Embed Size (px)
Citation preview

Theoretical characterization of two reaction pathways
for the intramolecular cyclization
of 2-(3-benzylaminopropanoylamino)benzamide
Christopher Knighta, M.C. Millettib,*
aDepartment of Chemistry, The Ohio State University, 110 West 18th Avenue, Columbus, OH 43210, USAbDepartment of Chemistry, Eastern Michigan University, 225 Mark Jefferson Building, Ypsilanti, MI 48197, USA
Received 7 December 2004; revised 8 March 2005; accepted 8 March 2005
Available online 28 April 2005
Abstract
During the investigation of the microwave synthesis of compounds related to benzodiazepines by Howard and co-workers, another
group of compounds was isolated. First believed to be bicyclic amidines, these compounds were later found to be 4-quinazolinones. In
this work, the potential energy surface of the precursor to the observed 4-quinazolinone is explored. The most stable arrangement is
found to be a linear conformation of the side chain with the amide group in the Z conformation. Using this structure as the starting
point, the reaction pathways for the intramolecular cyclization to form either the 4-quinazolinone or the bicyclic amidine are
characterized.
q 2005 Elsevier B.V. All rights reserved.
Keywords: Quinazolinone; Reaction pathway; Cyclization; Michael product; Hartree–Fock
1. Introduction
Benzodiazepines are a class of compounds which are
widely used as sedatives, e.g. valium (1). In the course of
synthetic work aimed at making analogues of huperzine A, a
small quantity of a compound, initially thought to have the
bicyclic amidine structure (2), was isolated [1]. The
structural relationship to one of the early benzodiazepines,
Librium (3), was interesting so the sequence, involving
reactions promoted by microwaves, was further investi-
gated. Once a larger sample of the product was obtained it
was shown not to be the bicyclic amidine 3 but rather the
isomeric 4-quinazolinone 5a (see Scheme 1). Since 2-alkyl-
4-quinazolinones also show interesting physiological activi-
ties, the scope of the methodology was investigated [2].
0166-1280/$ - see front matter q 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.theochem.2005.03.017
* Corresponding author. Tel.: C1 734 487 1183; fax: C1 734 487 1496.
E-mail address: [email protected] (M.C. Milletti).
N
NO
Ph
Cl
CH3
(1) Valium
N
N
NH2
PhO
(2) Amidine
N
NNHCH3
OPh
Cl
(3) Librium
In this paper, we report on the investigation of the
potential energy surfaces associated with the alternative
cyclizations of the precursor, the Michael product 2-(3-
benzylaminopropanoylamino)benzamide (4) as shown in
Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153
www.elsevier.com/locate/theochem

O
NH2
NH
O
R1
H
R2
NHBz
(4a-d)
O
NBz
N
NH2
R1
R2
(3a-d) O
NH
N
R1
H R2
NHBz
(5a-d)
Scheme 1.
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153144
Scheme 1 and demonstrate why the 4-quinazolinone is the
preferred product.
Fig. 1. Stacked and linear conformations for the Michael product.
2. Details of the calculations
Geometry optimization calculations using the Berny
algorithm [3] were carried out on all Michael products using
the Hartree–Fock method with a 6-31G basis set [4]. The
first optimization calculation (4a) was repeated using
density functional theory (B3LYP functionals) [5] using
the same basis set. Since the results from these two
calculations were equivalent, the Hartree–Fock method
was used for all calculations. Single point energy evalu-
ations were also repeated at the B3LYP/6-31G* level of
theory for critical points in the potential energy pathways
(6:PROD(A) and 6:PROD(B) in the quinazolinone path-
ways and 5:TS3, 5:TS(A) and 5:TS(B) in the bicyclic
amidine pathway).
In an effort to find the global minimum on the potential
energy surface for the Michael product, the relationship
between the angle of rotation around a variety of single
bonds and the energy of the molecule was investigated by
varying the dihedral angle in 108 increments from 0 to 3608
and calculating the total energy of the molecule at each step.
Transition states were located using linear (QST2) and
quadratic (QST3) synchronous transit algorithms. Reaction
path following calculations utilized the second order
method of Schlegel [6]. The structural information obtained
from the geometry optimization of the Michael product was
used as the input geometry for the reactant. Use of chemical
intuition and the proposed mechanism suggested stable
intermediates which would be minima along the potential
energy path. Frequency calculations insured that the
structure had in fact only one imaginary frequency. To
make sure that the correct transition structures had been
found, the structure was displaced slightly on both sides
from the center of the vibrational displacement vectors and
was optimized; this produced the minima originally used for
the QST2 and QST3 calculations. The process was repeated
in a step-wise fashion along the reaction path, thus
characterizing the entire mechanism. Intrinsic reaction
coordinates (IRC) calculations using the transition structure
were used to follow the paths leading to the two minima
associated with it, thus confirming the connection of two
minima and one maximum.
All calculations were performed on Dell Dimension
4300S (Pentium IV, 512 MB, 1.8 GHz), using GAUSSIAN
98W [7] and GaussView [8] software packages and on a
cluster of two Alpha processors using GAUSSIAN 94 [9].
3. Results and discussion
The first step in mapping out the reaction pathways was
to determine the best structure for the common reactants,
compounds 4a–d. Each one of this set of molecules has a
structure with many degrees of freedom; consequently,
finding a global minimum is not a trivial endeavor. Two
major structural variables were investigated: whether the
molecule is more stable as a linear extended structure or a
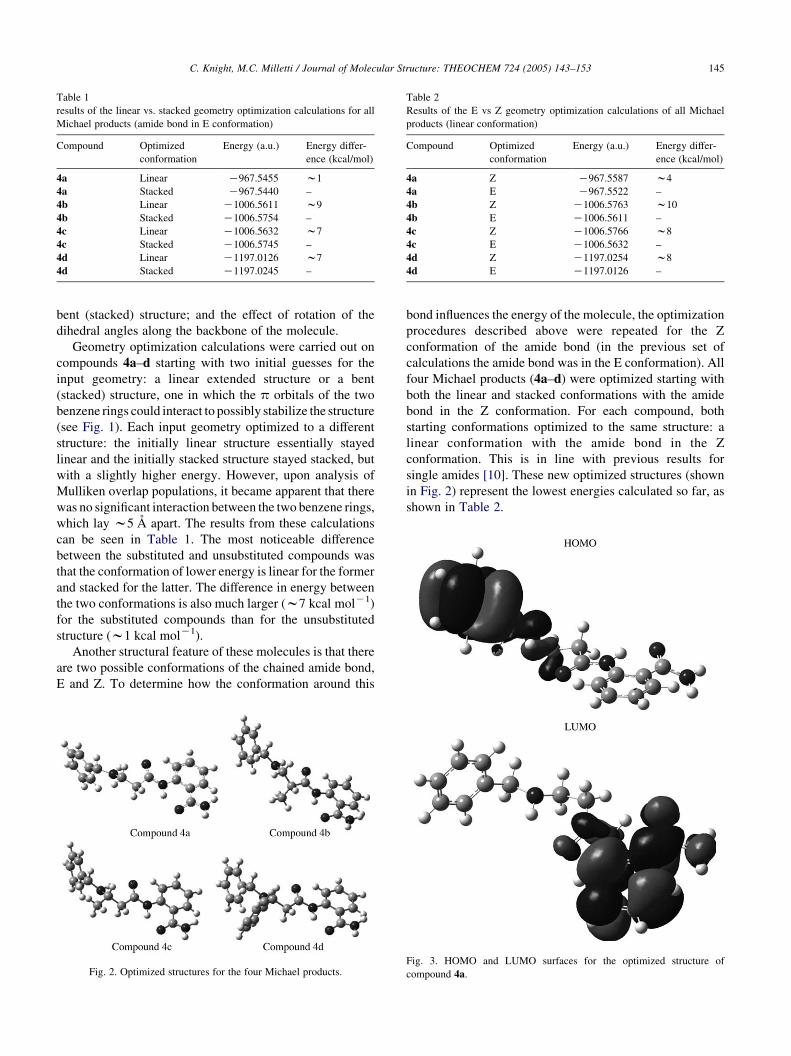
Table 1
results of the linear vs. stacked geometry optimization calculations for all
Michael products (amide bond in E conformation)
Compound Optimized
conformation
Energy (a.u.) Energy differ-
ence (kcal/mol)
4a Linear K967.5455 w1
4a Stacked K967.5440 –
4b Linear K1006.5611 w9
4b Stacked K1006.5754 –
4c Linear K1006.5632 w7
4c Stacked K1006.5745 –
4d Linear K1197.0126 w7
4d Stacked K1197.0245 –
Table 2
Results of the E vs Z geometry optimization calculations of all Michael
products (linear conformation)
Compound Optimized
conformation
Energy (a.u.) Energy differ-
ence (kcal/mol)
4a Z K967.5587 w4
4a E K967.5522 –
4b Z K1006.5763 w10
4b E K1006.5611 –
4c Z K1006.5766 w8
4c E K1006.5632 –
4d Z K1197.0254 w8
4d E K1197.0126 –
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153 145
bent (stacked) structure; and the effect of rotation of the
dihedral angles along the backbone of the molecule.
Geometry optimization calculations were carried out on
compounds 4a–d starting with two initial guesses for the
input geometry: a linear extended structure or a bent
(stacked) structure, one in which the p orbitals of the two
benzene rings could interact to possibly stabilize the structure
(see Fig. 1). Each input geometry optimized to a different
structure: the initially linear structure essentially stayed
linear and the initially stacked structure stayed stacked, but
with a slightly higher energy. However, upon analysis of
Mulliken overlap populations, it became apparent that there
was no significant interaction between the two benzene rings,
which lay w5 A apart. The results from these calculations
can be seen in Table 1. The most noticeable difference
between the substituted and unsubstituted compounds was
that the conformation of lower energy is linear for the former
and stacked for the latter. The difference in energy between
the two conformations is also much larger (w7 kcal molK1)
for the substituted compounds than for the unsubstituted
structure (w1 kcal molK1).
Another structural feature of these molecules is that there
are two possible conformations of the chained amide bond,
E and Z. To determine how the conformation around this
Fig. 2. Optimized structures for the four Michael products.
bond influences the energy of the molecule, the optimization
procedures described above were repeated for the Z
conformation of the amide bond (in the previous set of
calculations the amide bond was in the E conformation). All
four Michael products (4a–d) were optimized starting with
both the linear and stacked conformations with the amide
bond in the Z conformation. For each compound, both
starting conformations optimized to the same structure: a
linear conformation with the amide bond in the Z
conformation. This is in line with previous results for
single amides [10]. These new optimized structures (shown
in Fig. 2) represent the lowest energies calculated so far, as
shown in Table 2.
Fig. 3. HOMO and LUMO surfaces for the optimized structure of
compound 4a.
–1006.51
–1006.50
–1006.49
–1006.48
–1006.47
–1006.46
–1006.45
–1006.44
–1006.43
–1006.42
0 40 80 120
160
200
240
280
320
360
Angle
Ene
rgy
(a.u
.)
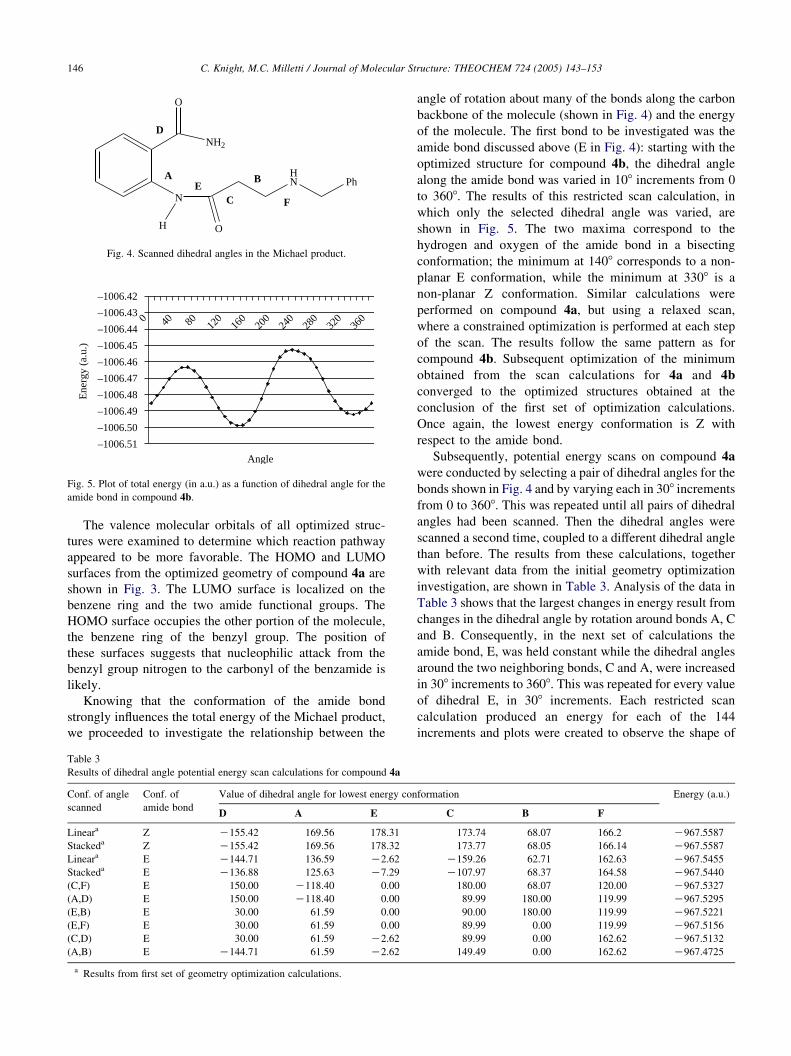
Fig. 5. Plot of total energy (in a.u.) as a function of dihedral angle for the
amide bond in compound 4b.
O
NH2
N
HN Ph
H O
A B
C
D
E
F
Fig. 4. Scanned dihedral angles in the Michael product.
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153146
The valence molecular orbitals of all optimized struc-
tures were examined to determine which reaction pathway
appeared to be more favorable. The HOMO and LUMO
surfaces from the optimized geometry of compound 4a are
shown in Fig. 3. The LUMO surface is localized on the
benzene ring and the two amide functional groups. The
HOMO surface occupies the other portion of the molecule,
the benzene ring of the benzyl group. The position of
these surfaces suggests that nucleophilic attack from the
benzyl group nitrogen to the carbonyl of the benzamide is
likely.
Knowing that the conformation of the amide bond
strongly influences the total energy of the Michael product,
we proceeded to investigate the relationship between the
Table 3
Results of dihedral angle potential energy scan calculations for compound 4a
Conf. of angle
scanned
Conf. of
amide bond
Value of dihedral angle for lowest energy con
D A E
Lineara Z K155.42 169.56 178.31
Stackeda Z K155.42 169.56 178.32
Lineara E K144.71 136.59 K2.62
Stackeda E K136.88 125.63 K7.29
(C,F) E 150.00 K118.40 0.00
(A,D) E 150.00 K118.40 0.00
(E,B) E 30.00 61.59 0.00
(E,F) E 30.00 61.59 0.00
(C,D) E 30.00 61.59 K2.62
(A,B) E K144.71 61.59 K2.62
a Results from first set of geometry optimization calculations.
angle of rotation about many of the bonds along the carbon
backbone of the molecule (shown in Fig. 4) and the energy
of the molecule. The first bond to be investigated was the
amide bond discussed above (E in Fig. 4): starting with the
optimized structure for compound 4b, the dihedral angle
along the amide bond was varied in 108 increments from 0
to 3608. The results of this restricted scan calculation, in
which only the selected dihedral angle was varied, are
shown in Fig. 5. The two maxima correspond to the
hydrogen and oxygen of the amide bond in a bisecting
conformation; the minimum at 1408 corresponds to a non-
planar E conformation, while the minimum at 3308 is a
non-planar Z conformation. Similar calculations were
performed on compound 4a, but using a relaxed scan,
where a constrained optimization is performed at each step
of the scan. The results follow the same pattern as for
compound 4b. Subsequent optimization of the minimum
obtained from the scan calculations for 4a and 4b
converged to the optimized structures obtained at the
conclusion of the first set of optimization calculations.
Once again, the lowest energy conformation is Z with
respect to the amide bond.
Subsequently, potential energy scans on compound 4a
were conducted by selecting a pair of dihedral angles for the
bonds shown in Fig. 4 and by varying each in 308 increments
from 0 to 3608. This was repeated until all pairs of dihedral
angles had been scanned. Then the dihedral angles were
scanned a second time, coupled to a different dihedral angle
than before. The results from these calculations, together
with relevant data from the initial geometry optimization
investigation, are shown in Table 3. Analysis of the data in
Table 3 shows that the largest changes in energy result from
changes in the dihedral angle by rotation around bonds A, C
and B. Consequently, in the next set of calculations the
amide bond, E, was held constant while the dihedral angles
around the two neighboring bonds, C and A, were increased
in 308 increments to 3608. This was repeated for every value
of dihedral E, in 308 increments. Each restricted scan
calculation produced an energy for each of the 144
increments and plots were created to observe the shape of
formation Energy (a.u.)
C B F
173.74 68.07 166.2 K967.5587
173.77 68.05 166.14 K967.5587
K159.26 62.71 162.63 K967.5455
K107.97 68.37 164.58 K967.5440
180.00 68.07 120.00 K967.5327
89.99 180.00 119.99 K967.5295
90.00 180.00 119.99 K967.5221
89.99 0.00 119.99 K967.5156
89.99 0.00 162.62 K967.5132
149.49 0.00 162.62 K967.4725
O
H2N
N
H
O
HN
PhN
N
O
H
NH
Ph
3
2
3
2
1
+ H2O
1
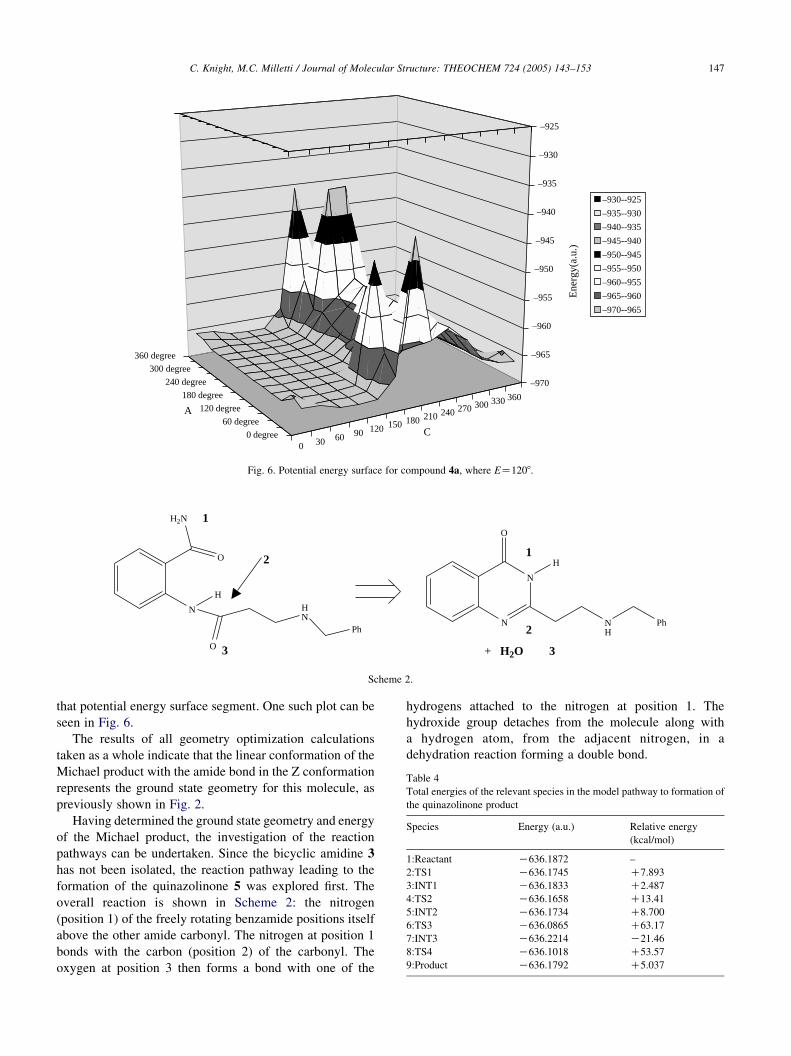
Scheme 2.
0 30 60 90 120 150 180 210 240 270 300 330 360
0 degree
60 degree
120 degree
180 degree
240 degree
300 degree
360 degree
–970
–965
–960
–955
–950
–945
–940
–935
–930
–925
Ene
rgy(
a.u.
)
–930--925
–935--930
–940--935
–945--940
–950--945
–955--950
–960--955
–965--960
–970--965
A
C
Fig. 6. Potential energy surface for compound 4a, where EZ1208.
Table 4
Total energies of the relevant species in the model pathway to formation of
the quinazolinone product
Species Energy (a.u.) Relative energy
(kcal/mol)
1:Reactant K636.1872 –
2:TS1 K636.1745 C7.893
3:INT1 K636.1833 C2.487
4:TS2 K636.1658 C13.41
5:INT2 K636.1734 C8.700
6:TS3 K636.0865 C63.17
7:INT3 K636.2214 K21.46
8:TS4 K636.1018 C53.57
9:Product K636.1792 C5.037
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153 147
that potential energy surface segment. One such plot can be
seen in Fig. 6.
The results of all geometry optimization calculations
taken as a whole indicate that the linear conformation of the
Michael product with the amide bond in the Z conformation
represents the ground state geometry for this molecule, as
previously shown in Fig. 2.
Having determined the ground state geometry and energy
of the Michael product, the investigation of the reaction
pathways can be undertaken. Since the bicyclic amidine 3
has not been isolated, the reaction pathway leading to the
formation of the quinazolinone 5 was explored first. The
overall reaction is shown in Scheme 2: the nitrogen
(position 1) of the freely rotating benzamide positions itself
above the other amide carbonyl. The nitrogen at position 1
bonds with the carbon (position 2) of the carbonyl. The
oxygen at position 3 then forms a bond with one of the
hydrogens attached to the nitrogen at position 1. The
hydroxide group detaches from the molecule along with
a hydrogen atom, from the adjacent nitrogen, in a
dehydration reaction forming a double bond.
1.94
O
N
N
H
O CH2CH3
H H
N
H
O CH2CH3
O
N
H
H
N
H
O CH2CH3
O
N
H
H
N
O
N
H
H
H
O
CH2CH3
N
O
N
H
H
H
O
CH2CH3
N
O
N
H
H
H
O
CH2CH3N
O
H
N
H
O
CH2CH3
H
N
O
H
N
H
O
CH2CH3
H
N
O
N
H
CH2CH3
+H2O
1:START 2:TS1 3: INT1 4:TS2
5:INT26:TS37:INT3
8:TS4 9:PROD
1.02
3.06
1.200.98
2.3
1.48
1.49
1.29
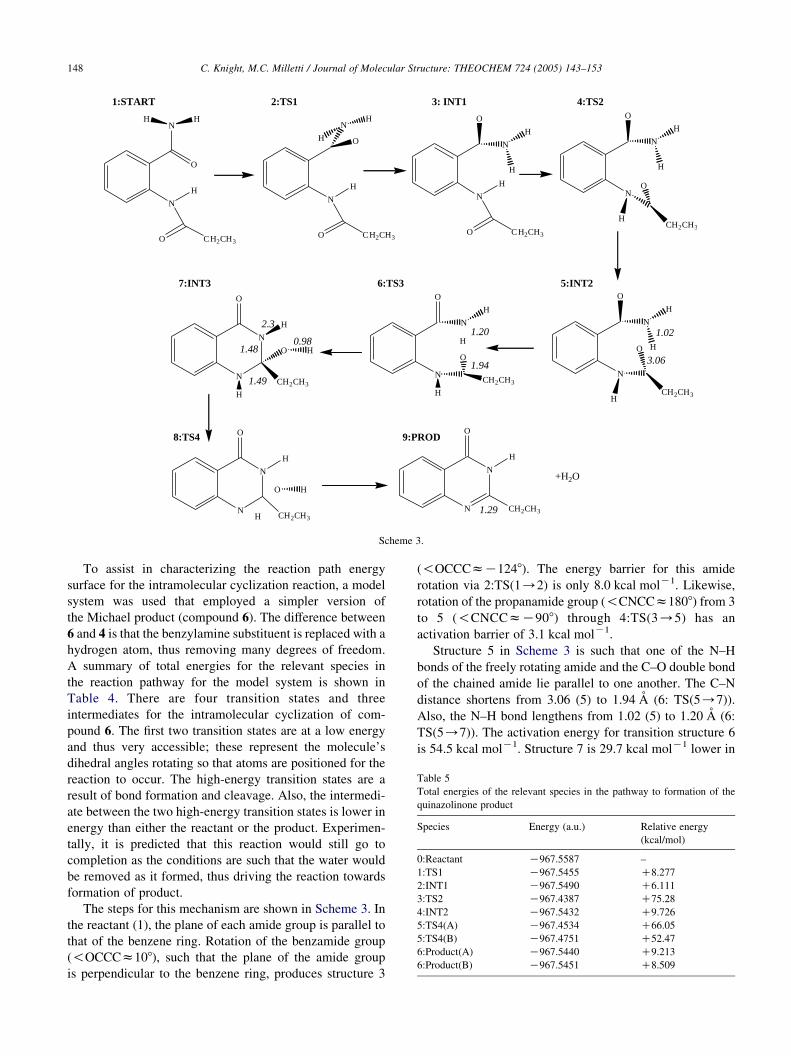
Scheme 3.
Table 5
Total energies of the relevant species in the pathway to formation of the
quinazolinone product
Species Energy (a.u.) Relative energy
(kcal/mol)
0:Reactant K967.5587 –
1:TS1 K967.5455 C8.277
2:INT1 K967.5490 C6.111
3:TS2 K967.4387 C75.28
4:INT2 K967.5432 C9.726
5:TS4(A) K967.4534 C66.05
5:TS4(B) K967.4751 C52.47
6:Product(A) K967.5440 C9.213
6:Product(B) K967.5451 C8.509
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153148
To assist in characterizing the reaction path energy
surface for the intramolecular cyclization reaction, a model
system was used that employed a simpler version of
the Michael product (compound 6). The difference between
6 and 4 is that the benzylamine substituent is replaced with a
hydrogen atom, thus removing many degrees of freedom.
A summary of total energies for the relevant species in
the reaction pathway for the model system is shown in
Table 4. There are four transition states and three
intermediates for the intramolecular cyclization of com-
pound 6. The first two transition states are at a low energy
and thus very accessible; these represent the molecule’s
dihedral angles rotating so that atoms are positioned for the
reaction to occur. The high-energy transition states are a
result of bond formation and cleavage. Also, the intermedi-
ate between the two high-energy transition states is lower in
energy than either the reactant or the product. Experimen-
tally, it is predicted that this reaction would still go to
completion as the conditions are such that the water would
be removed as it formed, thus driving the reaction towards
formation of product.
The steps for this mechanism are shown in Scheme 3. In
the reactant (1), the plane of each amide group is parallel to
that of the benzene ring. Rotation of the benzamide group
(!OCCCz108), such that the plane of the amide group
is perpendicular to the benzene ring, produces structure 3
(!OCCCzK1248). The energy barrier for this amide
rotation via 2:TS(1/2) is only 8.0 kcal molK1. Likewise,
rotation of the propanamide group (!CNCCz1808) from 3
to 5 (!CNCCzK908) through 4:TS(3/5) has an
activation barrier of 3.1 kcal molK1.
Structure 5 in Scheme 3 is such that one of the N–H
bonds of the freely rotating amide and the C–O double bond
of the chained amide lie parallel to one another. The C–N
distance shortens from 3.06 (5) to 1.94 A (6: TS(5/7)).
Also, the N–H bond lengthens from 1.02 (5) to 1.20 A (6:
TS(5/7)). The activation energy for transition structure 6
is 54.5 kcal molK1. Structure 7 is 29.7 kcal molK1 lower in
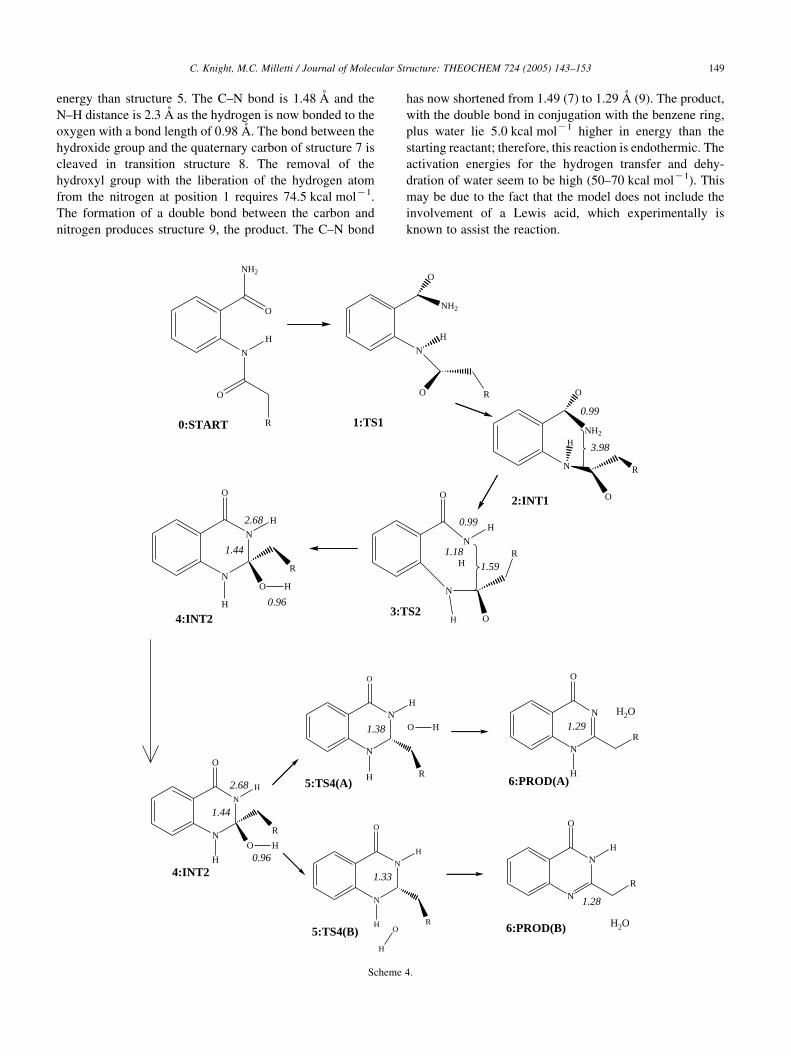
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153 149
energy than structure 5. The C–N bond is 1.48 A and the
N–H distance is 2.3 A as the hydrogen is now bonded to the
oxygen with a bond length of 0.98 A. The bond between the
hydroxide group and the quaternary carbon of structure 7 is
cleaved in transition structure 8. The removal of the
hydroxyl group with the liberation of the hydrogen atom
from the nitrogen at position 1 requires 74.5 kcal molK1.
The formation of a double bond between the carbon and
nitrogen produces structure 9, the product. The C–N bond
NH2
O
N
H
O
R0:START 1:TS1
3:T4:INT2
N
N
O
H
R
H
2.68
1.44
0.96
4:INT2
N
N
O
H
R
O H
O H
H
2.68
1.44
0.96
5:TS4(A)
N
N
O
H
1.38
5:TS4(B)
N
N
O
O
H
H
1.33
Scheme
has now shortened from 1.49 (7) to 1.29 A (9). The product,
with the double bond in conjugation with the benzene ring,
plus water lie 5.0 kcal molK1 higher in energy than the
starting reactant; therefore, this reaction is endothermic. The
activation energies for the hydrogen transfer and dehy-
dration of water seem to be high (50–70 kcal molK1). This
may be due to the fact that the model does not include the
involvement of a Lewis acid, which experimentally is
known to assist the reaction.
NH2
O
N
H
O R
NH2
O
N
H
O
R
2:INT1
0.99
3.98
N
O
N
H O
R
S2
0.99
1.59
H
H1.18
O H
H
R
H
R
6:PROD(A)
N
N
O
R
H
1.29
6:PROD(B)
N
N
O
H
R
1.28
H2O
H2O
4.

C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153150
After completion of the reaction surface for the model
system, those structures were used as starting points for the
system of interest. A summary of total energies for the
relevant species is shown in Table 5 and the reaction
mechanism in Scheme 4.
The most obvious difference between the model system
and the actual reaction is that the latter has fewer
intermediates and transition states. The model system
required an additional transition state and intermediate to
position atoms to initiate bond formation and breakage. This
is probably due to the differences in the structures of the
reactants. The reaction pathway describing the intramole-
cular cyclization of the Michael product to the quinazoli-
none has two intermediates and three transition states. The
largest activation barrier was found to be 75.3 kcal molK1,
which corresponds to the transition state involving closure
of the ring with formation of the C–N bond.
In structure 0, both of the amide groups lie almost
parallel with the benzene ring. Rotation of the benzamide
group (!OCCCz248) and the substituted propanamide
group (!CNCCz1708) gives rise to 2. The amide groups,
through the transition state, position themselves almost
perpendicular to the benzene ring. In structure 2, the
benzamide group (!OCCCz458) has rotated so that the
nitrogen atom is above the plane of the benzene ring.
The propanamide (!CNCCz608) is positioned such that
the oxygen of the carbonyl is above the plane of the ring. The
energy barrier to transition state 1:TS (0/2) is 8.3 kcal -
molK1. This single transition state is a significant difference
between the model system and the Michael product system:
in the model system two separate transition states were
required to position the amide groups, whereas the Michael
product only required a single transition state. Structure 2 is
one in which an N–H bond of the benzamide group and the
C–O bond of the propanamide are parallel; looking down the
C–O bond, the N–H bond appears E with the nitrogen behind
the carbon and the hydrogen behind the oxygen. In this
step, the C–N distance shortens from 3.98 (2) to 1.59 A (3:TS
(2/4)), while the N–H distance lengthens from 0.99 (2) to
1.18 A (3:TS (2/4)). The activation energy for transition
state 3:TS (2/4) is 75.3 kcal molK1. Structure 4 is
N
N
O
H
H
OH
NH Ph
4:INT3
Scheme
9.7 kcal molK1 higher in energy than the reactant (0). The
C–N bond is now 1.44 A (4) and the N–H distance is 2.68 A
(4) as the hydrogen is now bonded to the oxygen with a bond
length of 0.96 A (4). This new six-membered ring is nearly
planar, with the quaternary carbon protruding from the plane
and the hydroxyl group located on the front face of the
molecule.
Dehydration of structure 4 can occur in one of two ways.
In both instances, the hydroxyl group detaches from the
quaternary carbon of structure 4. This hydroxide ion can
now bond with a hydrogen atom from either nitrogen,
producing water and one of two isomeric 4-quinazolinones.
Bond formation with the hydrogen attached to the nitrogen
in the 3 position goes through transition state 5a:(TS(4/6a)). Structure 6a, the isomer with the double bond in
conjugation with the carbonyl, lies 9.2 kcal molK1 higher in
energy than the starting reactant. The C–N bond shortens
from 1.38 (5a) to 1.29 A (6a) with formation of the double
bond. The more stable isomer is formed through transition
state 5b:(TS(4/6b), which is 13.6 kcal molK1 more stable
than transition state 5a. Structure 6b, the isomer, where the
double bond is in conjugation with the benzene ring, is more
stable than structure 6a (by 0.7 kcal molK1 at the HF/6-31G
level of theory and by 11.8 kcal molK1 at the B3LYP/6-
31G* level of theory). The C–N bond shortens from 1.33
(5b) to 1.28 A (6b) with formation of the double bond.
Having completed the analysis of the 4-quinazolinone
reaction pathway, the pathway to the bicyclic amidine was
investigated. Since this compound has not been isolated to
date, it is not clear which pathway would lead to its formation.
One possibility is that structure 4 in the 4-quinazolinone
reaction pathway (Scheme 4) is a branching point that can
also lead to the amidine. In view of the high activation energy
associated with the dehydration reaction, it is possible that
compound 4 may exist long enough to allow for nucleophilic
attack on the carbonyl carbon by the benzylamine nitrogen,
leading to the eight-membered bicyclic product. This overall
reaction scheme is shown in Scheme 5 and the reaction
mechanism is detailed in Scheme 6.
The results of this set of calculations are summarized in an
energy plot of the reaction pathway (shown in Fig. 7). Starting
N
N
O Ph
NH2
+ H2O
(3a)
5.
N
N
O
H
H
O N
Ph
H
HN
N
O
H
H
O H
N
H
Ph
N
N
O
H
H
O H
N
HPh
N
N
O
H
H
OH
NH
Ph
N
NH
O
H
OH
N
H
Ph
2.79
4.39
1.230.99
3.36
4.48
N
N
O
H
H
O
H
H
N
Ph
N
N
O
H
H
O
H
H
N
Ph
N
N
O
H
O
H
H
N
Ph
H
N
N
O
H
H
O
H
H
N
Ph
N
N
O
H
H
O
H
H
N
Ph1.35
1.261.54
1.391.461.40
0.952.50
1.38
1.37
1.58
1.24
0.99 1.45
2.96
3.44
1.27
O
N
N
NH
H
H
Ph
O
H
1.34
1.032.27
O
N
N
N H
H
Ph
H2O
4:INT2 5:TS3
6:INT3
7:TS48:INT4
9:TS5 10:INT511:TS6
12:INT613:TS7
14:INT7
15:INT8
Scheme 6.
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153 151
0
20
40
60
80
100
REACTION PATH
RE
LA
TIV
E E
NE
RG
Y(k
cal/m
ol)
Unconj Quin
Conj Quin
Amidine
Fig. 8. A comparison of relative energies of relevant species for the two
pathways.
0
12
3
4
5
6
78
9
10
11
12
13
14
15
0
10
20
30
40
50
60
70
80
90
100
REACTION PATH
RE
LA
TIV
E E
NE
RG
Y (
kcal
/mol
)
Fig. 7. Relative energies of relevant species for the pathway to formation of
the amidine product.
C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153152
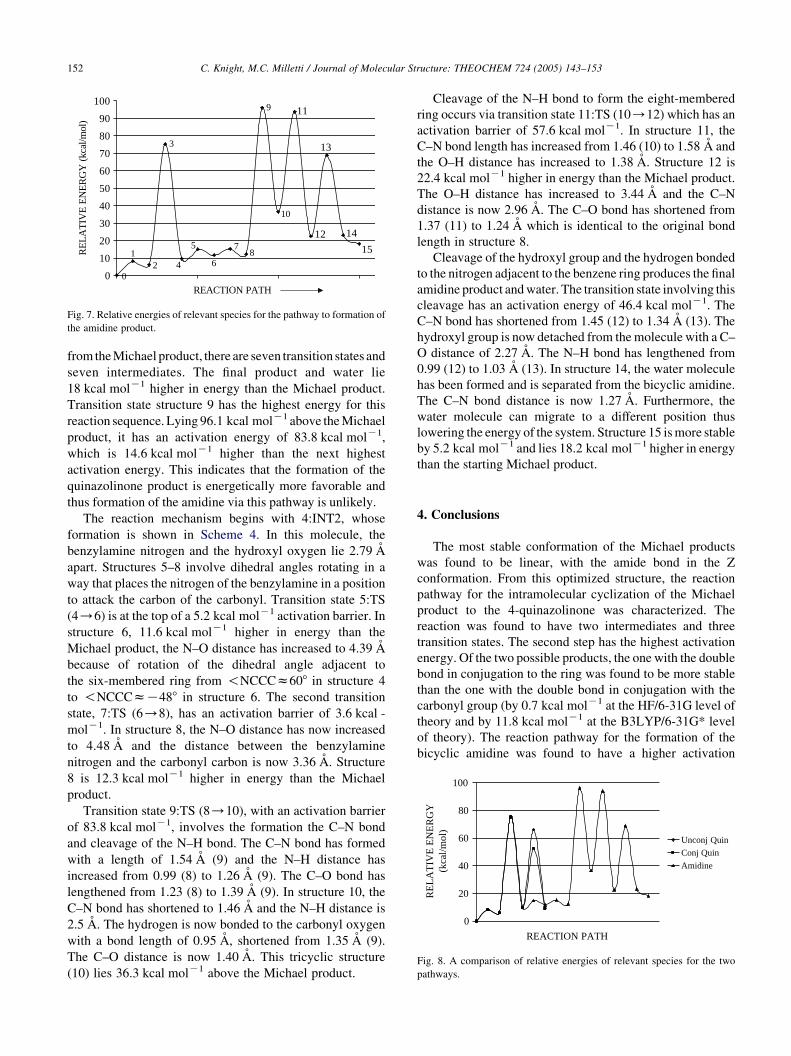
from the Michael product, there are seven transition states and
seven intermediates. The final product and water lie
18 kcal molK1 higher in energy than the Michael product.
Transition state structure 9 has the highest energy for this
reaction sequence. Lying 96.1 kcal molK1 above the Michael
product, it has an activation energy of 83.8 kcal molK1,
which is 14.6 kcal molK1 higher than the next highest
activation energy. This indicates that the formation of the
quinazolinone product is energetically more favorable and
thus formation of the amidine via this pathway is unlikely.
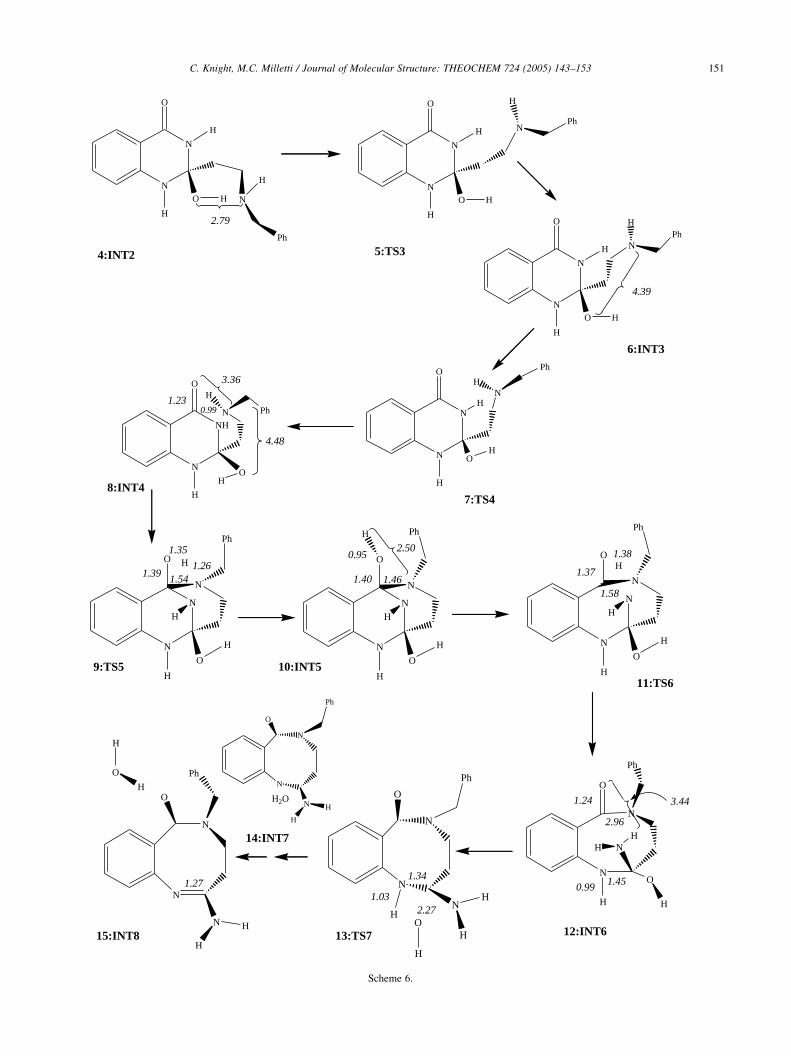
The reaction mechanism begins with 4:INT2, whose
formation is shown in Scheme 4. In this molecule, the
benzylamine nitrogen and the hydroxyl oxygen lie 2.79 A
apart. Structures 5–8 involve dihedral angles rotating in a
way that places the nitrogen of the benzylamine in a position
to attack the carbon of the carbonyl. Transition state 5:TS
(4/6) is at the top of a 5.2 kcal molK1 activation barrier. In
structure 6, 11.6 kcal molK1 higher in energy than the
Michael product, the N–O distance has increased to 4.39 A
because of rotation of the dihedral angle adjacent to
the six-membered ring from !NCCCz608 in structure 4
to !NCCCzK488 in structure 6. The second transition
state, 7:TS (6/8), has an activation barrier of 3.6 kcal -
molK1. In structure 8, the N–O distance has now increased
to 4.48 A and the distance between the benzylamine
nitrogen and the carbonyl carbon is now 3.36 A. Structure
8 is 12.3 kcal molK1 higher in energy than the Michael
product.
Transition state 9:TS (8/10), with an activation barrier
of 83.8 kcal molK1, involves the formation the C–N bond
and cleavage of the N–H bond. The C–N bond has formed
with a length of 1.54 A (9) and the N–H distance has
increased from 0.99 (8) to 1.26 A (9). The C–O bond has
lengthened from 1.23 (8) to 1.39 A (9). In structure 10, the
C–N bond has shortened to 1.46 A and the N–H distance is
2.5 A. The hydrogen is now bonded to the carbonyl oxygen
with a bond length of 0.95 A, shortened from 1.35 A (9).
The C–O distance is now 1.40 A. This tricyclic structure
(10) lies 36.3 kcal molK1 above the Michael product.
Cleavage of the N–H bond to form the eight-membered
ring occurs via transition state 11:TS (10/12) which has an
activation barrier of 57.6 kcal molK1. In structure 11, the
C–N bond length has increased from 1.46 (10) to 1.58 A and
the O–H distance has increased to 1.38 A. Structure 12 is
22.4 kcal molK1 higher in energy than the Michael product.
The O–H distance has increased to 3.44 A and the C–N
distance is now 2.96 A. The C–O bond has shortened from
1.37 (11) to 1.24 A which is identical to the original bond
length in structure 8.
Cleavage of the hydroxyl group and the hydrogen bonded
to the nitrogen adjacent to the benzene ring produces the final
amidine product and water. The transition state involving this
cleavage has an activation energy of 46.4 kcal molK1. The
C–N bond has shortened from 1.45 (12) to 1.34 A (13). The
hydroxyl group is now detached from the molecule with a C–
O distance of 2.27 A. The N–H bond has lengthened from
0.99 (12) to 1.03 A (13). In structure 14, the water molecule
has been formed and is separated from the bicyclic amidine.
The C–N bond distance is now 1.27 A. Furthermore, the
water molecule can migrate to a different position thus
lowering the energy of the system. Structure 15 is more stable
by 5.2 kcal molK1 and lies 18.2 kcal molK1 higher in energy
than the starting Michael product.
4. Conclusions
The most stable conformation of the Michael products
was found to be linear, with the amide bond in the Z
conformation. From this optimized structure, the reaction
pathway for the intramolecular cyclization of the Michael
product to the 4-quinazolinone was characterized. The
reaction was found to have two intermediates and three
transition states. The second step has the highest activation
energy. Of the two possible products, the one with the double
bond in conjugation to the ring was found to be more stable
than the one with the double bond in conjugation with the
carbonyl group (by 0.7 kcal molK1 at the HF/6-31G level of
theory and by 11.8 kcal molK1 at the B3LYP/6-31G* level
of theory). The reaction pathway for the formation of the
bicyclic amidine was found to have a higher activation

C. Knight, M.C. Milletti / Journal of Molecular Structure: THEOCHEM 724 (2005) 143–153 153
barrier than the quinazolinone pathway (by 14.6 kcal molK1
at the HF/6-31G level of theory and by 33.0 kcal molK1 at the
B3LYP/6-31G* level of theory). This agrees with experi-
mental observations, since the products isolated are the
energetically more favorable 4-quinazolinones. The reaction
path energy diagrams for the 4-quinazolinone and bicyclic
amidine are compared in Fig. 8.
Acknowledgements
We are grateful to Dr Arthur Howard for suggesting the
problem and many helpful discussions.
References
[1] P. Selveraj, A.S. Howard, unpublished results.
[2] C. Knight, Honors Undergraduate Thesis, Eastern Michigan Univer-
sity, 2003.
[3] H.B. Schlegel, J. Comput. Chem. 3 (1982) 214.
[4] (a) R. Ditchfield, W.J. Hehre, J.A. Pople, J. Chem. Phys. 54 (1971)
724;
(b) P.C. Hariharan, J.A. Pople, Theor. Chim. Acta 28 (1973) 213;
(c) P.C. Hariharan, J.A. Pople, Mol. Phys. 27 (1974) 209;
(d) M.S. Gordon, Chem. Phys. Lett. 76 (1980) 163;
(e) R.C. Binning Jr., L.A. Curtiss, J. Comput. Chem. 11 (1990)
1206.
[5] (a) C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785;
(b) A.D. Becke, Phys. Rev. A 38 (1988) 3098;
(c) B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett. 157
(1989) 200;
(d) A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
[6] (a) C. Peng, P.Y. Ayal, H.B. Schlegel, M.J. Frisch, J. Comput. Chem.
17 (1996) 49;
(b) C. Peng, H.B. Schlegel, Isr. J. Chem. 33 (1994) 449.
[7] M.J. Frisch, et al., GAUSSIAN 98, Revision A.7, Gaussian Inc., 1998.
[8] A.B. Nielson, A.J. Holder, GAUSSVIEW, v2.0, Gaussian Inc., 1997.
[9] M.J. Frisch, et al., GAUSSIAN 94, Gaussian Inc., 1992–1995.
[10] N.L. Allinger, E.L. Elliel (Eds.), Topics in Stereochemistry, Wiley,
New York, 1967.





























![Supporting Information Catalyzed C-H Activation Strategy … · 2019-04-30 · Supporting Information Intramolecular Cyclization of Imidazo[1,2-a]pyridines via a Silver Mediated](https://img.pdfslide.us/doc/110x75/5e3becfe3772c21a8722d017/supporting-information-catalyzed-c-h-activation-strategy-2019-04-30-supporting.jpg)