Embed Size (px)
Citation preview

The vagus nerve as a modulator of intestinal inflammation
The vag
us n
erve as a modulator ofi
ntestin
al inflam
mation
Esm
erij van d
er Zan
den
Esmerij van der Zanden

The vagus nerve as a modulator ofintestinal inflammation
Esmerij van der Zanden
van de Zanden.indb 1 23-5-2011 11:43:40

The vagus nerve as a modulator of intestinal inflammationThesis, University of Amsterdam, The Netherlands
ISBN: 978-90-9026193-5Cover: Confocal microscopy of macrophages (red) and cholinergic nerve fibers
(green) around the myenteric plexus of rat ileum. Lay-out: Chris Bor, Medical photography and Illustration, Academic Medical
Center Amsterdam, The NetherlandsPrinted by: Printservice-Ede
Copyright © E.P.M. van der Zanden, [email protected], Amsterdam, The NetherlandsAll right are reserved. No part of this thesis may be reproduced or transmitted in any form or by any means without permission of the author or the publishers of the included scientific papers.
The research described in this thesis was performed at the Department of Gastroenterology and Hepatology, AMC, Amsterdam
The printing of this thesis was financially supported by AMC, Nederlandse Vereniging voor Gastroenterologie (NVGE), Section Experimental Gastroenterology of the NVGE, Olympus Nederland B.V., Tramedico B.V., Ferring B.V., Dr. Falk Pharma Benelux B.V. Abbott Immunology
van de Zanden.indb 2 23-5-2011 11:43:40

The vagus nerve as a modulator ofintestinal inflammation
ACADEMISCH PROEFSCHRIFT
ter verkrijging van de graad van doctoraan de Universiteit van Amsterdamop gezag van de Rector Magnificus
prof. dr. D.C. van den Boomten overstaan van een door het college voor promoties ingestelde
commissie in het openbaar te verdedigen in de Agnietenkapelop donderdag 23 juni 2011, te 14:00 uur
door
Esmerij Petronella Maria van der Zandengeboren te De Lier
van de Zanden.indb 3 23-5-2011 11:43:40

Promotiecommissie
Promotores: Prof. dr. G.E.E. Boeckxstaens
Co-promotor: Dr. W.J de Jonge
Overige leden: Dr. G.R. van den Brink Prof. dr. E. Fliers Prof. dr. T. van der Poll Prof. dr. M. Schemann Prof. dr. H. Spits Prof. dr. P.P. Tak
Faculteit der Geneeskunde
van de Zanden.indb 4 23-5-2011 11:43:40

Voor mijn vader
van de Zanden.indb 5 23-5-2011 11:43:40

Table of contents
Chapter 1 Introduction and outline of the thesis 9
Chapter 2 The vagus nerve as a modulator of intestinal inflammation 17
Chapter 3 Stimulation of the vagus nerve attenuates macrophage activity by activating the JAK-2-STAT-3 signaling pathway
39
Chapter 4 Deciphering STAT3 signaling in cholinergic inhibition of inflammation
63
Chapter 5 Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor α4β2
77
Chapter 6 Cholinergic agonists can interact with immunomodulatory actions of neuropeptides VIP and SP
99
Chapter 7 Effects of smoking on nicotinic AChR expression and susceptibility to cholinergic immunomodulation in human monocytes
111
Summary and conclusions 131
Nederlandse samenvatting 141
List of contributing authors 151
List of publications 155
Dankwoord 159
Curriculum vitae 166
van de Zanden.indb 7 23-5-2011 11:43:40

1CHAPTER
Introduction and outline of the thesis
van de Zanden.indb 9 23-5-2011 11:43:40

11
Introduction and outline
The aim of this thesis is to further unravel the working mechanism of the so-called ‘cholinergic anti-inflammatory pathway’, specifically in intestinal inflammation. The gut homes our largest collection of microbes, with up to 1012 organisms packed together per milliliter of luminal content. Therefore, especially in the intestine, inflammatory reactions need to be tightly regulated since exaggerated inflammation may lead to tissue damage and morbidity, while at the same time the tissue needs to be protected from invading pathogens. The control of inflammation is realized by two major mechanisms: self-controlling innate immune mechanisms and brain-derived immunoregulatory output. Evidence is mounting that the parasympathetic nervous system comprised by the vagus nerve is a potent player in neuro-immune inflammation. The afferent vagus system is known to detect and subsequently regulate the inflammatory response via activation of the hypothalamic pituitary adrenal axis. However, more recent evidence reveals that efferent vagus nerve cholinergic activity exerts quite potent immuno-modulatory potential as well1.
A decade ago, Borovikova et al reported that acetylcholine, the principle neurotransmitter of the vagus nerve, can attenuate pro-inflammatory cytokine release in macrophages1. Moreover, they demonstrated that electrical stimulation of the vagus nerve attenuates the systemic inflammatory response to endotoxin. This implies an immune regulating role for the vagus nerve, and is referred to as ‘the cholinergic anti-inflammatory pathway’. In line, in several experimental animal models of inflammatory disease, such as ischaemia-reperfusion injury2, post-operative ileus3, haemorrhagic shock 4, peritonitis5, pancreatitis6, and experimental arthritis7, activation of the cholinergic nervous system inhibits inflammatory processes and ameliorates disease. Especially in the gastrointestinal tract, which is under strict control of the vagus nerve8,9, vagus nerve stimulation has been extensively studied as a novel approach to treat intestinal inflammatory conditions. Chapter 2 is a review in which the role of the vagus nerve as a regulator of intestinal inflammation is comprehensively discussed.
Acetylcholine, the principle neurotransmitter released by the vagus nerve, can exert its anti-inflammatory effect via binding to nicotinic acetylcholine receptors (nAChRs), which are expressed on macrophages and other immune cells10, 11. Wang et al published that acetylcholine or nicotine specifically interact with α7 cholinergic receptors to inhibit TNFα production12, but the exact intracellular mechanism remained unclear. In chapter 3, we further investigated via which intracellular signaling pathways nicotine exerts its anti-inflammatory effect on peritoneal macrophages. Moreover, we corroborated our in vitro findings using an in vivo mouse model of postoperative ileus. We conclude that stimulation of the vagus nerve attenuates macrophage activation by activating the intracellular JAK2-STAT3 signaling pathway. In chapter 4, we further analyzed the role of the JAK2-STAT3 pathway in the anti-inflammatory potential of nAChR activation.
van de Zanden.indb 11 23-5-2011 11:43:41

12
Chapter 1
The immune system of the gut faces the challenge of discriminating between self and non-self in order to elicit a proper response against pathogens, but must at the same time tolerate mutual beneficial organisms and food antigens. Antigen presenting cells such as macrophages and dendritic cells are thought to be crucial in maintaining this balance. Intestinal macrophages are the first phagocytic cells of the immune system to interact with microorganisms that have breached the epithelium. Smythies et al showed that human resident intestinal macrophages are rather phagocytes than cytokine producers13. Therefore, in chapter 5, we examined whether the anti-inflammatory potential of vagus nerve activity in intestinal inflammation solely rests on reduced macrophage cytokine production, or if other important macrophage functions such as endocytosis and phagocytosis are affected. Furthermore, we examined the nicotinic acetylcholine receptor type and signaling pathways involved in these processes. Finally, we studied the effects of vagal signaling on phagocytosis and uptake in vivo.
In chapter 6, we tried to analyze how vagus nerve activity can modulate immune cells in vivo. In chapter 3, we showed that cholinergic nerve fibers are in close anatomical apposition to macrophages in the small intestine. However, since the half-life of acetylcholine is very short, one may question if acetylcholine that is released at the vagal termini actually reaches the immune cells in a quantity that could explain the in vitro effects. We hypothesized that, next to the direct anti-inflammatory effects of vagus nerve derived acetylcholine on nAChR on tissue macrophages, vagus nerve stimulation could affect immune cells via post-ganglionic mechanisms that lead to the release of alternative neurotransmitters, such as neuropeptides. Vasoactive intestinal polypeptide (VIP) and substance P (SP) are neuropeptides that are abundantly expressed in the gut, and elicit important immunomodulatory functions in the intestine14-16. Therefore, in chapter 6, we investigated whether vagus nerve released acetylcholine negatively regulates macrophage reactivity directly, or by modulating the responses to co-released VIP or SP.
All previous data were conducted in cell lines, mouse macrophages and experimental mouse models. Data from human studies considering cholinergic immunomodulation are limited. Is has been long known that cigarette smoking is an important environmental factor in ulcerative colitis, as smoking appears to have a protective effect in the development of disease and reduces its severity17. However, clinical trials of nicotine treatment in ulcerative colitis have shown variable outcomes18. As previous data suggest that nicotinic receptor α7 may specifically participate in the inflammatory response of monocytes12, in chapter 7, we evaluated whether smoking, or repeated nicotine exposure affected nAChR α7 monocyte expression, and whether this renders human monocytes more susceptible to cholinergic immune-modulation. Luyer et al demonstrated in a hemorrhagic shock rat model, that high fat enteral nutrition could inhibit the inflammatory response by way of efferent vagal fibers19.
van de Zanden.indb 12 23-5-2011 11:43:41

13
Introduction and outline
Hence, the second aim of chapter 7 was to establish the effect of oral olive oil diet on human whole blood LPS-induced cytokine production.
van de Zanden.indb 13 23-5-2011 11:43:41

14
Chapter 1
REFERENCE LIST
1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
2. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, Tracey KJ. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg 2002;36:1231-1236.
3. The FO, Boeckxstaens GE, Snoek SA, Cash JL, Bennink R, Larosa GJ, van den Wijngaard RM, Greaves DR, De Jonge WJ. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 2007;133:1219-1228.
4. Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, Squadrito G, Minutoli L, Bertolini A, Marini R, Adamo EB, Venuti FS, Squadrito F. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation 2003;107:1189-1194.
5. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der PT. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis 2005;191:2138-2148.
6. van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, Tracey KJ, van der PT. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006;130:1822-1830.
7. van Maanen MA, Lebre MC, van der Poll T, Larosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-122.
8. Altschuler SM, Ferenci DA, Lynn RB, Miselis RR. Representation of the cecum in the lateral dorsal motor nucleus of the vagus nerve and commissural subnucleus of the nucleus tractus solitarii in rat. J Comp Neurol 1991;304:261-274.
9. Altschuler SM, Escardo J, Lynn RB, Miselis RR. The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology 1993;104:502-509.
10. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 2007.
11. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999;266:17-20.
12. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
13. Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 2005;115:66-75.
14. De la FM, Delgado M, Gomariz RP. VIP modulation of immune cell functions. Adv Neuroimmunol 1996;6:75-91.
15. O’Connor TM, O’Connell J, O’Brien DI, Goode T, Bredin CP, Shanahan F. The role of substance P in inflammatory disease. J Cell Physiol 2004;201:167-180.
16. Renzi D, Pellegrini B, Tonelli F, Surrenti C, Calabro A. Substance P (neurokinin-1) and neurokinin A (neurokinin-2) receptor gene and protein expression in the healthy and inflamed human intestine. Am J Pathol 2000;157:1511-1522.
17. Lakatos PL, Szamosi T, Lakatos L. Smoking in inflammatory bowel diseases: good, bad or ugly? World J Gastroenterol 2007;13:6134-6139.
van de Zanden.indb 14 23-5-2011 11:43:41

15
Introduction and outline
18. van der Zanden EP, Boeckxstaens GE, De Jonge WJ. The vagus nerve as a modulator of intestinal inflammation. Neurogastroenterol Motil 2009;21:6-17.
19. Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 2005;202:1023-1029.
van de Zanden.indb 15 23-5-2011 11:43:41

2CHAPTER
The vagus nerve as a modulator of intestinal inflammation
Esmerij P. van der Zanden Guy E. Boeckxstaens Wouter J. de Jonge
Neurogastroenterology and Motility 2009 Jan;21(1):6-17.
van de Zanden.indb 17 23-5-2011 11:43:41

18
Chapter 2
ABSTRACT
The cholinergic nervous system attenuates the production of proinflammatory cytokines and inhibits inflammatory processes. Hence, in animal models of intestinal inflammation, such as post-operative ileus and DSS-induced colitis, vagus nerve stimulation ameliorates disease activity. On the other hand, in infectious models of microbial peritonitis, vagus nerve activation seemingly acts counteractive; it impairs bacterial clearance and increases mortality. It is originally indicated that the key mediator of the cholinergic anti-inflammatory pathway, acetylcholine, inhibits cytokine release directly via the α7 nicotinic acetylcholine receptor (nAChR) expressed on macrophages. However, more recent data also point towards the vagus nerve as an indirect modulator of innate inflammatory processes, exerting its anti-inflammatory effects via postganglionic modulation of immune cells in primary immune organs. This review discusses advances in the possible mechanisms by which the vagus nerve can mediate the immune response, as well as the role of nAChR activation and signaling on macrophages and other immune cells.
van de Zanden.indb 18 23-5-2011 11:43:41

19
The vagus nerve a modulator of intestinal inflammation
VAGUS MODULATION OF IMMUNE RESPONSES
The innate immune system is pivotal in the first response to invading pathogens or tissue trauma. When challenged, the host needs an adequate inflammatory reaction but also needs to prevent collateral damage to tissues due to excessive systemic spread of inflammation and release of inflammatory mediators. Hence, regulation of the acute inflammatory response is important, and regulatory mechanisms are required to accomplish this. Decades ago, the sympathetic nervous system was already identified as a ‘hard-wired’ counter-regulatory mechanism that can locally regulate immune responses1. Besides the sympathetic nervous system, the parasympathetic nervous system, comprised by the vagus nerve (the largest nerve in the body) is increasingly recognized as a potent player in neuro-immune inflammation. The vagus nerve, in addition to its classically assigned function of controlling heart rate, hormone secretion, gastrointestinal peristalsis, and digestion, may also be involved in control of immune responses to commensal flora and dietary components. The vagus nerve is a mixed nerve composed of 90% afferent and 10% efferent fibers. The afferent vagus system is known to regulate the inflammatory response via activation of the hypothalamic pituitary adrenal axis. However, more recent evidence reveals that efferent vagus nerve cholinergic activity exerts quite potent immuno-modulatory potential as well2.
The vagus nerve transmits signals by releasing acetylcholine (ACh), its principal neurotransmitter, at its peripheral nerve endings. ACh activates nicotinic acetylcholine receptors (nAChRs), ligand-gated ion channels on neuronal cells3. In this function, ACh is historically referred to as a neurotransmitter. Immune cells that have been shown to be especially sensitive to modulation by vagus nerve activity are macrophages (the main class of innate immune cells)2. Macrophages express nAChRs and potently respond to ACh. This was corroborated by our observation of close anatomical association between cholinergic nerve fibers and enteric macrophages4. These data suggest that the classical neurotransmitter ACh also functions as neuro-immune cytokine, providing a molecular basis for the purported “neuro-immune axis” between the brain and immune system. The initial experiments to show the role of the parasympathetic nervous system in the regulation of the innate immune response were performed in a rat model of experimental sepsis2. In these experiments is was shown that surgical dissection of the vagus nerve enhanced pro-inflammatory cytokine production and accelerated the development of septic shock, whereas electrical stimulation of the efferent vagus nerve prevented systemic inflammation and reduced lethality2. Subsequently, in several studies it was demonstrated that activation of the cholinergic nervous system ameliorated disease in animal models of ischemia–reperfusion injury5, hemorrhagic shock6, and experimental arthritis7, pancreatitis8, peritonitis9 and DSS-colitis10. It is hypothesized that the vagus nerve exerts anti-inflammatory effects through the interaction of its principal
van de Zanden.indb 19 23-5-2011 11:43:41

20
Chapter 2
neurotransmitter ACh with acetylcholine receptors expressed on macrophages. The group of Tracey2 have originally showed that in vitro, ACh inhibited the endotoxin-induced release of pro-inflammatory cytokines in human macrophages2. Since ACh signals through nicotinic or muscarinic receptors, selective agonists and antagonists have been used to identify the receptors involved in the immunomodulatory effects of ACh. Nicotine was as efficient as ACh in inhibiting pro-inflammatory cytokine production in macrophages, indicating that the anti-inflammatory effects of ACh on immune cells are mediated through nicotinic receptors, rather than muscarinic receptors. nAChR are pentameric ligand-gated ion channels that can consist of a large number of different subunits (α1-α9, α8, β1-β4, γ , δ and ε)11 and it is reported that the nAChR α7 subtype, which is expressed on immune cells, is essential in mediating the anti-inflammatory effect of ACh12.
From the pioneering work of Tracey13 it is postulated that the cholinergic anti-inflammatory pathway may act as part of a anti-inflammatory reflex arch, in which the presence of proinflammatory cytokines in the periphery activates vagus afferents, resulting in a vagus efferent firing subsequently leading to an attenuation of cytokine release from macrophages via nAChR α7. On the other hand, recent data indicate that the efferent arm of the cholinergic anti-inflammatory pathway may, at least in part be mediated via post-ganglionic events14. Here we review the recent reports on the possible pathways via which vagus nerve activity can exert its anti-inflammatory effects, with specific focus on intestinal disease. Moreover, the role of nAChR expressed on macrophages as well as other immune cells in intestinal and peritoneal tissue in the immunomodulatory effects of cholinergic signaling are highlighted.
VAGUS NERVE SIGNALING IN INTESTINAL INFLAMMATION
Neuronal tracing studies reveal that efferent vagus nerve fibers innervate the small intestine and proximal colon of the gastro-intestinal (GI) tract15,16, leaving the possibility that cholinergic activity may modulate immune cells residing in, or recruited to, the densely innervated bowel wall. Therefore, vagus nerve stimulation, or the use of applied cholinergic agonists targeting distinct nAChR subtypes, has been studied extensively as a novel approach to treat intestinal inflammatory disease in several animal models.
One of the GI-disorders in which vagus nerve stimulation has been shown to ameliorate disease is post-operative ileus (POI)4,17. POI is a commonly occurring post-operative complication that results from manipulation of the bowel during abdominal surgery, and is characterized by a transient hypomotility of the GI tract. With ~22 million surgical procedures being performed annually in the US (data derived from hospital discharge records between 1980 and 199318), and with all of these procedures
van de Zanden.indb 20 23-5-2011 11:43:41

21
The vagus nerve a modulator of intestinal inflammation
causing some degree of POI, treatment strategies that can accelerate the recovery from POI represent an important unmet clinical need19. In rodent models of POI, intestinal manipulation leads to leukocyte influx into the muscularis externa, resulting in delayed gastric emptying20 and impaired small intestinal transit21,22. Electrical stimulation of the vagus nerve can reduce recruitment of neutrophils and restore gastric emptying in mice4,17. The anti-inflammatory effect attained with electrical vagus nerve stimulation can be mimicked by AR-R17779, which specifically targets the nAChR α7 subunit17.
Ingestion of dietary fat stimulates the production of cholecystokinin (CCK), which is a characteristic hormone released during ingestion to trigger several digestive functions including exocrine pancreas secretion, and activation of afferent vagus nerve signals to induce satiety. Interestingly, a recent study indicated that CCK, released as a result of high-fat enteral nutrition, inhibited hemorrhagic shock-induced TNFα and interleukin-6 release23. This anti-inflammatory effect of CCK release is mediated by the vagus nerve because surgical or chemical vagotomy abrogates the anti-inflammatory effect of high fat diet and CKK23. Along the same line, in mouse models of pancreatitis8, vagotomy exacerbates inflammation, and this effect is counteracted by pretreatment with nicotine or GTS-12, another selective nAChR α7 agonist8. These results demonstrate an important role for the nAChR α7 subunit in mediating the ‘cholinergic anti-inflammatory effect’. Correspondingly, in experimental models of acute colitis, the vagus nerve seems to possess regulatory properties in inflammatory responses. Several studies show that nicotine administration attenuates disease in TNBS and DSS colitis models, although fairly high doses of nicotine are required24,25. Ghia et al.10 demonstrate that acute colitis is more severe in vagotomized mice and in mice treated with the nAChR antagonist hexamethonium. Conversely, nicotine treatment resulted in reduction of the inflammatory response, independent of vagus nerve intactness, indicating that cholinergic signaling can be protective in animal models of experimental colitis. In addition, vagotomized nAChR α7 knock-out (KO) mice display more severe colitis than wild type (WT) mice, and nicotine pretreatment only attenuates disease activity in WT mice26, pointing towards a role for the nAChR α7 in this process. However, in another study, experimental colitis is aggravated in nAChR α5 subunit-deficient mice27, suggesting that not only the nAchR α7, but also other nAChR subunits can participate in the vagus modulation of colitis in mice. Finally, in IL10 KO mice, that develop colitis spontaneously, nicotine administration resulted in reduced colitis but enhanced jejunal inflammation28. Overall, cholinergic activation can reduce inflammation and disease activity in various animal models of intestinal inflammation, likely via a mechanism involving activation of nAChRα7 subtype, although this receptor may not be the sole nAChR involved.
van de Zanden.indb 21 23-5-2011 11:43:41

22
Chapter 2
THE VAGUS NERVE AND CHOLINERGIC EFFECTS ON GUT EPITHELIUM
It is now well established that cholinergic enteric neurons participate in epithelial transport as well as mucosal immune defense. The intestinal epithelium is continuously exposed to a plethora of luminal antigens. The human gut harbors an estimate of 1014
microbes of 400 different species in the digestive tract29, and the intestinal immune system has to fight invading pathogens while remaining tolerant to the beneficial flora and the many encountered food antigens. Under healthy conditions, specialized cells such as M-cells or CX3CR positive dendritic cells protruding through the epithelial layer of normal mucosa or Peyer’s patch, act as gatekeepers to the mucosal immune system30. However, penetration of the mucosal barrier by luminal antigens does occur under pathological conditions, and regulatory mechanisms of epithelial permeability are a key factor in the balance between immunosurveillance and inflammation of the gut. For example during episodes of stress, inflammation or trauma, impairment of the epithelial barrier function is increasingly acknowledged as a key perpetuating factor in the pathogenesis of inflammatory bowel disease (IBD), food allergy and celiac disease31. Many hypotheses exist on the regulatory mechanisms behind these permeability changes32, but interestingly, several studies indicate that cholinergic nerve activity plays a significant role in gut permeability.
Although theoretically, stress is associated with a strong sympathetic nervous system response, studies in rodents have revealed that both acute and chronic exposure to stress can increase epithelial permeability33 via cholinergic mechanisms. First of all, stress-susceptible rats have lower activity of cholinesterase in intestinal mucosa than less susceptible rats, leading to higher levels of mucosal ACh34, which may account for altered epithelial barrier function in stress-susceptible rats34. Second, the cholinergic muscarinic receptor antagonist atropine abolishes stress-induced epithelial barrier damage in rats, where nicotinic antagonists have no effect. This suggests that the cholinergic effects on epithelial barrier function are mediated via muscarinic, rather than nicotinic acetylcholine receptors. In stripped rat ileal epithelium mounted in Ussing chambers, cholinergic stimulation increases epithelial transport by disrupting tight junction integrity and induces the uptake of intact protein via endocytosis, which can be counteracted by atropine35. In line, in jejunal mouse tissue, muscarinic receptor activation increases epithelial permeability to macromolecules via enhanced apical endocytosis36. This is probably mediated via activation of muscarinic 3 receptors on epithelial cells and subsequent activation of phospholipase A2 and cyclooxygenase metabolites36. In contrast to rat ileum35, tight junction integrity is not affected by cholinergic signaling in mouse jejunum36, which can probably be explained by species differences or variable pharmacological conditions. In rabbit jenunum, vagus nerve stimulation increases intestinal epithelial
van de Zanden.indb 22 23-5-2011 11:43:41

23
The vagus nerve a modulator of intestinal inflammation
permeability, resulting in the passage of serum proteins into the lumen, possibly by opening tight junctions and paracellular pathways37.
On the other hand, other animal studies show that vagus nerve activity can be protective in maintaining gut barrier function under pathological conditions. Hemorrhagic shock results in gut barrier failure leading to translocation of endotoxin and bacteria. Luyer et al demonstrate that administration of high-fat nutrition, leading to the release of CCK inhibits bacterial translocation, reduces disruption of the tight junctions and attenuates TNFα and IL-6 production in hemorrhagic shock rat model38. High fat nutrition failed to reduce the inflammatory response in VGX mice23, indicating that this effect required CCK-induced activation of the vagus nerve. The observed maintenance of epithelial barrier integrity after vagus nerve stimulation may be a direct effect of cholinergic signaling, or indirect, via the reduced pro-inflammatory cytokine release39.
Altogether, the epithelial barrier function is affected by cholinergic signaling, presumably via activation of muscarinic receptors expressed on epithelial cells. Differential effects of cholinergic signaling on tight junction integrity are reported. It is important to consider that altered barrier function may not necessarily be indicative of pathophysiology, but could also be a physiologically adaptive response to increase luminal antigen sampling.
THE VAGUS NERVE MODULATING THE RESPONSE TO PERITONEAL BACTERIAL INFECTION AND CLEARANCE
The function of vagus nerve firing has been well established in models of sterile inflammation, in which vagus nerve stimulation attenuates inflammation by dampening immune cell activation. However, in infectious diseases, such as bacterial peritonitis, or bacterial sepsis, the host defense is a delicate balance between pro-inflammatory pathways aimed at the rapid elimination of bacteria and anti-inflammatory responses to prevent systemic inflammation. Therefore, the role of cholinergic modulation of the immune response in microbial infection and bacterial clearance is an important topic of interest.
In mice that underwent cecal ligation and puncture (CLP), causing lethal peritonitis induced by a polymicrobial infection, nicotine administration attenuates clinical symptoms of sepsis and improves survival40. These effects can be contributed to acute reduction of HMGB-1 and pro-inflammatory cytokines, rather than effects on bacterial outgrowth as mice received nicotine 24hrs after CLP. Moreover, to mimic the clinical scenario, these mice received antibiotics shortly after CLP, obscuring the potential effect of cholinergic signaling on bacterial outgrowth. In a septic peritonitis model induced by ip injection of E.Coli, vagotomy exaggerates, whereas nicotine reduces pro-inflammatory cytokine release, neutrophil influx and liver damage9. Interestingly,
van de Zanden.indb 23 23-5-2011 11:43:41

24
Chapter 2
nicotine treatment impairs bacterial clearance and significantly enhances mortality during this E.Coli induced peritonitis9. In line with this, in vitro studies indicate that nicotine can significantly impair antimicrobial activity of macrophages41. In contrast to nicotine, unilateral vagotomy does not affect bacterial outgrowth and survival in E.Coli induced peritonitis9. Bilateral vagotomy however, tested in a polymicrobial colon ascendens stent peritonitis model, has no effect on bacterial outgrowth, but does result in significantly increased mortality42.
Mice that are deficient for the nAChR α7 subunit had enhanced neutrophil recruitment in early infection with E.Coli in comparison to WT mice43. These results are in accordance with the anti-inflammatory properties of cholinergic nAChR α7 signaling12. However, 20 hrs after infection, nAChR α7 KO mice displayed an accelerated bacterial clearance compared to WT mice43. As a result of reduced bacterial loads, α7 nAChR KO mice had reduced numbers of infiltrating neutrophils and lower circulating cytokine levels at that time point43. These data suggest that stimulation of cholinergic α7 nAChRs reduces early neutrophil migration to the site of infection, finally resulting in a reduction of bacterial clearance and decreased survival. Although vagus nerve stimulation can reduce excessive inflammation and acute reaction to sepsis, vagus nerve firing and nAChR α7 receptor activation can have a detrimental effect on host defense against bacteria.
HOW DOES VAGUS NERVE STIMULATION MODULATE THE IMMUNE RESPONSE IN VIVO?
Several lines of evidence indicate that vagus nerve stimulation can inhibit immune cell activation and modulate inflammation via its peripheral release of ACh. Many reports point towards the macrophage nAChR α7 as an essential player in mediating the anti-inflammatory effect of ACh4,8,12,17,44-47. Specifically, nicotine exerts anti-inflammatory effects on human macrophages that can be counteracted by specific nAChR α7 antagonists or anti-sense oligonucleotides12. In addition, nAChR α7 KO mice display enhanced TNF production compared to WT mice in an endotoxemia model, which cannot be counteracted by vagus nerve stimulation12. In various animal models of inflammation, nAChR α7 agonists ARR17779 and GTS-21 ameliorate disease8,17,48. These data point towards the nAChR α7 as a crucial player in cholinergic modulation of inflammation.
However, it remains to be elucidated if ACh released from vagus nerve termini actually reaches the immune cells, and if so, in what quantities. Given the short half-life of ACh, cholinergic modulation of immune cell activation most likely requires close contact. Although macrophages are found in close anatomical apposition to cholinergic fibers in rat small intestine4, there is currently no evidence that parasympathetic neurons indeed innervate macrophages. As the vagus nerve
van de Zanden.indb 24 23-5-2011 11:43:42

25
The vagus nerve a modulator of intestinal inflammation
mainly synapses with neurons of the enteric nervous system, it is very likely that the cholinergic terminals shown in proximity of macrophages may belong to enteric rather than vagus nerve fibers.
In this regard, recent data indicate that the spleen may play a role in effectuating the anti-inflammatory effects of vagus nerve activity, as electrical stimulation of the vagus nerve fails to attenuate serum TNF levels in splenectomized mice treated with endotoxin49. This implies that the parasympathetic nervous system may regulate systemic inflammation by modulating immune cells in the spleen. Huston et al. show that vagus nerve stimulation fails to regulate splenic TNF production in α7nAChR-deficient mice, and splenocytes from α7 KO mice do not respond to in vitro stimulation with ACh49. In another study it was demonstrated that nerve endings are in close apposition to macrophages in the spleen, but interestingly, these nerve fibers, found in apposition to the TNF-secreting macrophages are catecholaminergic, not cholinergic14. The authors propose that ACh released by the vagus nerve does not reach the spleen directly, but acts on α7 nAChR at the level of the ganglia of the celiac-superior mesenteric ganglion to modulate splenic nerve function14. Hence, the vagus nerve via this ganglion, modulates adrenergic input to the spleen (via the n. splenicus), resulting in the release of catecholamines that stimulate adrenergic receptors on splenic macrophages and attenuate LPS-induced TNF production14
Whether or not the vagus nerve innervates the spleen directly, or indirectly, is currently under debate. Rosas-Ballinas et al.14 base their conclusion that the spleen does not receive direct vagus nerve input on the observation that ACh, choline acetyltransferase and the vescicular ACh transporter, are absent from splenic nerve terminals. Although this conclusion may seem justified, Buijs et al50 show that the absence of the classically assigned vagus neurotransmitter ACh, or the ACh metabolizing enzymes in the spleen, does not directly imply the lack of direct input from the vagus. Indeed, they demonstrate that in rats, the spleen is directly innervated by the vagus nerve50. Their results corroborate earlier observations regarding parasympathetic innervation of the liver. In parallel, ACh metabolizing enzymes are absent from the liver, although this organ has shown to be vagusly innervated51-53.
Given the recently purported role of the spleen in mediating the anti-inflammatory effect of vagus nerve activity, the question arises as to what is the role of macrophage nAChR α7 residing in the peritoneal or intestinal compartment is in this process. The interesting recent data on the role of the spleen in vagus nerve immune modulation put the role of nAChR α7 receptors on macrophages in a new perspective: although in vitro nicotine modulates macrophages function via nAChR α7, the in vivo effects of vagus nerve stimulation may rely on nAChR α7 on neurons rather than macrophages. Further research is required to assess the physiological mechanism by which the vagus nerve can modulate immune responses and whether this modulation is indeed the result of direct affects of ACh exposure to immune cells or whether vagus innervation of primary or secondary lymphoid organs plays a role (Figure 1).
van de Zanden.indb 25 23-5-2011 11:43:42

26
Chapter 2
WHICH nAChR IS INVOLVED IN THE VAGUS MODULATION OF IMMUNE CELLS?
The anti-inflammatory effects of nAChR activation on macrophages have previously been solely attributed to activation of the nAChR α712. However, some reports indicate that vagus activity may also convey its anti-inflammatory effect via distinct nAChRs expressed in macrophages. Mastunaga et al41 and others54 have shown expression of α4β2, but not α7 nAChR in alveolar macrophages. Likewise, we have failed to detect α7 nAChR transcripts in certain mouse macrophages types (unpublished 2008). Further analysis of potential α7 nAChR protein in these macrophages is hampered by the fact that commercially available α7 nAChR antisera seem not specific and stain an 57kD protein in brain homogenates from wildtype as well as α7 nAChR -/- mice55. Most previous studies, including our own4, have ascribed the effects of ACh and nicotine on peritoneal macrophage cytokine production solely to activation of
Figure 1. Hypothetical scheme of the mechanism via which vagus nerve activity can modulate immune cells in vivo. Vagal efferent fibres, that originate in the brainstem, release acetylcholine upon physiological or electrical stimulation. Vagal nerve stimulation may affect immune cells via direct release of acetylcholine, or via post-ganglionic mechanisms involving alternative neurotransmitters. This activates a connecting interneuron, resulting in the release of acetylcholine or other neurotransmitters, such as neuropeptides or catecholamines. Acetylcholine can act on nAChR which are broadly expressed on different types of immune cells, such as tissue macrophages. Possibly, neuropeptides and catecholamines acting on their specific receptors may be important as well. Receptor activation can lead to reduced pro-inflammatory cytokine production, abrogated microbial killing and enhanced phagocytosis. This skews immune cells to a more anergic phenotype, which can be beneficial in disorders that are characterized by an aberrant immune response.
van de Zanden.indb 26 23-5-2011 11:43:43

27
The vagus nerve a modulator of intestinal inflammation
the α7 nAChR, although expression of alternative nAChR subtypes on macrophages has been described54,56. Accordingly, we recently observed that α7 specific agonists are less effective in reducing pro-inflammatory cytokine production as compared to nicotine17. Surprisingly, α7 nAChR blockers are effective in counteracting nicotinic effects on pro-inflammatory cytokine production. These observations may question the selectivity of commonly used blockers αBgt and MLA for α7 nAChR. In fact, both blockers have been shown to bear affinity for other nAChR subunits, including α1, α6, α9, α10 and β2, as well57.
CHOLINERGIC MODULATION OF IMMUNE CELLS IN THE GUT
Several immune cells express various nAChR subtypes44, and other nAChR subtypes than nAChR α7 may play a more prominent role than originally assumed. Therefore, even though it is not clear how ACh released by the vagus nerve can directly interact with immune cells in vivo, it is plausible that different types of immune cells are sensitive to ACh. Here, we briefly describe effects of nAChR activation on macrophages, dendritic cells and mast cells.
Macrophages and monocytesIn response to inflammatory signals, monocytes can migrate from the bloodstream into the tissue and differentiate into macrophages. Macrophages play a fundamental role in early recognition of pathogens and the most important macrophage functions are ingestion of bacteria and debris, killing, and secretion of inflammatory mediators. Most research about cholinergic modulation of the immune response has focused on macrophages, and indeed macrophages are very responsive to ACh and nicotine. Macrophages and monocytes express the nAChR α2-α7, α9, α10 and β2-β454 , although the expression pattern is dependent on the type of macrophage and the tissue were it resides. In addition, in human monocytes and monocyte-derived macrophages, several splice variants and two different isoforms of α7 nAChR are detected. Six mRNA splice variants of the α7 gene have been described in human brain as well as leukocytes58-61, though it is uncertain whether any of these transcripts are processed to functional protein61. Interestingly, the human α7-nAChR has been described to be partially duplicated on this chromosome. Exons 5 to 10 of the gene have been duplicated in a “tail-to-head” orientation and this partially duplicated gene is combined with four novel exons (A to D) to comprise a new gene, the “hybrid alpha7” or the “cholinergic receptor family with sequence similarity 7A” (CHRFAM7A)61. Although it is reported that this gene is transcribed as a 45 kD protein (i.e. in human leukocytes)61, it remains
van de Zanden.indb 27 23-5-2011 11:43:43

28
Chapter 2
unclear whether this hybrid transcript is appropriately translated and processed to form a functional receptor.
In macrophages and monocytes of various species, nicotine alters inflammatory properties. Several studies describe nicotinic inhibition of the secretion of pro-inflammatory mediators, such as TNF, IL-6, IL1β, HMGB-1 and PGE22,4,40. In monocytes, nicotine not only abrogates production of pro-inflammatory cytokines, but shifts the response to a IL-10 dominant anti-inflammatory profile62, while studies in macrophages report no difference in IL-10 production2,63. Transcript levels of inflammatory mediators remain unchanged, suggesting a post-transcriptional effect of nicotine. Nicotine suppresses expression of CD14 and toll-like receptor 4 (TLR4) on monocytes, shifting the cells to a ‘deactivated state’ which can explain the nicotinic modulation of LPS-induced cytokine production63. In contrast, one study reports that nicotine increases transcript levels and production of TNF, IL-1Beta and iNOS64, while another shows that nicotine stimulates iNOS expression and NO65 production in mouse peritoneal macrophages, thus inducing inflammation.
Data on the effects of nicotine on macrophage-mediated functions, such as phagocytosis and bacterial killing, are limited. Matsunaga et al41 demonstrate that antimicrobial activity of alveolar macrophages to Legionella pneumophila infection is suppressed by nicotine. Some decades earlier, it was shown that nicotine partially inhibits endocytosis, pinocytosis, uptake and intracellular degradation and phagocytosis by macrophages66-68. This can partly be explained by the fact that nicotine accumulates in the lysosomes which impaires the digestive capacity69. Nevertheless, we have strong evidence that nicotine augments phagocytosis by intestinal macrophages, while pro-inflammatory cytokine release and NF-κB activation are decreased (van der Zanden et al, submitted). In conclusion, most reports show that nicotine attenuates pro-inflammatory cytokine release by macrophages and enhances phagocytosis, but inhibits antimicrobial killing (Figure 1).
Dendritic cellsMouse dendritic cells express nAChR α2, α5, α6, α7, β2 and β4 subunits56. Immature DCs that mature in a nicotinic environment manifest lower endocytic and phagocytic activities. Mature DC’s that are exposed to nicotine produce decreased levels of IL-12 and display reduced ability to induce T Cell responses70-72. In contrast, other studies reveal that nicotine activates DCs and augment their capacity to perform endocytosis, stimulate T-cell proliferation, and produce IL-1273,74. These different findings may be due to the exposure time and dose of nicotine used, or the maturation status of the dendritic cells at the time of assay.
These observations raise the possibility that some of the immunomodulatory effects of vagus nerve stimulation may be partly mediated by altered DC function.
van de Zanden.indb 28 23-5-2011 11:43:43

29
The vagus nerve a modulator of intestinal inflammation
Mast cells Although best known for their role in allergy, mast cells play an important immune protective role as well, being intimately involved in wound healing and defense against pathogens. Mast cells express the nAChR α3, α5, α7, α9 and α1075.76 and appear to make intimate contact with afferent vagus fibers in the small intestinal mucosa75. In human mucosal mast cells, ACh inhibits histamine release. However, this seems to be species specific, since in rats ACh stimulates mast cell degranulation77,78. Host sensitization status may also affect the response of mast cells to ACh, as sensitization to a specific allergen makes rat mast cells more sensitive to ACh-induced histamine release78 . In conclusion, cholinergic activation has broad effects on immune cell function. In animal models of intestinal inflammation, vagus nerve signaling may attenuate inflammation activity not exclusively via inhibition of macrophages but also other immune cells, such as dendritic cells and mast cells can be affected.
NICOTINIC ACETYLCHOLINE RECEPTOR SUBCELLULAR SIGNALING
The exact intracellular mechanism via which ACh exerts its effects on immune cells has been investigated in several studies. Most studies in immune cells focus on activation and subcellular signaling of the α7 nAChR. nAChR α7 receptor signaling on neuronal cells is mediated by ion channel fluxes due to ACh binding3, although the α7 nAChR can also activate alternative signaling pathways. In rat microglia, nicotine exposure elicits a transient increase in intracellulair Ca2+ levels79. This increase in intracellular Ca2+ may involve phosphoinositide 3-kinases (PI3K) and phospho-lipase C (PLC) activation and subsequent Ca2+ release from intracellular Ca2+ stores79, which can finally lead to Ca2+ depletion and cell deactivation. However, unpublished data from our group indicate that no intracellular Ca2+ flux was observed after nicotine exposure in mouse peritoneal macrophages, although nAChR signaling was present (experiments performed at David Greaves’ laboratory, Oxford University). Some studies link nAChR α7 to PI3K activation, possibly via phosphorylation of Akt62,80 and activation of Janus Kinase 2 (Jak2)81. In line with this observation , nAChR α7 activation in macrophages leads to recruitment and phosphorylation of Jak2 and subsequent activation of the Signal Transducer and Activator of Transcription 3 (STAT3) protein82. STAT3 is a negative regulator of inflammation, contributing to the anti-inflammatory effects of IL-1082. STAT3 does not inhibit transcription of pro-inflammatory cytokines directly82. Presumably, other signaling pathways are involved. Studies in macrophages indicate that nAChR α7 activation reduces NF-kB activation, resulting in decreased pro-inflammatory cytokine production40. This
van de Zanden.indb 29 23-5-2011 11:43:43

30
Chapter 2
reduction of NF-κB activation can be explained via induced STAT3 phosphorylation, as phosphorylated STAT3 has been shown to interact with NF-κB p6583 and thus inhibit p65 translocation to the nucleus, resulting in inhibition of NF-κB transcriptional activity. In monocytes and various other cell types, nicotine induces expression of COX-2 and the synthesis of one of its major products, prostaglandine E2 (PGE2). PGE2 is able to elicit an increase in cyclic AMP levels and protein kinase A (PKA) activity, leading to reduced adhesion molecule expression, cytokine production and lymphocyte proliferation84. Other studies show modulation of mitogen activated protein kinases (MAPK), which are involved in various cellular activities, upon AChR activation63.
In conclusion, nAChR α7 activation triggers various signaling mechanisms that most likely interact with each other to achieve immunomodulatory effects. However, the precise mechanism requires further investigation.
CLINICAL IMPLICATIONS OF THE ANTI-INFLAMMATORY PROPERTIES OF VAGUS NERVE SIGNALING
The finding that nicotine inhibits activation of immune cells, together with the observation that vagus nerve signaling or specific nAChR α7 agonists attenuate disease in several inflammatory animal models, implies that therapeutic agents modifying cholinergic signalling might be beneficial in humans.
Cigarette smoking is an important environmental factor in inflammatory bowel disease (IBD), but most strikingly smoking has differential effects in ulcerative colitis (UC) and Crohn’s disease (CD). While smoking increases the risk of developing CD and worsens its course, epidemiological studies of smokers in UC point out that smoking appears to have a protective effect in the development of this disease and reduces its severity85. The exact explanation for this discrepancy is far from clear, but it certainly adds to the current belief that UC and Crohn’s disease are two different disease entities. About 90% of UC patients are non-smokers. Patients with a history of smoking acquire their disease after they have stopped smoking44. Patients who smoke intermittently often experience improvement in their colitis symptoms during the periods when smoking86,87. In ex-smokers, onset is nearly always after quitting smoking.
However, clinical trials using nicotine for the treatment of UC have provided different results. Transdermal nicotine appears to be superior to placebo for the induction of remission in patients with UC, but no significant advantage for transdermal nicotine therapy compared to standard medical therapy was found. Moreover, adverse events associated with transdermal nicotine are significant which will limit its use in patients88. However, to avoid side effects caused by nicotine, more specific nAChR agonists are designed. Partial selective nAChR α7 and α4β2 agonists are already being
van de Zanden.indb 30 23-5-2011 11:43:43

31
The vagus nerve a modulator of intestinal inflammation
tested in patients with neuronal disorders, since both receptor subtypes have shown to mediate improvement in attention, learning and working memory89. The use of specific alpha7 nicotinic agonists is expected to bear potential as a maintenance therapy for active UC. Such selective nicotinic agonists were originally designed to mimic the cognitive effects of nicotine in patients with neurological disorders while avoiding the toxicity of nicotine. The most characterized specific alpha7 nAChR-agonists are GTS21 (3-[(2,4-dimethoxy)benzylidene]-anabaseine), 4OHGTS (3-(4-hydroxy,2-methoxybenzylidene) anabaseine), ARR17779 ((-)-spiro[1-azabicyclo[2.2.2]octane -3,5’-oxazolidin-2’-one]), CAP55, Exo2 (exo-2-(2-pyridyl)-7-azabicyclo[2.2.1] heptane), and PNU-282987 ([N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydro-chloride])90,91. Among these, the most characterized is GTS-21, a partial α7 nAChR agonist that also affects α4β2 nAChR91, is well tolerated in patients with schizophrenia, Alzheimer disease and in healthy volunteers44. Unlike trials using nicotine, patients tolerated doses of up 450 mg/day of GTS21 well, and there were no clinically significant differences in adverse events between the treatment groups. Besides these selective agonists, recent evidence indicates that centrally acting cholinergic drugs used in the treatment of Alzheimer disease can modulate peripheral immune responses and would therefore be interesting to explore92.
Future studies are needed to perform larger clinical trials and determine whether the cognitive potential of nicotinic agonists are based on their binding to neuronal receptors or whether their anti-inflammatory potential in immune and glia may contribute to their therapeutic potential in neurological disorders. In addition to the use of specific cholinergic agonists, vagus nerve stimulation could be a potential therapeutic asset in the treatment of patients with inflammatory diseases. Interestingly, in patients with drug-resistant epilepsy and depression, vagus nerve stimulation is already in use as a new adjunctive therapy. A pulse generator transmits impulses to the left vagus nerve via an implantable electrode. Overall, vagus nerve stimulation has shown better control of seizures or depression, with marginal side effects93. Moreover, it has been demonstrated that a noninvasive method of transcutaneous vagus stimulation, which has shown to improve survival in a mouse model of polymicrobial sepsis94, is feasible in healthy young and elderly subjects95. As the vagus nerve does not innervate the distal colon and rectum, the areas usually affected in IBD patients, vagus nerve stimulation may not be the first therapeutic choice in targeting IBD. Nevertheless, vagus nerve activity can regulate disease in animal models10, possibly clarified by the role of the spleen in exerting the anti-inflammatory effect of vagus nerve signaling or by changes in autonomic (para)sympathetic balance96,97.
van de Zanden.indb 31 23-5-2011 11:43:43

32
Chapter 2
CONCLUDING REMARKS
The hypothesis that vagus nerve stimulation, via the release of ACh, ameliorates inflammation solely via down-regulation of tissue macrophage reactivity and cytokine release via nAChR α7 receptors, seems more complicated than originally thought. It is likely that in vivo, more complex mechanisms play a role, including a variety of different (immune) cell types, neurotransmitters and ACh receptors that converge in the cholinergic down-regulation of inflammatory responses (Figure 1). Irrespectively, it is firmly established that electrical stimulation of the vagus nerve can attenuate inflammation in several animal models.
In conclusion, results obtained in a wide range of in vitro and in vivo models of inflammation imply that therapeutic agents targeting the cholinergic anti-inflammatory pathway can be an important asset in the treatment of immune disorders in human. However, the challenge is to define a specific nAChR agonist with highest anti-inflammatory potential and least side effects. Future studies are needed to explore the protective effects of these methods in the treatment of inflammatory disorders in humans.
van de Zanden.indb 32 23-5-2011 11:43:43

33
The vagus nerve a modulator of intestinal inflammation
REFERENCE LIST
1. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 2000;52:595-638.
2. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
3. Vidal C. Nicotinic receptors in the brain. Molecular biology, function, and therapeutics. Mol Chem Neuropathol 1996;28:3-11.
4. De Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844-851.
5. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, Tracey KJ. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg 2002;36:1231-1236.
6. Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, Squadrito G, Minutoli L, Bertolini A, Marini R, Adamo EB, Venuti FS, Squadrito F. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation 2003;107:1189-1194.
7. Saeed SA, Simjee RU, Mahmood F, Rahman NN. Dual inhibition of platelet-activating factor and arachidonic acid metabolism by ajmaline and effect on carrageenan-induced rat paw oedema. J Pharm Pharmacol 1993;45:715-719.
8. van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, Tracey KJ, van der PT. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006;130:1822-1830.
9. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der PT. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis 2005;191:2138-2148.
10. Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006;131:1122-1130..
11. Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 1999;51:397-401.
12. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
13. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 2007;117:289-296.
14. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A 2008.
15. Altschuler SM, Ferenci DA, Lynn RB, Miselis RR. Representation of the cecum in the lateral dorsal motor nucleus of the vagus nerve and commissural subnucleus of the nucleus tractus solitarii in rat. J Comp Neurol 1991;304:261-274.
16. Altschuler SM, Escardo J, Lynn RB, Miselis RR. The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology 1993;104:502-509.
van de Zanden.indb 33 23-5-2011 11:43:43

34
Chapter 2
17. The FO, Boeckxstaens GE, Snoek SA, Cash JL, Bennink R, Larosa GJ, van den Wijngaard RM, Greaves DR, De Jonge WJ. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 2007;133:1219-1228.
18. Greenwood-Van MB. Emerging drugs for postoperative ileus. Expert Opin Emerg Drugs 2007;12:619-626.
19. Prasad M, Matthews JB. Deflating postoperative ileus. Gastroenterology 1999;117:489-492.
20. De Jonge WJ, van den Wijngaard RM, The FO, ter Beek ML, Bennink RJ, Tytgat GN, Buijs RM, Reitsma PH, van Deventer SJ, Boeckxstaens GE. Postoperative ileus is maintained by intestinal immune infiltrates that activate inhibitory neural pathways in mice. Gastroenterology 2003;125:1137-1147.
21. Kalff JC, Carlos TM, Schraut WH, Billiar TR, Simmons RL, Bauer AJ. Surgically induced leukocytic infiltrates within the rat intestinal muscularis mediate postoperative ileus. Gastroenterology 1999;117:378-387.
22. Turler A, Schnurr C, Nakao A, Togel S, Moore BA, Murase N, Kalff JC, Bauer AJ. Endogenous endotoxin participates in causing a panenteric inflammatory ileus after colonic surgery. Ann Surg 2007;245:734-744.
23. Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 2005;202:1023-1029.
24. Eliakim R, Karmeli F, Rachmilewitz D, Cohen P, Fich A. Effect of chronic nicotine administration on trinitrobenzene sulphonic acid-induced colitis. Eur J Gastroenterol Hepatol 1998;10:1013-1019.
25. Sykes AP, Brampton C, Klee S, Chander CL, Whelan C, Parsons ME. An investigation into the effect and mechanisms of action of nicotine in inflammatory bowel disease. Inflamm Res 2000;49:311-319.
26. Ghia JE, Blennerhassett P, Park AJ, Deng YK, Cornell R, Collins SM. Critical role of the alpha-7 nicotinic acetylcholine receptor in the development of colitis. Gastroenterology 2008;134:A98.
27. Orr-Urtreger A, Kedmi M, Rosner S, Karmeli F, Rachmilewitz D. Increased severity of experimental colitis in alpha 5 nicotinic acetylcholine receptor subunit-deficient mice. Neuroreport 2005;16:1123-1127.
28. Eliakim R, Fan QX, Babyatsky MW. Chronic nicotine administration differentially alters jejunal and colonic inflammation in interleukin-10 deficient mice. Eur J Gastroenterol Hepatol 2002;14:607-614.
29. Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 1977;31:107-133.
30. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005;307:254-258.
31. DeMeo MT, Mutlu EA, Keshavarzian A, Tobin MC. Intestinal permeation and gastrointestinal disease. J Clin Gastroenterol 2002;34:385-396.
32. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427-434.
33. Saunders PR, Kosecka U, McKay DM, Perdue MH. Acute stressors stimulate ion secretion and increase epithelial permeability in rat intestine. Am J Physiol 1994;267:G794-G799.
34. Saunders PR, Hanssen NP, Perdue MH. Cholinergic nerves mediate stress-induced intestinal transport abnormalities in Wistar-Kyoto rats. Am J Physiol 1997;273:G486-G490.
35. Bijlsma PB, Kiliaan AJ, Scholten G, Heyman M, Groot JA, Taminiau JA. Carbachol, but not forskolin, increases mucosal-to-serosal transport of intact protein in rat ileum in vitro. Am J Physiol 1996;271:G147-G155.
36. Cameron HL, Perdue MH. Muscarinic acetylcholine receptor activation increases transcellular transport of macromolecules across mouse and human intestinal epithelium in vitro. Neurogastroenterol Motil 2007;19:47-56.
37. Greenwood B, Mantle M. Mucin and protein release in the rabbit jejunum: effects of bethanechol and vagal nerve stimulation. Gastroenterology 1992;103:496-505.
van de Zanden.indb 34 23-5-2011 11:43:43

35
The vagus nerve a modulator of intestinal inflammation
38. Luyer MD, Buurman WA, Hadfoune M, Jacobs JA, Konstantinov SR, Dejong CH, Greve JW. Pretreatment with high-fat enteral nutrition reduces endotoxin and tumor necrosis factor-alpha and preserves gut barrier function early after hemorrhagic shock. Shock 2004;21:65-71.
39. Han X, Fink MP, Delude RL. Proinflammatory cytokines cause NO*-dependent and -independent changes in expression and localization of tight junction proteins in intestinal epithelial cells. Shock 2003;19:229-237.
40. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004;10:1216-1221.
41. Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol 2001;167:6518-6524.
42. Kessler W, Traeger T, Westerholt A, Neher F, Mikulcak M, Muller A, Maier S, Heidecke CD. The vagal nerve as a link between the nervous and immune system in the instance of polymicrobial sepsis. Langenbecks Arch Surg 2006;391:83-87.
43. Giebelen IA, Le Moine A, van den Pangaart PS, Sadis C, Goldman M, Florquin S, van der PT. Deficiency of alpha7 cholinergic receptors facilitates bacterial clearance in Escherichia coli peritonitis. J Infect Dis 2008;198:750-757.
44. De Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 2007;151:915-929.
45. Giebelen IA, van Westerloo DJ, Larosa GJ, de Vos AF, van der PT. Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock 2007;27:443-447.
46. Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, Hudson L, Lin X, Patel N, Johnson SM, Chavan S, Goldstein RS, Czura CJ, Miller EJ, Al Abed Y, Tracey KJ, Pavlov VA. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med 2008;14:567-574.
47. Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, Levi J, Parrish WR, Rosas-Ballina M, Czura CJ, Larosa GJ, Miller EJ, Tracey KJ, Al Abed Y. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 2007;35:1139-1144.
48. Giebelen IA, van Westerloo DJ, Larosa GJ, de Vos AF, van der PT. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock 2007;28:700-703.
49. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006;203:1623-1628.
50. Buijs RM, van d, V, Garidou ML, Huitinga I, Escobar C. Spleen vagal denervation inhibits the production of antibodies to circulating antigens. PLoS ONE 2008;3:e3152.
51. Schafer MK, Eiden LE, Weihe E. Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. II. The peripheral nervous system. Neuroscience 1998;84:361-376.
52. Sakaguchi T, Liu L. Hepatic branch vagotomy can block liver regeneration enhanced by ursodesoxycholic acid in 66% hepatectomized rats. Auton Neurosci 2002;99:54-57.
53. Buijs RM, La Fleur SE, Wortel J, Van Heyningen C, Zuiddam L, Mettenleiter TC, Kalsbeek A, Nagai K, Niijima A. The suprachiasmatic nucleus balances sympathetic and parasympathetic output to peripheral organs through separate preautonomic neurons. J Comp Neurol 2003;464:36-48.
54. Galvis G, Lips KS, Kummer W. Expression of nicotinic acetylcholine receptors on murine alveolar macrophages. J Mol Neurosci 2006;30:107-108.
van de Zanden.indb 35 23-5-2011 11:43:44

36
Chapter 2
55. Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De BM, Schroder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem 2007;102:479-492.
56. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 2007.
57. Whiteaker P, Marks MJ, Christensen S, Dowell C, Collins AC, McIntosh JM. Synthesis and characterization of 125I-alpha-conotoxin ArIB[V11L;V16A], a selective alpha7 nicotinic acetylcholine receptor antagonist. J Pharmacol Exp Ther 2008;325:910-919.
58. Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese C, Freedman R, Leonard S. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics 1998;52:173-185.
59. Gault J, Hopkins J, Berger R, Drebing C, Logel J, Walton C, Short M, Vianzon R, Olincy A, Ross RG, Adler LE, Freedman R, Leonard S. Comparison of polymorphisms in the alpha7 nicotinic receptor gene and its partial duplication in schizophrenic and control subjects. Am J Med Genet B Neuropsychiatr Genet 2003;123:39-49.
60. Severance EG, Zhang H, Cruz Y, Pakhlevaniants S, Hadley SH, Amin J, Wecker L, Reed C, Cuevas J. The alpha7 nicotinic acetylcholine receptor subunit exists in two isoforms that contribute to functional ligand-gated ion channels. Mol Pharmacol 2004;66:420-429.
61. Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA. Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol 2002;126:86-98.
62. Rehani K, Scott DA, Renaud D, Hamza H, Williams LR, Wang H, Martin M. Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim Biophys Acta 2007.
63. Hamano R, Takahashi HK, Iwagaki H, Yoshino T, Nishibori M, Tanaka N. Stimulation of alpha7 nicotinic acetylcholine receptor inhibits CD14 and the toll-like receptor 4 expression in human monocytes. Shock 2006;26:358-364.
64. Lau PP, Li L, Merched AJ, Zhang AL, Ko KW, Chan L. Nicotine induces proinflammatory responses in macrophages and the aorta leading to acceleration of atherosclerosis in low-density lipoprotein receptor(-/-) mice. Arterioscler Thromb Vasc Biol 2006;26:143-149.
65. Chen YC, Shen SC, Lin HY, Tsai SH, Lee TJ. Nicotine enhancement of lipopolysaccharide/interferon-gamma-induced cytotoxicity with elevating nitric oxide production. Toxicol Lett 2004;153:191-200.
66. Schwartz SL, Evans DE, Lundin JE, Bond JC. Inhibition of pinocytosis by nicotine. J Pharmacol Exp Ther 1972;183:370-377.
67. Schwartz SL. Interaction of nicotine and other amines with the endocytic and exocytic functions of macrophages. Fed Proc 1976;35:85-88.
68. Thyberg J, Nilsson J. Effects of nicotine on endocytosis and intracellular degradation of horseradish peroxidase in cultivated mouse peritoneal macrophages. Acta Pathol Microbiol Immunol Scand [A] 1982;90:305-310.
69. Thyberg J, Hedin U, Stenseth K, Nilsson J. Effects of nicotine on the fine structure of cultivated mouse peritoneal macrophages. Acta Pathol Microbiol Immunol Scand [A] 1983;91:23-30.
70. Guinet E, Yoshida K, Nouri-Shirazi M. Nicotinic environment affects the differentiation and functional maturation of monocytes derived dendritic cells (DCs). Immunol Lett 2004;95:45-55.
71. Nouri-Shirazi M, Tinajero R, Guinet E. Nicotine alters the biological activities of developing mouse bone marrow-derived dendritic cells (DCs). Immunol Lett 2007;109:155-164.
72. Takahashi HK, Iwagaki H, Hamano R, Kanke T, Liu K, Sadamori H, Yagi T, Yoshino T, Tanaka N, Nishibori M. The immunosuppressive effects of nicotine during human mixed lymphocyte reaction. Eur J Pharmacol 2007;559:69-74.
van de Zanden.indb 36 23-5-2011 11:43:44

37
The vagus nerve a modulator of intestinal inflammation
73. Aicher A, Heeschen C, Mohaupt M, Cooke JP, Zeiher AM, Dimmeler S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation 2003;107:604-611.
74. Gao FG, Wan dF, Gu JR. Ex vivo nicotine stimulation augments the efficacy of therapeutic bone marrow-derived dendritic cell vaccination. Clin Cancer Res 2007;13:3706-3712.
75. Williams RM, Berthoud HR, Stead RH. Vagal afferent nerve fibres contact mast cells in rat small intestinal mucosa. Neuroimmunomodulation 1997;4:266-270.
76. Sudheer PS, Hall JE, Donev R, Read G, Rowbottom A, Williams PE. Nicotinic acetylcholine receptors on basophils and mast cells. Anaesthesia 2006;61:1170-1174.
77. Reinheimer T, Mohlig T, Zimmermann S, Hohle KD, Wessler I. Muscarinic control of histamine release from airways. Inhibitory M1-receptors in human bronchi but absence in rat trachea. Am J Respir Crit Care Med 2000;162:534-538.
78. Masini E, Fantozzi R, Conti A, Blandina P, Brunelleschi S, Mannaioni PF. Immunological modulation of cholinergic histamine release in isolated rat mast cells. Agents Actions 1985;16:152-154.
79. Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, Andra M, Matsubayashi H, Sakai N, Kohsaka S, Inoue K, Nakata Y. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 2006;83:1461-1470.
80. Cheng PY, Lee YM, Law KK, Lin CW, Yen MH. The involvement of AMP-activated protein kinases in the anti-inflammatory effect of nicotine in vivo and in vitro. Biochem Pharmacol 2007;74:1758-1765.
81. Shaw S, Bencherif M, Marrero MB. Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against Abeta-(1-42) amyloid. J Biol Chem 2002;277:44920-44924.
82. Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A 2005;102:8686-8691.
83. Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 2002;367:97-105.
84. Takahashi HK, Iwagaki H, Hamano R, Yoshino T, Tanaka N, Nishibori M. Effect of nicotine on IL-18-initiated immune response in human monocytes. J Leukoc Biol 2006;80:1388-1394.
85. Lakatos PL, Szamosi T, Lakatos L. Smoking in inflammatory bowel diseases: good, bad or ugly? World J Gastroenterol 2007;13:6134-6139.
86. Van Assche G., Vermeire S, Rutgeerts P. Medical treatment of inflammatory bowel diseases. Curr Opin Gastroenterol 2005;21:443-447.
87. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA, . Transdermal nicotine for active ulcerative colitis. N Engl J Med 1994;330:811-815.
88. McGrath J, McDonald JW, Macdonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev 2004;CD004722.
89. Cincotta SL, Yorek MS, Moschak TM, Lewis SR, Rodefer JS. Selective nicotinic acetylcholine receptor agonists: potential therapies for neuropsychiatric disorders with cognitive dysfunction. Curr Opin Investig Drugs 2008;9:47-56.
90. Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov 2005;4:673-684.
91. Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, Burnett AL. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology 2003;28:542-551.
92. Pavlov VA, Ochani M, Gallowitsch-Puerta M, Ochani K, Huston JM, Czura CJ, Al Abed Y, Tracey KJ. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci U S A 2006;103:5219-5223.
van de Zanden.indb 37 23-5-2011 11:43:44

38
Chapter 2
93. Shafique S, Dalsing MC. Vagus nerve stimulation therapy for treatment of drug-resistant epilepsy and depression. Perspect Vasc Surg Endovasc Ther 2006;18:323-327.
94. Huston JM, Gallowitsch-Puerta M, Ochani M, Ochani K, Yuan R, Rosas-Ballina M, Ashok M, Goldstein RS, Chavan S, Pavlov VA, Metz CN, Yang H, Czura CJ, Wang H, Tracey KJ. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007;35:2762-2768.
95. Fallgatter AJ, Ehlis AC, Ringel TM, Herrmann MJ. Age effect on far field potentials from the brain stem after transcutaneous vagus nerve stimulation. Int J Psychophysiol 2005;56:37-43.
96. Ghia JE, Blennerhassett P, Collins SM. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J Clin Invest 2008;118:2209-2218.
97. Mouzas IA, Pallis AG, Kochiadakis GE, Marketou M, Chlouverakis GI, Mellisas J, Vardas PE, Kouroumalis EA. Autonomic imbalance during the day in patients with inflammatory bowel disease in remission. Evidence from spectral analysis of heart rate variability over 24 hours. Dig Liver Dis 2002;34:775-780.
van de Zanden.indb 38 23-5-2011 11:43:44

3CHAPTER
Stimulation of the vagus nerve attenuates macrophage activity by activating the JAK-2-STAT-3 signaling pathway
Wouter J. de JongeEsmerij P. van der ZandenFrans O. ThéMaarten F. BijlsmaDavid J. van WesterlooRoelof J. BenninkHans-Rudolf BerthoudSatoshi UematsuShizuo AkiraRené MJGJ. van den WijngaardGuy EE. Boeckxstaens
Nature Immunology. 2005 Aug;6(8):844-51.
van de Zanden.indb 39 23-5-2011 11:43:44

40
Chapter 3
ABSTRACT
Acetylcholine released by efferent vagus nerves inhibits macrophage activation. Here we show that the anti-inflammatory action of nicotinic receptor activation in peritoneal macrophages was associated with activation of the transcription factor STAT3. STAT3 was phosphorylated by the tyrosine kinase Jak2 that was recruited to the alpha7 subunit of the nicotinic acetylcholine receptor. The anti-inflammatory effect of nicotine required the ability of phosphorylated STAT3 to bind and transactivate its DNA response elements. In a mouse model of intestinal manipulation, stimulation of the vagus nerve ameliorated surgery-induced inflammation and postoperative ileus by activating STAT3 in intestinal macrophages. We conclude that the vagal anti-inflammatory pathway acts by alpha7 subunit-mediated Jak2-STAT3 activation.
van de Zanden.indb 40 23-5-2011 11:43:44

41
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
INTRODUCTION
The innate immune response has been increasingly recognized as being under substantial neuronal control1. For example, acetylcholine or nicotine effectively attenuates the activation of macrophages2. This so-called ‘cholinergic anti-inflammatory pathway’ is characterized by a nicotine dose−dependent decrease in the production of proinflammatory mediators, including high-mobility group box 1 proteins3, tumor necrosis factor (TNF), interleukin 1β (IL-1β), IL-6 and IL-18 (ref 2), by macrophages stimulated with endotoxin. Consistently, stimulation of the efferent vagus nerve dampens macrophage activation in rodent models of endotoxemia and shock1,2. Two nicotinic acetylcholine receptor (nAChR) subtypes are involved in the nicotine-induced decrease in proinflammatory cytokine production by stimulated human and mouse macrophages: the α7 homopentamer expressed by monocyte-derived human and mouse macrophages4, and the α4β2 heteropentamer expressed by alveolar macrophages5. Activation of the α7 homopentamer nAChR inhibits transactivational activity of the transcription factor NF-ĸB p65 (ref 3). However, the subcellular mechanism explaining the deactivating effect of acetylcholine on macrophages has remained unknown.
Here we evaluated the involvement of the transcription factor STAT3 in this process, because STAT3 is a potential negative regulator of inflammatory responses6,7. STAT3 and the tyrosine kinase Jak2, which phosphorylates STAT3, are required for both IL-6 receptor (IL-6R) and IL-10R signaling. IL-6 contributes to the progression of many inflammatory diseases, whereas IL-10 is an anti-inflammatory cytokine that suppresses the activation of macrophages. IL-6R signaling is inhibited by the Src homology 2 domain protein SOCS3, whose expression is induced by STAT3 activation8,9. SOCS3 binds to the glycoprotein 130 (gp130) subunit of the IL-6R, leading to inhibited activation of STAT3 by IL-6R ligands8,9. Consistent with that finding, in LPS-stimulated macrophages deficient in SOCS3, IL-6R ligands induce a sustained STAT3 activation, which leads to the reduced production of proinflammatory cytokines such as TNF10.
Here we demonstrate that nicotine exerts its anti-inflammatory effect on peritoneal macrophages via Jak2 and STAT3 signaling in vitro and in vivo. In isolated peritoneal macrophages, nicotine activated nAChRs, leading to phosphorylation of STAT3 via Jak2. Jak2 was recruited to the α7 subunit of the nAChR and was phosphorylated after nicotine binding. We further studied the effect of cholinergic inhibition of macrophage activity in vivo on the occurrence of post-surgical intestinal inflammation in a mouse model of postoperative ileus11,12. Postoperative ileus is characterized by general hypomotility of the gastrointestinal tract and delayed gastric emptying13 and is a pathological condition commonly noted after abdominal surgery with intestinal manipulation. This condition is the result of inflammation of the intestinal muscularis due to activation of resident macrophages14,15 that are triggered
van de Zanden.indb 41 23-5-2011 11:43:44

42
Chapter 3
by bowel manipulation12. We show here that perioperative stimulation of the vagus nerve prevented manipulation-induced inflammation of the intestinal muscularis externa and ameliorated postoperative ileus. The effectiveness of stimulation of the vagus nerve in reducing intestinal inflammation depended on STAT3 activation in macrophages in the intestinal muscularis. Hence, our data demonstrate the molecular pathway responsible for cholinergic inhibition of macrophage activation and suggest that stimulation of the vagus nerve or administration of cholinergic agents may be effective anti-inflammatory therapy for the treatment of postoperative ileus and other inflammatory diseases.
MATERIALS AND METHODS
Reagents and antibodies. Nicotine, Hexamethonium, α-Bungarotoxin, Methyl-lycaconitine citrate, (+)-Tubocurarine chloride hydrate, dihydro-β-erythroidine, AG490, cyclohexamin, actinomycin-D, and monoclonal rat anti-β2 were from Sigma-Aldrich. Polyclonal rabbit antibodies against Jak-2, phosphorylated Jak-2 (PY1007-8
Jak-2), Socs-3 and α7 were obtained from Abcam (Cambridge, UK), goat polyclonal anti-actin, rabbit polyclonal anti-Stat-1, and anti-Stat-3 were from Santa Cruz Biotechnology (Santa Cruz, California), and rabbit polyclonal against phosphorylated Stat-1 and Stat-3 (PY705) were from Cell Signaling Technology (Beverly, Maryland). ELISAs for IL-6, IL-10, MIP-1α, MIP-2 and TNF were from R&D Systems, (Minneapolis, Michigan).
Cell culture and transient transfection. Resident peritoneal macrophages were harvested from Balb/C mice by flushing the peritoneal cavity with 5 mL of ice-cold Hank’s Balanced Salt Solution containing 10 U/mL heparin. Peritoneal cells (1*106 per cm2) were plated in RPMI medium supplemented with 10% FCS and macrophages were left to adhere for 2 h in a humidified atmosphere at 37 °C with 5% CO2. Cells were washed and remaining macrophages were left for 16-20 h. Subsequently, cells were pre-incubated with the appropriate concentration of nicotine for 15 min, followed by LPS (1-100 ng/mL) challenge for 3 h. NachR blockers were added 30 min before nicotine, and no toxicity was observed after 4 h incubation of any blocker as assessed by tryphan blue exclusion test. Cells were lysed 30 min following nicotine/LPS exposure for immunoblotting. Transfection of peritoneal macrophages was performed using Effectine reagent (Qiagen, Cambridge, UK) according to the manufacturer’s instructions. A CMV driven Renilla luciferase reporter plasmid was co-transfected to assess transfection efficiency. The pCAGGS-neo expression vectors encoding wild type hemagluttinin (HA)-tagged Stat-3, or the dominant negative mutant HA-Stat-3D cDNA 17 were kindly provided by Drs I. Touw and T. Hirano. In HA-Stat-3D, glutamic acids 434 and 435 were replaced by alanines 17. Following
van de Zanden.indb 42 23-5-2011 11:43:44

43
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
transfection, cells were selected using neomycin (2.0 mg/mL; Sigma-Aldrich) for 16 h, washed, and treated with nicotine-LPS 24 h after transfection. Transfection was verified by immunoblotting using HRP-tagged anti-HA rabbit polyclonal antibodies (Abcam). For siRNA transfection, a siRNA oligonucleotide specific to Socs-3 (ID 160220; Ambion) was transfected using RNAiFect (Qiagen) according to the manufacturer’s instructions. A FITC labeled control random RNA oligo nucleotide (Ambion) was co-transfected to allow optimalisation of transfection efficiency.
Immunoblotting. Cells were scraped in 50 µL of ice-cold lysis buffer containing 150 mM NaCl, 0.5% Triton X-100, 5 mM EDTA, and 0.1% SDS. Samples were taken up in 50 µL sample buffer (125 mM Tris-HCl pH 6.8, 2% SDS, 10% βz-mercaptoethanol, 10% glycerol, and 0.5 mg/mL bromophenol blue), loaded onto SDS-PAGE gels and blotted onto PVDF membranes (Millipore). Membranes were blocked in TBS/0.1% Tween-20 (TBST) containing 5% non-fat dry milk and incubated overnight with appropriate antibodies in TBST/1% BSA. HRP-conjungated secondary antibodies were visualized using Lumilite plus (Boehringer-Mannheim, Germany).
Immunoprecipitation. Peritoneal macrophages (1*106 per cm2) were scraped in lysis buffer (20 mM Tris-HCl pH 7.6, 2.5 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5% sodiumdeoxycholate, 10% glycerol, 1 mM Na3VO4, 50 mM NaF, 1 µg/mL aproptinine, 1 µg/mL leupeptine, and 1 mM PMSF), sonicated for 10 s and centrifuged at 14000 x g at 4 °C for 20 min. Lysates, preabsorbed with 20 µL Protein A/G (Sigma-Aldrich), were incubated overnight with the appropriate antibodies, and immunoprecipitated with 40 µL of Protein A/G. Alternatively, immunoprecipitation was carried out using the TrueBlot™ system (eBioscience, San Diego, CA) according to the manufacturer’s instructions. Immunoprecipitates were recovered by centrifugation, washed in ice-cold wash buffer (TBS, 0.1% Triton X-100, 1 mM PMSF), taken up in sample buffer (125 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, and 0.5 mg/mL bromophenol blue), and immunoblotted as described above.
Surgical procedures. Mice (female BalB/C) were used at 15-20 weeks of age. IL-6 and IL-10 deficient mice and their respective C57Bl wild types were obtained from Jackson Laboratories (Maine, USA). LysM-Cre Stat-3fl/fl and Stat-3fl/fl mice were maintained at Osaka University, Japan. Abdominal surgery with intestinal manipulation was performed as described elsewhere 12. Mice (n=10-12) were divided in 4 groups: 1-undergoing control surgery of only laparotomy (L), 2-laparotomy followed by intestinal manipulation (IM) combined with sham preparation of the cervical area, or 3-L- or 4-IM in combination with electrical stimulation of the vagus nerve. IM consisted of manipulation from the distal duodenum to the cecum during 5 min using sterile moist cotton applicators. At 3 or 24 h after surgery, mice were killed by cervical dislocation. Small intestine was removed, flushed, and fixed in ice-cold 100% ethanol
van de Zanden.indb 43 23-5-2011 11:43:44

44
Chapter 3
for preparation of whole mounts. Small intestinal muscularis strips were prepared by pinning freshly isolated intestinal segments in ice-cold PBS and removal of mucosa facing upwards. Muscle strips were snap-frozen in liquid nitrogen until analysis.
Electric stimulation of the vagal nerve. Stimulation of the vagus nerve was essentially performed as described previously 2. The left cervical nerve was prepared free from the carotid artery and ligated with 6-0 silk suture. The distal part of the ligated nerve trunk was placed between a bipolar platinum electrode unit. In part of the experiments, the vagus nerve was transected, and the distal part stimulated. Voltage stimuli (5Hz, 2ms, 1 or 5 V) were applied for 5 min before-, and 15 min following the intestinal manipulation protocol described above. In sham VNS control mice the cervical skin was opened and left for 20 min. covered by moist gaze.
Local hexamethonium application. Local blockade of nicotinic receptors in the ileum was performed as follows: in anaesthetized mice (n=7) a midline laparotomy was performed, and 6 cm of ileum proximal to the cecum was carefully externalized and placed in a sterile preheated tube. The segment was continuously flushed with a preheated (37 °C) solution of hexamethonium (10-4 M in 0.9% NaCl), or vehicle for 20 min. Temperature of intestinal tissue was monitored using a thermal probe. Leakage of hexamethonium solution into the peritoneal cavity was strictly avoided. After incubation, the hexamethonium solution was removed, the ileal segment was washed three times with 0.9% NaCl, and included in the manipulation protocol.
Measurement of gastric emptying. Gastric emptying of a semi-liquid, non-caloric test meal (0.5% methylcellulose) containing 10Mq 99Tc was determined by scintigraphic imaging as described previously 42.
Quantification of leukocyte accumulation at the intestinal muscularis.Myeloperoxidase (MPO) activity in ileal muscularis tissue was assayed as a
measure of leukocyte infiltration as described12, 23. Whole mounts of ethanol-fixed ileal muscularis were prepared and stained for MPO activity as described12, 23.
RT-PCR. Total RNA from tissue was isolated using Trizol (Invitrogen, Carlsbad, CA), treated with Dnase, and reverse transcribed. The resulting cDNA (0.5 ng) was subjected to Light Cycler PCR (CYBR Green Fast start polymerase; Roche, Mannheim, Germany) for 40 cycles. Primers used were: TNF: As 5-AAAGCATGATCCGCGACGT-3 and Sen 5-TGCCACAAGCAGGAATGAGAA-3; MIP-2: As 5-AGTGAACTGCGCTGTCAATGC-3 and Sen 5-GCAAAC TTTTTGACCGCCCT-3; Socs-3 As 5-ACCTTTCTTATCCGCGACAG-3and Sen 5’-TGCACCAGCTTGAGTACACAG-3’; and GAPDH As 5’- ATGTGTCC GTCGTGGATCTGA-3’ and Sen 5’-ATGCCTGCTTCACCACCTTCT-3’. PCR products were quantified using a linear regression method on the Log(fluorescence) per cycle number data43, and expressed as percentage of GAPDH transcripts for
van de Zanden.indb 44 23-5-2011 11:43:44

45
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
each sample. For qualification, resulting PCR products were analyzed on an ethidium bromide-stained 2.5% agarose gel.
Immunohistochemistry. For double-labeling of macrophages and cholinergic fibers, rats were anesthetized with pentobarbital sodium (90mg/kg, ip) and transcardially perfused with heparinized saline (20 U/ml), followed by ice-cold 4% phosphate-buffered (pH 7.4) paraformaldehyde. Gastric and intestinal tissue were extracted and postfixed in the same fixative for a minimum of 2 hr. Tissue was cryoprotected overnight in 18% sucrose and 0.05% sodium azide in 0.01M phosphate-buffered saline (PBS). Twenty-micron flat-sections and 25 µm cross-sections of corpus and mid ileum were cryostat cut and processed in PBS. Sections were pretreated with 0.5% sodium borohydride in PBS and subsequently blocked in donkey normal serum. Monoclonal mouse anti-rat ED2 (Serotec, Raleigh, NC), and polyclonal goat anti-Vesicular Acetylcholine Transporter (VAChT; Chemicon, Temecula, CA) were diluted in 0.1% gelatin and 0.05% sodium azide in PBS with 0.5% Triton X-100 (PBST) and incubated for 20 hr at room temperature or for 48 hr at 4oC. Secondary antibodies used were Cy-3-conjugated donkey anti-mouse (Jackson ImmunoResearch, West Grove, PA) for ED2 and Cy-2-conjugated donkey anti-goat (Jackson ImmunoResearch, West Grove, PA) for VAChT in PBST. Sections were mounted in 100% glycerol with the addition of 5% n-propyl gallate as an antifade agent.
In vivo labeling of mouse phagocytes was performed by i.p. injection of 20 µg Alexa546-labeled Dextran particles (Mw 10,000; Molecular Probes, Sunnydale, CA) 24 h before surgery. One hour following surgery, anaesthetized mice were perfused with 10 mL of ice-cold 0.9% NaCl containing 1mM Na3VO4, followed by 20 mL of ice-cold 4 % formaldehyde solution, pH 7.4. Intestinal tissue was isolated, fixed overnight in 4% formaldehyde, dehydrated and embedded in paraffin. Six mm sections were cut and immunostained for PY705-Stat-3 using polyclonal rabbit antibodies (Cell Signaling Technologies) and biotin-labeled anti-rabbit antibodies according to the manufacturer’s instructions. Biotin was visualized using 3-amino-9-ethyl carbazole (Sigma, St Louis, MO) as chromogen, followed by counterstaining using haematoxylin. Alternatively, Alexa488-streptavidin (Molecular probes) with DaPi nuclear counterstain was used for analysis by confocal microscopy.
RESULTS
Nicotine activates STAT3 in macrophages. To study the cellular response of macrophages to nicotinic receptor activation, we isolated peritoneal macrophages from mice and investigated the effect of nicotine on LPS-induced cytokine production. Nicotine reduced the LPS-induced release of TNF, MIP-2 and IL-6 but not IL-10 in a dose-dependent way (fig.1a), consistent with published reports on
van de Zanden.indb 45 23-5-2011 11:43:44
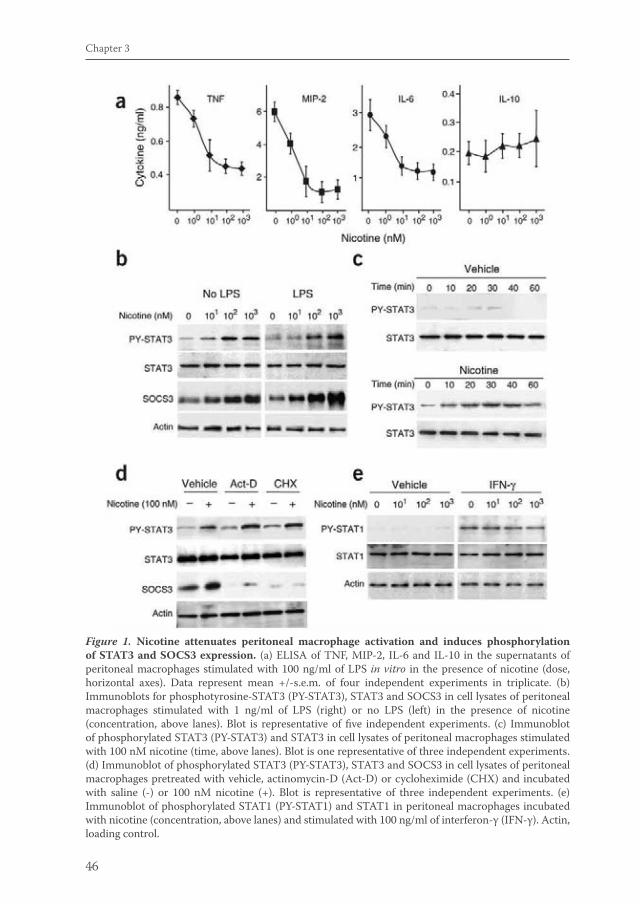
46
Chapter 3
Figure 1. Nicotine attenuates peritoneal macrophage activation and induces phosphorylation of STAT3 and SOCS3 expression. (a) ELISA of TNF, MIP-2, IL-6 and IL-10 in the supernatants of peritoneal macrophages stimulated with 100 ng/ml of LPS in vitro in the presence of nicotine (dose, horizontal axes). Data represent mean +/-s.e.m. of four independent experiments in triplicate. (b) Immunoblots for phosphotyrosine-STAT3 (PY-STAT3), STAT3 and SOCS3 in cell lysates of peritoneal macrophages stimulated with 1 ng/ml of LPS (right) or no LPS (left) in the presence of nicotine (concentration, above lanes). Blot is representative of five independent experiments. (c) Immunoblot of phosphorylated STAT3 (PY-STAT3) and STAT3 in cell lysates of peritoneal macrophages stimulated with 100 nM nicotine (time, above lanes). Blot is one representative of three independent experiments. (d) Immunoblot of phosphorylated STAT3 (PY-STAT3), STAT3 and SOCS3 in cell lysates of peritoneal macrophages pretreated with vehicle, actinomycin-D (Act-D) or cycloheximide (CHX) and incubated with saline (-) or 100 nM nicotine (+). Blot is representative of three independent experiments. (e) Immunoblot of phosphorylated STAT1 (PY-STAT1) and STAT1 in peritoneal macrophages incubated with nicotine (concentration, above lanes) and stimulated with 100 ng/ml of interferon-γ (IFN-γ). Actin, loading control.
van de Zanden.indb 46 23-5-2011 11:43:44

47
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
the anti-inflammatory effect of nicotine on human and mouse monocyte-derived macrophages2,4,5.
Given the crucial function of STAT3 in anti-inflammatory responses6,7, we hypothesized that activation of STAT3 and its gp130-binding regulatory protein SOCS3 may be involved in the anti-inflammatory effect of nicotine. Consistent with that hypothesis, we found that nicotine treatment activated STAT3 as well as SOCS3 in resting and LPS-stimulated primary peritoneal macrophages in a dose- and time-dependent way (fig. 1b,c). Nicotine activated STAT3 directly, as phosphorylation of STAT3 was not affected by the protein synthesis inhibitors actinomycin D and cycloheximide (Fig.1d). In contrast, interferon-γ-induced STAT1 activation was not effected by nicotine (Fig. 1e). Thus, nicotine reduced the production of proinflammatory cytokines and activated STAT3 as well as SOCS3 in stimulated macrophages.
Deactivation by nicotine requires STAT3 transactivationWe next sought to determine whether the anti-inflammatory effect of nicotine depended on nuclear transactivation of phosphorylated STAT3. We overexpressed a dominant negative form of STAT3 (STAT3D) in primary peritoneal macrophages. Dimerized STAT3D is altered in its ability to bind DNA response elements and induce transcription of target genes16,17. Nicotine failed to reduce LPS-induced TNF release in LPS-stimulated macrophages transfected with STAT3D but not those transfected with the STAT3 wild-type construct (Fig. 2a). Thus, the nicotine-induced inhibition of TNF release is dependent on STAT3 DNA transactivation. To evaluate whether SOCS3 expression is crucial to the nicotinic anti-inflammatory effect, we abrogated SOCS3 expression in peritoneal macrophages with SOCS3-specific small interfering RNA (Fig. 1b). SOCS3 expression was substantially decreased in response to nicotine (less than 10% of that expressed in control transfected cells), whereas STAT3 activation was not affected (transfection efficiency was more than 90%; Fig. 2b). In macrophages with reduced SOCS3, however, nicotine was still able to decrease endotoxin-induced production of IL-6 (data not shown) and TNF in a concentration-dependent way, although the reduction was less pronounced than that in control transfected cells (Fig. 2c). Thus, blockade of STAT3 transactivation counteracted the anti-inflammatory effects of nicotine, whereas blockade of SOCS3 expression did not. These results indicate that SOCS3 expression is not strictly required for the reduction in macrophage TNF release by nicotine.
STAT3 phosphorylation depends on α7 nAChR activation To determine whether STAT3 activation by nicotine was mediated by nAChR, we pretreated cells with nAChR antagonists. The nonselective antagonists hexamethonium and d-tubocurarine prevented the STAT3 phosphorylation induced by nicotine (Fig. 3a). In addition, the α7 nAChR−selective antagonists α-bungarotoxin
van de Zanden.indb 47 23-5-2011 11:43:44
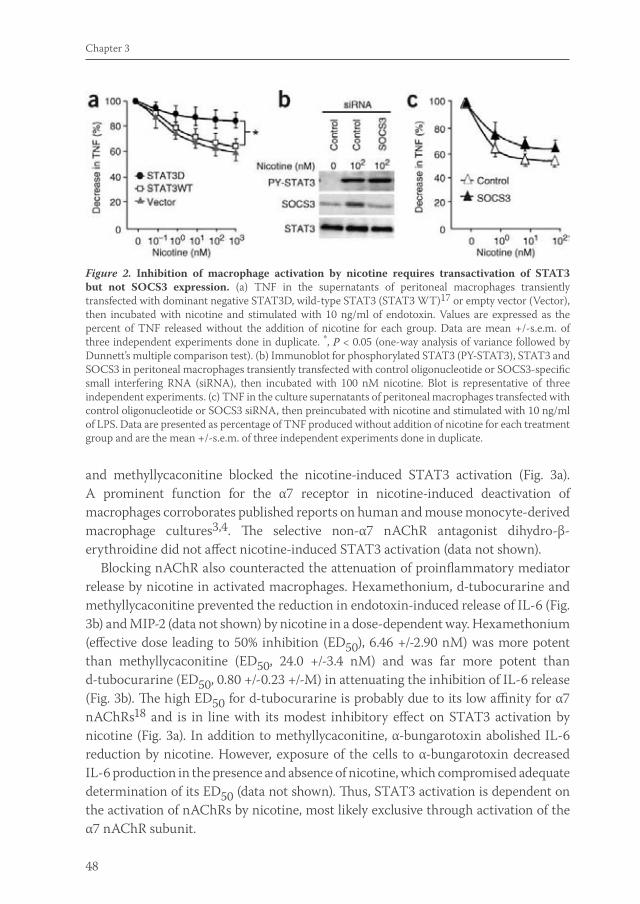
48
Chapter 3
and methyllycaconitine blocked the nicotine-induced STAT3 activation (Fig. 3a). A prominent function for the α7 receptor in nicotine-induced deactivation of macrophages corroborates published reports on human and mouse monocyte-derived macrophage cultures3,4. The selective non-α7 nAChR antagonist dihydro-β-erythroidine did not affect nicotine-induced STAT3 activation (data not shown).
Blocking nAChR also counteracted the attenuation of proinflammatory mediator release by nicotine in activated macrophages. Hexamethonium, d-tubocurarine and methyllycaconitine prevented the reduction in endotoxin-induced release of IL-6 (Fig. 3b) and MIP-2 (data not shown) by nicotine in a dose-dependent way. Hexamethonium (effective dose leading to 50% inhibition (ED50), 6.46 +/-2.90 nM) was more potent than methyllycaconitine (ED50, 24.0 +/-3.4 nM) and was far more potent than d-tubocurarine (ED50, 0.80 +/-0.23 +/-M) in attenuating the inhibition of IL-6 release (Fig. 3b). The high ED50 for d-tubocurarine is probably due to its low affinity for α7 nAChRs18 and is in line with its modest inhibitory effect on STAT3 activation by nicotine (Fig. 3a). In addition to methyllycaconitine, α-bungarotoxin abolished IL-6 reduction by nicotine. However, exposure of the cells to α-bungarotoxin decreased IL-6 production in the presence and absence of nicotine, which compromised adequate determination of its ED50 (data not shown). Thus, STAT3 activation is dependent on the activation of nAChRs by nicotine, most likely exclusive through activation of the α7 nAChR subunit.
Figure 2. Inhibition of macrophage activation by nicotine requires transactivation of STAT3 but not SOCS3 expression. (a) TNF in the supernatants of peritoneal macrophages transiently transfected with dominant negative STAT3D, wild-type STAT3 (STAT3 WT)17 or empty vector (Vector), then incubated with nicotine and stimulated with 10 ng/ml of endotoxin. Values are expressed as the percent of TNF released without the addition of nicotine for each group. Data are mean +/-s.e.m. of three independent experiments done in duplicate. *, P < 0.05 (one-way analysis of variance followed by Dunnett’s multiple comparison test). (b) Immunoblot for phosphorylated STAT3 (PY-STAT3), STAT3 and SOCS3 in peritoneal macrophages transiently transfected with control oligonucleotide or SOCS3-specific small interfering RNA (siRNA), then incubated with 100 nM nicotine. Blot is representative of three independent experiments. (c) TNF in the culture supernatants of peritoneal macrophages transfected with control oligonucleotide or SOCS3 siRNA, then preincubated with nicotine and stimulated with 10 ng/ml of LPS. Data are presented as percentage of TNF produced without addition of nicotine for each treatment group and are the mean +/-s.e.m. of three independent experiments done in duplicate.
van de Zanden.indb 48 23-5-2011 11:43:45
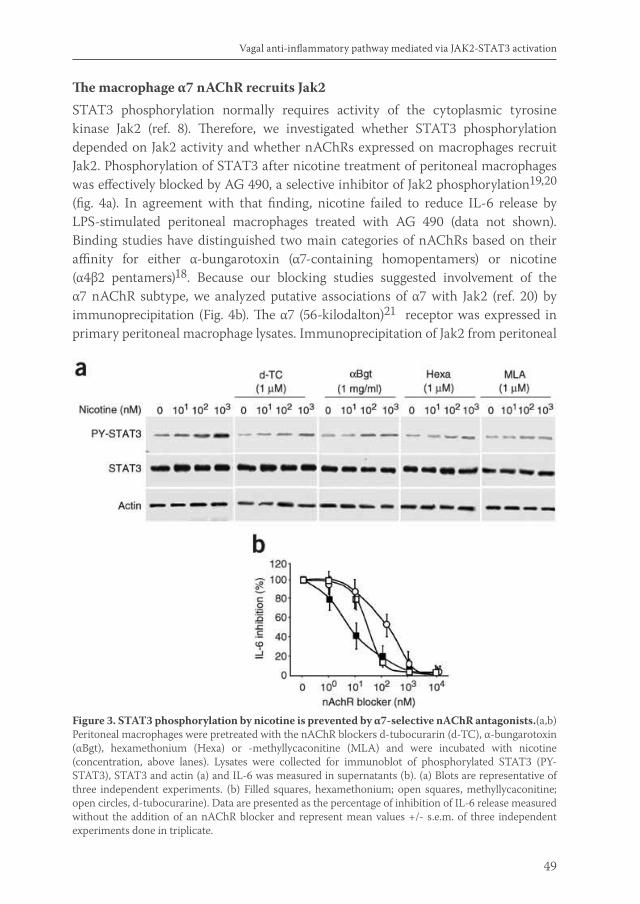
49
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
The macrophage α7 nAChR recruits Jak2STAT3 phosphorylation normally requires activity of the cytoplasmic tyrosine kinase Jak2 (ref. 8). Therefore, we investigated whether STAT3 phosphorylation depended on Jak2 activity and whether nAChRs expressed on macrophages recruit Jak2. Phosphorylation of STAT3 after nicotine treatment of peritoneal macrophages was effectively blocked by AG 490, a selective inhibitor of Jak2 phosphorylation19,20 (fig. 4a). In agreement with that finding, nicotine failed to reduce IL-6 release by LPS-stimulated peritoneal macrophages treated with AG 490 (data not shown). Binding studies have distinguished two main categories of nAChRs based on their affinity for either α-bungarotoxin (α7-containing homopentamers) or nicotine (α4β2 pentamers)18. Because our blocking studies suggested involvement of the α7 nAChR subtype, we analyzed putative associations of α7 with Jak2 (ref. 20) by immunoprecipitation (Fig. 4b). The α7 (56-kilodalton)21 receptor was expressed in primary peritoneal macrophage lysates. Immunoprecipitation of Jak2 from peritoneal
Figure 3. STAT3 phosphorylation by nicotine is prevented by α7-selective nAChR antagonists.(a,b) Peritoneal macrophages were pretreated with the nAChR blockers d-tubocurarin (d-TC), α-bungarotoxin (αBgt), hexamethonium (Hexa) or -methyllycaconitine (MLA) and were incubated with nicotine (concentration, above lanes). Lysates were collected for immunoblot of phosphorylated STAT3 (PY-STAT3), STAT3 and actin (a) and IL-6 was measured in supernatants (b). (a) Blots are representative of three independent experiments. (b) Filled squares, hexamethonium; open squares, methyllycaconitine; open circles, d-tubocurarine). Data are presented as the percentage of inhibition of IL-6 release measured without the addition of an nAChR blocker and represent mean values +/- s.e.m. of three independent experiments done in triplicate.
van de Zanden.indb 49 23-5-2011 11:43:45

50
Chapter 3
macrophage cell lysates showed a weak association of Jak2 with the α7 receptor after culture in the absence of nicotine. To investigate whether Jak2 is recruited to the nAChR and is phosphorylated after binding of its ligand, we preincubated cells with nicotine. Nicotine exposure increased the amount of α7 nAChR detected in Jak2 and phosphorylated Jak2 immunoprecipitates (fig. 4b). To further demonstrate that Jak2 is phosphorylated after nAChR activation, we pretreated cells with the Jak2 phosphorylation blocker AG 490 before adding nicotine. Cells treated with AG 490 had reduced phosphorylated Jak2 in α7 immunoprecipitates, whereas Jak2 recruitment to the α7 receptor was not affected (Fig. 4b). The latter finding demonstrates that Jak2 is recruited and phosphorylated after nicotine binding.
Stimulation of the vagus nerve ameliorates inflammationWe next evaluated whether activation of nAChR on macrophages would attenuate intestinal inflammation in vivo. We assessed the effect of stimulation of the vagus nerve on the inflammation that follows intestinal manipulation in our mouse model11, because this immune response is associated with the activation of macrophages12,22. We electrically stimulated the left cervical vagus nerve during intestinal manipulation surgery and investigated the effects on muscular inflammation and gastric emptying 24 h later (Fig. 5). Consistent with published findings11,23, intestinal manipulation of mice resulted in a delayed gastric emptying compared with that of mice that underwent only laparotomy, indicative of the development of postoperative ileus11 (gastric retention after 60 min, 14.5% +/-2.7% for laparotomy and 43.0 +/-6.7% for intestinal manipulation). However, stimulation of the vagus nerve prevented the intestinal manipulation−induced gastroparesis 24 h after surgery (gastric retention, 25.2% +/-3.2%;Fig. 5). Notably, stimulation of the vagus nerve in itself may alter gastric emptying during the vagus stimulation protocol24. However, we found that stimulation of the vagus nerve did not affect basal gastric emptying 24 h after surgery (gastric retention, 15.7% +/-3.6%; Fig 5). The last finding demonstrates that normalization of gastric emptying after stimulation of the vagus nerve was not a direct effect on gastric motility but resulted from reduced inflammation of the manipulated bowel segment11.
We next analyzed muscularis tissue for granulocytic infiltrates by measuring myeloperoxidase activity in muscularis tissue homogenates and quantifying cellular infiltrates (Supplementary figure 1). The intestinal manipulation−induced inflammation of the muscularis externa in mice that received stimulation of the vagus nerve was reduced in a voltage-dependent way compared with that of mice that received intestinal manipulation plus sham stimulation. Prior vagotomy of the proximal end of the stimulated vagus nerve did not affect these results (data not shown), indicating that the anti-inflammatory effect of stimulation of the vagus nerve was not dependent on the activation of central nuclei, which confirms published
van de Zanden.indb 50 23-5-2011 11:43:45
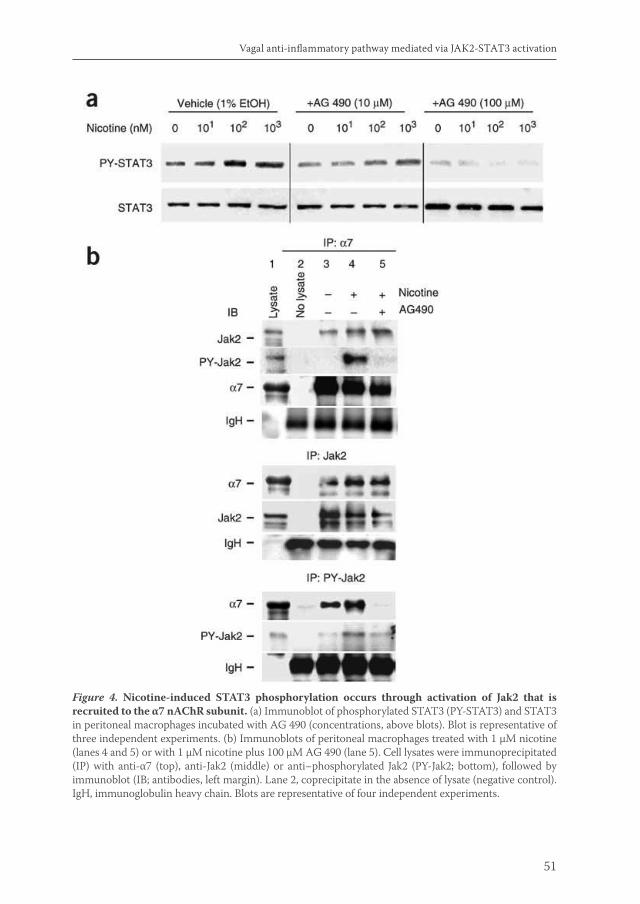
51
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
Figure 4. Nicotine-induced STAT3 phosphorylation occurs through activation of Jak2 that is recruited to the α7 nAChR subunit. (a) Immunoblot of phosphorylated STAT3 (PY-STAT3) and STAT3 in peritoneal macrophages incubated with AG 490 (concentrations, above blots). Blot is representative of three independent experiments. (b) Immunoblots of peritoneal macrophages treated with 1 μM nicotine (lanes 4 and 5) or with 1 μM nicotine plus 100 μM AG 490 (lane 5). Cell lysates were immunoprecipitated (IP) with anti-α7 (top), anti-Jak2 (middle) or anti−phosphorylated Jak2 (PY-Jak2; bottom), followed by immunoblot (IB; antibodies, left margin). Lane 2, coprecipitate in the absence of lysate (negative control). IgH, immunoglobulin heavy chain. Blots are representative of four independent experiments.
van de Zanden.indb 51 23-5-2011 11:43:45

52
Chapter 3
reports2. We next incubated intestinal segments with the nicotinic receptor blocker hexamethonium before intestinal manipulation combined with stimulation of the vagus nerve. In intestinal segments treated with hexamethonium, stimulation of the vagus nerve failed to prevent inflammation, in contrast to incubation with vehicle (supplementary figure 1), demonstrating that the anti-inflammatory effect of vagus stimulation acted through local activation of nicotinic receptors.
Stimulation of the vagus nerve activates STAT3 in vivoTo further investigate whether macrophages mediated the anti-inflammatory effect of stimulation of the vagus nerve, we analyzed the expression of transcripts of macrophage-derived inflammatory mediators in muscularis tissue 3 h after surgery. Stimulation of the vagus nerve reduced the expression of Cxcl2 mRNA (Fig. 6a,b) and Ccl3 mRNA (data not shown) but did not notably alter the expression of Tnf transcripts in muscularis tissue, confirming earlier reports2,4. However, when we analyzed peritoneal lavage fluid for the presence of macrophage inflammatory mediators 3 h after intestinal manipulation, we found that stimulation of the vagus nerve significantly reduced the secretion of TNF, IL-6, MIP-2 (Fig. 6c) and MIP-1α (data not shown) in the peritoneal cavity. This reduction was not due to enhanced expression of IL-10, as stimulation of the vagus nerve was similarly potent in reducing intestinal manipulation−induced inflammation in IL-10-deficient mice (Fig. 6d). Moreover, the peritoneal IL-10 in wild-type mice did not reach the limit of detection (31 pg/ml) at 1, 3 or 6 h after intestinal manipulation (data not shown). Expression of Socs3 (Fig. 6a) but not Socs1 (data not shown) was increased in muscularis tissue after stimulation of the vagus nerve even in mice that underwent this stimulation without manipulation of the bowel.
Given the short half-life of acetylcholine, cholinergic regulation of macrophage activation most likely requires that cholinergic nerves be in close proximity to intestinal macrophages. To investigate this, we immunohistochemically double-labeled vesicular acetylcholine transporter−positive vagal efferent fibers and macrophages in rat intestinal musclaris tissue. Macrophages were in close proximity to nerve terminals in the myenteric plexus in the ileum (Fig. 7a) and circular muscle of gastric corpus (data not shown). Hence, acetylcholine released from efferent nerve terminals could easily reach macrophages in the nanomolar concentration range.
To verify that the enhanced SOCS3 expression reflected increased STAT3 activation in vivo, we immunohistochemically analyzed intestinal tissues for the presence of phosphorylated STAT3 in mice that underwent control laparotomy surgery, intestinal manipulation alone or intestinal manipulation plus stimulation of the vagus nerve (Fig. 7b,c). We found phosphorylated STAT3−positive nuclei in mice that underwent control laparotomy (Fig. 7b). Intestinal manipulation resulted in the appearance of phosphorylated STAT3−positive cells adhering to the serosal site of the bowel wall,
van de Zanden.indb 52 23-5-2011 11:43:45
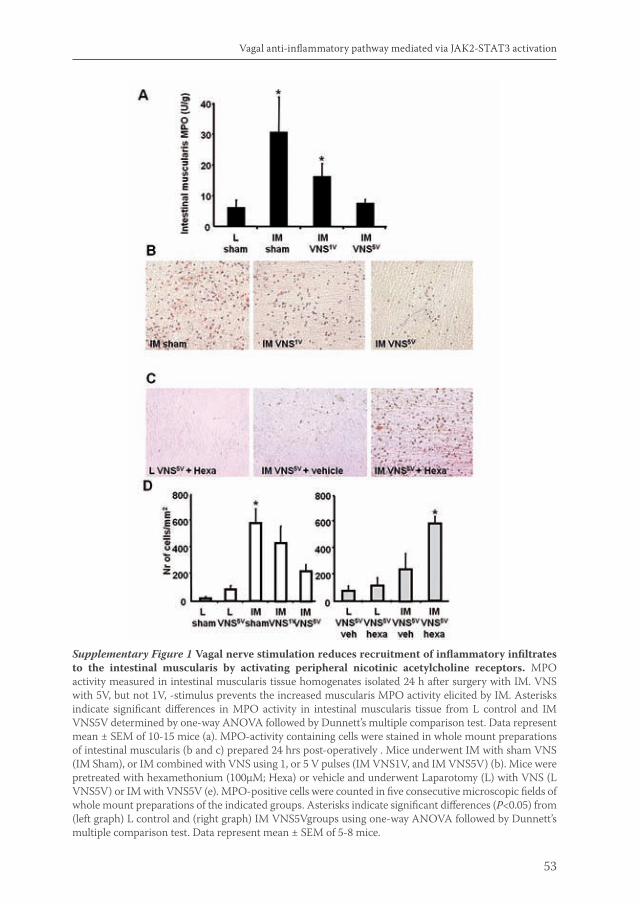
53
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
Supplementary Figure 1 Vagal nerve stimulation reduces recruitment of inflammatory infiltrates to the intestinal muscularis by activating peripheral nicotinic acetylcholine receptors. MPO activity measured in intestinal muscularis tissue homogenates isolated 24 h after surgery with IM. VNS with 5V, but not 1V, -stimulus prevents the increased muscularis MPO activity elicited by IM. Asterisks indicate significant differences in MPO activity in intestinal muscularis tissue from L control and IM VNS5V determined by one-way ANOVA followed by Dunnett’s multiple comparison test. Data represent mean ± SEM of 10-15 mice (a). MPO-activity containing cells were stained in whole mount preparations of intestinal muscularis (b and c) prepared 24 hrs post-operatively . Mice underwent IM with sham VNS (IM Sham), or IM combined with VNS using 1, or 5 V pulses (IM VNS1V, and IM VNS5V) (b). Mice were pretreated with hexamethonium (100μM; Hexa) or vehicle and underwent Laparotomy (L) with VNS (L VNS5V) or IM with VNS5V (e). MPO-positive cells were counted in five consecutive microscopic fields of whole mount preparations of the indicated groups. Asterisks indicate significant differences (P<0.05) from (left graph) L control and (right graph) IM VNS5Vgroups using one-way ANOVA followed by Dunnett’s multiple comparison test. Data represent mean ± SEM of 5-8 mice.
van de Zanden.indb 53 23-5-2011 11:43:48
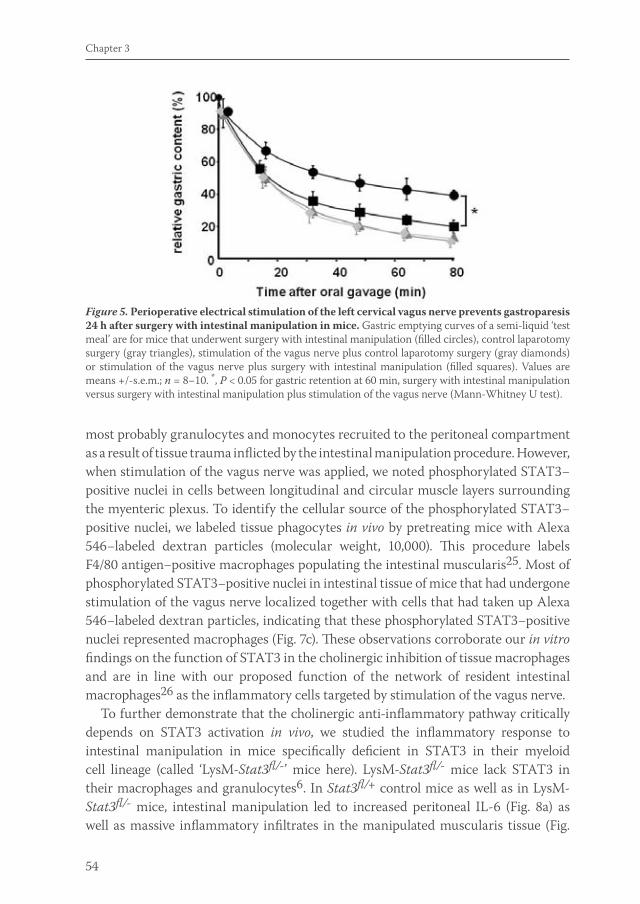
54
Chapter 3
most probably granulocytes and monocytes recruited to the peritoneal compartment as a result of tissue trauma inflicted by the intestinal manipulation procedure. However, when stimulation of the vagus nerve was applied, we noted phosphorylated STAT3−positive nuclei in cells between longitudinal and circular muscle layers surrounding the myenteric plexus. To identify the cellular source of the phosphorylated STAT3−positive nuclei, we labeled tissue phagocytes in vivo by pretreating mice with Alexa 546−labeled dextran particles (molecular weight, 10,000). This procedure labels F4/80 antigen−positive macrophages populating the intestinal muscularis25. Most of phosphorylated STAT3−positive nuclei in intestinal tissue of mice that had undergone stimulation of the vagus nerve localized together with cells that had taken up Alexa 546−labeled dextran particles, indicating that these phosphorylated STAT3−positive nuclei represented macrophages (Fig. 7c). These observations corroborate our in vitro findings on the function of STAT3 in the cholinergic inhibition of tissue macrophages and are in line with our proposed function of the network of resident intestinal macrophages26 as the inflammatory cells targeted by stimulation of the vagus nerve.
To further demonstrate that the cholinergic anti-inflammatory pathway critically depends on STAT3 activation in vivo, we studied the inflammatory response to intestinal manipulation in mice specifically deficient in STAT3 in their myeloid cell lineage (called ‘LysM-Stat3fl/-’ mice here). LysM-Stat3fl/- mice lack STAT3 in their macrophages and granulocytes6. In Stat3fl/+ control mice as well as in LysM-Stat3fl/- mice, intestinal manipulation led to increased peritoneal IL-6 (Fig. 8a) as well as massive inflammatory infiltrates in the manipulated muscularis tissue (Fig.
Figure 5. Perioperative electrical stimulation of the left cervical vagus nerve prevents gastroparesis 24 h after surgery with intestinal manipulation in mice. Gastric emptying curves of a semi-liquid ‘test meal’ are for mice that underwent surgery with intestinal manipulation (filled circles), control laparotomy surgery (gray triangles), stimulation of the vagus nerve plus control laparotomy surgery (gray diamonds) or stimulation of the vagus nerve plus surgery with intestinal manipulation (filled squares). Values are means +/-s.e.m.; n = 8−10. *, P < 0.05 for gastric retention at 60 min, surgery with intestinal manipulation versus surgery with intestinal manipulation plus stimulation of the vagus nerve (Mann-Whitney U test).
van de Zanden.indb 54 23-5-2011 11:43:48
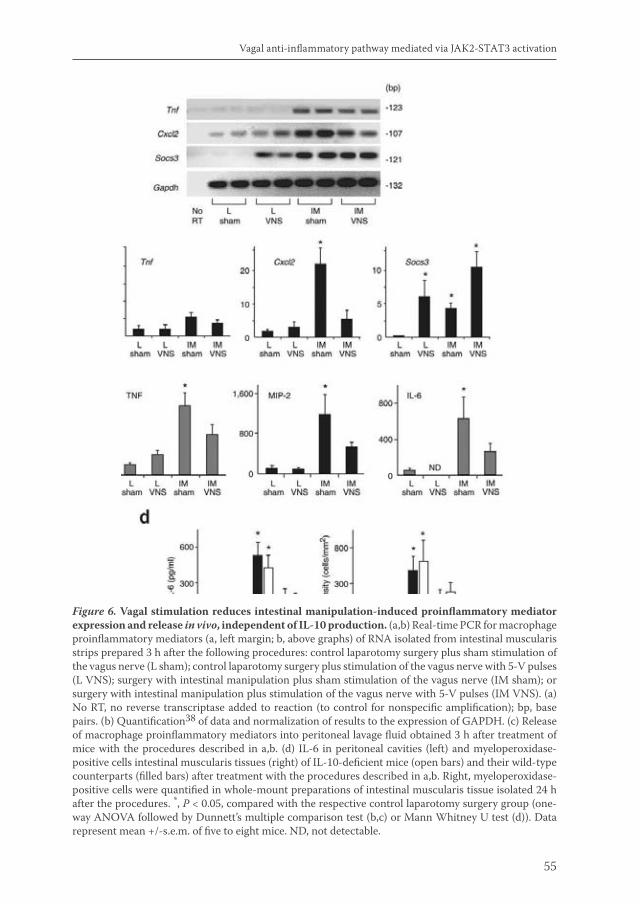
55
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
Figure 6. Vagal stimulation reduces intestinal manipulation-induced proinflammatory mediator expression and release in vivo, independent of IL-10 production. (a,b) Real-time PCR for macrophage proinflammatory mediators (a, left margin; b, above graphs) of RNA isolated from intestinal muscularis strips prepared 3 h after the following procedures: control laparotomy surgery plus sham stimulation of the vagus nerve (L sham); control laparotomy surgery plus stimulation of the vagus nerve with 5-V pulses (L VNS); surgery with intestinal manipulation plus sham stimulation of the vagus nerve (IM sham); or surgery with intestinal manipulation plus stimulation of the vagus nerve with 5-V pulses (IM VNS). (a) No RT, no reverse transcriptase added to reaction (to control for nonspecific amplification); bp, base pairs. (b) Quantification38 of data and normalization of results to the expression of GAPDH. (c) Release of macrophage proinflammatory mediators into peritoneal lavage fluid obtained 3 h after treatment of mice with the procedures described in a,b. (d) IL-6 in peritoneal cavities (left) and myeloperoxidase-positive cells intestinal muscularis tissues (right) of IL-10-deficient mice (open bars) and their wild-type counterparts (filled bars) after treatment with the procedures described in a,b. Right, myeloperoxidase-positive cells were quantified in whole-mount preparations of intestinal muscularis tissue isolated 24 h after the procedures. *, P < 0.05, compared with the respective control laparotomy surgery group (one-way ANOVA followed by Dunnett’s multiple comparison test (b,c) or Mann Whitney U test (d)). Data represent mean +/-s.e.m. of five to eight mice. ND, not detectable.
van de Zanden.indb 55 23-5-2011 11:43:48
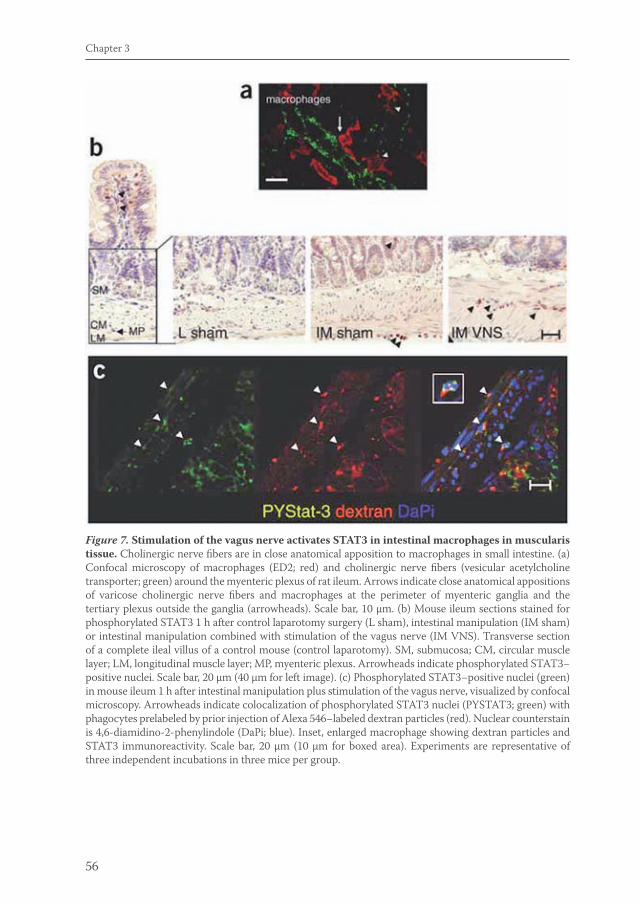
56
Chapter 3
Figure 7. Stimulation of the vagus nerve activates STAT3 in intestinal macrophages in muscularis tissue. Cholinergic nerve fibers are in close anatomical apposition to macrophages in small intestine. (a) Confocal microscopy of macrophages (ED2; red) and cholinergic nerve fibers (vesicular acetylcholine transporter; green) around the myenteric plexus of rat ileum. Arrows indicate close anatomical appositions of varicose cholinergic nerve fibers and macrophages at the perimeter of myenteric ganglia and the tertiary plexus outside the ganglia (arrowheads). Scale bar, 10 μm. (b) Mouse ileum sections stained for phosphorylated STAT3 1 h after control laparotomy surgery (L sham), intestinal manipulation (IM sham) or intestinal manipulation combined with stimulation of the vagus nerve (IM VNS). Transverse section of a complete ileal villus of a control mouse (control laparotomy). SM, submucosa; CM, circular muscle layer; LM, longitudinal muscle layer; MP, myenteric plexus. Arrowheads indicate phosphorylated STAT3−positive nuclei. Scale bar, 20 μm (40 μm for left image). (c) Phosphorylated STAT3−positive nuclei (green) in mouse ileum 1 h after intestinal manipulation plus stimulation of the vagus nerve, visualized by confocal microscopy. Arrowheads indicate colocalization of phosphorylated STAT3 nuclei (PYSTAT3; green) with phagocytes prelabeled by prior injection of Alexa 546−labeled dextran particles (red). Nuclear counterstain is 4,6-diamidino-2-phenylindole (DaPi; blue). Inset, enlarged macrophage showing dextran particles and STAT3 immunoreactivity. Scale bar, 20 μm (10 μm for boxed area). Experiments are representative of three independent incubations in three mice per group.
van de Zanden.indb 56 23-5-2011 11:43:49

57
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
8b). Notably, however, stimulation of the vagus nerve reduced peritoneal IL-6 and intestinal inflammation in Stat3fl/+ control mice but failed to do so in LysM-Stat3fl/- mice. These data support the critical function of STAT3 activation in the cholinergic anti-inflammatory pathway in vivo.
DISCUSSION
The cholinergic anti-inflammatory pathway represents a physiological system for controlling macrophage activation and inflammation in sepsis models1. Its working mechanism ultimately involves the prevention of NF-ĸB p65 activity3 after α7 nAChR activation4, but the exact cellular mechanism has remained unclear. Here we have demonstrated that nicotine acts on macrophages via the recruitment of Jak2 to the α7 nAChR and activation of Jak2, thereby initiating the anti-inflammatory STAT3 and SOCS3 signaling cascade. Notably, recruitment of Jak2 to the α7 nAChR subunit has also been described in neuronal PC12 cells exposed to nicotine, as part of a neuroprotective mechanism against β-amyloid-induced apoptosis20. Our results in resident peritoneal macrophages were consistent with our in vivo data, as we found activation of STAT3 in intestinal macrophages in response to stimulation of the vagus nerve in mice, which indicates activation of STAT3 induced by acetylcholine derived from vagal efferents.
Figure 8. Stimulation of the vagus nerve fails to reduce inflammation in LysM-Stat3fl/- mice.IL-6 was measured in peritoneal lavage fluid (a) and myeloperoxidase-positive inflammatory infiltrates were quantified in muscularis tissues (b) of Stat3fl/+ control mice (filled bars) or LysM-Stat3fl/- mice (open bars) treated with control laparotomy surgery (L sham), intestinal manipulation (IM sham) or intestinal manipulation plus stimulation of the vagus nerve (IM VNS). (a) Peritoneal lavage fluid was collected 3 h after the procedures. (b) Infiltrates were quantified in whole-mount preparations 24 h after the surgical procedures. Data are mean +/-s.e.m.; n = 3−4. *, P < 0.05, compared with the respective control laparotomy surgery group (Mann-Whitney U test).
van de Zanden.indb 57 23-5-2011 11:43:49

58
Chapter 3
Activation of the STAT3 cascade after nAChR ligation is fully consistent with the observed inhibition of proinflammatory cytokine release by macrophages, because STAT3 is a negative regulator of the inflammatory response6,27. In our studies, the anti-inflammatory effect of nicotine on macrophages required DNA binding and transactivation of STAT3, as nicotine failed to inhibit TNF production in macrophages overexpressing STAT3 altered in its in DNA-binding capacity17. Likewise, activation of STAT3 is required for the anti-inflammatory properties of IL-10 (refs. 8, 28) and the IL-10-induced attenuation of cytokine production and proliferation28. In addition, STAT3 phosphorylation is required for IL-6-induced growth arrest and differentiation29.
SOCS3 specifically disables STAT3 phosphorylation via IL-6R but does not interfere with IL-10R signaling9, 10, 30. Conditional knockout mice specifically lacking SOCS3 in their macrophages (LysM-Socs3fl/-) show resistance to endotoxemia, explained by the anti-inflammatory effect of sustained STAT3 activation through IL-6R ligands10. Regardless of that finding, our results have indicated that the enhanced expression of SOCS3 did not contribute to the anti-inflammatory effect of nAChR activation, as blockade of SOCS3 expression did not prevent the anti-inflammatory action of nicotine. Hence, the anti-inflammatory effect of cholinergic activation in macrophages rests mainly on enhanced STAT3 rather than SOCS3 activation.
We have shown that STAT3 was activated by nicotine directly and that involvement of enhanced signaling via IL-10R here was unlikely, as we found the macrophage deactivation induced by stimulation of the vagus nerve to be similarly effective in IL-10-deficient mice. Moreover, nicotine-induced STAT3 activation could be prevented by nAChR blockers. Our observations suggest that the molecular route exerting the anti-inflammatory effect of nAChR activation mimics the signaling pathway of IL-10R without the requirement of IL-10 itself. That hypothesis is supported by our finding and those of another study2 that, consistent with the action of IL-10 (ref. 31), nicotine does not alter TNF mRNA expression but decreases the release of TNF protein. Furthermore, LysM-Stat3fl/- mice have a phenotype resembling that of IL-10-deficient mice6. Nicotine-induced inhibition of the release of high-mobility group box 1 in mouse RAW264.7 macrophages is associated with inhibition of NF-ĸB p65 transcriptional activity3. Our finding that nicotine repressed macrophage activity via STAT3 may very well explain that observation, as IL-10−STAT3 (ref. 28) signaling blocks NF-ĸB DNA-binding32, 33, possibly through direct interaction of dimerized STAT3 with the p65 subunit34.
We have shown here that recruitment of inflammatory infiltrates induced by bowel manipulation and the resulting symptoms of postoperative ileus were reduced substantially by stimulation of the vagus nerve. Our results have shown strict cholinergic control of macrophage activation in vivo, which may be substantiated by the observation that cholinergic (vesicular acetylcholine transporter−positive) nerve fibers are situated in close proximity to resident macrophages in intestinal myenteric
van de Zanden.indb 58 23-5-2011 11:43:49

59
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
plexus. At first glance, our data may seem contradictory to the outcome of earlier attempts to treat postoperative ileus using cholinergic agents such as neostigmine, which had only limited success35. That lack of efficacy could be explained by the fact that the inflammatory process had already been fully accomplished by the time these agents were administered, leaving the activation of inhibitory neural pathways11 unaffected. Our results indicate that nicotinic receptor activation before or during surgery prevents postoperative intestinal inflammation and will certainly be a promising strategy for treating postoperative ileus. Notably, vagus nerve stimulators are clinically approved devices for the treatment of epilepsy and depression36. In conclusion, we have shown here that inhibition of macrophage activation via the cholinergic anti-inflammatory pathway is brought about via Jak2-STAT3 signaling. Our data may aid in further development of therapeutic strategies for modifying the cholinergic anti-inflammatory pathway to treat various inflammatory conditions.
van de Zanden.indb 59 23-5-2011 11:43:49

60
Chapter 3
REFERENCE LIST
1. Tracey, K.J. The inflammatory reflex. Nature 420, 853-9 (2002).
2. Borovikova, L.V. et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458-62 (2000).
3. Wang, H. et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med (2004).
4. Wang, H. et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384-8 (2003).
5. Matsunaga, K., Klein, T.W., Friedman, H. & Yamamoto, Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol 167, 6518-24 (2001).
6. Takeda, K. et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10, 39-49 (1999).
7. Welte, T. et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A 100, 1879-84 (2003).
8. Levy, D.E. & Lee, C.K. What does Stat3 do? J Clin Invest 109, 1143-8 (2002).
9. Kubo, M., Hanada, T. & Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat Immunol 4, 1169-76 (2003).
10. Yasukawa, H. et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol 4, 551-6 (2003).
11. de Jonge, W.J. et al. Postoperative ileus is maintained by intestinal immune infiltrates that activate inhibitory neural pathways in mice. Gastroenterology 125, 1137-47 (2003).
12. Kalff, J.C. et al. Intra-abdominal activation of a local inflammatory response within the human muscularis externa during laparotomy. Ann Surg 237, 301-15. (2003).
13. Livingston, E.H. & Passaro, E.P., Jr. Postoperative ileus. Dig Dis Sci 35, 121-32 (1990).
14. Bauer, A.J. & Boeckxstaens G.E. Mechanisms of postoperative ileus. Neurogastroenterol. Motil. 16, 54-60 (2004)
15. Mikkelsen, H.B. Macrophages in the external muscle layers of mammalian intestines. Histol Histopathol 10, 719-36 (1995).
16. Shimozaki, K., Nakajima, K., Hirano, T. & Nagata, S. Involvement of STAT3 in the granulocyte colony-stimulating factor-induced differentiation of myeloid cells. J Biol Chem 272, 25184-9 (1997).
17. de Koning, J.P. et al. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 19, 3290-8 (2000).
18. Lukas, R.J. et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51, 397-401 (1999). 19. Meydan, N. et al. Inhibition of acute lymphoblastic leukemia by a Jak-2 inhibitor. Nature 379, 645-648 (1996)
20. Shaw, S., Bencherif, M. & Marrero, M.B. Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against Abeta-(1-42) amyloid. J Biol Chem 277, 44920-4 (2002).
21. Drisdel, R.C. & Green, W.N. Neuronal alpha-bungarotoxin receptors are alpha7 subunit homomers. J Neurosci 20, 133-9 (2000).
22. Kalff, J.C. et al. Surgically induced leukocytic infiltrates within the rat intestinal muscularis mediate postoperative ileus. Gastroenterology 117, 378-87. (1999).
23. de Jonge, W.J. et al. Mast cell degranulation during abdominal surgery initiates postoperative ileus in mice. Gastroenterology 127, 535-45 (2004).
van de Zanden.indb 60 23-5-2011 11:43:49

61
Vagal anti-inflammatory pathway mediated via JAK2-STAT3 activation
24. Takahashi, T. & Owyang, C. Vagal control of nitric oxide and vasoactive intestinal polypeptide release in the regulation of gastric relaxation in rat. J Physiol (Lond) 484, 481-92 (1995).
25. Ozaki, H. et al. Isolation and characterization of resident macrophages from the smooth muscle layers of murine small intestine. Neurogastroenterol Motil 16, 39-51 (2004).
26. Mikkelsen, H.B. Macrophages in the external muscle layers of mammalian intestines. Histol. Histopathol. 10, 719-736 (1995(
27. Wang, T. et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10, 48-54 (2004).
28. Williams, L.M., Ricchetti, G., Sarma, U., Smallie, T. & Foxwell, B.M. Interleukin-10 suppression of myeloid cell activation - a continuing puzzle. Immunology 113, 281-92 (2004).
29. Akira, S. et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63-71 (1994).
30. Lang, R. et al. SOCS3 regulates the plasticity of gp130 signaling. Nat. Immunol. 4, 546–550 (2003).
31. Kontoyiannis, D. et al. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. Embo J 20, 3760-70 (2001).
32. Wang, P., Wu, P., Siegel, M.I., Egan, R.W. & Billah, M.M. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem 270, 9558-63 (1995).
33. Schottelius, A.J., Mayo, M.W., Sartor, R.B. & Baldwin, A.S., Jr. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem 274, 31868-74 (1999).
34. Yu, Z., Zhang, W. & Kone, B.C. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 367, 97-105 (2002).
35. Longo, W.E. & Vernava, A.M., 3rd. Prokinetic agents for lower gastrointestinal motility disorders. Dis Colon Rectum 36, 696-708 (1993).
36. George, M.S. et al. Vagus nerve stimulation. A potential therapy for resistant depression? Psychiatr Clin North Am 23, 757-83 (2000).
37. Bennink, R.J. et al. Validation of gastric-emptying scintigraphy of solids and liquids in mice using dedicated animal pinhole scintigraphy. J Nucl Med 44, 1099-104 (2003).
38. Ramakers, C., Ruijter, J.M., Deprez, R.H. & Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339, 62-6 (2003).
van de Zanden.indb 61 23-5-2011 11:43:50

4CHAPTER
Deciphering STAT3 signaling in cholinergic inhibition of inflammation
Esmerij P. van der ZandenGeber peňaGuy E. BoeckxstaensLuis UlloaWouter J. de Jonge
Published in modified version in Eur J Immunol 2010 Sep;40(9):2580-9
van de Zanden.indb 63 23-5-2011 11:43:50

64
Chapter 4
ABSTRACT
We previously reported that stimulation of the vagus nerve attenuates macrophage responses by activating the intracellular Jak2-STAT3 signaling pathway. Here, we further analyzed the potential of STAT3 to modulate TNF responses using STAT silencing strategies, pharmacological blockade, and dominant negative STAT3 constructs. We reveal that deletion of STAT3 using siRNA strategies augments the TNF production by endotoxin, confirming that STAT3 is a negative regulator of the immune response. On the other hand, pharmacological blockade of JAK2-STAT3 phosphorylation attenuates LPS-induced TNF production and NF-kB activation in macrophages. Expression of a mutant form that allows STAT3 tyrosine phosphorylation, but not STAT3 DNA binding, prevents the anti-inflammatory potential of nicotine. STAT3 inhibition specifically prevents the activation of the p65RelA/p50NF-kB1 pathway without affecting alternative other NF-kB signaling routes. These data suggest that nicotinic inhibition of inflammation in macrophages is dependent on STAT3 DNA binding and STAT3 protein, rather than STAT3 phosphorylation. Hypothetically, unphosphorylated STAT3 (U-STAT) can be important in mediating the anti-inflammatory effect of nAChR activation, via binding to NF-kB20 and inhibition of NF-kB-activation of TNF transcription.
van de Zanden.indb 64 23-5-2011 11:43:50

65
Deciphering STAT3 signaling in cholinergic immune-modulation
INTRODUCTION
Vagal activity has been implicated in the negative regulation of inflammatory reactions via the peripheral release of acetylcholine. Vagus nerve activity either via electrical stimulation or ablation of vagal output, has been shown to affect the disease course in experimental animal models of inflammatory conditions such as sepsis1, post-operative ileus2, colitis3, peritonitis4 and arthritis5. This concept is based on the assumption that the vagus nerve, via peripheral release of the neurotransmitter acetylcholine (Ach), can regulate the immune response through activation of nicotinic acetylcholine receptors (nAChR) expressed on immune cells. At present, two nicotinic acetylcholine receptor (nAchR) subtypes have been put forward to be involved in this process: the nAChRα7 homopentamer6, and the α4β2 hetero pentamer7.
This so called ‘cholinergic anti-inflammatory pathway’ is characterized by a nicotine dose-dependent decrease in the production of pro-inflammatory mediators, including TNF, MIP2, IL-6 and HMGB1 by macrophages2. In conjunction, nAChR activation has shown to inhibit NF-κB transcriptional activity8. Besides an effect on inflammatory properties, the cholinergic nervous system also affects more professional macrophage functions such as endo- and phagocytosis of bacteria and particles7. Nicotine has already been used in clinical trials for inflammatory disorders such as ulcerative colitis, but the therapeutic potential of this mechanism is hampered by the collateral toxicity of nicotine3. Identification and specific targeting of the intracellular signaling pathway involved in the anti-inflammatory effect of nAChR activation would increase the translational potential of this mechanism to a great extent.
We previously evaluated the involvement of the janus kinase 2/signal transducer and activator of transcription 3 (JAK2-STAT3) pathway in mediating cholinergic anti-inflammatory effects in macrophages2. Mice, that specifically lack expression of STAT3 in their in macrophages and neutrophils, are highly susceptible to endotoxin shock and develop chronic enterocolitis10. Furthermore, production of inflammatory cytokines from STAT3-deficient macrophages is dramatically augmented in response to lipopolysaccharide10. These findings indicate that STAT3 is a potential negative regulator of inflammation.
In JAK2-STAT3 signaling, cytokine receptor activation induces tyrosine phosphorylation activation of JAK2, which leads to the phosphorylation of STAT3. Upon phosphorylation, STAT3 dimerizes and translocates to the nucleus to bind specific DNA sequences, to activate genes involved in diverse functions such as cell cycle progression, apoptosis and inflammation4. STAT3 activation is required for both IL-10 receptor (IL-10R) and IL-6 receptor (IL-6R) signaling5. IL6 receptor signaling is negatively regulated through the suppressor of cytokine signaling protein SOCS3, whose expression is induced by STAT3 activation11,6. We previously showed that nicotine induced STAT3 phosphorylation and SOCS-3 activation and failed to decrease TNF production in macrophages transfected with an inactive form of
van de Zanden.indb 65 23-5-2011 11:43:50

66
Chapter 4
STAT32. Moreover, stimulation of the vagus nerve failed to reduce inflammation in LysM-Stat3fl/- mice2. These data demonstrated that nicotine exerts its anti-inflammatory effect on peritoneal macrophages via Jak2-STAT3 signaling.
Here, we further analyze the role of the JAK2-STAT3 pathway in the anti-inflammatory potential of nAChR activation. The immunological implications of the JAK2-STAT3 pathway are analyzed in NF-kB signaling and cytokine production from macrophages. Our results confirm that Jak2-STAT3 signaling is involved in mediating cholinergic anti-inflammatory responses and add to the previous findings in STAT3 conditional knockout mice. However, these data show that this appears to be independent of STAT3 phosphorylation, and suggest there might be a role for unphopsphorylated STAT3 in this process.
MATERIALS AND METHODS
Chemicals and reagents. AG490, Stattic, LPS and nicotine were purchased from Sigma- Aldrich (Zwijndrecht, the Netherlands). AG490 was dissolved in ethanol stock 20mM; and LPS dissolved in PBS stock 1 mg/ml (GIBCO, Invitrogen, CA). Anti-NFkB-p65, anti-actin and anti-stat3 antibodies were from Cell Signaling and HRP-conjugated anti-rabbit from DakoCytomation.
Cell Culture. Murine RAW264.7 cells (ATCC, Middlesex, UK) were grown in RMPI-1640 medium with antibiotics and L-glutamine (Gibco, Breda, The Netherlands), supplemented with 10% heat-inactivated fetal bovine serum at 37ºC in a humidified incubator with 5% CO2.
Stabile transfection. RAW264.7 macrophages were stably transfected with a NF-κB luciferase reporter construct (Clontech, Mountain View, CA) in which a PDNA3.1(+) derived neomycin resistance TK cassette was inserted (referred to as pNF-kBneo-luc). Transfection was performed using electroporation. Briefly, 2*106 cells were resuspended in 100µL Nucleofector V reagent (Amaxa Biosystems Inc.) with 2 µg of DNA, mixed and electroporated using the Amaxa nucleofection device according to manufacturer’s instructions. Immediately after transfection, cells were cultured in RPMI 1640/10% FCS o/n. Transfected cells were selected in the culture medium supplemented with 1000ug/ml neomycine for 14 days. Resistant cells were subcloned and clones were cultured up to 20 passages with RPMI containing neomycine.
Transient transfections. The pCAGGS-neo expression vectors encoding hemagglutinin-tagged dominant negative mutants STAT3D, STAT3F or the empty expression vector (EV) were provided by I. Touw (Erasmus University, Rotterdam, The Netherlands). RAW264.7 cells were transfected with STAT3F, STAT3D, STAT3EV
van de Zanden.indb 66 23-5-2011 11:43:50

67
Deciphering STAT3 signaling in cholinergic immune-modulation
reporter constructs using Jet PEI transfection reagent (PolyTransfection), according to the manufacturer’s instructions. For shRNA transfection, Macrophages were nucleofected using materials supplied in the Amaxa Cell Line Nucleofector Kit V (Lonza Inc). Briefly, 1 x106 cells were centrifuged and suspended with 100 ul of Cell Line Nucleofector Solution V, in an Amaxa-certified cuvette, using 0.25 to 2 nmol/ml Non-Targeting DHARMACON siCONTROL (CAT# D-001206-13) or siGENOME SMART pool STAT3 (Cat# M-003544-00) and the program V-001 (AMAXA Biosystem nucleofector) for high transfection efficiency.
Cell stimulation. For experiments, cells were transferred to 48-well suspension plates (Greiner-Bio, Alphen aan de Rijn, The Netherlands) at 2.5 x105 cells/well. After overnight incubation, the medium was removed and replaced with RPMI containing 1% serum. Cells were pretreated with AG490, stattic or nicotine at the concentrations indicated for 30 minutes, washed and subsequently stimulated with LPS for 3 h. Medium was harvested three hours after LPS stimulation and TNF levels were analyzed using the TNF ELISA kit from R&D systems Inc (Abingdon, UK). For NF-kB luciferase measurement, the medium was removed 3 hours after LPS stimulation; the cells were washed three times with ice-cold PBS and lysed with Passive Lysis Buffer supplied in the LuciferaseTM Reporter Assay Kit (Promega Corporation, Madison, WI ), the lysate was assayed for luciferase activity according to the manufacturer’s instructions. Specific NF-kB protein binding to DNA was analyzed using the TransAM DNA-Binding ELISA (Active Motif; Cambridge, MA) following the manufacturer’s instructions.
Immunoblotting. Immunoblotting was performed routinely as described2. Nuclear and cytosol extractions were prepared using the NucBuster Protein Extraction Kit (Novagem), according to the manufacturer’s instructions.
Statistical Analyses. All data in the figures and text are expressed as mean ± standard error (SEM). Statistical analyses were performed using the non-parametric Mann-Whitney U test. A probability value (P) of less than 0.05 was considered significant.
RESULTS
Previous studies reveal that the anti-inflammatory potential of nicotine requires STAT3 activation by α7nAChR2. First, we analyzed the specificity of this pathway by using the α7nAChR-knockout and littermate wild-type mice. Nicotine inhibits LPS-induced serum TNF levels in both wild-type and α7nAChR-knockout mice by over 50% with a statistically similar efficiency (Fig.1A). These results suggest that the
van de Zanden.indb 67 23-5-2011 11:43:50
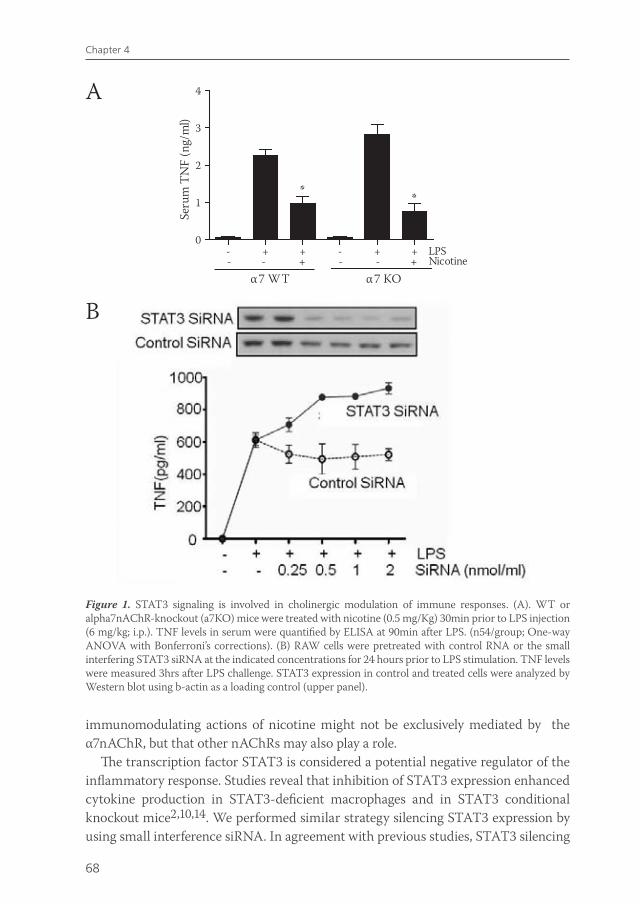
68
Chapter 4
immunomodulating actions of nicotine might not be exclusively mediated by the α7nAChR, but that other nAChRs may also play a role.
The transcription factor STAT3 is considered a potential negative regulator of the inflammatory response. Studies reveal that inhibition of STAT3 expression enhanced cytokine production in STAT3-deficient macrophages and in STAT3 conditional knockout mice2,10,14. We performed similar strategy silencing STAT3 expression by using small interference siRNA. In agreement with previous studies, STAT3 silencing
Figure 1. STAT3 signaling is involved in cholinergic modulation of immune responses. (A). WT or alpha7nAChR-knockout (a7KO) mice were treated with nicotine (0.5 mg/Kg) 30min prior to LPS injection (6 mg/kg; i.p.). TNF levels in serum were quantified by ELISA at 90min after LPS. (n54/group; One-way ANOVA with Bonferroni’s corrections). (B) RAW cells were pretreated with control RNA or the small interfering STAT3 siRNA at the indicated concentrations for 24 hours prior to LPS stimulation. TNF levels were measured 3hrs after LPS challenge. STAT3 expression in control and treated cells were analyzed by Western blot using b-actin as a loading control (upper panel).
A
B
- + + - + +0
1
2
3
4
- - + - - +LPSNicotine
* *
α7 WT α7 KO
Seru
m T
NF
(ng/
ml)
van de Zanden.indb 68 23-5-2011 11:43:50

69
Deciphering STAT3 signaling in cholinergic immune-modulation
in peritoneal RAW macrophages enhances LPS-induced TNF responses significantly (Fig. 1B). These results confirm that STAT3 is a negative regulator of the immune response.
Pharmacological inhibition of the JAK2-STAT3 pathway blunts NF-kB activation Cytokine receptor activation induces tyrosine phosphorylation and subsequent activation of JAK2, leading to phosphorylation of STAT3. This is demonstrated in figure 2a, as endotoxin induced STAT3 phosphorylation in tyrosine, which is proportional to endotoxin concentration (Fig. 2A). To assess the role of JAK2-STAT3 signaling in modulating the LPS-induced inflammatory response in macrophages, we pharmacologically blocked JAK2 activation using the tyrosine kinase inhibitor AG490. The efficiency of the JAK2 inhibitor was confirmed with STAT3 phosphorylation. JAK2 inhibition with AG490 prevents LPS-induced STAT3 tyrosine (Y705) phosphorylation in a concentration dependent manner without affecting the STAT3 serine (S727) phosphorylation (Fig. 2B). In figure 2C, it is shown that JAK2 inhibition using AG490, blunts LPS-induced NF-kB transactivation in a dose-dependent way at a maxima activity of ~50% inhibition at 25µM. In line, JAK2 inhibition with AG490 reduces LPS-induced TNF production in RAW264.7 cells (data not shown). These results demonstrate that pharmalogical inhibition of the JAK2-STAT3 signaling pathway reduces inflammatory responses in macrophages.
STAT3 phosphorylation is not involved in cholinergic modulation of TNF release Above data reveal that deletion of STAT3 using siRNA strategies augments the TNF production by endotoxin. On the other hand, pharmacological blockade of JAK2-STAT3 signaling attenuates LPS-induced TNF production and NF-kB activation in macrophages. We previously reported that the JAK2-STAT3 pathway is involved in mediating the anti-inflammatory action of nicotinic receptor activation in peritoneal macrophages2. Next, we sought to determine whether nAChR activation modulates the immune response via STAT3 phosphorylation, or via STAT3 DNA binding. To establish the role of STAT3 in the immune-modulatory actions of nicotine, we used the dominant negative constructs STAT3F, STAT3D and the empty vector. RAW264.7 cells were transfected with the mutant forms STAT3D or STAT3F, or with the empty vector as a control. STAT3F has the tyrosine 705 mutated by phenylalanine to prevent STAT3 tyrosine phosphorylation and subsequent dimerization, whereas STAT3D has the glutamic residues 434 and 435 replaced by alanine to allow tyrosine phosphorylation and dimerization but preventing STAT3 binding to DNA15. Nicotine significantly reduced TNF production in peritoneal macrophages transfected with STAT3F or Empty Vector (Fig. 3). However, this anti-inflammatory
van de Zanden.indb 69 23-5-2011 11:43:50
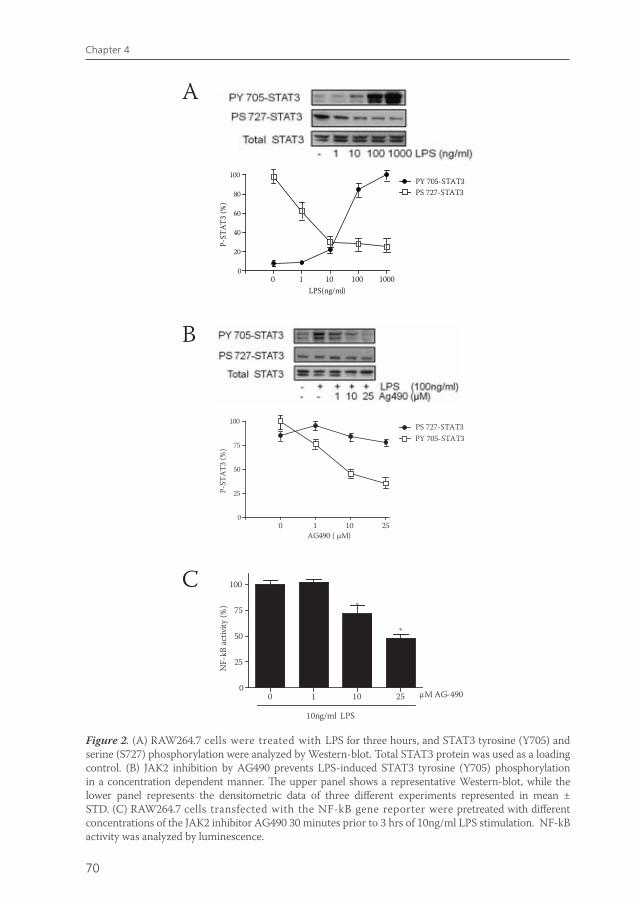
70
Chapter 4
Figure 2. (A) RAW264.7 cells were treated with LPS for three hours, and STAT3 tyrosine (Y705) and serine (S727) phosphorylation were analyzed by Western-blot. Total STAT3 protein was used as a loading control. (B) JAK2 inhibition by AG490 prevents LPS-induced STAT3 tyrosine (Y705) phosphorylation in a concentration dependent manner. The upper panel shows a representative Western-blot, while the lower panel represents the densitometric data of three different experiments represented in mean ± STD. (C) RAW264.7 cells transfected with the NF-kB gene reporter were pretreated with different concentrations of the JAK2 inhibitor AG490 30 minutes prior to 3 hrs of 10ng/ml LPS stimulation. NF-kB activity was analyzed by luminescence.
A
B
0
20
40
60
80
100PY 705-STAT3PS 727-STAT3
0 1 10 100 1000LPS(ng/ml)
P-ST
AT3
(%)
0
25
50
75
100
PY 705-STAT3PS 727-STAT3
0 1 10 25AG490 ( µM)
P-ST
AT3
(%)
0 1 10 250
25
50
75
100
10ng/ml LPS
*
*
µM AG-490
NF-
kB a
ctivi
ty (%
)
C
van de Zanden.indb 70 23-5-2011 11:43:51
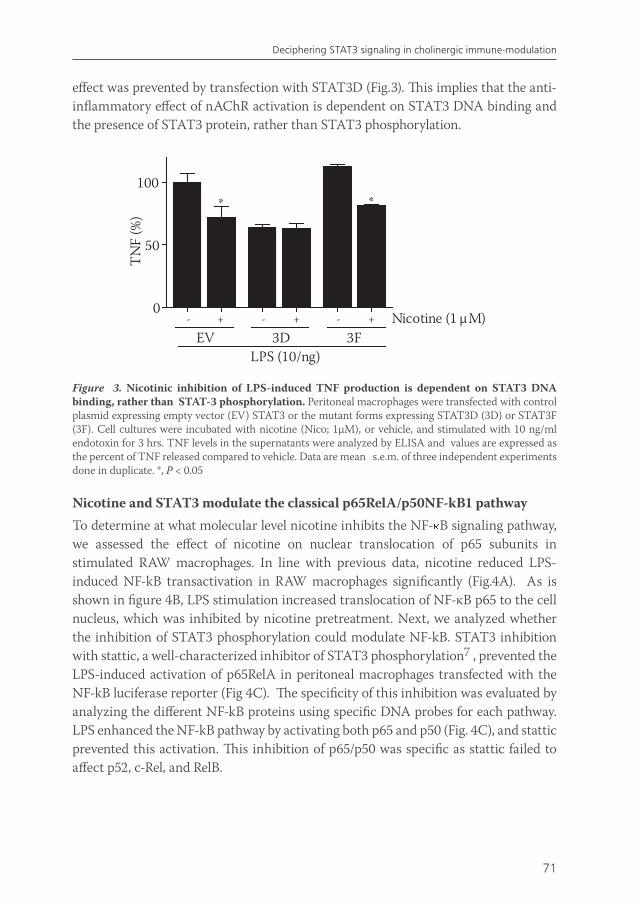
71
Deciphering STAT3 signaling in cholinergic immune-modulation
effect was prevented by transfection with STAT3D (Fig.3). This implies that the anti-inflammatory effect of nAChR activation is dependent on STAT3 DNA binding and the presence of STAT3 protein, rather than STAT3 phosphorylation.
- + - + - +0
50
100*
Nicotine (1 µM)EV 3D 3F
LPS (10/ng)
TN
F (%
)
* *
Figure 3. Nicotinic inhibition of LPS-induced TNF production is dependent on STAT3 DNA binding, rather than STAT-3 phosphorylation. Peritoneal macrophages were transfected with control plasmid expressing empty vector (EV) STAT3 or the mutant forms expressing STAT3D (3D) or STAT3F (3F). Cell cultures were incubated with nicotine (Nico; 1µM), or vehicle, and stimulated with 10 ng/ml endotoxin for 3 hrs. TNF levels in the supernatants were analyzed by ELISA and values are expressed as the percent of TNF released compared to vehicle. Data are mean s.e.m. of three independent experiments done in duplicate. *, P < 0.05
Nicotine and STAT3 modulate the classical p65RelA/p50NF-kB1 pathwayTo determine at what molecular level nicotine inhibits the NF- B signaling pathway, we assessed the effect of nicotine on nuclear translocation of p65 subunits in stimulated RAW macrophages. In line with previous data, nicotine reduced LPS-induced NF-kB transactivation in RAW macrophages significantly (Fig.4A). As is shown in figure 4B, LPS stimulation increased translocation of NF-κB p65 to the cell nucleus, which was inhibited by nicotine pretreatment. Next, we analyzed whether the inhibition of STAT3 phosphorylation could modulate NF-kB. STAT3 inhibition with stattic, a well-characterized inhibitor of STAT3 phosphorylation7 , prevented the LPS-induced activation of p65RelA in peritoneal macrophages transfected with the NF-kB luciferase reporter (Fig 4C). The specificity of this inhibition was evaluated by analyzing the different NF-kB proteins using specific DNA probes for each pathway. LPS enhanced the NF-kB pathway by activating both p65 and p50 (Fig. 4C), and stattic prevented this activation. This inhibition of p65/p50 was specific as stattic failed to affect p52, c-Rel, and RelB.
van de Zanden.indb 71 23-5-2011 11:43:51
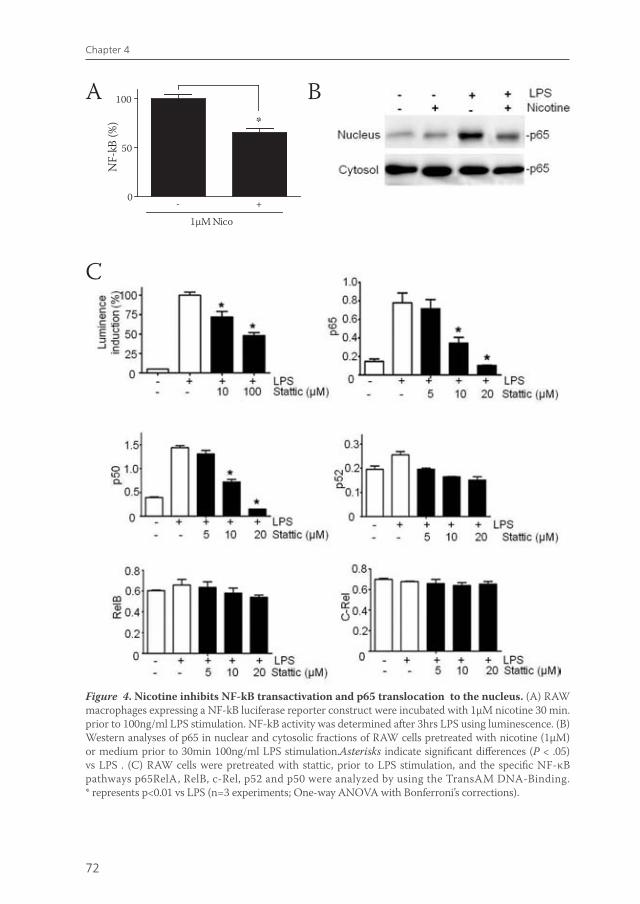
72
Chapter 4
Figure 4. Nicotine inhibits NF-kB transactivation and p65 translocation to the nucleus. (A) RAW macrophages expressing a NF-kB luciferase reporter construct were incubated with 1µM nicotine 30 min. prior to 100ng/ml LPS stimulation. NF-kB activity was determined after 3hrs LPS using luminescence. (B) Western analyses of p65 in nuclear and cytosolic fractions of RAW cells pretreated with nicotine (1µM) or medium prior to 30min 100ng/ml LPS stimulation.Asterisks indicate significant differences (P < .05) vs LPS . (C) RAW cells were pretreated with stattic, prior to LPS stimulation, and the specific NF-κB pathways p65RelA, RelB, c-Rel, p52 and p50 were analyzed by using the TransAM DNA-Binding. * represents p<0.01 vs LPS (n=3 experiments; One-way ANOVA with Bonferroni’s corrections).
A B
0
50
100
*
- +
1µM Nico
NF-
kB (%
)
C
van de Zanden.indb 72 23-5-2011 11:43:52

73
Deciphering STAT3 signaling in cholinergic immune-modulation
DISCUSSION
The JAK2-STAT3 pathway is an essential modulator of the immune response and plays also an important role in cell proliferation, migration, differentiation and apoptosis. However, the implications of this pathway in inflammatory disorders have remained elusive. We previously reported that the JAK2-STAT3 pathway is involved in mediating the anti-inflammatory action of nicotinic receptor activation in peritoneal macrophages2. Nicotine induced STAT3 phosphorylation and SOCS-3 activation and did not decrease TNF production in macrophages transfected with an inactive form of STAT32. Moreover, stimulation of the vagus nerve failed to reduce inflammation in mice LysM-Stat3fl/- mice2.
The present study reveals that pharmacological inhibition of JAK2-STAT3 activation using AG490 prevents NF-kB transactivation and TNF production in peritoneal macrophages. In contrast, several studies demonstrate that genetic deletion of STAT3 increases inflammatory responses. Stat3 deficient macrophages secrete large amounts of cytokines including TNFa, IL-1, IL-6, and IL-12 in response to inflammatory stimuli. In addition, LysMcre/Stat3flox/− mice, which were designed for cell type-specific STAT3 disruption in macrophages and neutrophils, are born normal, but are very susceptible to endotoxemia10 and sepsis17, and develop lethal chronic inflammatory bowel disease at ~20 weeks age10. Two factors appear to contribute to the higher inflammatory responses of these mice. First, STAT3-depletion sustains early inflammatory responses by inhibiting IL-10 signaling. The inhibitory activity of IL-10 on LPS-induced production of pro-inflammatory cytokines from STAT-3 deficient macrophages was completely blocked. Second, nontreated lysMcre/Stat3flox/- mice are characterized by macrophages which exhibit an constitutively activated phenotype. Similar to the results in conditional knockout mice, our results with siRNA confirm that STAT3 silencing enhances inflammatory responses.
The finding that pharmacological inhibition of the JAK2-STAT3 pathway reduces inflammation seems to be contradictory to the STAT3 silencing data. Possibly, the JAK2 inhibitor AG490 affects other signaling pathways as well. Indeed, it is shown that AG490 not only inhibits JAK2, but also JAK3 activity, STAT5a and 5b signaling18. These data suggest that AG490, especially at high dosages, can inhibit other signaling pathways than JAK2-STAT3 signaling alone.
We previously revealed that nicotine exerts its anti-inflammatory effects on macrophages via JAK2 mediated phosphorylation of STAT3. However, the interpretation of these data is not straightforward, because phosphorylation of STAT3 is fluctuating in time. Moreover, it appears that the use of immunoblotting as a readout for STAT3 activation does not correlate well with the response at the level of gene expression8. Therefore, the use of dominant negative constructs is a more reliable tool to determine the role of STAT3 phosphorylation in mediating the cholinergic anti-inflammatory effect. In RAW264.7 macrophages, we show that
van de Zanden.indb 73 23-5-2011 11:43:52

74
Chapter 4
nicotinic reduction of TNF activation does not depend on STAT3 phosphorylation, since nicotine still reduces TNF production in STAT3F transfected cells. In contrast, transfection with STAT3D does prevent the anti-inflammatory potential of nicotine. In STAT3D macrophages, glutamic residues 434 and 435 are replaced by alanine, that way inhibiting the DNA-binding activity of STAT by making dimers unable to bind the target DNA19, while tyrosine phosphorylation and dimerization are allowed. This suggests that nicotinic inhibition of the inflammatory response is dependent on STAT3 DNA binding. However, we clearly showed that nicotine inhibits p65 translocation to the nucleus (Fig. 4B), which makes it more likely that STAT3 exerts its effect in the cytosol, rather than in the nucleus. Moreover, our results indicate that pharmacological inhibition of STAT3 prevents the activation of the p65RelA/p50NF-kB pathway without affecting the other NF-kB proteins including RelB, c-Rel and p52NF-kB2. In normal conditions, after JAK2 phoshporylates STAT3, it dimerizes and translocates into the nucleus. Together, our data indicate a role for unphosphorylated STAT3 (U-STAT) in mediating the anti-inflammatory effect of nicotine. In conjunction with this hypothesis, inhibition of STAT3 tyrosine phosporylation by using STAT3F constructs can lead to higher levels of unphosphorylated U-STAT3. Recent studies indicate that U-STAT3 can bind to NF-κB in competition with IκBα, ultimately resulting in reduced NF-kB transactivation and cytokine production20. This mechanism is completely abolished with inhibition of STAT3 expression leading to the enhanced activation of NF-κB observed in STAT3-knockout mice. Additional work is required to further explore this role of USTAT3 in the cholinergic anti-inflammatory response.
Identification of intracellular signaling pathways involved in mediating the anti-inflammatory effect of cholinergic agonists is important to specifically target the pathway and avoid toxic side effects. Our results confirm that JAK2-STAT3 signaling is required for nicotine induced reduction in NF-kB activation and cytokine production in peritoneal macrophages. However, STAT3 phosphorylation seems to be of less significance in this process than previously thought.
Furthermore, our results suggest that targeting JAK2-STAT3 signaling may provide pharmacologic advantages for the treatment of inflammatory disorders. However, the JAK2-STAT3 signaling pathway is a complex system which can act both pro-and anti-inflammatory, is tightly regulated and not fully understood yet9. Characteristic examples of different responses are the differential expression of STAT3 isoforms in different cell types. Therefore, further examination is needed to establish the potential of targeting JAK2-STAT3 in restraining inflammation.
van de Zanden.indb 74 23-5-2011 11:43:52

75
Deciphering STAT3 signaling in cholinergic immune-modulation
REFERENCES
1. Borovikova LV, Ivanova S, Zhang M et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-62.
2. De Jonge WJ, van der Zanden EP, The FO et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844-51.
3. Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006;131:1122-30.
4. van Westerloo DJ, Giebelen IA, Florquin S et al. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006;130:1822-30.
5. van Maanen MA, Lebre MC, van der PT et al. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-22.
6. van der Zanden EP, Snoek SA, Heinsbroek SE et al. Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology 2009;137:1029-39, 1039.
7. van der Zanden EP, Boeckxstaens GE, De Jonge WJ. The vagus nerve as a modulator of intestinal inflammation. Neurogastroenterol Motil 2009;21:6-17.
8. De Jonge WJ, van der Zanden EP, The FO et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844-51.
9. Takeda K, Clausen BE, Kaisho T et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999;10:39-49.
10. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol 2007;178:2623-29.
11. Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol 2003;4:1169-76.
12. Levy DE, Lee CK. What does Stat3 do? J Clin Invest 2002;109:1143-48.
13. Welte T, Zhang SS, Wang T et al. STAT3 deletion during hematopoiesis causes Crohn‘s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A 2003;100:1879-84.
14. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006;13:1235-42.
15. Hui L, Yao Y, Wang S et al. Inhibition of Janus kinase 2 and signal transduction and activator of transcription 3 protect against cecal ligation and puncture-induced multiple organ damage and mortality. J Trauma 2009;66:859-65.
16. de Koning JP, Soede-Bobok AA, Ward AC et al. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 2000;19:3290-3298.
17. Matsukawa A, Takeda K, Kudo S, Maeda T, Kagayama M, Akira S. Aberrant inflammation and lethality to septic peritonitis in mice lacking STAT3 in macrophages and neutrophils. J Immunol 2003;171:6198-205.
18. Kirken RA, Erwin RA, Taub D et al. Tyrphostin AG-490 inhibits cytokine-mediated JAK3/STAT5a/b signal transduction and cellular proliferation of antigen-activated human T cells. J Leukoc Biol 1999;65:891-99.
19. Nakajima K, Yamanaka Y, Nakae K et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J 1996;15:3651-58.
20. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 2007;21:1396-408.
van de Zanden.indb 75 23-5-2011 11:43:52

5CHAPTER
Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor alpha4beta2
Esmerij P van der ZandenSusanne A SnoekSigrid E HeinsbroekOana I StanisorCaroline VerseijdenGuy E BoeckxstaensMaikel P PeppelenboschDavid R GreavesSiamon GordonWouter J de Jonge
Gastroenterology. 2009 Sep;137(3):1029-39, 1039.
van de Zanden.indb 77 23-5-2011 11:43:52

78
Chapter 5
ABSTRACT
Background and Aims: the vagus nerve negatively regulates macrophage cytokine production via the release of acetylcholine (ACh) and activation of nicotinic acetylcholine receptors (nAChR). In various models of intestinal inflammation, vagus nerve efferent stimulation ameliorates disease. Given the actively constrained cytokine responses of intestinal macrophages, we explored the effect of nAChR activation on endo- and phagocytosis by macrophages residing in the peritoneal and mucosal compartment.
Methods: the phagocytic uptake by intestinal and peritoneal macrophages was measured by FACS analysis, and the nAChR involved was determined by pharmacological blockade, shRNA-assisted gene knockdown and the use of specific nAChR KO mice. The effect of electrical vagus nerve stimulation on epithelial translocation and macrophage uptake of luminal particles was studied in mice.
Results: in isolated intestinal and peritoneal macrophages, nAChR activation enhanced endocytosis and phagocytosis. This effect was mediated via stimulated recruitment of GTPase Dynamin-2 to the forming phagocytic cup. These effects involve nAChR α4/β2, rather than nAChR α7. Despite enhanced bacterial uptake, acetylcholine reduced NF-κB activation and pro-inflammatory cytokine production, while stimulating anti-inflammatory IL10 production. Vagus nerve stimulation in mice altered mucosal immune responses by augmenting epithelial transport and uptake of luminal bacteria by lamina propria macrophages.
Conclusions: acetylcholine enhances phagocytic potential while inhibiting immune reactivity via nAChR α4/β2 in mouse macrophages. Hence, vagus nerve efferent activity may stimulate surveillance in the intestinal mucosa and peritoneal compartment.
van de Zanden.indb 78 23-5-2011 11:43:52

79
Vagal activity enhances macrophage phagocytosis
INTRODUCTION
In the gastro-intestinal tract, the vagus nerve regulates motility and digestive functions mostly via the peripheral release of the parasympathetic neurotransmitter acetylcholine (ACh) that activates nicotinic acetylcholine receptors (nAChRs). However, vagal activity has also immune-regulatory properties. While the afferent vagus system is known to regulate the inflammatory response via the hypothalamic pituitary adrenal axis, efferent vagus nerve activity possesses immuno-modulatory potential as well. Borovikova et al1 have revealed the potency of the vagus nerve to inhibit TNF production by macrophages after systemic endotoxin2. Peritoneal and PBMC-derived macrophages express nAChRs, and nAChR activation has been shown to inhibit NF-κB transcriptional activity3,4 and pro-inflammatory cytokine production4,5. In conjunction, electrical vagus nerve stimulation has been shown to ameliorate disease in animal models of inflammatory conditions such as post-operative ileus4,6, colitis7, peritonitis8,9, and hemorrage10. An immune regulating role for the cholinergic nervous system may be particularly evident in intestinal tissue, given the dense cholinergic innervation, and the abundant number of resident macrophages that populate the intestinal mucosa and muscularis externa, of which some closely associate with cholinergic fibers6.
Cholinergic inhibition of pro-inflammatory cytokine production by macrophages has been firmly established1,3,6. However, besides an effect on cytokine secretion, the cholinergic nervous system may also affect more professional macrophage functions such as endo- and phagocytosis of bacteria and particles. Especially in the intestinal compartment macrophages may rather function as phagocytes that, along with dendritic cells, form critical effectors in the surveillance of luminal antigens11,12. Hence, the question arises whether the anti-inflammatory effect of vagus nerve activity in intestinal inflammation solely rests on reduced macrophage cytokine production, or whether the vagus nerve also regulates other macrophage functions important in host defense. This is supported by the observation that vagus nerve activity affects bacterial clearance and mortality in various mouse models of infectious disease8,13,14. Therefore, we studied the effect of nAChR activation on endo- and phagocytosis by macrophages residing in the peritoneal and mucosal compartment.
We show here that nAChR α4/β2, rather than α7, activation enhances the phagocytic potential in mouse macrophages. Despite enhanced phagocytosis, the activation of NF-κB activity and pro-inflammatory cytokine production is inhibited. In conjunction, we also demonstrate that in mice, electrical stimulation of the vagus nerve increases the epithelial permeability for luminal bacteria and stimulates phagocytosis by F4/80+CD11b+ macrophages residing in the intestinal mucosa. Taken together, our data suggest that vagus nerve activity can enhance macrophage bacterial uptake via activation of the nAChR α4/β2, while reducing inflammatory cell activation.
van de Zanden.indb 79 23-5-2011 11:43:52

80
Chapter 5
METHODS
Reagents and antibodies. (-)-Nicotine, (+)-Nicotine, Acetylcholine, α-Bugarotoxin, Methylcaconitine, 2,2,6,6-Tetramethylpiperidin-4-yl heptanoate, Dihydro-β-erythroidine, 3-Methyl-5-[(2S)-1-methyl-2-pyrrolidinyl]isoxazole, Zymosan A from S. cerevisiae were from Sigma-Aldrich (Zwijndrecht, the Netherlands). Polyclonal antibodies against phosphorylated and unphosporylated Akt and PI3K, p38 and anti-NFkB-p65 were from Cell Signaling. Anti-α4 nAChR was purchased from Santa Cruz Biotechnology; anti-β2 nAChR subunit (MAb clone 270) and anti-Dynamin-2 (clone 3457) were from Abcam (Cambridge, UK). Dil-AcLDL was obtained from (Invitrogen). Rat anti mouse monoclonal CD11b, F4/80 and CD11c were purchased from BDBiosciences (Franklin Lakes, NJ). FITC (Sigma-Aldrich), labelling of 1*109 heat-killed E. faecium was performed in 0.1 M NaHCO3 pH9.0 for 1h at 37°C (in shaking waterbath).
Mammalian cell culture. Macrophages from an immortalized spleen macrophage cell line (Mf4/415) were cultured in RPMI 1640/10% FCS. Primary peritoneal cells (PMF) from BALB/c, C57/Bl6 WT mice, nAChR alpha7-/- or nAChRbeta2-/- mice (kindly provided by Dr. G.LaRosa and Dr. U. Maskos respectively) were collected by lavage 4 days after ip. injection of 1ml of Biogel solution and cultured in OptiMEM (Invitrogen).
Phagocytosis assays. For phagocytosis assays, cells were plated to 80 % confluence in 24 well suspension plates and pretreated with cholinergic agonists for 20min. Subsequently, cells were challenged with FITC-labeled Zymosan particles(20particles/cell) for 10min or other time-points where indicated. Cells were washed by PBS, final washing was in PBS/EDTA/lidocaine and cells were harvested by scraping. After fixation in 2% PFA, bead uptake was determined by flow cytometry (FACS Calibur, Becton Dickinson).
Fc Receptor-mediated phagocytosis of RBC. Sheep RBC (300 μl of a 10% suspension; Cappel/ICN) were opsonized with 1:1000 diluted rabbit-anti-sheep RBC IgG (Cappel/ICN). Opsonized RBCs were allowed to bind to Mf4/4 or peritoneal macrophages at a 1/80 cell to RBC ratio at 37°C. After 15min, slides were exposed to ice-cold water for 15 sec to lyse RBCs that were not internalized, washed in PBS and fixed.
Cell stimulations. Peritoneal macrophages were incubated with nicotinic agonists at the concentration indicated for 45 minutes and stimulated with Zymosan(10particles/cell) for 6hrs. After treatment, the medium was removed; the cells washed three times with ice-cold PBS, and medium and cell lysates were measured by ELISA.
van de Zanden.indb 80 23-5-2011 11:43:52

81
Vagal activity enhances macrophage phagocytosis
RT-PCR of total RNA. To determine nAChR expression on MF4/4 cells and PMF, total RNA was isolated using the RNeasy Mini kit (Qiagen), treated with DNase, and reverse transcribed. The resulting cDNA was subjected to Light Cycler polymerase chain reaction(Roche) for 35 cycles. Primer sequences are provided in supplementary data.
Immunostaining and imaging. Immune-histochemistry on intestinal sections was performed as described earlier6. For confocal microscopy, MF4/4 were grown on glass slides (Nuncbrand), pretreated with nicotine, challenged with FITC-labeled Zymosan and phagocytosis was allowed for 5min. For a detailed description of the protocols and antibodies used, see supplementary methods.
shRNA transfection. 2*106 MF4/4 cells were transfected by electroporation with 2 μg shRNA expression plasmid (psilencerTM-CMV4.1neo; Ambion, Austin, TX) according to the manufacturer’s instructions (570 V, 50 μs, Amaxa; Gaithersburg, MD). shRNA sequences are given in the supplementary method section.
Immunoblotting. Immunoblotting was performed as previously described6. Plasma membrane was extracted using the cell compartment kit (Qiagen), according to the manufacturer’s instructions.
Isolation of lamina propria macrophages. Lamina propria macrophages were isolated after neutral protease digestion (a modified protocol as described previously16) and Magnetic bead-assisted Cell Sorting (MACS; Miltenyi Biotec Inc., Auburn, CA). A detailed description of the procedure is provided in the supplementary methods. After isolation, cells were taken up in RPMI/1%FCS at 107 cells/mL, left for 2 h, allowed to phagocytose FITC labeled heat-killed E.faecium at 37°C during 1h, finally washed and analyzed by FACS.
In vivo uptake assay and electrical vagus nerve stimulationAnimals. Male C57/Bl6 mice (Harlan Nederland, Horst, The Netherlands), 12 to 15 weeks old, were kept under environmentally controlled conditions (light on from 8:00 AM till 8:00 PM; water and rodent non-purified diet ad libitum; temperature 20°C-22°C; 55% humidity). Experiments were performed according to the guidelines of the Ethical Animal Research Committee of the University of Amsterdam. Mice were used after a 7 days adaptation period.Electrical vagus nerve stimulation. Mice were anesthetized by i.p. injection of a mixture of Fentanyl Citrate / Fluanisone (Hypnorm; Janssen, Beerse, Belgium) and Midazolam (Dormicum; Roche, Mijdrecht, The Netherlands). VNS was performed as
van de Zanden.indb 81 23-5-2011 11:43:52

82
Chapter 5
described previously6. In short: the right cervical vagal branch was prepared free from the carotid artery and ligated with 6-0 silk suture. The part distal from the ligation was attached to an electrode and 5V stimuli with a frequency of 5Hz, duration of 2ms10 were applied for 5 min. In sham mice the cervical skin was opened and left for 30 min. covered by moist gauze. After a 30 min. recovery period, the in vivo uptake assay was initiated as described below.In vivo uptake. Surgical procedures were performed under sterile conditions. Mice underwent a laparotomy and an ileum segment 3-10 cm proximal from the caecum was opened and its lumen rinsed with pre-warmed (37°C) oxygenated Krebs buffer. The ileum segment was filled with 1-2 mL of buffer containing FITC-labeled E.faecium bacteria and Alexa546-labeled Dextran particles (10,000Mw, Mol. Probes), and clamped at both sides. After 30 min. the clamped intestinal segment was removed, washed in PBS and processed for immune-histochemistry. Bacterial translocation. Mice were killed 18 h after surgery and mesenteric lymph nodes (MLN) were harvested under aseptic conditions. Lymph nodes were weighed, placed in a tube containing 300 µl of ice-cold Luria-Bertani (LB) broth, homogenized with a sterile grinder and plated onto blood agar plates in aerobic and anaerobic conditions. After 48 h of incubation at 37ºC, the number of colony forming units (CFU) per g lymph node was assessed.
Ussing chamber experiments. Segments of tissue of the distal small intestine were opened, cut and immersed in Krebs’ buffer (115 mM NaCl, 1.25 mM CaCl2, 1.2 mM MgCl2, 2.0 mM KH2PO4, and 25 mM NaHCO3 at pH 7.35). Within 15 minutes the tissue was mounted in Ussing chambers (World Precision Instruments, Berlin, Germany) with serosal and mucosal areas exposed to 2 ml of circulating oxygenated Krebs buffer (containing 10 mM glucose) maintained at 37°C. Isc (in μA/cm2), a measure of net active ion transport, was recorded by a computer connected a voltage-clamp system. After 15 minutes, HRP was added to the luminal buffer at a final concentration of 10µM. Samples (300μl) were taken at the serosal side and replaced with fresh buffer. After 30 minutes, nicotine (1µM), Carbachol (1 µM) or buffer was added to the serosal side. The enzymatic activity of HRP was measured using tetramethyl-benzidine (TMB) (Biosource Europe, Nivelles, Belgium) as a substrate.
Statistics. Statistical analysis was performed using SPSS 12.02 software (SPSS Inc. Chicago, Ill, USA). The Friedman’s two-way analysis of variance was used to explore multiple dependent value assays. If the Friedman’s analysis was significant, individual values compared to the 0 nM concentration were tested with a Mann-Whitney U test. P-values <0.05 were considered statistically significant and results were expressed as mean ± SEM.
van de Zanden.indb 82 23-5-2011 11:43:52

83
Vagal activity enhances macrophage phagocytosis
RESULTS
Cholinergic agonists enhance phagocytosis in macrophagesWe first tested whether ACh, the main neurotransmitter of the vagus nerve, or nicotine, that activates nAChRs, affected macrophage endo- and phagocytosis. In PMF (Fig. 1A-D), and spleen macrophage Mf4/4 cells (not shown), ACh as well as nicotine significantly (p<0.05) enhanced phagocytosis in a time-dependent (Fig. 1B) and dose-dependent (fig. 1C) manner as analyzed by FACS and fluorescence microscopy. The enhanced phagocytosis induced by nicotine was not based on enhanced particle binding to the cells, as no increase in phagocytosis was observed when cells were incubated on ice instead of 37°C (Fig. 1A, B).
To assess whether the cholinergic stimulation of phagocytosis was receptor pathway specific, we compared the effect of nicotine on the uptake of heat-killed E.faecium, opsonised sheep red blood cells (RBC), Zymosan, and on the endocytosis of acetylated low-density lipoprotein (AcLDL) by PMF (Fig. 1D) and Mf4/4 macrophages (not shown). Zymosan uptake depends on Dectin-1, IgG-opsonized RBC serve as cargo for Fc receptor-mediated phagocytosis, while AcLDL endocytosis is dependent on the scavenger receptor binding. However, nicotine stimulated uptake via all these pathways to a similar extent, indicating that nicotinic enhancement of phagocytosis involves a general pathway in phagocytosis rather than a specific effect on receptor expression. Moreover, further analysis confirmed that expression of Dectin-1, CD11b/CD18, or scavenger receptor A was not affected in peritoneal macrophages after a 6h treatment with 1µM of nicotine (results not shown).
Cholinergic agonists stimulate phagocytosis via the α4/β2 nAChR rather than α7 nAChR We next sought to establish which nAChR mediated the cholinergic effects on macrophage phagocytosis. The nAChR α7 has been implicated in the inhibition of cytokine production and NF-κB activity by nicotinic receptor activation in macrophages (a.o. in4,5 ). In accordance with these results, nicotine inhibited the activation of NF-κB induced by Zymosan (4and results not shown), inhibited Zymosan-induced TNFα and MIP-2 production and induced IL-10 production (Fig. 2A). This effect was not only blocked by nAChRα7 blockers Bungarotoxin (Bgt) and Methyllicaconitine (MLA), but also by nAChR α4/β2 antagonist dihydro-β-erythroidine (DHβE) (Fig. 2B). In PMF of nAChRα7 knock-out mice, nicotine failed to reduce TNF production, confirming earlier reports(5and results not shown).
Surprisingly, specific blockers of α7 nAChRs failed to block the nicotinic increase in phagocytosis in peritoneal macrophages, in fact, MLA pretreatment further augmented nicotinic stimulation of phagocytosis. On the other hand, 2,2,6,6-tetramethylpiperidin-4-yl heptanoate (TMPH), a blocker of nAChR with low
van de Zanden.indb 83 23-5-2011 11:43:52
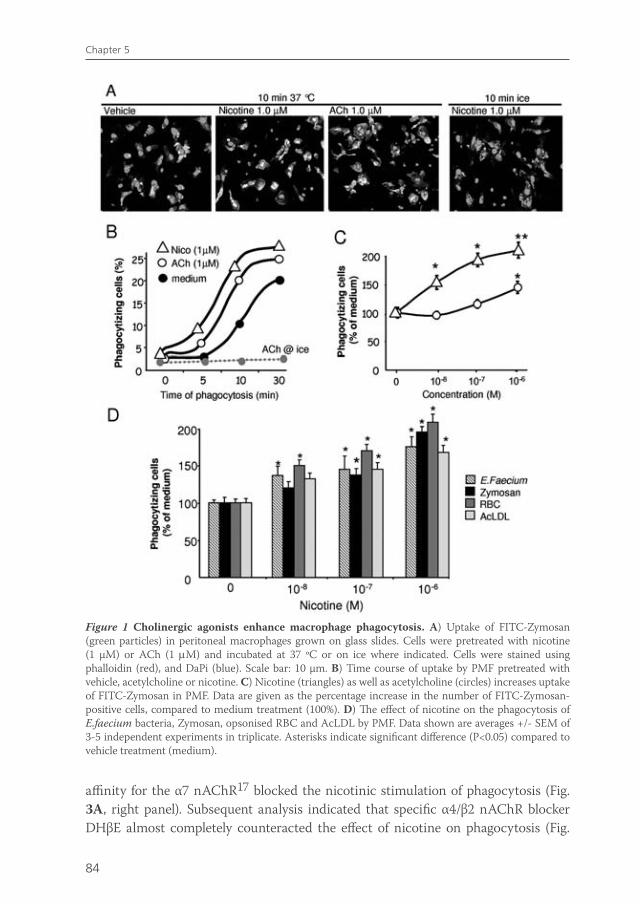
84
Chapter 5
affinity for the α7 nAChR17 blocked the nicotinic stimulation of phagocytosis (Fig. 3A, right panel). Subsequent analysis indicated that specific α4/β2 nAChR blocker DHβE almost completely counteracted the effect of nicotine on phagocytosis (Fig.
Figure 1 Cholinergic agonists enhance macrophage phagocytosis. A) Uptake of FITC-Zymosan (green particles) in peritoneal macrophages grown on glass slides. Cells were pretreated with nicotine (1 µM) or ACh (1 µM) and incubated at 37 ºC or on ice where indicated. Cells were stained using phalloidin (red), and DaPi (blue). Scale bar: 10 µm. B) Time course of uptake by PMF pretreated with vehicle, acetylcholine or nicotine. C) Nicotine (triangles) as well as acetylcholine (circles) increases uptake of FITC-Zymosan in PMF. Data are given as the percentage increase in the number of FITC-Zymosan-positive cells, compared to medium treatment (100%). D) The effect of nicotine on the phagocytosis of E.faecium bacteria, Zymosan, opsonised RBC and AcLDL by PMF. Data shown are averages +/- SEM of 3-5 independent experiments in triplicate. Asterisks indicate significant difference (P<0.05) compared to vehicle treatment (medium).
van de Zanden.indb 84 23-5-2011 11:43:53
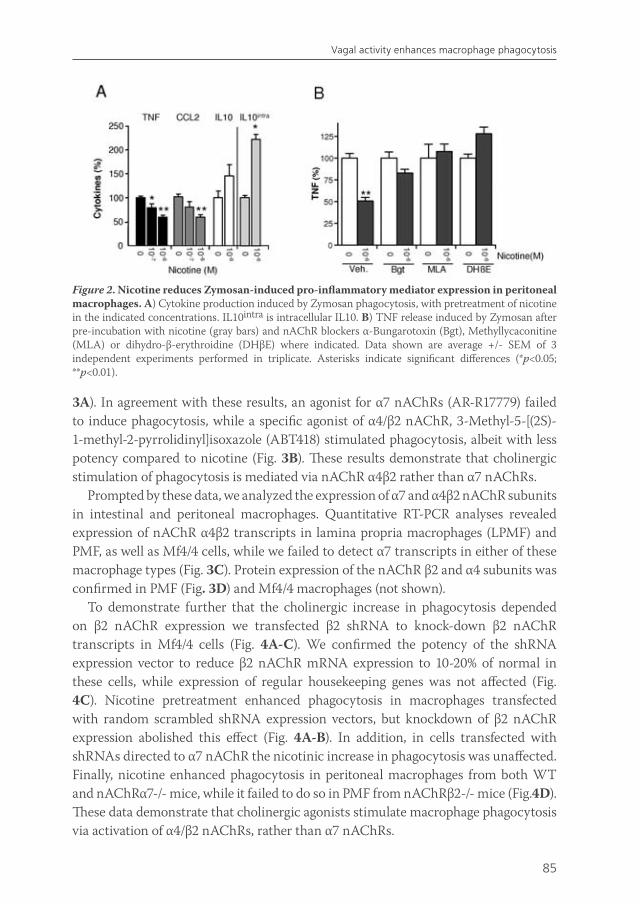
85
Vagal activity enhances macrophage phagocytosis
Figure 2. Nicotine reduces Zymosan-induced pro-inflammatory mediator expression in peritoneal macrophages. A) Cytokine production induced by Zymosan phagocytosis, with pretreatment of nicotine in the indicated concentrations. IL10intra is intracellular IL10. B) TNF release induced by Zymosan after pre-incubation with nicotine (gray bars) and nAChR blockers α-Bungarotoxin (Bgt), Methyllycaconitine (MLA) or dihydro-β-erythroidine (DHβE) where indicated. Data shown are average +/- SEM of 3 independent experiments performed in triplicate. Asterisks indicate significant differences (*p<0.05; **p<0.01).
3A). In agreement with these results, an agonist for α7 nAChRs (AR-R17779) failed to induce phagocytosis, while a specific agonist of α4/β2 nAChR, 3-Methyl-5-[(2S)-1-methyl-2-pyrrolidinyl]isoxazole (ABT418) stimulated phagocytosis, albeit with less potency compared to nicotine (Fig. 3B). These results demonstrate that cholinergic stimulation of phagocytosis is mediated via nAChR α4β2 rather than α7 nAChRs.
Prompted by these data, we analyzed the expression of α7 and α4β2 nAChR subunits in intestinal and peritoneal macrophages. Quantitative RT-PCR analyses revealed expression of nAChR α4β2 transcripts in lamina propria macrophages (LPMF) and PMF, as well as Mf4/4 cells, while we failed to detect α7 transcripts in either of these macrophage types (Fig. 3C). Protein expression of the nAChR β2 and α4 subunits was confirmed in PMF (Fig. 3D) and Mf4/4 macrophages (not shown).
To demonstrate further that the cholinergic increase in phagocytosis depended on β2 nAChR expression we transfected β2 shRNA to knock-down β2 nAChR transcripts in Mf4/4 cells (Fig. 4A-C). We confirmed the potency of the shRNA expression vector to reduce β2 nAChR mRNA expression to 10-20% of normal in these cells, while expression of regular housekeeping genes was not affected (Fig. 4C). Nicotine pretreatment enhanced phagocytosis in macrophages transfected with random scrambled shRNA expression vectors, but knockdown of β2 nAChR expression abolished this effect (Fig. 4A-B). In addition, in cells transfected with shRNAs directed to α7 nAChR the nicotinic increase in phagocytosis was unaffected. Finally, nicotine enhanced phagocytosis in peritoneal macrophages from both WT and nAChRα7-/- mice, while it failed to do so in PMF from nAChRβ2-/- mice (Fig.4D). These data demonstrate that cholinergic agonists stimulate macrophage phagocytosis via activation of α4/β2 nAChRs, rather than α7 nAChRs.
van de Zanden.indb 85 23-5-2011 11:43:53
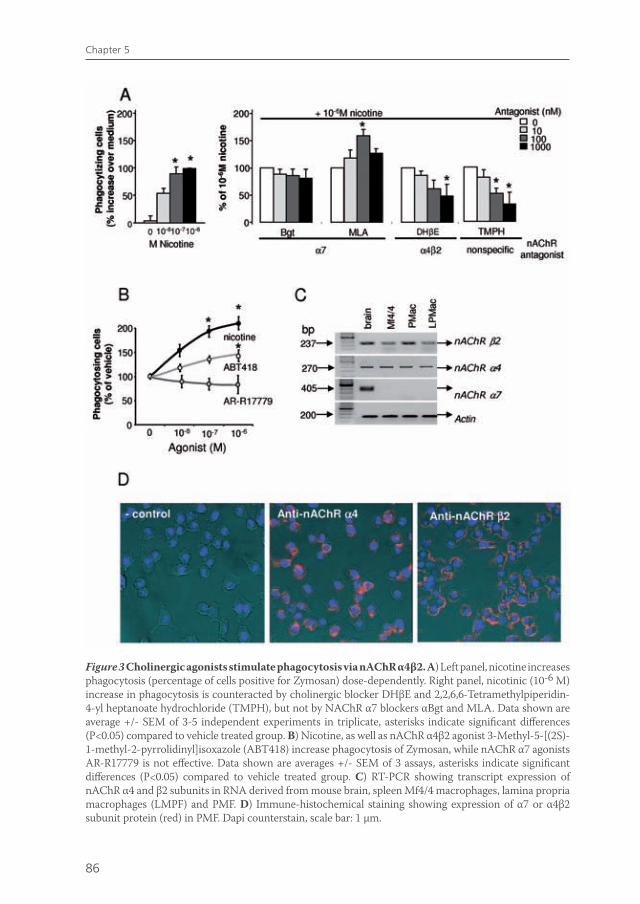
86
Chapter 5
Figure 3 Cholinergic agonists stimulate phagocytosis via nAChR α4β2. A) Left panel, nicotine increases phagocytosis (percentage of cells positive for Zymosan) dose-dependently. Right panel, nicotinic (10-6 M) increase in phagocytosis is counteracted by cholinergic blocker DHβE and 2,2,6,6-Tetramethylpiperidin-4-yl heptanoate hydrochloride (TMPH), but not by NAChR α7 blockers αBgt and MLA. Data shown are average +/- SEM of 3-5 independent experiments in triplicate, asterisks indicate significant differences (P<0.05) compared to vehicle treated group. B) Nicotine, as well as nAChR α4β2 agonist 3-Methyl-5-[(2S)-1-methyl-2-pyrrolidinyl]isoxazole (ABT418) increase phagocytosis of Zymosan, while nAChR α7 agonists AR-R17779 is not effective. Data shown are averages +/- SEM of 3 assays, asterisks indicate significant differences (P<0.05) compared to vehicle treated group. C) RT-PCR showing transcript expression of nAChR α4 and β2 subunits in RNA derived from mouse brain, spleen Mf4/4 macrophages, lamina propria macrophages (LMPF) and PMF. D) Immune-histochemical staining showing expression of α7 or α4β2 subunit protein (red) in PMF. Dapi counterstain, scale bar: 1 µm.
van de Zanden.indb 86 23-5-2011 11:43:54
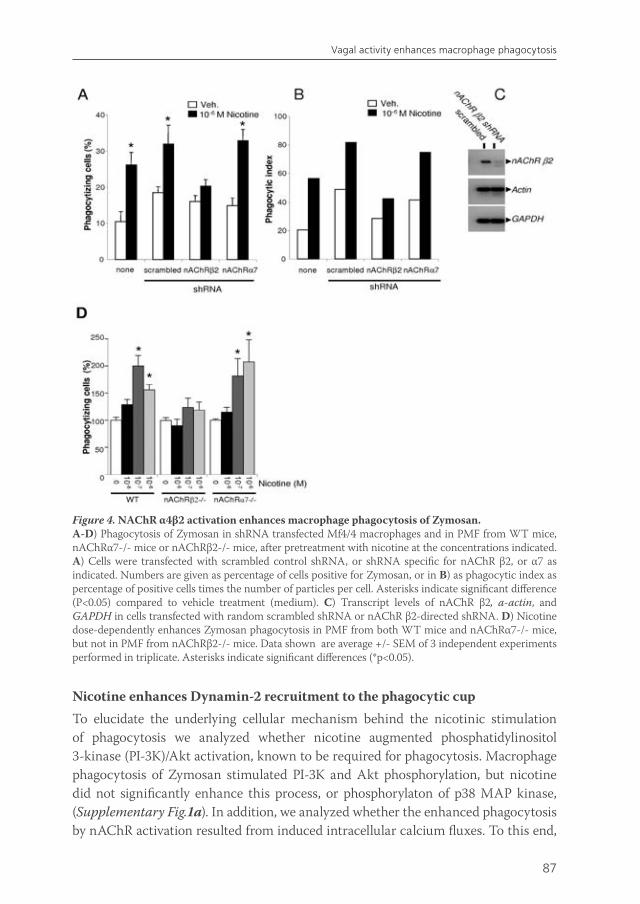
87
Vagal activity enhances macrophage phagocytosis
Nicotine enhances Dynamin-2 recruitment to the phagocytic cupTo elucidate the underlying cellular mechanism behind the nicotinic stimulation of phagocytosis we analyzed whether nicotine augmented phosphatidylinositol 3-kinase (PI-3K)/Akt activation, known to be required for phagocytosis. Macrophage phagocytosis of Zymosan stimulated PI-3K and Akt phosphorylation, but nicotine did not significantly enhance this process, or phosphorylaton of p38 MAP kinase, (Supplementary Fig.1a). In addition, we analyzed whether the enhanced phagocytosis by nAChR activation resulted from induced intracellular calcium fluxes. To this end,
Figure 4. NAChR α4β2 activation enhances macrophage phagocytosis of Zymosan. A-D) Phagocytosis of Zymosan in shRNA transfected Mf4/4 macrophages and in PMF from WT mice, nAChRα7-/- mice or nAChRβ2-/- mice, after pretreatment with nicotine at the concentrations indicated. A) Cells were transfected with scrambled control shRNA, or shRNA specific for nAChR β2, or α7 as indicated. Numbers are given as percentage of cells positive for Zymosan, or in B) as phagocytic index as percentage of positive cells times the number of particles per cell. Asterisks indicate significant difference (P<0.05) compared to vehicle treatment (medium). C) Transcript levels of nAChR β2, a-actin, and GAPDH in cells transfected with random scrambled shRNA or nAChR β2-directed shRNA. D) Nicotine dose-dependently enhances Zymosan phagocytosis in PMF from both WT mice and nAChRα7-/- mice, but not in PMF from nAChRβ2-/- mice. Data shown are average +/- SEM of 3 independent experiments performed in triplicate. Asterisks indicate significant differences (*p<0.05).
van de Zanden.indb 87 23-5-2011 11:43:55
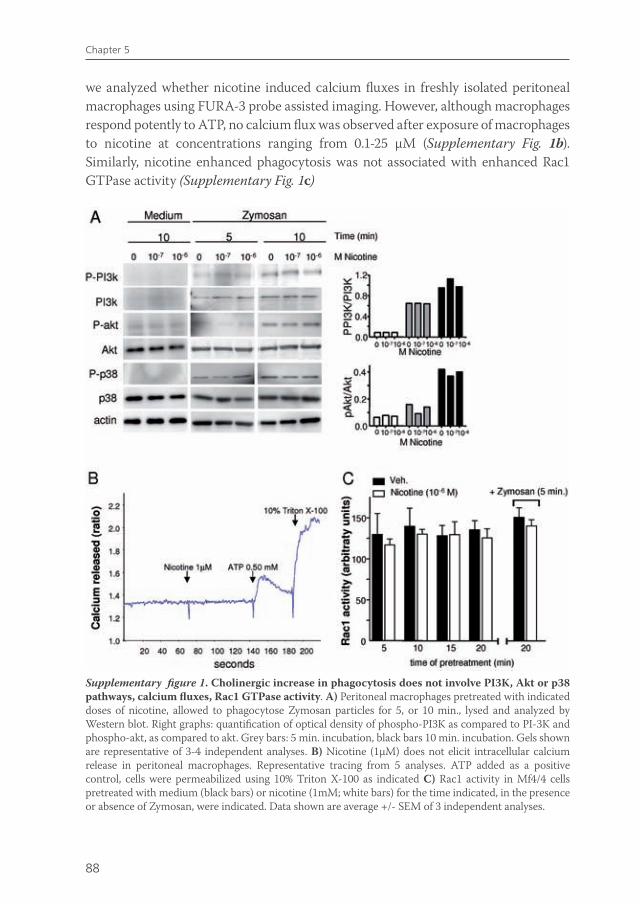
88
Chapter 5
we analyzed whether nicotine induced calcium fluxes in freshly isolated peritoneal macrophages using FURA-3 probe assisted imaging. However, although macrophages respond potently to ATP, no calcium flux was observed after exposure of macrophages to nicotine at concentrations ranging from 0.1-25 μM (Supplementary Fig. 1b). Similarly, nicotine enhanced phagocytosis was not associated with enhanced Rac1 GTPase activity (Supplementary Fig. 1c)
Supplementary figure 1. Cholinergic increase in phagocytosis does not involve PI3K, Akt or p38 pathways, calcium fluxes, Rac1 GTPase activity. A) Peritoneal macrophages pretreated with indicated doses of nicotine, allowed to phagocytose Zymosan particles for 5, or 10 min., lysed and analyzed by Western blot. Right graphs: quantification of optical density of phospho-PI3K as compared to PI-3K and phospho-akt, as compared to akt. Grey bars: 5 min. incubation, black bars 10 min. incubation. Gels shown are representative of 3-4 independent analyses. B) Nicotine (1µM) does not elicit intracellular calcium release in peritoneal macrophages. Representative tracing from 5 analyses. ATP added as a positive control, cells were permeabilized using 10% Triton X-100 as indicated C) Rac1 activity in Mf4/4 cells pretreated with medium (black bars) or nicotine (1mM; white bars) for the time indicated, in the presence or absence of Zymosan, were indicated. Data shown are average +/- SEM of 3 independent analyses.
van de Zanden.indb 88 23-5-2011 11:43:56
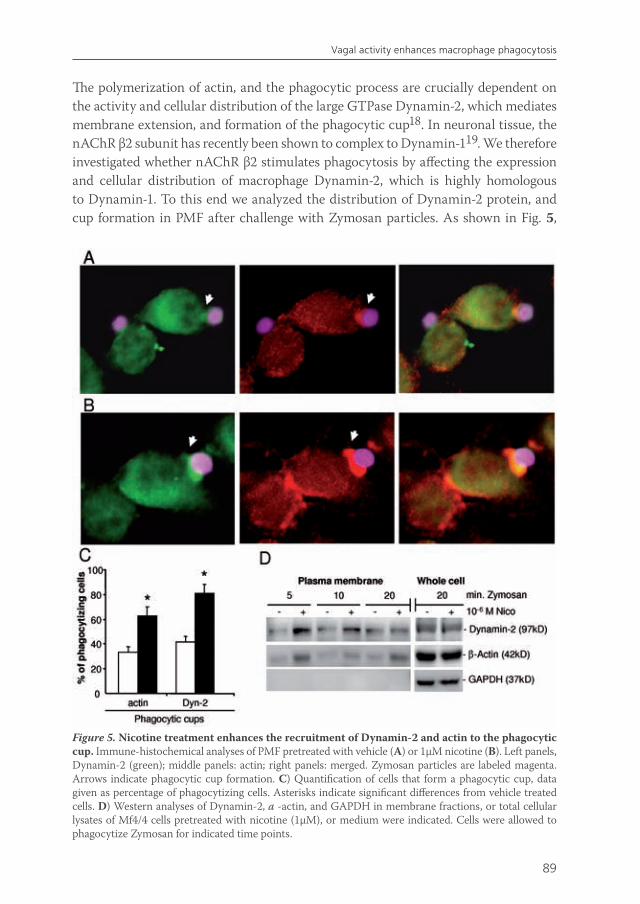
89
Vagal activity enhances macrophage phagocytosis
The polymerization of actin, and the phagocytic process are crucially dependent on the activity and cellular distribution of the large GTPase Dynamin-2, which mediates membrane extension, and formation of the phagocytic cup18. In neuronal tissue, the nAChR β2 subunit has recently been shown to complex to Dynamin-119. We therefore investigated whether nAChR β2 stimulates phagocytosis by affecting the expression and cellular distribution of macrophage Dynamin-2, which is highly homologous to Dynamin-1. To this end we analyzed the distribution of Dynamin-2 protein, and cup formation in PMF after challenge with Zymosan particles. As shown in Fig. 5,
Figure 5. Nicotine treatment enhances the recruitment of Dynamin-2 and actin to the phagocytic cup. Immune-histochemical analyses of PMF pretreated with vehicle (A) or 1µM nicotine (B). Left panels, Dynamin-2 (green); middle panels: actin; right panels: merged. Zymosan particles are labeled magenta. Arrows indicate phagocytic cup formation. C) Quantification of cells that form a phagocytic cup, data given as percentage of phagocytizing cells. Asterisks indicate significant differences from vehicle treated cells. D) Western analyses of Dynamin-2, a -actin, and GAPDH in membrane fractions, or total cellular lysates of Mf4/4 cells pretreated with nicotine (1µM), or medium were indicated. Cells were allowed to phagocytize Zymosan for indicated time points.
van de Zanden.indb 89 23-5-2011 11:43:57
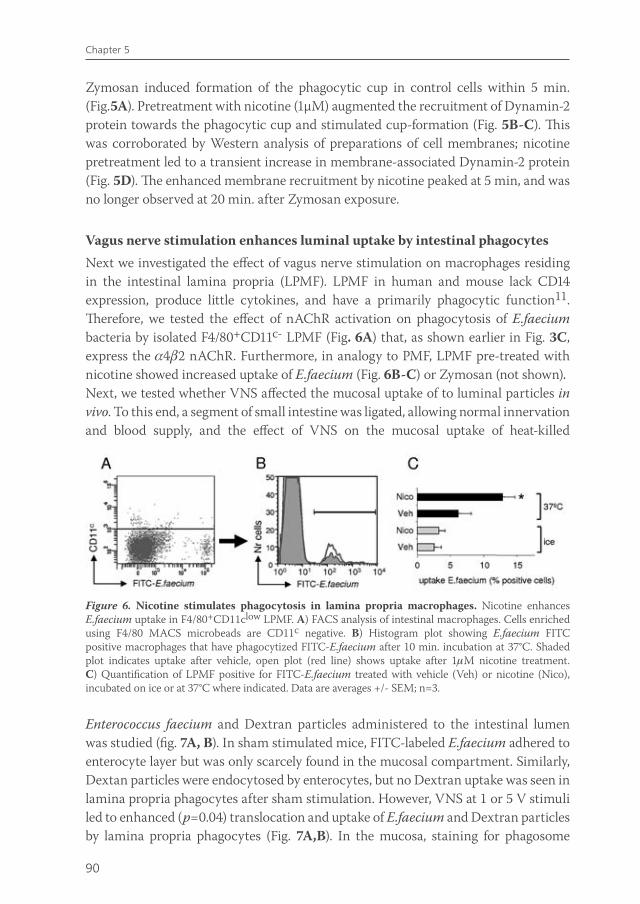
90
Chapter 5
Zymosan induced formation of the phagocytic cup in control cells within 5 min. (Fig.5A). Pretreatment with nicotine (1µM) augmented the recruitment of Dynamin-2 protein towards the phagocytic cup and stimulated cup-formation (Fig. 5B-C). This was corroborated by Western analysis of preparations of cell membranes; nicotine pretreatment led to a transient increase in membrane-associated Dynamin-2 protein (Fig. 5D). The enhanced membrane recruitment by nicotine peaked at 5 min, and was no longer observed at 20 min. after Zymosan exposure.
Vagus nerve stimulation enhances luminal uptake by intestinal phagocytesNext we investigated the effect of vagus nerve stimulation on macrophages residing in the intestinal lamina propria (LPMF). LPMF in human and mouse lack CD14 expression, produce little cytokines, and have a primarily phagocytic function11. Therefore, we tested the effect of nAChR activation on phagocytosis of E.faecium bacteria by isolated F4/80+CD11c- LPMF (Fig. 6A) that, as shown earlier in Fig. 3C, express the 42 nAChR. Furthermore, in analogy to PMF, LPMF pre-treated with nicotine showed increased uptake of E.faecium (Fig. 6B-C) or Zymosan (not shown). Next, we tested whether VNS affected the mucosal uptake of to luminal particles in vivo. To this end, a segment of small intestine was ligated, allowing normal innervation and blood supply, and the effect of VNS on the mucosal uptake of heat-killed
Figure 6. Nicotine stimulates phagocytosis in lamina propria macrophages. Nicotine enhances E.faecium uptake in F4/80+CD11clow LPMF. A) FACS analysis of intestinal macrophages. Cells enriched using F4/80 MACS microbeads are CD11c negative. B) Histogram plot showing E.faecium FITC positive macrophages that have phagocytized FITC-E.faecium after 10 min. incubation at 37°C. Shaded plot indicates uptake after vehicle, open plot (red line) shows uptake after 1M nicotine treatment. C) Quantification of LPMF positive for FITC-E.faecium treated with vehicle (Veh) or nicotine (Nico), incubated on ice or at 37°C where indicated. Data are averages +/- SEM; n=3.
Enterococcus faecium and Dextran particles administered to the intestinal lumen was studied (fig. 7A, B). In sham stimulated mice, FITC-labeled E.faecium adhered to enterocyte layer but was only scarcely found in the mucosal compartment. Similarly, Dextan particles were endocytosed by enterocytes, but no Dextran uptake was seen in lamina propria phagocytes after sham stimulation. However, VNS at 1 or 5 V stimuli led to enhanced (p=0.04) translocation and uptake of E.faecium and Dextran particles by lamina propria phagocytes (Fig. 7A,B). In the mucosa, staining for phagosome
van de Zanden.indb 90 23-5-2011 11:43:58
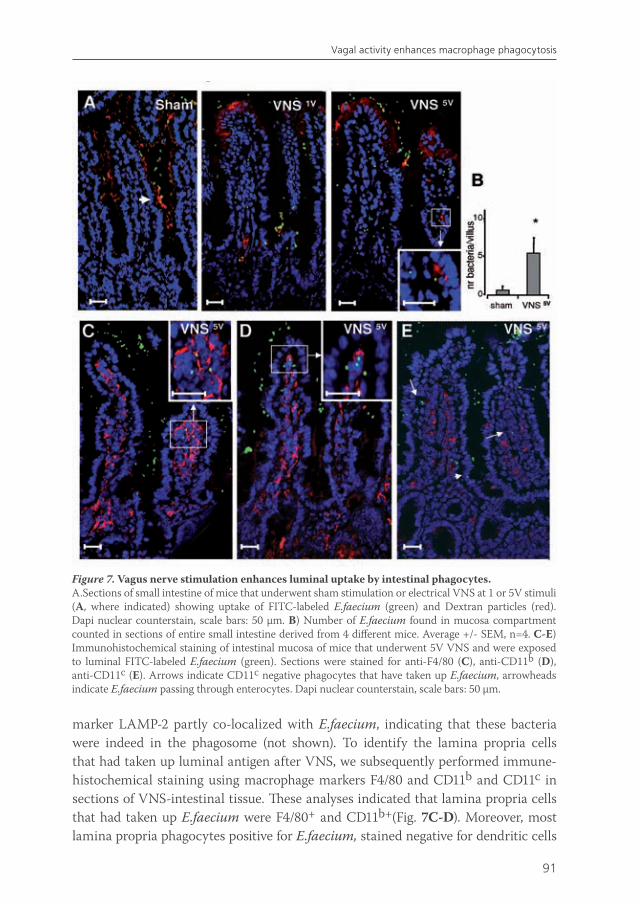
91
Vagal activity enhances macrophage phagocytosis
Figure 7. Vagus nerve stimulation enhances luminal uptake by intestinal phagocytes.A.Sections of small intestine of mice that underwent sham stimulation or electrical VNS at 1 or 5V stimuli (A, where indicated) showing uptake of FITC-labeled E.faecium (green) and Dextran particles (red). Dapi nuclear counterstain, scale bars: 50 µm. B) Number of E.faecium found in mucosa compartment counted in sections of entire small intestine derived from 4 different mice. Average +/- SEM, n=4. C-E) Immunohistochemical staining of intestinal mucosa of mice that underwent 5V VNS and were exposed to luminal FITC-labeled E.faecium (green). Sections were stained for anti-F4/80 (C), anti-CD11b (D), anti-CD11c (E). Arrows indicate CD11c negative phagocytes that have taken up E.faecium, arrowheads indicate E.faecium passing through enterocytes. Dapi nuclear counterstain, scale bars: 50 µm.
marker LAMP-2 partly co-localized with E.faecium, indicating that these bacteria were indeed in the phagosome (not shown). To identify the lamina propria cells that had taken up luminal antigen after VNS, we subsequently performed immune-histochemical staining using macrophage markers F4/80 and CD11b and CD11c in sections of VNS-intestinal tissue. These analyses indicated that lamina propria cells that had taken up E.faecium were F4/80+ and CD11b+(Fig. 7C-D). Moreover, most lamina propria phagocytes positive for E.faecium, stained negative for dendritic cells
van de Zanden.indb 91 23-5-2011 11:43:59

92
Chapter 5
marker CD11c (Fig. 7E), indicating that bacteria were taken up by LPMF rather than dendritic cells.
The increased uptake of luminal particles after VNS may also be the result of modulation of the epithelial barrier. Enhanced cholinergic activity has been implicated in changes in mucosal barrier function20,21. To evaluate whether the enhanced mucosal uptake of luminal bacteria after VNS resulted from enhanced epithelial transport, we mounted intestinal tissue in Ussing chambers to measure the flux of HRP passing the bowel wall via para- or transcellular routes. As shown in Fig. 8A, basal flux of HRP was not altered in intestinal tissue derived from mice that had undergone VNS5V 2h earlier. However, when control tissue was mounted and nicotine, or carbachol (an agonist for cholinergic receptors) was added to the mucosal compartment, a transient (30min.) increase in HRP flux towards the serosal layer was observed (Fig. 8A). No enhanced flux was observed after subsequent washout of nicotine (not shown). These observations indicate that activation of cholinergic receptors in the intestinal tissue induces a transiently enhanced mucosal passage of luminal bacteria, in agreement with the role of ACh in stress-induced epithelial permeability22,23.
We next evaluated whether VNS also led to an enhanced drainage of phagocytosed bacteria to mesenteric lymph nodes. To this end, we determined whether VNS increased the number of Colony Forming Units (CFUs) cultured from MLN. As shown in Fig 8B, stimulation of the vagus nerve led to a significant increase (aerobic p=0.04; anaerobic p=0.018) in the number of CFUs cultured as compared to sham stimulated mice, confirming that stimulation of vagal activity enhances mucosal uptake and drainage of luminal bacteria.
Figure 8. Cholinergic activity reduces the epithelial barrier function. A) Translocation of HRP over the intestinal mucosa measured in Ussing chambers. Nicotine (1µM) or Carbachol (1µM) added to the chambers increases HRP flux to the serosal compartment. Data are average +/- SEM of 8 preparations from 4 mice. B) The number of bacteria cultured from mesenteric lymph node (MLN) after cervical stimulation of the vagus nerve (VNS), compared to sham stimulation. Data shown are average +/- SEM, n=5. Asterisks indicate significant differences (*p<0.05; **p<0.01). .
van de Zanden.indb 92 23-5-2011 11:43:59
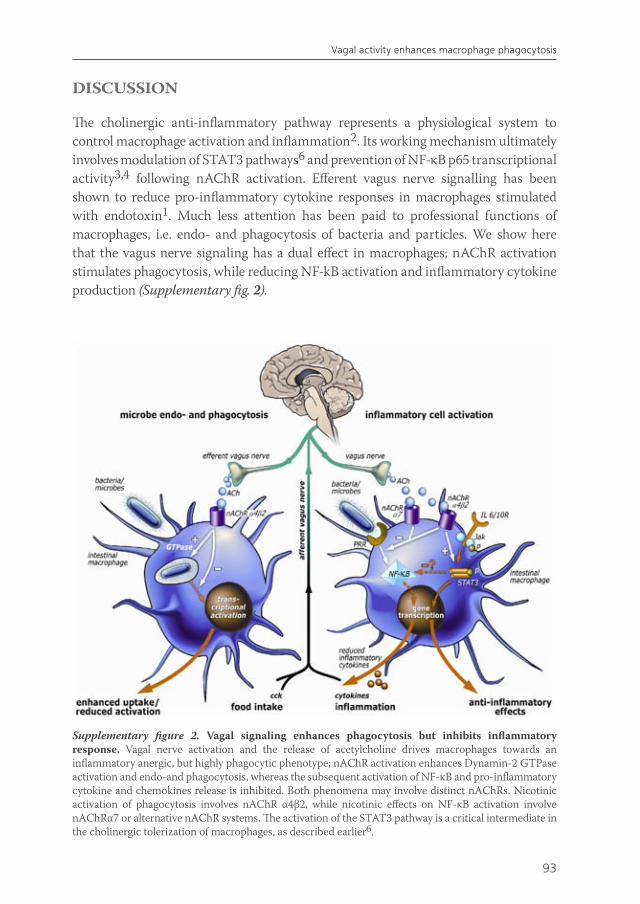
93
Vagal activity enhances macrophage phagocytosis
Supplementary figure 2. Vagal signaling enhances phagocytosis but inhibits inflammatory response. Vagal nerve activation and the release of acetylcholine drives macrophages towards an inflammatory anergic, but highly phagocytic phenotype; nAChR activation enhances Dynamin-2 GTPase activation and endo-and phagocytosis, whereas the subsequent activation of NF-κB and pro-inflammatory cytokine and chemokines release is inhibited. Both phenomena may involve distinct nAChRs. Nicotinic activation of phagocytosis involves nAChR α4β2, while nicotinic effects on NF-κB activation involve nAChRα7 or alternative nAChR systems. The activation of the STAT3 pathway is a critical intermediate in the cholinergic tolerization of macrophages, as described earlier6.
DISCUSSION
The cholinergic anti-inflammatory pathway represents a physiological system to control macrophage activation and inflammation2. Its working mechanism ultimately involves modulation of STAT3 pathways6 and prevention of NF-κB p65 transcriptional activity3,4 following nAChR activation. Efferent vagus nerve signalling has been shown to reduce pro-inflammatory cytokine responses in macrophages stimulated with endotoxin1. Much less attention has been paid to professional functions of macrophages, i.e. endo- and phagocytosis of bacteria and particles. We show here that the vagus nerve signaling has a dual effect in macrophages; nAChR activation stimulates phagocytosis, while reducing NF-kB activation and inflammatory cytokine production (Supplementary fig. 2).
van de Zanden.indb 93 23-5-2011 11:44:01

94
Chapter 5
Our data demonstrate that vagus activity conveys its anti-inflammatory effect via distinct nAChRs expressed in macrophages. The anti-inflammatory effects of nAChR activation on macrophages have previously been solely attributed to activation of the nAChR α75,6. Although previous data imply a role for α7 nAChR in modulation of NF-κB activation4, we clearly demonstrate that the nAChR α4β2, rather than α7, mediates stimulation of phagocytosis. Matsunaga et al24 and others25 have shown expression of α4β2, and no α7 nAChR in alveolar macrophages. Likewise, we failed to demonstrate α7 nAChR transcripts in intestinal, Mf4/4 spleen or peritoneal macrophages. Further analysis of potential α7 nAChR protein in these macrophages was hampered by our observation that commercially available α7 nAChR antisera are not specific and stain an approximately 57kD protein in brain homogenates from wildtype as well as α7 nAChR -/- mice, in agreement with earlier reports26. Surprisingly,α7 nAChR blockers were effective in counteracting nicotinic effects on NF-κB activity as well as cytokine production. These data indicate that the selectivity of commonly used blockers αBgt and MLA for α7 nAChR may be questioned, and both blockers have shown to bear affinity for other nAChR subunits, including α1, α6, α9, α10 and β2, as well27. Alternatively, the β2 nAChR blocker DHβE used in the current study also bears affinity for other nAChRs. Together, our results indicate that ACh induces phagocytosis via α4β2 nAChR, but seemingly activates different nAChR systems on macrophages to achieve inflammatory anergy.
Our data reveal the potency of vagus nerve activity to stimulate macrophage phagocytosis via the large GTPase Dynamin-2. In macrophages, Dynamin-2 plays a crucial role in exocytosis of endomembranes at the site of phagocytosis, and phagosome formation28. Our data indicate that the vagal modulation of phagocytosis involves nAChRβ2 mediated modulation of Dynamin-2 activity and cellular distribution, rather than PI3K, Akt, Rac1 activation or calcium signaling pathways. Formation of the phagosome and subsequent phagocytosis is the result of cellular remodeling of actin filaments, a process that requires Dynamin GTPase activity18. Interestingly, in mouse cholinergic neurons, the nAChR β2 subunit has recently been shown to complex to the GTPase Dynamin-1, which is highly homologous to the macrophage Dynamin-2. Although we do not know how the nAChR β2-mediated cellular redistribution of the Dynamin is brought about, it is tempting to speculate that in macrophages, in analogy to cholinergic neurons, nAChR β2 activation modulates Dynamin-2 protein distribution directly. The recruitment of Dynamin to membrane receptors was initially thought to be mainly required for receptor internalization29. However, the internalization of nAChRs has been shown not to involve Dynamin30 and our results demonstrate that ACh mediated Dynamin-2 activation supports the phagocytic potential. The exact cellular mechanism by which nAChR β2 mediates Dynamin-2 cellular distribution is currently under investigation.
Intestinal macrophages co-operate with dendritic cells to form the first line of defense in the intestinal mucosa. We show here that vagus nerve activity assists in
van de Zanden.indb 94 23-5-2011 11:44:01

95
Vagal activity enhances macrophage phagocytosis
surveillance of luminal antigen uptake by inducing a transient increase in epithelial permeability and augmenting the uptake of luminal bacteria by mucosal macrophages. Our data confirm the intimate involvement of the autonomous nervous system in the maintenance of mucosal barrier function23,31,32. Our current finding of the vagus nerve regulating the mucosal barrier function is in agreement with earlier reports of cholinergic regulation of enterocyte endocytosis and intestinal epithelial permeability20, although epithelial signaling is thought to be mediated via non-neuronal ACh production21. The cholinergic modulation of the epithelial barrier function was not explained by altered tight junction protein expression, as levels of occludin and claudin-2 were not affected in intestinal tissue from mice that underwent vagus nerve stimulation (results not shown). A possible alternative explanation for the transient reduction of barrier function after VNS is that stimulation of vagal activity, under normal physiological conditions, leads to a reduction in mean arterial blood pressure, a parameter that is shown to mediate tight junction integrity33. Hence, although vagal activation preserves barrier function under pathological conditions such as ischemia, via its anti-inflammatory actions, our data indicate that enhanced vagus nerve activity under physiological conditions can allow transient passage of antigens, possibly to assist in routine surveillance. The physiological relevance of this system is currently under investigation.
van de Zanden.indb 95 23-5-2011 11:44:01

96
Chapter 5
REFERENCE LIST
1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
2. Tracey KJ. The inflammatory reflex. Nature 2002;420:853-859.
3. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004;10:1216-1221.
4. The FO, Boeckxstaens GE, Snoek SA, Cash JL, Bennink R, Larosa GJ, van den Wijngaard RM, Greaves DR, De Jonge WJ. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 2007;133:1219-1228.
5. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
6. De Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844-851.
7. Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006;131:1122-1130.
8. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der PT. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis 2005;191:2138-2148.
9. Giebelen IA, van Westerloo DJ, Larosa GJ, de Vos AF, van der PT. Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock 2007;27:443-447.
10. Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 2005;202:1023-1029.
11. Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 2005;115:66-75.
12. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005;307:254-258.
13. Kessler W, Traeger T, Westerholt A, Neher F, Mikulcak M, Muller A, Maier S, Heidecke CD. The vagal nerve as a link between the nervous and immune system in the instance of polymicrobial sepsis. Langenbecks Arch Surg 2006;391:83-87.
14. Giebelen IA, Le Moine A, van den Pangaart PS, Sadis C, Goldman M, Florquin S, van der PT. Deficiency of alpha7 cholinergic receptors facilitates bacterial clearance in Escherichia coli peritonitis. J Infect Dis 2008;198:750-757.
15. Desmedt M, Rottiers P, Dooms H, Fiers W, Grooten J. Macrophages induce cellular immunity by activating Th1 cell responses and suppressing Th2 cell responses. J Immunol 1998;160:5300-5308.
16. Smith PD, Smythies LE, Mosteller-Barnum M, Sibley DA, Russell MW, Merger M, Sellers MT, Orenstein JM, Shimada T, Graham MF, Kubagawa H. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol 2001;167:2651-2656.
17. Papke RL, Buhr JD, Francis MM, Choi KI, Thinschmidt JS, Horenstein NA. The effects of subunit composition on the inhibition of nicotinic receptors by the amphipathic blocker 2,2,6,6-tetramethylpiperidin-4-yl heptanoate. Mol Pharmacol 2005;67:1977-1990.
van de Zanden.indb 96 23-5-2011 11:44:01

97
Vagal activity enhances macrophage phagocytosis
18. Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 1999;190:1849-1856.
19. Kabbani N, Woll MP, Levenson R, Lindstrom JM, Changeux JP. Intracellular complexes of the beta2 subunit of the nicotinic acetylcholine receptor in brain identified by proteomics. Proc Natl Acad Sci U S A 2007;104:20570-20575.
20. Greenwood B, Mantle M. Mucin and protein release in the rabbit jejunum: effects of bethanechol and vagal nerve stimulation. Gastroenterology 1992;103:496-505.
21. Groot J, Bijlsma P, Van Kalkeren A, Kiliaan A, Saunders P, Perdue M. Stress-induced decrease of the intestinal barrier function. The role of muscarinic receptor activation. Ann N Y Acad Sci 2000;915:237-246.
22. Gareau MG, Jury J, Perdue MH. Neonatal maternal separation of rat pups results in abnormal cholinergic regulation of epithelial permeability. Am J Physiol Gastrointest Liver Physiol 2007;293:G198-G203.
23. Soderholm JD, Perdue MH. Stress and gastrointestinal tract. II. Stress and intestinal barrier function. Am J Physiol Gastrointest Liver Physiol 2001;280:G7-G13.
24. Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol 2001;167:6518-6524.
25. Galvis G, Lips KS, Kummer W. Expression of nicotinic acetylcholine receptors on murine alveolar macrophages. J Mol Neurosci 2006;30:107-108.
26. Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De Biasi M, Schroder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem 2007;102:479-492.
27. Whiteaker P, Marks MJ, Christensen S, Dowell C, Collins AC, McIntosh JM. Synthesis and characterization of 125I-alpha-conotoxin ArIB[V11L;V16A], a selective alpha7 nicotinic acetylcholine receptor antagonist. J Pharmacol Exp Ther 2008;325:910-919.
28. Huynh KK, Grinstein S. Phagocytosis: dynamin’s dual role in phagosome biogenesis. Curr Biol 2008;18:R563-R565.
29. van Koppen CJ, Kaiser B. Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther 2003;98:197-220.
30. Kumari S, Borroni V, Chaudhry A, Chanda B, Massol R, Mayor S, Barrantes FJ. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J Cell Biol 2008;181:1179-1193.
31. Savidge TC, Newman P, Pothoulakis C, Ruhl A, Neunlist M, Bourreille A, Hurst R, Sofroniew MV. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 2007;132:1344-1358.
32. Soderholm JD, Yates DA, Gareau MG, Yang PC, MacQueen G, Perdue MH. Neonatal maternal separation predisposes adult rats to colonic barrier dysfunction in response to mild stress. Am J Physiol Gastrointest Liver Physiol 2002;283:G1257-G1263.
33. Derikx JP, van Waardenburg DA, Thuijls G, Willigers HM, Koenraads M, van Bijnen AA, Heineman E, Poeze M, Ambergen T, van Ooij A, van Rhijn LW, Buurman WA. New Insight in Loss of Gut Barrier during Major Non-Abdominal Surgery. PLoS ONE 2008;3:e3954.
van de Zanden.indb 97 23-5-2011 11:44:01

6CHAPTER
Cholinergic agonists interact with immunomodulatory actions of neuropeptides VIP and SP
Esmerij P. van der ZandenLaurens J. NijhuisCaroline VerseijdenKlaus MichelMichael SchemannGuy E. BoeckxstaensWouter J. de Jonge
Manuscript in preparation
van de Zanden.indb 99 23-5-2011 11:44:01

100
Chapter 6
ABSTRACT
INTRODUCTION Vagus nerve activity ameliorates intestinal inflammation. Its mechanisms may involve attenuation of pro-inflammatory cytokine production by macrophages (MF) and inhibition of inflammatory processes via the release of acetylcholine (ACh). However, vagus nerve stimulation may also modulate the release of neuropeptides with immune-modulatory potential, such as substance P (SP) and vasoactive intestinal polypeptide (VIP). Here, we questioned whether vagus nerve released ACh may relay its anti-inflammatory potential via modulation of release of VIP or SP.
AIMS & METHODS: Electrical vagal nerve stimulation (VNS) was performed using bipolar electrodes, after which intestinal tissue was analyzed for expression of VIP and VIP receptors VPAC1-2, as well as Substance P and its receptor NK-1. In vitro, peritoneal mouse MF were pretreated with Ach/nicotine (30min, 10µM) and/or VIP or SP (30min, 10µM), followed by 3hrs LPS challenge (100ng/mL). Cytokine levels were determined using ELISA, NF-kB activity was measured in RAW MF stably transfected with NF-kB luciferase reporter construct. Calcium measurements were performed using Fluo-4-AM.
RESULTS: VNS modulated expression levels of VIP and SP in ileum and colonic tissue. Expression of VIP and its receptors VPAC1 and VPAC2 was induced by VNS, whereas on the other hand, VNS resulted in a gradual decline in SP expression. In vitro, VIP inhibited LPS-induced TNF production in RAW MF down to 52.4+/-4.2% of LPS alone. When ACh and VIP (10 μM) were co-administered, LPS induced TNF production was decreased almost down to unstimulated levels (25±4.2% of LPS alone). On the other hand, SP enhanced NF-kB transcriptional activity and TNF production in RAW MF. ACh reduced TNF production and NF-kB activation in MF significantly, whereas co-application of nicotine and SP attenuated LPS induced NF-kB activation and TNF production even further, down to 57.2±5.1% and 52.4±3.0% respectively compared to substance P treatment alone. Nicotine pretreatment showed similar results. Intracellular calcium was not affected using either treatment.
CONCLUSION: Cholinergic agonists reduce TLR4 activation on peritoneal macrophages a mechanism that involves nAChRs, and interference with VIP and SP pathways. These data suggest that the vagus nerve anti-inflammatory effect may be amplified via modulation of neuropeptide expression.
van de Zanden.indb 100 23-5-2011 11:44:01

101
Role of neuropeptides in the vagus nerve anti-inflammatory effect
INTRODUCTION
Electrical stimulation of vagal efferent fibers inhibits inflammatory processes in various experimental animal models of inflammatory disease, such as sepsis1, rheumatoid arthritis2, post-operative ileus3 and DSS-colitis4. In vitro data reveal that activation of nicotinic acetylcholine receptors (nAChR) on macrophages can attenuate pro-inflammatory cytokine release1 and enhance phagocytosis5. These findings might explain the anti-inflammatory effect of activation of the vagus nerve. However, it remains to be elucidated if acetylcholine (ACh) released from vagus nerve termini actually reaches the immune cells, and if so, in what quantities. Given the short half-life of ACh, cholinergic modulation of immune cell activation most likely requires close contact and there is currently no strong evidence that parasympathetic neurons indeed innervate macrophages. Therefore, it is likely that, next to the ‘classical’ direct anti-inflammatory effect of acetylcholine on tissue macrophages, vagus nerve stimulation affects immune cells via post-ganglionic mechanisms involving alternative neurotransmitters, such as neuropeptides or catecholamines.
In septic models, the protective effect of vagus nerve stimulation has been ascribed to modulation of splenic release of catecholamines6. Alternatively, in the gastrointestinal tract, the vagus nerve mainly synapses with neurons of the enteric nervous sytem (ENS) and vagus nerve activation leads to the release of several neuropeptides at the nerve endings in the intestine. Neuropeptides are neuronal signaling molecules, that diffuse into surrounding tissues and bind to their corresponding receptors, affecting nearby muscle, epithelium, endothelium, and immune cells. Besides the known functions that neuropeptides exhibit in the gastrointestinal tract (GI tract), such as secretion of salivary, gastric fluids, intestinal fluids and electrolytes, neuropeptides are increasingly appreciated as modulators of the immune response.
Substantial evidence shows that substance P and vasoactive intestinal peptide (SP and VIP), two neuropeptides that are abundantly expressed in the gut, have important neuroimmunomodulatory properties in the intestine. Increased SP expression is observed in both tissue and nerve fibers in the colons of patients with UC7 and substance P receptor antagonists attenuate disease activity in a DSS-colitis rat model8. In vitro data reveal that substance P enhances NF-kB transactivation and chemokine and cytokine responses in murine macrophages9. In contrast, vasoactive intestinal peptide (VIP), displays potent anti-inflammatory properties, including inhibition of leukocyte migration and stimulation of IgA production by B lymphocytes10. Moreover, VIP administration reduces clinical symptoms and cytokine profiles in mice models of experimental colitis11. Hence, substance P and VIP can modulate the intestinal immune response, however, the exact mechanism via which these neuropeptides exert their immunomodulatory effects is unknown. It is a complex system, in which secretion and action of a single neuropeptide can be influenced by other neuropeptides, neurotransmitters, cytokines, hormones and drugs.
van de Zanden.indb 101 23-5-2011 11:44:01

102
Chapter 6
The present study shows that VNS modulates expression levels of VIP and SP in ileum and colonic tissue. In vitro, substance P enhances NF-kB transactivation and TNF production, whereas VIP reduces inflammation in peritoneal macrophages. Cholinergic agonists nicotine and acetylcholine prevent the pro-inflammatory effects of SP, while the anti-inflammatory effects of VIP are enhanced. Taken together, our data suggest that acetylcholine, released upon vagus nerve stimulation, can affect immunomodulatory actions of neuropeptides VIP and SP.
MATERIALS AND METHODS
General reagents. (-)-Nicotine, Acetylcholine, LPS, VIP and substance P were from Sigma-Aldrich (Zwijndrecht, the Netherlands). ELISA kits for TNF were from R&D Systems ( Abingdon, UK).
Mice. For electrical vagal nerve stimulation, 12- to 15-week-old female BALB/c mice (Harlan Nederland) were kept in environmentally controlled conditions (light on from 08:00 to 20:00; water and rodent nonpurified diet ad libitum; temperature, 20–22 °C; 55% humidity). All animal experiments were approved by the local animal experimental committees.
Electrical vagal nerve stimulation. Mice were anesthetized by intraperitoneal injection of a mixture of fentanyl citrate and fluanisone (Hypnorm; Janssen) and midazolam (Dormicum; Roche). The vagus nerve was stimulated as described12 . The right cervical vagal branch was prepared free from the carotid artery and was ligated with 6-0 silk suture. The part distal from the ligation was attached to a bipolar electrode and 5 V of stimulation with a frequency of 5 Hz and a duration of 2 ms was applied for 15 min. In sham-operated mice, the cervical skin was opened and was left for 15 min covered by moist gauze. Groups of three mice were electrically stimulated and ileum and colonic tissue were collected at 4, 8 and 24 h after stimulation.
NF-κB activity assay. Peritoneal mouse RAW264.7 macrophages (ATCC, Middlesex, UK) were stably transfected with a NF-κB luciferase reporter construct (Clontech, Mountain View, CA) in which a PDNA3.1(+) derived neomycin resistance TK cassette was inserted. Transfection was performed using electroporation. In short, 2*106
RAW264.7 cells were scraped and resuspended in 100µL Nucleofector V reagent (Amaxa Biosystems Inc.) with 2 µg of highly purified NF-kB-luciferase construct, mixed carefully and transferred to a 0.2-cm Amaxa electroporation cuvette. The cuvette was placed in the Amaxa nucleofection device and nucleofected according to manufacturer’s instructions using program D-32. Immediately after transfection, cells were cultured in RPMI 1640/10% FCS o/n. Transfected cells were selected in
van de Zanden.indb 102 23-5-2011 11:44:01

103
Role of neuropeptides in the vagus nerve anti-inflammatory effect
the culture medium supplemented with neomycine (1000µg/ml) for 14 days. Resistent cells were subcloned and clones were cultured up to 20 passages with RPMI containing 1000ug/ml neomycine. Clones stably expressing the NF-kB reporter construct were used.
Cell culture and stimulation. Murine RAW264.7 cells (ATCC, Middelsex, UK) and RAW cells stably expressing the NF-kB luciferase reporter construct were grown in RMPI-1640 medium with antibiotics and L-glutamine (Gibco, Breda, The Netherlands), supplemented with 1% heat-inactivated fetal bovine serum at 37ºC in a humidified incubator with 5% CO2. For experiments, cells were transferred to 48-well suspension plates (Greiner-Bio, Alphen aan de Rijn, The Netherlands) at 2.5 x105
cells/well. After o/n incubation, the medium was removed and replaced with RPMI containing 1% serum. Cells were pretreated with vehicle, nicotine or acetylcholine 30 minutes prior to SP or VIP treatment. VIP or SP were applied for another 30min, cells were subsequently stimulated with 100ng/ml LPS for 3 hours. TNF levels were analyzed using the TNF sandwich ELISA kit. For NF-kB luciferase measurement, the medium was removed after 3 hrs. of LPS stimulation; the cells were washed three times with ice-cold PBS and lysed with Passive Lysis Buffer supplied in the LuciferaseTM Reporter Assay Kit (Promega Corporation, Madison, WI), the lysate was assayed for luciferase activity according to the manufacturer’s instructions.
Ca2+ measurement. RAW264.7 cells were loaded with the fluorescenct dye 10 μM Fluo-4-AM (Invitrogen, Darmstadt, Germany) and 2.5 mM probenecid (Sigma-Aldrich) in HEPES buffer for 20 min at RT. Cells were washed three times to remove the dye and perfused with HEPES solution (+2.5mM probenecid) for another 20 min. ATP, nicotine, acetylcholine, VIP and SP (all 10µM) were applied with rapid ejections using a picospritzer. The dye was excited with a HC482/35 filter and fluorescence was monitored through a HC529/28 filter. Calcium fluxes were measured until 20 sec after application of ATP, nicotine, ACh, VIP or SP.
Statistical analyses. Data are expressed as mean ± standard error (SEM). Statistical analyses were performed using the non-parametric Mann-Whitney U test. A probability value (P) of less than 0.05 was considered significant.
RESULTS
VNS modulates VIP and SP expression To assess whether VNS can modulate VIP or SP receptor expression in vivo, we applied VNS for 15 minutes and collected mucosal and muscle tissue from the ileum
van de Zanden.indb 103 23-5-2011 11:44:01

104
Chapter 6
and colon 4, 8, or 24 hrs after VNS. Using QPCR, we determined expression levels of VIP, or its main receptors VPAC-1 and -2. VPAC1 is constitutively expressed in both stimulated and unstimulated macrophages, whereas VPAC2 is only expressed in activated macrophages. Moreover, we tested the effect of VNS on the expression of Substance P and its receptor NK-1. Figure 1A shows that, after an initial drop in VIP expression (4 hrs after VNS), VIP expression is enhanced 24 hrs after VNS stimulation in ileum. In colon, VNS significantly enhances VIP expression after 4 hrs. On the other hand, SP expression is gradually reduced by VNS in ileum and colonal tissue. In figure 1B, it is demonstrated that VNS enhances expression of VPAC1 in mucosal colon tissue after 4 hrs. Besides that, VNS does not alter expression levels of VIP receptors VPAC1-2 or SP receptors NK-1 in colonic or ileum tissue at other timepoints. In duodenal and jejunum tissue, no differences in expression levels of VIP, SP and its receptors upon VNS were observed (data not shown).
ACh, nicotine and VIP reduce NF-kB activity and cytokine release in a cumulative mannerNext we studied the potential of VIP and ACh/nicotine to modulate MF activity in vitro; To test the immunomodulatory capacity of VIP on MF, RAW macrophages were treated with VIP for 30 minutes before 3 hrs LPS challenge. VIP inhibited LPS-induced TNF production and NF-kB activation in a dose dependent fashion in RAW macrophages, down to resp. 27+/-6.2% and 52.4+/-4.2% of control release (Fig 2A and 2B). ACh or nicotine pretreatment (30 min) attenuated LPS-induced TNF production down to 59±4.5% and 60.6±4.5% confirming previous studies (Fig 2C). Interestingly, when ACh and VIP, or nicotine and VIP, were co-administered, LPS induced TNF production was decreased almost down to unstimulated levels (ACh+VIP: 25±4.2%; nicotine+VIP 23.2±3.1% of LPS) (Fig 2C).
The pro-inflammatory activity of SP is neutralized by cholinergic agonistsSubsequently, we examined whether ACh or nicotine could alter immuno-modulatory actions of SP. Cells were treated with different concentrations of SP 30 minutes prior to 3 hours LPS (100ng/ml) challenge. SP enhanced TNF-production and NF-kB activity dose-dependently in macrophages, up to resp. 132±7.4% and 135±6.3% of LPS (Fig. 3A and 3B). To assess whether ACh or nicotine affects SP induced increase in NF-kB transactivation and TNF release, we pretreated cells with ACh or nicotine for 30 minutes prior to substance P (30min) and LPS challenge (3hrs). ACh reduced LPS induced TNF production and NF-kB activation in macrophages down to 63±3.0% and 69±4.0% of LPS treated cells. Nicotine attenuated NF-kB activation and TNF production to the same extent, down to 75,9+/-5.3% and 69.5+/-3.3% (Fig. 3B). However, co-application of SP with ACh attenuated LPS induced TNF production and NF-kB activation even further, down to 53,2±5.1% and 44±3.0% respectively compared
van de Zanden.indb 104 23-5-2011 11:44:01
105
Role of neuropeptides in the vagus nerve anti-inflammatory effect
Figure 1. Modulation of VIP and SP expression upon VNS Electrical VNS was performed using bipolar electrodes for 15 minutes and after 4, 8 or 24hrs, mucosal and muscle tissue of the ileum (left panel) and colon (right panel) were collected. (A) Tissue was analyzed using QPCR for VIP (grey lines; open dots) and SP (black lines; closed squares) expression levels. In Figure 1B, expression levels of VPAC1 (black closed triangles), VPAC-2 (grey rhombus) and NK-1 (open squares) were depicted. All values are corrected for B-actin and expressed as % of sham stimulated tissue. Experiments were performed in triplicate. Asterisks indicate significant differences (P<.05) vs SHAM stimulation.
to substance P treatment alone. In conjunction, when cells were treated with SP and nicotine, LPS-induced TNF production and NF-KB activation were decreased to 57,2±5.1% and 52,4±6.6% as compared to SP alone (Fig. 3C and 3D). These data suggest that cholinergic agonists can neutralize the pro-inflammatory actions of the neuropeptide SP.
0 5 10 15 200
50
100
150
Muscle
Timepoint
% S
HA
M
Ileum
0 5 10 15 200
100
200
300
Mucosa
Timepoint
% S
HA
M
Colon
0 5 10 15 200
100
200
300VIPSP
*
Timepoint
0 5 10 15 200
100
200
300
Mucosa
Timepoint
% S
HA
M
0 5 10 15 200
100
200
300
VPAC1VPAC2NK-1
Timepoint0 5 10 15 20
0
50
100
150
200
Muscle
Timepoint
% S
HA
M
0 5 10 15 200
50
100
150
*
SPVIP
Timepoint
0 5 10 15 200
200
400
600
VPAC-2VPAC-1
NK-1
*
Timepoint
A
B
van de Zanden.indb 105 23-5-2011 11:44:02
106
Chapter 6
- - - VIP VIP VIP (100
50
100
*
*
** **
- Nico ACh - Nico ACh (10 µM)
*
µM)
TN
F (%
) **
0 1 10 100 10000
25
50
75
100
****
**T
NF
(%)
nM VIP
LPS (100ng/ml)
0 0 1 10 100 10000
50
100
* ** *
LPS (100ng/ml)
* * *
nM VIP
NF-
kB (%
)
LPS (100ng/ml)
Figure 2. VIP and ACh/nicotine reduce inflammatory mediators in a cumulative manner. RAW macrophages were incubated with a dose range of VIP (0-1000nM) for 30min prior to 3hrs LPS stimulation (100ng/ml). NF-kB activity was determined using luminescence and TNF production with ELISA. TNF levels (A) or NF-kB activity (B) were measured after 3hrs. and depicted as % of vehicle treated cells . Data are mean s.e.m. of three independent experiments done in triplicate. (C) Cells were pretreated with 10µM ACh/nicotine for 30min, then stimulated with 10µM VIP, followed by 100ng/ml LPS challenge. TNF levels were measured after 3hrs. Data are mean s.e.m. of three independent experiments done in triplicate. *, P < 0.05
Figure 3. Co-application of cholinergic agonists with SP neutralizes pro-inflammatory actions of SP RAW cells were incubated with SP (0-1000nM) for 30min, followed by 3hrs LPS stimulation (100ng/ml). (A) TNF levels or (B) NF-kB levels were determined and represented as % of vehicle treated cells. (C and D) Cells were pretreated with 10µM ACh/nicotine for 30min, then stimulated with 10µM SP, prior to 3hrs of 100ng/ml LPS challenge. (C) TNF production was measured with ELISA and NF-kB (D) was determined using luminescence. Data are mean s.e.m. of three independent experiments done in duplicate. *, P < 0.05
A
B
C
0 0 0.1 1 100
50
100
150*
***
µM SPLPS (100ng/ml)
TN
F(%
)
0 0.01 0.1 1 100
50
100
150 * ***
µM SP
LPS (100ng/ml)
NF-
kB (%
)
0
50
100
150
** **
**
** **
- - - SP SP SP - Nico ACh - Nico ACh
TN
F(%
)
LPS(100ng/ml)
(10 µM)(10 µM)
0
50
100
150 *
* ** ******
- - --- Nico ACh Nico ACh
SP SP (10µM)(10µM)
LPS (100ng/ml)
NF-
kB (%
)
A
C
B
D
van de Zanden.indb 106 23-5-2011 11:44:02

107
Role of neuropeptides in the vagus nerve anti-inflammatory effect
Calcium measurementsTo elucidate the cellular mechanism behind the immuno-modulatory actions of ACh, VIP and SP on macrophages, we analyzed whether modulation of SP and VIP pathways by ACh was reflected by altered cellular calcium flux. However, we found that the effects of ACh, VIP and SP, are independent of calcium signaling pathways, as cells respond potently to adenosine triphosphate (ATP), but calcium fluxes were not affected by exposure to ACh, VIP or SP alone, or in combination.
ATP ATP+ACh ACh
VIPSP
SP+ACh VIP+ACh
Figure 4. Exposure to ACh, VIP, or SP, alone or in combination, does not affect calcium fluxes. RAW264.7 cells were loaded with Fluo-4-AM and probenecid in HEPES buffer. ATP, ACh, VIP and substance P (all 10µM) , alone or in combination were applied with rapid ejections and calcium fluxes were measured until 20 sec application of either of the substances. Data are representitive of four independent experiments.
DISCUSSION
Intestinal immune homeostasis requires strictly controlled regulatory mechanisms. Evidence is mounting that these factors can be modulated by signals emerging from the nervous system. For instance, the parasympathetic and sympathetic pathways can restrain inflammatory responses and affect innate immune cell reactivity. Vagus nerve stimulation ameliorates inflammation via actylcholine. However, vagus nerve stimulation may also affect immune cells via postganglionic mechanisms involving alternative neurotransmitters, such as neuropeptides. For decades, neuropeptides are recognized as modulators of the immune system that can play a crucial role in the pathogenesis of inflammation7-11. Here, we show that VNS can affect expression of neuropeptides in the gut and in vitro, cholinergic activation affects immuno-modulatory actions of neuropeptides.
In vivo, VNS has shown to ameliorate disease in diverse inflammatory mouse models. However, in vitro, cholinergic agonists reduce LPS-induced TNF production in macrophages only to a moderate extent that can not entirely explain the in vivo effect of VNS. Therefore, we reasoned that neuropeptides, which are abundantly present in the intestine, could play a role in the the anti-inflammatory effects observed after VNS. We showed that VNS modulated expression of VIP and SP in ileum and colonal tissue.
van de Zanden.indb 107 23-5-2011 11:44:03

108
Chapter 6
Since the cholinergic anti-inflammatory effects have been extensively studied in macrophages, we performed our in vitro experiments using peritoneal mouse macrophages. These experiments clearly display that co-application of VIP and ACh or nicotine reduces inflammation in RAW macrophages in a cumulative manner. However, it is unclear if the VNS-induced modulation of VIP receptors is macrophage mediated. Presumably, other immune cells can be affected as well. For example, it is reported that DC isolated from the PP exhibit increased levels of VPAC1 and VPAC2 mRNA as compared to peripheral DC’s, indicating that these PP DC are susceptible to modulation by VIP16. Therefore, our future prospects are to investigate if besides macrophages, other immune cells such as DC, are involved in the anti-inflammatory actions of ACh and VIP.
The mechanism via which vagus nerve released ACh can co-operate with VIP to reduce inflammation is still unclear. Suprisingly, calcium fluxes were unaffected by application of ACh, nicotine, VIP or SP. Nevertheless, we and others previously showed that ACh does induce kinase signaling17 , therefore, we would have expected that ACh modulated calcium flux. Presumably, the cell culture system used was not suitable for measurement of gradual changes in calcium flux, or the experiment set-up was not sensitive enough.
Delgado et al demonstrated that the anti-inflammatory effect of VIP on RAW cells is mediated via two intracellular pathways: affecting both NF-kB binding and the composition of the cAMP responsive element binding complex (CREB/c-Jun)18. The working mechanism of ACh ultimately involves modulation of STAT3 pathways6 and prevention of NF-κB p65 transcriptional activity19, following nAChR activation. Possibly, VIP and ACh act synergistic in the inhibition of nuclear translocation of NF-kB. Alternatively, the cross-talk between VIP and ACh could be extracellular instead of intracellular. This is supported by the finding that, in isolated parasympathetic neurons of rat ganglia, VIP selectively increased the affinity of nAChR for their agonists, thereby potentiating ACh-evoked whole cell currents in rat cholinergic neurons20 . Otherwise, it is possible that ACh modulates VIP and SP receptor expression which renders cells more susceptible to immuno-modulation. Finally, studies in rodents have demonstrated that VIP, SP and ACh can be locally produced by inflammatory cells such as macrophages, lymphocytes, and dendritic cells. Possibly, the nAChR-mediated effects of ACh could be potentiated in presence of locally released VIP.
We show here that VNS modulates VIP and SP expression in the gut, and in vitro, cholinergic agonists neutralize the pro-inflammatory SP effects and further enhance anti-inflammatory VIP actions in intestinal macrophages. These data suggest that the anti-inflammatory effects of cholinergic activation in vivo may be mediated not only via direct ACh binding to nAChR, but also via modulation of co-released neuropeptides SP and VIP.
van de Zanden.indb 108 23-5-2011 11:44:03

109
Role of neuropeptides in the vagus nerve anti-inflammatory effect
REFERENCE LIST
1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
2. van Maanen MA, Lebre MC, van der PT, Larosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-122.
3. The FO, Boeckxstaens GE, Snoek SA, Cash JL, Bennink R, Larosa GJ, van den Wijngaard RM, Greaves DR, De Jonge WJ. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 2007;133:1219-1228.
4. Ghia JE, Blennerhassett P, Park AJ, Deng YK, Cornell R, Collins SM. Critical role of the alpha-7 nicotinic acetylcholine receptor in the development of colitis. Gastroenterology 2008;134:A98.
6. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A 2008.
7. Goode T, O’Connell J, Anton P, Wong H, Reeve J, O’Sullivan GC, Collins JK, Shanahan F. Neurokinin-1 receptor expression in inflammatory bowel disease: molecular quantitation and localisation. Gut 2000;47:387-396.
8. Stucchi AF, Shofer S, Leeman S, Materne O, Beer E, McClung J, Shebani K, Moore F, O’Brien M, Becker JM. NK-1 antagonist reduces colonic inflammation and oxidative stress in dextran sulfate-induced colitis in rats. Am J Physiol Gastrointest Liver Physiol 2000;279:G1298-G1306.
9. Sun J, Ramnath RD, Zhi L, Tamizhselvi R, Bhatia M. Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol 2008;294:C1586-C1596.
10. De la FM, Delgado M, Gomariz RP. VIP modulation of immune cell functions. Adv Neuroimmunol 1996;6:75-91.
11. Abad C, Martinez C, Juarranz MG, Arranz A, Leceta J, Delgado M, Gomariz RP. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn’s disease. Gastroenterology 2003;124:961-971.
12. De Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844-851.
13. Fodor M, Pammer C, Gorcs T, Palkovits M. Neuropeptides in the human dorsal vagal complex: an immunohistochemical study. J Chem Neuroanat 1994;7:141-157.
14. Dai XL, Triepel J, Heym C. Immunohistochemical localization of neuropeptide Y in the guinea pig medulla oblongata. Correlation with VIP and DBH. Histochemistry 1986;85:327-334.
15. Bjorck S, Fahrenkrug J, Jivegard L, Svanvik J. Release of immunoreactive vasoactive intestinal peptide (VIP) from the gallbladder in response to vagal stimulation. Acta Physiol Scand 1986;128:639-642.
16. Massacand JC, Kaiser P, Ernst B, Tardivel A, Burki K, Schneider P, Harris NL. Intestinal bacteria condition dendritic cells to promote IgA production. PLoS ONE 2008;3:e2588.
18. Delgado M, Pozo D, Martinez C, Leceta J, Calvo JR, Ganea D, Gomariz RP. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit endotoxin-induced TNF-alpha production by macrophages: in vitro and in vivo studies. J Immunol 1999;162:2358-2367.
19. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004;10:1216-1221.
20. Liu DM, Cuevas J, Adams DJ. VIP and PACAP potentiation of nicotinic ACh-evoked currents in rat parasympathetic neurons is mediated by G-protein activation. Eur J Neurosci 2000;12:2243-2251.
van de Zanden.indb 109 23-5-2011 11:44:03

7CHAPTER
Effects of smoking on nicotinic AChR expression and susceptibility to cholinergic immunomodulation in human monocytes
EP Van der ZandenF HilbersC VerseijdenRM van den WijngaardL UlloaGE BoeckxstaensWJ de Jonge
Manuscript Submitted
van de Zanden.indb 111 23-5-2011 11:44:03

112
Chapter 7
ABSTRACT
Smoking and nicotine positively affect the disease course in ulcerative colitis. As previous data suggest that nicotinic receptor CHRNA7 activation reduces inflammatory responses in monocytes, we evaluated whether smoking could affect monocyte CHRNA7 expression. Repeated nicotine exposure up-regulated CHRNA7 expression on THP-I monocytes and in conjunction, in a pilot study, CHRNA7 was only detectable in isolated blood monocytes of smokers. The duplicated CHRNA7 variant gene CHRFAM7A was ubiquitously expressed on human monocytes and was not affected by nicotine exposure, while CHRNB2 was not detectable. In a volunteer study (n=11), we next aimed to demonstrate that smoking rendered blood monocytes more susceptible to cholinergic immuno-modulation, either by application of nicotine or by administration of fat nutrition. Pro-inflammatory cytokine secretion was inhibited by nicotinic receptor activation in THP-I monocytes, but this response was not consistently seen in blood monocytes from smoking individuals.
van de Zanden.indb 112 23-5-2011 11:44:03

113
nAChR expression and cholinergic immune-modulation in human monocytes
INTRODUCTION
It is increasingly recognized that the vagus nerve has immuno-modulating properties via activation of cholinergic receptors on non-neuronal cells. Cholinergic signaling attenuates pro-inflammatory cytokine release and improves clinical outcome in experimental animal models of inflammatory disease, such as sepsis 1 , pancreatitis 2, post-operative ileus 3 and rheumatoid arthritis 4 . It has been proposed that electric stimulation of the efferent vagus nerve leads to the release of acetylcholine, which could bind to nicotinic acetylcholine receptors (nAChRs) that are broadly expressed on different types of immune cells 5,6, resulting in attenuation of pro-inflammatory cytokine release in monocytes and macrophages 7. However, more recent results indicate that the vagus nerve and cholinergic agonists inhibit systemic inflammation by activating the noradrenergic splenic nerve 8. Earlier studies have indicated that the anti-inflammatory effect of ACh is mediated through the alpha7 nicotinic acetylcholine receptor (CHRNA7) expressed on the surface of primary human monocytes 9. Besides the CHRNA7, a partially duplicated hybrid form of this gene was identified (CHRFAM7A) in which exon 5 to 10 of the alpha7 genes have been duplicated in a “tail to head” organization and combined with four novel exons (A to D) to comprise a new gene 10. The CHRFAM7A gene transcript is expressed in human monocytes has been identified in brain and human monocyte cell lines 11. This CHRFAM7A mRNA is transcribed to 45 kDa protein which lacks a nicotine binding domain, and hence it is unclear whether it is part of a functional nicotinic receptor 12. However, recent work indicates that its expression is transcriptionally regulated by LPS responsive genes and may contribute to the cholinergic regulation of the immune response 13. To date, the physiological and immunological implications of this receptor remain unknown.
Importantly, as most of the above work considering cholinergic inhibition of inflammation has been done in vitro and in experimental mouse models, the relevance of cholinergic immunmodulation to human disease remains sketchy. In one prominent human inflammatory disease, ulcerative colitis, cholinergic immunomodulation may play a significant role and provide therapeutic advantages for the treatment of the disorder. It is widely acknowledged that nicotine and smoking have a protective effect in the development of ulcerative colitis and reduces its severity 14-19. As a consequence of this clinical observation, nicotine patches and nicotine enemas have been successfully tested in patients with ulcerative colitis, but showed no advantage as compared to standard therapy 20. However, the protective mechanism and its clinical implications remain unknown.
We reasoned that the protective effect of smoking in UC patients could be due to preferential activation of the ‘cholinergic anti-inflammatory pathway’ in smokers via enhanced exposure to nicotine in tobacco smoke. Human immune cells display a variation in nAChR expression due to genetic or environmental factors 21-23. One of
van de Zanden.indb 113 23-5-2011 11:44:03

114
Chapter 7
these environmental factors resulting in altered nAChR expression could be cigarette smoking. In brain regions, it is already demonstrated that long-term consumption of tobacco elicits nAChR upregulation 24-26. Hence, in this study, we hypothesized that regular exposure to nicotine in tobacco smoke, affects CHRNA7 and CHRFAM7A expression on human monocytes. Aims of the study were to determine whether priming to nicotine, either by cigarette smoking or by in vitro nicotine exposure, altered nAChR expression levels on human monocytes. Moreover, we determined whether repeated nicotine exposure rendered human monocytes more susceptible to cholinergic immunomodulation.
We show here that pre-exposure with nicotine as well as tobacco smoking specifically elevates nAChR CHRNA7 expression on human monocytes. However, this nicotine priming or cigarette smoking does not consistently affect LPS-induced TNF production in human monocytes or whole blood samples. In addition, we demonstrate that human peripheral blood monocytes show a large intra-individual variation in response to nAChR activation or high fat nutrition, which has been put forward as a modulator of the cholinergic anti-inflammatory pathway 27.
MATERIALS AND METHODS
Cell culture and cell treatment. The human acute monocytic leukemia cell line THP-I was cultured in RPMI 1640 (Sigma Aldrich), supplemented with 1% fetal bovine serum (FBS), 100units/ml penicillin, 100µg/ml streptomycin and 2mM L-glutamine. Cells were maintained at a recommended density between 2×105/ml and 1×106/ml at 37 °C in 5% CO2. Before experiments, cells were seeded at a density of 1×106/ml in 12-Well plates. Cells were incubated with different doses of nicotine (0, 1µM, 10µM) at the time-points indicated, followed by vehicle or 10ng/ml LPS challenge for two hrs. Medium was collected and TNF levels were measured using sandwich ELISA (R&D Systems, Minneapolis, Michigan).
RNA isolation and Polymeric Chain Reaction. For RNA isolation, 1×106 THP-I or 2*106 whole blood isolated monocytes were centrifuged (8’ 1500rpm), washed in ice-cold PBS and total RNA was extracted using the RNeasy minikit (Qiagen, Venlo, the Netherlands), according to the provided protocol. RNA concentration was determined using a nanodrop ND-1000 spectrophotometer (Thermo scientific). Subsequently, samples were treated with DNase, and reverse transcribed. Amplification from 100 ng of RNA was carried out using Reddymix (Thermoscientific) in a thermocycler with 30 cycles of denaturation at 94º for 30 seconds, annealing at 60º for 30 s, and elongation for 30 s at 72ºC. The bands were visualised on an ethidium bromide-stained 2.5% agarose gel. For lightcycler analysis, The complementary DNA (cDNA; 100ng) was subjected to Light Cycler polymerase
van de Zanden.indb 114 23-5-2011 11:44:03

115
nAChR expression and cholinergic immune-modulation in human monocytes
chain reaction (Roche, Woerden, the Netherlands) for 35 cycles at 60°C. Following primers were used: CHRNA7, forward: 5’-CCCAAGTGGACCAGAGTCAT-3’ reverse: 5’-GCCACACACTACCCCAGAGT-3’. CHRFAM7A, forward: 5’- GGAGGTGAGGGGAAGATGTC -3’, reverse: 5’- CAGGTCCTGCTGACTCAGGT -3’. CHRNB2 forward: 5’- TGGGAAGATTATCGCCTCAC -3’, reverse: 5v- AGACC ACGGCATTGGAATAG -3’. B2M was used as a reference gene.
Volunteers olive oil study. 11 healthy volunteers (6 male, 5 female; aged 25-35) were recruited. All volunteers refrained from caffeine/alcohol-containing beverages and food for at least 12 hrs before blood collection. At 8.30 a.m., 60ml of venous blood was drawn from the cubital vein into sodium heparine tubes (Vacutainer system, BD biosciences, Plymouth, UK). Fifteen minutes afterwards, 50ml (44g) of olive oil (extra virgin) was orally administered to the volunteers in a single dose. Thirty minutes after
intake, another 60ml of venous blood was drawn. Characteristics of the 11 healthy subjects considering gender, age and smoking habits are represented in table 1.
Whole blood assay. Whole-blood was diluted 1:1 in RPMI 1640 medium supplemented with antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin, (Sigma-Aldrich, Mannheim, Germany)) and L-glutamine. The blood was incubated in 96 wells plates (total volume 200µL/Well) and pretreated with nicotine (0-1000nM) or vehicle. Thirty minutes after nicotine pretreatment, whole blood samples were stimulated with 10ng/ml LPS for 4h. TNF levels were determined using sandwich ELISA. Monocyte isolation. For the isolation of monocytes of the eleven healthy volunteers, 50ml of anti-coagulated blood was diluted with buffered saline (PBS). Aliquots (25ml) of this suspension were layered over an equal volume of Ficoll-Hypaque (GE.
Table 1: characteristics healthy subjectsCharacteristics of the eleven healthy volunteers that participated in the study considering gender, age and smoking. Four of the eleven healthy volunteers (subject 1-4) were regular cigarette smokers.
Gender Age Smoking
1 F 29 +2 F 30 +3 M 29 +4 M 33 +5 F 22 -6 M 27 -
7 M 26 -
8 F 25 -
9 M 29 -
10 F 31 -
11 M 30 -
van de Zanden.indb 115 23-5-2011 11:44:03

116
Chapter 7
Healthcare, Little Chalfont, U.K.) and gradients were subjected to centrifugation (800xg for 20min at RT). PBMC were washed three times in sterile PBS, after which they were counted. CD14+ cells were positively separated by high-gradient magnetic sorting using the MIDI-magnetic cell sorting (MACS) and Clini-MACS techniques according to the protocol provided by the manufacturer (Miltenyi Biotec, Bergisch Gladbach, Germany). In short, PBMC were resuspended in MACS buffer (0.5% BSA, 2mM EDTA in PBS) at a density of 12,5 × 107 cells/mL, CD14 MicroBeads (20 μl/107 cells) were added and the samples were kept at 4 °C for 15 min. Subsequently, cells were washed in 10 mL buffer by centrifugation (8 min, 300 ×g, 4 °C), resuspended in buffer (500µL/108 cells) and transferred to LS columns placed in the magnetic field of the MACS separator. CD14+ positive cells were selected by adhesion to the magnetized column, supernatant containing unselected cells was discarded, the labeled and positively enriched cells were eluted after removal of the columns from the magnetic device.
Statistical analysis. Statistical analysis was performed in Prism (Graphpad) version 4. The Friedman 2-way analysis of variance was used to explore multiple dependent value assays. If the Friedman analysis was significant, individual values compared with the saline were tested with a Mann-Whitney U test. P values < .05 were considered statistically significant, and results were expressed as mean ± SEM.
RESULTS
Nicotine significantly reduces TNF production in human monocytesTo determine whether nicotine reduced TNF production in human monocytes, we pre-incubated the human monocyte THP-I cell-line with 1 or 10 µM of nicotine, prior to 3hrs of LPS challenge (10ng/ml). In line with previous data28, nicotine caused a concentration dependent inhibition of the LPS-induced TNF production in THP-1 cells . At the highest concentration of 10µM nicotine, TNF production was significantly reduced from 100,0 ± 5.4% down to 68,3 ± 3,4% of vehicle (Fig. 1).
Nicotine priming induces CHRNA7 expression in human monocytes As is shown in figure 2A, THP-I cells express both CHRNA7 and CHRFAM7A although the expression if the CHRFAM7A is 7 fold higher than the CHRNA7 expression. CHRNA7 expression levels are low, but not undetectable, which is in contrast with earlier studies 29 that reported a total absence of CHRNA7 expression in THP-1 cells.
To relate the CHRNA7 transcriptional expression pattern with the anti-inflammatory effect of nicotine exposure in smokers, we next hypothesized that
van de Zanden.indb 116 23-5-2011 11:44:04
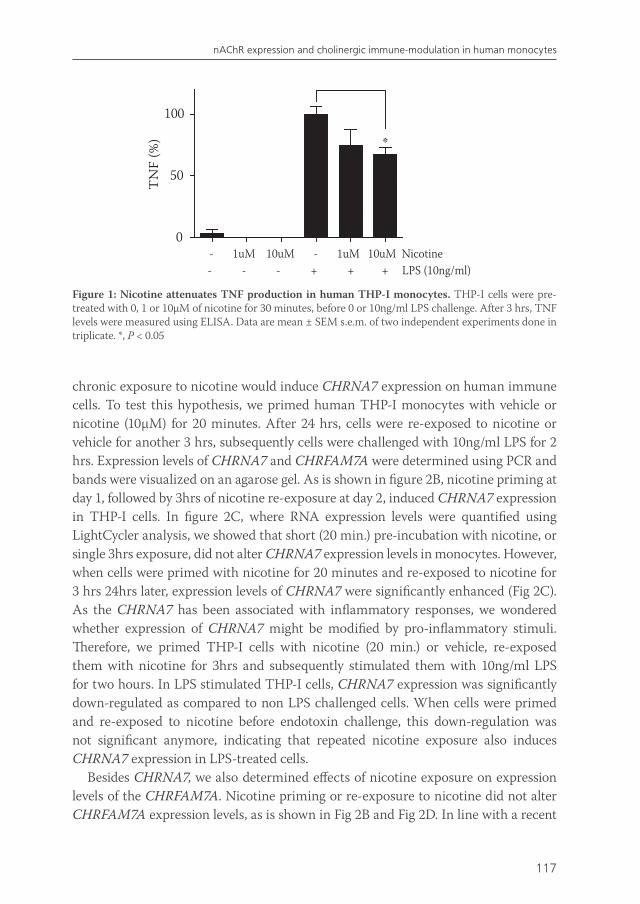
117
nAChR expression and cholinergic immune-modulation in human monocytes
chronic exposure to nicotine would induce CHRNA7 expression on human immune cells. To test this hypothesis, we primed human THP-I monocytes with vehicle or nicotine (10µM) for 20 minutes. After 24 hrs, cells were re-exposed to nicotine or vehicle for another 3 hrs, subsequently cells were challenged with 10ng/ml LPS for 2 hrs. Expression levels of CHRNA7 and CHRFAM7A were determined using PCR and bands were visualized on an agarose gel. As is shown in figure 2B, nicotine priming at day 1, followed by 3hrs of nicotine re-exposure at day 2, induced CHRNA7 expression in THP-I cells. In figure 2C, where RNA expression levels were quantified using LightCycler analysis, we showed that short (20 min.) pre-incubation with nicotine, or single 3hrs exposure, did not alter CHRNA7 expression levels in monocytes. However, when cells were primed with nicotine for 20 minutes and re-exposed to nicotine for 3 hrs 24hrs later, expression levels of CHRNA7 were significantly enhanced (Fig 2C). As the CHRNA7 has been associated with inflammatory responses, we wondered whether expression of CHRNA7 might be modified by pro-inflammatory stimuli. Therefore, we primed THP-I cells with nicotine (20 min.) or vehicle, re-exposed them with nicotine for 3hrs and subsequently stimulated them with 10ng/ml LPS for two hours. In LPS stimulated THP-I cells, CHRNA7 expression was significantly down-regulated as compared to non LPS challenged cells. When cells were primed and re-exposed to nicotine before endotoxin challenge, this down-regulation was not significant anymore, indicating that repeated nicotine exposure also induces CHRNA7 expression in LPS-treated cells.
Besides CHRNA7, we also determined effects of nicotine exposure on expression levels of the CHRFAM7A. Nicotine priming or re-exposure to nicotine did not alter CHRFAM7A expression levels, as is shown in Fig 2B and Fig 2D. In line with a recent
Figure 1: Nicotine attenuates TNF production in human THP-I monocytes. THP-I cells were pre-treated with 0, 1 or 10µM of nicotine for 30 minutes, before 0 or 10ng/ml LPS challenge. After 3 hrs, TNF levels were measured using ELISA. Data are mean ± SEM s.e.m. of two independent experiments done in triplicate. *, P < 0.05
0
50
100
- 1uM 10uM - 1uM 10uM Nicotine - - - + + + LPS (10ng/ml)
*T
NF
(%)
van de Zanden.indb 117 23-5-2011 11:44:04
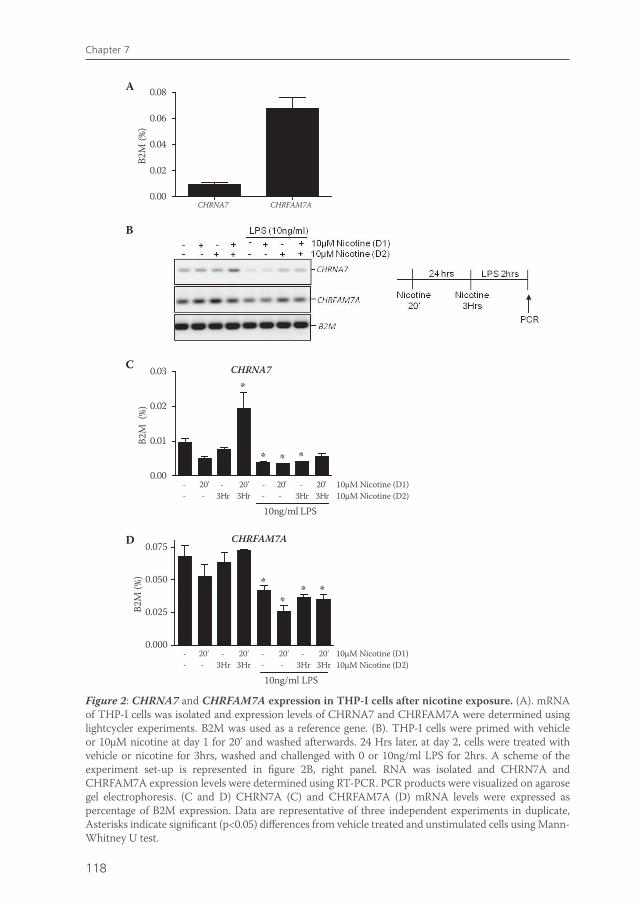
118
Chapter 7
Figure 2: CHRNA7 and CHRFAM7A expression in THP-I cells after nicotine exposure. (A). mRNA of THP-I cells was isolated and expression levels of CHRNA7 and CHRFAM7A were determined using lightcycler experiments. B2M was used as a reference gene. (B). THP-I cells were primed with vehicle or 10µM nicotine at day 1 for 20’ and washed afterwards. 24 Hrs later, at day 2, cells were treated with vehicle or nicotine for 3hrs, washed and challenged with 0 or 10ng/ml LPS for 2hrs. A scheme of the experiment set-up is represented in figure 2B, right panel. RNA was isolated and CHRN7A and CHRFAM7A expression levels were determined using RT-PCR. PCR products were visualized on agarose gel electrophoresis. (C and D) CHRN7A (C) and CHRFAM7A (D) mRNA levels were expressed as percentage of B2M expression. Data are representative of three independent experiments in duplicate, Asterisks indicate significant (p<0.05) differences from vehicle treated and unstimulated cells using Mann-Whitney U test.
0.000
0.025
0.050
0.075
- 20' - 20' - 20' - 20' 10µM Nicotine (D1) - - 3Hr 3Hr - - 3Hr 3Hr 10µM Nicotine (D2)
10ng/ml LPS
CHRFAM7A
**
* *
B2M
(%)
0.00
0.01
0.02
0.03 CHRNA7
*
* * *
- 20' - 20' - 20' - 20' 10µM Nicotine (D1) - - 3Hr 3Hr - - 3Hr 3Hr 10µM Nicotine (D2)
10ng/ml LPS
B2M
(%
)
CHRNA7 CHRFAM7A0.00
0.02
0.04
0.06
0.08
B2M
(%)
A
C
D
B
van de Zanden.indb 118 23-5-2011 11:44:05
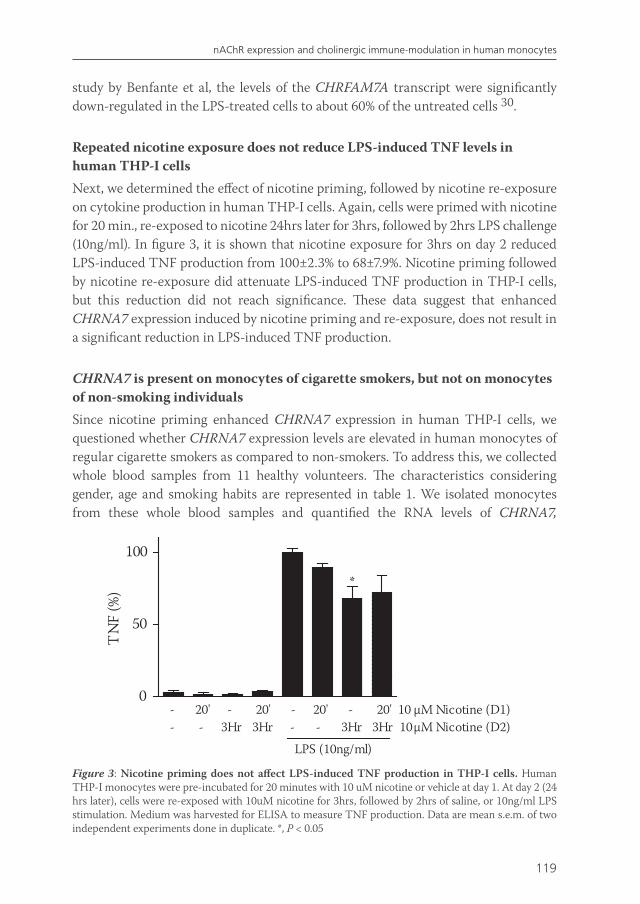
119
nAChR expression and cholinergic immune-modulation in human monocytes
study by Benfante et al, the levels of the CHRFAM7A transcript were significantly down-regulated in the LPS-treated cells to about 60% of the untreated cells 30.
Repeated nicotine exposure does not reduce LPS-induced TNF levels in human THP-I cells Next, we determined the effect of nicotine priming, followed by nicotine re-exposure on cytokine production in human THP-I cells. Again, cells were primed with nicotine for 20 min., re-exposed to nicotine 24hrs later for 3hrs, followed by 2hrs LPS challenge (10ng/ml). In figure 3, it is shown that nicotine exposure for 3hrs on day 2 reduced LPS-induced TNF production from 100±2.3% to 68±7.9%. Nicotine priming followed by nicotine re-exposure did attenuate LPS-induced TNF production in THP-I cells, but this reduction did not reach significance. These data suggest that enhanced CHRNA7 expression induced by nicotine priming and re-exposure, does not result in a significant reduction in LPS-induced TNF production.
CHRNA7 is present on monocytes of cigarette smokers, but not on monocytes of non-smoking individuals Since nicotine priming enhanced CHRNA7 expression in human THP-I cells, we questioned whether CHRNA7 expression levels are elevated in human monocytes of regular cigarette smokers as compared to non-smokers. To address this, we collected whole blood samples from 11 healthy volunteers. The characteristics considering gender, age and smoking habits are represented in table 1. We isolated monocytes from these whole blood samples and quantified the RNA levels of CHRNA7,
Figure 3: Nicotine priming does not affect LPS-induced TNF production in THP-I cells. Human THP-I monocytes were pre-incubated for 20 minutes with 10 uM nicotine or vehicle at day 1. At day 2 (24 hrs later), cells were re-exposed with 10uM nicotine for 3hrs, followed by 2hrs of saline, or 10ng/ml LPS stimulation. Medium was harvested for ELISA to measure TNF production. Data are mean s.e.m. of two independent experiments done in duplicate. *, P < 0.05
0
50
100
- 20' - 20' - 20' - 20' 10 µM Nicotine (D1) - - 3Hr 3Hr - - 3Hr 3Hr 10µM Nicotine (D2)
LPS (10ng/ml)
*
*
TN
F (%
)
van de Zanden.indb 119 23-5-2011 11:44:05
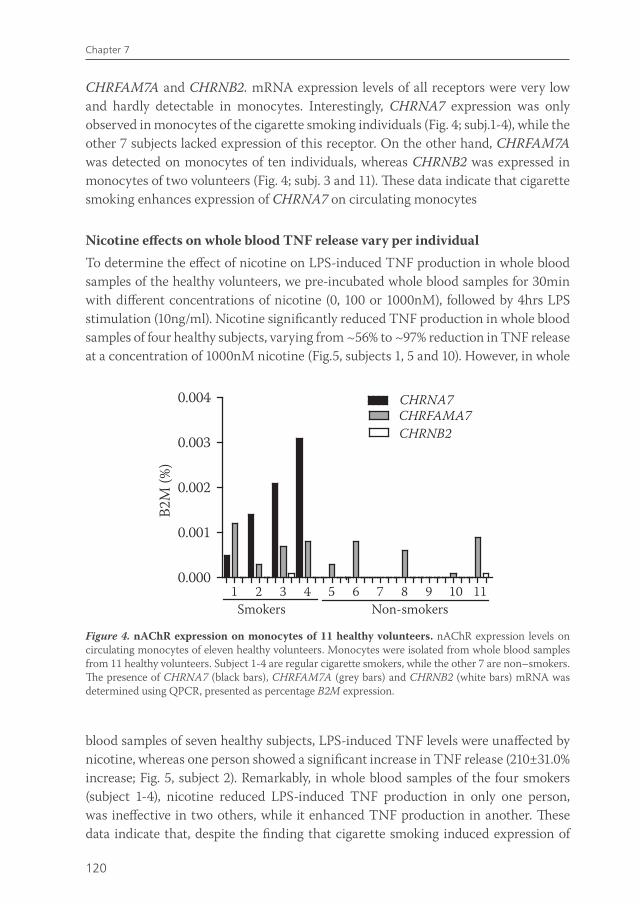
120
Chapter 7
CHRFAM7A and CHRNB2. mRNA expression levels of all receptors were very low and hardly detectable in monocytes. Interestingly, CHRNA7 expression was only observed in monocytes of the cigarette smoking individuals (Fig. 4; subj.1-4), while the other 7 subjects lacked expression of this receptor. On the other hand, CHRFAM7A was detected on monocytes of ten individuals, whereas CHRNB2 was expressed in monocytes of two volunteers (Fig. 4; subj. 3 and 11). These data indicate that cigarette smoking enhances expression of CHRNA7 on circulating monocytes
Nicotine effects on whole blood TNF release vary per individualTo determine the effect of nicotine on LPS-induced TNF production in whole blood samples of the healthy volunteers, we pre-incubated whole blood samples for 30min with different concentrations of nicotine (0, 100 or 1000nM), followed by 4hrs LPS stimulation (10ng/ml). Nicotine significantly reduced TNF production in whole blood samples of four healthy subjects, varying from ~56% to ~97% reduction in TNF release at a concentration of 1000nM nicotine (Fig.5, subjects 1, 5 and 10). However, in whole
Figure 4. nAChR expression on monocytes of 11 healthy volunteers. nAChR expression levels on circulating monocytes of eleven healthy volunteers. Monocytes were isolated from whole blood samples from 11 healthy volunteers. Subject 1-4 are regular cigarette smokers, while the other 7 are non–smokers. The presence of CHRNA7 (black bars), CHRFAM7A (grey bars) and CHRNB2 (white bars) mRNA was determined using QPCR, presented as percentage B2M expression.
blood samples of seven healthy subjects, LPS-induced TNF levels were unaffected by nicotine, whereas one person showed a significant increase in TNF release (210±31.0% increase; Fig. 5, subject 2). Remarkably, in whole blood samples of the four smokers (subject 1-4), nicotine reduced LPS-induced TNF production in only one person, was ineffective in two others, while it enhanced TNF production in another. These data indicate that, despite the finding that cigarette smoking induced expression of
1 2 3 4 5 6 7 8 9 10 110.000
0.001
0.002
0.003
0.004CHRFAMA7CHRNA7
CHRNB2
Smokers Non-smokers
B2M
(%)
van de Zanden.indb 120 23-5-2011 11:44:06
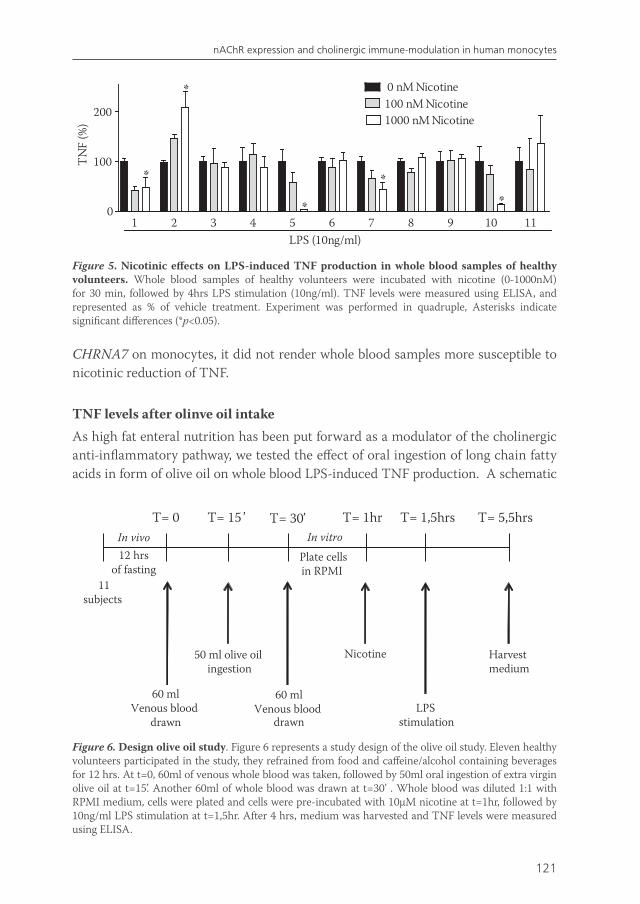
121
nAChR expression and cholinergic immune-modulation in human monocytes
CHRNA7 on monocytes, it did not render whole blood samples more susceptible to nicotinic reduction of TNF.
TNF levels after olinve oil intake As high fat enteral nutrition has been put forward as a modulator of the cholinergic anti-inflammatory pathway, we tested the effect of oral ingestion of long chain fatty acids in form of olive oil on whole blood LPS-induced TNF production. A schematic
Figure 5. Nicotinic effects on LPS-induced TNF production in whole blood samples of healthy volunteers. Whole blood samples of healthy volunteers were incubated with nicotine (0-1000nM) for 30 min, followed by 4hrs LPS stimulation (10ng/ml). TNF levels were measured using ELISA, and represented as % of vehicle treatment. Experiment was performed in quadruple, Asterisks indicate significant differences (*p<0.05).
1 2 3 4 5 6 7 8 9 10 110
100
200
*
*
* **
LPS (10ng/ml)
0 nM Nicotine100 nM Nicotine1000 nM Nicotine
TN
F (%
)
Figure 6. Design olive oil study. Figure 6 represents a study design of the olive oil study. Eleven healthy volunteers participated in the study, they refrained from food and caffeine/alcohol containing beverages for 12 hrs. At t=0, 60ml of venous whole blood was taken, followed by 50ml oral ingestion of extra virgin olive oil at t=15’. Another 60ml of whole blood was drawn at t=30’ . Whole blood was diluted 1:1 with RPMI medium, cells were plated and cells were pre-incubated with 10µM nicotine at t=1hr, followed by 10ng/ml LPS stimulation at t=1,5hr. After 4 hrs, medium was harvested and TNF levels were measured using ELISA.
T= 0
60 mlVenous blood
drawn
T= 15 ’
50 ml olive oil ingestion
T= 30’
12 hrsof fasting
11 subjects
Nicotine
T= 1hr
LPS stimulation
T= 1,5hrs T= 5,5hrs
Harvest medium
60 mlVenous blood
drawn
Plate cells in RPMI
In vitroIn vivo
van de Zanden.indb 121 23-5-2011 11:44:07
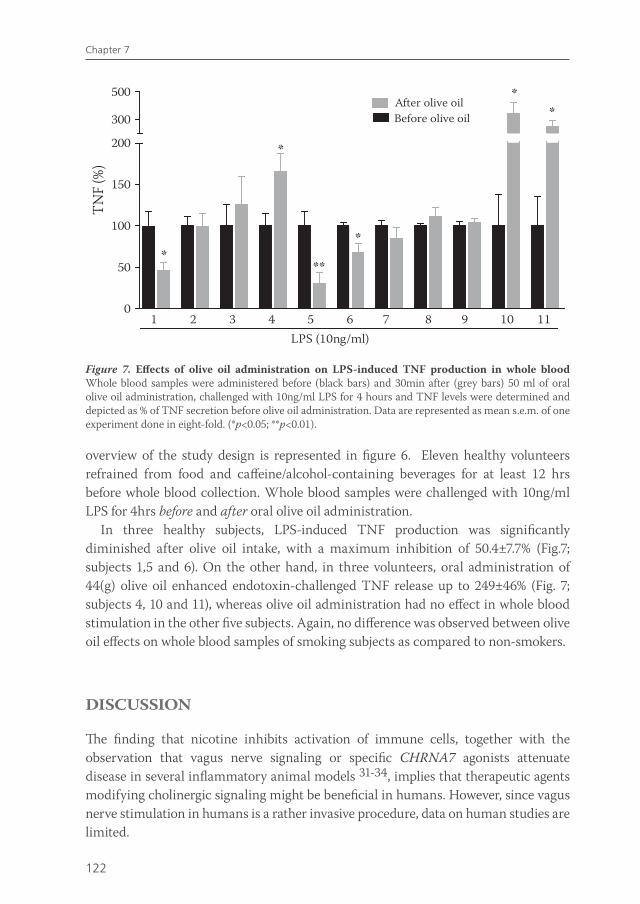
122
Chapter 7
overview of the study design is represented in figure 6. Eleven healthy volunteers refrained from food and caffeine/alcohol-containing beverages for at least 12 hrs before whole blood collection. Whole blood samples were challenged with 10ng/ml LPS for 4hrs before and after oral olive oil administration.
In three healthy subjects, LPS-induced TNF production was significantly diminished after olive oil intake, with a maximum inhibition of 50.4±7.7% (Fig.7; subjects 1,5 and 6). On the other hand, in three volunteers, oral administration of 44(g) olive oil enhanced endotoxin-challenged TNF release up to 249±46% (Fig. 7; subjects 4, 10 and 11), whereas olive oil administration had no effect in whole blood stimulation in the other five subjects. Again, no difference was observed between olive oil effects on whole blood samples of smoking subjects as compared to non-smokers.
DISCUSSION
The finding that nicotine inhibits activation of immune cells, together with the observation that vagus nerve signaling or specific CHRNA7 agonists attenuate disease in several inflammatory animal models 31-34, implies that therapeutic agents modifying cholinergic signaling might be beneficial in humans. However, since vagus nerve stimulation in humans is a rather invasive procedure, data on human studies are limited.
Figure 7. Effects of olive oil administration on LPS-induced TNF production in whole blood Whole blood samples were administered before (black bars) and 30min after (grey bars) 50 ml of oral olive oil administration, challenged with 10ng/ml LPS for 4 hours and TNF levels were determined and depicted as % of TNF secretion before olive oil administration. Data are represented as mean s.e.m. of one experiment done in eight-fold. (*p<0.05; **p<0.01).
0
50
100
150
200
300
500
**
*
**
**
1 2 3 4 5 6 7 8 9 10 11LPS (10ng/ml)
TNF
(%)
After olive oilBefore olive oil
van de Zanden.indb 122 23-5-2011 11:44:07

123
nAChR expression and cholinergic immune-modulation in human monocytes
Cigarette smoking is an important environmental factor in ulcerative colitis (UC). Ulcerative colitis patients with a history of smoking usually acquire their disease after they have stopped smoking 35-37 and patients who smoke intermittently often experience an improvement in their colitis symptoms during the periods when they smoke 38-40 . The reason behind the protective effect of smoking in UC remains obscure. The effects of smoking and nicotine are numerous and since the pathogenesis of inflammatory bowel disease is only partially understood, any discussion on the possible mechanisms can only be speculative. Potentially, nicotine in cigarette smoke can restrain the immune response in UC via activation of nicotinic acetylcholine receptors (nAChRs) on immune cells, which may explain its protective effect. Given the purported role of CHRNA7 in mediating the cholinergic anti-inflammatory pathway 41, we hypothesized that chronic nicotine exposure induced CHRNA7 expression on human monocytes.
In this study, we demonstrated that repeated nicotine exposure up-regulated CHRNA7 expression on THP-I monocytes. In conjunction, in a volunteer study, CHRNA7 was only detectable on monocytes of smoking individuals, while it was lacking on monocytes of non-smokers. These data suggest that enhanced CHRNA7 monocyte expression by repeated nicotine exposure could be important in the protective effect of smoking in UC. However, enhanced CHRNA7 expression did not render human monocytes more susceptible to nicotinic immune-modulation. Seemingly, the CHRNA7 is not the only mediator effectuating the cholinergic anti-inflammatory effect and another nAChR might be involved. For example, although it remains unclear whether the CHRFAM7A is appropriately translated and processed to a functional receptor, the finding that CHRFAM7A is down-regulated in monocytes by LPS challenge suggests that it could play a role in inflammation. Benfante et al speculated that receptors of mixed subunits of CHRNA7 and CHRFAM7A are formed that can modulate the signaling potential of immune cells to respond to ACh released from the vagus nerve during infection 42.
In human subjects, we failed to demonstrate a direct relation between CHRNA7 expression and TNF release. In this respect, it should be noted that alternative mechanisms may play a role vagal immuno-modulation in vivo. Recent data indicate for instance that the spleen may play a role in effectuating the anti-inflammatory effects of vagus nerve activity, as electrical vagus nerve stimulation fails to attenuate serum TNF levels in splenectomized mice treated with endotoxin 43. The authors propose that ACh released by the vagus nerve does not reach the spleen directly, but acts on CHRNA7 at the level of the ganglia of the celiac-superior mesenteric plexus to modulate splenic nerve function. They suggest that attenuation of TNF production by spleen macrophages induced by vagus nerve stimulation is mediated by norepinephrine released from splenic nerve endings 44 . These data put the role of the CHRNA7 in a new perspective; although in vitro ACh modulates macrophage function via CHRNA7, the in vivo effects of vagus nerve stimulation may rely on CHRNA7 on neurons, rather
van de Zanden.indb 123 23-5-2011 11:44:07

124
Chapter 7
than on monocytes or macrophages. This implies that, although nAChR expression on circulating monocytes in humans is rather low, cholinergic activation could still display in vivo immuno-modulating functions via neuronal nAChRs 45.
We tested the effect of nicotine pretreatment on TNF production by endotoxin-stimulated whole blood samples of 11 healthy volunteers. We choose to work with whole blood samples since this was the closest to the in vivo situation, and cytokines produced from whole-blood were found to be strongly correlated with monocytic cytokines 46. Nicotine dose-dependently reduced TNF production in 4 of 11 human whole blood samples, whereas in one sample, TNF release was enhanced. Other studies report that in other cell types, such as peripheral blood derived human macrophages 47 and mononuclear cells 48 , nicotine reduced TNF production in all samples. On the other hand, Kox et al 49 published that nicotine attenuated TNF release equally on PBMCs, monocytes and human whole blood samples, but only at a high dosage of 1 mM. These differences may be due to differential nAChR expression, since it is demonstrated that nAChR expression on human leukocytes may vary due to genetic or environmental factors 50,,51. Indeed, we observed that CHRNA7, CHRFAM7A and CHRNB2 expression varied between individuals. However, expression of all nAChRs tested was extremely low and hard to detect. Moreover, we found no correlation between nAChR expression levels and nicotine effects.
Activating the so called ‘cholinergic anti-inflammatory pathway’ via electrical vagus nerve stimulation, or the use of applied cholinergic agonists targeting distinct nAChR subtypes, could be a potential therapeutic asset in the treatment of inflammatory disorders in human. However, more physiological applied high-fat enteral nutrition can activate this ‘cholinergic anti-inflammatory pathway’ as well. High-fat enteral nutrition, sensed in the gastrointestinal tract, activates the parasympathetic nervous system, and leads to inhibition of the inflammatory response by way of efferent vagal fibers 52. In addition, enteral nutrition with long chain fatty acids in form of olive oil suppresses LPS-induced TNF release of macrophages in the gut wall in mice (Jacob et al; DDW2009; 1013). Moreover, extra virgin olive oil enriched diet (10%) showed protective effects in experimental mouse models of DSS-colitis and endotoxic shock 53,54. In our study, olive oil administration reduced LPS-induced TNF production in whole blood samples of four healthy subjects, while it enhanced TNF production in three other volunteers. However, although it is presumed that olive oil stimulates the vagus nerve, we did not have a proper read out for vagus nerve activity in our study. There is evidence that exposure to fat for only three minutes is sufficient to elicit a pancreatic polypeptide response 55. Secretion of pancreatic polypeptide, a polypeptide secreted by pancreatic peptide cells in the endocrine pancreas, is regulated by cholinergic stimulation 56. In the future, measurement of pancreatic polypeptide serum levels would be indicative of vagus nerve activity. On the other hand, it is questionable whether acetylcholine released from vagus nerve termini upon olive oil administration, actually reaches immune cells in the blood. However, olive oil
van de Zanden.indb 124 23-5-2011 11:44:07

125
nAChR expression and cholinergic immune-modulation in human monocytes
administration did alter TNF production in whole blood samples of seven volunteers, indicating that olive oil can modulate the immune response in whole blood. If this is indeed vagus nerve mediated, and what causes the individual variation, needs to be further established.
In conclusion, we demonstrated that chronic exposure to nicotine enhances CHRNA7 expression on circulating monocytes in humans, in vitro and in vivo. However, this CHRNA7 upregulation does not render human monocytes more susceptible to cholinergic immune-modulation. Results obtained in a wide range of in vitro and in vivo models of inflammation imply that therapeutic agents targeting the cholinergic anti-inflammatory pathway can be an important asset in the treatment of immune disorders in human. Our data indicate that there might be an individual variation in response to future therapeutic agents modifying the cholinergic anti-inflammatory pathway in humans.
van de Zanden.indb 125 23-5-2011 11:44:07

126
Chapter 7
REFERENCE LIST
1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
2. van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, Tracey KJ, van der PT. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006;130:1822-1830.
3. The FO, Boeckxstaens GE, Snoek SA, Cash JL, Bennink R, Larosa GJ, van den Wijngaard RM, Greaves DR, De Jonge WJ. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 2007;133:1219-1228.
4. van Maanen MA, Lebre MC, van der PT, Larosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-122.
5. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 2007.
6. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999;266:17-20.
7. van der Zanden EP, Snoek SA, Heinsbroek SE, Stanisor OI, Verseijden C, Boeckxstaens GE, Peppelenbosch MP, Greaves DR, Gordon S, De Jonge WJ. Vagus Nerve Activity Augments Intestinal Macrophage Phagocytosis via Nicotinic Acetylcholine Receptor alpha4beta2. Gastroenterology 2009;137:1029-1039.
8. Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdes-Ferrer SI, Patel NB, Chavan S, Al Abed Y, Yang H, Tracey KJ. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med 2009;15:195-202.
9. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
10. Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese C, Freedman R, Leonard S. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics 1998;52:173-185.
11. Benfante R, Antonini RA, De PM, Gotti C, Clementi F, Locati M, Fornasari D. Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol 2011;230:74-84.
12. Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA. Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol 2002;126:86-98.
13. Benfante R, Antonini RA, De PM, Gotti C, Clementi F, Locati M, Fornasari D. Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol 2011;230:74-84.
14. Birrenbach T, Bocker U. Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm Bowel Dis 2004;10:848-859.
15. Ingram JR, Rhodes J, Collins PW, Williams GT, Newcombe RG, Thomas GA. Plasma fibrinogen in ulcerative colitis: the effect of disease activity and nicotine therapy in a randomised controlled trial. Dig Liver Dis 2005;37:832-837.
van de Zanden.indb 126 23-5-2011 11:44:07

127
nAChR expression and cholinergic immune-modulation in human monocytes
16. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA,. Transdermal nicotine for active ulcerative colitis. N Engl J Med 1994;330:811-815.
17. Thomas GA, Rhodes J, Green JT, Richardson C. Role of smoking in inflammatory bowel disease: implications for therapy. Postgrad Med J 2000;76:273-279.
18. Sandborn WJ. Nicotine therapy for ulcerative colitis: a review of rationale, mechanisms, pharmacology, and clinical results. Am J Gastroenterol 1999;94:1161-1171.
19. van der HF, Dijkstra A, Weersma RK, Albersnagel FA, van der Logt EM, Faber KN, Sluiter WJ, Kleibeuker JH, Dijkstra G. Effects of active and passive smoking on disease course of Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 2009;15:1199-1207.
20. McGrath J, McDonald JW, Macdonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev 2004;CD004722.
21. Wongsriraksa A, Parsons ME, Whelan CJ. Characterisation of nicotine receptors on human peripheral blood mononuclear cells (PBMC). Inflamm Res 2009;58:38-44.
22. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999;266:17-20.
23. De Rosa MJ, Esandi MC, Garelli A, Rayes D, Bouzat C. Relationship between alpha 7 nAChR and apoptosis in human lymphocytes. J Neuroimmunol 2005;160:154-161.
24. Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol 2000;61:75-111.
25. Perry DC, vila-Garcia MI, Stockmeier CA, Kellar KJ. Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther 1999;289:1545-1552.
26. Sykes AP, Brampton C, Klee S, Chander CL, Whelan C, Parsons ME. An investigation into the effect and mechanisms of action of nicotine in inflammatory bowel disease. Inflamm Res 2000;49:311-319.
27. Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 2005;202:1023-1029.
28. Sykes AP, Brampton C, Klee S, Chander CL, Whelan C, Parsons ME. An investigation into the effect and mechanisms of action of nicotine in inflammatory bowel disease. Inflamm Res 2000;49:311-319.
29. Benfante R, Antonini RA, De PM, Gotti C, Clementi F, Locati M, Fornasari D. Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol 2011;230:74-84.
30. Benfante R, Antonini RA, De PM, Gotti C, Clementi F, Locati M, Fornasari D. Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol 2011;230:74-84.
31. The FO, Boeckxstaens GE, Snoek SA, Cash JL, Bennink R, Larosa GJ, van den Wijngaard RM, Greaves DR, De Jonge WJ. Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 2007;133:1219-1228.
32. van Maanen MA, Lebre MC, van der PT, Larosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-122.
33. van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, Tracey KJ, van der PT. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006;130:1822-1830.
34. De Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 2005;6:844-851.
van de Zanden.indb 127 23-5-2011 11:44:07

128
Chapter 7
35. Ingram JR, Rhodes J, Collins PW, Williams GT, Newcombe RG, Thomas GA. Plasma fibrinogen in ulcerative colitis: the effect of disease activity and nicotine therapy in a randomised controlled trial. Dig Liver Dis 2005;37:832-837.
36. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA, . Transdermal nicotine for active ulcerative colitis. N Engl J Med 1994;330:811-815.
37. Thomas GA, Rhodes J, Green JT, Richardson C. Role of smoking in inflammatory bowel disease: implications for therapy. Postgrad Med J 2000;76:273-279.
38. Birrenbach T, Bocker U. Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm Bowel Dis 2004;10:848-859.
39. Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA, . Transdermal nicotine for active ulcerative colitis. N Engl J Med 1994;330:811-815.
40. Sandborn WJ. Nicotine therapy for ulcerative colitis: a review of rationale, mechanisms, pharmacology, and clinical results. Am J Gastroenterol 1999;94:1161-1171.
41. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
42. Benfante R, Antonini RA, De PM, Gotti C, Clementi F, Locati M, Fornasari D. Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol 2011;230:74-84.
43. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006;203:1623-1628.
44. Vida G, Pena G, Deitch EA, Ulloa L. {alpha}7-Cholinergic Receptor Mediates Vagal Induction of Splenic Norepinephrine. J Immunol 2011;186:4340-4346.
45. Vida G, Pena G, Deitch EA, Ulloa L. {alpha}7-Cholinergic Receptor Mediates Vagal Induction of Splenic Norepinephrine. J Immunol 2011;186:4340-4346.
46. Damsgaard CT, Lauritzen L, Calder PC, Kjaer TM, Frokiaer H. Whole-blood culture is a valid low-cost method to measure monocytic cytokines - a comparison of cytokine production in cultures of human whole-blood, mononuclear cells and monocytes. J Immunol Methods 2009;340:95-101.
47. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
48. Hamano R, Takahashi HK, Iwagaki H, Yoshino T, Nishibori M, Tanaka N. Stimulation of alpha7 nicotinic acetylcholine receptor inhibits CD14 and the toll-like receptor 4 expression in human monocytes. Shock 2006;26:358-364.
49. Kox M, van Velzen JF, Pompe JC, Hoedemaekers CW, van der Hoeven JG, Pickkers P. GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem Pharmacol 2009.
50. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 2007.
51. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999;266:17-20.
52. Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 2005;202:1023-1029.
van de Zanden.indb 128 23-5-2011 11:44:07

129
nAChR expression and cholinergic immune-modulation in human monocytes
53. Sanchez-Fidalgo S, Villegas I, Cardeno A, Talero E, Sanchez-Hidalgo M, Motilva V, arcon de la LC. Extra-virgin olive oil-enriched diet modulates DSS-colitis-associated colon carcinogenesis in mice. Clin Nutr 2010;29:663-673.
54. Leite MS, Pacheco P, Gomes RN, Guedes AT, Castro-Faria-Neto HC, Bozza PT, Koatz VL. Mechanisms of increased survival after lipopolysaccharide-induced endotoxic shock in mice consuming olive oil-enriched diet. Shock 2005;23:173-178.
55. Simonian HP, Kresge KM, Boden GH, Parkman HP. Differential effects of sham feeding and meal ingestion on ghrelin and pancreatic polypeptide levels: evidence for vagal efferent stimulation mediating ghrelin release. Neurogastroenterol Motil 2005;17:348-354.
56. Schwartz TW. Pancreatic polypeptide: a hormone under vagal control. Gastroenterology 1983;85:1411-1425.
van de Zanden.indb 129 23-5-2011 11:44:08

8CHAPTER
Summary and conclusions
van de Zanden.indb 131 23-5-2011 11:44:08

133
Summary and conclusions
The goal of this thesis was to further unravel the working mechanism of the so-called ‘cholinergic anti-inflammatory pathway’, specifically in intestinal inflammation. The pioneering work of Tracey and colleagues has demonstrated that the vagus nerve, via its neurotransmitter acetylcholine (ACh), has an important role in limiting the inflammatory response1. Subsequently, several studies have demonstrated that cholinergic activation ameliorates disease in a range of animal models such as ischaemia-reperfusion injury2, hemorrhagic shock3, peritonitis4, DSS-colitis5 and rheumatoid arthritis6.
In this thesis, we focused on the role of cholinergic modulation in intestinal inflammation. The gastro-intestinal tract is under strict control of the vagus nerve7,8, rendering it susceptible to cholinergic immuno-modulation. Moreover, especially in the intestine, which homes our largest collection of microbes, tight regulation of immune responses to discriminate between self and non-self is crucial. An imbalance of this process has consequences and may lead to disease. An example of this is inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis (UC), which is caused by an inappropriate and exaggerated mucosal immune response to constituents of the gut flora in genetically predisposed individuals. Chapter 2 gives an overview of the current knowledge regarding cholinergic modulation of intestinal inflammation. This review discusses advances in the possible mechanisms via which the vagus nerve can mediate the immune response, as well as the role of nAChR activation and signaling on macrophages and other immune cells. Moreover, the clinical implications of the anti-inflammatory properties of vagus nerve signaling are discussed.
JAK2-STAT3 signaling in cholinergic modulation of the immune response
The ‘cholinergic anti-inflammatory pathway’ is characterized by a dose-dependent decrease in the production of proinflammatory mediators via activation of nicotinic acetylcholine receptors (nAChR) on macrophages1 . Many reports point towards the macrophage nAChR α7 as an essential player in mediating the anti-inflammatory effect of ACh9, 10-12. Wang et al showed that activation of the nAChR α7 inhibits transcriptional activity of the transcription factor NF-KBp659,13, resulting in reduced cytokine production. In chapter 3, we evaluated the involvement of the transcription factor STAT3 in this process, since STAT3 is a potential negative regulator of inflammatory responses14,15. We demonstrated that nicotine exerts its anti-inflammatory effect on peritoneal macrophages via nAChR α7-Jak2 and STAT3 signaling in vitro. In vivo, we tested the involvement of STAT3 in the cholinergic anti-inflammatory pathway in an animal model of postoperative ileus (POI). POI is characterized by general hypomotility of the gastrointestinal tract and delayed gastric
van de Zanden.indb 133 23-5-2011 11:44:08

134
Chapter 8
emptying and is a pathological condition commonly noted after abdominal surgery16. This condition is the result of inflammation of the intestinal muscularis layer due to activation of resident macrophages that are triggered by bowel manipulation17. In an animal model, postoperative ileus was improved substantially by stimulation of the vagus nerve. Moreover, we found activation of STAT3 in intestinal macrophages in response to stimulation of the vagus nerve, which indicates activation of STAT3 induced by acetylcholine derived from vagal efferents. The finding that macrophage activation is under strict neuronal control may be substantiated by the observation that cholinergic nerve fibers are in close proximity to resident macrophages in intestinal myenteric plexus.
In conclusion, in chapter 3, we have shown that inhibition of macrophage activity via the cholinergic anti-inflammatory pathway is brought about via Jak2-STAT3 signaling. We speculated that nicotine repressed macrophage activity via direct interaction of dimerized STAT3 with the p65 subunit18.
In chapter 4, the role of the JAK2-STAT3 pathway in the anti-inflammatory effect of nAChR activation was further analyzed. We studied the potential of STAT3 to modulate TNF responses using STAT silencing strategies, pharmacological blockade, and dominant negative STAT3 constructs. Two dominant negative STAT3 constructs were used: STAT3D, which prevents STAT3 binding to DNA, and STAT3F, which prevents STAT3 phosphorylation and subsequent dimerization19.
Nicotine reduced TNF production in RAW cells transfected with STAT3F, but failed to do so in cells transfected with STAT3D. Hence, in contrast to our findings in chapter 3, we demonstrated that nicotinic inhibition of inflammation in macrophages is dependent on STAT3 DNA binding and STAT3 protein, rather than STAT3 phosphorylation. Hypothetically, unphosphorylated STAT3 (U-STAT) can be important in mediating the anti-inflammatory effect of nAChR activation, via binding to NF-kB20 and inhibition of NF-kB-activation of TNF transcription.
In conclusion, chapter 3 and 4 identify a novel molecular pathway involved in the vagal modulation of macrophage activity, and indicate that JAK2-STAT3 targeting may aid in further development of therapeutic strategies to modify the ‘cholinergic anti-inflammatory pathway’.
Cholinergic modulation of phagocytosis
Cholinergic inhibition of pro-inflammatory cytokine production by macrophages has been firmly established. However, especially in the intestinal compartment, macrophages may rather function as phagocytes that, along with dendritic cells, form critical effectors in the surveillance of luminal antigens. Therefore, it may be questioned if the anti-inflammatory effect of vagus nerve activity in intestinal inflammation exclusively rests on reduced macrophage cytokine production, or
van de Zanden.indb 134 23-5-2011 11:44:08

135
Summary and conclusions
whether the vagus nerve also regulates other macrophage functions important in host defense. In chapter 5, we explored the effect of nAChR activation on more professional macrophage functions, such as endo- and phagocytosis by macrophages residing in the peritoneal and mucosal compartment. We demonstrated that nAChR activation enhanced endocytosis and phagocytosis in intestinal and peritoneal macrophages. This effect was mediated via enhanced recruitment of dynamin-221 to the phagocytic cup. The anti-inflammatory effects of nAChR activation on macrophages have previously been attributed to activation of the nAChRα79. Interestingly, we clearly showed that cholinergic agonists induced phagocytosis via nAChR α4β2, rather than the α7 nAChR. Despite enhanced phagocytosis, acetylcholine reduced NF-kB activation and pro-inflammatory cytokine production, while stimulating anti-inflammatory IL10 production. In vivo, vagus nerve stimulation enhanced luminal uptake by intestinal phagocytes. Moreover, nAChR activation in intestinal tissue induced a transiently enhanced mucosal passage of luminal bacteria. In line, stimulation of vagus activity enhanced mucosal uptake and drainage of luminal bacteria.
In conclusion, in chapter 5 we show that acetylcholine has a dual effect in macrophages, it stimulates phagocytosis via nAChR α4β2 activation, while reducing NF-κB activation and inflammatory cytokine production. Vagus nerve activity induces a transient increase in epithelial permeability and augments the uptake of luminal bacteria by mucosal macrophages. That way, vagus nerve activity assists in surveillance in the intestinal mucosa and peritoneal compartment.
Vagus nerve modulation of immune cells in vivo
In chapter 6, we tried to analyze how vagus nerve activity can modulate immune cells in vivo. We examined whether vagus activation restrains the immune response not only via the release of acetylcholine, but also via alternative neurotransmitters such as neuropeptides, released via post-ganglionic mechanisms. VIP and substance P are neuropeptides that are abundantly expressed in the gut and display important immunomodulatory functions. Vagus nerve stimulation altered expression levels of VIP and SP in mouse intestinal tissue. Co-administration of acetylcholine and VIP decreased LPS-induced production almost down to un-stimulated levels. In addition, substance P enhanced NF-kB transcriptional activity and TNF production in peritoneal macrophages, but when Substance P and acetylcholine were applied together, the pro-inflammatory actions of substance P were neutralized by acetylcholine. The immunomodulatory effects of ACh, nicotine, VIP and SP were independent of calcium signaling pathways.
In summary, VNS modulates VIP and SP expression in the intestine, and in vitro, cholinergic agonists can affect VIP and SP immuno-modulatory actions. These data
van de Zanden.indb 135 23-5-2011 11:44:08
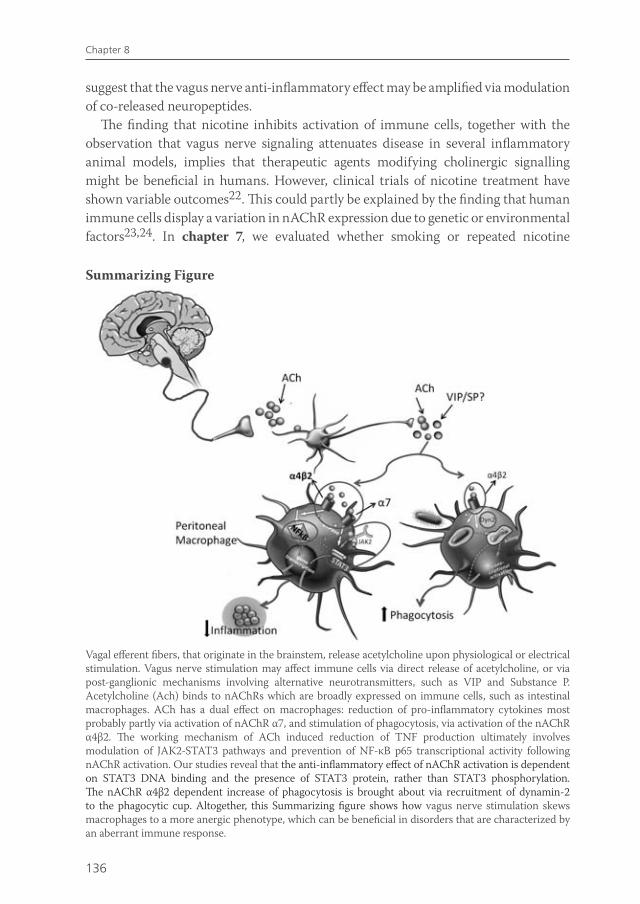
136
Chapter 8
suggest that the vagus nerve anti-inflammatory effect may be amplified via modulation of co-released neuropeptides.
The finding that nicotine inhibits activation of immune cells, together with the observation that vagus nerve signaling attenuates disease in several inflammatory animal models, implies that therapeutic agents modifying cholinergic signalling might be beneficial in humans. However, clinical trials of nicotine treatment have shown variable outcomes22. This could partly be explained by the finding that human immune cells display a variation in nAChR expression due to genetic or environmental factors23,24. In chapter 7, we evaluated whether smoking or repeated nicotine
Vagal efferent fibers, that originate in the brainstem, release acetylcholine upon physiological or electrical stimulation. Vagus nerve stimulation may affect immune cells via direct release of acetylcholine, or via post-ganglionic mechanisms involving alternative neurotransmitters, such as VIP and Substance P. Acetylcholine (Ach) binds to nAChRs which are broadly expressed on immune cells, such as intestinal macrophages. ACh has a dual effect on macrophages: reduction of pro-inflammatory cytokines most probably partly via activation of nAChR α7, and stimulation of phagocytosis, via activation of the nAChR α4β2. The working mechanism of ACh induced reduction of TNF production ultimately involves modulation of JAK2-STAT3 pathways and prevention of NF-κB p65 transcriptional activity following nAChR activation. Our studies reveal that the anti-inflammatory effect of nAChR activation is dependent on STAT3 DNA binding and the presence of STAT3 protein, rather than STAT3 phosphorylation. The nAChR α4β2 dependent increase of phagocytosis is brought about via recruitment of dynamin-2 to the phagocytic cup. Altogether, this Summarizing figure shows how vagus nerve stimulation skews macrophages to a more anergic phenotype, which can be beneficial in disorders that are characterized by an aberrant immune response.
Summarizing Figure
van de Zanden.indb 136 23-5-2011 11:44:08

137
Summary and conclusions
exposure could affect expression of nAChRα7, dupα7 or β2 on human monocytes. We demonstrated that repeated nicotine exposure up-regulated nAChRα7 expression in a human monocyte cell line. In conjunction, in a pilot study, nAChRα7 was only detectable in monocytes of smoking individuals. nAChR dupα7 was ubiquitously expressed on human monocytes, while nAChRβ2 was not detectable. However, the nitotine induced up-regulation of nAChR α7 in human monocytes did not render cells more susceptible to cholinergic immune-modulation, either via nicotine application or via administration of olive oil.
The Summarizing Figure is a model of the mechanism via which vagus nerve activity modulates intestinal macrophage function, according to the results obtained in this thesis.
Therapeutic options and future perspectives Results obtained in a wide range of in vitro and in vivo models of inflammation imply that therapeutic agents targeting the ‘cholinergic anti-inflammatory pathway’ can be an important asset in the treatment of immune disorders in human. In vivo, cholinergic activation can be accomplished in several ways. The best known, although not the most selective way, is by cigarette smoking.
In inflammatory bowel disease, cigarette smoking is an important environmental factor, but has differential effects in ulcerative colitis (UC) and Crohn’s disease (CD). While smoking increases the risk of developing CD, it appears to have a protective effect in the development of UC and reduces its severity25. However, clinical trials using nicotine for the treatment of UC have showed no significant advantage for transdermal nicotine therapy compared to standard therapy, while nicotine did show more side effects22 . Therefore, the challenge is to define a specific nAChR agonist with highest anti-inflammatory potential and least side effects. Partial selective nAChR α7 and α4β2 agonists are already being tested in patients with neuronal disorders, since both receptor subtypes have shown to mediate improvement in attention, learning and working memory26. The most characterized nAChR-agonist is GTS-21, a partial α7 nAChR agonist that also affects α4β2 nAChR27, is well tolerated humans. In vitro, this agonist has shown to diminish production of pro-inflammatory mediators in mouse and human immune cells28,29. In a recent study on the effects of GTS-21 on the innate response during human endotoxemia in 14 non-smoking individuals, there were no differences in the LPS-induced cytokine response between the GTS-21 and placebo-treated groups30.
Targeting the nAChRs using specific agonists, requires exact knowledge of which nAChR is involved in the anti-inflammatory effects of cholinergic activation. These effects have previously been only attributed to activation of the nAChR α79. Nevertheless, nAChRα7 was not present on monocytes of non-smoking individuals (chapter 7), in conjunction we failed to detect α7 nAChR transcripts in certain mouse
van de Zanden.indb 137 23-5-2011 11:44:08

138
Chapter 8
macrophage types (chapter 5). Further analysis of potential α7 nAChR protein in these macrophages is hampered by the fact that commercially available α7 nAChR antisera seem not specific and stain an 57kD protein in brain homogenates from wildtype as well as α7 nAChR -/- mice31. In line, nicotine reduced TNF production in α7 WT, as well as in α7 KO mice (chapter 4). Accordingly, we observed that α7 specific agonists were less effective in reducing pro-inflammatory cytokine production as compared to nicotine (chapter 5). Altogether, these observations imply that the acetylcholine and nicotine effects on peritoneal macrophage cytokine production should not be exclusively attributed to nAChR α7 activation, but maybe also due to activation of alternative nAChR subtypes. Therefore, the use of selective agonists targeting other nAChRs than the α7 receptor could be successful, especially specific agonists of the nAChRα4β2, which is required for cholinergic activation of phagocytosis.
In addition to the use of specific cholinergic agonists, vagus nerve stimulation itself could be a potential therapeutic asset in the treatment of patients with inflammatory diseases. Interestingly, in patients with drug-resistant epilepsy and depression, vagus nerve stimulation is already in use as a new adjunctive therapy. Furthermore, high-fat enteral nutrition, sensed in the gastrointestinal tract, has shown to activate the parasympathetic nervous system. However, as the vagus nerve does not innervate the distal colon and rectum, the areas usually affected in IBD patients, vagus nerve stimulation may not be the first therapeutic choice in targeting IBD. Nevertheless, vagus nerve activity can regulate disease in animal models5, possibly clarified by the role of the spleen in exerting the anti-inflammatory effect of vagus nerve signaling32,33.
The finding that nicotine inhibits activation of immune cells, together with the observation that vagus nerve signaling or specific nAChR agonists attenuate disease in several inflammatory animal models, implies that therapeutic agents modifying cholinergic signalling might be beneficial in humans. However, one should keep in mind that targeting the cholinergic anti-inflammatory pathway in humans could be less straightforward than originally thought, as there might be an individual variation in response to future therapeutic agents modifying the cholinergic anti-inflammatory pathway in humans.
Overall, in this thesis, we further identified the working mechanism of the ‘cholinergic anti-inflammatory pathway ’ to alleviate specific targeting of this pathway, in order to increase the translational potential.
Our data may aid in the development of therapeutic strategies to modify the cholinergic anti-inflammatory pathway, in order to treat various inflammatory conditions.
van de Zanden.indb 138 23-5-2011 11:44:08

139
Summary and conclusions
REFERENCE LIST
1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
2. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, Tracey KJ. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg 2002;36:1231-1236.
3. Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, Squadrito G, Minutoli L, Bertolini A, Marini R, Adamo EB, Venuti FS, Squadrito F. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation 2003;107:1189-1194.
4. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der PT. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis 2005;191:2138-2148.
5. Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006;131:1122-1130.
6. van Maanen MA, Lebre MC, van der Poll T, Larosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-122.
7. Altschuler SM, Ferenci DA, Lynn RB, Miselis RR. Representation of the cecum in the lateral dorsal motor nucleus of the vagus nerve and commissural subnucleus of the nucleus tractus solitarii in rat. J Comp Neurol 1991;304:261-274.
8. Altschuler SM, Escardo J, Lynn RB, Miselis RR. The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology 1993;104:502-509.
9. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
10. Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, Levi J, Parrish WR, Rosas-Ballina M, Czura CJ, Larosa GJ, Miller EJ, Tracey KJ, Al Abed Y. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 2007;35:1139-1144.
11. Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, Hudson L, Lin X, Patel N, Johnson SM, Chavan S, Goldstein RS, Czura CJ, Miller EJ, Al Abed Y, Tracey KJ, Pavlov VA. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med 2008;14:567-574.
12. De Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 2007;151:915-929.
13. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004;10:1216-1221.
14. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999;10:39-49.
15. Levy DE, Lee CK. What does Stat3 do? J Clin Invest 2002;109:1143-1148.
16. Prasad M, Matthews JB. Deflating postoperative ileus. Gastroenterology 1999;117:489-492.
17. De Jonge WJ, van den Wijngaard RM, The FO, ter Beek ML, Bennink RJ, Tytgat GN, Buijs RM, Reitsma PH, van Deventer SJ, Boeckxstaens GE. Postoperative ileus is maintained by intestinal immune infiltrates that activate inhibitory neural pathways in mice. Gastroenterology 2003;125:1137-1147.
van de Zanden.indb 139 23-5-2011 11:44:08

140
Chapter 8
18. Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 2002;367:97-105.
19. de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, Lowenberg B, Touw IP. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 2000;19:3290-3298.
20. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 2007;21:1396-1408.
21. Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 1999;190:1849-1856.
22. McGrath J, McDonald JW, Macdonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev 2004;CD004722.
23. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 2007.
24. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999;266:17-20.
25. Lakatos PL, Szamosi T, Lakatos L. Smoking in inflammatory bowel diseases: good, bad or ugly? World J Gastroenterol 2007;13:6134-6139.
26. Cincotta SL, Yorek MS, Moschak TM, Lewis SR, Rodefer JS. Selective nicotinic acetylcholine receptor agonists: potential therapies for neuropsychiatric disorders with cognitive dysfunction. Curr Opin Investig Drugs 2008;9:47-56.
27. Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, Burnett AL. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology 2003;28:542-551.
28. Giebelen IA, van Westerloo DJ, Larosa GJ, de Vos AF, van der PT. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock 2007;28:700-703.
29. Kox M, van Velzen JF, Pompe JC, Hoedemaekers CW, van der Hoeven JG, Pickkers P. GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem Pharmacol 2009.
30. Kox M, Pompe JC, Gordinou de Gouberville MC, van der Hoeven JG, Hoedemaekers CW, Pickkers P. Effects of the alphaalpha7nAChR Agonist GTS-21 on the Innate Immune Response in Humans. Shock 2011.
31. Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De BM, Schroder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem 2007;102:479-492.
32. Ghia JE, Blennerhassett P, Collins SM. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J Clin Invest 2008;118:2209-2218.
33. Mouzas IA, Pallis AG, Kochiadakis GE, Marketou M, Chlouverakis GI, Mellisas J, Vardas PE, Kouroumalis EA. Autonomic imbalance during the day in patients with inflammatory bowel disease in remission. Evidence from spectral analysis of heart rate variability over 24 hours. Dig Liver Dis 2002;34:775-780.
van de Zanden.indb 140 23-5-2011 11:44:08

Nederlandse samenvatting
van de Zanden.indb 141 23-5-2011 11:44:09

143
Nederlandse samenvatting
In dit proefschrift werd onderzocht hoe de nervus vagus ontstekingen in de darm kan beïnvloeden. Het pionierswerk van Tracey en collega’s heeft gedemonstreerd dat de nervus vagus, via zijn neurotransmitter acetylcholine, een belangrijke rol speelt bij het remmen van ontstekingsreacties1. Tevens hebben meerdere studies aangetoond dat cholinerge activatie ziekte vermindert in verschillende diermodellen zoals ischemie-reperfusie schade2, hemorrhagische shock3, peritonitis4, DSS-colitis5 en reumatoïde artritis6.
Het doel van dit proefschrift was om het werkingsmechanisme van de zogenaamde ‘cholinerge anti-inflammatoire pathway’ in darmontstekingen verder te ontrafelen. Het maag-darmstelsel staat onder strikte controle van de nervus vagus7,8, waardoor het gevoelig is voor immuun-modulatie door deze zenuw. Vooral in de darm, waar onze grootste verzameling microben huist en er goed gediscrimineerd moet worden tussen lichaamsvreemd en lichaamseigen, is strikte regulatie van de immuunreactie cruciaal. Wanneer dit proces uit balans is kunnen er ziekten optreden. Dit is bijvoorbeeld het geval in inflammatoire darmziekten (IBD), zoals de ziekte van Crohn en Colitis Ulcerosa, ziekten die veroorzaakt worden door een te sterke mucosale immuunreactie tegen onderdelen van de darmflora in genetisch vatbare personen. Hoofdstuk 2 geeft een overzicht van de huidige kennis omtrent cholinerge modulatie van ontstekingen in de darm. Dit review beschrijft de mogelijke manieren waarop de nervus vagus de immuun respons kan reguleren. Tevens wordt de rol van activatie van de nicotinerge acetylcholine receptor (nAChR) en daaropvolgende signaleringsroutes in macrofagen en andere immuuncellen belicht. Tenslotte worden de mogelijke klinische implicaties van nervus vagus signalering besproken.
JAK2-STAT3 signalering in de ‘cholinerge anti-inflammatoire pathway’De ‘cholinerge anti-inflammatoire pathway’ wordt gekarakteriseerd door een verminderde productie van pro-inflammatoire cytokines via activatie van nAChRs op macrofagen1. Activatie van nAChRs kan in vivo tot stand komen door nervus vagus stimulatie, in vitro door het toedienen van cholinerge agonisten zoals acetycholine en nicotine. De nAChR bestaat uit verschillende onderdelen, subunits genoemd. De meeste studies wijzen de nAChR α7 subunit aan als essentiële schakel in het ontstekingsremmende effect van acetylcholine of nicotine9, 10-12. Wang et al hebben laten zien dat activatie van de nAChR α7 de transcriptiefactor NF-kBp65 remt, wat resulteert in een verminderde pro-inflammatoire cytokine productie9,13. In hoofdstuk 3 hebben we geprobeerd uit te vinden via welke intra-cellulaire signaleringsroute acetycholine en nicotine hun effect bereiken. Wij hebben de betrokkenheid van de transcriptiefactor STAT3 in dit proces onderzocht, omdat STAT3 in potentie een negatieve regulator is van ontstekingsreacties14, 15. Wij toonden aan dat in vitro, nicotine en acetylcholine hun anti-inflammatoire effect op peritoneale macrofagen via nAChR α7-Jak2-STAT3 signalering bewerkstelligen. In vivo, hebben we de rol
van de Zanden.indb 143 23-5-2011 11:44:09

144
Chapter 7
van STAT3 in de ‘cholinerge anti-inflammatoire pathway’ getest in een diermodel van post-operatieve ileus (POI). POI wordt gekarakteriseerd door een verminderde darmwerking en vertraagde maagontlediging en is een pathologische conditie die normaal gezien wordt na abdominale chirurgie16. Deze aandoening is het resultaat van ontsteking van het spierweefsel van de darm, ontstaan doordat residente macrofagen geactiveerd worden door manipulatie van de darm17. In een diermodel verbeterde POI aanzienlijk door stimulatie van de nervus vagus. Stimulatie van deze zenuw leidde tot STAT3 activatie in intestinale macrofagen, wat aangeeft dat acetylcholine, afkomstig van efferente zenuwvezels van de nervus vagus, STAT3 activeert. De bevinding dat macrofagen activiteit onder strikte neuronale controle staat wordt versterkt door de observatie dat cholinerge zenuwvezels en residente macrofagen zeer dichtbij elkaar liggen in de myenterische plexus. Concluderend hebben wij in hoofdstuk 3 laten zien dat remming van macrofagen activiteit via de ‘cholinerge anti-inflammatoire pathway’ wordt bewerkstelligd via JAK2-STAT3 signalering. Theoretisch zou het mogelijk zijn dat nicotine macrofagen activiteit remt via directe interactie van gedimeriseerd STAT3 met de NF-kB p65 subunit18.
In hoofdstuk 4, werd de rol van de JAK2-STAT3 signaleringsroute in het anti-inflammatoire effect van nAChR activatie verder onderzocht. Dit hebben we gedaan door middel van STAT3 silencing, farmacologische antagonisten en dominant negatieve STAT3 constructen. Hierbij werden twee dominant negatieve constrcuten gebruikt: STAT3D, dat STAT3 binding aan het DNA tegengaat, en STAT3F, dat STAT3 fosforylatie en dimerisatie voorkomt19. Nicotine remde TNF productie in peritoneale macrofagen getransfecteerd met STAT3F, maar niet in cellen getransfecteerd met STAT3D. Dus, in tegenstelling tot onze bevindingen in hoofdstuk 3, zagen we dat het ontstekingsremmende effect van nicotine in macrofagen meer afhankelijk was van STAT3 DNA binding en de aanwezigheid van STAT3 eiwit, dan van STAT3 fosforylatie. Hypothetisch, kan ongefosforyleerd STAT3 (U-STAT3) belangrijk zijn in het ontstekingsremmende effect van nicotine, via binding aan NF-kB en remming van TNF transcriptie20.
In conclusie, wordt er in hoofdstuk 3 en 4 een nieuwe moleculaire route geïdentificeerd die betrokken is bij het anti-inflammatoire effect van cholinerge activiteit in macrofagen. Dit geeft aan dat medicijnen die ingrijpen op de JAK2-STAT3 signaleringsroute in de toekomst mogelijk belangrijk kunnen zijn in de behandeling van ontstekingsziekten.
Cholinerge activiteit en fagocytoseActivatie van het cholinerge systeem kan de productie van pro-inflammatoire cytokines door macrofagen remmen. Echter, zeker in de darm, functioneren macrofagen meer als fagocyten die, samen met dendritische cellen, kritische bewakers zijn in het toelaten van luminale antigenen. Daarom is het de vraag of het ontstekingsremmende
van de Zanden.indb 144 23-5-2011 11:44:09

145
Nederlandse samenvatting
effect van nervus vagus stimulatie in de darm exclusief rust op verminderde cytokine productie door macrofagen. Mogelijk beïnvloedt de nervus vagus ook andere belangrijke macrofaag functies. In hoofdstuk 5, hebben we het effect van nAChR activatie op endo- en fagocytose van peritoneale en mucosale macrofagen bestudeerd. Het bleek dat nAChR activatie endocytose en fagocytose in intestinale en peritoneale macrofagen stimuleert. Dit effect werd teweeggebracht door rekrutering van dynamin-221 naar de ‘fagocyterende cup’. De anti-inflammatoire effecten van nAChR activatie werden eerder toegeschreven aan nAChR α7 activatie9. Wij hebben duidelijk laten zien dat de toegenomen fagocytose door cholinerge agonisten via de nAChR α4β2 verliep, en niet via α7. Ondanks deze toename in fagocytose, reduceerde acetylcholine NF-kB activatie en pro-inflammatoire cytokine productie, terwijl de productie van het anti-inflammatoire IL-10 toenam. In vivo, leidde nervus vagus stimulatie tot verhoogde opname van luminale antigenen door intestinale fagocyten. Hierbij leidde nAChR activatie in darm weefsel tot een verhoogde passage van luminale bacteriën over de mucosa en een verhoogde drainage van luminale bacteriën. Concluderend wordt in hoofdstuk 5 aangetoond dat acetylcholine een tweeledig effect heeft in macrofagen, het stimuleert fagocytose via nAChR α4β2 activatie, terwijl het NF-kB activatie en productie van ontstekingsmediatoren remt. Op die manier draagt nervus vagus stimulatie bij aan de surveillance in het intestinale en peritoneale compartiment.
Modulatie van immuun cellen door de nervus vagus in vivoIn hoofdstuk 6, hebben we geprobeerd te analyseren hoe nervus vagus activiteit immuun cellen in vivo kan beïnvloeden. Wij onderzochten of nervus vagus activiteit de immuunreactie mogelijk niet alleen via de uitscheiding van acetylcholine remt, maar misschien ook via alternatieve neurotransmitters zoals neuropeptides. Vasoactive intestinal polypeptide (VIP) en substance P (SP) zijn neuropeptides die uitgebreid tot expressie komen in de darm en zij hebben belangijke immuunmodulerende functies. Stimulatie van de nervus vagus veranderde de expressie van VIP en SP in darm weefsel van muizen . In vitro, leidde het tegelijkertijd toedienen van Ach en VIP tot een verlaagde LPS-geinduceerde TNF productie, bijna tot aan ongestimuleerde waarden. SP verhoogde NF-kB transcriptionele activiteit en TNF productie in macrofagen, maar wanneer SP en Ach tegelijk werden toegediend, neutraliseerde nicotine de pro-inflammatoire effecten van SP. Deze immunomodulerende effecten van acetylcholine, nicotine, VIP en SP waren onafhankelijk van calcium signalering. Samenvattend hebben we in hoofdstuk 6 aangetoond dat nervus vagus stimulatie VIP en SP expressie in de darm verandert, terwijl cholinerge agonisten in vitro de immunomodulerende acties van VIP en SP beïnvloeden. Deze data wekken de suggestie dat het anti-inflammatoire effect van de nervus vagus in de darm versterkt zou kunnen worden door gelijktijdig vrijkomende neuropeptiden.
van de Zanden.indb 145 23-5-2011 11:44:09
146
Chapter 7
De bevinding dat nicotine activatie van immuun cellen remt, samen met de observatie dat nervus vagus activiteit ziekte tegengaat in verschillende diermodellen, impliceert dat therapeutische middelen die de op de ‘cholinergic anti-inflammatoire pathway’ aangrijpen, bruikbaar zouden kunnen zijn in mensen. Desalniettemin hebben klinische trials waarin nicotine als behandeling getest werd, verschillende uitkomsten laten zien22. Dit zou deels verklaard kunnen worden door de bevinding dat humane immuun cellen een gevarieerde nAChR expressie hebben, veroorzaakt door genetische of omgevingsfactoren23, 24. In hoofdstuk 7, hebben we onderzocht of roken of herhaaldelijk blootstelling aan nicotine de expressie van nAChR α7, dupα7 of β2 op humane monocyten kan veranderen. Wij demonstreerden dat herhaaldelijke nicotine blootstelling nAChR α7 expressie verhoogt in een humane monocyten cellijn. Tevens was nAChR α7 in een pilot studie alleen aantoonbaar op monocyten van rokers. Aan de andere kant werden cellen niet gevoeliger voor cholinerge immuunmodulatie door deze toegenomen nAChR α7 expressie.
Dit samenvattend figuur is een model van het mechanisme waarmee nervus vagus activiteit de functie van darm macrofagen beїnvloedt, zoals is gebleken uit de resultaten in dit proefschrift.
Efferente vezels van de nervus vagus, die ontspringen uit de hersenstam, scheiden acetylcholine uit als gevolg van fysiologische of electrische stimulatie. Stimulatie van de nervus vagus kan immuun cellen beїnvloeden via directe vrijstelling van acetylcholine of via post-ganglionaire mechanismen waarbij alternatieve
van de Zanden.indb 146 23-5-2011 11:44:09

147
Nederlandse samenvatting
neurotransmitters betrokken zijn zoals VIP en SP. Acetylcholine bindt op nAChRs die tot expressie komen op immuun cellen zoals intestinale macrofagen. Acetylcholine heeft een tweeledig effect op macrofagen: aan de ene kant reduceert het de productie van pro-inflammatoire cytokines meest waarschijnlijk deels via nAChR α7 activatie, aan de andere kant stimuleert het fagocytose via activatie van de nAChR α4β2. Acetylcholine remt TNF prodcutie uiteindelijk via JAK2-STAT3 signalering. Uit onze studies blijkt dat het anti-inflammatoire effect van nAChR activatie meer afhankelijk is van STAT3 DNA binding en STAT3 eiwit, dan van STAT3 fosforylatie. De nAChR α4β2 afhankelijke toename in fagocytose wordt bewerkstelligd door recrutering van Dynamin-2 naar de ‘fagocyterende cup’. Samenvattend laat dit figuur zien hoe nervus vagus stimulatie macrofagen naar een meer ‘inactief’ fenotype duwt, wat van nut kan zijn in ziektes die gekarakteriseerd worden door een ontregelde immuunreactie.
Therapeutische opties en toekomstperspectieven.Resultaten uit veel in vitro en in vivo onstekingsmodellen impliceren dat therapeutische middelen die op de ‘cholinerge anti-inflammatoire pathway’ aangrijpen in de toekomst belangrijk zouden kunnen zijn bij de behandeling van immuunziekten. In vivo kan cholinerge activiteit op meerdere manieren worden verkregen. De meest bekende, maar niet de meest selectieve manier, is door het roken van sigaretten. In inflammatoire darmziekten is roken een belangrijke omgevingsfactor, het heeft echter verschillende effecten in colitis ulcerosa en in de ziekte van Crohn. Terwijl roken het risico op het ontwikkelen van de ziekte van Crohn verhoogt, lijkt het een beschermend effect te hebben in de ontwikkeling van colitis ulcerosa en het vermindert de ernst van de ziekte25. Desalniettemin hebben klinische trials naar nicotine als behandeling van colitis ulcerosa geen significant voordeel aangetoond vergeleken met standaard therapie, terwijl nicotine wel meer bijwerkingen gaf22. Juist om deze reden is het een uitdaging om een specifieke nAChR agonist te ontwikkelen met de sterkste anti-inflammatoire effecten en de minste bijwerkingen. Gedeeltelijk selectieve nAChR α7 en α4β2 agonisten zijn al getest in patiënten met neuronale aandoeningen26. De best gekarakteriseerde nAChR agonist GTS-21, een partiële α7 agonist die ook werkt op de α4β2 nAChR27, wordt goed getolereerd in mensen. In vitro, bleek deze agonist de productie van pro-inflammatoire cytokines door immuun cellen van muizen en mensen te verminderen28, 29. In een recente studie naar de effecten van GTS-21 op de afweerreactie tijdens endotoxemie in 14 niet-rokende vrijwilligers, werden geen verschillen gevonden in de cytokine respons tussen de GTS-21 en placebo groep30.
Om werkzame nAChR agonisten te ontwikkelen, is exacte kennis nodig over welke nAChRs betrokken zijn bij het cholinerge anti-inflammatoire effect. Deze effecten zijn eerder alleen toegeschreven aan activatie van de nAChR α79. Desondanks was de nAChR α7 niet aanwezig op monocyten van niet rokende vrijwilligers (hoofdstuk 7) en tevens slaagden wij er niet in om α7 nAChR transcript in bepaalde muis
van de Zanden.indb 147 23-5-2011 11:44:09

148
Chapter 7
macrofagen aan te tonen (hoofdstuk 5). Verdere analyse van nAChR α7 eiwit in deze macrofagen wordt belemmerd door het feit dat commercieel verkrijgbare α7 antibodies niet specifiek blijken te zijn, omdat deze niet alleen een band aankleuren in brein homogenaten van wildtypes maar ook in brein van α7 nAChR Knock-out (KO) muizen31. Hierbij komt dat nicotine zowel in wildtype, als in α7 KO muizen TNF productie reduceerde (hoofdstuk 5). Deze observaties impliceren dat de effecten van acetylcholine en nicotine op peritoneale macrofagen niet exclusief moeten worden toegeschreven aan nAChR α7 activatie, maar mogelijk ook aan andere nAChR subtypes. Daarom kan het gebruik van selectieve agonisten voor andere nAChR subunits dan de α7 subunit succesvol zijn, met name agonisten voor de nAChR α4β2, het receptor subtype dat essentieel blijkt te zijn voor activatie van fagocytose. Naast het gebruik van specifieke cholinerge agonisten zou nervus vagus stimulatie zelf een potentiële therapeutische behandeling kunnen zijn bij ontstekingsziekten. In patiënten met therapieresistente epilepsie en depressie, wordt nervus nagus stimulatie al gebruikt als nieuwe adjuvante therapie. Daarnaast is gebleken dat vette voeding het parasympathische zenuwstelsel kan activeren.
De bevinding dat nicotine activiteit van immuun cellen remt, samen met de observatie dat nervus vagus stimulatie of specifieke nAChR agonisten ziekte in meerdere inflammatoire diermodellen tegengaan, impliceert dat therapeutische behandelingen die zich richten op cholinerge signalering, voordelig kunnen zijn in mensen. Toch moet men in gedachten houden dat het ontwikkelen van medicijnen die aangrijpen op de ‘cholinerge anti-inflammatoire pathway’ gecompliceerd kan zijn, omdat er in mensen individuele verschillen kunnen zijn in de reactie op deze middelen.
Samenvattend, hebben we in dit proefschrift het werkingsmechansime van de ‘cholinerge anti-inflammatoire pathway’ verder geïdentificeerd. Onze data kunnen nieuwe aangrijpingspunten bieden voor de ontwikkeling van toekomstige medicijnen tegen de behandeling van darmontstekingen, zoals colitis ulcerosa.
van de Zanden.indb 148 23-5-2011 11:44:09

149
Nederlandse samenvatting
REFERENCE LIST
1. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000;405:458-462.
2. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, Tracey KJ. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg 2002;36:1231-1236.
3. Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, Squadrito G, Minutoli L, Bertolini A, Marini R, Adamo EB, Venuti FS, Squadrito F. Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation 2003;107:1189-1194.
4. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der PT. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis 2005;191:2138-2148.
5. Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology 2006;131:1122-1130.
6. van Maanen MA, Lebre MC, van der Poll T, Larosa GJ, Elbaum D, Vervoordeldonk MJ, Tak PP. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum 2009;60:114-122.
7. Altschuler SM, Ferenci DA, Lynn RB, Miselis RR. Representation of the cecum in the lateral dorsal motor nucleus of the vagus nerve and commissural subnucleus of the nucleus tractus solitarii in rat. J Comp Neurol 1991;304:261-274.
8. Altschuler SM, Escardo J, Lynn RB, Miselis RR. The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology 1993;104:502-509.
9. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003;421:384-388.
10. Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, Levi J, Parrish WR, Rosas-Ballina M, Czura CJ, Larosa GJ, Miller EJ, Tracey KJ, Al Abed Y. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 2007;35:1139-1144.
11. Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, Hudson L, Lin X, Patel N, Johnson SM, Chavan S, Goldstein RS, Czura CJ, Miller EJ, Al Abed Y, Tracey KJ, Pavlov VA. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med 2008;14:567-574.
12. De Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 2007;151:915-929.
13. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004;10:1216-1221.
14. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999;10:39-49.
15. Levy DE, Lee CK. What does Stat3 do? J Clin Invest 2002;109:1143-1148.
16. Prasad M, Matthews JB. Deflating postoperative ileus. Gastroenterology 1999;117:489-492.
17. De Jonge WJ, van den Wijngaard RM, The FO, ter Beek ML, Bennink RJ, Tytgat GN, Buijs RM, Reitsma PH, van Deventer SJ, Boeckxstaens GE. Postoperative ileus is maintained by intestinal immune infiltrates that activate inhibitory neural pathways in mice. Gastroenterology 2003;125:1137-1147.
van de Zanden.indb 149 23-5-2011 11:44:09

150
Chapter 7
18. Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 2002;367:97-105.
19. de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, Lowenberg B, Touw IP. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 2000;19:3290-3298.
20. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 2007;21:1396-1408.
21. Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 1999;190:1849-1856.
22. McGrath J, McDonald JW, Macdonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev 2004;CD004722.
23. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci 2007.
24. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett 1999;266:17-20.
25. Lakatos PL, Szamosi T, Lakatos L. Smoking in inflammatory bowel diseases: good, bad or ugly? World J Gastroenterol 2007;13:6134-6139.
26. Cincotta SL, Yorek MS, Moschak TM, Lewis SR, Rodefer JS. Selective nicotinic acetylcholine receptor agonists: potential therapies for neuropsychiatric disorders with cognitive dysfunction. Curr Opin Investig Drugs 2008;9:47-56.
27. Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, Burnett AL. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology 2003;28:542-551.
28. Giebelen IA, van Westerloo DJ, Larosa GJ, de Vos AF, van der PT. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock 2007;28:700-703.
29. Kox M, van Velzen JF, Pompe JC, Hoedemaekers CW, van der Hoeven JG, Pickkers P. GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem Pharmacol 2009.
30. Kox M, Pompe JC, Gordinou de Gouberville MC, van der Hoeven JG, Hoedemaekers CW, Pickkers P. Effects of the alphaalpha7nAChR Agonist GTS-21 on the Innate Immune Response in Humans. Shock 2011.
31. Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De BM, Schroder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem 2007;102:479-492.
van de Zanden.indb 150 23-5-2011 11:44:09

List of contributing authors
van de Zanden.indb 151 23-5-2011 11:44:09

153
List of contributing authors
Shizuo AkiraDepartment of Host Defense, Research Institute for Microbial Diseases, Osaka University, Suita
Roelof J. BenninkNuclear Medicine, Academic Medical Center, Amsterdam, The Netherlands
Hans-Rudolf BerthoudNeurobiology of Nutrition Laboratory, Pennington Biomedical Research Center, Louisiana State University, Baton Rouge, LA, USA
Maarten F. BijlsmaLaboratory of Experimental and Internal Medicine, Academic Medical Center, Amsterdam, The Netherlands
Guy E.E. BoeckxstaensDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The NetherlandsDept. of Gastroenterology, University Hospital of Leuven, Catholic University of Leuven, Leuven, Belgium
David R Greaves Sr William Dunn School of Pathology, University of Oxford, Oxford, UK
Siamon Gordon Sr William Dunn School of Pathology, University of Oxford, Oxford, UK
Sigrid E. HeinsbroekDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
Francisca HilbersDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
Wouter J. de JongeDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
Klaus MichelLehrstuhl für Humanbiologie, Technische Universität München, Germany
Laurens J. Nijhuis Dept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
Geber PeñaLaboratory of Immunity and Infection, Department of Surgery, UMDNJ-New Jersey Medical School, Newark, NJ, USA
Maikel P PeppelenboschDep. of Cell Biology, University of Groningen, Groningen
Michael SchemannLehrstuhl für Humanbiologie, Technische Universität München, Germany
Susanne A. SnoekDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
van de Zanden.indb 153 23-5-2011 11:44:09

154
Oana I. Stanisor Dept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
Frans O. TheDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
Satoshi UematsuDepartment of Host Defense, Research Institute for Microbial Diseases, Osaka University, Suita, Japan
Luis UlloaLaboratory of Immunity and Infection, Department of Surgery, UMDNJ-New Jersey Medical School, Newark, NJ, USA
Caroline Verseijden Dept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
David J. van WesterlooDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands Laboratory of Experimental and Internal Medicine, Academic Medical Center, Amsterdam, The Netherlands
Rene MJGJ. van den WijngaardDept. of Gastroenterology & Hepatology, Academic Medical Center, Amsterdam, The Netherlands
van de Zanden.indb 154 23-5-2011 11:44:10

List of publications
van de Zanden.indb 155 23-5-2011 11:44:10

157
List of publications
Peña G, Cai B, Liu J, van der Zanden EP, Deitch EA, de Jonge WJ, Ulloa L. Unphosphorylated STAT3 modulates alpha 7 nicotinic receptor signaling and cytokine production in sepsis. European Journal of Immunology. 2010;40(9):2580-9
Snoek SA, Verstege MI, van der Zanden EP, Deeks N, Bulmer DC, Skynner M, Lee K, Te Velde AA, Boeckxstaens GE, de Jonge WJ. Selective alpha7 nicotinic acetylcholine receptor agonists worsen disease in experimental colitis. British Journal of Pharmacology. 2010;160(2):322-33.
van der Zanden EP, Snoek SA, Heinsbroek SE, Stanisor OI, Verseijden C, Boeckxstaens GE, Peppelenbosch MP, Greaves DR, Gordon S, De Jonge WJ.Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology. 2009 Sep;137(3):1029-39, 1039.e1-4.
van Maanen MA, Stoof SP, van der Zanden EP, de Jonge WJ, Janssen RA, Fischer DF, Vandeghinste N, Brys R, Vervoordeldonk MJ, Tak PP. The alpha7 nicotinic acetylcholine receptor on fibroblast-like synoviocytes and in synovial tissue from rheumatoid arthritis patients: a possible role for a key neurotransmitter in synovial inflammation.Arthritis Rheumatism. 2009;60(5):1272-81
Van Der Zanden EP, Boeckxstaens GE, de Jonge WJ. The vagus nerve as a modulator of intestinal inflammation. Neurogastroenterology and Motility. 2009 Jan;21(1):6-17.
De Jonge WJ, The FO, van der Zanden EP, van den Wijngaard RM, Boeckxstaens GE.Inflammation and gut motility; neural control of intestinal immune cell activation.J Pediatric Gastroenterology Nutrition. 2005 Sep;.
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE.Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nature Immunology 2005;6(8):844-51
van de Zanden.indb 157 23-5-2011 11:44:10

Dankwoord
van de Zanden.indb 159 23-5-2011 11:44:10

161
Dankwoord
Na vier jaar nu het leukste, maar ook een van de moeilijkste stukken om te schrijven. Ik heb met enorm veel plezier op verschillende afdelingen gewerkt en één ding is zeker: zonder hulp van velen was dit proefschrift nooit tot stand gekomen. Ik wil iedereen die op welke wijze dan ook heeft bijgedragen aan het tot stand komen van dit proefschrift hartelijk bedanken! In het bijzonder de volgende mensen:
Dr. de Jonge, beste Wouter, het voelt als de dag van gisteren dat wij ons eerste kennismakingsgesprekje hadden. Je vertelde met zoveel passie en enthousiasme over het onderzoek, dat ik niet lang hoefde te twijfelen over mijn stageplek. Dat is in al die jaren niet veranderd. Ik heb heel veel bewondering voor je wetenschappelijke inzicht, gedrevenheid en optimisme. Als ik weleens bij de pakken neer zat vanwege negatieve data, hoefde ik maar even langs je kamer te lopen om er vol goede moed weer uit te komen. Daarnaast was het ook erg gezellig, je was altijd in voor een onzingesprekje en vol´flauwe grappen. Je groep blijft groeien en ik denk dat mede door jouw begeleiding de sfeer binnen ons cluppie zo goed is. Ik wil je ontzettend bedanken voor alles wat je me hebt geleerd de afgelopen jaren en voor de mooie tijd die ik gehad heb!
Professor Dr. Boeckxstaens, beste Guy. Bedankt dat je mijn promotor bent. Ik heb veel geleerd van je kritische wetenschappelijke blik en kennis van de neurogastroenterologie. Daarnaast was het ook altijd goed pintjes drinken… Hartelijk dank voor de mogelijkheden die je me de afgelopen jaren hebt geboden.
Graag wil ik Dr. G.R. van den Brink, Prof. Dr. E. Fliers, Prof. Dr. T. van der Poll, Prof. Dr. H. Spits en Prof. Dr. P.P. Tak bedanken voor de kritische beoordeling van mijn proefschrift en jullie bereidheid plaats te nemen in mijn promotiecommissie. Dear Prof. Dr M. Schemann, it is an honour for me that you are willing to take part in my committee. I’d like to thank you and Klaus for assisting me with the calcium experiments in your lab.
Collega s van het CEMM, waar ik mijn eerste schreden zette als onderzoekster, hartelijk dank voor jullie hulp en advies bij blotjes, ELISA s en alle andere mogelijke technieken…en natuurlijk de fantastische labdagen. Het voelde vanaf het begin af aan als een warm bad. Speciaal wil ik mijn lieve kamergenootjes bedanken, Maarten, Joppe en Marcello. Heerlijk om even tegen jullie aan te kunnen kletsen en jullie verhalen aan te horen. Van stackers tot Salp15 tot opera, altijd wat te beleven op kamertje 1! En…na een tussenbiertje pipetteer je toch net iets beter… Daarnaast wil ik Kees en Alex bedanken, jullie waren altijd beschikbaar voor advies en nuttige tips. Kees, bedankt voor het deelnemen in mijn promotiecommissie. Francesca en Tatjana, bedankt voor jullie gezelligheid en de leuke etentjes. Marjolein, α7- partner-in-crime, echt veel nuttigs bespraken we niet tijdens onze koffiedates-met-labboek, maar gezellig was het wel! Tenslotte moet ik zeker mijn vrijwilligers niet vergeten, overal vandaan geplukt
van de Zanden.indb 161 23-5-2011 11:44:10

162
om de ene keer olijfolie te drinken (!) en dan weer liters bloed af te staan, altijd zonder morren…ontzettend bedankt. Lieve mensen van de (kinder)motiliteit: Olle, Breg, Hanneke, Olaf, Tammie, Marloes, Roel, Michiel, Maartje, Rosa, Claire, Babette, Wout, Olivia, Aaltje, Ramona: bedankt voor de gezellige tijd, de GAP borrels met ´hole in one , het delen van de werksores, de geweldige motilititeitsuitjes met de windmachine in Antwerpen met stip op 1… Noor, ook al raakten we elkaar soms kwijt op de fiets, ik heb zeer goede herinneringen aan onze reisjes naar Amerika!
In het allerlaatste jaar maakten wij de overstap naar het ALC, later Tytgatinstituut. Ik wil alle collega’s daar bedanken dat jullie me wegwijs hebben gemaakt in het lab. Mona, ontzettend bedankt voor de hulp bij de laatste, zeer stress-volle, loodjes.
En natuurlijk mijn derde ‘honk’, de Peyer’s patch… Laurens, vagusbroeder, bedankt voor je gezelligheid, relaxedheid en relativeringsvermogen. Een zeer belangrijke eigenschap met twee die stresskippen als Suus en ik naast je. Sjoerd, het heeft wat bloed zweet en tranen(!) gekost maar goed dat de LAFA studie toch zo’n mooie bestemming heeft gekregen! Marleen, bedankt voor je hulp met experimenten en heel veel succes in je verdere carrière. Ronnie, erg jammer dat we maar kort samengewerkt hebben, maar wat heerlijk om een mede-boer-zoekt-vrouw-kijker naast me te hebben. Shobit, José and Lea, thanks for the good atmosphere! Dames uit het kippenhok, Sophie en Oana, ik heb me altijd goed vermaakt met jullie grappige verhalen tijdens lunches en borrels. Caroline, Regina en Frannie, wat moest ik zonder jullie… Erg fijn om goede analisten aan je zij te hebben die altijd meedenken. Heel erg bedankt voor jullie hulp! Esther, Sander, Jan, Sigrid, Anje en Cathy, dank voor jullie hulp en nuttige input tijdens de werkbesprekingen. René, ontzettend veel dank voor je adviezen en het kritisch lezen van mijn manuscripten.
Klinische AIO’s: Koen, Dennie, Kirsten, Tessa, Thomas, Karampie, Frans, Wouter, Wytske, Roos, Renée, Fred, Lorenza, Simone, Charlotte, Taeco, Annikki, Joep, David, Femme en Emma en ‘nieuwe’ garde, bedankt voor de onvergetelijke congressen, borrels en feestjes. Ik denk niet dat ik ooit nog met 9 collega’s in 1 hot-tub zal belanden… We mogen trots zijn op zo’n gezellige groep, ik hoop dat we in de toekomst nog vaak zullen borrelen en elkaar hopelijk weer als collega’s mogen begroeten. Natuurlijk ook de ‘ Leiden-gang’, Marjolijn, Rutger en Jarom, bedankt voor de biertjes, steun op het basale vlak en gezelligheid. Lieve vrienden en vriendinnen, in het bijzonder José, Anne en Noortje, heel veel dank voor de broodnodige afleiding tijdens deze afgelopen jaren. Zeker de laatste tijd ben ik niet de attentste en gezelligste geweest, maar ik beloof: daar komt verbetering in!
van de Zanden.indb 162 23-5-2011 11:44:10

163
Dankwoord
Lieve Willemijn, Doppie, ik ben erg blij dat ik jou heb ontmoet tijdens mijn promotie. Vanaf de eerste minuut wist ik dat ik het goed met je zou kunnen vinden en dat bleek inderdaad te kloppen. Ik heb genoten van onze eeuwige biertjes, feestjes, roddels en buitenlandse avonturen. Hopelijk gaan er nog veel volgen. Ik ben heel blij dat jij mijn paranimf wilt zijn.
Lieve Susan, Susie, wat is het heerlijk om 4 jaar te werken met een van je beste vriendinnen aan je zijde. Alles is te bespreken en je voelt je nooit alleen. Echt een luxepositie, besef ik me nu ik elke maand weer opnieuw in mijn eentje ergens moet beginnen. Bedankt voor je steun, je gezelligheid en het feit dat je naast me staat bij de verdediging.
Lieve mama, dank voor je onvoorwaardelijke steun. Je hebt altijd achter mijn keuzes gestaan ook was je het er misschien stiekem niet zo mee eens. Zonder jou was ik nooit zover gekomen! Lieve Marjolein, Rogier en Tineke en ‘ ukkies’ Renske en Norrie: bedankt voor de leuke momenten samen en de gezellige afleiding.
Lieve, lieve Rogier. Wat fantastisch dat ik 3 jaar geleden mijn grote liefde tegen het lijf ben gelopen, zomaar, op de gangen van het AMC…met jou is het leven gewoon leuker! Het was erg fijn dat we in hetzelfde schuitje zaten, maar ik kijk er nu ontzettend naar uit om niet meer elk weekend laptop-aan-laptop te zitten... Bedankt voor alles, en volgens mij zijn wij het levende bewijs dat klinisch en basaal onderzoek heel goed samengaan!
van de Zanden.indb 163 23-5-2011 11:44:10

165
Curriculum Vitae
Esmerij van der Zanden is geboren op 24 december 1979 te De Lier. Zij behaalde haar gymnasiumdiploma in 1998 aan het Stanislascollege te Delft. In datzelfde jaar startte zij met de studie Medische Biologie aan de Universiteit van Amsterdam (UvA), waarvan zij in 2005 het doctoraalexamen behaalde. In 2000 begon Esmerij tevens met haar studie Geneeskunde aan de UvA. Haar wetenschappelijk stage verrichtte zij op de afdeling Maag-, Darm- en Leverziekten en het Center of Experimental Medicine (CEMM) van het Academisch Medisch Centrum te Amsterdam, onder begeleiding van dr. W.J. de Jonge en prof. dr. G.E. Boeckxstaens. Tijdens deze stage deed zij onderzoek naar de rol van de nervus vagus in darmontstekingen. Deze stage heeft zich uitgebreid tot een promotieonderzoek, waarmee zij in 2005 is begonnen. De resultaten van dit onderzoek staan beschreven in dit proefschrift. In 2009 is Esmerij gestart met haar co-schappen, zij hoopt eind 2011 haar arts-examen te behalen. Zij woont samen in Amsterdam met haar vriend Rogier Voermans.
Curriculum Vitae
van de Zanden.indb 165 23-5-2011 11:44:10