Embed Size (px)
Citation preview

T H E N E X T G E N E R A T I O N I B D D I A G N O S T I C T E S T
The Synergistic Role of Serology, Genetics,and Inflammation in the Diagnosis of
INFLAMMATORY BOWEL DISEASE

INSIDE FRONT COVER(BLANK)

1
multiple imaging techniques are available to aid in the diagnosis of IBD,
many are associated with shortcomings from the standpoint of
performance. Computed tomographic enterography (CTE) is able to
detect clinically occult inflammatory and penetrating disease, but has
been reported to have less sensitivity than capsule endoscopy (CE) in
detecting ileal inflammation in patients with non-stricturing disease.
CTE sensitivity in detecting active colon inflammation has been estimated
at 74%. Magnetic resonance enterography (MRE) is associated with
suboptimal images in some patients. Like CTE, its ability to detect
inflammation is variable, with sensitivity ranging from 55% to 62%.
CE has the sensitivity to detect mucosal inflammation of the small bowel,
but its use can be complicated by capsule retention due to stricturing
disease and may lead to surgical intervention for a retained capsule.9,10
In patients with suspected or established CD, the diagnostic yield of CE
obtained from case reports, retrospective reviews, and prospective
evaluations ranges from 28% to 71%. Ileocolonoscopy is considered the
best test for detection of inflammation of the colon and has the added
benefit of allowing the collection of biopsy samples for pathologic
review. Biopsy results may not prove definitive, however, and the invasive
nature of the procedure remains a consideration, especially in the
pediatric age group. Ileocolonoscopy is associated with a small risk
of perforation (0.03% to 0.15%), as reported in a recent review of nine
large clinical series, each of which contained more than 30,000 cases.11
The need for an adjunctive and comprehensive testing tool is underscored
by the challenges related to diagnosing IBD and differentiating CD
from UC using a single modality, and with the knowledge that has
been gained regarding the known derangements present in IBD (eg,
inflammation, immune response to enteric bacteria, genetic factors
leading to IBD susceptibility).
Distinguishing Between CD and UC
Differentiating CD from UC is important for its implications in
selecting treatment and in the timing and type of surgery that may
ultimately be required. However, this differentiation remains a
challenge. A recent systematic evaluation by the Swiss IBD Study
Group in nearly 1600 patients with diagnoses of CD, UC, or
indeterminate colitis (IC) found that diagnostic delay was significantly
longer for CD than UC (median 9 vs 4 mo; P< .001).12 The intervals
between first symptoms to physician visit (P= .002), physician visit to
diagnosis (P < .001), and first symptoms to IBD diagnosis (P< .001)
were all significantly longer for patients with CD than for patients with
UC.12 On multivariate regression analysis, the presence of ileal
disease and age less than 40 years at diagnosis were found to be
independent risk factors for long diagnostic delay (>24 mo) in
patients with CD.12 In patients with UC, use of nonsteroidal anti-
inflammatory drugs was related to a prolonged diagnostic delay
(>12 mo) at a trend-level association (P= .093).12
Introduction
Inflammatory bowel disease (IBD) is a chronic and relapsing
idiopathic disorder of the gastrointestinal (GI) tract that is characterized
by mucosal inflammation and marked by recurrent diarrhea and
abdominal pain.1 While the pathogenesis of IBD is not completely
understood, current thinking is that it arises from complex interactions
involving the immune system, enteric commensal bacteria, and
genetic factors.2,3 IBD is divided into two main subtypes of IBD —
Crohn’s disease (CD) and ulcerative colitis (UC). While presenting
symptoms may be suggestive of one or the other entity, there can be
a large overlap of GI symptoms, associated physical findings, and
extraintestinal manifestations that are found in both UC and CD that
make it difficult to differentiate the two disorders,4 and thus there is a
medical need for adjunctive testing tools to aid clinicians with these
diagnostic challenges. This monograph outlines some of the
limitations of current IBD diagnostic testing modalities and describes
a newly available serologic, genetic, and inflammation biomarker
panel that may prove to be beneficial in this process.
Diagnostic Challenges in IBD
Establishing the IBD Diagnosis
At present, there is no “gold standard” test to definitively diagnose
IBD.4 Traditionally, the diagnosis of IBD is based on a combination of
data obtained from the patient history and physical examination in
association with laboratory, endoscopic, histologic, and radiographic
investigations. In adult and pediatric patients, presenting symptoms of IBD
are related to both disease location and extent of intestinal inflammation.5
However, even when symptoms suggest a diagnosis of IBD, they are
often too nonspecific to result in a definitive diagnosis, such as in the
case of symptom overlap with functional bowel disorders6 or infectious
colitides.4 This overlap frequently delays an accurate diagnosis of IBD.6
Despite the lack of a pathognomonic test for IBD, laboratory values
provide useful information in the diagnostic process. Hematologic
parameters (increased leukocyte and thrombocyte counts), elevated
levels of acute phase reactants (C-reactive protein [CRP] and
erythrocyte sedimentation rate), and the presence of fecal markers
(leukocytes, lactoferrin, calprotectin) may be associated with active
intestinal inflammation,7,8 although they do not identify an exact
underlying cause. As a result of the limited negative predictive value
of laboratory values, the differential diagnosis requires additional
testing to rule out alternative disorders including other inflammatory
bowel diseases such as those related to infectious agents, medications,
radiation, or ischemia; other intestinal disorders (eg, celiac disease,
microscopic colitis); or irritable bowel syndrome (IBS).7
Establishing the diagnosis of IBD generally relies on the use of
invasive testing such as imaging studies and endoscopy. Although

2
Differentiating CD from UC can be difficult in patients whose disease
manifestations are exclusively colonic in nature and who lack typical
endoscopic or histologic findings, such as in patients with extensive
acute, severe colitis.13 While the main pathologic criterion for the diagnosis
of CD is the presence of granulomas, these may not be universally
evident in tissue samples obtained either at surgery or at biopsy. A
retrospective chart review including 102 patients with CD by Wolfson
and colleagues found granulomas in only 45% to 58% of surgically
resected specimens examined.14 An even smaller percentage (as low
as 2% to over 20%) is reported to be found in biopsy samples.15
Evaluation of Populations at Risk
Genetic susceptibility has been implicated in the pathogenesis of
IBD.2 Twin studies carried out in Sweden and Denmark revealed UC
concordance rates of 6.3% to 18.2% in monozygotic twins and 3.9%
to 4.5% in dizygotic twins.16,17 In these same studies, concordance in
CD was high for monozygotic twins (58.3%) but not for dizygotic
twins (0%).16,17 Furthermore, multiple investigations have shown that
first-degree relatives of patients with IBD are at an increased risk of
developing IBD. In a 2004 review of such studies, Russell and Satsangi
reported that the risk of developing CD was highest in siblings of CD
probands, where the relative risk was 30 to 42, whereas the overall
relative risk for CD in any first-degree relative was 10 to 35.18 The
relative risk of developing UC in first-degree relatives of UC probands
ranged from 10 to 15.18
Results of family and twin studies suggesting a contribution of
genetic factors to IBD prompted the search for susceptibility genes in
the 1990s. Initial efforts in genetic linkage studies and candidate gene
approaches in IBD were slow and gave rise to “susceptibility gene”
replication studies.
In 2000, efforts to sequence the human genome were followed by the
discovery of more than 10 million single nucleotide polymorphisms
(SNPs), which are DNA sequence variations that occur when a single
nucleotide (A, T, C, or G) in the genome sequence is altered.19 Many
SNPs have no effect on cell function, but scientists believe others could
predispose people to disease or influence their response to a drug.19
Together, these scientific advances made possible a new type of
research effort—the genome-wide association study (GWAS). In a
GWAS, the distribution of SNPs is determined in large populations
with and without a specific disease. By determining which SNPs
co-occur with disease symptoms, researchers can make a statistical
estimate of the level of risk associated with each SNP. This cutting-
edge research has resulted in the discovery of many susceptibility
genes in IBD. Furthermore, genes that confer susceptibility to one or
the other constituent diseases—either CD or UC—have also been
elucidated, providing a useful tool for diagnostic differentiation.
Evolution of IBD Testing: PROMETHEUS Laboratories, Inc.
Biomarkers that differentiate IBD from functional and other bowel
disorders, and that can distinguish UC from CD, have the potential to
improve the diagnostic process and minimize complications resulting
from invasive diagnostic procedures. These biomarkers may be
especially advantageous in a pediatric population.
Initially, two serologic markers—ASCA (anti-Saccharomyces
cerevisiae antibodies) and pANCA (perinuclear antineutrophil
cytoplasmic antibodies) — showed utility in diagnosing IBD and
differentiating between UC and CD.20 ASCA is more prevalent in CD than
in UC and healthy controls,21 and pANCA has more specificity for UC.20
However, some overlap was noted in that about one fourth of CD patients
will express pANCA,22 and ASCA sensitivity is suboptimal for a definitive
diagnosis of CD despite its high specificity. Moreover, pANCA is not
highly sensitive for UC because of its overlap in expression in UC-like
CD.20 The low sensitivity of ASCA or pANCA in IBD limits their clinical
utility in that a negative test result can rule out neither CD nor UC.23
Whereas positivity for ASCA and/or pANCA may help assign IC patients
a definitive diagnoses of CD or UC, a large percentage of IC patients
(85.1%) have been found to be seronegative for both markers.24
Efforts to improve the sensitivity of serologic testing in IBD have
led to the discovery and validation of new biomarkers and the
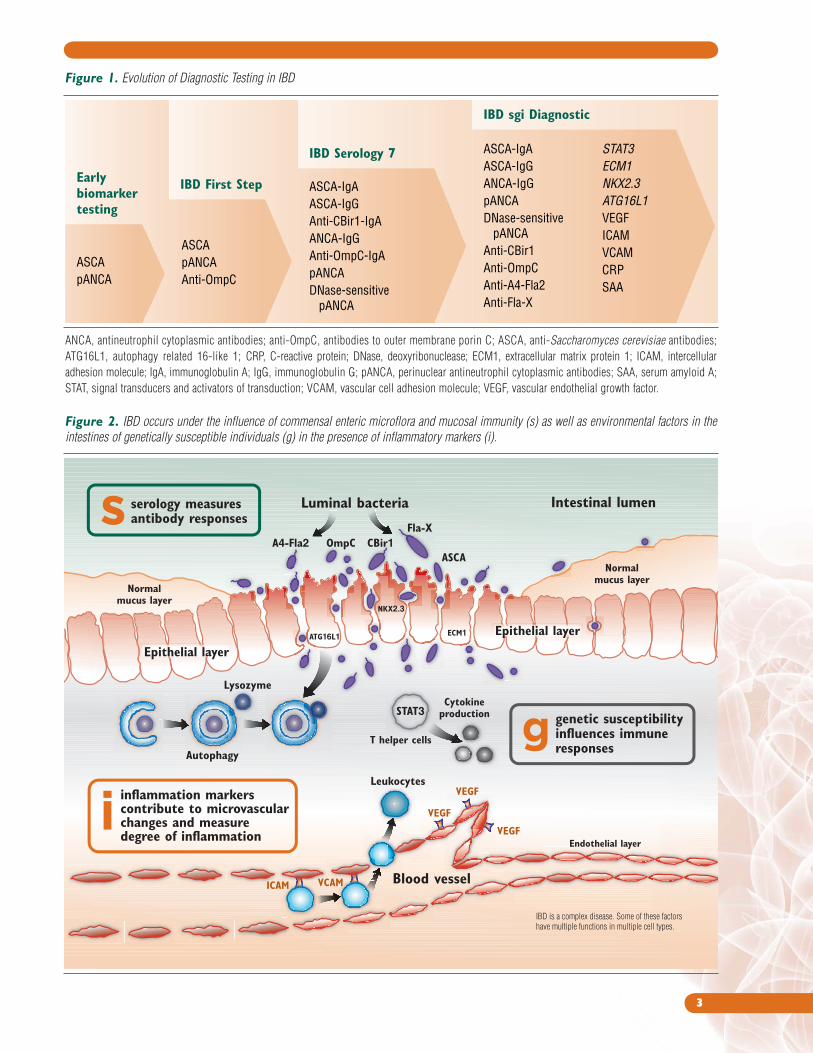
evolution of diagnostic test panels (Figure 1).
The IBD First Step (PROMETHEUS Laboratories, Inc., San Diego, CA)
improved the sensitivity of assays for ASCA and pANCA and added a
new biomarker, anti-OmpC (antibody to outer membrane porin C), to
increase sensitivity for CD.20 The next generation of this test, IBD
Serology 7, was augmented with the recently discovered biomarker
anti-CBir1 and incorporated the use of a Smart Diagnostic Algorithm
to interpret the overall (versus individual) biomarker interaction
results, thus providing a more accurate assessment than previous
serologic assays.20
In efforts to continue to improve the performance of testing for
IBD, as well as the ability to better differentiate between CD and
UC, PROMETHEUS Laboratories has now developed the novel
and innovative PROMETHEUS IBD sgi Diagnostic test. Designed as
an adjunctive test for the workup of suspected IBD and for the
differentiation of CD and UC in known IBD, the PROMETHEUS IBD sgi
Diagnostic test contains three major classes of biomarkers —serologic,
genetic, and inflammatory — that correspond to the complex
multidimensional nature of IBD (Figure 2). Genetic markers chosen to
be included in the PROMETHEUS IBD sgi Diagnostic test have relevance
to various physiologic aspects known to be compromised or impaired in
IBD, thus leading to disease susceptibility. Specifically these processes
include defective bacterial handling/autophagy, dysregulated signaling
pathways, and impaired epithelial barrier function.
3
Figure 1. Evolution of Diagnostic Testing in IBD
Earlybiomarkertesting
ASCA pANCA
ASCA pANCAAnti-OmpC
ASCA-IgAASCA-IgGAnti-CBir1-IgAANCA-IgGAnti-OmpC-IgApANCADNase-sensitive pANCA
ASCA-IgAASCA-IgGANCA-IgGpANCADNase-sensitive pANCAAnti-CBir1 Anti-OmpC Anti-A4-Fla2Anti-Fla-X
IBD sgi Diagnostic
IBD Serology 7
IBD First Step
STAT3ECM1NKX2.3ATG16L1VEGFICAMVCAMCRPSAA
Figure 2. IBD occurs under the influence of commensal enteric microflora and mucosal immunity (s) as well as environmental factors in the
intestines of genetically susceptible individuals (g) in the presence of inflammatory markers (i).
ANCA, antineutrophil cytoplasmic antibodies; anti-OmpC, antibodies to outer membrane porin C; ASCA, anti-Saccharomyces cerevisiae antibodies;
ATG16L1, autophagy related 16-like 1; CRP, C-reactive protein; DNase, deoxyribonuclease; ECM1, extracellular matrix protein 1; ICAM, intercellular
adhesion molecule; IgA, immunoglobulin A; IgG, immunoglobulin G; pANCA, perinuclear antineutrophil cytoplasmic antibodies; SAA, serum amyloid A;
STAT, signal transducers and activators of transduction; VCAM, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor.

4
The text that follows describes the specific serologic, genetic, and
inflammatory markers that make up the PROMETHEUS IBD sgi
Diagnostic test.
Serologic Markers
Serologic markers are important in CD because their expression
represents the host response to translocation of intestinal pathogens
into the bloodstream as a result of breakdown of the gut mucosal
barrier. Investigations to uncover the underlying cause of the abnormal
intestinal inflammatory reaction that characterizes IBD have led to the
discovery of antibodies that are present specifically in the blood of
patients with CD or UC.25 Initially, research focused on ASCA, which
was associated with CD, and pANCA, which was associated with UC.
Given the heterogeneity of IBD, it soon became clear that these
biomarkers would not lead to a diagnosis of IBD in all patients who
have the disorder, as about 30% are negative for both ASCA and
pANCA immune responses.26 Additionally, it was noted that a subset of
CD patients have seroreactivity to pANCA and phenotypically have
“UC-like CD.” Continued research expanded the number of serologic
markers that are associated with IBD and/or have the capacity to better
differentiate CD and UC. The resulting set of serologic markers that now
compose the PROMETHEUS IBD sgi Diagnostic test are detailed below.
Yeasts
ASCA-IgA and -IgG
ASCA (anti-Saccharomyces cerevisiae antibodies) are antibodies
directed against mannose sequences from phosphopeptidomannan
comprising the cell wall of the baker’s and brewer’s yeast Saccharomyces
cerevisiae.21 Candida albicans has also been shown to express ASCA
epitopes on mannoproteins similar to that found in S cerevisiae.27
ASCA has been shown to correlate with C albicans colonization in
healthy relatives of patients with CD but not in CD patients themselves.28
The prevalence of ASCA positivity is highest in CD patients, ranging
from 35% to 76%, but also occurs in 5% to 15% of patients with UC
and in up to 5% of healthy controls.29,30
Bacterial Proteins
Anti-OmpC
OmpC (outer membrane porin C) is a bacterial antigen originally
isolated from Escherichia coli.31 This antigen results in
immunoglobulin G and A (IgG and IgA) antibody responses (anti-
OmpC)30 in 37% to 55% of adults with CD.32-34 In a cohort of
children with CD, 17% were found to have a seropositive response
to anti-OmpC.35 In another evaluation of antibody responses in
children and young adults (age ≤24.0 yr) with CD or UC, anti-OmpC
positivity was found in 25% of patients with CD, 11% of patients with
UC, and 5% of healthy controls.36 While anti-OmpC as a stand-alone
marker lacks sensitivity in CD and UC, the addition of this marker to
a panel that includes ANCA, ASCA-IgG, and ASCA-IgA improves
sensitivity for IBD. Indeed, a four-marker diagnostic panel including
anti-OmpC identified CD in 65% of children and UC in 74% of children,
with a 94% specificity.36
Anti-CBir1
The human intestinal tract is colonized by a vast assortment of
commensal microbial species, while also being occasionally exposed to
bacteria that are potentially pathogenic. Subsets of both commensal and
pathogenic bacteria are motile as a result of the expression of one or more
flagella. The primary structural component of bacterial flagella is the
35-50 kDa protein flagellin. The region of the flagellin molecule involved
in its polymerization to flagella is highly conserved among various
bacteria. Flagellin has been found to be a major target of both innate37
and adaptive immune responses38,39 that are associated with IBD.
In 2004, Lodes and colleagues used serologic expression cloning
to search for bacterial antigens relevant to IBD and identified flagellins
as among the immunodominant antigens of the microbiota.38 The specific
flagellin expression clone, CBir1, was subsequently noted to induce a
strong antibody response (anti-CBir1) in four genetically distinct
mouse models of IBD.38 In humans, these researchers noted significantly
higher levels of anti-CBir1 in patients with CD than in patients with UC
and controls, suggesting that this response might be useful in the
diagnosis of IBD and in the differentiation of CD and UC.38 Further
research by Targan and colleagues found that anti-CBir1 was expressed
in 50% to 55% of patients with CD.40 Controlling for the previously
described microbial antigens ASCA (-IgG and -IgA), anti-OmpC, and
anti-I2 (antibody to the I2 bacterial sequence found in Pseudomonas
fluorescens), these researchers found that anti-CBir1 was independently
associated with CD (P<.001) and its expression level was unrelated to
any one of the other four individual antimicrobial antibodies associated
with CD; as a result, anti-CBir1 reactivity defined another subpopulation
of CD patients that was pathophysiologically distinct.40 Anti-CBir1
expression was also found in approximately 50% of CD patients who
did not express ASCA; 40% to 44% of patients with CD whose sole
antibody response was to pANCA were found to express anti-CBir1
compared with only 4% of patients with UC who were otherwise solely
positive for pANCA.40 Furthermore, anti-CBir1 expression was found
in 40% of patients who lacked a serologic response to any of the
other antigens studied.40 More recently, Markowitz and colleagues
investigated immune responses in 705 children, age 0 to 15 years, with
CD. This group found that 52% were seronegative for pANCA, ASCA-
IgA and -IgG, and anti-OmpC.35 This percentage of seronegative
patients fell to 27% when assessment of anti-CBir1 was added
(P < .0001), thus identifying a significant subset of children with CD
who were otherwise seronegative.35

5
(ie, DNase-sensitive pANCA) appears to be a dominant characteristic
of UC-specific pANCA, distinguishing the disorder from primary
sclerosing cholangitis and type 1 autoimmune hepatitis.45
Genetic Markers
GWAS methodology has revolutionized research into IBD.46 The
GWAS approach is very effective in the detection of relatively common
gene variants and forms the basis for fine-mapping of identified genes
and research into their function. This has increased the knowledge of
the various signaling pathways involved in the disorder(s) and also
has implications for the development of future treatments. Genes that
have been definitively associated with IBD, CD, or UC are listed in
Figure 3 and the list is expanding rapidly.
ATG16L1
Defective bacterial handling has become one of the most consistent
focuses in the study of CD pathogenesis.46 One gene that is related to
the process of autophagy, ATG16L1 (autophagy related 16-like 1 gene),
has been associated with CD, autophagy has many physiologic roles
in health and disease, including acting as a “housekeeping” mechanism
by which cells digest parts of their own cytoplasm for removal or
providing an innate immune mechanism by which the host can
combat intracellular bacteria.47
ATG16L1 was first identified by Hampe and colleagues as a
susceptibility gene for CD in a GWAS of 19,779 non-synonymous single
nucleotide polymorphisms (SNPs) present in 735 patients with CD and
in 368 controls.48 Non-synonymous SNPs are SNPs that have different
alleles that encode different amino acids.49 Analysis showed that the
marker rs2241880 encoded a threonine-to-alanine substitution at amino
acid position 300 (T300A), which was correlated with the incidence of
CD in one British and two German studies of CD.48 No correlation
between the rs2241880 marker and UC was noted. Cummings and
colleagues50 attempted to replicate the findings of Hampe and
colleagues48 in a cohort of 648 CD cases, 677 UC cases, and 1190
controls that were well characterized. This group found that carriage
of the G risk allele of rs2241880 was strongly associated with CD
(P=2.33 × 10-7; odds ratio [OR] 1.45 [1.25–1.67]), thus reproducing
the Hampe nsSNP GWAS findings. A recent meta-analysis of 25 studies
(published up to June 1, 2009)51 evaluating the association of ATG16L1
and IBD found a positive association between the T300A polymorphism
and susceptibility to CD (OR 1.32; 95% confidence interval [CI]
1.26–1.39; P< .00001). ATG16L1 was also associated with the risk of
childhood-onset CD but not childhood-onset UC.51
The same polymorphism was recently evaluated in a Spanish cohort
comprising 557 CD and 425 UC patients compared with 672 ethnically
matched controls52 and was found to be strongly associated with
susceptibility to CD (OR 1.62) but showed no association with UC.
Anti-Fla-X and Anti-A4-Fla2
In their research, Lodes and colleagues cloned a second reactive
flagellin (Fla-X) that was highly homologous to CBir1 (83.5%
similarity at the NH2 conserved domain) and, similar to the findings
with CBir1, strong immune reactivity directed toward the NH2 terminal
end of Fla-X was noted in both a colitic mouse model and in patients
with CD.38 The level of response to Fla-X observed in sera from
patients with CD was significantly higher than that found in sera from
normal controls and patients with UC. Given that more than 50% of
the UC sera tested came from patients with a Truelove and Witts
severity index indicative of moderate to active disease (ie, >7), these
antiflagellin responses have potential not only in the diagnosis of IBD
but also in the discrimination of UC and CD.38
Since prior phylogenic analyses have linked the Clostridium
phylogenetic cluster XIVa to the origin of flagellins.41 Duck and colleagues
investigated this cluster and characterized a number of flagellated
bacteria from it.42 These investigators found that the A4 bacterial strain
expresses a flagellin, A4-Fla2, which has a very similar amino acid
sequence to the flagellin Fla-X43 to which patients with CD have
demonstrated seropositivity.38 Subsequently, Schoepfer and colleagues44
assessed seroreactivity to A4-Fla2 and Fla-X in a population of well-
characterized CD (n=252) or UC (n=53) patients and in healthy controls
(n=43). Seropositivity to A4-Fla2 and Fla-X was found in 59% and 57%
of patients with CD, respectively, 6% each in patients with UC, and 0%
and 2% of healthy controls, respectively.44 The serologic response
generated against either flagellin in patients with CD was most frequently
IgG, being found in 94% of those positive for anti-A4-Fla2 and in 93%
of those positive for anti-Fla-X. An IgA response was found in < 20% of
CD patients for either flagellin.44 No correlation was found between
disease duration and serologic response to A4-Fla2 or Fla-X. This
research has underscored the dominance of flagellin as an antigen in CD.
Given the slightly higher rate of seroreactivity to Fla-X and A4-Fla2 than
to CBir1 noted in CD patients, these two flagellins may enhance the
ability to diagnose CD over and above assessment of anti-CBir1.
Autoantibodies
ANCA, pANCA, and DNase-sensitive pANCApANCA (perinuclear antineutrophil cytoplasmic antibodies) are
autoantibodies directed against a component of neutrophil granules.30
Distinct subgroups of ANCAs have been noted with indirect
immunofluorescence based on their individual staining pattern.29 The
use of immunofluorescence has revealed a perinuclear pattern of
staining (pANCA) in association with many different diseases including
vasculitides and collagenous and eosinophilic colitides.23 The pANCA
staining pattern has also been found in 30% to 83% of patients with UC,
6% to 20% of patients with CD, and up to 2.5% of healthy controls.29,30
Loss of this antigenic response after DNase digestion of neutrophils

6
STAT3
The STAT (signal transducers and activators of transcription)-
Janus kinase pathway controls signal transduction between cell surface
receptors and the nucleus. STAT3, an inducible DNA binding protein that
binds to the interleukin (IL)-6 responsive element within the promoters
of hepatic acute phase protein genes,58 is activated by a wide variety of
cytokines and growth factors.59 Upon activation, STAT3 is phosphorylated,
resulting in dimerization; the dimer migrates to the nucleus where it
induces the expression of genes that have roles in many biologic
functions including cell growth, antiapoptosis and proapoptosis, cell
motility, negative feedback (suppression of cytokine production),
regulatory cytokine production, and antibacterial activity.59-62
It is well established that STAT3 is necessary for the signaling
of proinflammatory cytokines such as IL-6, which is involved in
the pathogenesis of both multiple sclerosis and IBD.63 STAT3 is
also important for multiple aspects of the biology of Th17 cells,
which, in turn, are critically important as mediators of a number of
human inflammatory diseases.64 In mouse models, STAT3 plays
distinct roles in both innate and acquired immunity. STAT3-mediated
activation of acquired immune response plays a pathogenic role in
NKX2.3
Signaling pathways that are critical to normal mammalian gut
development are dysregulated in IBD. NKX2.3 (NK2 transcription
factor-related, locus 3) is a member of the NKX family of
homeodomain-containing transcription factors implicated in cell
type specification and maintenance of differentiation in several
types of tissue.46 NKX2.3 is expressed in small blood vessel
endothelial cells and intestinal lamina propia mesenchymal cells.46
Evidence from mouse studies suggests that abnormal expression
of NKX2.3 alters the migration of antigen-responsive lymphocytes
in the gut and influences the inflammatory response in the
intestines.53 Susceptibility for CD was demonstrated with the
rs10883365 variant of NKX2.3 in multiple GWAS studies in which
ORs between 1.3 and 1.5 were observed.53-55 Additionally, in the first
non-synonymous SNP scan in UC, Fisher and colleagues found an
association between the rs10883365 NKX2.3 variant and UC
(P = 3.3×104).56 Meggyesi and colleagues later found this association
in a group of Czech and Hungarian IBD patients (increased risk for
UC, P= .003 after Bonferroni correction; OR 1.36; 95% CI 1.13–
1.63),57 demonstrating that NKX2.3 represents an IBD locus.
Figure 3. Genes Associated with Inflammatory Bowel Disease, Crohn’s Disease, and Ulcerative Colitis
(Adapted with permission from Thompson and Lees .68)
UC
IBD
CD
ECM1IL10IFN-gammaIL22FCGR2AIL2/IL21IL17REL/PIM3CAPN10/GPR35
LAMB1/SCL26A3OTUD3/PLA2G2EARPC2HNF4-alpha CDH1IL2613q121p36
CARD9CEP72/TPPP13q13SMURF1/KPNA7IFN-gamma/IL26/IL22
ATG16L1NOD2C11orf30ZNF365CCR61q246q217p12
13q14ICOSLGGPX4GCKRSLC22A23NLRP3PTPN22LRRK2
1q326q258q2418q11ITLN1ORMDL3CCL2/CCL7/CCL11/CCL8
STAT3NKX2.3IL23RIL12/p40JAK23p21/MST1REL/PUS10ZMIZ1MTMR3
IRGMORMDL321q21MHC*IL18RAPLYRM4CDKAL1TNFRSF6B (DCR3)PSMG1
CCNYIL27IBD5PUS10TNFSF15 (TL1A)19q13HORMAD2/MTMR3PTGER4
*Colonic disease

7
gut tissues causing gut inflammation and injury.74,75 Inflammatory
characteristics shared between patients with IBD and those with
other chronic immune disturbances include immune activation,
the infiltration of leukocytes into tissues, and an increased
vascular density.75
Intercellular Adhesion Molecule 1 and
Vascular Cell Adhesion Molecule 1
Dense inflammatory infiltrates, including monocytes,
lymphocytes, and neutrophils, characterize IBD and are seen in
distinct distributions in both CD and UC.76 Infiltration of these cells
is stimulated both by various cytokines and by the interactions
that occur between adhesion molecules expressed on inflammatory
cells in the circulation and their integrin receptors on localized
target cells.77
Intercellular adhesion molecule 1 (ICAM-1) and vascular cell
adhesion molecule 1 (VCAM-1) are cytokine-inducible cell surface
glycoproteins that belong to the immunoglobulin supergene
family.78 Soluble ICAM and VCAM are also detected in serum and
are believed to arise from proteolytic cleavage of the cell surface
molecules. ICAM-1 is expressed at low levels on endothelial,
epithelial, and other cells as well as on lymphocytes and
monocytes.76 The transcription of ICAM-1 is upregulated by
proinflammatory cytokines.79,80 In inflamed tissues, increased
expression of ICAM-1 on endothelial cells promotes the
recruitment of inflammatory cells expressing its ligands (any
substance that binds specifically and reversibly to another
chemical entity to form a larger complex), leukocyte function
antigen-1, or macrophage activation complex-1, thus propagating
the inflammatory process. In the rat, interferon-γ– induced
expression of ICAM-1 has been noted to be higher in deep
mucosa/submucosa and muscle layer microcirculations than in
the superficial mucosa—layers of the gut involved in the
characteristic pathology of CD—where it may contribute to
leukocyte-endothelial interactions.81
The expression of VCAM-1 is found on activated endothelial
cells, macrophages, dendritic cells, and fibroblasts. Ligands for
VCAM-1 include the integrins α4/β4 (very late antigen 4) and
α4/β7, which are present on monocytes, lymphocytes, and
eosinophils. Upon binding to its ligands, VCAM-1 modulates
leukocyte adhesion to endothelial cells and migration to sites of
inflammation.82,83
Jones and colleagues investigated circulating concentrations
of ICAM-1 and VCAM-1 in 43 patients with IBD (22 with CD and
21 with UC) compared with healthy controls (n=90).77 Circulating
colitis by enhancing survival of pathogenic T cells and by
inducing tumor necrosis factor alpha (TNF-α). In contrast,
STAT3-mediated activation of innate response contributes to the
suppression of colitis by enhancing the mucosal repair and
induction of mucin production.65
Using GWAS, Barrett and colleagues found that the rs744166
variant of STAT3 was associated with CD susceptibility.66 This
association was confirmed in a Spanish cohort of patients reported by
Cenit and colleagues.67 This group analyzed polymorphisms in the
STAT3 region (rs3809758/rs744166/rs1026916/rs12948909). The
haplotype conformed by the risk alleles of each polymorphism was
significantly associated with both CD (P= .005; OR 1.25; 95% CI
1.06–1.46) and UC (P= .002; OR 1.19; 95% CI 1.02–1.38).67
ECM1
One of the most intriguing concepts that has emerged regarding
the genetics of UC has been its association with epithelial barrier
genes.68 Intercellular junctions between intestinal epithelial cells play
a crucial gatekeeping role in the gut by allowing the extraction of
needed nutrients while keeping pathogens out of the systemic
circulation. These tight junctions determine overall intestinal
permeability. In IBD, epithelial permeability is enhanced and is a
predictor of clinical relapse.69,70
In the first non-synonymous SNP scan for UC, Fisher and
colleagues were able to identify a previously unknown
susceptibility locus, ECM1, in UC.56 ECM1 is an epithelial barrier
gene that encodes the glycoprotein ECM1 (extracellular matrix
protein 1), which is expressed in the small and large intestines
where it interacts with the basement membrane and inhibits
matrix metalloproteinase 9. ECM1 also strongly activates nuclear
factor-κB (NF-κB) signaling, which is a key component in a variety
of regulatory pathways.71 Expression of ECM1 is upregulated in
esophageal squamous cell carcinoma and colorectal cancer.72
While an association with the ECM1 variant has been found for UC,
it was not observed when formally tested in an adequate sample of
CD subjects, implying that ECM1 may provide a specific
susceptibility association for UC alone.73
Inflammatory Markers
The presence of intestinal inflammation is a primary criterion
for making the diagnosis of IBD. Under normal conditions, the
intestinal mucosa is in a state of “controlled” inflammation that is
regulated by a delicate balance between proinflammatory factors
and anti-inflammatory factors. In IBD, immune response activation
causes the release of inflammatory cytokines (eg, TNF-α) and
growth factors (eg, vascular endothelial growth factor [VEGF]) into

8
median levels of ICAM-1 were significantly higher in patients
with active CD vs controls (273 vs 168 ng/mL; P < .003). Median
circulating VCAM-1 concentrations were significantly higher in
active vs inactive UC patients (165 vs 117 U/mL; P < 005) and
were also significantly elevated compared with VCAM-1
concentrations in patients with active CD (124 U/mL; P < .02)
and in healthy controls (50 U/mL; P < .0001).77 It should be noted
that increased levels of ICAM and VCAM are not specific for
IBD and can be found in other inflammatory, infectious, and
neoplastic diseases.84
Vascular Endothelial Growth Factor
Angiogenesis, the growth of new blood vessels from preexisting
vasculature, is important in embryogenesis, tissue growth, wound
healing, and ovulation. Angiogenesis plays a key role in pathologic
processes as well, including cancer, ischemic cardiovascular
disease, diabetic retinopathy, and in chronic inflammatory diseases
such as IBD.85-87
VEGFs represent a family of factors that mediate angiogenesis.
Stimulation of angiogenesis is known to occur as a result of hypoxia
or mechanical shear stress and stretch. Inflammation that underlies
the pathology of IBD may promote angiogenesis in a number of
ways. Many inflammatory cytokines upregulated in IBD are
proangiogenic including IL-17 and TNF-α.88,89 Inflamed tissue,
such as that seen in IBD, is often hypoxic,90 and this hypoxia can
induce angiogenesis through upregulation of factors such as VEGF,
basic fibroblast growth factor-1, TNF-α, and hypoxia-inducible
factor-1.91,92 Neovascularization may also be stimulated by
extravasated fibrinogen.93 Angiogenic factors produced by
macrophages, mast cells, lymphocytes, and fibroblasts can stimulate
growth of blood vessels.91 Additionally, shear stress on the
endothelium related to increased blood flow itself may stimulate
angiogenesis.94,95 Several studies have demonstrated that circulating
levels of VEGF are elevated in patients with IBD compared with
healthy or disease controls and that VEGF levels correlate with
disease activity in IBD.96-101
C-Reactive Protein
CRP, predominantly produced in liver cells, is one of at least
40 proteins that participate acutely in the inflammatory response.
Normally, the amount of CRP produced by hepatocytes is low
(<1 mg/L), but following an acute stimulus such as inflammation, the
production of CRP is rapidly increased under the influence of IL-6,
TNF-α, and IL-1β.102 While the function of CRP in vivo is not
completely understood, CRP is a useful marker for detecting infections
and inflammation due to its rapid rise and short half-life (~19 h)
and for assessing the effect of therapy on the underlying disease,
as resolution of the stimulus triggering its production normalizes
CRP levels.102,103
Shine and colleagues were the first to show that a CRP
increase can be used to differentiate IBD from functional bowel
disorders. In 82 patients with chronic abdominal symptoms, 19
were diagnosed with CD, 22 with UC, and 41 with a functional
bowel disorder. All of the 19 patients with CD and 59% of the 22
patients with UC showed increases in CRP compared with none
of the 41 patients with functional symptoms.104 Schoepfer and
colleagues found that CRP had a 64% sensitivity and a 92%
specificity in discriminating IBD (n=64) from IBS (n=30).105 A much
larger study performed by Henriksen and colleagues in Norway
provided CRP data on 454 patients with UC and 200 patients
with CD, which was measured at diagnosis, with follow-up at 1
and 5 years.103 Patients with CD were noted to have a stronger mean
CRP response when compared with those with UC at diagnosis
(CD 51 mg/L vs UC 18 mg/L; P< .001), at 1 year (CD 51 mg/L vs
UC 18 mg/L; P< .001) and at 5 years (CD 13 mg/L vs UC 6 mg/L;
P< .001) after diagnosis. Additionally, these investigators found
that CRP levels at the time of diagnosis were related to the extent
of disease in patients with UC, but no such association was found
in CD.103
Serum Amyloid A
Serum amyloid A (SAA) is a family of four proteins, including
the acute phase proteins SAA1 and SAA2, whose primarily hepatic
expression is markedly increased in response to a variety of inflammatory
stimuli,106,107 much like CRP. As SAA has a physiologic level that is
10 times higher than that for CRP, it may allow for easier detection
of slight elevations.108 Its longer half-life (24 hours) compared with
that of CRP (19 hours) may make SAA more sensitive as an acute
phase reactant. In contrast to differential CRP levels that have been
seen in CD versus UC (see above), Niederau and colleagues found
that SAA levels were similarly elevated in patients with both forms
of IBD.109 Jijon and colleagues have demonstrated that SAA activates
the NF-κB signaling pathway, highlighting the proinflammatory
capacity of SAA.110 NF-κB is known to be one of the major regulatory
components involved in the dysregulation of cytokine production
and signaling mechanisms by intestinal epithelial cells, lymphocytes,
and macrophages that has been implicated in the pathogenesis
of IBD.111
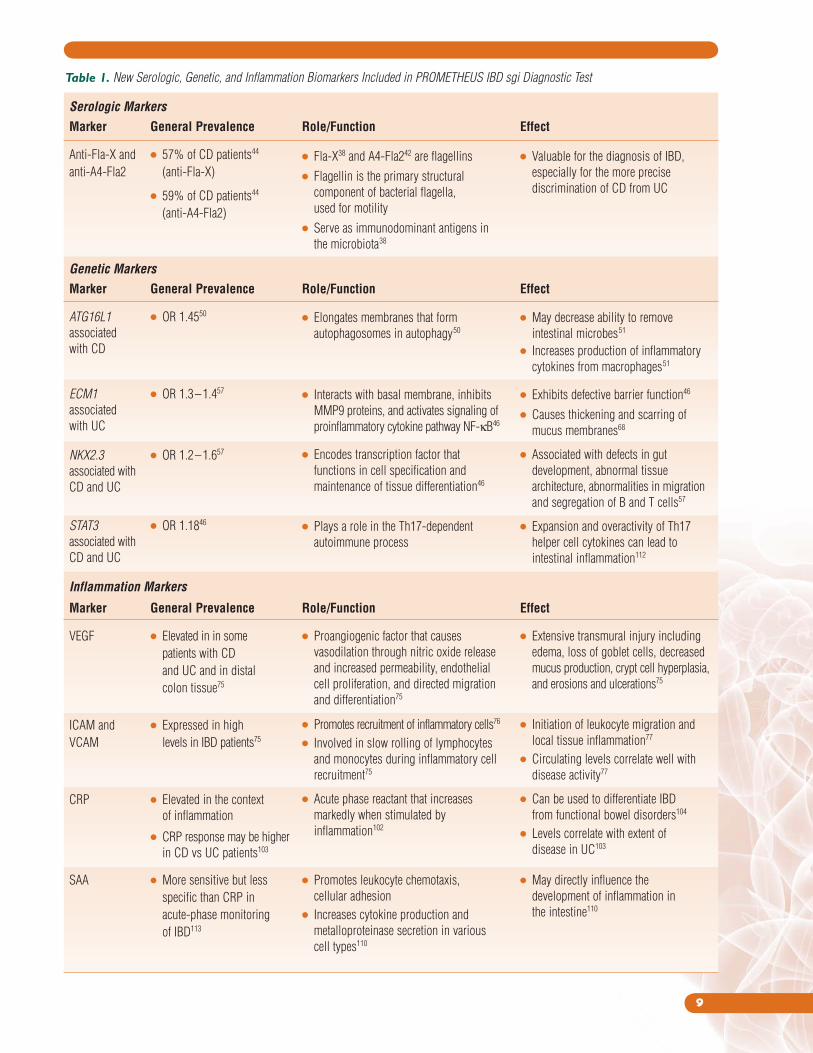
A summary of the new serologic, genetic, and inflammatory
biomarkers that have been included in the new PROMETHEUS IBD sgi
Diagnostic test can be found in Table 1.
9
Table 1. New Serologic, Genetic, and Inflammation Biomarkers Included in PROMETHEUS IBD sgi Diagnostic Test
Serologic Markers
Marker General Prevalence Role/Function Effect
Anti-Fla-X and ● 57% of CD patients44
anti-A4-Fla2 (anti-Fla-X)
● 59% of CD patients44
(anti-A4-Fla2)
Genetic Markers
Marker General Prevalence Role/Function Effect
ATG16L1 ● OR 1.4550
associated
with CD
ECM1 ● OR 1.3–1.457
associated
with UC
NKX2.3 ● OR 1.2–1.657
associated with
CD and UC
STAT3 ● OR 1.1846
associated with
CD and UC
Inflammation Markers
Marker General Prevalence Role/Function Effect
VEGF ● Elevated in in some
patients with CD
and UC and in distal
colon tissue75
ICAM and ● Expressed in high
VCAM levels in IBD patients75
CRP ● Elevated in the context
of inflammation
● CRP response may be higher
in CD vs UC patients103
SAA ● More sensitive but less
specific than CRP in
acute-phase monitoring
of IBD113
● Fla-X38 and A4-Fla242 are flagellins
● Flagellin is the primary structural
component of bacterial flagella,
used for motility
● Serve as immunodominant antigens in
the microbiota38
● Valuable for the diagnosis of IBD,
especially for the more precise
discrimination of CD from UC
● Elongates membranes that form
autophagosomes in autophagy50
● Interacts with basal membrane, inhibits
MMP9 proteins, and activates signaling of
proinflammatory cytokine pathway NF-κB46
● Encodes transcription factor that
functions in cell specification and
maintenance of tissue differentiation46
● Plays a role in the Th17-dependent
autoimmune process
● May decrease ability to remove
intestinal microbes51
● Increases production of inflammatory
cytokines from macrophages51
● Exhibits defective barrier function46
● Causes thickening and scarring of
mucus membranes68
● Associated with defects in gut
development, abnormal tissue
architecture, abnormalities in migration
and segregation of B and T cells57
● Expansion and overactivity of Th17
helper cell cytokines can lead to
intestinal inflammation112
● Proangiogenic factor that causes
vasodilation through nitric oxide release
and increased permeability, endothelial
cell proliferation, and directed migration
and differentiation75
● Promotes recruitment of inflammatory cells76
● Involved in slow rolling of lymphocytes
and monocytes during inflammatory cell
recruitment75
● Acute phase reactant that increases
markedly when stimulated by
inflammation102
● Promotes leukocyte chemotaxis,
cellular adhesion
● Increases cytokine production and
metalloproteinase secretion in various
cell types110
● Extensive transmural injury including
edema, loss of goblet cells, decreased
mucus production, crypt cell hyperplasia,
and erosions and ulcerations75
● Initiation of leukocyte migration and
local tissue inflammation77
● Circulating levels correlate well with
disease activity77
● Can be used to differentiate IBD
from functional bowel disorders104
● Levels correlate with extent of
disease in UC103
● May directly influence the
development of inflammation in
the intestine110
10
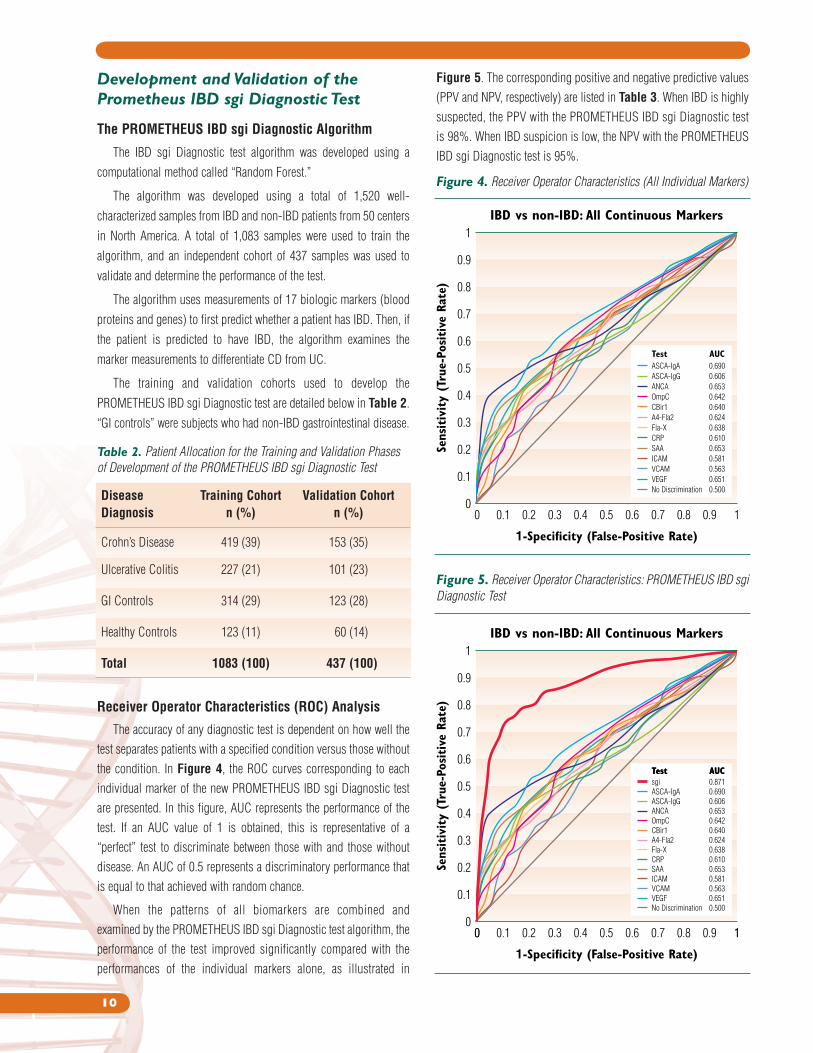
Figure 5. The corresponding positive and negative predictive values
(PPV and NPV, respectively) are listed in Table 3. When IBD is highly
suspected, the PPV with the PROMETHEUS IBD sgi Diagnostic test
is 98%. When IBD suspicion is low, the NPV with the PROMETHEUS
IBD sgi Diagnostic test is 95%.
Figure 4. Receiver Operator Characteristics (All Individual Markers)
1-Specificity (False-Positive Rate)
IBD vs non-IBD: All Continuous Markers
Sens
itiv
ity
(Tru
e-Po
sitive
Rat
e)
1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
00 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
AUCTest0.690
0.606
0.653
0.642
0.640
0.624
0.638
0.610
0.653
0.581
0.563
0.651
0.500
ASCA-IgA
ASCA-IgG
ANCA
OmpC
CBir1
A4-Fla2
Fla-X
CRP
SAA
ICAM
VCAM
VEGF
No Discrimination
Table 2. Patient Allocation for the Training and Validation Phases
of Development of the PROMETHEUS IBD sgi Diagnostic Test
Disease Training Cohort Validation Cohort
Diagnosis n (%) n (%)
Crohn’s Disease 419 (39) 153 (35)
Ulcerative Colitis 227 (21) 101 (23)
GI Controls 314 (29) 123 (28)
Healthy Controls 123 (11) 60 (14)
Total 1083 (100) 437 (100)
Development and Validation of the Prometheus IBD sgi Diagnostic Test
The PROMETHEUS IBD sgi Diagnostic Algorithm
The IBD sgi Diagnostic test algorithm was developed using a
computational method called “Random Forest.”
The algorithm was developed using a total of 1,520 well-
characterized samples from IBD and non-IBD patients from 50 centers
in North America. A total of 1,083 samples were used to train the
algorithm, and an independent cohort of 437 samples was used to
validate and determine the performance of the test.
The algorithm uses measurements of 17 biologic markers (blood
proteins and genes) to first predict whether a patient has IBD. Then, if
the patient is predicted to have IBD, the algorithm examines the
marker measurements to differentiate CD from UC.
The training and validation cohorts used to develop the
PROMETHEUS IBD sgi Diagnostic test are detailed below in Table 2.
“GI controls” were subjects who had non-IBD gastrointestinal disease.
Receiver Operator Characteristics (ROC) Analysis
The accuracy of any diagnostic test is dependent on how well the
test separates patients with a specified condition versus those without
the condition. In Figure 4, the ROC curves corresponding to each
individual marker of the new PROMETHEUS IBD sgi Diagnostic test
are presented. In this figure, AUC represents the performance of the
test. If an AUC value of 1 is obtained, this is representative of a
“perfect” test to discriminate between those with and those without
disease. An AUC of 0.5 represents a discriminatory performance that
is equal to that achieved with random chance.
When the patterns of all biomarkers are combined and
examined by the PROMETHEUS IBD sgi Diagnostic test algorithm, the
performance of the test improved significantly compared with the
performances of the individual markers alone, as illustrated in
Figure 5. Receiver Operator Characteristics: PROMETHEUS IBD sgi
Diagnostic Test
1-Specificity (False-Positive Rate)
Sens
itiv
ity
(Tru
e-Po
sitive
Rat
e)
1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
00 1
AUCTest0.871sgi
AUCTest
0.690
0.606
0.653
0.642
0.640
0.624
0.638
0.610
0.653
0.581
0.563
0.651
0.500
ASCA-IgA
ASCA-IgG
ANCA
OmpC
CBir1
A4-Fla2
Fla-X
CRP
SAA
ICAM
VCAM
VEGF
No Discrimination
IBD vs non-IBD: All Continuous Markers
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1

11
Table 3. Positive and Negative Predictive Values for the
PROMETHEUS IBD sgi Diagnostic Test
Prevalence, % IBD, %
Positive 15 56
predictive value (PPV)
50 88
85 98
Negative 15 95
predictive value (NPV)
50 77
85 37
Conclusion
The diagnosis of IBD and the differentiation between CD and UC
can pose a number of challenges in the clinical setting. Invasive
radiographic and endoscopic diagnostic investigations are not
without complications, and there is a subset of patients for whom
these tests do not result in a definitive diagnosis. Diagnostic delays
may adversely affect the management of IBD and illustrate the need for
diagnostic accuracy. Ongoing research into the underlying
pathophysiology of IBD has helped to identify more markers,
including serologic, genetic, and inflammatory markers, that may
prove to be a useful, noninvasive adjunctive diagnostic tool when IBD
is suspected. Helping to provide actionable diagnostic information
has the potential to allow for personalized treatment planning and
better patient outcomes.

12

13
18. Russell RK, Satsangi J. IBD: a family affair. Best Pract Res Clin
Gastroenterol. 2004;18(3):525-539.
19. Human genome project information: SNP fact sheet. U.S.
Department of Energy Genome Program. Available at: http://
www.ornl.gov/sci/techresources/Human_Genome/faq/snps.
shtml. Accessed: September 27, 2011.
20. Seidman EG. Emerging prognostic markers in IBD. Gastroenterol
Hepatol. 2007;3(12 Suppl 36):5-6.
21. Sendid B, Colombel JF, Jacquinot PM, et al. Specific antibody
response to oligomannosidic epitopes in Crohn’s disease. Clin
Diagn Lab Immunol. 1996;3(2):219-226.
22. Vasiliauskas EA, Plevy SE, Landers CJ, et al. Perinuclear
antineutrophil cytoplasmic antibodies in patients with Crohn’s
disease define a clinical subgroup. Gastroenterology. 1996;110(6):
1810-1819.
23. Reese GE, Constantinides VA, Simillis C, et al. Diagnostic precision
of anti-Saccharomyces cerevisiae antibodies and perinuclear
antineutrophil cytoplasmic antibodies in inflammatory bowel
disease. Am J Gastroenterol. 2006;101(10):2410-2422.
24. Joossens S, Reinisch W, Vermeire S, et al. The value of serologic
markers in indeterminate colitis: a prospective follow-up study.
Gastroenterology. 2002;122(5):1242-1247.
25. Dubinsky M. What is the role of serological markers in IBD?
Pediatric and adult data. Dig Dis. 2009;27(3):259-268.
26. Dubinsky MC, Hanauer SB, [section editor]. Emerging strategies in
the use of IBD-related serologic markers (in Advances in IBD [Q&A
section]). Gastroenterology & Hepatology. 2008;4(8):1-3.
27. Standaert-Vitse A, Jouault T, Vandewalle P, et al. Candida albicans
is an immunogen for anti-Saccharomyces cerevisiae antibody
markers of Crohn’s disease. Gastroenterology. 2006;130(6):
1764-1775.
28. Standaert-Vitse A, Sendid B, Joossens M, et al. Candida albicans
colonization and ASCA in familial Crohn’s disease. Am J
Gastroenterol. 2009;104(7):1745-1753.
29. Sandborn WJ. Serologic markers in inflammatory bowel disease:
state of the art. Rev Gastroenterol Disord. 2004;4(4):167-174.
30. Peyrin-Biroulet L, Standaert-Vitse A, Branche J, Chamaillard M.
IBD serological panels: facts and perspectives. Inflamm Bowel Dis.
2007;13(12):1561-1566.
31. Cohavy O, Bruckner D, Gordon LK, et al. Colonic bacteria express
an ulcerative colitis pANCA-related protein epitope. Infect Immun.
2000;68(3):1542-1548.
32. Arnott ID, Landers CJ, Nimmo EJ, et al. Sero-reactivity to microbial
components in Crohn’s disease is associated with disease severity
and progression, but not NOD2/CARD15 genotype. Am J
Gastroenterol. 2004;99(12):2376-2384.
33. Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance
evidenced by Crohn’s disease-associated immune responses to auto-
and microbial antigens. Gastroenterology. 2002;123(3):689-699.
34. Mow WS, Vasiliauskas EA, Lin YC, et al. Association of antibody
responses to microbial antigens and complications of small bowel
Crohn’s disease. Gastroenterology. 2004;126(2):414-424.
References
01. Matricon J, Barnich N, Ardid D. Immunopathogenesis of
inflammatory bowel disease. Self Nonself. 2010;1(4):299-309.
02. Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;
347(6):417-429.
03. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of
inflammatory bowel disease. Nature. 2007;448(7152):427-434.
04. Sands BE. From symptom to diagnosis: clinical distinctions among
various forms of intestinal inflammation. Gastroenterology. 2004;
126(6):1518-1532.
05. Ruemmele FM, Targan SR, Levy G, et al. Diagnostic accuracy of
serological assays in pediatric inflammatory bowel disease.
Gastroenterology. 1998;115(4):822-829.
06. Seidman EG. Recent advances in the diagnosis and treatment of
pediatric inflammatory bowel disease. Curr Gastroenterol Rep.
2000;2(3):248-252.
07. Lichtenstein GR, Hanauer SB, Sandborn WJ, and the Practice
Parameters Committee of the American College of Gastroenterology.
Management of Crohn’s disease in adults. Am J Gastroenterol.
2009;104(2):465-483.
08. Nikolaus S, Schreiber S. Diagnostics of inflammatory bowel
disease. Gastroenterology. 2007;133(5):1670-1689.
09. Fletcher JG, Fidler JL, Bruining DH, Huprich JE. New concepts in
intestinal imaging for inflammatory bowel diseases. Gastroenterology.
2011;140(6):1795-1806.
10. Cheifetz AS, Kornbluth AA, Legnani P, et al. The risk of retention of
the capsule endoscope in patients with known or suspected Crohn’s
disease. Am J Gastroenterol. 2006;101(10):2218-2222.
11. Lohsiriwat V. Colonoscopic perforation: incidence, risk factors,
management and outcome. World J Gastroenterol. 2010;16(4):
425-430.
12. Vavricka SR, Spigaglia SM, Rogler G, et al. Systematic evaluation of
risk factors for diagnostic delay in inflammatory bowel disease.
Inflamm Bowel Dis. 2011; epub ahead of print.
13. Pezim ME, Pemberton JH, Beart RW, Jr., et al. Outcome of
"indeterminant" colitis following ileal pouch-anal anastomosis. Dis
Colon Rectum. 1989;32(8):653-658.
14. Wolfson DM, Sachar DB, Cohen A, et al. Granulomas do not affect
postoperative recurrence rates in Crohn’s disease. Gastroenterology.
1982;83(2):405-409.
15. Papadakis KA, Tabibzadeh S. Diagnosis and misdiagnosis of
inflammatory bowel disease. Gastrointest Endosc Clin North Am.
2002;12(3):433-449.
16. Orholm M, Binder V, Sorensen TI, et al. Concordance of inflammatory
bowel disease among Danish twins. Results of a nationwide study.
Scand J Gastroenterol. 2000;35(10):1075-1081.
17. Tysk C, Lindberg E, Jarnerot G, Floderus-Myrhed B. Ulcerative
colitis and Crohn’s disease in an unselected population of
monozygotic and dizygotic twins. A study of heritability and the
influence of smoking. Gut. 1988;29(7):990-996.

14
35. Markowitz J, Kugathasan S, Dubinsky M, et al. Age of diagnosis
influences serologic responses in children with Crohn’s disease: a
possible clue to etiology? Inflamm Bowel Dis. 2009;15(5):714-719.
36. Zholudev A, Zurakowski D, Young W, et al. Serologic testing with
ANCA, ASCA, and anti-OmpC in children and young adults with
Crohn’s disease and ulcerative colitis: diagnostic value and
correlation with disease phenotype. Am J Gastroenterol. 2004;
99(11):2235-2241.
37. Zeng H, Carlson AQ, Guo Y, et al. Flagellin is the major
proinflammatory determinant of enteropathogenic Salmonella.
J Immunol. 2003;171(7):3668-3674.
38. Lodes MJ, Cong Y, Elson CO, et al. Bacterial flagellin is a dominant
antigen in Crohn disease. J Clin Invest. 2004;113(9):1296-1306.
39. Sitaraman SV, Klapproth JM, Moore DA, III, et al. Elevated flagellin-
specific immunoglobulins in Crohn’s disease. Am J Physiol
Gastrointest Liver Physiol. 2005;288(2):G403-G406.
40. Targan SR, Landers CJ, Yang H, et al. Antibodies to CBir1 flagellin
define a unique response that is associated independently with
complicated Crohn’s disease. Gastroenterology. 2005;128(7):
2020-2028.
41. Collins MD, Lawson PA, Willems A, et al. The phylogeny of the
genus Clostridium: proposal of five new genera and eleven new
species combinations. Int J Syst Bacteriol. 1994;44(4):812-826.
42. Duck LW, Walter MR, Novak J, et al. Isolation of flagellated bacteria
implicated in Crohn’s disease. Inflamm Bowel Dis. 2007;13(10):
1191-1201.
43. Schoepfer AM, Schaffer T, Seibold-Schmid B, et al. Antibodies to
flagellin indicate reactivity to bacterial antigens in IBS patients.
Neurogastroenterol Motil. 2008;20:1110-1118.
44. Schoepfer AM, Schaffer T, Mueller S, et al. Phenotypic associations
of Crohn’s disease with antibodies to flagellins A4-Fla2 and Fla-
X, ASCA, p-ANCA, PAB, and NOD2 mutations in a Swiss Cohort.
Inflamm Bowel Dis. 2009;15(9):1358-1367.
45. Vidrich A, Lee J, James E, et al. Segregation of pANCA antigenic
recognition by DNase treatment of neutrophils: ulcerative colitis,
type 1 autoimmune hepatitis, and primary sclerosing cholangitis.
J Clin Immunol. 1995;15(6):293-299.
46. Lees CW, Satsangi J. Genetics of inflammatory bowel disease:
implications for disease pathogenesis and natural history. Expert
Rev Gastroenterol Hepatol. 2009;3(5):513-534.
47. Deretic V. Links between autophagy, innate immunity, inflammation
and Crohn’s disease. Dig Dis. 2009;27(3):246-251.
48. Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association
scan of nonsynonymous SNPs identifies a susceptibility variant for
Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207-211.
49. SNP class definition. National Center for Biotechnology
Information; U.S. National Library of Medicine. Available at:
http://www.ncbi.nlm.nih.gov/books/NBK44488/. Accessed:
September 27, 2011.
50. Cummings JR, Cooney R, Pathan S, et al. Confirmation of the role
of ATG16L1 as a Crohn’s disease susceptibility gene. Inflamm
Bowel Dis. 2007;13(8):941-946.
51. Cheng JF, Ning YJ, Zhang W, et al. T300A polymorphism of
ATG16L1 and susceptibility to inflammatory bowel diseases: a
meta-analysis. World J Gastroenterol. 2010;16(10):1258-1266.
52. Palomino-Morales RJ, Oliver J, Gomez-Garcia M, et al. Association
of ATG16L1 and IRGM genes polymorphisms with inflammatory
bowel disease: a meta-analysis approach. Genes Immun.
2009;10(4):356-364.
53. Wellcome Trust Case Control Consortium. Genome-wide association
study of 14,000 cases of seven common diseases and 3,000 shared
controls. Nature. 2007;447(7145):661-678.
54. Yamazaki K, Takahashi A, Takazoe M, et al. Positive association of
genetic variants in the upstream region of NKX2-3 with Crohn’s
disease in Japanese patients. Gut. 2009;58(2):228-232.
55. Weersma RK, Stokkers PC, Cleynen I, et al. Confirmation of multiple
Crohn’s disease susceptibility loci in a large Dutch-Belgian cohort.
Am J Gastroenterol. 2009;104(3):630-638.
56. Fisher SA, Tremelling M, Anderson CA, et al. Genetic determinants
of ulcerative colitis include the ECM1 locus and five loci implicated
in Crohn’s disease. Nat Genet. 2008;40(6):710-712.
57. Meggyesi N, Kiss LS, Koszarska M, et al. NKX2-3 and IRGM
variants are associated with disease susceptibility to IBD in Eastern
European patients. World J Gastroenterol. 2010;16(41):5233-5240.
58. Wegenka UM, Buschmann J, Lutticken C, et al. Acute-phase
response factor, a nuclear factor binding to acute-phase response
elements, is rapidly activated by interleukin-6 at the posttranslational
level. Mol Cell Biol. 1993;13(1):276-288.
59. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling
pathway. J Cell Sci. 2004;117(Pt 8):1281-1283.
60. Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and
development of chronic enterocolitis in mice devoid of Stat3 in
macrophages and neutrophils. Immunity. 1999;10(1):39-49.
61. Wolk K, Kunz S, Witte E, et al. IL-22 increases the innate immunity
of tissues. Immunity. 2004;21(2):241-254.
62. Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine
signaling pathways. Annu Rev Immunol. 2000;18:143-164.
63. Adamson AS, Collins K, Laurence A, O’Shea JJ. The Current STATus
of lymphocyte signaling: new roles for old players. Curr Opin
Immunol. 2009;21(2):161-166.
64. Chen Z, Laurence A, O’Shea JJ. Signal transduction pathways and
transcriptional regulation in the control of Th17 differentiation.
Semin Immunol. 2007;19(6):400-408.
65. Sugimoto K. Role of STAT3 in inflammatory bowel disease. World J
Gastroenterol. 2008;14(33):5110-5114.
66. Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association
defines more than 30 distinct susceptibility loci for Crohn’s disease.
Nat Genet. 2008;40(8):955-962.
67. Cenit MC, Alcina A, Marquez A, et al. STAT3 locus in inflammatory
bowel disease and multiple sclerosis susceptibility. Genes Immun.
2010;11(3):264-268.
68. Thompson AI, Lees CW. Genetics of ulcerative colitis. Inflamm
Bowel Dis. 2011;17(3):831-848.

15
086. Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;
9(6):653-660.
087. Carmeliet P. Angiogenesis in life, disease and medicine. Nature.
2005;438(7070):932-936.
088. Polzer K, Baeten D, Soleiman A, et al. Tumour necrosis factor
blockade increases lymphangiogenesis in murine and human
arthritic joints. Ann Rheum Dis. 2008;67(11):1610-1616.
089. Heidenreich R, Rocken M, Ghoreschi K. Angiogenesis drives
psoriasis pathogenesis. Int J Exp Pathol. 2009;90(3):232-248.
090. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J
Med. 2011;364(7):656-665.
091. Qu Z, Liebler JM, Powers MR, et al. Mast cells are a major source
of basic fibroblast growth factor in chronic inflammation and
cutaneous hemangioma. Am J Pathol. 1995;147(3):564-573.
092. Taylor PC, Sivakumar B. Hypoxia and angiogenesis in rheumatoid
arthritis. Curr Opin Rheumatol. 2005;17(3):293-298.
093. Hatton MW, Southward SM, Legault KJ, et al. Fibrinogen catabolism
within the procoagulant VX-2 tumor of rabbit lung in vivo:
Effluxing fibrin(ogen) fragments contain antiangiogenic activity.
J Lab Clin Med. 2004;143(4):241-254.
094. Cullen JP, Sayeed S, Sawai RS, et al. Pulsatile flow-induced
angiogenesis: role of G(i) subunits. Arterioscler Thromb Vasc Biol.
2002;22(10):1610-1616.
095. Schaper W, Scholz D. Factors regulating arteriogenesis. Arterioscler
Thromb Vasc Biol. 2003;23(7):1143-1151.
096. Bousvaros A, Leichtner A, Zurakowski D, et al. Elevated serum
vascular endothelial growth factor in children and young adults
with Crohn’s disease. Dig Dis Sci. 1999;44(2):424-430.
097. Di Sabatino A, Ciccocioppo R, Armellini E, et al. Serum bFGF and
VEGF correlate respectively with bowel wall thickness and
intramural blood flow in Crohn’s disease. Inflamm Bowel Dis.
2004;10(5):573-577.
098. Duenas Pousa I, Mate Jimenez J, Salcedo Mora X, et al. Analysis
of soluble angiogenic factors in Crohn’s disease: a preliminary
study. Gastroenterol Hepatol. 2007;30(9):518-524.
099. Griga T, Tromm A, Spranger J, May B. Increased serum levels of
vascular endothelial growth factor in patients with inflammatory
bowel disease. Scand J Gastroenterol. 1998;33(5):504-508.
100. Kanazawa S, Tsunoda T, Onuma E, et al. VEGF, basic-FGF, and
TGF-beta in Crohn’s disease and ulcerative colitis: a novel
mechanism of chronic intestinal inflammation. Am J Gastroenterol.
2001;96(3):822-828.
101. Pousa ID, Mate J, Salcedo-Mora X, et al. Role of vascular
endothelial growth factor and angiopoietin systems in serum of
Crohn’s disease patients. Inflamm Bowel Dis. 2008;14(1):61-67.
102. Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD:
useful, magic, or unnecessary toys? Gut. 2006;55(3):426-431.
103. Henriksen M, Jahnsen J, Lygren I, et al. C-reactive protein: a
predictive factor and marker of inflammation in inflammatory
bowel disease. Results from a prospective population-based study.
Gut. 2008;57(11):1518-1523.
69. Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability
predicts relapse in inactive Crohn disease. Scand J Gastroenterol.
2000;35(11):1163-1169.
70. Wyatt J, Vogelsang H, Hubl W, et al. Intestinal permeability and the
prediction of relapse in Crohn’s disease. Lancet. 1993;341(8858):
1437-1439.
71. Matsuda A, Suzuki Y, Honda G, et al. Large-scale identification and
characterization of human genes that activate NF-κB and MAPK
signaling pathways. Oncogene. 2003;22(21):3307-3318.
72. Wang L, Yu J, Ni J, et al. Extracellular matrix protein 1 (ECM1) is
over-expressed in malignant epithelial tumors. Cancer Lett.
2003;200(1):57-67.
73. Anderson CA, Massey DC, Barrett JC, et al. Investigation of Crohn’s
disease risk loci in ulcerative colitis further defines their molecular
relationship. Gastroenterology. 2009;136(2):523-529.
74. Sands BE, Kaplan GG. The role of TNF-α in ulcerative colitis. J
Clin Pharmacol. 2007;47(8):930-941.
75. Chidlow JH, Jr., Shukla D, Grisham MB, Kevil CG. Pathogenic
angiogenesis in IBD and experimental colitis: new ideas and
therapeutic avenues. Am J Physiol Gastrointest Liver Physiol.
2007;293(1):G5-G18.
76. Rijcken E, Krieglstein CF, Anthoni C, et al. ICAM-1 and VCAM-1
antisense oligonucleotides attenuate in vivo leucocyte adherence
and inflammation in rat inflammatory bowel disease. Gut.
2002;51(4):529-535.
77. Jones SC, Banks RE, Haidar A, et al. Adhesion molecules in
inflammatory bowel disease. Gut. 1995;36(5):724-730.
78. Simmons D, Makgoba MW, Seed B. ICAM, an adhesion ligand of
LFA-1, is homologous to the neural cell adhesion molecule NCAM.
Nature. 1988;331(6157):624-627.
79. Myers CL, Wertheimer SJ, Schembri-King J, et al. Induction of
ICAM-1 by TNF-α, IL-1β, and LPS in human endothelial cells after
downregulation of PKC. Am J Physiol. 1992;263(4 Pt 1):
C767-C772.
80. Haraldsen G, Kvale D, Lien B, et al. Cytokine-regulated expression
of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and
vascular cell adhesion molecule-1 (VCAM-1) in human microvascular
endothelial cells. J Immunol. 1996;156(7):2558-2565.
81. Perry MA, Phillipson M, Holm L. Transmural gradient of leukocyte-
endothelial interaction in the rat gastrointestinal tract. Am J Physiol
Gastrointest Liver Physiol. 2005;289(5):G852-G859.
82. Shimizu Y, Newman W, Tanaka Y, Shaw S. Lymphocyte
interactions with endothelial cells. Immunol Today. 1992;13(3):
106-112.
83. Panes J, Granger DN. Leukocyte-endothelial cell interactions:
molecular mechanisms and implications in gastrointestinal disease.
Gastroenterology. 1998;114(5):1066-1090.
84. Mustjoki S, Alitalo R, Elonen E, et al. Intercellular adhesion
molecule-1 in extravasation of normal mononuclear and leukaemia
cells. Br J Haematol. 2001;113(4):989-1000.
85. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other
disease. Nat Med. 1995;1(1):27-31.

16
104. Shine B, Berghouse L, Jones JE, Landon J. C-reactive protein as
an aid in the differentiation of functional and inflammatory bowel
disorders. Clin Chim Acta. 1985;148(2):105-109.
105. Schoepfer AM, Trummler M, Seeholzer P, et al. Discriminating IBD
from IBS: comparison of the test performance of fecal markers,
blood leukocytes, CRP, and IBD antibodies. Inflamm Bowel Dis.
2008;14(1):32-39.
106. Malle E, de Beer FC. Human serum amyloid A (SAA) protein: a
prominent acute-phase reactant for clinical practice. Eur J Clin
Invest. 1996;26(6):427-435.
107. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate
acute-phase reactant. Eur J Biochem. 1999;265(2):501-523.
108. Yamada T. Serum amyloid A (SAA): a concise review of biology,
assay methods and clinical usefulness. Clin Chem Lab Med. 1999;
37(4):381-388.
109. Niederau C, Backmerhoff F, Schumacher B, Niederau C.
Inflammatory mediators and acute phase proteins in patients with
Crohn’s disease and ulcerative colitis. Hepatogastroenterology.
1997;44(13):90-107.
110. Jijon HB, Madsen KL, Walker JW, et al. Serum amyloid A activates
NF-κB and proinflammatory gene expression in human and murine
intestinal epithelial cells. Eur J Immunol. 2005;35(3):718-726.
111. Atreya I, Atreya R, Neurath MF. NF-∫B in inflammatory bowel
disease. J Intern Med. 2008;263(6):591-596.
112. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med.
2009;361(21):2066-2078.
113. Chambers RE, Stross P, Barry RE, Whicher JT. Serum amyloid
A protein compared with C-reactive protein, alpha 1-antichymotrypsin
and alpha 1-acid glycoprotein as a monitor of inflammatory bowel
disease. Eur J Clin Invest. 1987;17(5):460-467.

INSIDE BACK COVER(BLANK)

9410 Carroll Park Drive
San Diego, CA 92121
888-423-5227
858-824-0896 fax
www.prometheuslabs.com
Prometheus diagnostic services provide important information to aid in the diagnosis
and management of certain diseases and conditions.
How this information is used to guide patient care is the responsibility of the physician.
PROMETHEUS and the Link Design are trademarks or registered trademarks of Prometheus Laboratories Inc.
© 2011 Prometheus Laboratories Inc. All rights reserved. CD10037 09/11
Prometheus products, services, and technology are covered by one or more
US patents and patents pending. For more information, see www.prometheuslabs.com.





























