Embed Size (px)
Citation preview

Rt?v.1965
NUCLEARSCIENCESERIES
National Academy of Sciences-National Research Council
Pubtished $3y:
United States Atomic Energ Commission

COMMITTEEON NUCLEARSCIENCE
S.K.Allison,Chairman f?.D. Evans,Vice ChairmanUniversityofChicago Mass.InstituteofTechnology
LewisSlack,SecretaryNationalResearchCouncil
E. C.Anderson BerndKahnLos Alamos Sci.Laboratory TaftSanitaryEngineeringCenter
N. E. Ballou JerryB.Marion
U.S.NavalRadiologicalDefenseLaboratory UniversityofMaryland
MartinJ.BergerNationalBureauofStandards
C.J.BorkowskiOak RidgeNatl.Laboratory
RobertG. CochranA & M CollegeofTexas
HerbertGoldsteinColumbiaUniversity
PaulC.AebersoldAtomicEnergyCommission
R. L.PlatzmanArgonneNationalLaboratory
ErnestC.PollardPennsylvaniaStateUniversity
KatherineWayOak RidgeNationallaboratory
GeorgeW. WetherillUniversityofCalifornia(LosAngeles)
LIAISONMEMBERS
RalphG.AllenAirForceOfficeofScientificReeearch
J.HowardMcMillenNationalScienceFoundation
SUBCOMMITTEEON NAOIOCHEMISTRY
N.E. Ballou,ChairmanU.S.NavalRadiologicalDefenseLaboratory
G. R. ChoppinFloridaStateUniversity
H. M. ClarkRensselaerPolytechnicInstitute
R. M. DiamondLawrenceRadiationLaboratory
W. E. NervikLawrenceRadiationLaboratory
J.M. NielsenGeneralElectricCompany (Richfand)
G.D. O’KelleyOak RidgeNationalLaboratory
R.P.SchumanAtomicEnergyDivisionPhillipsPetroleumCompany (IdahoFalls)
A.W. Fairhall E.P. SteinbergUniversityofWashington ArgonneNationalLaboratory
JeromeHudie D. N. SundermanBrookhavenNationalLaboratory BattelleMemorialInstitute
J.D. Knight J.W. WinchesterLos Alamos ScientificLaboratory MassachusettsInstituteofTechnology

Radiochemistry of Selenium
V. J. Molinski
and
G. W. Leddicotte
Ismed,November1966
Union Crmbide Co@oration
Mining and Metals Division
Research Center
Tuxedo, New Yoyk
Subcommitteeon RadiOchemiStry
NationalAcademy ofSciences— NationalResearch Councfl
PrintedinUSA. Price$1.00.AvailablefromtbeClearinghoussforFederalScientificandTechnicalInfmmaticqNationalBureauofStandards,U. S.DepnrunentofCommerce,Springfield,Virginia22151

FOREWORD
The Subcommitteeon Radiochemistry is one of a number ofsubcommittees working under the Comni.tteeon Nuclear Sciencewithin the National Academy of Sciences - National Researchcouncil. Its members represent government, industrial, anduniversity laboratories in the areas of radlochemistry andnuclear chemistry. Support for the activities of this andother subcommittees of the Committee on Nuclear Science isprovided by a grant from the National Science Foundation.
The subcommittee has concerned itself wi’thpreparation ofpublications, encouraghg and supporting ac%ivlties in nucleareducation, sponsoring symposia on selected current topics inradiochemistry and nuclear chemistry, and investigating specialproblems as they arise. A series of monographs on the radio-chemi.sta?yof?essentiallyall %he elementsand on radiochemicaltechniquesis being published. Initiationand encouragementof publication of articles on nuclear education In %rioussubjec% areas of chemistry have occurred, and development andimprovement of certain education activities (e.g., laboratoryand demonstration experiments with radioactivity] have beenencouraged and assisted. Radioactive Contamination of reagentsand materials has been investigated and specific recommendationsmade.
This series of monographshas resulted from the need forcomprehensivecompilationsof radiochemicaland nuclear chemicalinformation. Each monograph collectsin one volume the pertinentinformationrequiredfor radiochemicalwork with an individualelement or with a specializedtechnique. ‘IkeU. S. Atomic EnergyCommissionhas sponsoredthe printing of the series.
Commentsand suggestionsfor ~her publicationsandactivitiesof value to persons working with radioactivityarewelccmedby the Subcommittee.
N. E. lhllOU, ClwuknanSubcommitteeon Mdiochemistry
iii

INTRODUCTION
This monograph on the radiochemistmy of selenium is oneseries covering %he rad%ochemis%ry of essentially all the
elements. It Ls a revised and expanded version of an earliermonograph. In it are included reviews of nuclear and chemicalproperties of selenium, discussionsof methods of sample disso-lution and of separationreactions,descriptionsof couutingtechniques,and a compilationof radiochemicalseparationprocedures~or seleniumas found in the literature.
As new informationaccumulateson chemicaland nuclearpropertiesof seleniumand on separationand measurementtech-niques, considerationwill be given to further revision OI?thismonograph. Consequently as additional information hcomesavailable in Ixsthpublished and unpublished form, readers areencouraged to bring it.to the a’thent%onof the author forpossible inclusion i.nfuture editions of %his monograph.
iv

CONTENTS
I.
II.
III.
rv.
v.
VI.
GENERAL REIFHENCESON THEOF SEMNTO’M . . . . . . .
INORMNIC Am M’?ALY!HCALCHEMISTRY. . . . . . . . . . . . . . . . . . . . . 1
GENERAL REFERENCESCONTAININGINI’ORMK!ZONON THE RAIIBCHEMISTRYOF SELENIUM . . . . . . . . . . . . . . . . . . . . ...2
TAELEOF ISOT~S OF SELENIUM . . . . . . . . . . . . . . . . ...2
IE4TURES OF SELENIUM CHEKK3TRYOF CKCEF INTEREST !IUTHE
1.
2.
3.
4.
5.
6.
7.
8.
9.
RADIOCHEMIST.
INE30DUCTION
THE NmrALIrc
m Oxmzs .
. . . . . . . . . . . . . . . . . . . . . . . . . 6
. . . . . . . . . . . . . . . . . . . . . . . . . 6
SANE . . . . . . . . . . . . . . . . . . . ...6
. . . . . . . . . . . . . . . . . . . . . . . . . 6
0= DATZONSTA3XS . . . . . . . . . . . . . . . . . . ... . . . 8
Sacs.. . . . . . . . . . . . . . . . . . . . . . . . . . .
COMPLEX IONS . . . . . . . . . . . . . . . . . . . . . . . .
cEEtuTEs. . . . . . . . . . . . . . . . . . . . . . . . . .
voLAmLEcoMPoLmnx. . . . . . . . . . . . . . . . . . . . .
PRINCIPALANALYTICALM3THOIJ5. . . . . . . . . . . . . . . .
SELENIUMSEPAFWIONREAC!ITONS . . . . . . . . . . . . . . . . . .
1. SEPARATIONEYPRECIPITJWCON. . . . . . . . . . . . . . . . .
2. SEPAEtATZONBY SOLVENTEXZ’RACTTON. . . . . . . . . . . . . .
3. SEPARMZONBYVOLATILITY. . . . . . . . . . . . . . . . . .
4. ELECI’ROLYTICSEE’ARATION.. . . . . . . . . . . . . . . . . .
5. SEFAMTIONBY IONEXCHKNGE.HHCNS. . . . . . . . . . . . . .
6. SEPARATIONEY PAPER CHROMATOGFWWY . . . . . . . . . . . . .
DISSOLUTIONOF SAMPLES CON’IMININGFZLR%WM. . . . . . . . . . . .
1. MEmsmonms. . . . . . . . . . . . . . . . . . . . . .
2. SALTS. . . . . . . . . . . . . . . . . . . . . . . . . . . .
3. oFm. . . . . . . . . . . . . . . . . . . . . . . . . . . .
4. BIOCHEMICALSAMPLES. . . . . . . . . . . . . . . . . . . . .
5. 0-. . . . . . . . . - . .. . .. . . . . . . . . . ..
v
8
10
10
U
J-1
32
12
u16
16
16
17
17
18
18
18
18
18

VII.
VIII.
lx
x.
Xt.
HKZANXAND PRWAU1210NS. . . . . . . . . . . . . . . . . . . . .
co~TEcHNIms. . . . . . . . . . . . . . . . . . . . . . ●
SEPARATIONPROCEIY.JRESlWR SELEWOM . . . . . . . . . . . . . . . .
INIJEXOF SEPARATIONPROCEDURES. . . . . . . . . . . . . . . . . .
KEFEKENCES. . . . . . . . . . . . . . . . . : . . . . . . . . . .
LIST OF TABLES
I. TABLE OF TEE ISOTOPES OF SELEWUM. . . . . . . . . . . . . . . .
11. CHEMICALANDPSYSICAL PROPERTIESOF ~ . . . . . . . . . . .
III . VOLUBILITY OF SFLENIUMCOMPOUNES. . . . . . . . . . . . . . . . .
Iv. SOLWENT EXCRACTTON SYSTEMS FOR SELENTW . . . . . . . . . . . . .
v. CALCULATEDWECIFTC SATURATIONMZCTVITICESFOR SELIWUM RADIONUCIJXEWPRODUCED BYT!HERMALNEUTRON CKPrUR3. . . . . . . . . . . . . . . . . . . . .
LIST OF FIGURES
1. PARTIAL CHMKCOFTHENUCLIEES SHOWING~ . . . . . . . . . .
2. GAMMA-RAY SPECTRUMOF 7.1-HOUR Sea= . . . . . . . . . . . . . . .
3. GAMMA-FWiSPECITIUMOFl.21-D.&iSe75. . . . . . . . . . . . . . . .
4. GAMMA-RAYSPECTRUMOF 17.5-SEC Se- . . . . . . . . . . . . . . .
5. GAbWA-RM SPE~OF56.&MIN Seam . . . . . . . . . . . . . . .
6. Gw-MsPEcIcurJd oF25-~N Se- . . . . . . . . . . . . . . . .
I-0
19
26
27
46
.3
.7
9
14
20
.5
21
22
23
24
25
vi

Radiochemistry of Selenium
1.
2.
3.
5.
6.
7.
8.
V. J. Molineki
and
G. W. Leddicotte
Union Cmbide Corporation
Mining and Metals Dim’sion
Research Center
Tuxedo, New York
r. GENERALREFERENCESOH INORGANICAND ANALYTICcBIM13TRYOF SELEMIUM
J. W. Keller, A C rehensiveT!reatiseon Inor anic and Theoretical(Lon~% Green and Co., Inc., New fork, 1960), VO1. x,
=2.
W. F. Hlllebrand,G. E. F. LundeU, H. A. Bfi@t, ~d J. I. ~f~,APPlied Inorganic Analysis (JohnWiley and Sons, Inc., New York,1955), Chapt. 19, p. 327-338.
W. R. Schoeller end A. R. Powell, The Analysisof .~,n~ralsand Oresof the Rarer Elements (Hsf’nerPublishingCompany,New York, 1955)~Chapt.XIX, p. 8-254.
Robert C. Brasted, ComprehensiveInorganicChemlst~, Sulfux,Selenium,TelluriuuI,Polonium and O~g en (D. Van Nostrand Co., Inc.,Princeton,New Jersey, 1%1 ).
H. Remy, Treatise on InorganicChemlst~ (Elsevier,Amsterdam,1956),Vol. I, p. 741-752.
J. ~einberg, W. J. Argersinger,and E. Griswold,InorUanic Chemistry
(Heath, Boston, 1960), P. 434-455.
C, L. Wilson and D. W. Wilson, Comprehenslve Analytical ChemistryElsevier, Amaterdmz, 1959).
G. Chariot and D. Bezier, @antltatlve Inorganic ATIalysis (John Wileyand Sons, Inc., New York, 1957).
1

9.
10.
11.
1.
2.
3.
4.
5.
6.
N. V. Sidgwick, The Cheml.cslElements and Their Cmp2uudB (UniversityPress, oxford, 1951) .
B. S. HopkinB, Chapters in the ChemiEtryof the Less FamiliarElements(StipesPublishingCo., Illinois,1939), Chapt. 19.
Selenium amd Tellurium Abstracts,prepared for the Selenium-TelJ.uriumDevelopmentAssociation,Inc. by BattelleMemorial Institute,1959-1964.
II. CXNEML FEIERENCESCONTAININGINFORMAITOIVON THE RADIOCHSTRY OF SELENIUM
w. w.
G. W.
G. W,
H. L.
Meinke, “Nucleonics”,hal. Chein.~, 104R (1960).
Leddlcotte,“NucleonIce”,Anal, Chem.~, 143R (1962).
Leddicotte,“Nucleonlcs”,Anal. them.~, 419R (lg64).
Finstin and J, Miskel, “RadiochemicalSeparationTechniques”,hnual Review of Nuclear Sciencej, 269 (1955);
H. J. M. Bowen and D. Gibbons,RadiosctivationAn- 16 (oxfordUniversityPress, Herd, 1963).
G, W. Leddicotte,cd., “The Radiochemi8tryof Selenium”,Natl. Acad.Sci. - Natl. Research Council Rept. NAS-NS-3030 (1961).
III. TABLE OF THE ISOTUPES OF SEUKHJM
The radioactivenuclldea of seleniumof interestin the radiochemiatryof
8eleniumare given in Table I. This table has been ccunpiledfrcm information
appearingin reporteby Strominger,et al.(1) (2)
and in the Nuclear Data Sheets .
ltLgure1 shows the seleniumportion of the chart of Nuclides(3) includingthe
adjacent elements.

TABLE I
TA8LE OF ISOTOPES OF SELENIUM
Isotopic Type of Pa-title
- * ‘f-life = w
Reactions
se70 &m B+ As75(d,7n)Se70
%71 4.5 m ~+ p: 3.4 &75 (ti, 6n]se717, 0.16
8.4 a EC,~+ 7, 0.046 A.e75(d,5n)Se72
se7m 44m ~+ p: 1.7 AS75(d,k )Se7m;7, 0.88 73m.*74(y, n)Se ,
Se74 (n,2n )Se73m
*73 7,1 h ~:EC ~; 1.29,1.65 As75(d,k) Se7m;y, 0.36,0.066
Se74(y,n)Se7s;
Se74(n,2n)Se7=
~74 0.87$
a?? 5 121 d EC 7, 0.265,0.136 Se74(n,y)Se75;no 9+ 0,28 ,0.405
As75(p,n)Se75;
As75(d,2n)Se75
se7e 9.@
*77 7.584
se- 17.5 s IT Y, 0.162 Se7s(n,7)Se77m;
U fission
~7e 23.52$
*7em3.9 m IT y, 0.096 Se7e (n,7)Se7m;
Se=(n,2n)Se7m;
Br7g(n,p)Se7m;
U fission
3
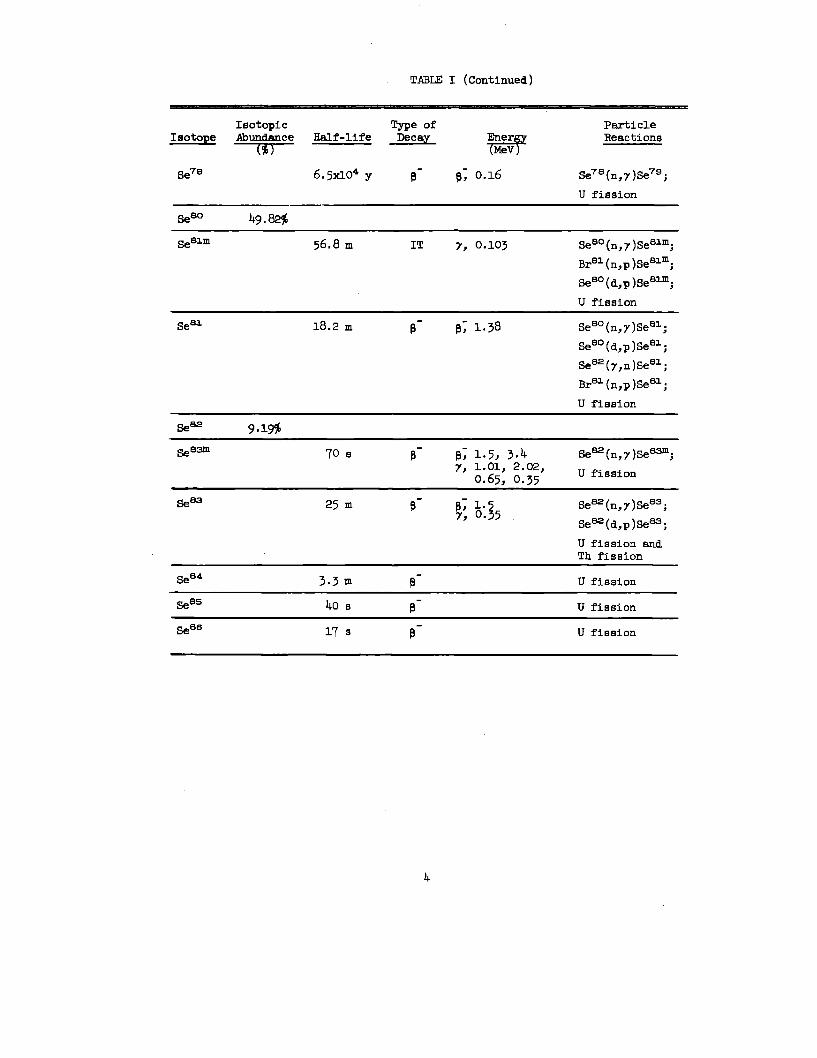
TABLE I (Continued)
Iflotoplc Type of Particle
= w ‘f-life ~
Ener
H
ReactionsMeV
se7e 6.5x104 y B- P; 0.16 Se7e(n,7)Se7g;
U fission
*SO 49,82$
~Slm 56,8 m IT y, 0.103 SeeO(n,~)Seslm;Slm
Brel(n,p)Se ;
SeeO(d,p)Seam;
U fission
Se.sl 18.2 m B- P; 1.38 Sem(n,7)Seal;
SeeO(d,p)SeaL;
Se=(7,n)See1;
BraL(n,p)Seel;
U fission
.&2 9.1*
@3m 70 s B- ~; 1.5, 3.4 S3m.*=(n,7)Se ,7, 1.01, 2.02,
0,65, 0,35U fission
*B3 25 m B- p; 1.3
Se=(n,7)Se=;7, 0. 5
Se=(d,p)Se=;
U fission andTh fission
3.3 m B- U fission
40 s B- U fission
@6 17 s @- U fission
4

U
FIGURE 1: Partial chart of the nuclides showing selenium.

IV. F!EJ4TUFCZSOF SELENIUM CWSTRYOF CHG3F INTEWISTTO TBE RADIO-ST
1. INTROIXJCTTON
Selenium is found associatedwith sulfur aud sulfides. The usual sources
are the native sulfidessuch ss p~te, chalcop~ite, and zinc blende. Flue
dusts from the roastingof seleniferousores, and the sludge formed in the
lead chambersused In sulftudc-acidmanufacturingprocesses are also highly
enriched sources. Seleniumin these material~ is usually brought into solu-
tion by treatmentwith sulfuric acid and sodiuq nitrate. In this treatment,
It is convertedinto seleniousacid (%SeOS) and selenic acid (H&eOa) and
fintiy precipitatedas elementalseleniumwhen sulfux dioxide is p-seal
through the solution.
Some of the chemlcsland physical propertiesof elementalseleniumare
summarizedin Table 11. Data have been com@led from publicationsby Hopkins(4)
and by Latimer(5).
2. TSE METALLIC STA!171
Selenium,like sulfur, exists in at least three allotropicforms, or
modifications: crystsJllne,metalllc, and amorphous. Cmly the crystalline
and metallic fores have been well ch=acterized. The crystallineform ( a
loose powder ccmposedof at least two varietiesof red, monoclinic,c~stals)
is prep=ed by crystfiization from carbon disulfide,in which it is soluble.
Gray, metallic seleniumis the most stablemodificationand forms hexagonal
crystals. It is relativelyinsolublein carbon dlaulfide,is a good conduc-
tor of electricity,and ia less active chemicallythan the other fores. Gray
seleniumis preparedby heating sny of the other forms at temperaturesof
frcm 200 to 230”c. Amorphousseleniumis obtained in two forms: (1) red
powder produced by reducinga selenitein an acid solutionwith sulfur diox-
ide; end (2) vitreous or glassy selenium,black in color and describedSE
a super-cooledllquid. Both amorphousfomns are soluble in carbon disulfide.
Seleniumwill burn with a characteristicodor, and will combinedirectly
with hydrogen,oxygen, the hslogens, and many other metals. It is not attacked
by non-oxidizingacids; but nhen heated, it will dissolvein concentratedsul-
furic acid, nitric acid, or caustic alJralis.Nitric acid oxidizes seleniumto
H&eO~.
3. TBE O~DES
The most stable ccmpoundof Se (and its only well-definedoxide) is sele-
nium diotide (Se02) produced by burning selenim in oxygen or sir, Pure Se02
is composedof brilliantwhite needles, It sublimesreadilybut can be melted
6

TABLE II
CHEMICALAND PHYSICALPROPERTIESOF SELENIUM
Atomic Weight
SpecificGravity (solid)
Color (solid)
Melting Point
Boiling Point
Atomic Volume (approx.)
Heat 01’Fozmation, ~X (Csls.)
Atomic R8diuS
Ionization Potentials
Ion volts—
I 9.751II 21.514III 30,079IV 42,902v 73.1C9
78.96
4.26-4.79
dark red
170-217°C
688°c
MC
-25.1
1.5 Augstrom
OxidationPotentials (volts)at 25’c
(a) Acid solution
E&e 0.40 Se -0.74 H#e03 -1.15 Se04=— .
(b) Base solution
%= 0.92 se 0.366 Seoa= -0.05 seed=
without decomposition. In the vapor state, it has a yellow-greencolor. Sele-
nium diotidewill dissolvein water, concentratedsul-c acid, or alcohol.
It can combinewith hydrogen chlorideto form Se02.2HCl,and will form similar
double compoundswith other substances. It can easily be reduced to elemental
selenium.
The characteristicbuznlng odor of seleniumis reportedby Berzelius(6) to
be due to the formationof a suboxide,SeO. It is also reportedthat a mitiure
of SeOz and SeOg is producedwhen seleniumreacts”withoxygen In an electrical
discharge, Seleniumtriotide is decomposedTn water to form selenic acid, and
is stable to 185”c, at which temperatureit decomposesto oxygen and SeOz.
7

4. OIZDATIONSTATES
Selenium cam form compounds havl~ oxidation states of -2 (the aelenidea),
+4 (the selenites),and +6 (the selenates). Polyaelenides,such as Nasez end
NaSe3 are known, but they are leaa stable than the polysulfides.
Selenium IB electronegativelybivalent towards hydrogen and the metals.
In its electropoaitlvebehavior,seleniumhas a valence of +4 in compounds
formedwith most elements;but in combinationwith fluorineit has a valence
of +6. In general, the compoundsof selenium,sulfur, and tellurlum,behave
similarlyand are analogousto each other. The principaldifferenceis in
the lower stabilityof the selenium (and tellurium)compounds,which are
easily decomposedto the element by heat or reducing agents.
SALTS5.
~.1 Soluble Salts
Selenium dioxide 18 a true anhydride,five parts disaoltingin one
part of water. In combinationwith water, Se02 will form seleniousacid
(H&o, ). The alkali selenites.(Li2Se03,Na&e03, &Se03, Rb2Se03j
Cs#3e03) are all soluble in water, as are the acid selenitesor hydro-
selenltesof the type MHSe03, Table III lists some of the more inpor-
tant
5.2
soluble compoundsof selenium.
InsolubleSalts
The selenate compounds of lead, barium and calcium, like the corres-
ponding sulfates, -e insoluble; however, they are leas stable to heat
than the sulfates.
Hydrogen seletidecan form both acid selenides (M%Se) and neutral
selenides (&zSe). The alkali and alkalineearth metals can combine
with additionalseleniumto form polyselenides(~lSex) in the same way
that these metals form polysulfideswith sulfur. Pure alkali selenides
are colorless. Heavy metal selenidesprepared by the action of hydrogen
selenide on solutionsof the heavy metal salts are more or leas strongly
colored. Like the heavy metal sulfides,they exe insolublein water,
and some are insolublein acids.
Selenium can combinedirectlywith nitrogen to form seleniumnitride
(Se~4), which is insolublein water, and only slightlysoluble in benzene,
acetone,acids, and carbon disulfide.
Selenium combineswith sulfur to fomn seleniummonosulfide, (SeS)
and seleniumtisulfide (SeS2). Seleniummonosulfideis insolublein
water and ether, but very soluble in carbon disulfide. Seleniumdisulfide
8
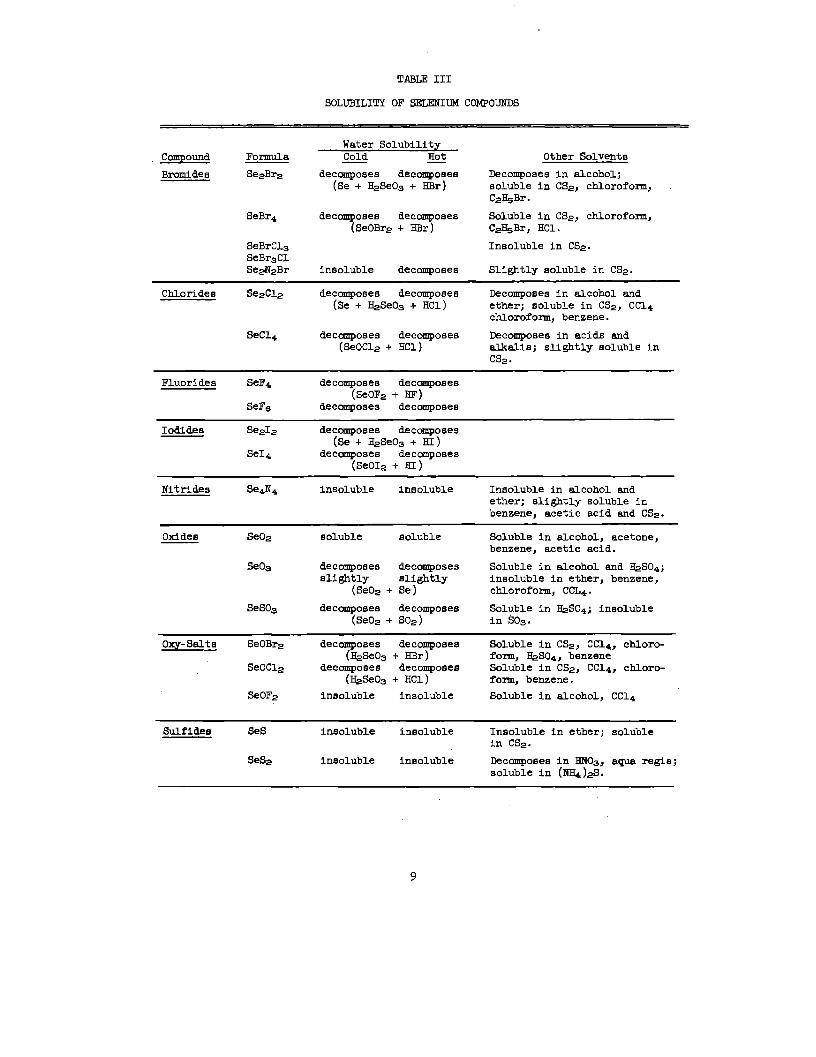
TABLEIII
VOLUBILITYOF SELENIUMCOMPOUNDS
Water VolubilityCompOund Formula Cold Hot— Other Solvent8
Bromide@ Se2Br2 decomposes decomposes Decomposes In alcohol;(Se + H#e03 + HBr) solublein CS2, chloroform,
C&.Br.
SeBr4 deccmpoaea decomposes Soluble in CS2, chloroform,(SeOBr2+ HBr) C2~Br, HC1.
SeBrC13 Insolublein CSZ.SeBr3ClSe$2Br inBoluble decompoEe6 Slightly solublein CS2.
Chlorides Se2.C12 decomposes decompoaem Decomposesin alcoholand(Se+ H#e03 + HC1) ether;solublein CS2,CC14
chloroform,benzene.
SeC14 decomposes decompoae8 Decomposes in acids and(SeOC12+ HC1) alkelia; slightly soluble In
CS2.
Fluorides SeF4 decompoae8 decomposes(*OF.+ SF)
SeF8 dec~oses– dec~oses
Iodides Ss212 decomposes decomposes(Se+ H#e03+ BI)
SeI~ decomposes decomposes(SeOI=+ =)
Nitfldes se4N4 insoluble insoluble Insoluble in alcohol andether; slightly soluble Inbenzene, acetic acid end CS2.
Oxides Se02 soluble soluble Soluble in alcohol, acetone,benzene, acetic acid.
Seog decomposes decomposes Soluble in alcohol and fiS04;slightly sligktly insoluble in ether, benzene,
(SeO~+ Se) chloroform, CCL4.
seso~ deCOnIp08eE decomposes Soluble in ~S04; ineoluble(Se02+ S02) in SOS.
Oxy-sslts SeOBr2 decomposes decomposes Soluble in CS2, CC14, chloro-(H#eO~+ HBr) form, B@34, benzene
SeOC12 decomposes decomposes Soluble in CS2, CC14, chloro-(H@e03+ HC1) form, benzene.
Seol?z insoluble insoluble Soluble in alcohol, CC14
Sulfides SeS insoluble insoluble Insoluble in ether; solublein CS2.
SeS2 inBoluble insoluble Decm.poses in BN03, aqua regia;soluble in (NH4)fi.
9

ia Insoluble
acid. Table
of selenium.
“Handboakof
Tn water amd will decompose only in aqua regia or nitric
III lists Borneof the more Important Insoluble compounds
Additional information on solubilities appe-s in the
Chemistry and Physics” (7),
5.3 Non-isotopicCarriers
In some cases, a hfgh specific activityselenium sourcemust be pre-
pared. A non-isotopiccarrier, chemicallysimilar to selenium,must be
used rather than inactive selenium. Telluriummeets these requirements
aud can be used to sep=ate small quantitiesof radioactiveselenium
High-purity (99.999$)tellufiumia availablecmmnercially. Total helo-
gena, sulfur, and seleniumdo not exceed 5 ppm by chemicalanalysis.
Garrison,et al.(8)
re’potieda procedure in which they separatedradio-
active seleniumfrom an eraenic target using tellurouaacid, precipi-
tating telluriumwith sulfur diotide. They reported that 95* of the
selenium c=fes, and can then be aeperatedfrom telluriumby distil-
lation.
5.4 Coprecipitation
Microgrmn quantitiesof selenium can be collectedby coprecipita-
tion on ferric hydrotide. The weight of the iron present shouldbe 30
times that of the selenium(9). The Iron Is precipitatedwith an excess
of ammoniumhydroxide. A method(lo)for separatingsmall amounts of
selenite and selenateions ia baaed on the copreclpitationof the sele-
nite ion with iron hydrotideat pH 8.4-8.6. Under these conditions,
the selenateion Is not coprecipitated.
6. COMPLEX IONS
Several of the selenium halogen compounds can form complex acids by
adding on hydrogen halide ions. For example,SeBr4 will combinewith H&
to form hexabromoaelenicacid (B#eBre). Hexabromoselenatesand hexachloro-
selenates have
ccmplex acids.
7. CHELATES
been formed by the reactionof al12mli
Their aolubilitiesresemblethose of
halide salts upon these
the selenates.
Selenium forma a chelate In an acid solutionwith sodium dlethldithio-
c=bamate that can be extracted (pH 3) with ethyl acetate‘U)W). The c~-
plex can be extractedinto carbon tetrachloride(13)
, end telluriumis also
extracted.
10

Bode WI. Aanswold(’4)reportedthe quantitativeextraction of the SeIV
chelate of dlet~ldltblophosphatefrom 10~ HCl into CC4, only 40$ of the
te~urium being extracted.
che*(’5) has shown that a 5$ aqueouB dlanrlnobezitinehydrochloride
solutionvIIJ.form a seleniumcomplex In a fotic acid-watersystem that can
be extractedinto toluene at pH 6-7. AlthoughFe+=, CU+2, @5 and other oxi-
danta Interferes,the Fe+g and CU+2 interferencescan be eliminatedby EDT.A
completingat pH 2-3. TelluzYumforms a cmuplex that is extractedbut doet3
not Interferein the colorlmetricanalysis.
8. VOIJICILECcwourms
The distillationof neutral or acid alkali selenldeswith potassium alkyl
sulfatesproduces sJ@l selenldessew (whereR=the alkyl group) and alkyl
selenomercaptansSeFH. The a.lkylseletidesare volatile liquidswith pungent,
repulsiveodors. They readily combinewith, or add on, halogens or oxygen
to fomn such compoundsas (C41S)&eC12 or (C2%)@e0. In addition,they can
add on alkyl iodl.desto form such compoundsas eJ.kylselenonium, e.g.
(C2~)@eI. These compoundsam strong bases and, when treatedwith moist
silver otide, form the correspondinghydroxide.
9. PFLIIKXPAL AIwlYTrcAL m.!EHoDS
9.1 Gravimetric
The gravimetricdeterminationof seleniumis csrrled out byweigb-
ing the element after it has been precipitatedby reductionin strong
acid solutionwith either sulfur dioxide or hydro~lemine hydrochloride(16).
,.2 vohnnetric(’6)
9.2.1 Selenic acid is reducedby hydrochloricor hydrobrmnic
acid. The halogen is caught in a Bolutionof potassiumiodide and
titratedwith arsenlteand thiosulfate.
9.2.2 Seleniousacid is reducedby hydriodic acid followedby
titrationof the liberated Iodinewith thiosulfate.
9.2.3 Seleniousacid in hydrochloricacid solutionIs reduced
by a brown excess of sodium thiosulfate. The excess thiosulfate
Is then titratedwith stand- iodine solution.
9.2.4 2eleniousacid is oxldlzedby an excess of standardper-
manganate solutionin sulfuricacid solution. The excess permauga-
nate Is titratedwith ferrous sulfate or oxalic acid solution.
n

9.3 Colorlmetric
9.3,1 3,3’-rn~nObe~iUne reacts with Se to form an intenee
yellow compound (piazselenol).~eng(15) reporteda procedure for
determiningtraces of Se by extractingthis ccmpoundinto toluene
and measuring the color apectrophotcanetricaldyat a wave length of
420 m.
9.3.2 2,3-DiaminonaphthaLenealso reactswith Se to fomn a
piaaselenolwhich 1s a reddishprecipitate. It may be extracted
into toluene and measured spectrophotometricallyat a wave length
of 380 UP(17).
9.3.3 Seleniousacid liberatesiodine quantitativelyby ofida-
tion of iodide ion. The blue color is developedwith a cadmium-
iodide starch reagent and measued spectrophot~etricallyat a
wave length of 615 mI(18).
v. SELENIUMSKPARA’ITON~ACTIONS
1. SEPARATIONBY P~CIPITATION
Selenium ia separatedfrom moat elementsby the use of reducingagents
in acid solutions. EH.llebrand,et al..(16)
report that Se (and Te) can be
initiallyseparatedfrcnnmost elementsby reductionwith S02 in 3.7 to 4,~
HC1 solution. Gold, Pd, and small mounts of Sb, Bi, Cu, and other elements
are also reduced under the same conditons. Gold can be separatedby filtra-
tion after the mixed metal precipitatehas been digestedfor some times in a
dilute HN03 solution. The seleniumC= then be separatedby precipitation
with E&03 or hydroxylamlnehydrochloride.
Quadrivalentseleniumor telltium can be separatedfrom other elements
by saturatingau acid solutionwith H#(19),
Sexivalentseleniumand tellu-
rium can be completelyprecipitatedfrom 122 HC1 solution at room tempera-
tul-e(20).Sulfides of the copper subgroupmay interfereunder the same con-
ditions.
In their studies,Seath and Besmiah(21)
end Noakes(22)showed that
nitrates of CU+2 and Au+= interferedin the reductionof Se (or Te) to ele-
mental Se (or Te). The nitrateswere removedby evaporationwith HCl and
NaCl, and CU+2 and Au+= were separatedbefore the reductionwith hydroqui-none(23,24).
Selenium (and tellurium)may be separatedfrom many elementsby reduc-
tion of an acid solutionwith stannous chloride. Selenium and telluriumare
precipitatedand can be sep=ated by a brcminatedHCl dissolutionof the
metals followedby a precipitationof Se with S&(25)0
12

Bode(26) has shown that a separationof Se from Te can be effected in a
51 HC1 solutionusing a solutionof tetraphenylarsoniumchloride.Telluium+8
will precipitateand bromide, iodide, fluoride,nitrate,Mo+e, and tie will
interfere.
Ripen-Tilici(27)Snd SPQCU(28) have shown that Se can be detetined as
lead selenate. To obtain this form, Se must be oxidizedto the hexavalent
statewith *02 and precipitatedby a lead salt solution.
2. SEPARATIONBY SOLVENTEXTRACTION
Solvent extractionmethods, used as wparation methods for other analy-
sis techniques,can often be adapted for use in radiochemistry. They can be
quite useful in separatingthe desired radionuclidefrcm .&sampleby either
a carrier-freeor carrier rsdiochemist~ method. Morrison and Freiser(29)
have reviewedthe applicationsof ion-associationand chelate-complexsys-
tems to the detehnation of most of the elements. Some of these are
applicablesa separationprocesses in the radiochemistryof selenium.
Table IV gives a few of the solvent extractionsystems that could be
used for the separationof selenium. Few epecificsolvenbextraction
methods for seleniumhave been developed,but many could be adapted.
2.1 Ion-AssociationSystems
Kitahar.(3°)has reportedthat Sn+2 and Sn+4 can be completely
extractedwhile Se+4 (md As+’, %+’, mdMo+’) iS 01dyp3.I’ti-y
extracted (about L%) from HF solutions of varying concentrations by
ethyl ether and a 4:1 volume ratio of organic-to-aqueous phase.
Using equal volumes of IIFand ethyl ether, Bock and Herman(31) have
shown that only Nb+5, Ta+5, and Re+7 were extractedin amounts greater
than 5*; Se+4 (aawell as Sn+2, Sn+4, As+3, As+5, Te+4, Ge+4, ~5, @3,
@?, h!o+eaud Sb+3) was only partially extracted. However, the extract-
ions of Se+4 and the other ions Incresaedwith increasingHF concentr-
tion. These two studies show differentextractionvalues; however, these
differencesmay be due to the manner in which the metel fluorideswere(32)
prep-ed. Stevensonand Hicks report that Se+e (sswell aa Te+8,
Fe+3, Ga+3, sb+5, and Aa‘s) canbe quantitativelyextractedbytiis~
propyl ketone from anlneral acid-hydrofluoticacid (6E HC1 - O.% HF)
aqueous system. Se+4 (aawell as Sb+3, As+5 and Te+4) is only slightly
extractedunder the8e conditions. In this same study, it was shown that
Se+4 (and Te+e ~d Ta+5) co~d be extractedby diisopropylketone frcnna
6~ H2s04 - O.bg m’ Systemp
At least 3C$ Se+4 can be etiractedfrom a mixture of metal bromides
13

mHLE Iv
SoLsm’rmcmoN sYs-JEMs FOR slImNIuM *
OxidationSkate
IV
IV
VI
Iv
IV
IV
r?
rv
IV
rv
Iv
4.6ME?
20M HI?
@ Hcl-o.g HF
4-% HBr
I.METJog
HC1
30-35$HBr
Na-tiethyldlthio-c-bamate
Pi=selenol-complex of3,3 ~tinobenzitinehydrochloride
Complex of 2,3diamino-naphthalene
Complex withtoluene-3-~dlthiol and10~ HCl
ethyl ether
ethyl ether
diimpropylketone
ethyl ether
o.ca-o.6MdibutylphophoricBCid
methylisobutylketone(4-methyl-2-pentanone)
di-iaobutylketone(2,6-dlmethyl-~heptanone)
ethyl acetate
t oluene
toluene
mixtureethylenechloride- ccl~
%Extraction
4.0
12.9
100
31
<5
100
90
100
100
Reference
30
31
32
33
34
35
36
11,12,13
15
100 17
100 37
* All of theee ❑ol.ventextraction system - also applicable to tellurium.
The Na-dietbyldithiocarbamatesystem is the only practical method for separating
Te from Se. Tellurium can be extracted from thiB ayatem into carbon tetrachlor-
ide atpH 8.5 - 8.7; selenium ia not extracted,
14

(~ to @ E&) into ethyl ether(33). AU+3, Ga+3, In+3, T1+3, Sb+5, Sn+2,
Sn+4, and Fe+= qU8ntit8tiVely extract from thie ByBtem; As+3, sb+3 and
Mo+e extract slightly;Cu+2 and Zn+2 do not extract.
Scsdden and Ballou(34)
have shown that leas than 5$ Se+4 (and Te+4)
till extract Into O.@ or O.@ di-n-butylphosphoricacid (DBPA)frm a
1# HN03 solution. A 1:1 orgamlc-lio-aqueouavolume ratio was used In
these studies. Yttrium, Sn, Mo, Nb, Ta, Zr, and In me extractedIn
concentration v~ng from 5 to more than 95$ fram the same system.
Selenium (esweu as Tej Fe, Ge, Sn, As, and Sb) can be quantit-
tively extracted into methyl iBobutyl ketone I’rcnnvarious concentratlon8
of HC1(35). This study determined the approximate ranges of extract-
ability for metallic Baits of the~e and other elementB.
Kuroda(%) used a HBz=dllsobutylketone Bystem for the rapid radlo-
chemicel separationof Se7sm. Selenium (8s well aa As, Sn, Hg and Au)
can be quantitativelyextrhctedinto dilsobutylketone frmn 30 to 35$
HBr. The interferingelements can be strippedby shakingwithED’TA
~d sodium t-rate, or with sodium hydroxide solution.
2.2 Chelate Complex Systems
Selenim can be extractedfrcm an acid solution (PH 3) as the
Bodlum diethyldlthiocarbamatesalt with ethyl acetate (U,M) .
Se+4 (andTe+4) will also forma complexin an acid solu~~ (pH 5-6)
by use of O.~ solutionof Bodium diethyldithiocarbamate . The
conplex can be extractedinto CC14. The addition of a 5$ solutionof
EDIM to the s stem assists in masking out other extractableelements.,hew(15$ haB 8hown that a 5$ aqueous diaminobenzidlnehydro-
chloridesolutiontill fozm a piazselen@ cmnplex in a fomnic acid-
water systm that can be extractedat pH 6-7 by toluene. Fe+3, Cu+2,
@5j and other ofidantsinterfere;however, the Fe+3 and Cu+2 inte~
ferences can be eliminatedbyEDTA ccxuplexingat pH 2-3. Vanadium
(V) otidizes diaudnobenzidlnepreventingthe fomaticm of the cmn-
plex, but traces of vanadium have Httle effect. Tellurlumis
extractedbut does not interferecolorimetricalJ.y.
Lott, et al.(17) used a similar 8yetem for extractingSe. A l?
aqueoue (0.~ HC1) solution of 2,3-dieminonaphthalene was used to
complex Se; and the complex was extracted, at pH 1.5 to 2.5, into
toluene. hlmking agents were used to prevent interferences.
Watkinson(37) etimcted Se from strong HC1 solutions (QO~) 8s
the Se (IV) complexwith
lene chlorideand carbon
with toluene-3,&dithiol
toluene-3,~dithiol into a mixture of ethy-
tetrachloride. Elements that also react
are Te (~), Pd, Au, W (VI), andRe (VII).
15

3. SEPARMICONBY VOLATUITY
Selenium (or tellurium)in the selenide (or telltide ) fom can be
sepwated fran metalm whose chlortdes- nonvolatileby pasdng C12 gas
into a hot aqueous solution(38)
. Hydrochloricacid gas csnbe used
instead of C~ to sep=ate either”Se+4or Se+e. Selenim can be 8epua-
ted from telluriumby a distillationmethod in which HC1 gas is passed
into a H#04 solution. The distillateis caught in cold water, and
Se+e 18 precipitatedby adding S02. Either Se+4 or Se+e can be quanti-
tatively~eparated fran te~urium by passing C02 gas into a l@Tobromlc-(40)
phosphoricacid solution . Selenium o~chloride and SeOBr2 ( as well
as TeOC~ and TeOBr2) are volatile from 61 HC1(41)
0, 6~ ~(@-44)SOk
tiong at temperaturesabove 100”C. Arsenic, antimony,tti and germanium
are also volatileunder these conditions.
4, ELlmImLrmc sEPARA!tToN
Electroseparationand electrogravlmetrymethods for seleniumme not
extensivelyreported. Only a few electroanalysistechniquesexist for the
determinationof seleniumby amperometryand coulometry;however,a consi-
deration of these ml t be profitablefor use in rediochemistry. For example,
Lingane and NiedrachF +4 with CUpfic COPper45)report on the titrationof Se
in an mnmonicslmedium. Se+4 is reduced to Se+2, and cupric selenideis
only slightly soluble in thi6 medium. The potentialof the droppingelec-
trode is such that the tetreazdnocupricions and the eeleniteions are
reducible;its value will increasewhen excess cupric ion is added.
and swift(~)
Rowley
have used a coulometrictitrationof thiosulfatefor the
determinationof Se+4. The back-titrationprocedureswere followed. In one,
en excess of iodide is added to a strong seleniousacid solutionand iodine
iB liberatedand reducedwith sodium thiosulfatesolution,the excess thio-
sulfatebeing detemined by coulometrictitration. In the second procedure,
excess thiosulfateis added so that the selenlousacid is reduced to sele-
nopentathionate(SeS40e),and the excess thiosulfateis dete?.minedby coulo-
metric titrationwith iodine. More accurateresults (YO.1~for severalhun-
dred micrograms)were obtainedby use of the secondmethod.
5. HEFAB-ATIONBY ION EXCHANGE RESINS
Kraus aud Nelson(47) report that selenite (Se+4)and telluriteshow
good adsorptionon an anion resin column as chloride complexes. As en+4 ha de it poesible tO Sepwate ‘t ‘rmexample,this behavior of Se
As+a ~d Br- by use Of solutions of various N&Cl concentrations. AS+3
ie removedby elutionwith O.01# ~Cl; Se+4 with 0.5CljNI&Cl; and Br
16

with 5.OFJ~C1. Attebury,et al.(49)
, and Aoki(’g) report that se can
be sep=ated from Te by adsorptionupon an anion resfi column from ~
HC1, A mlxlmre of ~ HC1 and ~ NH&NS was used as the eluant.
Lederer and Kertes(50)
used seleniteend tellu%te ions in a study
concernedwith the use of a Ibwex-re,ein-lmpre&ate& paper. Optimum con-
ditions for wepsxationwere calculatedby consideringthe relationship
of the ion adsorptionwith the pH of the aqueou6 acid used as elusnt.
6. SEPARATIONBY PAPER CHROMMIWRAPHY
Levi and Banon(5’)have used paper
of BI=o, pb=o, Po, Se, and Te in ~Os
ture was used as the solvent and a good
obtdned. Crouthameland Gatrousis(52)
chrczaatographyto separatemixtures
BOlUtiOn8. A butanol-propanolmix-
separatlonof each elementwea
also report on a similar separation
of Se, Te, Po, and Bi by peqer chromatography. Specific separationsof Se
(ss se+4) from teU&5~ (SS Te+4) in HCl, ~03, and HBr have been studied
by Bursta12, et al. , Lederer(54), md Weatherley(55). Solvents such
as mixtures of butanol-methanol,butanol-water,and butanol-HClwere uOed
to effect the separation.
VI. DISSOLU’ITONOF SAMPLES CONTAININGSELENIUM
When dissolvingselenium-contairihgseanpleO,it is necessaryto use
techniquesthat will minimize the 10BS of Se by volatilization(56)
. Sele-
nium is easily volatilizedas Se02S2HClfrom strong HC1 solutionsby
heating. Very little Se is lost by heating dilute HCl solutionsat
temperat~s” below 100”C. The presence of alJmli salts do not prevent the
volatilizationof Se. The volatilemonochlorideof selenium (Se2C12)can
be formed in strong HC1 solutionsby the action of reducing agents. The
neductionof Se Bhouldbe performed rapidly,with an excess of reducing
agent, to reduce the Se to the element. No loss of Se occurs if sulfuric
acid or perchloricacid solutionsof seleniousacid - evaporatedto dense
fulnes(s~)unlees there are chlotidesor bromidespresent.
Some selenidesattack platinum during a fusion process. The fusion
can be done with N@2 or a mixture of N&C03 and KN@ in a nickel or zir-
conium crucible. Fbion with KCN may cause sane loss due to volatili~ation.
The addition of inactive selenium cexrier to the mixture before dis-
solution begins wild assist in achieving an exchmge of the radlonuclide
with the carrier atom. The chmical states of the hradlated seleniumand
the carrier seleniumshould be the same dting the dissolutionprocedm
so that any losses that occur will be accountedfor in the chemicalyield
determinationof the added inactive carrier.
17

1.
For
may
and
MEmLs AND O.xJIIEs
Acid dissolution18 TeC~ ded for the asBay of metals and oxides.
exszuple,coppermetal can be dissolvedin nitric acid; iron and steels
be dissolvedin HN03, HC1 and HC104; lead muat be fused with N~COg
Na#OS in a nickel crucible. Insolubleoxidesmaybe fuse~=~th N@Oz
or sodium carbonateand potassim nitrate in a nickel crucible’>”).
2. QXG!rs
Nitric acid
that no selenium
3. =
Ore samples
and HCl will decmpose most selts. C= should be taken
is volatilized.
can be fused with N@2 or a ml- of sodium carbonate.and sodium nitrate in an iron or nickel crucible(59J. Some ores can be
decomposedwith 1:3 BN03 followedby funbg with s~ic acid to remove
BN03. Ores containingselenidescan be deccmrposedby heating at dull red-
ness h a current of C~ and catchingthe volatile chloridesof seleniumin
1:1 dilute HC1.
4. BIOCHEMICALSAMPLES
Biochemicalsamples can be &ccrmposedby destroyingthe organicmatter
with a 1:1 aahing mixture of nitric end perchloricacids(60). Tissues can
be decaposed by fusing with N@@ (61). SmsJl amounts of Se in vegetation,
grains, and anhal matter have been z=eleaaedfrom these materialsby dis-
tillingwith HSr(62). Organic or biologicalsamples cam be decomposedIn a
SchUnige~type ~gen-combustion flask; the gases are absorbedin a HC1
solution containinga selenium carrier. Gorsuch(63), in a series of ra&Lo-
chemicalinvestigationsconcernedwith the recoveryof trace elementsfrom
organicmatter, has evaluateda number of dissolutionmethcds for recover-
ing trace Se frmn similarmaterials. The results of these studies showed
that Se was lost by volatilization.
5. mm
Glass samples
acida(64). Rubber
and Sll@r.
may be taken into solutionwith hydrofluoricand nitric
is deccnnposedby a fusionwith a mixture of Ns20z, NaNOs,
VTI . HAZAFUM
No matter what method is used
AND PRECAUTIONS
to decomposea sample, adequatesafety
18

precautionsshouldbe followed. The toxicologyof most elementalcompounds
has been reportedby Pieters and Creyghton(65)
, and It should be consulted
for infonuatlonon handling selenium-containingmaterials safely. Due to
the volatilenature of some of the selenium compounti,radiochemicalRepa-
rations requiringheat should be carried out in a well-ventilatedhood.
Safety practices are always importantin rSdiOChemi6try. The disch=ge
of radioactivityby explosionor evolutioninto a laboratoryarea can be
hazardousand can result in wide-spre.%iradioactivecontamination. Thus,
some souxce of informationon safe-handlingpractices in processingradio-
active samples shouldbe consultedbefore a rsdiochemlcalanalysisIs
undertaken. One such source is the Oak Ridge NationalLaboratory’sMsster
AnalyticalMsmual(66). Other similar sources of informationexist and
shouldbe consulted.
VIII. COUNTINGTECHNIQUES
The analysisof smple materials containingseleniumrsdionuclidesmay
be completedeither by a direct (non-destmctive)measurementof the radio-
activityof the particularradionuclideor by obttiningthe radionuclldein
some form by radiochemicallyprocessingthe radioactives.mple. The choice
of techniquedepends upon the seleniumradionuclidesbeing measured, and
such characteristicsas half-life,type of radiationemitted, and the ener&y
of the radiations. An excellentsource for countingtechniquesis the mono-
graph by OrKelly entitled,Detectionand Measurement of Nuclear Radiation(67).
Table I shows the nuclear characteristicsof each of the known radio-
active isotopeaof selenium. Those usually encountered by the radiochemist
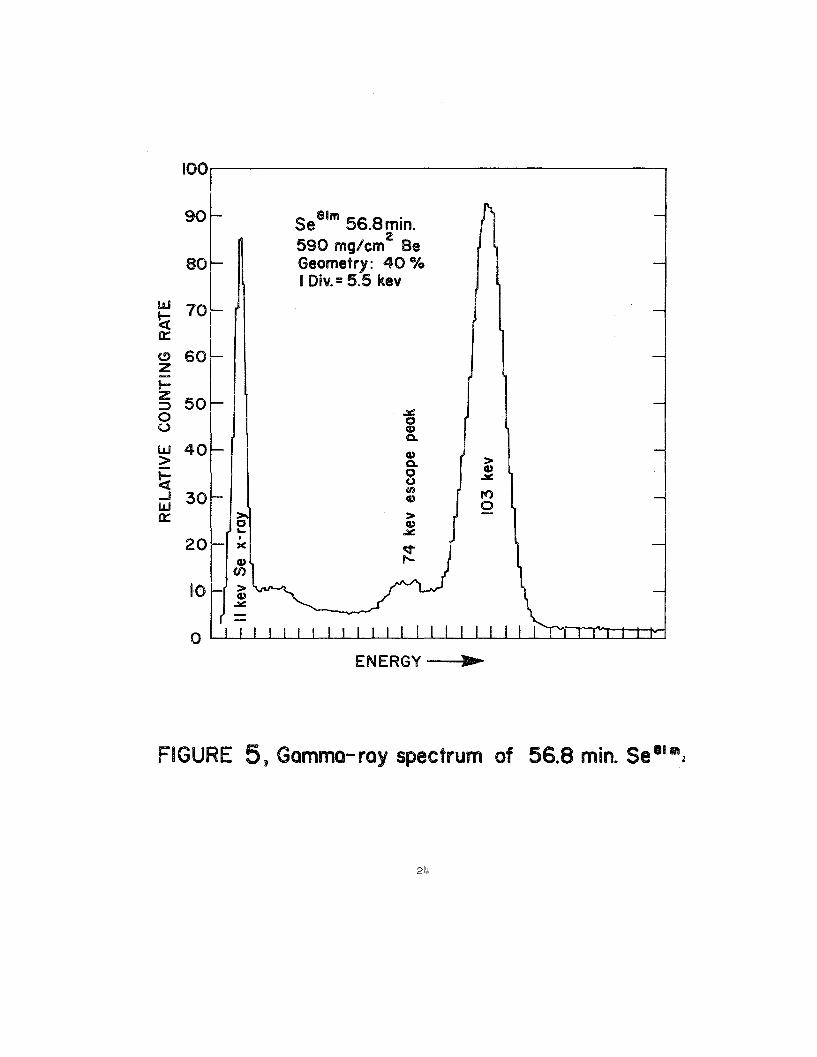
are Se75 (121 d), Se77m (17.5 see), Seem (56.8m), Sesl (18.2m) sndse=
(25 m). These isotopes are produced as a result of a number of nuclear
reactions on stable isotopes, either of selenium or other elements. Se=m
decays to Seel by ismnerlc transition; Se= decays further to stable Br81
by the endssion of 1.3@-MeVbeta radiation. Se= decayswith the half life
of Seelm (56.8m), stice the two states are intransigentequllibtium.“The
measurementof Se= must be completedby beta counting,using 4 rror 2 n
Geiger-MuelJeror proportional-betacounters.
The other isotopeadecay by means of gamma radiationsand can be measured
with a gamma scintillationcounter or gsmna scintillationspectrometer. Figures
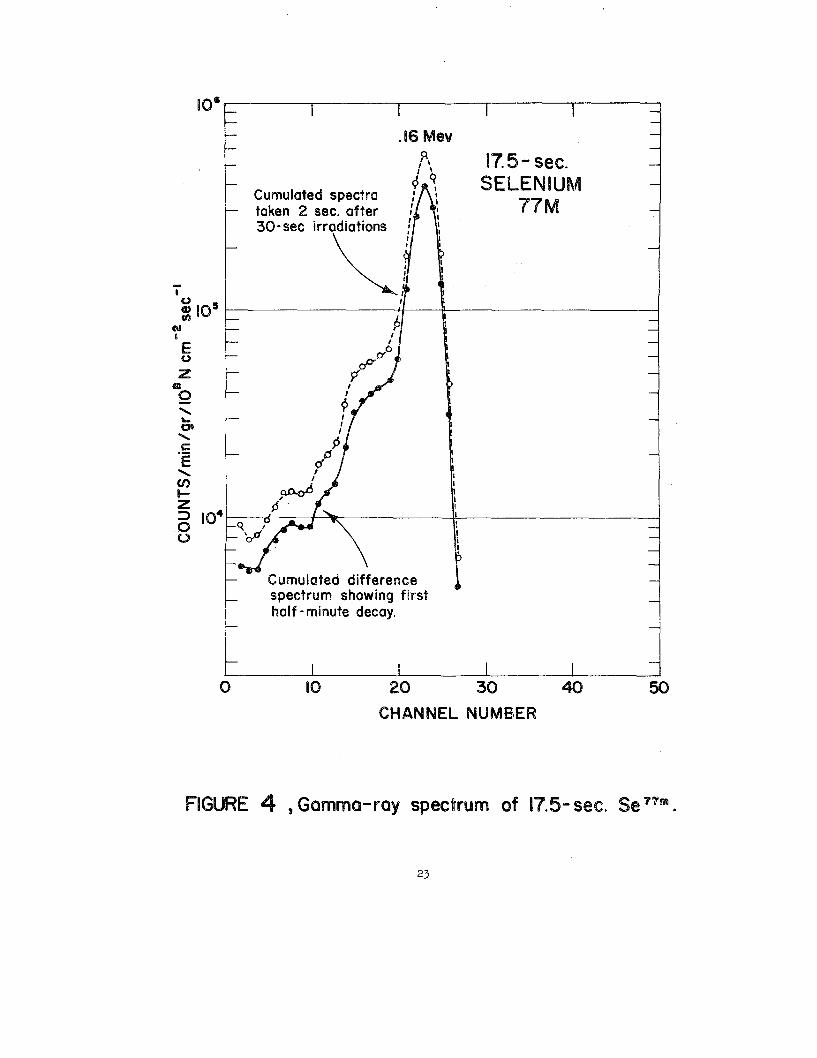
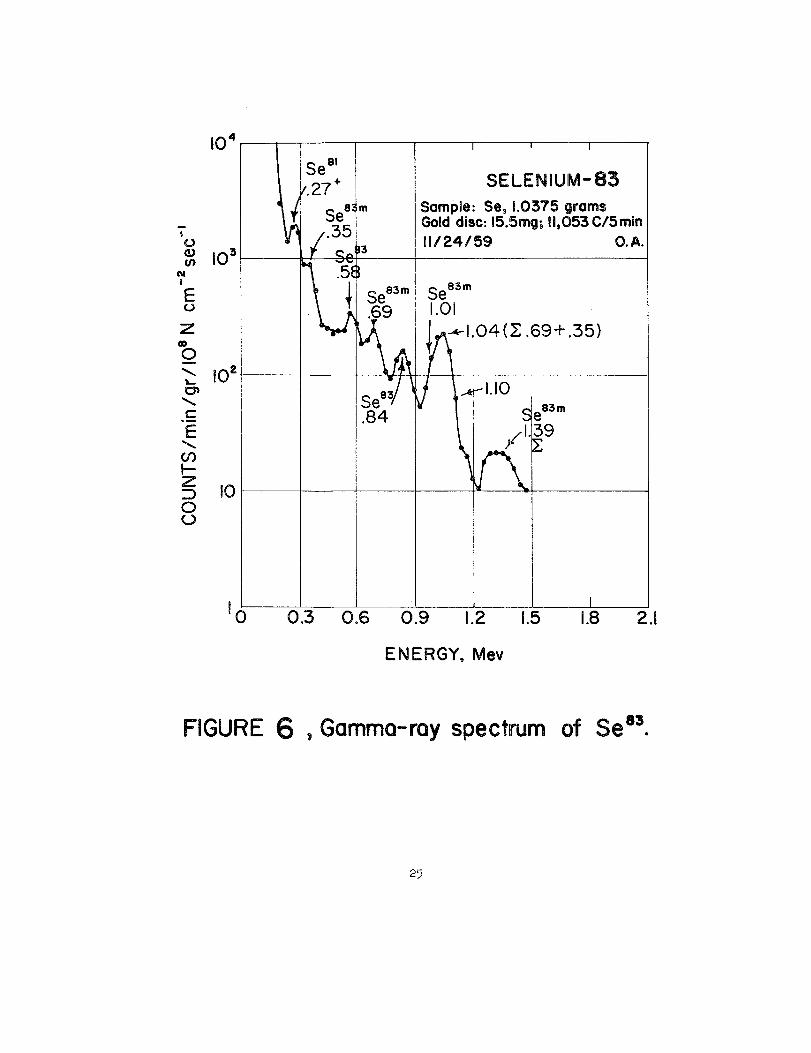
2 through 6 give gamma-rayspectra of the ss7s(87), Se7s(87),~mn(~), See1rn(89
~~ sem(90) radioisotopes.
Although a number of radionuclidesof seleniumare formed by radiative
capture in stable selenlunisotopes,L21-day Se75 and 3.g-minuteSe79m have
been most extensivelyused as the analyticalindicatorsfor the activation
analysis of selenium. However, these are not the most sensitiveindicators
19

for a seleniummalyais. Table V is a tabulationof the calculatedspeciflc-
saturationactivitiesof the nuclides formed by radiativethemsl-neutron
capture in the stable selenium isotopes. Activationcross sectionswere
ccm@led frm Informatim appearingin reportsby Eughes and Harvey(Q) ~
by Weigman(70). Krsmer, etal~m) discuss the metastableisaners of eele-
nium, emd show that the greatest sensitivitycan be obtainedby using l~.5-
second Se7m as the radioactiveindicator.
Seleniumm, a metastableisomer of stible Se=, decays with a half-
life of 17.5 secondswith the emission of 0.160-Mevgamma ~. Its
short ~-life has precludedthe use of Se- in an activationanalytical
scheme in most laboratories. In fact, there appear to be only two refer-
ences(61)~) that report quantitativeseleniumanalyaesbased onthia
particularnuclide. With rapid pneumatic-tubesyatems, s~les carIbe
transferredfram the irradiationfacillty to the gamma scintillation
counter inafew6econd3. OkS&(~) haE recentlyreported on the use of
gamma spectrometryto detect and &tennine t@ radioactivityof Se-
(17.5-see.) in nondestructiveanalysesof neutron-imsdiated sulfur,
ammonium cmrpounds,end ores for trace selenium. Putman and Taylor(72’73)
have reporteda techmlquefor ualng Se75 (121 d), for detenuining trace
selenium in glass. ‘I’@ made measurements of irradiated sauple & glass,
with and without selenlum, simultaneously, with two IieZ(Tl)scintillation
TABLE V
CALCULATEDSPECIFICSATURATIONACTIVITIESFOR SELENIUM
RADIONUCLICESPROEUCED BY THERMAL NEU’ITONCAPTURE
‘~ecificStablell&lll@
*74
*7e
%7 s
Se*
*8O
*S2
sea2
Isotopic
T0.87
9.02
23.52
49.&2
4$),82
9.19
9.19
Radio-nuelide
*75
Se77m
Se7m
Seslm
*81
Half-Life
l’ld
17.5s
3.9m
56.&tI
18.2m
70s
25m
ActivationCros8-section
(bma )
26
22
0.40
0.030
0,5
0.050
0.004
SaturationActivity (a)
1.7 x lCP
1.5 x 1o11
7.2 Xld
1.1 x ld
1.9 x ldo
3.5 Xlos
2.8 x 107
(a) dis/sec.g, baaed on a themsl flux of 1 Xldg neutrons/cm2. 8ec.
20
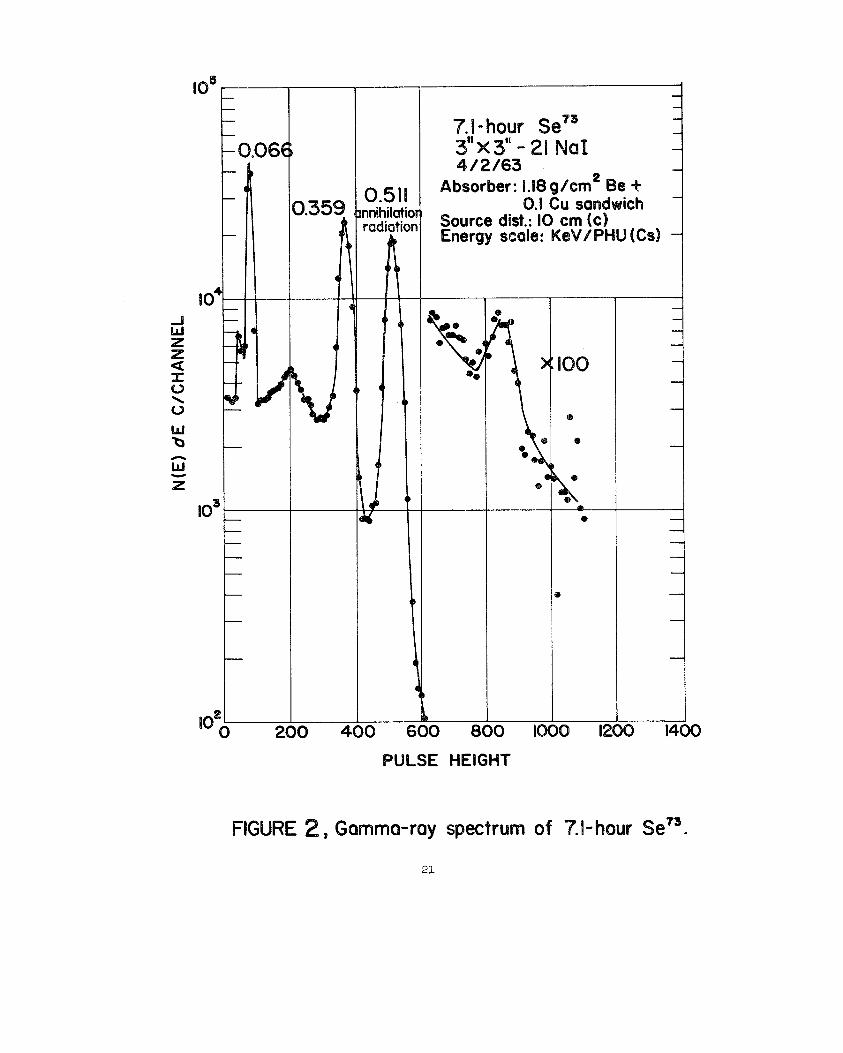
0.51!mihilotio
7’.1-hour sir:“KJ”;21 NaI :
Absorber: 1.!8 g/cm2 Be+0.1 Cu sandwich -
Source dist.: 10 cm (c)Energy scale: KeV/PHU (Cd -
%
\
.4e
e.
I
-Lo 800 Kx )0 12
PULSE HEIGHT
10 1400
FIGURE 2, Gommwmy spectrum of ‘?.l-hour Se73.
————
—
—
—
2(
FIGURE
(
—
.
.2$33, .:
.410)
6)
4
64
l-----0 400 6’
-i-
—
—
—
—
—
121- day Se753“X3*’- 2 N(ll
5/’19/58Absorber: 2CX3rng/cxnz -source C!istfmce: IOcm(c) -Eh3rgy scale: ~ lKev/PHU
PULSE HEIGHT
3, GQmmQ-rQyspectrum of
22
TI
)0 1200
121 -day Se?s.
—
I
.16 Mew
—
—
$“!
11
,y%#;II
~. IE
—,d I
~q’d tI\&
— Cumulated difference— spectrum showing first W
1- half - minute decay.
f
$7’.!5-sec.SELENIUM
77M
I I
23
se “m 56.8 fTtifl.590 mglcm2 Be(horne?ry: 40701W.= !5.5 kev
ENERGY ---+--
FIGURE !5, Gammo-ray spectrum of 56.8 min.SeaB%
24
0A4Iu
KY
!02
10
LOc
n
3
Se831n
.69
+se83.84
FIGURE 6,
3 0.6 c
I I 1
SELENIUM-83Wr@e: Se, 1.0375 gramsGold disc: 15.5mg; !1,053 CX5mir
11/’24?’!59 0. A
Se83m
1.01
I
3 1.2 1.5 1.8 :
ENERGY, Nlev
GammQ-roy spectrwm of Sease
25

counters; the responseof the counterscan be subtractedto leave only the
Erion, et al.’74) (75)selenium-75activity. and Fineman,et al. have also
determinedtrace seleniumin sulfur and ore concentratesby a nondestruc-
tive analysismethod.
IX. SETJ!RATTONPROCEDURESFOR SELENIUM
The radiochemlcalproceduresdescribedin the folloVingpages have been
divided inta four.cat.egories.In each category,special informationregard-
ing the method used, aep=ation time, type of bcunhardment,etc., appems as
part of each procedure. Wheneverpossible, an evaluaticmof the procedure
is made concerningits use in the decontaminationof other radioactivespecies
from the radioactiveseleniumisotopes.
“Activation-analyaisprocedures”deacrib.evariouamethods used to deter-
mine microgram and aubmicrogrememounta of seleniumin various typea of amplea.
In the activation-analyaisprocedu=s cited, a carriertechniquewas used to
separatethe radionuclidefrcunthe rgioactive mixture. In the procedum,by
Kuroda(36)the c~erwas labelledwith Se75. The chemicalyield can be75 in the find sample wfth ‘hedetermined by comparing the peak =ea of Se
Se75 &ded with the Cufiero
Moat proceduresdescribingthe prep-ation of radioactivetracera use
carrier techniquesto aep=ate the radlonuclidefrom the targetmaterial.
In the prep=ation of smne radionuclides,however, it la neceaaaryto sepa-
rate them without any additionof c~er. These procedures appe=”under
a aepsrate classification: carrier-free separation. In addition to the
procedures cited, It should be poaaible to uBe ion exchange in the carrler-
free sepexationof radioactiveseleniumisotopes.
Procedureswe also includedfor the emlysia of fission-productacti-
vities in plant-process solutions.
26

x. INIEX OF SEPARATIONPROCEDURES
1. ACZEVA!TZOWANALYSIS PROCEDURES
1.1 Metals and Alloys, BiologicalMaterials,Waters . . . . . . . 28
1.2 Ore Concentrates,Slags,Waate Gase8. . . . . . . . . . . . . 32
lm3Stony Meteoritea. . . . . . . . . . . . . . . . . . . . ...33
1.4 Fertilizers,Tmnato Tissue, Huron Blood . . . . . . . . . . . 34
1.5 SildcateRocka, Sedhants . . . . . . . . . . . . . . . ...35
1.6platinum Met~. . . . . . . . . . . . . . . . . . . . . . . . 36
2. TRAcER-PREPARAmoriPROCEDURES
2.1 Frmn Arsenic. . . . . . . . . . . . . . . . . . . . . . . . . 38
2.2 FrcenBismuthMetal. . . . . . . . . . . . . . . . . . . . . . 38
2.311rom Uranyl Nitrate. . . . . . . . . . . . . . . . . . ...39
3. FISSION-PIMXXKTPROCEDURES
3.1 Ilssion-productSolutions . . . . . . . . . . . . . . . . . . 40
3.2 POIYphe~ls . . . . . . . . . . . . . . . . . . . . . . . . . 41
4. CARRDZR-= SEPARATION PROC3LIJRES
4.1 FYom Szilard-ChalmereReaction. . . . . . . . . . . . . . . . 42
4.2 horn IkuteronEambardmentof an Arsenic ~get. . . . . . . . 43
4.3 FromumilRll Fls6ion. . . . . . . . . . . . . . . . . . ...44
5. CARFCtERPREPARATIONAND S~TION. . . . . . . . . . . . . . 45
27

1.1 Metals aud Alloye, Biolo@cal Materials, Waters (76,77-79)
Procedm used in: Activationaualyeis
Method: Volatilityand precipitation
ITucle= banb~t: *74 (n,y)s875
*so(n, y)*U
S& (n,y)Seel
Procedureby: Leddicotte,G..W.’76)
Chemicalyield of carrier 75-W%
.&pLmationtime: 1.5 hours
I&gree of purification: Btudlemwith radioactivetracersof Aa, W, Sn, Sb, Ha show thatdecontamrlnatiogiB better than108 for each
E@paent required: neutron source and stmdardI_ehoratoqequi~nt
1.1.1 Irradiaticmof Smuple Materials
I“madlatebmoun euwunlm of test (Note1) and comparator
(0.C$25to 0.030 g of Se metal to neeze,et0.1 lug)samples (Note2)
in a neutron flux of at leaat 5 x @l n/c#. sec for 16 hours.
(Note3). Use mall quartz tubes, polyethylenebottles,.or alu-
minuM foil to contadn the s.mples..
1.1.2 Preparatim of Cmp aratar Smnplea
a. After the irradiation,quantitativelytmazmfer”the
cmparator omples (Note2) to a MM-ml volmetric flack, dis-
solve it in a mall meaeured volume”of 6M BHOg, then dilute the
solutionto 100 ml with water. Mx well, ueing stie-hendmng
practicesfor radioactivemateriels.
b. Pipet a 1.OCHul aliquot of this solutioninto a
second 10WDI. volumetricfleak; dilute to volume with water
and ndx well.
c. Pipet a 1.O&nl aliquot of this solutioninta a 125-ml
@ems distillationflack. By meane of a volumetricpipet, add to
the mme distillationflack 2.~ ml of a standardcarrier solution
of known Se concentration(Note4). Also add 1 ml each of hold-
back carriersof k, Cu, Co, Fe, IVa,and Sr (Notes5 end 6). Con-
tinue with Pti 1.1.4 helm.
28

1.1.3 PrepWatlon of Test Ssmples
.9. If the sample is a metel or tioy, quantitat~vely
tramfer the irradiatedtest portion frcnzthe qu.&tz tube or slu-
minum wrap to a 125-ml glass distillationflask, and then add, by
means of a volumetricpipet, to the same distillationflask 2.00 ml
of standard cexrler solutionof known Se concentration. AhO add
1 ml each of holdback caxriersof Aa, Cu, Co, Fe, Na and Sr (Notes
5 and 6). To this tixture, add dropwiseenough concentratedmine-
ral acid to dissolve the sample completely. If neceasaxy,heat the
mixture to effect solution. Continuewith Part 1.1.4 below.
b. If the sample is an aqueous liquid,pipet an aliquot
of the irradiatedportion into a 125-ml glass distillationflask.
By means of a volumetricpipet,”add to the seinedistillationflask
2.CO ml of a standardcarrier solutionof known Se concentration.
Also add 1 ml each of holdback carriersof As, Cu, Co, Fe, Na, and
Sr (Notes5 and6). Continuewith Part 1.1.4 below.
c. If the sample is biologicalmaterial (plant ash, tissue
ash, bdy fltid, etc.) or if it is an organic liquid (a petrochemi-
cal or one of its derivatives),pipet a luwwn aliquot, or quantita-
tively transfer the total amount of solid, to a 125-ml glass dis-
tillation flask. Then add (by means of a volumetric pipet) to the
same distillation flask 2.00 ml of a standti carrier solution of
known Se concentration. Also add 1 ml each of holdback cerrlers
of As, Cu, Co, Fe, Na and Sr (Notes 5 and 6). To this mixture add
3 nil of ~0, 1 ml of @ HC1, ,md 15 ml of BN03-HC104mixture. ~SO,
put 2 or 3 glass beads into the flask, then heat the solutlonat a
nderate temperature(60”C)until all of the HNOg is removed and
HC104 condenseson the neck of the flask (Note7). Digest the mix-
ture in this manner frcun1 to 2 hours. Remove the flask from the
hot plate, cool it aud continuewikh Pert 1.1.4 below.
1,1.4 RadiochemicalSeparationof Selenium
a. Add 10 ml of concentratedHC1 and 10 ml of 48$ HBr to
the distillationflask and place a 50-ml glass centrifugetube at
the end of the distillatetube. Add 10
solution)to the centrifugetube. cool
an ice bath.
b. Connect the condenserhead
bubbling alr through the mixture in the
ml of water (or WmLli
the tube by placing it in
to en air flow end begin
distillationflask. Heat
29

the flask (an open fl=e can be used) to boiling. Continue distil-
lation process for at least 5 minutes (Note8).
c. Remove the centrifugetibe containingthe distillate
fractidn from the ice bath, then neutralizethe solutionby adding
concentrated~OH dropwiae (Note 9).
d. Acidify the solutionwith 2 ml of cone. HCL. Add 4 ml
of @ H@33 solution, Stir the mixture until the aelenioua acid is
reduced to elemental Se. Centrifuge the mixture; tiscti the super-
natant liquid.
e. wash the precipitate once with 15 ml of hot water.
Centrifuge; discard the wash liquid. Add a 10-ml volume of hot
water to the tube and stir.
f. Rllter off the Se metal thr&gh a weighed filter paper
(MunktellsNo. 00 or Whatman No. k?) held in a Hirsch funnel;wash
the precipitatetwice with 10-ml portions each of &O, 95$ ethyl
alcohol, and ether.
~. Dry the precipitatefor 30 tinutea in a dqlng oven
at llO”C (Note10). Weigh the Se metal precipitate (Note11) and
filter paper on am analyticalbalance. Then mount the precipitate
for the refkloactititymeaaurements.
1.1.5 Measurementof the Radioactivityfrcm Se75 (and/orSe= or
Se-) and Calculationof Inactive Selenium Content of the
originalSSrQle
a. The analyses involvingthe meaeurement of Sem must be
completedby beta counting. S-e= m a ha,lf-ufe of 10.2 tiutea
end decays to stible Bra only by the emission of 1.3&Mev beta ra-
diations. These mesaurementa can be made with a Geige~Mueller
counter.
The analyses involvingthe measurementof Se75 and Se-must
be cczzpletedbygamma couuting. Se75 has a half-lifeof 121 days
and dec~ by means of electron capture and @ma radiations of
various ener~es (chiefly, o.u1, 0.136, 0.27, 0.28, 0.405 b,
plus others). Se- has a half-lifeof 57 minutes and decays to
Sea (18.2m) by isomeric transitionwith the emissim of O.10>Mev
gamma radiations. These measurementscan be made by means of a
~ scintmatim comter or a gwma scintillationspectrometer.
In the use of the latter, the @or gamma radiationsare measured.
b. FolJowingthe -oactive messurements,the observed
radioactivityis correctedfor loss of carrier during the experi-
ment, half-life of the seleniumisotopemeasured, and the weights
30

of the tei3tand cmqxrator semples. A comparisonof these cor-
rected radioactivitiesbecmnes a measure of the stable selenium
content of the test ssmple:
Percent stable Se in test =sample
NOTES:
1. At least O.lGg
CorrectedSe radioactivity: in test smupleOorrectedSe radioactivity: in cmnparator
ssmple
portions of solid samples (metals,aUoys,
tissue, etc.) shouldbe used. Liquid ssnples (water,body fluids,
etc.) should be 1 to 20 ml.
2, Compsxatorsample may be either Se metal or a solution of
a Se cmpound. If a solutionis used, an aliquot of the irmdi-
ated solution containingat least 20 micrograms of Se can be
pipetted tirectlyinto the distillationflask.
3. The sensitivityof the method is such that 2 x l~s gm
of Se can be determined. The sensitivitycan be improvedby
use of higher neutron fluxes.
4. As seleniou2acid (E&3eO~),Se+4
10 mg/ml.
5. Holdback carriersshould be tie
of desired elementalspecies.
concentration
up to contain
6. Solutionsof ions of other elementsmay also be added
e.aholdback carrLers.
7. If the biologicalmaterials is a fatty substance,a Wet
digestionwith HN09-&S04 mdtiure should be used to effect a
solution.
8. Telltium, As, Sb or Ge can follow,wholly or in part,
through this distillationprocedure. In most of the materiels
analyaedby this method, the radioactivecontaminantswere in
minor concentrations,or completelyelhdmated, in the reduc-
tion of Se to elementalform.
9. The Br2 color of the solutiondisappearsduring neutrali-
zation.
10. If the Se= or
p=t of the procedure
measurements.
Sem radioactivityis to be measured, this
cam be completedafter the radioactivity
31

Il. By ccmpexingthe final weight of the Se-metalprecipi-
tate obtainedhere with the theoreticalyield expectedfor the
amount of Se carrieradded, it is poaaible to determinethe
chemicalyield of the expertient. The chemicalyield correc-
tion is then used to determinethe emoumt of Se75 (end/orSee’
or Semm) recoveredduring the separationprocess.
1.2 Ore Concentrate, SISRS,Waste Gases(75)
Procedureused in: ActivationanalyBis
Method: Volatility and precipitation
Type of bombardment: Se74(n,7)Se75
Procedureby: Fineman, et al.(75)
Chemicalyield of curler: 50-00$
separationthe: Several houm
Degree of purification: Excellent frmn other volatileimpurities
Equipment required: Neutron source and standardlaboratoryequipment
1.2.1 Procedure:
a. Irradiate samples (0.1 to 1 g) in a nuclear reactor
for 14 days at a flux of 4 x ldl n/cm2,sec.
b. Mix a weighed amount of Se (10-100 mg) thoroughly
with irradiatedsample and fuse the mixture in a nickel or iron
cruciblewith 25 grsonsof Na20z at 9OO-1OOO”C.
c. DLssolve cake In 50 ml of water. Cool solution and
neutralizewith “(1:1)H&Q. Add at least 50 ml more of concen-
trated &S04 to the solutionand evaporateto 100 uL. To separate
Si02, boil the solutionfor 15 minutes, then cool to 60 to 70°C.
Add 15 ml or 5$ gelatin solution and bubble air through the solu-
tion for 30 minutes. Add hot water (50 ml) to the solutionand
filter the Si02. Evaporate the filtrateto 100 ml, cool, end
then transferto a distillationflask.
d. Add 100 ml of 4~ HBr, contahing 7 ml Br2, to the
flask, and distill the mixture into 10 ml of water; collect 40-
50ml distillate.
32

1.3
e. Neutr.sJlzedistillatewith N&OH, add 10 ml HC1 and
1-2 g hydrazine sulfate. After a 12-hour digestion,filter,
wash with water and ethanol, and dry at 105’C; weigh and mount for
the radioactivitymeawuement.
NaI(Tl
manner
lyzed.
f. Measure the Se75 gmzaa radioactivityby use of a 3“ x 3“
scintillationcrystal.
g. Irradiateand process compmator s~les in a similar
to obtain the Se concentrationof the materialsbeing ana-
Stonv Meteorites(m)
Procedureused in:
Method:
me of nuclear bombardment:
Procedure by:
Chemicalyield of c=rier:
Sep=ation time:
Degree of purification:
Equipment required:
Activationanalysis
Fusion, precipitationandion exchange
Se74(n,7)Se75
Schindewolf’80)
Quantitative
Several hours
Decontaminationof >104 forAg, Ce, Co, Cs, Hg, Ir, RuSb, Sc, Ta, Zn, and Zr.
Neutron source and standardlaboratoryequipment
1.3.1 Procedure:
a. Irradiatepowde~d meteorite samplesweighing 0.050
to 0.20 grams.
b. Add 0.020 g each of Se and Te to the irradiatedsample
and fuse the mixture in a nickel cruciblewith 1-2 grams of NQ02.
c. After cooling,dissolvethe cake in 6~ HC1 and boil
the mixture. Reduce Se and Te to elementalform with S02 gas.
a. Dissolveprecipitatein aqua regla and make the solu-
tion basic with N&OH. Scavenge impuritieswith Fe(OH)3.
e. Acidify the solutionwith HC1 and then adsorb Se and
Te on a Dcwex-1 (100-200mesh) anion-exchangeresin column. Sele-
nium can be sepuated from Te by eluting it from the columnwith
3 to ~-columnvolumes of 31 HC1.
33

1.4
f. heat the acid solutionwith S02 gas to reduce Se to
the elementalfem. Collectprecipitateby filtration,weigh, and
mount for the radioactivitymeasurement.
g. Measure the Se75 gsnma rsdioactititywith a 3“ x 3“
NaI(TL) scintillationcrystal.
h. Irradiateand proces8 comparatorsamples in a similar
manner to obtain the Se concentrationof the meteorite.
Fertilizer, Tomato Tissues, Human Blood(60)
Procedureused in: Activation
Method: Volatility
analysia
and precipitation
Type of nuclem bomb~nt: Sem(n,y)Se=
Procedureby: Bowen and Cawse(60)
Chemicalyield of carrier: 7@
Separationtime: 40 minutes
Ikgree of purification: Decontaminationfrom sxsenic,bromine,manganese, sodiumand zinc was found to besatisfactory
Equipment required: Neutron source and standardlaboratoryequipnent
1.4.1 Procedure:
a. Irradiate samples in a nuclear reactor for 18 minutes
at a flux of l&2 n/cm2.sec.
b. After irradiationtransfer the sample to a beaker con-
taining 10 ml of asblngmixture (1:1 16~ HNOg-7@ HC104) and-25 mg
each of As, P, Mn, and Te carrier solutions.
c. Heat solution until organic is destroyedand fumes of
perchloricacid evolve.
d. Transfer acid solutionto distillationflask with 5 ml
each of HC1 and HBr.
gas,
e. Collect
Distill for 4 ndnutes using N2 as a cwrier
distillatein 50-ml centrifuge
5 ml of HC1, 10 ml water, O.lml of
Shell Chemical Company) and 2 drops
34
Teepol solution
of Mn carrier.
tube conteini~
(wettingagent,

f. Pass sulfur dioxide through solutionfor 1 minute end
centrifugethe seleniumprecipitate.
8. Dissolve the precipitatein about 0.25 ml of bTT@.
Add ten ml of hot water, 0.1 ml of ~02 and 5 ml of HC1, and pre-
cipitatethe Se again with S02.
h. After separationby centrlfugation,wash the precipi-
tate with water and acetone,and transferto a weighed eihnnlnum
counting tray with acetone.
1. After the sample is C&y, count the beta-actititywith
an end-windowGeiger counter. Check radiochemicalpurity by com-
paring decay curve~ of samples and standards over several half-
lives.
il- After counting,weigh the seleniumto obtain the chemi-
cal yield of the carrier.
k. Irradiatecomparatorssn@es and process in a simila
manner to obtain the Se concentrationof the materialsbeing ana-
lyse~.
Procedureused in:
Method:
Type of nuclear bombardment:
Procedure by:
Chemical yield of carrier:
Separationtime:
Ikgree of purification:
Equ@ment required:
1.5.1 Procedure:
Activationanalysis
Solvent extraction
Se7e(n,7)Se7em
Kuroda, R. reportedby
Mei*e(36)
. 6%
10 minutes
Good except for iodine radioactivity
Neutron source and standardlaboratoryequipment
a. Irradiatel-g sample in nuclear reactor for 10 minutes
at a neutron flux of 2 x ld2 n/cm2.sec.
b. Fuse the sample with 6 g of N*02
Se curier labeledwith a lumwn amount of Se75
35
containing 30 mg of
(121 d).

c. Cool the cruciblein an ice bath; put the crucibleinto
a beaker; and add successively10 ml of &O, 10 ml of formic acid,
35 ml of HC1 end 75 ml of KBr.
d. Filter off the precipitatedsodium salts.
e. Add a few grams of hydroxylemlnehydrocblorldeand
1~ ti of 2,6-dimet~l-4-heptEmoneto the filtrateand stir vigor-
ouslywith a mechanicalstirrer.
f. W-h the organic phase with 50ml of water (Note1).
g. Wash the organicphase again with 20 ml of 5% tartaric
acid and then with 40 ml of 5$ ELXCAsolution containingsmall Smounta
of ~ NeOH. Diacmiwashinga.
h. ~lute the organicphase to 15 ml with the extractant
and measure the Se7- gamma activity.
i. Chemicalyield is determinedby compting the peak area
of Se75 -n the orgdc phaae from ample with that of se75 in a known
amount of carier.
NOTE :
1. If the two layera do not aeperate quickly,add small
amounta of formic acid.
PL5LthumMetal(84)
Procedureused in:
Methcii:
Type of nuclear bombardment:
Procedureby:
Chemicalyield of carrier:
Separationtime:
1.6
Activation~yais
Ion exchangeaad precipitation
Se74(n,~)Se7s
Morris and Killick(84)
. 8@
Severalhours
Degree of purification:
Equipmentneeded:
1.6.1 Procedure:
a. Irradiate
@a at a flux of l&2
Separationfactor of seleniumfrom telluriumof greater than104.
Staatid
aemple (041 g) in nuclear reactor for E?
n/cnF.sec.
36

b. Trarmfer sample to a flask attachedto a water-cooled
reflux condenserthat contains10 mg of Te and 15 mg of Se carriers.
Wash samples into flaak with ~ HNOS and add 5 ml of ~ HC1 and
dissolvethe platinm by wamhg at 600C on a water bath.
c. Remove HN03 by addjng ~ HC1 and heating. Cool, aud
transfer to a centrifuge tube. Dilute to 35 JIJ with water. Place
in an ice bath and add 1 g N&Cl to precipitate (~)2 PtCle. Allow
to stand 10 minutes and centrifuge.
d. Separate supernateinto cleea centrifugetube. Pass
S02 rapidly through solctionfor 2 to 3 minutes. Centrifugeand
va9h with water.
e. Dissolve Se and Te in ndnbnnn amount of ~ llNOg. Add
2 mg of Pd carrier and dilute to 20 ml with &.O. Heat to boiling.
Neutralizewith ~ NaOH .md add 1 ml more of the reagent. Add 1 to
2 mg Fe+s carrier dropwisewith stirring. Centrifugeand discard
the precipitationof Fe(OH)S.
f. Add 1 ml of 6~ EIN03to supernateand then add, with
stirring,1 ml of 1$ solution of dimethylglyoximein 95$ C2HSOH.
Allow to stand 10 minutes and filter through a Whatman No. 41
paper. Evaporatethe filtrateto dryness, adding a few drops
of lC$ KI during the evaporation;then dissolvewith 5 ml of
12M HClc—
g. Pass the solutionthrough a deaciditeFF (100-203
mesh, preequilibratedwith la HC1) anion-exchangecolumn (equiva-
lent to Resin Dowex 1-x8), 5 cm in len@h and 1 cm in diameter.
Elute with 20 ml of 31jHC1 into a 50-ml glass centrifugetube.
h. Add 20 ml of l% HC1 to the eluate in a centrifuge
tube. Paas S02 through the solutionto precipitatethe Se and
centrifuge, Wash with water and dissolve in minimum quantity
of @ HIT03. Evaporate to dryness. Add 1 ml of 6M NaOH, then—
20 ml of @ HC1, and boil for 15 minutes. After the addition
of 10 ml of 2C$ solutionof hydroxylsminehydrochloride,centri-
fuge, then discard supemate. Wash the Se with water and then
with C2HSOH. Transfer the precipitatewith a little C2H50H to
a weighed aluminum countingtray, and dry at 100”C. Weigh as
Se and determinethe chemicalyield.
37

2.1
2.2
Arsenic
Procedureused h:
Method:
Type of nuclear
Procedureby:
bombardment:
Separationtime:
Chemicalyield of
Decontamination:
Equipment needed:
carrier:
Preparationof radioactivetracers
Volatill.tyand precipitation
194-Mev deuterom
H. Hopkins,Jr., reportedby
Mei*e(8’)
45 minutes
> 90%
RadiocheH&caUy pure by afactor of - 100
Standard
2.1.1 Procedure:
a. Dissolve
b. Add5mg
move excess HN03.
Aa metal in mintium 10~ HN03.
Se carrier, evaporate to near dryness to re-
c. Make up to 3 ml with ~ HC1, add N& OH,HCl until Se
darts to precipitatefrm hot Bolution.
a. Add 1 ml ~ ICI,heat 5 minutes, centrifugeoff mixture
of Se and 12.
em Dissolvewith a udnlnmm amount of fuming HTJOS,repeat
precipitation.
Bismuth Metal(81)
Procedu uged in:
Method:
Type of nuclear bombardment:
Procedureby:
Sepexationtime:
Chemicalyield of
Decontamination:
Equipmentneeded:
terrier:
Preparationof radioactivetraceri3
Volatilityaud precipitation
184” cyclotron (388-MeveLphas;3~Mev protone; 194-Mev deuterons)
Goeckermen,reportedby Mei*e(8’)
1 hour
- 9&
_ 104 from fission and spallationproducts
Stsnhrd
38

2.2.1
and Te,
still.
taining
disti~
Procedure:
a. To aliquot of BN03 solutionof target, add 10 mg Se
10 ml concentratedHBr, sad 0.5 ml liquid Brz in a glass
Uging en air stream, distill into a centrifugetube con-
5 ml saturatedBr2 water, which is cooled in en ice bath,
until only a >ml residue remains.
b. Keep at ice temperatureand reduce to Se (red)with
S02 or N&OH.HCl. Add aerosol and centrifuge.
c. Dissolve Se in a few drops of concentratedHN09, add
10 ml concentratedHC1 and reduce with S02 in am ice bath. Centri-
fuge with aerosol.
d. Repeat SeBr4 distillationand Se precipitationsas
often as necessuy for desiredpurity.
e. PrecipitateSe, filter, wash three times with 5 ml
~0, three times with 5 ml ethyl alcohol, three times with 5ml
ether, dry 10 minutes at llOUC. Weigh as Se.
2.3 Uranyl Nitrate(36)
Procedure uBed in:
Method:
Type of nuclear bombardment:
Procedureby:
Separationtime:
Chemicalyield of
Decontamination:
Equipmentneeded:
carrier:
2.3.1 Procedure:
Preparationof radioactivetracers
Solvent extraction
Neutron - 5min. atpower level of 1000 Kw.
Kuroda, reportedby Meinke(36)
19 min.
70%
Radiochemicallypure by gammaspectroscopy
Neutron source and stand-laboratoryequipment
a. Di8BOlVe 30-50 mg irradiateduanYl nitrate in 20 ti
of concentratedHBr containingw 1 mg of Te and 30 mg of Se carriers.
Pass air stream through the solutionfor 5 minutes to expel fission
gases.
39

b. Extract Se with ~ ml of 2,6-dlmethyl-&heptanonefor
@ sec. and discard aqueousphase,
c. Shake the organicphase for 30 secondswith 10 ti 10$
tartaric scid; repeatwith 10 ml of ~ NsOH. Dlscti aqueous
phases.
d. Wash the organicphase by shaking for 30 secondswith
30 ml of a solution5$ in EDTA and 5$ in sodium tartrate. Then
shake with 20 ml of water.
e. Strip Se from the organicphase by shaking for 20
seconds each with three 10-ml portions of C12-saturatedvater.
f. To the aqueousphase add a
and 6 ml of 12 NsOH md shake.
g. Add 20 drOpS Of COnC. HN03
few mg of iodide carder
to acidify the solution.
h. Add dropwise a 2* solutionof hydroxylamhe hydro-
chlorideuntil brown color of iodine appears.
i. Extract iodinewith successive5-ml portions of C-
bon tetrachlorideuntil the color of the organicphase disappe=s.
J. The organicphase Is discardedeach time and the radle
chemicalPurim of the aqueous phase is tested by gemma spectroscopy.
3.1 Fission-Roduct Solutions(82)
Procedm used in:
Method:
Type of nuclear
Procedureby:
bombardment:
Chemicalyield of c~er:
Sepmation the:
Degree of purification:
Equipment reqtired:
Determinationof fission-product activitiesin plantprocess solutions.
Volatility and precipitation
Uranium fission
Glendeninsnd Winsberg(83)
M
1 hour
Excellent
Standexdlaboratoryequip-ment
a. Place
still and add 2 ml
3.1.1 Procedure:
not more than 5 ml of seanple(Note1) in a glass
of Se csxrier =d 10 ml of cone. HBr. Distill
40

the Se into ~ HL of water contained in a 50-ml centrifugetube
placed in an ice bath. Continuethe distillationuntil no more
than 2 to 3 ml of Bolutlonremains in the distillationflask
(Note2).
b. Paas S02 rapidly (Note 3) through the distillate (in
an ice bath) until the red precipitateof Se is coagulated(2-3
minutes), centrifuge,and wash with 10 ml ofwater (Note 4).
c. Dissolve the Se by heatingwith 5 to 10 drops of cone.
SNO~; evaporatenearly to dryness; and take up In 10 UL of cone.
HC1. Place in an ice bath and precipitatethe Se with S02 as in
step b,
d. Repeat step c.
e. Transfer the Se with 5 to 10,ml of water onto a
weighed paper (Note 5) in a small Hirsch Ihumel and filterwith
suction. Wash three times with 5 ml of ethanol,and three times
with 5 ml of ether. Dry at 11O”C for ten minutes,weigh as elemen-
t~ se; and munt.
NOTES:
1. If the sample containsa large amount of nitrate, add
the Se carrier and boil down to about 1 niibefore distilling
with HBr.
2. The distillationof Se is practicallycompleteat this
point. The solutionshould not be taken to drynees.
3. A rapid stream of S02 hastens precipitationand aids
in coagulation.
4. A few drops of aerosol solutionprevents scum fomna-
tion and aids in centrifugation.
5. The filter paper is washed with ethanol end ether, and
dried under the conditionsof the procedurebefore weighing.
3.2 IrradiatedPolyphenyls(85)
Proceduremed in: Iktemination of fissionproduct activitiesin irradiatedpolyphenylB
Method: Volatility and precipitation
41

Type of nuclear bombardment: Uranium fission
Frocedum by: Felber and Koch(85)
Chemlcel yield of carrier: Wgojt
Ilsgreeof purification: Good
Equipment required: Stand-d laboratoryequipment
3.2.1 Procedure:
a. Aliquots of samples (not exceeding15 grams) and
carriersare dissolvedin cone. PN03, and perchloricacid is
added to deccunposethe organicmaterial. S-ampleis taken to
fumes of HC104.
b, Sulfur is sep=ated from the solutionby precipi-
tation of B8S04.
c. Seleniumis separatedby reductionwith S02. The
Se precipitateis dissolvedin cone. HBr containingsulfuric
and nitric acids. SeBr4 is distilledin the presence of air
into water. Seleniummetal is precipitatedby reductionwith
S02.
d. Selenium Is distilleda second time and the metal
Is repeatedlyprecipitated,and then weighed as the metal smd
the Se75 is counted.
4.1 Szilard-ChalmersReaction
Procedureused in: Carrier-freeseparationofSeleniumradioactivity
Radioelement(s)separated: Se=(25m), Se-(5~),
Se=(17m), and Se75(121d)
Procedureby: Gest and Edwexda(&)
4.1.1 Procedure:
a. One gram of ammonium Eelenite (Note1) irradiated
for 30 minutes.
b. After irradiationdissolve selenitein 31 HC1 and
extractwith CS2 (Notes2 end 3).
c. Evaporate CS2 fraction to dryness and the take up
Se in cone. HNO~.
42

N~S:
1. Anmonl.umseleniteprepmed by neutralizingSe02 solu-
tions with Ii&OH and crystallizingwith acetone.
2. 2C$ of the seleniumactivity faund in CS2 fraction.
3. In a second method, a 62 HC1 solution of the irradi-
ated selenltewas filtered through a fine sinte~d-glaas
filter. Much of the radioactivitywas depositedon the
filter. 28$ of the original aelenlumradioactivityremoved
by passing boiling cone. EUT09through the filter.
h.2 Ikuteronlkmbardmentof an Arsenic Tex@
ProcedureUBed in: Preparationof carrier-freeselenium
Method: Bombardmentof arsenic tiget
Type of bombardment: lg-Mev deuteronbombardmentwith 60” cyclotron
Time of sepantion: 3-4 hours
Chemicalyield: 80-95$
Procedureby: Gartison,Maxwell and Hamilton(8)
Ikgree of purification: At leaat factor of 103
Equipment required: Standard laboratoryequipment
4.2.1 Proceduxe:
a. Dissolve arsenic-powdertarget in aqua regla.
b. Add ~ HC1 to destroy excess HNOS and adjust acidity
to yJ.
C. AddlOmg of
S02; approtitely 95% of
d. Diesolvethe
tellurousacid
Se carries.
Te precipitate
and precipitateTe with
in cone. BIT03and repre-
cipitatefrom 31 HC1 in presence of As holdback caxrier.
e. Repeat Step d two more times.
f. Wash the Te precipitate,dissolve in minimum volume
of 16x HN03, and transfer to distillingflask.
g. Add 141 EBr dropwise whil-ea
is bubbled through the solution at 200eC.
in a water trap cooled w~th ice.
stream of N2 carrier gas
Collect the distill.ate
43

h. Remove the HBr with HN03 and evaporateto dryness on
40 mg of NaCl in the presence of excess EC1. Th&actltity, presu-
mably as aelenate,is quantitativelysoluble in 5 ml of water.
4.3 Uraulum FYssion
Procedureused in:
Type of bombardment!
Reikloelement(s)sep=ated:
Procedure by:
Eegree of purification:
Equipment required:
Prep=ation of carrier-freeseleniumradioactitities
Neutron source
Se7m(17.5 s), Se7m(3.9 m),
Se=m(57.5 s), See1(18.2m).
Kuroda, reportedbyhleitie(36)
Radiochemicallypure by gaunnaspectroscopy
Standard
4.3.1 Procedure:
a. Put irradiateduranyl nitrate (40-50mg) into a centri-
fuge tube. Add 2 or 3 drops of concentratednitric acid md 4ml of
water. Pass air streem through solutionfor 5 minutes.
b. Add- 1 mg of Te c~er and 20 ml of cone. HBr.
c. Extract seleniumwith 5 ml of 2,6-dimethyl-4-heptanone
for 40 seconds and discard aqueousphase.
d. Shake the organicphsae for 30 secondswith 10ml l@
tartsricacid; repeatwith 10 ml of lliNaOH. Discard aqueous
phases.
e. Wash the orgaaicphase by shaking for 30 secondswith
30 ml of a solution 5$ in EETA and 5$ in sodium tartrate,then
shakewith 20 ml of water.
f. Strip seleniumfrom the organicphase by shaking for
20 seconds each with three 10 ml portions of C12 - saturatedwater.
g, To the aquecrusphase add a few mg of iodide carrier
and 6 ml of lIJNaOH and shake.
h. Add 20 drops of cone. HN03 to acidify the solution.
i. Add dropwisea 2C$ solutionof hydroxyleminehydro-
chlorideuntil brown color of iodine appears.

J. Extract iodinewith successive5-ml portions of c-
bon tetrachlorideuntil the color of the organicphase d.isappeus.
k. The organicphase is discardedeach time, end the
mdiochemical purity of the aqueousphase is tested by gamma spec-
troscopy.
5. CARRIXR PIUZPARATIONAND STANDARDIZATION
1. Prepamti on
Dissolve 16.4 grams of seleniousacid in water and dilute to one
Ilter (1 ml ==lCkug
2. Standardization
Se).
Pipet 5 ml of carrier solutionInto a beaker. Add 100 ml of 5!
HC1. Add 10 ml of a 25$ hydroxyleminehydrochloridesolution. Heat
for 1-2 hours at 90°C. Filter through a pretreatedslntezwdglass
crucible, wash with water, and then with alcohol. Dry in an oven
105-110°Cfor 20 ndnutes. Cool and weigh as selenlummetal.
Mg S+l =wt. of Se metal
5ml
NOTES:
1. If the red selenium
the crystallinegrey-black
is overheatedit may convert
variety. This may result in
et
to
high results due to the occlusion of water and oxidation.
2. To prevent oxidationthe precipitatecan be dried
in an atxnosphereof carbon dioxide.
45

1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
I-2.
13.
14.
15.
Strominger,D., Hollander,J.
REF!EMNCES
M *J and Seaborg,G, T., “Table ofIsotopes,”“Rev:Mod. Physj ~ (2) p. 585-904 (1958).
Nuclear Data Sheets,Nat’1 Aced. Sci.-Nat’lResemch Council,Washington,D.C.
Chext of Nuclides,GeneralElectric Company, Schenectady,NewYork, December,71962 ).
Hopkins,B. S., ~ti%ch~”trx.— of the Less FamiliarElements, Chapter19, Stipes, Champaign,Illinoi~l~
Latimer,W. M., The CMdation Potentials,2nd Ed., p. 81-9,Prentice HaU, N~Y_2].
Berzelius,J. J., Acad. Han&l. Stockholm,~, 13 (1818).
Eodgman, C. D., Weaet, R. C., Shankland,R. S., and Selby, S. M.,Handbooks Chemist~ and Physics, 44th Ed., 1962-1963,chellliCSlRubber PublishingComp~, Cleveland, (1962).
GarrisonjW. M., MmWell, R. D., and Hamilton,J. G., USAEC ReportAECU-579 (UCRL-450)(1949).
Schoeller,W. R., and Powell, A. R., The Anal. is of Minerals and~&er, N. Y=,Ores of the Rarer Elements, Chapter~~ p.
m5T — —
All-UnionMining MetallurgicalScientificReseexch InOtituteofNon-FerrousMetals, ZavodskayaLab., 25, No. 6, 666, (1959).—
Goto, E. and Kakita, Y., Sci. Repts. Reaemch Insts. Toholm Univ.7A, 365, (1955),—
Chernikov,Y. A., and Dobkina,B. M., ZavodskayaLab., ~, 1143,(1949).
Eode, H., Z. Aual. Chem. 143, 182 (1954).—
Bode, H. and Arnawold,W c, Z. Anal. Chem. 185, 179-201 (1962),
Cheng, K. L., kml. Chem.28, 1738 (1956).
46

16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26,
27,
28.
29.
30.
31.
32.
33,
34.
35,
36.
37.
38.
39.
40.
41.
42.
Hillebrand,W. F., Lundell, G. E. F., Bright, H. A. end Hoffman, J. L.,
##~~kI~~IC AntiYaia,chapter 19, p. 327, John Wiley and Sons, Inc.,.
Lott, P. F., Cukor, P., Moriber, G. and Solga, J., Anal. C&m. ~, 1159(1963 ).
Snell, F. D., Snell, C. I. and Snell, C. A., CalorimetricMethods ofAnalysis,Vol. 11A, pp. 680, D. Van Nostrand Company,New-9~).
Taboury,M. F. and Gray, E., Compt.Rend. ~, 481 (1941).
Noyes, A. A. and Bray, T?.C., A System of QualitativeA&lyEIisfor theRarer Elements,p. 272-330,Macmillan,~- York (1937).
————
Seath, J. and Beamlsh,F. E., Ind. Eng. Chem., Anal. Ed. ~, 373 (1937).
Noakes, F, D. L., k~yst 76, 542 (1951).—
Beamish,F, E., Russell, J, J. and Seath, J., Ind. Eng. Chem., Anal. Ed.Q, 174 (193’7).
ltlllazzo,G., #real,~. Acta~, I-26(1949).
Schoeller,W. R., et al., op. cit., p. 237.
E@de, H., Z. Anal. Chem.=4, 100 (1951).
Ripan-Tilici,R., Z. Anal. Chem.~, 343 (1935).
Spacu, P., Bull. Sot. Chem.~, 159 (1936).
Morrisonj G. H. and Ereiser, H., Solvent Extraction AnalyticalC-hemlstry, John-Wileyaud Sons, New York (1957),
Kitahara, S., BuJ1. Inst., Phys. them. Research (Tokyo)~, 165 (1949).
Bock, R. and Herman, M., Z. Anorg, U. Allgem Chemie,~4, 288 (1956).
Stevenson,P. C. and Hicks, H. G., Anal. Chem.~, 1517 (1953).
Bock, R., Kuache, H. and Bock, E., Z. Anal. Chem.=8, 167 (1953).
Scadden,E, M. and Eallou, N. E., Anal. Chem. 25, 1602 (1953).—
Goto, H., Ksketa, Y. and Furukawa,T., Nippon Kagaku Zashohi 79,1513-20 (1958) (see UCRL-Trqns-541(L), 1960).
—
Msddock, R. S. andMeirdse,W. W., “ProgressReport No. n,” USAECPro~ect No. 7, ContractNo. AT(u-1)-70; TID-14131O (1962).
Watkinson,J. H., Aal. them. 32, 981 (1960).—
Hillebrand,W. F., et al., Op.d.t.,p. 333.
Lenher, V. and Ehdth, D.
Gooch, F. A. and Pierce,
Dolique,R. and Perahra,
Dudley, H. C. and Byers,
P,, Ind. Eng. Chem.,1-6,837 (1924).
A. W., fm. J. Se., ~, 181 (1896).
s.) Bull. Sot. Chh. Fr, ~, 44 (1946).
H. G., Ind. Eng, Chem. Anal. Ed. ~, 3 (1935).
47

43.
44,
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57,
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
McNutly, J. S., Center,F. J. and McIntosh, R. M., Anal. Chem. ~,123 (1951).
Lambert, J. M., Arthur,P. and Moorse, T. E., Anal. Chem.~, 1101(1951).
Lingane, J. J. and Niedrach,L. W., J. m. them. Sot. 71, 196 (1949).—
Rowley, K. and Swift, E. H., Anal. Chem. 27, 818 (1955).
Kraus, K. A. and Nelson, F., “Metal Sepsxationaby Anion Exchange,”AmericanSociety for TestihgMaterials,Philadelphia,SpecialTechnicalPublicationNo. 195, p. 27-57 (1958).
Attebury,R. W., L=sen, Q. V., and Boyd., G.E., Abstract,AmericanChemical Satiety,l18th Meeting, September,1950.
Aoki, F,, Bull. Chema Sot. Japan~, 480 (1953).
Lederer,M. and Kertes, S., Anal. Chim. Acta~, 122 (1956).
Levi, M. C. and Damn, J. USAEC Report No. NP-8653 (1959).
Crouthsmel,C. E., and Gatrousis,C., Talanta,~, 39-40 (1958).
Buretall,1?.H,, Davies, G. R., Linatead,R. P. and Wells, R. A.,J, Chem. Sot. ~, 516 (1950).
Lederer,M., Anal.
Weatherley, E. G.,
Hillebrand, W. F.,
Willard, B. H. and
Chim, Acta~, 142 (1955).
Andyst&, 404 (1956).
et al., op.cit.,p. 328.
Fenwick,J. Am. Chem. Sot. 45, 936 (1923).—
Scott, W. W,, StandardMethoda of ChemicalAnalysis,Vol. I, D. VanNostrand Co., 5th Ed., New YorkTl~. 787.
Hillebrand,W. F., et al., op.cit.,pp. 329.
Bowen, H, J. M., and Cawse,P. A., Andyst~, 721 (1963).
Maddock, R. S. and Meinke, W. W., Progress Report 8, p. 94, AECU-4438(1959).
Williams, K. T. and Lakin, H, W., Ind. Eng. Chem, Anal. Ed. ~, 409(1935).
Gorsuch, T. T., Jklaystg, 135-150 (1959).
Scott,w. w., op. cit., pp. 791.
Pieters, H. A. J. and Creyghton,J. W., Safet in the ChemicalLaboratoq, +-–AcademicPress, New York (1957 .
Leddicotte,G. W., Reynolds,S. A. and Corbin,L. T., Safety,MethodNo. 50150, ORNL Master AnalyticalManual, TID-7015,Section 5 (1960).
O’Kelley,G. D., Nuclew SciencesSeries,NAS-NS-3105,APtil, 1962.
48

68.
69.
70.
71.
72,
73.
74.
75.
76.
77.
78.
79.
80.
81.
Okada, M., Nature~, 594-595 (1960).
Hughes, D. J, and Harvey, J. A., “Neutron Cross Sections,”BrookhavenNationalLaboratory,Upton, New York, Report No. BNL-325 (1958).
Weigmann, H., z. Physik~, 549 (1962).
Kramer, Hi H., Molinski,V. J., Tilbury,R. S. end Wehl, W. H.,“ResearchIn ActivationAnalysis,”Report No. NYo-10,174 (1963).
Putman, J. L. and Taylor,W. H., Intern. J. App. Radiation and Isotopes,~, 315 (1957).
Putman, J. L. and Taylor, W. H., TYans. Sot. Glass Technolo~~, 84(1958).
Erion, W. E., Mott, W. E. and Shedlovsky,J, P., Trans. Nucl. Sot. ~,(1) 253 (1960).
Fineman, I., Ljunggren,K., Forsberg,H. G. and Erwsll, L. G., Int. J.#pp. Rad, and Isotopes~, 280-288 (1959).
Leddicotte,G. W., “Selenium,Neutron ActivationAnalysis (IsotopicCarrier,Distillation-Precipitation)Method,”Method No. 5-1176o,Oak Ridge NationalLaborato~Maater AnalyticalManual.
Blemchard,R. L., Leddicotte,G. W., U.S. Atomic Enerm Report No.ORWL-2620 (1959).
Blanchti, R. L,, Leddicotte,G. W., and Moeller, D. W., Proc. SecondUnited Nations. Conf. on Peaceful Uses of Atomic Energy, Geneva, 1958,28, 511 (1959).
Blanchard,R. L., Leddicotte,G. W. and Moeller, D. W., J. Amer. WaterWorks Assoc.~, 967 (1959).
Schindewolf,U., Geochim. et Cosmochim.Acta~, 134-8 (1960).
Meinke, W, W., ChemicalProceduresUsed in BombsxdmentWork at Berkeley,U.S. Atomdc Energy CommissionReport AECD-2738, (1949].
—. —.

MONOGRAPHS IN THE RADIOCHEMISTRY AND THETECHNfQUE SERIES
RADIOCHEMICAL
Copiesofthefollowingmonographsare availablefrom theClearinghouseforFedeml ScientificandTeohnioalfnforma-tlon,NationalBureauofStandards,U.S.DepartmentofCom-merce, Springfield,Va.
Alumtium and Gallium,NAS-NS-3032, $0.50
Americium and Curium, NAS-NS-3006,$0.75
Antimony,NAS-NS-3033, $0.50
Arserdc,NAS-NS-3002 (Rsv.)1965$1.00Astatine,NAS-NS-3012, $0.50
Barlurn,Calcium,and strontium,NAS-NS-3010,$1.25
Be@ium, NAS-NS-3013, $0.75
Cadrolum, NAS-NS-3001, $0.75Carbon, Nitrogen, and @gen, NAS-NS-
3019, $0.50Cesium, NAS-NS-3035, $0.75Chromium, NAs-Ns-3007, (Rsv.)1964 $0.7scobalt, NAS-NS-3041, $1.00Copper, NAS-NS-3027, $0.75Fluorine, Cblor5ne, Bromine, and Iodine,
NAS-NS-3005. $0.50Franoium, NAS-NS-3003, $0,50Germanium, NAS-NS-3043, $0,50Gold, NAS-NS-3036, $0,50Ind.ium, NAS-NS-3014, $0,50Iridium, NAS-NS-3045, $0.50Iron, NAS-NS-3017, $0,50Lead, NAS-NS-3040, $1.75Magnesium,,NAS-NS-3024, $0,50
_ese, ”NAS-NS-301B,$0.50Mercury, NAS-NS-3026, $0.50Molybdenum, NAS-NS-3009, $0.50
Nickel,NAS-NS-3051, $0,50Niobium and Tantalum,NAS-NS-3039, $0.75
Osmium, NAS-NS-3CM6, $0.50Palladium,NAS-NS-3052, $0.75Phosphome, NAS-NS-3056, $0.50
PIMnum, NAS-NS-3044, $0.50Polonium,NAS-NS-3037, $0.75Potassium,NAS-NS-3043, $0.50Protactinium,NAS-NS-3016, $1.00
Radium, NAS-NS-305’7,$2.25Rare Earths— Scandium,Yttrium,and Ac-tirdum,NAS-NS-3020, $3.00
Rare Gases, NAS-NS-3025, $0.75Rhenium, NAS-NS-302S, $0,50
Rhodium, NAS-NS-300S, (Rev.)1965$1.00
Rubidium, NAS-NS-3053, $0.50Ruthenium, NAS-NS-3029, $1.00Selenium, NAS-NS-3030, (Rsv.)1965 $1.00Sfliccm, NAS-NS-3049, $0.50Sflver, NAS-NS-3047, $0.75&xUum, NAS-NS-3055, $0.50Sulfur, NAS-NS-3054, $0.50Technetium, NAS-NS-3021, $0.60Tellurium, NAS-NS-3038, $0,50Thorhun, NAs-Ns-3004, $0.75Tin,NAS-NS-3023, $0.75TiWunl, NAS-NS-3034, $0.50Tranecurium Elements, NAS-NS-3031P
$0.50Tungsten, NAS-NS-3042, $0.50Umrdlml, NAS-NS-3050, $3.50Vanadium, NAS-NS-3022, $0.75Zblc, NAS-NS-3015, $0.75Ziroordum and Hafniurn, NAS-NS- 3011,
$0.50
Applications of Computers to Nuclear andRadiochemiatry, NAS-NS-3107, $2.50
Application of Instillation Techniques toBadiochemical Separations, NAS-NS-3108, $0.50
Detection and Measurement of Nuclear Ra-diation, NAS-NS-3105, $1,50
Liquid-liquid Extinction with Higb-molecular-weight Amines, NAS-NS-3101, $1.00
Low-level Radiochemical Sepamtions, NAS-NS-3103, $0,50
Paper Chromatographic and Electromigra-tion Techniques in FQdiochemistry, NAS-NS-3106, $0.50
Rapid Radiochemical Sepamtions, NAS-NS-31W, $1.25
Separations by Solvent Extnmtion with Tri-n-octylphospbine Oxtde, NAS-NS- 3102,$0.75









![[Henry Sidgwick] Methods of Ethics](https://img.pdfslide.us/doc/110x75/55cf8d1c5503462b139228ed/henry-sidgwick-methods-of-ethics.jpg)



















