Embed Size (px)
Citation preview

Chemical Physics 410 (2013) 66–70
Contents lists available at SciVerse ScienceDirect
Chemical Physics
journal homepage: www.elsevier .com/locate /chemphys
Theoretical study of the cooperativity in substituted dimethyl etherscomplexed with two water molecules. Red or blue shifts of the m(CH) vibrations?
Asit K. Chandra a, Thérèse Zeegers-Huyskens b,⇑a Department of Chemistry, North Eastern Hill University, Shillong 793022, Indiab Department of Chemistry, University of Leuven, 200F Celestijnenlaan, 3001 Heverlee, Belgium
a r t i c l e i n f o a b s t r a c t
Article history:Received 26 September 2012In final form 31 October 2012Available online 14 November 2012
Keywords:EthersInteraction with waterCooperativityCharge transfer
0301-0104/$ - see front matter � 2012 Elsevier B.V. Ahttp://dx.doi.org/10.1016/j.chemphys.2012.10.019
⇑ Corresponding author.E-mail address: [email protected]
The cooperative interactions in CH3OCH3, CH3OCH2F, CH3OCHF2, CH2FOCHF2 and CHF2OCHF2 complexedwith two H2O molecules are investigated using the B3LYP method with the 6-311++G(d,p) level. Thecalculations include the optimized geometries, the cooperative energies along with a natural bond orbital(NBO analysis). Cyclic complexes characterized by OwHw. . .O and CH. . .Ow hydrogen bonds are formed.The pairwise or two body interaction energies are computed. The results show that the OwHw. . .O inter-action energies increase with the proton affinity of the O atom of the ethers and that the CH. . .Ow inter-action energies increase with increasing acidity of the CH bond. This is in agreement with theintermolecular distances. The cooperative energy represents 15–20% of the total energy. When nF =2–4, blue shifts are predicted for the m(CH) vibration for the 1–1 complexes but red shifts are predictedfor the 1–2 complexes. The variation of the CH distances is explained by a competition between thevariation of the intramolecular hyperconjugation energies and the intermolecular hyperconjugationenergies. The charge transfer between the ether and water molecules is analyzed. When the negativecharge on one H2O molecule decreases, the positive charge on the other H2O molecule increases, indicat-ing a nice reciprocal effect.
� 2012 Elsevier B.V. All rights reserved.
1. Introduction
Cooperative interactions involving several molecules is animportant component of intermolecular interactions, particularlythose involving hydrogen bonds. Complexes involving two mole-cules are observed in diluted solution, in gases at low pressure orin low-temperature inert gases matrices. In concentrated solution,the interaction takes place between several molecules and theimportance of cooperativity has been recognized since a long time[1–10]. Quantitative aspects of cooperativity have been discussedin recent works [10–16]. Cooperativity can be positive or negative.Positive cooperativity occurs in hydrogen bonded trimers present-ing a cyclic pattern where each molecule is simultaneously donorand acceptor of hydrogen bond. A negative cooperativity is pre-dicted when two bonds of the same molecule act simultaneouslyas proton donor or proton acceptor.
In the present work, the complexes between dimethylether(CH3OCH3) and some of its fluorinated derivatives (CH3OCHF2,CH3 OCHF2, CH2FOCHF2, CHF2OCHF2) and two water moleculesare investigated by theoretical methods. These molecules werechosen in order to cover a broad range of acidity/basicity of the
ll rights reserved.
e (T. Zeegers-Huyskens).
interacting sites. The vibrational spectrum of the dimethyl ether-water complex has been investigated in low-temperature argonmatrices [17]. To the best of our knowledge, no experimental datahave been reported for the other systems.
The present work is arranged as follows. In the first part, thestructure of the complexes and the cooperative energies arediscussed. The second part deals with a NBO analysis, more specif-ically the effect of intra- and intermolecular hyperconjugations onbond lengths will be outlined. The effect of cooperativity on rele-vant vibrational frequencies will be discussed as well.
2. Computational methodology
Calculations of the properties of CH3OCH3, CH3OCH2F, CH3
OCHF2, CH2FOCHF2 and CHF2OCHF2 complexed with 2 water mol-ecules were carried out using the density functional B3LYP method[18] and the Gaussian suite of programs [19]. The basis set6-311++G(d,p) was invoked. For these systems, the B3LYP methodwas shown to give reliable interaction energies [16]. The computedinteraction energies were corrected for the basis set superpositionerrors [20].
The cooperativity in a molecular trimer containing A, B and Cmolecules is given by the three-body term DEcoop which can be de-fined as the difference between the total interaction energy E(ABC)

A.K. Chandra, T. Zeegers-Huyskens / Chemical Physics 410 (2013) 66–70 67
and the sum of the pairwise or two-body interaction energiesE2(AB), E2(BC), E2(AC)
DEcoop ¼ EðABCÞ � E2ðABÞ � E2ðBCÞ � E2ðACÞ: ð1Þ
Here, the E2 values correspond to the two-body contributions atthe trimer geometry calculated with the same basis set [21–22].
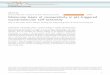
Fig. 1. B3LYP/6-311++G(d,p) optimized structures of CH3OCH3, CH3OCH2F, CH3-OCHF2, CH2FOCHF2 and CHF2OCHF2 complexed by one and two water molecules.
The harmonic vibrational frequencies were calculated to charac-terize the stationary points. No scaling factor was used. Theelectronic charges on individual atoms, orbital occupancies, hybrid-ization and second-order intra- or intermolecular hyperconjugationenergies were obtained by a NBO analysis [23].
3. Results and discussion
3.1. Structure of the complexes and cooperative energies
Fig. 1 shows the structure of the complexes between the investi-gated ethers and two water molecules. Let us notice that the ethersderivatives can adopt different conformations [24] and the most sta-ble conformer was chosen for our study. The structure of the 1–1complexes is shown for the comparison. Note than the structure ofthe 1–1 complexes has been calculated in an earlier work at theMP2/6-311++G(d,p) level [24]. At this level, the intermolecular dis-tances are almost the same as in the B3LYP/6-311++G(d,p) level ex-cept for the complex CHF2OCHF2 .1H2O giving shorter OAHA. . .O1
distance (2.549 Å).These optimized structures show that the 1–1 complexes of
CH3OCH3 and CH3OCH2F are stabilized by only the OAHA. . .O1hydrogen bonds but that the other 1–1 complexes are cyclic andcharacterized by OAHA. . .O1 and C3H7. . .OB hydrogen bonds. Forthe other fluorinated ethers, both the 1–1 and 1–2 complexes arecyclic. The intermolecular angles OAHA. . .O1 and C3H7. . .OB anglesare also markedly larger in the 1–2 than in the 1–1 complexes.
The total interaction energy, E(ABC) and the pairwise ortwo-body interaction energies, E2(AB) , E2(AC), E2(BC) are listed inTable 1. A, B and C refer to the ethers, the (H2O)A and (H2O)B mole-cules, respectively. The cooperative energies DEcoop calculated byEq. (1) are also indicated in Table 1. This Table also reports the protonaffinity (PA) of the O1 atom along with the deprotonation enthalpy(DPE) of the C3H7 bond taken from a previous work [24].
These results indicate that the interaction energies E2(AB) rang-ing from �4.92 and �2.06 kcal mol�1are ordered according the PAof the O1 atom which decreases from 189.1 to 154.2 kcal mol�1. Thisis also in line with the increasing OAHA. . .O1 distance from 1.810 to2.139 Å. It should be mentioned here that for the 1–1complexes, nocorrelation was found between the interaction energies and the PAof the O1 atom. This was explained by large variations of theOH. . .O1 angles and for this reason, the concept of ‘‘effective’’ PAwas introduced [24]. This is in line with the present results indicat-ing that in the 1–2 complexes, the OAHA. . .O1 angles are much largerand nearly constant (Fig. 1). Further, the E2(AC) values increase from�1.07 to�3.15 kcal mol�1 when the DPE of the C3H7 bond decreases.This is also in agreement with the shortening of the H7. . .OB dis-tances from 2.466 to 2.093 Å on going from the CH3 OCH3.2H2O sys-tem to the CHF2OCHF2.2H2O one. At last, the interaction energybetween the two water molecules remains almost constant(�4.60 kcal mol�1), in agreement with the nearly constant valuesof the intermolecular distances. The same conclusion can be drawnfor the interaction between fluorinated acetone derivatives and twowater molecules where the interaction energy between the twowater molecules remains almost constant (�4.50 kcal mol�1) [16].Let us notice that for the present systems, the cooperative energyvaries within small limits, between �1.77 and �2.25 kcal mol�1
and represents 15–20% of the total energy.
3.2. Properties of the C3H7 bonds. Distances and NBO analysis
The next step in our study is to discuss the properties of theC3H7 bond. Table 2 lists the C3H7 distances in the isolated mole-cules and the 1–1 and 1–2 water complexes. These results indicatethat the interaction with water results in a contraction of the C3H7

Table 1Binding energies at the B3LYP/6-311++G(d,p) level at the trimer geometry. EHB includes BSSE correction. Proton affinities of the ethers and DPE of the C3H7 bond [24] (inkcal mol�1).
system E(ABC) E2(AB) E2(AC) E2(BC) DECoop PA (O1) DPE (C3H7)
CH3OCH3.2H2O �12.69 �4.92 �1.07 �4.60 �2.10 189.1 410.5CH3OCH2F.2H2O �11.74 �3.82 �1.35 �4.63 �1.94 177.7 401.0CH3OCHF2.2H2O �11.51 �2.73 �1.90 �4.64 �2.24 169.1 385.1CH2FOCHF2.2H2O �11.26 �2.34 �2.01 �4.66 �2.25 159.2 375.7CHF2OCHF2.2H2O �11.63 �2.06 �3.15 �4.65 �1.77 154.2 366.7
#without ZPE correction.DECoop = EHB (trimer)�EHB(AB)�EHB(AC)�EHB(BC) [including BSSE correction but without ZPE correction].
68 A.K. Chandra, T. Zeegers-Huyskens / Chemical Physics 410 (2013) 66–70
bond and a blue shift of the m(C3H7) vibration in the 1–1 complexesand in the 1–2 complexes (nF = 0,1). However, when nF = 2–4, theinteraction with two water molecules interestingly results in anelongation of the C3H7 bond by 0.7 to 2.1 Å. Red shifts of them(C3H7) vibrations which are decoupled from the other m(CH)modes are predicted when nF = 3–4. The changes of the C3H7 bondlengths and the variation of the m(C3H7), vibrations are linearly cor-related [25]. Although IR intensities predicted by the B3LYP meth-od are not always reliable, blue shifts parallel a decrease of the IRintensities and red shifts an increase of these intensities. It must benoticed that blue shifts of the other m(CH) vibrations were pre-dicted for CH3OCH3 complexed with water [24,26]. As discussedabove, red shifts of the m(C3H7) vibrations are predicted for the1–2 complexes when nF = 2–4. Interestingly, the other m(CH) vibra-tional modes are also shifted. In the case of the CHF2OCHF2 2H2Osystem, the m(C2H4) vibration is blue-shifted by 11 cm�1 (from3138 to 3149 cm�1) and in the case of the CH2FOCHF2 2H2Osystem, the two m(C2H4H5) modes are both blue-shifted by5 cm�1 (from 3084 and 3156 cm�1 to 3089 and 3161 cm�1).
Blue shifts have been discussed in numerous works (see forexample Refs. [27–37]. Three factors, intra-and intermolecularhyperconjugation and rehybridization determine commonly red-or blue shifts of the formed hydrogen bonds [37]. We will try toanalyze these parameters for the present systems. For this purpose,we have considered the intermolecular (E2
inter) and intramolecular(E2
intra) hyperconjugation energies indicated in Table 2. Let us noticethat the third column of Table 2 refer to all the intramolecularshifts to the r⁄C3H7) orbitals. There are other large intramolecularshifts such as the LPF ? r⁄(O1C), LPO1 ? r⁄(CF) and LPF orLPO1 ? RY⁄C delocalizations in the isolated molecules as well in
Table 2Properties of the C3H7 bond: r(C3H7) distances (Å), E2
intra and E2inter hyperconju
intensiites (km lmol�1).
System r(C3H7)aE2
intra (DE2intra)
CH3OCH3 1.0903 3.28CH3OCH3 1H2O 1.0897(�0.6) 2.96(�0.32)CH3OCH3 2H2O 1.0896(�0.7) 2.60(�0.68)CH3OCH2F 1.0896 9.45CH3OCH2F 1H2O 1.0892(�0.4) 9.28(�0.17)CH3OCH2F 2H2O 1.0887(�0.9) 7.61(�1.84)CH3OCHF2 1.0883 16.99CH3OCHF2 1H2O 1.0875(�0.8) 16.20(�0.79)CH3OCHF2 2H2O 1.0890(+0.7) 13.53(�3.46)CH2FOCHF2 1.0877 17.23CH2FOCHF2 1H2O 1.0865(�1.2) 15.93(�1.30)CH2FOCHF2 2H2O 1.0888(+1.1) 13.70(�3.53)CHF2OCHF2 1.0870 17.17CHF2OCHF2 1H2O 1.0857(�1.3) 15.52(�1.65)CHF2OCHF2 2H2O 1.0891(+2.1) 13.51(�3.66)
a in parentheses, variation of the C3H7 bond length (mÅ) and of E2intra(kcal m
b in parentheses, IR intensities (km mol�1).c in parentheses, change of the m(C3H7) frequencies (cm�1) and variation of
the complexes. These charge transfers will no more be consideredhereafter. The E2
inter values refer to the LPOA ? r⁄(C3H7) delocaliza-tion in the 1–1 complexes and to the LPOB ? r⁄(C3H7) delocaliza-tion in the 1, 2 complexes. In a previous work [24], we haveconsidered for the 1–1 complexes only the intermolecular hyper-conjugation taking place from the LPs of the O1 atom to the r⁄
(C3H7) orbital. In order to obtain a more complete picture of thefactors determining the C3H7 bond length, we have consideredfor the present systems, not only this classical lone pair effectbut also other hyperconjugations to the r⁄(C3H7) orbital such asthe r(C1O2) ? r⁄(C3H7) hyperconjugation and in the fluorinatedderivatives, the RLPF ? r⁄(C3H7) hyperconjugation which alsoinfluence the C3H7 distances. For the CH2FOCHF2 2H2O complexas for example, the LPO1 ? r⁄C3H7, the r(C1O2) ? r⁄(C3H7) andRLPF ? r⁄(C3H7) hyperconjugation energies decrease by 0.87,0.21 and 2.45 kcal mol�1 with respect to the isolated molecule. Itis important to notice here that the third column of Table 2 referto ALL the intramolecular shifts to the r⁄(C3H7) orbitals. Theimportance of these delocalized interactions in cyclic ethers andother systems has been emphasized by Alabugin et al. [38,39].More details are given in S.I.1. Let us also notice that the r(C3H7)and r⁄ (C3H7) occupancies listed in S.I.2 are about the same inthe isolated molecules and in their complexes and do not markedlyinfluence the C3H7 bond length. The same remark also holds for thevariation of the hybridization of the C3(H7) atom; the %s-characterin the 1–2 systems increases by 1.3 to 2.2 %, independently of theelongation or the contraction of the bond.
A decrease of the E2intra hyperconjugation energy results in a
contraction of the C3H7 bond and an intermolecular charge transferto the r⁄(C3H7) orbital results in an increase of the C3H7 bond
gation energies(kcal mol�1),m(C3H7) vibrational frequencies (cm�1) and IR
E2inter
%sC3(H7) m (C3H7)
– 25.66 3112(33)b
– 25.82(+0.16) 3125(+13,�20)c
1.58 26.98(+1.32) 3132(+20,�32)27.39 3124 (30)
– 27.58(+0.19) 3134(+10,�11)2.45 29.0(+1.61) 3148(+24,�26)
29.59 3148(28)0.61 30.33(+0.74) 3166(+18,�22)4.42 31.48(+1.89) 3149(+1, �11)
29.88 3159(21)1.20 30.92(+1.04) 3181(+22,�19)4.99 31.96(+2.08) 3151(�8,+5)– 30.10 3168(18)1.89 31.36(+1.26) 3194(+26,�16)6.11 32.31(+2.21) 3146(�22,+32)
ol�1).
the IR intensities (km mol�1) in the complexes.

Table 4NBO parameters for the H2O molecules. Sum of the electronic charges on the A and Bmolecules, (me), Hyperconjugation energies from O lone pairs to r⁄(OH) orbitals(kcal mol�1).
System Rq(H2O)A Rq(H2O)B LPO1 ? r⁄(OAHA) LPOA ? r⁄(OBHB)CH3OCH3 1H2O �19.0 – 8.05 –CH3OCH3 2H2O 0 �20.0 10.60 11.05CH3OCH2F 1H2O �12.8 – 5.75 –CH3OCH2F 2H2O 2.8 �17.7 7.75 10.26CH3OCHF2 1H2O �6.0 – 2.28 –CH3OCHF2 2H2O 6.1 �12.7 5.75 10.64CH2FOCHF2 1H2O �1.2 – 0.65 –CH2FOCHF2 2H2O 9.1 �10.9 3.88 9.49CHF2OCHF2 1H2O 2.2 – 0.09a (0.14)b –CHF2OCHF2 2H2O 13.3 �8.2 1.97 9.56
a to r⁄(OAHA).b to r⁄(OAHA’).
A.K. Chandra, T. Zeegers-Huyskens / Chemical Physics 410 (2013) 66–70 69
length. Inspection of the results of Table 2 shows that, for the fiveinvestigated systems, there is no simple correlation between thevariation of the r(C3H7) distances and the variation of the valuesof the E2
intra (DE2intra) and E2
inter hyperconjugation energies. For theCH3OCH3 2H2O system where the C3H7 bond is contracted by0.7 mÅ, these values are respectively equal to �0.68 and1.58 kcal mol�1. For the CHF2OCHF2 2H2O system where the C3H7
bond shows the largest elongation of 2.1 Å, these values are respec-tively equal to �3.66 and 6.11 kcal mol�1, for all the complexes, theintermolecular charge transfer are larger than the intramolecularones. Let us notice that the relative variation of the intramoleculardelocalization energies with respect to the isolated molecules(DE2
intra/E2ð0Þintra) are about the same for all the 1–2 systems (0.2). A
red shift can be predicted when the E2inter values are at least
4 kcal mol�1.For the interaction between CHF3 and several proton acceptor,
the variation of the nF ? r⁄(CH) hyperconjugation was invokedin order to explain the change of the CH bond length [36]. In thiscase also, the results indicate the predominance of the intramolec-ular effects over the intermolecular ones. A linear equationbetween these two parameters was derived, the proton donor mol-ecule being constant for all the complexes.
It should be mentioned here that the interaction with two (ormore) water molecules with carbonyl derivatives may either causea transition from blue to red shifts or may amplify the blue shifts.The interaction of H2C@@O with two water molecules increases theblue shift of the m(CH2) vibrations. In contrast, when HFC@@O inter-acts with one water molecule, blue shift of the m(CH) vibration ispredicted; there is a transition from blue to red shifts when HFC@@Ointeracts with three water molecules [35]. This was predicted by amechanism involving a negative intramolecular coupling betweenthe CH and C@O bonds. In the present systems, however, complexformation with one or two water molecules results in a small elon-gation of the C3O1 bonds by ca 0.020 Å. even if the C3H7 bond iselongated. The results of Ref. [35] show that the cooperativitystrongly depends on the nature of the interacting molecules, in fullagreement with the data of the present work.
-10
-5
00 5 10 15
e)
3.3. Properties of the OH bonds. Distances and NBO analysis
Water also shows interesting cooperative properties. At first, wewill briefly discuss the values of the r(OH) distances and m(H2O)vibrational frequencies in the 1–1 and 1–2 systems which are listedin Table 3. As expected from the PA of the O1 atom, the r(OAHA)distances decrease and the intermolecular HA. . .O1 distances in-crease with the number of F atoms implanted on the ether deriva-tive. Cooperative interactions are reflected in the fact that ther(OAHA) distances are longer in the 1–2 than in the 1–1 complexes.
Table 3r(OH) distances (Å) in the 1–1 and 1–2 complexes and corresponding m(OH)vibrational frequencies (cm�1)a.
System r(OAHA) r(OBHB) m(H2O)A m(H2O)B
CH3OCH3 1H2O 0.9722 – 3889, 3660 –CH3OCH3 2H2O 0.9781 0.9756 3884, 3548 3890, 3610CH3OCH2F 1H2O 0.9690 – 3891, 3722 –CH3OCH2F 2H2O 0.9744 0.9748 3884, 3645 3893, 3600CH3OCHF2 1H2O 0.9663 – 3904, 3770 –CH3OCHF2 2H2O 0.9718 0.9743 3887, 3688 3893, 3620CH2FOCHF2 1H2O 0.9644 – 3914, 3799 –CH2FOCHF2 2H2O 0.9690 0.9744 3888, 3737 3894, 3624CHF2OCHF2 1H2O 0.9635 – 3919, 3811 –CHF2OCHF2 2H2O 0.9671 0.9745 3896, 3772 3893, 3625
In (H2O)2, r(OH) bonded: 0.9699 Å, r(H. . .O) = 1.933 Å, m(H2O) = 3892, 3706 cm�1,r⁄(OH) = 0.0148e.
a in H2O monomer, r(OH) = 0.9618 Å, m(H2O) = 3926, 3825 cm�1.
Further, the difference between the r(OAHA) values in the 1–1 and1–2 systems decreases from 5.9 to 3.6 mÅ. In agreement with thenearly constant value of the E2
BC interaction energies, the r(OBHB)and the intermolecular HA. . .OB distances vary within a small range.The vibrational m(OH) frequencies are in agreement with theseconsiderations. The differences between the ms(OAHA) frequenciesin the 1–1 and 1–2 complexes decrease from 112 to 39 cm�1 withthe number of F atoms. The ms(OBHB) frequencies remain approxi-mately constant.
Table 4 lists the sum of the electronic charges on the two watermolecules and the hyperconjugation energies LPO1 ? r⁄(OAHA)and LPOA ? r⁄(OBHB) which are important for the discussion. TheNBO charges on the OA, HA, OB and HB atoms are indicated in S.I.3.The polarity of the OAHA bond is larger in the 1–2 complexes. Inthe CH3OCHF2 1H2O, the NBO charges on the HA and OA atoms areequal to 0.474 and �0.938e; in the 1–2 complex, these charges are0.491 and �0.952e (S.I.3). Further, the smaller positive charges onthe HB atom are compensated by the larger negative charges on
-20
-15
Sig
ma
q(H
2O
)B(m
-25
-20
Sigma q(H2O)A (me)
Fig. 2. Rq(H2O)B as a function of Rq(H2O)A (me).

70 A.K. Chandra, T. Zeegers-Huyskens / Chemical Physics 410 (2013) 66–70
the OA atom. This is in line with the nearly constant value of theOBHB. . .OA binding energies as outlined in the previous sections.
The results of Table 4 show that in the 1–1 complexes, the chargetransfer takes place from the ethers to the (H2O)A molecule exceptin the tetrafluorinated ether where the charge transferoccurs in the reverse direction. This results from the decreasingOAHA. . .O1 interaction and the increasing C3H7. . .OA interaction, aspreviously outlined. This is in full agreement with the decreasingLPO1 ? r⁄(OAHA) hyperconjugation. Let us notice that in theCHF2OCHF2 1H2O system, the value of this hyperconjugation isnearly zero and that a small charge transfer takes place to theexternal O1H0A bond. The cooperativity effect also results in a largerLPO1 ? r⁄(OAHA) delocalization in the 1–2 complexes than in the1–1 complexes. The elongations of the OAHA and OBHB bonds inthe 1–1 and and the 1–2 complexes are linearly correlated to thehyperconjugation energies taking place from the O1 or OA atomsto the r⁄(OAHA) or r⁄(OBHB) orbitals [40]. Finally, our calculationsreveal that in the 1–2 complexes, the (H2O)A molecule is positivelycharged and the (H2O)B molecule negatively charged. As illustratedin Fig.2, when the negative charge on (H2O)B decreases, the positivecharge on (H2O)A increases , indicating a beautiful reciprocaleffect.
4. Conclusions
In this work, the interaction between dimethyl ether and someof its fluorinated derivatives with two water molecules (A and B)has been studied using the B3LYP method in conjunction withthe 6-311++G(d,p) basis set. The most important conclusionsobtained in this work can be summarized as follows:
1. In the optimized structures, the complexes are cyclic and stabi-lized by OAHA. . .O and CH. . .OB hydrogen bonds.
2. The pairwise or two-body interaction energies show that theOAHA. . .O interaction energies range between �2.06 and�4.93 kcal mol�1 and are ordered according the proton affinityof the O atom of the ether derivative. In turn, the CH. . .OB inter-action energies are comprised between �1.07 and�3.15 kcal mol�1 and increase with the acidity of the CH bonds.The energy of the OBHB. . .OA hydrogen bonds between the twowater molecules remains almost constant (�4.60 kcal mol�1).These conclusions are in line with the variations of the intermo-lecular distances. The cooperativity energy represents 15–20%of the total interaction energy.
3. In all the 1–1 complexes, the m(CH) vibrations are blue-shifted.Complex formation of the, tri- and tetra-fluorinated ethersresults in an elongation of the CH bond and a blue shift a them(CH) vibrations. These results are explained by a competitionbetween the intra - and intermolecular hyperconjugations. Aquantitative correlation is obtained between these two factorsand the variation of the CH bond length.
4. A further indication of the cooperativity is that the elongation ofthe OAHA bond, the red shifts of the m(OAHA) vibration and thehyperconjugation energies to the r⁄(OAHA) bonds are larger inthe 1–2 complexes than in the 1–1 ones. The charge transferbetween the molecules is discussed. In the 1–2 complexes,the (H2O)B molecules bear negative charges which decreasewith increasing positive charges on the (H2O)A molecules.
Note added in revision
After submission of the present work, a paper dealing with therelation between the shifts of the proton donor XH bond and the
occupation of both the r(XH) bonding and r⁄(XH) antibondingorbitals has appeared in the literature [41]. The correlationbetween bond length and bond order predicts much smaller elon-gations (or red shifts) of the CH bonds for the present systems. Forthe CH3OCHF2 2H2O system, a small contraction of 0.1 mÅ of theC3H7 bond was predicted by the theory based on bond order. Ourcalculations reveal an elongation of 0.7 mÅ.
Acknowledgments
A.K. Chandra thanks CSIR India for providing financial assis-tance through Project no. 01(2494)/11/EMR-II and ComputerCentre, NEHU for extending computational facilities. Th. Zeegers-Huyskens thanks the Department of Chemistry of the Universityof Leuven for computer facilities.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.chemphys.2012.10.019.
References
[1] H.S. Frank, W.-Y. Wen, Discuss. Faraday Soc. 24 (1957) 133.[2] P.L. Huyskens, J. Am. Chem. Soc. 99 (1977) 2576.[3] A. Karpfen, in: S. Scheiner (Ed.), Hydrogen Bonding, A Theoretical Perspective,
Oxford University Press, New York, USA, 1994.[4] H. Kleeberg, in: P.L. Huyskens, W.A.P. Luck, Th. Zeegers-Huyskens (Eds.),
Intermolecular Forces: An Introduction to Modern Methods and Results,Springer, Berlin, Germany, 1991.
[5] J.E. Koehler, W. Saenger, B. Lesyng, J. Comput. Chem. 8 (1987) 1090.[6] W.A.P. Luck, D. Klein, K. Rangsriwatananon, J. Mol. Struct. 416 (1997) 287.[7] Th. Zeegers-Huyskens, S.G. Pandai (Eds.), Recent Research Development in
Physical Chemistry vol. 2 (1998).[8] M. Masella, J. Chem. Phys. 110 (1999) 7245.[9] M. Rode, J. Sadlej, Chem. Phys. Lett. 342 (2001) 220.
[10] T. Kar, S. Scheiner, J. Phys. Chem. A 108 (2004) 9191.[11] W. Wang, A. Tian, J. Phys. Chem. A 109 (2005) 8035.[12] Q. Li, W. An, B. Gong, J. Cheng, J. Phys. Chem. A 111 (2007) 10166.[13] N. Dozova, L. Kim, J. Phys. Chem. A 111 (2007) 10055.[14] L. Qingzhong, N. Wang, Y. Zhivu, J. Mol. Struct. (THEOCHEM) 847 (2007) 68.[15] E.L. Angelina, N. Peruchena, J. Phys. Chem. A 115 (2011) 4701.[16] A.K. Chandra, Th. Zeegers-Huyskens, J. At. Mol. Opt. Phys. (2012).
101155:2012:754879.[17] A.J. Barnes, T.R. Beech, Chem. Phys. Lett. 94 (1983) 568.[18] A.D. Becke, J. Chem. Phys. 104 (1996) 1040.[19] M.J. Frisch, Gaussian 03, Revision D. 01, Wallingford, Conn., USA, 2004.[20] S.F. Boys, F. Bernardi, Mol. Phys. 19 (1970) 553.[21] S.S. Xantheas, J. Chem. Phys. 104 (1996) 8821.[22] M. Weimann, M. Farnik, M.A. Suhm, M.E. Alikhani, J. Sadlej, J. Mol. Struct. 790
(2006) 18.[23] A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88 (1988) 899.[24] S. Parveen, A.K. Chandra, Th. Zeegers-Huyskens, J. Phys. Chem. A 113 (2009)
6182.[25] The correlation is the following one: Dr(CH)(mÅ) = �0.075 Dm (CH)
(cm�1) + 0.61 (r = 0.985).[26] A. Karpfen, E.S. Kryachko, Chem. Phys. Lett. 431 (2006) 428.[27] Y. Gu, T. Kar, S. Scheiner, J. Am. Chem. Soc. 121 (1999).[28] P. Hobza, Z. Havlas, Chem. Rev. 100 (2000) 4253.[29] B.J. Van der Veken, W.A. Herrebout, R. Szostak, D.N. Shcherpkin, Z. Havlas, P.
Hobza, J. Am. Chem. Soc. 123 (2001) 12290.[30] E.S. Kryachko, Th. Zeegers-Huyskens, J. Phys. Chem. A 105 (2001) 7118.[31] A. Masunov, J.J. Dannenberg, R.H. Contreras, J. Phys. Chem. A105 (2001) 4737.[32] K. Hermansson, J. Phys. Chem. A 106 (2002) 4695.[33] X. Li, L. Liu, H.B. Schlegel, J. Am. Chem. Soc. 124 (2002) 9639.[34] I.V. Alabugin, M. Manoharan, S. Peabody, F. Weinhold, J. Am. Chem. Soc. 125
(2003) 5973.[35] A. Karpfen, E.S. Kryachko, J. Phys. Chem. A 111 (2007) 8177.[36] J. Joseph, E.D. Jemmis, J. Am. Chem. Soc. 129 (2007) 4620. references therein.[37] A.Y. Li, J. Chem. Phys. 126 (2007) 154102.[38] I.V. Alabugin, J. Org. Chem. 65 (2000) 3910.[39] I.V. Alabugin, T.A. Zeidan, J. Am. Chem. Soc. 124 (2002) 3175.[40] Dr(OH) (mÅ) = 1.11 [LPO1 ? r⁄(OH)] (kcal mol�1) r = 0.967.[41] P.-P. Zhou, W.-Y. Qiu, N.-Z. Jin, J. Chem. Phys. 137 (2012) 084311.