Embed Size (px)
Citation preview

DIAGNOSTIC METAGENOMICS
The Quest for the One True Test
Tanya Golubchik
Big Data Institute, Oxford UniversityWellcome Centre for Human Genetics, Oxford University

What is diagnostic metagenomics?
Pathogen identification

Pathogen identification: needle in a haystack1. Culture
– Routine practice for many bacteria, but what about viruses?– 40-50% of childhood meningitis in UK culture-negative (aseptic)
2. Organism-specific PCR– Extremely high specificity, not always desirable for diverse pathogens– Several PCRs required, one positive may mean no further testing is done– Order of testing can introduce bias– Multiplex PCR can save costs but hard to develop
3. Serological assays– Assays developed for flu, HIV, other viruses
4. Other molecular diagnostics– Immunoassays (ELISA), mass spec, etc good for some specific applications
5. “Let’s just sequence everything!”

1. Culture – Routine practice for many bacteria, but what about viruses?– 40-50% of childhood meningitis in UK culture-negative (aseptic)
2. Organism-specific PCR– Extremely high specificity, not always desirable for diverse pathogens– Several PCRs required, one positive may mean no further testing is done– Order of testing can introduce bias– Multiplex PCR can save costs but hard to develop
3. Serological assays– Assays developed for flu, HIV, other viruses
4. Other molecular diagnostics– Immunoassays (ELISA), mass spec, etc good for some specific applications
5. Metagenomics
Pathogen identification: needle in a haystack

There are two types of metagenomics:
(1) Hay classification
https://hackingmaterials.com/2013/11/11/why-hack-materials U.S. Marine Corps photo by Lance Cpl. James Purschwitz
(2) Needle detection

There are two types of metagenomics:
(1) Standard (2) TargetedFrom clinical sample (plasma, CSF, …):
Extract total DNA & RNA
Prepare sequencing library
Sequence reads
Classify by organism/strain
From clinical sample [ or culture ]:
Extract total DNA & RNA [ Deplete host material ] Prepare sequencing library [ Enrich for targets of interest ] Sequence reads
Classify by organism/strain

v
There are two types of metagenomics:
https://hackingmaterials.com/2013/11/11/why-hack-materials
• Great idea…– … when you are interested in the “hay”,
ie. predominant components of the sample
• The more you look, the more you find…– … but most of what you find is “hay”
• More likely to detect larger genomes– HIV genome: 0.01 Mb– S. aureus genome: 3 Mb– Human genome: 6500 Mb
• YES: Microbial community composition & abundance, biomarkers
• NO: Pathogen detection
(1) Hay classification

There are two types of metagenomics:
U.S. Marine Corps photo by Lance Cpl. James Purschwitz
(2) Needle detection
• Great idea…– … when you know (roughly) what
you’re looking for, and relative abundance is not important
• Adds extra steps to an already complex protocol…– … but greater sensitivity and specificity,
no longer looking for 1-2 reads in millions
• YES: Targeted diagnostics, amplification of low-abundance pathogens (includes most viruses)
• NO: Abundance quantification, finding novel pathogens
From clinical sample [ or culture ]:
Extract total DNA & RNA [ Deplete host material ] Prepare sequencing library [ Enrich for targets of interest ] Sequence reads
Classify by organism/strain
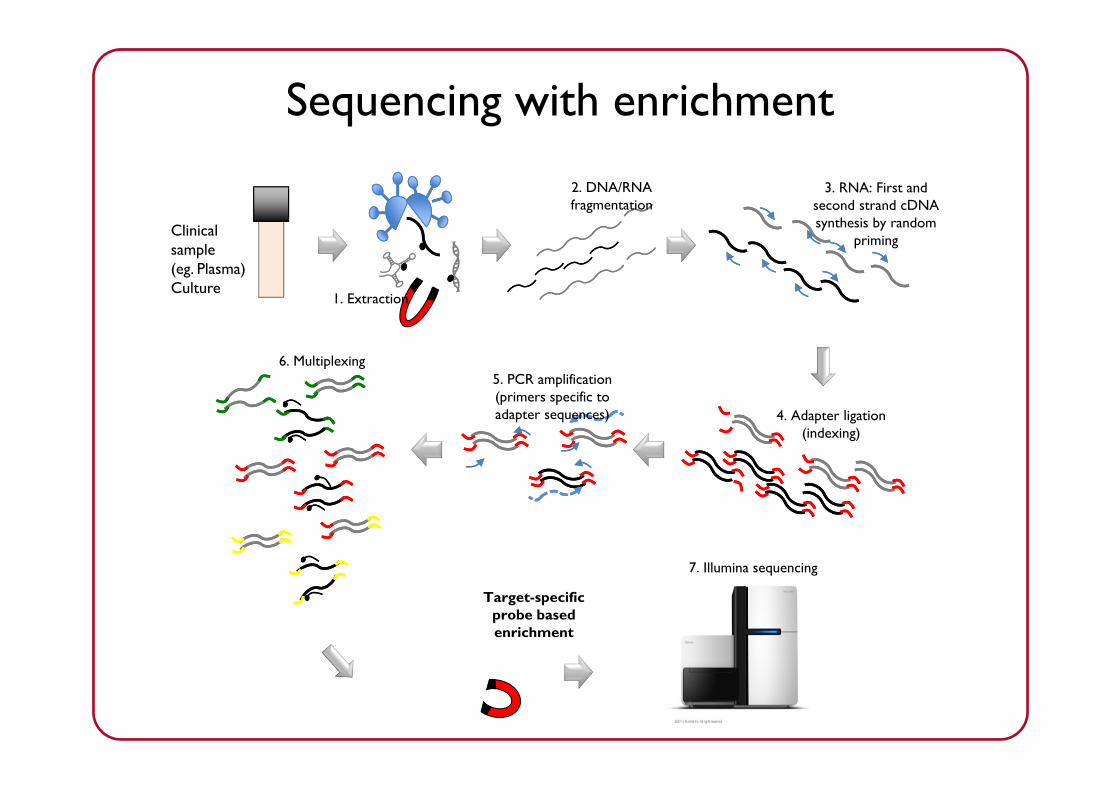
Make a libraryClinical sample (eg. Plasma)Culture
1. Extraction
2. DNA/RNA fragmentation
3. RNA: First and second strand cDNAsynthesis by random
priming
4. Adapter ligation (indexing)
5. PCR amplification (primers specific to adapter sequences)
6. Multiplexing
Target-specific probe based enrichment
7. Illumina sequencing
Sequencing with enrichment
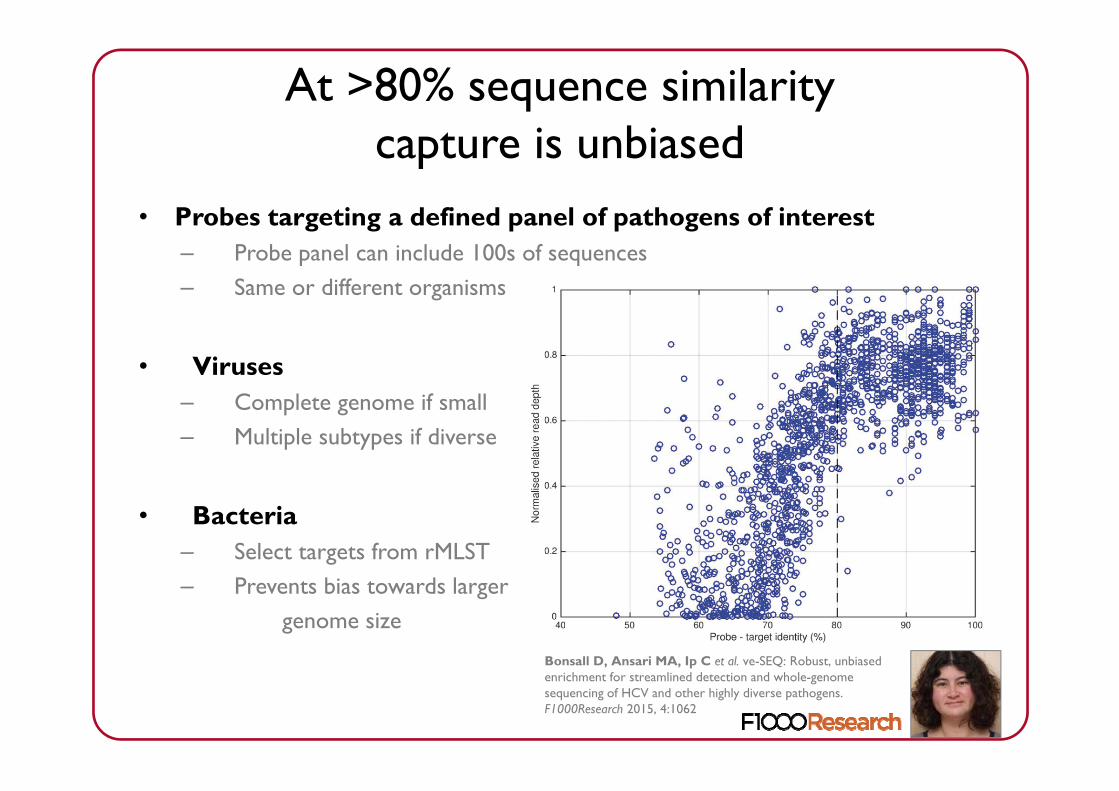
At >80% sequence similaritycapture is unbiased
• Probes targeting a defined panel of pathogens of interest– Probe panel can include 100s of sequences– Same or different organisms
• Viruses– Complete genome if small– Multiple subtypes if diverse
• Bacteria– Select targets from rMLST– Prevents bias towards larger
genome size
Bonsall D, Ansari MA, Ip C et al. ve-SEQ: Robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens. F1000Research 2015, 4:1062
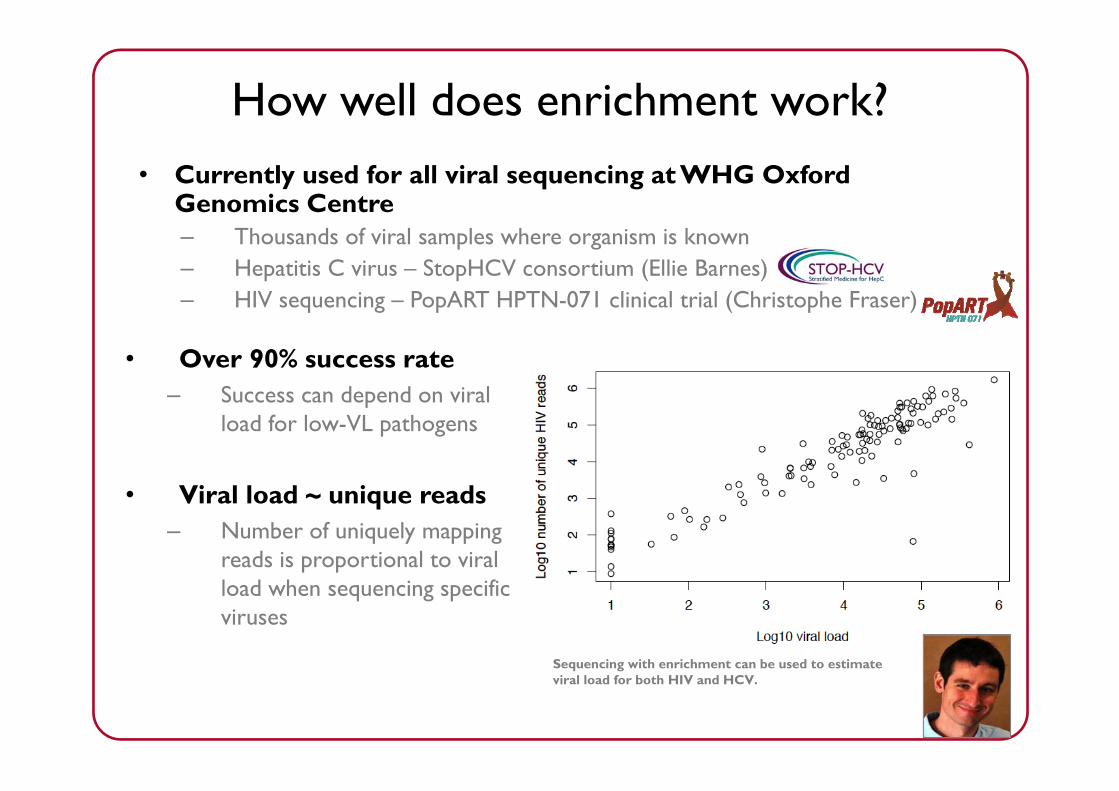
How well does enrichment work?• Currently used for all viral sequencing at WHG Oxford
Genomics Centre– Thousands of viral samples where organism is known– Hepatitis C virus – StopHCV consortium (Ellie Barnes)– HIV sequencing – PopART HPTN-071 clinical trial (Christophe Fraser)
• Over 90% success rate– Success can depend on viral
load for low-VL pathogens
• Viral load ~ unique reads– Number of uniquely mapping
reads is proportional to viral load when sequencing specific viruses
Sequencing with enrichment can be used to estimate viral load for both HIV and HCV.
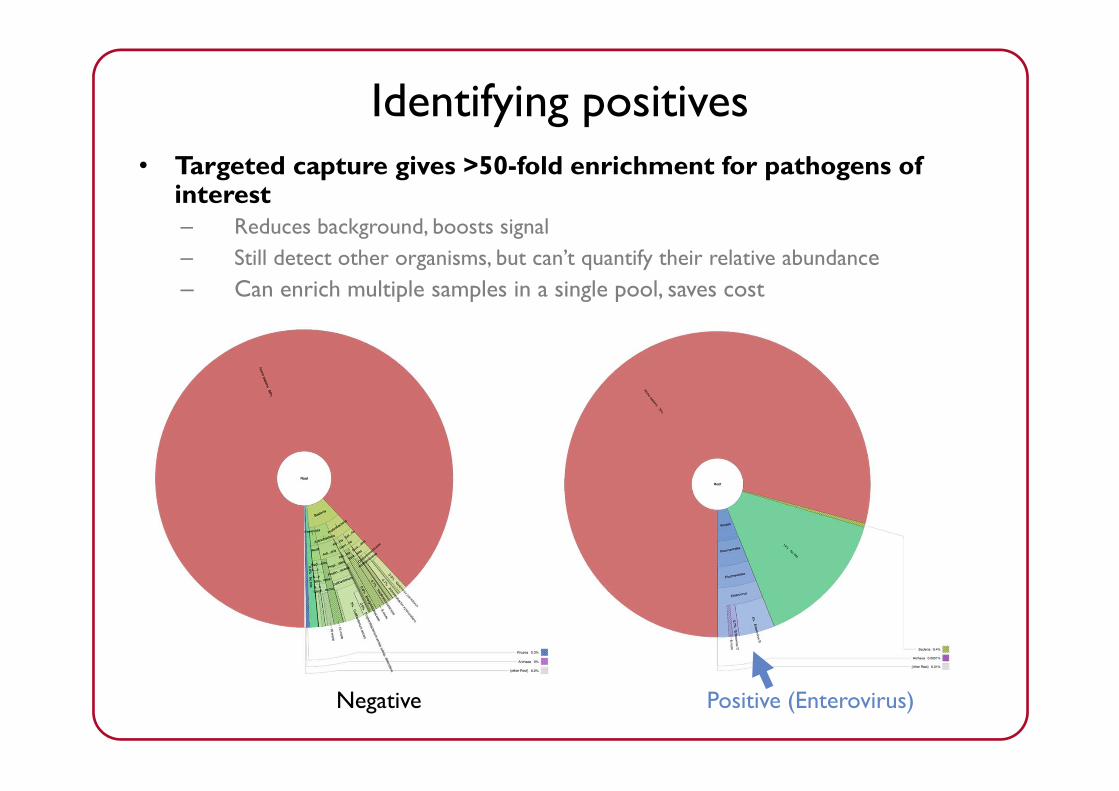
Identifying positives• Targeted capture gives >50-fold enrichment for pathogens of
interest– Reduces background, boosts signal– Still detect other organisms, but can’t quantify their relative abundance– Can enrich multiple samples in a single pool, saves cost
Negative Positive (Enterovirus)

True negatives vs true positivesAnother method: nuclease pre-treatment
• OSCAR project: Oxford Screening for CSF and Respiratory Viruses– Sequencing done at the Roslin Institute– Protocol includes DNAse & RNAse digestion before extraction
– Immense depth (40-60 mln reads per sample) – high cost & processing time– Most samples have <1 pathogen reads per million – ?significance
• Example CSF sample:– 95% human– 5% unknown– 0.002% bacterial– 0.002% viral
Taxonomic classification of reads from CSF sample VS067.
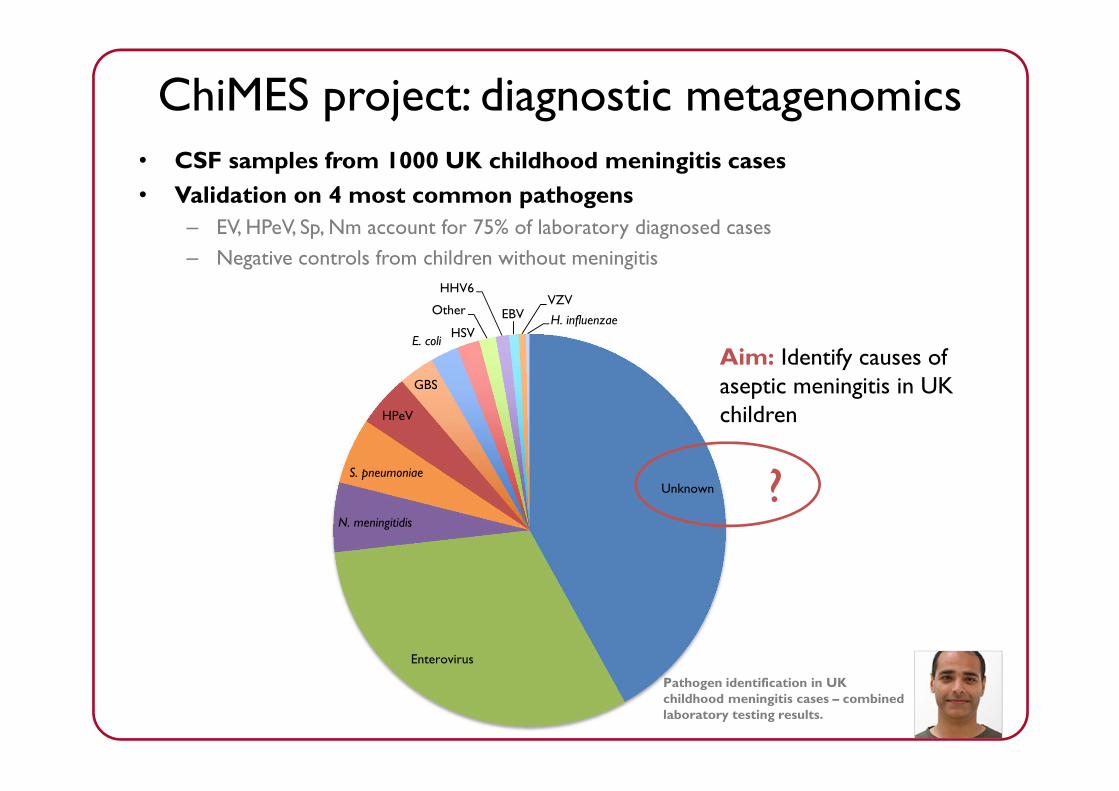
Reads per sampleChiMES project: diagnostic metagenomics• CSF samples from 1000 UK childhood meningitis cases• Validation on 4 most common pathogens
– EV, HPeV, Sp, Nm account for 75% of laboratory diagnosed cases– Negative controls from children without meningitis
Unknown
Enterovirus
N. meningitidis
S. pneumoniae
HPeV
GBS
E. coli HSV
Other
HHV6
EBVVZVH. influenzae
Pathogen identification in UK childhood meningitis cases – combined laboratory testing results.
?
Aim: Identify causes of aseptic meningitis in UK children

ChiMES project: probe panel development• Identifying likely suspects sooner rather than later
– Clinically relevant pathogens – Probes designed to cover pathogen diversity (10% cutoff, similar seqs clustered)
Bacteria1. Streptococcus pneumoniae2. Streptococcus pyogenes3. Streptococcus agalactiae4. Staphylococcus aureus5. Mycoplasma pneumoniae6. Legionella pneumophila7. Coxiella burnetii8. Escherichia coli 9. Klebsiella pneumoniae10. Klebsiella oxytoca11. Enterobacter cloacae12. Enterobacter aerogenes13. Serratia marcescens14. Haemophilus influenzae15. Haemophilus parainfluenzae16. Chlamydophila pneumoniae17. Chlamydia psittaci18. Pseudomonas aeruginosa19. Moraxella catarrhalis20. Acinetobacter baumannii21. Acinetobacter calcoaceticus22. Mycobacterium tuberculosis23. Stenotrophomonas maltophilia24. Bordetella pertussis25. Neisseria meningitidis26. Listeria monocytogenes27. Borrelia burgdorferi28. Treponema pallidum29. Leptospira (multiple spp)30. Bartonella henselae31. Brucella (multiple spp)
Viruses1. Mastadenovirus A/B/C/D/E/F/G complete2. Lassa mammarenavirus complete3. Lymphocytic chriomeningitis mammarenavirus complete4. California Encephalitis Virus complete5. Rift Valley Fever Virus complete6. Sandfly Fever Naples Virus complete7. Sandfly Fever Sicillian Virus complete8. Human Coronavirus HCoV-229E complete9. Human Coronavirus HCoV-NL63 complete10. Human Coronavirus HCoV-HKU1 Genotypes A, B, C complete11. MERS-Coronavirus complete12. Human Coronavirus HCoV-OC43 Genotypes A-E complete13. SARS-Coronavirus complete14. Dengue Fever Virus Genotype 1/2/3/4 complete15. Japanese Encephalitis Virus - All Genotypes complete16. Murray Valley Encephalitis Virus - All Genotypes complete17. St. Louis Encephalitis Virus - All Genotypes complete18. West Nile Virus - All Genotypes complete19. Tick-borne Encephalitis Virus - All Genotypes complete20. Yellow Fever Virus - All Genotypes complete21. HHV1 / Herpes Simplex Virus Type 1 (HSV-1) partial22. HHV2 / Herpes Simplex Virus Type 2 (HSV-2) partial23. HHV3 / Varicella-Zoster Virus (VZV) partial24. HHV4 / Epstein-Barr Virus (EBV) partial25. HHV5 / Human Cytomegalovirus (HCMV) partial26. HHV6A/B / Human Herpesvirus 6A/B partial27. HHV7 / Human Herpesvirus 7 partial28. HHV8 / Kaposi's Sarcoma Herpesvirus (KSHV) partial29. Influenza A/B/C virus (multiple genotypes)30. Human Parainfluenza virus 1/2/3/4a/4b/5 complete31. Mumps virus (multiple genotypes) complete32. Measles virus (multiple genotypes) complete
33. sosuga virus complete34. hendra virus complete35. henipavirus – B/M complete36. Respiratory syncytial virus – A/B complete37. Human metapneumovirus complete38. Primate erythroparvovirus 1 complete39. Primate tetraparvovirus 1 complete40. Human bocavirus 1 complete41. Human Parechovirus 1-8 complete42. Parechovirus B complete43. Enterovirus A/B/D complete44. Rhinovirus A/B/C complete45. Rhinovirus B complete46. Rhinovirus C complete47. Cardiovirus A complete48. Cardiovirus B complete49. Rubella virus complete50. Hepatitis A complete51. Rosavirus 2 complete52. Salivirus A complete53. Salivirus FHB complete54. JC polyomavirus complete55. BK polyomavirus complete56. Rotavirus A/B/C complete57. Rhabdovirus 5 - European Bat Lyssavirus 1 (EBLV2) complete58. Rhabdovirus 6 - European Bat Lyssavirus 2 (EBLV1) complete59. Rhabdovirus 7 - Austrailian Bat Lyssavirus(es) complete60. Rhabdovirus 1 - Rabies complete61. Rhabdovirus 4 - Duvenhage complete62. Rhabdovirus 2 - Lagos Bat virus complete63. Rhabdovirus 3 - Mokola virus complete64. Equine Encephalitis Virus (multiple) complete
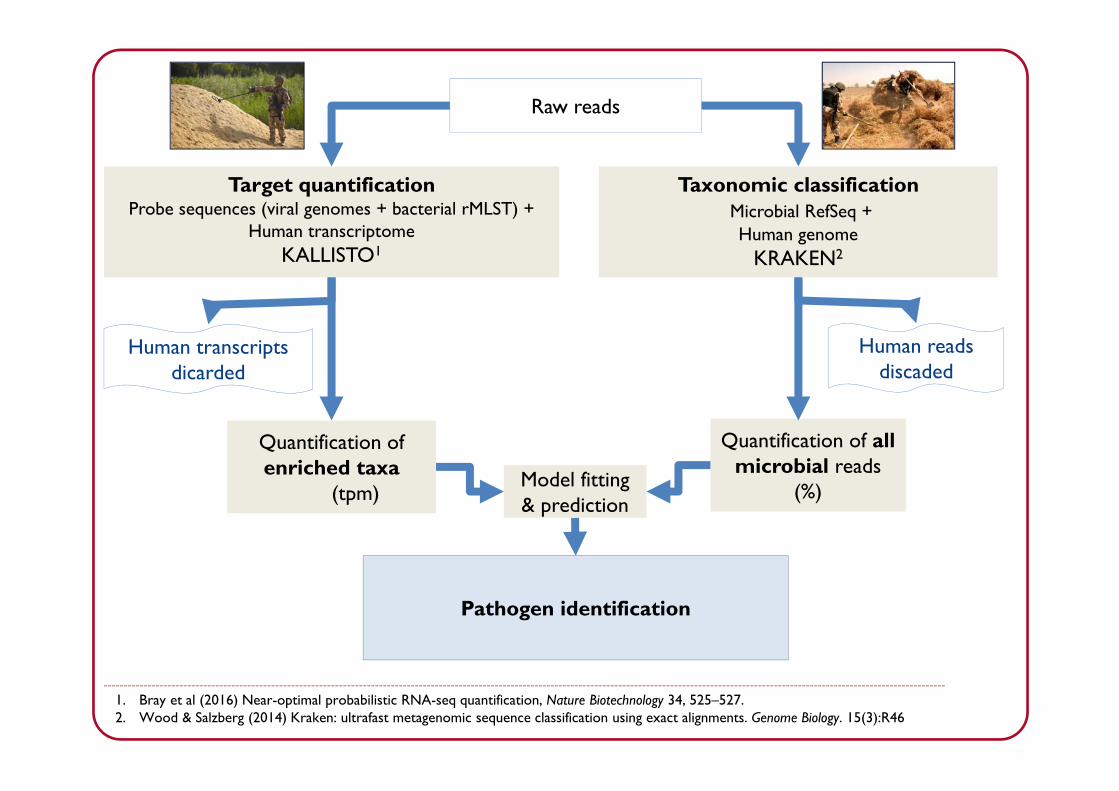
Raw reads
Taxonomic classificationMicrobial RefSeq + Human genome
KRAKEN2
Target quantificationProbe sequences (viral genomes + bacterial rMLST) +
Human transcriptomeKALLISTO1
Human reads discaded
Quantification of all microbial reads
(%)
Human transcripts dicarded
Quantification of enriched taxa
(tpm)Model fitting & prediction
1. Bray et al (2016) Near-optimal probabilistic RNA-seq quantification, Nature Biotechnology 34, 525–527.2. Wood & Salzberg (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology. 15(3):R46
Pathogen identification
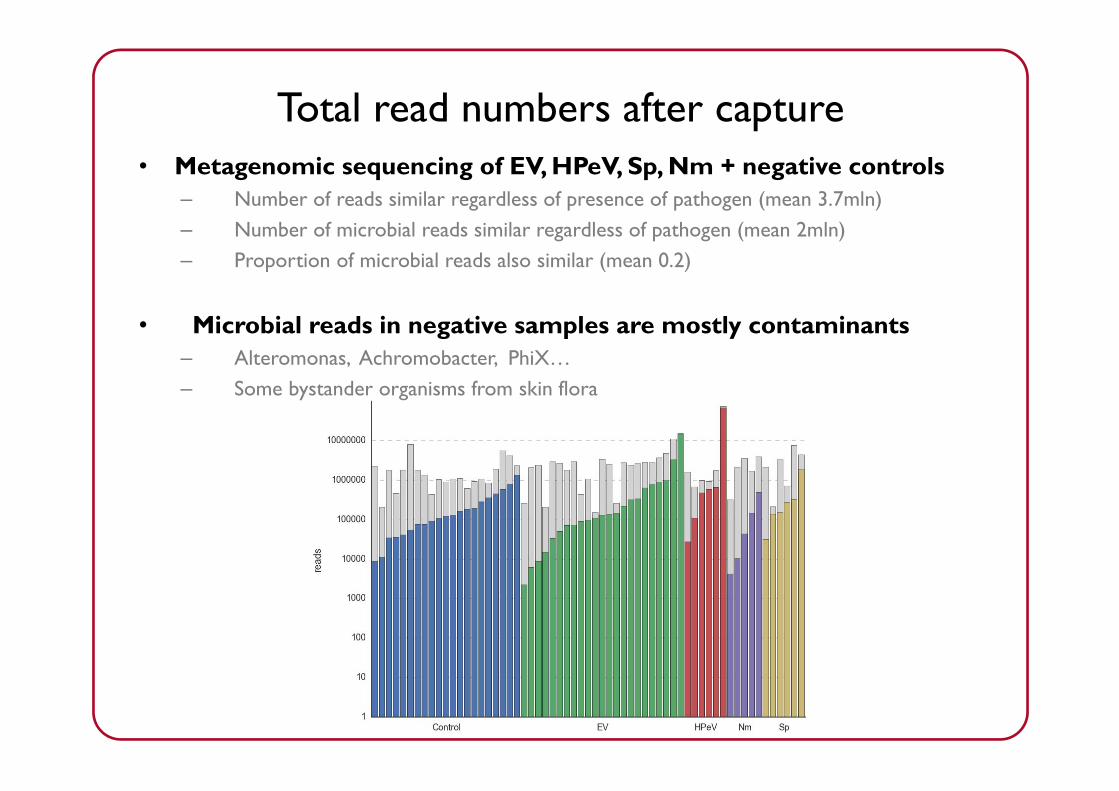
Reads per sample
• Metagenomic sequencing of EV, HPeV, Sp, Nm + negative controls– Number of reads similar regardless of presence of pathogen (mean 3.7mln)– Number of microbial reads similar regardless of pathogen (mean 2mln)– Proportion of microbial reads also similar (mean 0.2)
• Microbial reads in negative samples are mostly contaminants– Alteromonas, Achromobacter, PhiX…– Some bystander organisms from skin flora
Total read numbers after capture
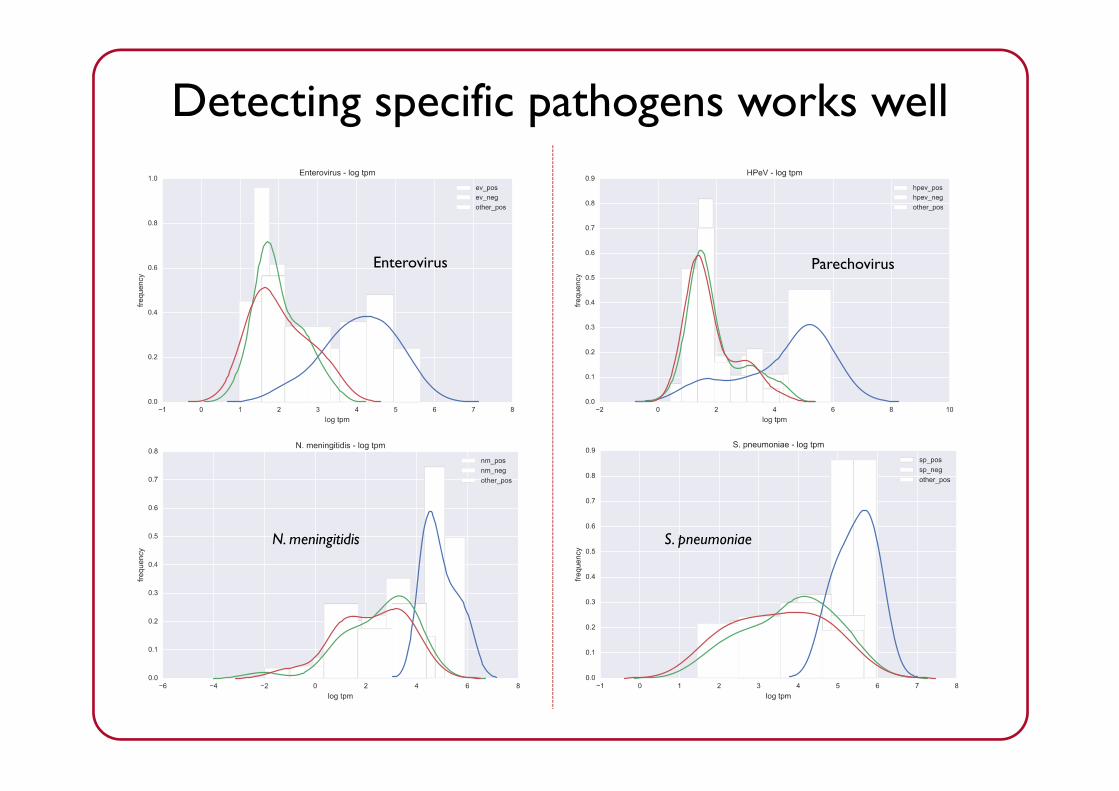
Enterovirus Parechovirus
N. meningitidis S. pneumoniae
Detecting specific pathogens works well
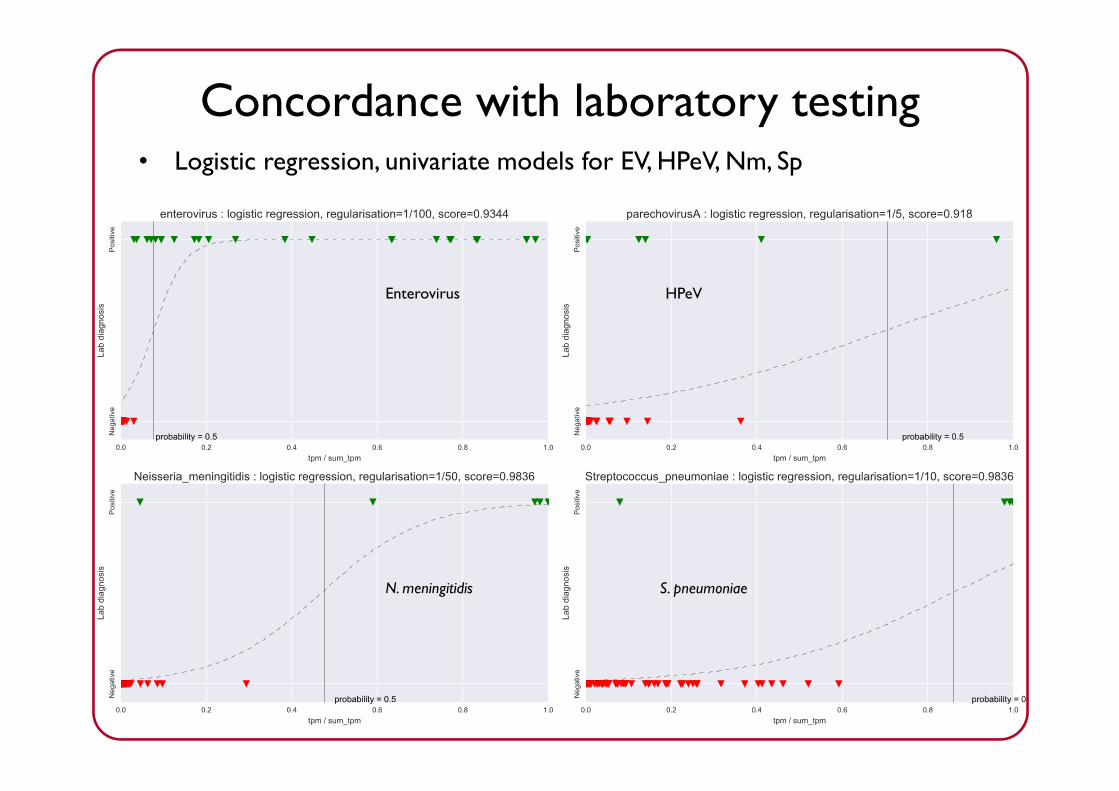
Enterovirus HPeV
N. meningitidis S. pneumoniae
• Logistic regression, univariate models for EV, HPeV, Nm, Sp
Concordance with laboratory testing
Can we confidently identify single-pathogen infection?
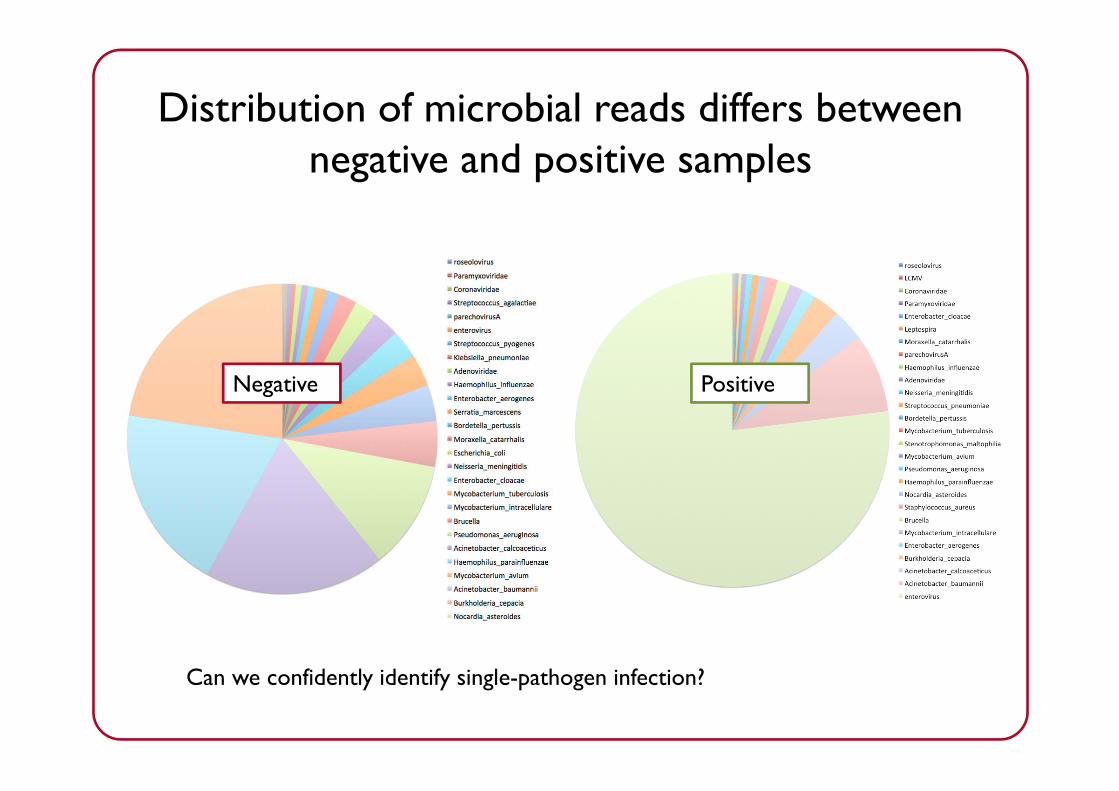
Negative Positive
Distribution of microbial reads differs between negative and positive samples
Samples coloured by lab result
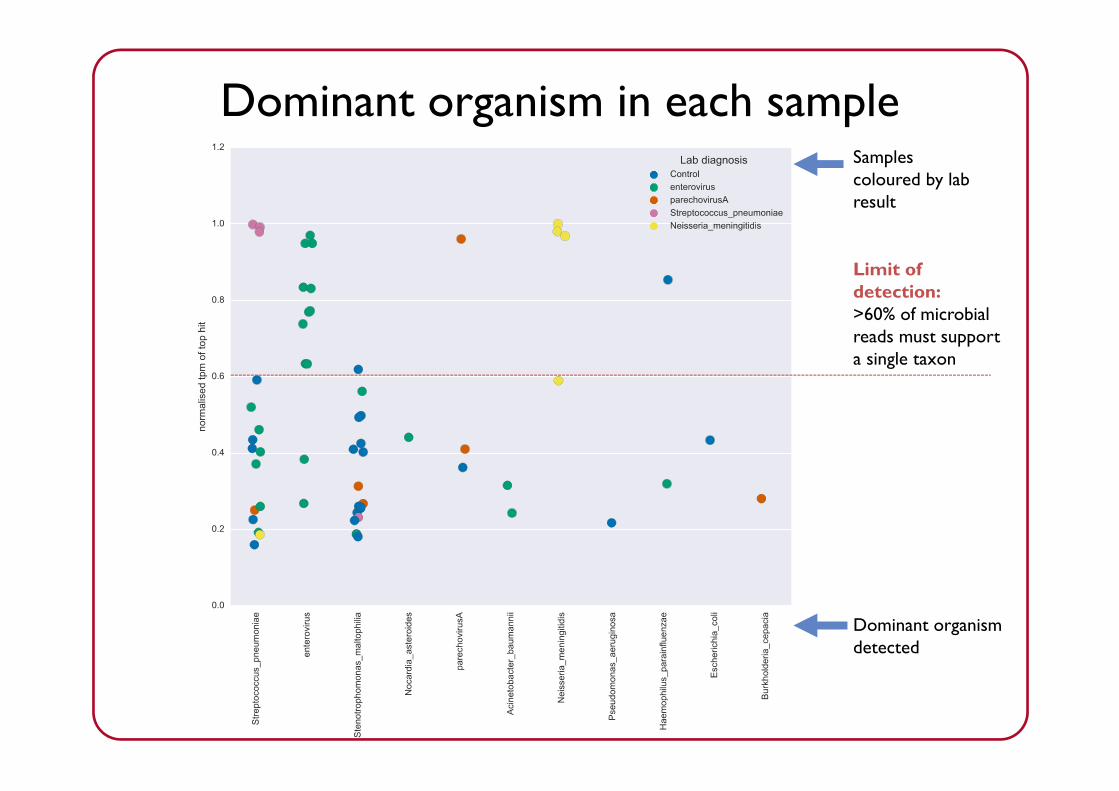
Dominant organism detected
Dominant organism in each sample
Limit of detection:>60% of microbial reads must support a single taxon
“Other”: anything not likely to be clinically significant
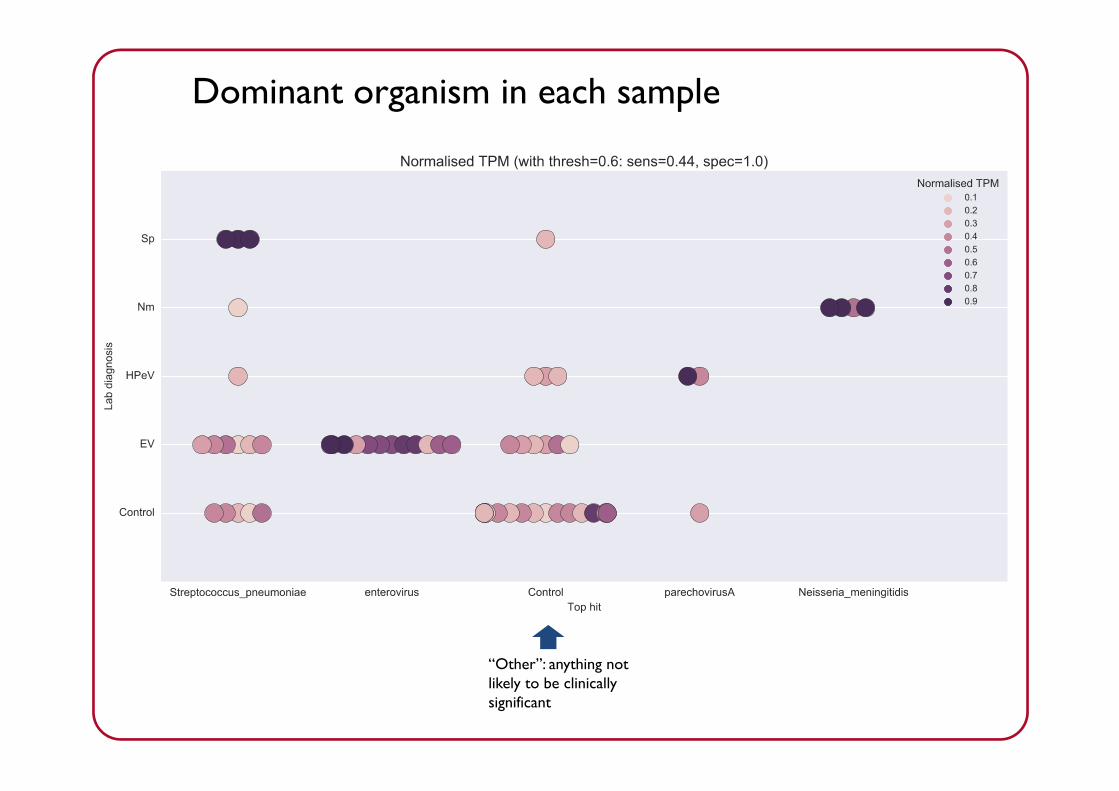
Dominant organism in each sampleDominant organism in each sample
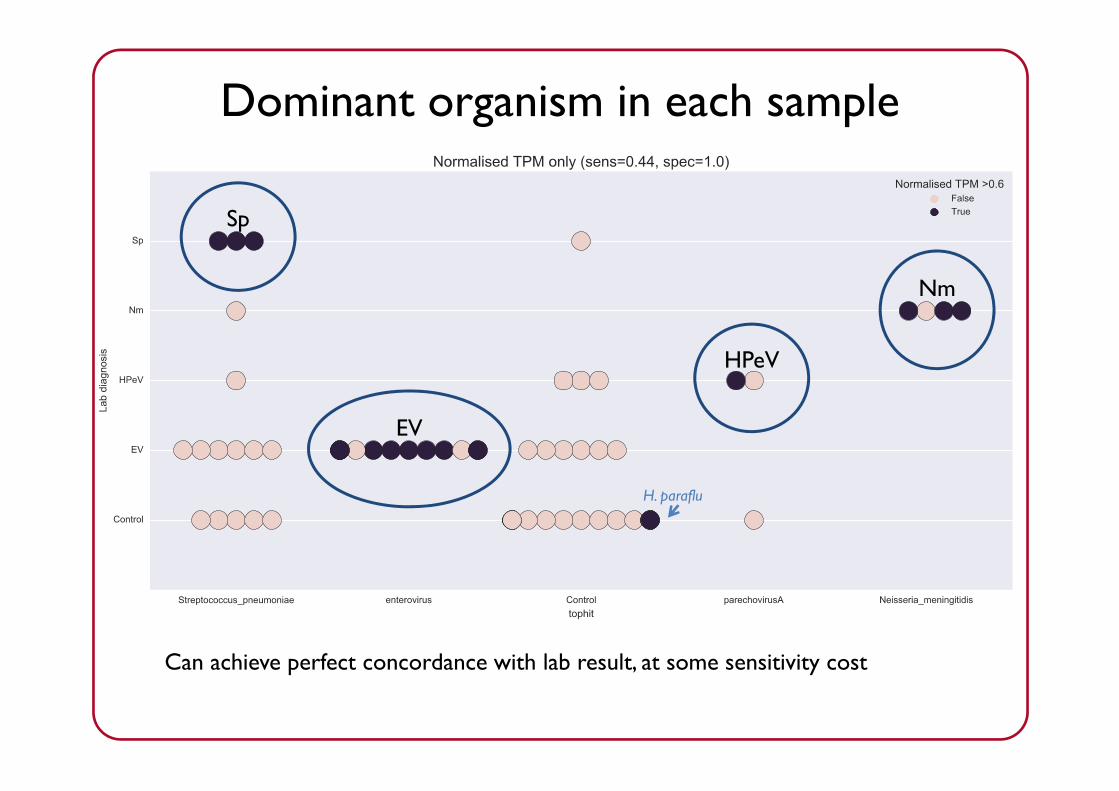
Can achieve perfect concordance with lab result, at some sensitivity cost
Nm
HPeV
Sp
EV
H. paraflu
Dominant organism in each sample

What’s needed for a “one true test”?1. Unbiased enrichment
– Method of choice for Oxford Viromics initiative in WHG• StopHCV consortium, PopART HIV clinical trial
– Unbiased capture of sequences with > 80% similarity to probes
2. Sensitivity – how much sequencing do we need to do?– Probe design is important– More pathogens: greater sensitivity– Can include entire microbial RefSeq if required– Viruses: can sequence samples with viral load <104 & quantify viral load
3. Cost?– Upfront cost of IDT or SureSelect probe panel can be reduced by designing
probe panels across related projects

Thank youOxford Viromics WHGRory BowdenDavid BonsallMariateresa de CesareAzim AnsariCamilla IpHubert SlawinskiAmy TrebesPaolo PiazzaDavid BuckCyndi GohIvo Elliott
OSCAR studyPhilippa MatthewsPeter SimmondsAnna McNaughtonColin Sharp (Roslin)
Viral SequencingEllie Barnes & STOPHCV collaboratorsChristophe Fraser & PopART HPTN-071
collaborators
Oxford Dept. Zoology & Peter Medawar InstitutePaul KlenermanOliver PybusJames Iles
UK Childhood Meningitis and Encephalitis Study (UK-ChiMES) Rory BowdenAndrew PollardManish Sadarangani
SamplesOVG: Annabel Coxon, Gretchen Meddaugh
Probe panel & method developmentAzim AnsariIvo ElliottCyndi Goh
Peter Medawar laboratoryAnthony Brown
WHG laboratoryMariateresa de CesareHubert Slawinski
SequencingAmy TrebesPaolo PiazzaDavid BuckHigh-Throughput Genomics WHG




























