged reaction times. The
conversion after 300 min was the highest over H-MCM-41
followed by Beta-75. In the H-Beta catalyst series, an optimum
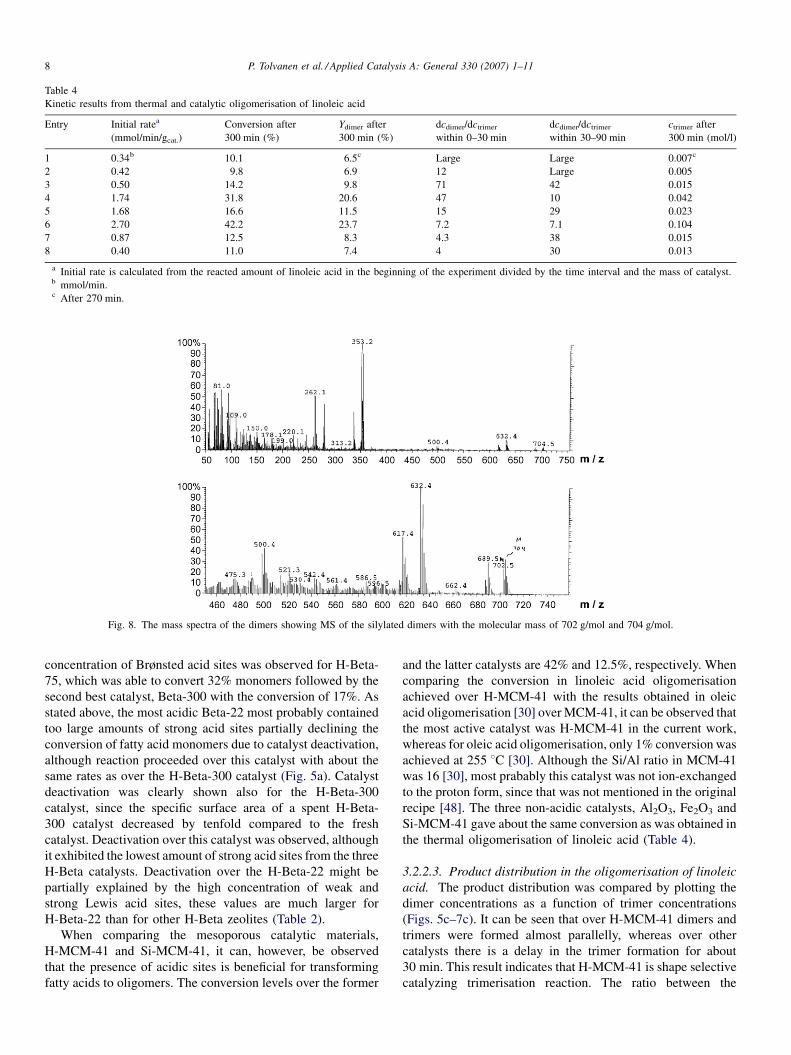
Table 4
Kinetic results from thermal and catalytic oligomerisation of linoleic acid
Entry Initial ratea
(mmol/min/gcat.)
Conversion after
300 min (%)
Ydimer after
300 min (%)
dcdimer/dctrimer
within 0–30 min
dcdimer/dctrimer
within 30–90 min
ctrimer after
300 min (mol/l)
1 0.34b 10.1 6.5c Large Large 0.007c
2 0.42 9.8 6.9 12 Large 0.005
3 0.50 14.2 9.8 71 42 0.015
4 1.74 31.8 20.6 47 10 0.042
5 1.68 16.6 11.5 15 29 0.023
6 2.70 42.2 23.7 7.2 7.1 0.104
7 0.87 12.5 8.3 4.3 38 0.015
8 0.40 11.0 7.4 4 30 0.013
a Initial rate is calculated from the reacted amount of linoleic acid in the beginning of the experiment divided by the time interval and the mass of catalyst.b mmol/min.c After 270 min.
Fig. 8. The mass spectra of the dimers showing MS of the silylated dimers with the molecular mass of 702 g/mol and 704 g/mol.
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–118
concentration of Brønsted acid sites was observed for H-Beta-
75, which was able to convert 32% monomers followed by the
second best catalyst, Beta-300 with the conversion of 17%. As
stated above, the most acidic Beta-22 most probably contained
too large amounts of strong acid sites partially declining the
conversion of fatty acid monomers due to catalyst deactivation,
although reaction proceeded over this catalyst with about the
same rates as over the H-Beta-300 catalyst (Fig. 5a). Catalyst
deactivation was clearly shown also for the H-Beta-300
catalyst, since the specific surface area of a spent H-Beta-
300 catalyst decreased by tenfold compared to the fresh
catalyst. Deactivation over this catalyst was observed, although
it exhibited the lowest amount of strong acid sites from the three
H-Beta catalysts. Deactivation over the H-Beta-22 might be
partially explained by the high concentration of weak and
strong Lewis acid sites, these values are much larger for
H-Beta-22 than for other H-Beta zeolites (Table 2).
When comparing the mesoporous catalytic materials,
H-MCM-41 and Si-MCM-41, it can, however, be observed
that the presence of acidic sites is beneficial for transforming
fatty acids to oligomers. The conversion levels over the former
and the latter catalysts are 42% and 12.5%, respectively. When
comparing the conversion in linoleic acid oligomerisation
achieved over H-MCM-41 with the results obtained in oleic
acid oligomerisation [30] over MCM-41, it can be observed that
the most active catalyst was H-MCM-41 in the current work,
whereas for oleic acid oligomerisation, only 1% conversion was
achieved at 255 8C [30]. Although the Si/Al ratio in MCM-41
was 16 [30], most prabably this catalyst was not ion-exchanged
to the proton form, since that was not mentioned in the original
recipe [48]. The three non-acidic catalysts, Al2O3, Fe2O3 and
Si-MCM-41 gave about the same conversion as was obtained in
the thermal oligomerisation of linoleic acid (Table 4).
3.2.2.3. Product distribution in the oligomerisation of linoleic
acid. The product distribution was compared by plotting the
dimer concentrations as a function of trimer concentrations
(Figs. 5c–7c). It can be seen that over H-MCM-41 dimers and
trimers were formed almost parallelly, whereas over other
catalysts there is a delay in the trimer formation for about
30 min. This result indicates that H-MCM-41 is shape selective
catalyzing trimerisation reaction. The ratio between the
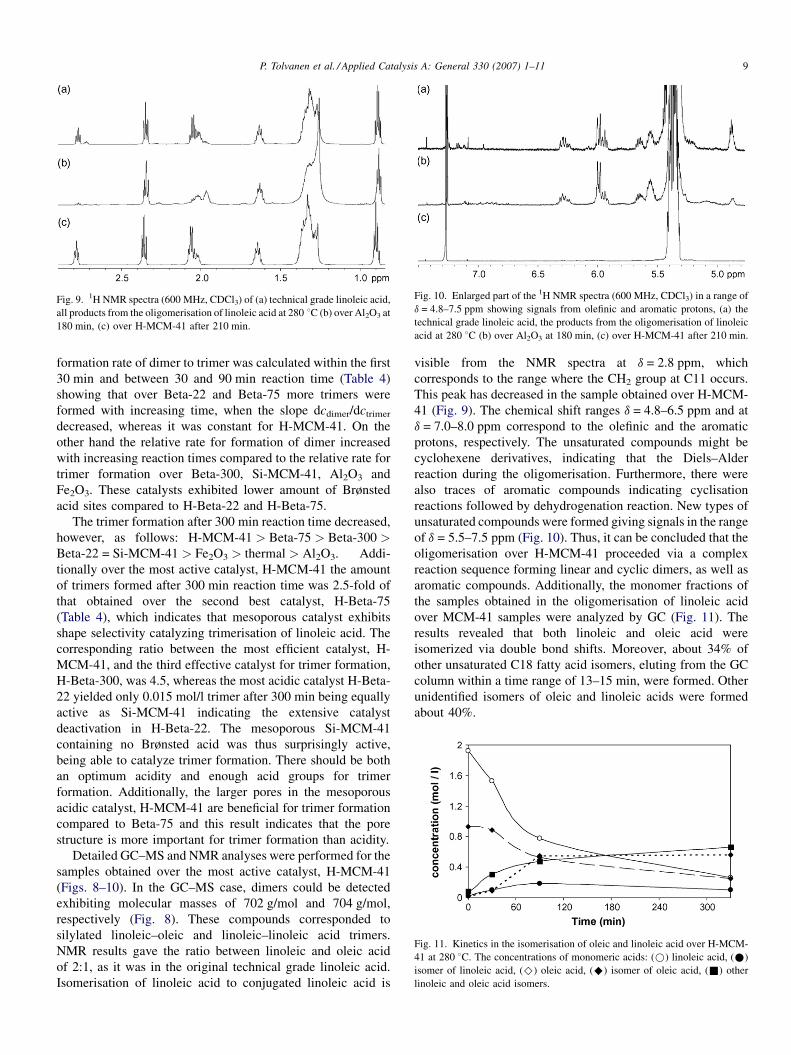
Fig. 9. 1H NMR spectra (600 MHz, CDCl3) of (a) technical grade linoleic acid,
all products from the oligomerisation of linoleic acid at 280 8C (b) over Al2O3 at
180 min, (c) over H-MCM-41 after 210 min.
Fig. 10. Enlarged part of the 1H NMR spectra (600 MHz, CDCl3) in a range of
d = 4.8–7.5 ppm showing signals from olefinic and aromatic protons, (a) the
technical grade linoleic acid, the products from the oligomerisation of linoleic
acid at 280 8C (b) over Al2O3 at 180 min, (c) over H-MCM-41 after 210 min.
Fig. 11. Kinetics in the isomerisation of oleic and linoleic acid over H-MCM-
41 at 280 8C. The concentrations of monomeric acids: (*) linoleic acid, (*)
isomer of linoleic acid, (^) oleic acid, (^) isomer of oleic acid, (&) other
linoleic and oleic acid isomers.
P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–11 9
formation rate of dimer to trimer was calculated within the first
30 min and between 30 and 90 min reaction time (Table 4)
showing that over Beta-22 and Beta-75 more trimers were
formed with increasing time, when the slope dcdimer/dctrimer
decreased, whereas it was constant for H-MCM-41. On the
other hand the relative rate for formation of dimer increased
with increasing reaction times compared to the relative rate for
trimer formation over Beta-300, Si-MCM-41, Al2O3 and
Fe2O3. These catalysts exhibited lower amount of Brønsted
acid sites compared to H-Beta-22 and H-Beta-75.
The trimer formation after 300 min reaction time decreased,
however, as follows: H-MCM-41 > Beta-75 > Beta-300 >Beta-22 = Si-MCM-41 > Fe2O3 > thermal > Al2O3. Addi-
tionally over the most active catalyst, H-MCM-41 the amount
of trimers formed after 300 min reaction time was 2.5-fold of
that obtained over the second best catalyst, H-Beta-75
(Table 4), which indicates that mesoporous catalyst exhibits
shape selectivity catalyzing trimerisation of linoleic acid. The
corresponding ratio between the most efficient catalyst, H-
MCM-41, and the third effective catalyst for trimer formation,
H-Beta-300, was 4.5, whereas the most acidic catalyst H-Beta-
22 yielded only 0.015 mol/l trimer after 300 min being equally
active as Si-MCM-41 indicating the extensive catalyst
deactivation in H-Beta-22. The mesoporous Si-MCM-41
containing no Brønsted acid was thus surprisingly active,
being able to catalyze trimer formation. There should be both
an optimum acidity and enough acid groups for trimer
formation. Additionally, the larger pores in the mesoporous
acidic catalyst, H-MCM-41 are beneficial for trimer formation
compared to Beta-75 and this result indicates that the pore
structure is more important for trimer formation than acidity.
Detailed GC–MS and NMR analyses were performed for the
samples obtained over the most active catalyst, H-MCM-41
(Figs. 8–10). In the GC–MS case, dimers could be detected
exhibiting molecular masses of 702 g/mol and 704 g/mol,
respectively (Fig. 8). These compounds corresponded to
silylated linoleic–oleic and linoleic–linoleic acid trimers.
NMR results gave the ratio between linoleic and oleic acid
of 2:1, as it was in the original technical grade linoleic acid.
Isomerisation of linoleic acid to conjugated linoleic acid is
visible from the NMR spectra at d = 2.8 ppm, which
corresponds to the range where the CH2 group at C11 occurs.
This peak has decreased in the sample obtained over H-MCM-
41 (Fig. 9). The chemical shift ranges d = 4.8–6.5 ppm and at
d = 7.0–8.0 ppm correspond to the olefinic and the aromatic
protons, respectively. The unsaturated compounds might be
cyclohexene derivatives, indicating that the Diels–Alder
reaction during the oligomerisation. Furthermore, there were
also traces of aromatic compounds indicating cyclisation
reactions followed by dehydrogenation reaction. New types of
unsaturated compounds were formed giving signals in the range
of d = 5.5–7.5 ppm (Fig. 10). Thus, it can be concluded that the
oligomerisation over H-MCM-41 proceeded via a complex
reaction sequence forming linear and cyclic dimers, as well as
aromatic compounds. Additionally, the monomer fractions of
the samples obtained in the oligomerisation of linoleic acid
over MCM-41 samples were analyzed by GC (Fig. 11). The
results revealed that both linoleic and oleic acid were
isomerized via double bond shifts. Moreover, about 34% of
other unsaturated C18 fatty acid isomers, eluting from the GC
column within a time range of 13–15 min, were formed. Other
unidentified isomers of oleic and linoleic acids were formed
about 40%.

P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–1110
Iron oxide was able to catalyze trimer formation. The
concentration of trimers at 330 min reaction time was, however,
only 0.01 mol/l over this material. A part of Fe2O3 dissolved in
the reaction mixture, since the colour of the mixture was red
after centrifugation. These results are not surprising, since it is
known from litterature [49] that dissolved homogeneous iron
species together with potassium iodide can catalyze oligomer-
isation of fatty acids. Moreover, montmorillonite catalyzing
oligomerisation of fatty acids [25], contains Fe2O3.
Aluminium oxide was not a very efficient catalyst in the
oligomerisation of linoleic acid. This result is in accordance
with literature [30], according to which only below 1% dimers
of oleic acid was formed at 355 8C over g-Al2O3. The viscosity
of the sample obtained from the oligomerisation of linoleic acid
over Al2O3 after 180 min reaction time was, however, very
high. Due to the high viscosity of the samples further
investigations with NMR spectroscopy were carried out with
two samples, obtained over H-MCM-41 after 210 min and from
Al2O3 after 180 min. This further investigation with NMR
spectroscopy was performed, since the samples analyzed with
SEC were filtered prior to the analysis (see Experimental),
while in the NMR studies, the samples were not treated prior to
the analysis. The NMR spectra of linoleic acid along with
samples treated with Al2O3 and H-MCM-41 are displayed in
Fig. 10. It is clearly visible from the NMR spectra that there are
no organic higher polymers formed over Al2O3. The high
viscosity in the sample obtained from Al2O3 is thus not due to
formation of organic higher polymers, indicating that the SEC-
analyses of these samples are correct. The high viscosity could
thus originate, most probably, from dissolved aluminium ions,
which can interact with organic molecules. The dissolved
aluminium species were confirmed to be present in the
centrifuged reaction mixture by laser ablation ICP–MS
technique in the oligomerisation of linoleic acid with Al2O3.
4. Conclusions
Thermal and catalytic oligomerisation kinetics of linoleic acid
was studied in a batch reactor at 280 8C under inert atmosphere.
The products were analyzed by size exclusion chromatography,
NMR and GC–MS techniques. In thermal oligomerisation, the
conversion level was about 25% of the conversion level obtained
over the most efficient catalyst, H-MCM-41 within 300 min
reaction time. In catalytic oligomerisation three different H-Beta
zeolites with varying acidities, as well as mesoporous acidic and
nonacidic materials, namely H-MCM-41 and Si-MCM-41, were
studied. Additionally, the performances of these catalysts were
compared with the performance of g-Al2O3 and Fe2O3. The
initial oligomerisation rates did not correlate either with the
catalyst acidity or with the pore sizes of the catalysts. The highest
initial rate over H-Beta zeolites was achieved over H-Beta-75
exhibiting an optimum amount of acidic sites.
Acknowledgements
This work is part of activities at the Abo Akademi Process
Chemistry Centre of Excellence Programmes (2000–2011)
financed by the Academy of Finland. The authors acknowledge
Mr. Teemu Heikkila at the University of Turku, Department of
Physics, who performed XRD-analysis and Dr Paul Ek at Abo
Akademi University, who carried out ICP–MS analysis.
Appendix A
Since the response for the dimer fraction was not linear, it
should be corrected by using a correction factor with soya oil as
external standard. The response for soybean oil should be the
same as for the dimer. The correction factor could be related to
the ratio between the area of the dimer peak to the area of the
monomer peak (Table 1) as follows. In order to calculate cf the
real concentration (creal) was divided with the concentration
achieved by SEC (cSEC) and multiplied by a new correction
factor cf2, which explained the difference between the different
samples having different initial concentrations:
cf ¼creal
cSEC
� cf2; cf2 ¼csample;x
1:26(1)
The value 1.26 in Eq. (1) is the total concentration in the
selected reference sample (mg/ml), which was related to
the other samples. This correction factor cf2 describes the
difference between the reference sample and the sample. This
difference is inevitable, since there are no direct ways to weigh
very accurately these oils, the precision being one droplets
accuracy. After multiplying the correction factor with the peak
areas of the dimer and trimer, respectively, the values were
converted to molar concentrations (mol/l). The final correction
curve is given in Fig. 3a via applying exponential regression to
the following equation:
cf dim ¼ 3:3908
�Adimer
Atotal
��0:3329
(2)
Additionally, some simplifications were applied due to
significant challenges with SEC-analysis. Instead of treating all
the compounds of the system separately, only monomers,
dimers and trimers were analysed separately and the mass
balance was calculated as follows:
mmonomer;0 ¼ mmonomer þ mdimer þ mtrimer (3)
where m denotes the mass. Taking into account stoichiometry
the molar amounts (n) are
nmonomer;0 ¼ nmonomer þ 2ndimer þ 3ntrimer (4)
References
[1] 6th ed., Ullmann Ullmann’s Encyclopedia of Industrial Chemistry, vol.
13, Wiley–VCH Verlag GmbH & Co., Weinheim, 2003.
[2] R.W. Johnson, E. Fritz (Eds.), Fatty Acids in Industry, Marcel Dekker,
New York, 1988, p. 667.
[3] L. Kuprina, Gidroliznaya i Lesokhimicheskaya Promyshlennost 3 (1983)
23.
[4] P. Patrick, U.S. Patent 3,157,629 (1964).
[5] B. Cicel, P. Komadel, M. Nigrin, Coll. Czech. Chem. Commun. 57 (1992)
1666.
[6] W. Schwarz, J. Zajic, J. Brat, Fett Wiss. Technol. 95 (1993) 253.

P. Tolvanen et al. / Applied Catalysis A: General 330 (2007) 1–11 11
[7] H. Mohring, G. Spiteller, Fett Wiss. Technol. 92 (1990) 126.
[8] W. Link, G. Spiteller, Fett Wiss. Technol. 92 (1990) 135.
[9] W. Link, G. Spiteller, Fett Wiss. Technol. 94 (1992) 9.
[10] H. Mohring, G. Spiteller, Fett Wiss. Technol. 94 (1992) 41.
[11] H. Mohring, G. Spiteller, Fett Wiss. Technol. 94 (1992) 241.
[12] R. Adelhardt, G. Spiteller, Fett Wiss. Technol. 95 (1993) 85.
[13] R. Brutting, G. Spiteller, Fett Wiss. Technol. 95 (1993) 193.
[14] R. Brutting, G. Spiteller, Fett Wiss. Technol. 96 (1994) 361.
[15] R. Brutting, G. Spiteller, Fett Wiss. Technol. 96 (1994) 445.
[16] J. Brat, W. Schwarz, J. Zajfc, Fett Wiss. Technol. 97 (Sonderausgabe 2)
(1995) 513.
[17] M.J.A.M. Den Otter, Fette Seifen Anstrichmittel 72 (1970) 667.
[18] M.J.A.M. Otter, Fette Seifen Anstrichmittel 72 (1970) 875.
[19] M.J.A.M. Den Otter, Fette Seifen Anstrichmittel 72 (1970) 1056.
[20] H.W.G. Heynen, W.H.M. van Opstal, M.J.A.M. den Otter, Fette Seifen
Anstrichmittel 74 (1972) 677.
[21] A.A. Bagaev, G.I. Tsarev, A.I. Kirianov, Izv. Vyss. Uchebn. Zaven, Lesn.
Z. 3 (1979) 78.
[22] A.K. Sen Gupta, H. Scharmann, Fette Seifen Anstrichmittel 70 (1968) 86.
[23] A.K. Sen Gupta, H. Scharmann, Fette Seifen Anstrichmittel 70 (1968)
153.
[24] D.H. Wheeler, J. White, J. Am. Oil’s Chem. Soc. 44 (1967) 298.
[25] D.H. Wheeler, A. Milun, F. Linn, J. Am. Oil Chem. Soc. 47 (1970) 242.
[26] R.A. Korus, T.L. Mousetis, J. Am. Oil Chem. Soc. 61 (1984) 537.
[27] D.H. McMahon, J. Am. Oil Chem. Soc. 51 (1974) 522.
[28] P. Vuzvusova, Plovdivski Universitet Paisii Khilendarski 26 (1989) 71.
[29] K. Ishihara, JP patent 2001278837 (2001).
[30] R.M. Koster, M. Bogert, B. Leeuw, J. Mol. Catal. A: Chem. 134 (1998)
159.
[31] A.F. Elsasser, L.A. McCargar, US Patent 6,187,903 (2001).
[32] J.K. Chowdhury, A.C. Chakraborty, A.J. Majumber, J. Indian Chem. Soc.
12 (1935) 441.
[33] B.S. Zmachimskii, A.N. Trofimov, E. Ryabova, A.M. Chashchin, Gidro-
liznaya i Lesokhimicheskaya Promyshlennost 1 (1991) 17.
[34] H. Stage, Fette Seifen Anstrichmittel 77 (1975) 174.
[35] H. Stage, Fette Seifen Anstrichmittel 75 (1973) 160.
[36] H. Stage, Fette Seifen Anstrichmittel 80 (1978) 257.
[37] H.G. Arlt Jr., US Patent 3,367,952 (1968).
[38] N. Laane, M. Colja, G.A. Graham, C. Beindorff, K.D. Haase, J. Klooster-
man, Rec. Trav. Chim. Pays-Bas 110 (1991) 195.
[39] K.D. Haase, A.J. Heynen, N.C.M. Laane, Proc. Jpn. Oil Chem. Soc. 1
(1988) 510.
[40] C.T. Kresge, M.E. Leonowicz, W.J. Roth, J.C. Vartuli, US Patent
5,098,684 (1992).
[41] A. Bernas, P. Laukkanen, N. Kumar, P. Maki-Arvela, J. Vayrynen,
E. Laine, B. Holmbom, T. Salmi, D.Yu. Murzin, J. Catal. 210 (2002)
354.
[42] P. Maki-Arvela, N. Kumar, V. Nieminen, R. Sjoholm, T. Salmi, D.Yu.
Murzin, J. Catal. 225 (2004) 155.
[43] D. Kubicka, N. Kumar, P. Maki-Arvela, M. Tiitta, V. Niemi, T. Salmi,
D.Yu. Murzin, J. Catal. 222 (2004) 65.
[44] C.A. Emeis, J. Catal. 141 (1993) 347.
[45] E.M. Sulman, V.V. Alferov, Yu.Yu. Kosivtsov, A.I. Sidorov, O.S. Minis-
kov, A.E. Afanasiev, H. Kumar, D. Kubicka, J. Agullo, T. Salmi, D. Yu.
Murzin, Development of method of low temperature peat pyrolysis on the
pyrolysis on the basis of aluminosilicate catalytic system, Chem. Eng. J.,
in press.
[46] B. de Surville, Compt. Rend. 235 (1952) 724.
[47] D.A. Afonin, A.A. Bagaev, G.I. Tsarev, A.I. Kiprianov, Izv. Vyss. Uchebn.
Zaven, Lesn. Z. 4 (1984) 89.
[48] J.S. Beck, J.C. Vartuli, W.J. Roth, M.E. Leonowicz, C.T. Kresge, K.D.
Schmitt, C.T.-W. Chu, D.H. Olson, E.W. Sheppard, S.B. McCullen, J.B.
Higgins, J.L.J. Schlenker, J. Am. Chem. Soc. 114 (1992) 10834.
[49] K. Matsuo, S. Kawamura, T. Toshine, US Patent 4,271,066 (1981).





























