Embed Size (px)
Citation preview

The
Nat
ion
al O
rgan
izat
ion
fo
r R
are
Dis
ord
ers
NORD Guides for Physicians
The Physician’s Guide to
Lipodystrophy Disorders
Visit website at:nordphysicianguides.org/Lipodystrophies/

For more information about NORD’s programs and services, contact:National Organization for Rare Disorders (NORD)PO Box 1968Danbury, CT 06813-1968Phone: (203) 744-0100Toll free: (800) 999-NORDFax: (203) 798-2291Website: www.rarediseases.org Email: [email protected]
NORD’s Rare Disease Database and Organizational Database may be accessed at www.rarediseases.org.
Contents ©2012 National Organization for Rare Disorders®

1
What are Lipodystrophy Disorders?Lipodystrophies are disorders characterized by selective loss of adipose tissue(bodyfat)fromvariousregionsofthebody.Theextentoffatlosscanrange from very small areas on one part of the body to near total absence of adipose from the entire body. Some patients may have only cosmetic problems while others may also have severe metabolic complications. The magnitude of fat loss determines the severity of metabolic complications related to insulin resistance, such as diabetes mellitus, high levels of serum triglycerides and fatty liver. The two major types of lipodystrophies are inherited (familial or genetic lipodystrophies) or secondary to various types ofillnessesordrugs(acquiredlipodystrophies).Geneticlipodystrophiesaremonogenic disorders caused by mutations (alterations or blips) in a gene. Several genes responsible for inherited lipodystrophies have been identified. Some physicians also use the term “lipoatrophy” for these disorders.
Inherited LipodystrophiesIn the last 15 years, considerable progress has been made in understanding the molecular basis of many subtypes of inherited lipodystrophy. Congenital generalizedlipodystrophy(CGL)andfamilialpartiallipodystrophy(FPL)arethe two main subtypes of inherited lipodystrophy; the other subtypes are extremelyrare.
Congenitalgeneralizedlipodystrophy(CGL)isarareautosomalrecessivedisorder characterized by near total absence of body fat from birth. Homozygous or compound heterozygous mutations in four genes, 1-acylglycerol-3-phosphate-O-acyltransferase2(AGPAT2),Berardinelli-Seipcongenitallipodystrophy2(BSCL2),caveolin1(CAV1),andPolymeraseIandtranscriptreleasefactor(PTRF;alsoknownascavin)areassociatedwiththefoursubtypesCGL1,CGL2,CGL3,andCGL4,respectively.

2
Familialpartiallipodystrophy(FPL)ismostlyinheritedasanautosomaldominant condition caused by heterozygous mutations in genes, such as, laminA/C(LMNA),peroxisomeproliferator-activatedreceptorgamma(PPARG),v-AKTmurinethymomaoncogenehomolog2(AKT2)andperilipin1(PLIN1).Recently,apatientwithautosomalrecessiveFPLhasbeenidentifiedwithahomozygousmutationincelldeath–inducingDffa-likeeffector C (CIDEC).
Mandibuloacraldysplasia(MAD)associatedlipodystrophyisanautosomalrecessiveconditionwithskeletalmanifestationsandpartialorgeneralizedlipodystrophy. Homozygous or compound heterozygous mutations in the LMNAandzincmetalloproteinase(ZMPSTE24)geneshavebeenlinkedtoMAD.
ThegeneticbasisofSHORTsyndrome,neonatalprogeroidsyndrome(NPS)and some other rare types of lipodystrophy remains to be elucidated.
Congenital Generalized Lipodystrophy (CGL)
CGLisarareautosomalrecessivedisorderinwhichneartotalabsenceof the adipose tissue is usually evident from the birth. It was originally describedbyBerardinelli(1954)andSeip(1959),andsincethen,approximately300caseshavebeenreported.ThediagnosisofCGLisusuallymadeatbirthorsoonafter.Assumingthatonly1in4patientsarereported in the published literature, the estimated worldwide prevalence is about 1 in 10 million.
Clinical Features
PatientswithCGLhaveextrememuscularityatbirththatisduetoneartotalloss of body fat. They grow at an accelerated rate during early childhood, havemarkedlyincreasedappetite,andmayhaveslightlyenlargedhands,feetandmandible,knownas,acromegaloidfeatures.Nearlyallpatientshaveumbilicalherniaorenlargementoftheumbilicus.Patientsalsopresentwithacanthosisnigricans(darkvelvetypigmentationoftheskin)intheaxilla,neck,andgroinandsometimesevenoverthetrunk,hands,knees,elbowsandankles.Liverenlargementduetoexcessfatdepositionisusuallynoticedduringinfancy.Afewpatientsdevelopcirrhosis(liverdamage)anditscomplicationslateroninlife.Manypatientsalsodevelopsplenicenlargement.Infemales,itiscommontoseemildhirsutism(excesshairon the body especially on the upper lip and chin), enlargement of clitoris, irregularmenstrualperiodsandevenlackofmenstruationandpolycystic

3
ovaries.Mostaffectedwomenareunabletoconceive,however,afewpatientshavehadsuccessfulpregnancies.Affectedmenusuallyhavenormal reproductive ability.
Metaboliccomplicationsassociatedwithinsulinresistancesuchasimpairedglucose tolerance, diabetes, and hypertriglyceridemia are evident at a youngageandareoftendifficulttocontrol.Extremehypertriglyceridemiamay result in recurrent episodes of acute pancreatitis. Of the four distinct subtypesofCGL,type1andtype2arethemostcommon.Table1showsvariousdistinguishingfeaturesofeachsubtypeofCGL.
Molecular Basis
CGL1 locus: AGPAT2
Located on the long arm of chromosome 9 (9q34) is the gene that encodesfortheenzymeAGPAT2whichisresponsibleforthesynthesisof
Clinical features CGL1 CGL2 CGL3 CGL4 (AGPAT2) (BSCL2) (CAV1 (PTRF)
Loss of metabolically active adipose tissue +++ +++ ++ ++
Loss of mechanical adipose tissue - + - -
Bonemarrowfat - - + +
Mildintellectualdisability - + - -
Acanthosisnigricans +++ +++ ++ +/-
Hepatomegaly + + + +
Diabetes mellitus + + + +
Hypertriglyceridemia + + + +
Hyperinsulinemia + + + +
Cardiomyopathy - + - +
Cardiac arrhythmias and sudden death - - - +
Congenital myopathy - - - +
* – * absent; *+* present.
Table 1: Clinical Features of Various Types of Congenital Generalized Lipodystrophy (CGL)
CGL subtype
(gene)

4
triglycerides(fat)andphospholipids.Mutations(alterations)inthisgenemayresultinlipodystrophyeitherduetolackoftriglyceridesynthesisordueto abnormal adipocyte (fat cell) function.
CGL2 locus: BSCL2
Locatedonthelongarmofchromosome11(11q13)isthegeneBSCL2that encodes a protein, Seipin. Studies in yeast have suggested that Seipin may play a role in fusion of lipid droplets. Recent data also suggest that it plays a role in adipocyte differentiation.
CGL3 locus: CAV1
TheCAV1geneislocatedonchromosome7q31.Caveolin-1isexpressedin abundance in caveolae, invagination on cell membranes of adipocytes. Caveolae play a role in lipid droplets formation by bringing lipids and phospholipids from outside to inside of the cell.
CGL4 locus: PTRF
PTRFgenelocatedonchromosome17encodespolymeraseIandtranscriptreleasefactor(PTRF;alsoknownascavin).Thegeneislocatedonchromosome17.PTRFisahighlyabundantcaveolaecomponentandplaysacriticalroleinthebiogenesisofcaveolae.PTRFregulatestheexpressionofcaveolins 1 and 3 and may also contribute to lipid droplet formation.
Other loci: unknown
SomepatientswithCGL(lessthan20percent)havemutationsinnoneofthefourknowngenes,suggestingthatadditionallociandotherdistinctpathways are involved.
Familial Partial Lipodystrophy (FPL)
FPLisarareautosomaldominantdisorderwhichischaracterizedbyvariablelossofbodyfatfromtheextremitiesaswellasfromthetruncalregion.Individuals, both males and females, of several generations can be affected. The chance of transmission from an affected parent to offspring is 50%. MostofthereportedpatientshavebeenofEuropeanorigin;however,patientsofAfrican-AmericanandIndianoriginhavebeennoted.
Clinical Features
Patientswithfamilialpartiallipodystrophy(FPL)havereducedsubcutaneous(sc)fatinthelimbsaswellasinthetruncalregionsbutmayhaveexcessscfatdepositioninnonlipodystrophicregions(e.g.neck,face,andintra-

5
abdominal regions). The phenotype can be easily recognized in affected women; however, affected men are often more difficult to diagnose clinically, as many normal men are also quite muscular. The diagnosis should be suspected in patients who show signs of insulin resistance early in life manifested by acanthosis nigricans or polycystic ovarian syndrome (menstrual irregularity, hirsutism) and early onset of diabetes and severe hypertriglyceridemia.SeveraldistinctsubtypesofFPLhavebeenreportedandthemoleculargeneticbasisoffivedistinctsubtypesisknown.ThemostcommonsubtypeisFPL,Dunniganvariety(FPLDorFPL,type2)whichisduetomissenselaminA/C(LMNA)mutations.Itisalsothemostwellcharacterized disorder. These patients have normal body fat distribution during early childhood, but around the time of puberty, sc fat from the extremitiesandtrunkisprogressivelylost.Somepatientsatthesametimegainexcessfatontheface,chin(‘doublechin’),andneck(‘Cushingoidappearancewithbuffalohump’).Acanthosisnigricans,andhepatomegalydue to steatosis are common and 20-25% of affected females have hirsutism (increased body hair), menstrual abnormalities, and polycystic ovaries (enlarged ovaries) are observed. Women are more severely affected with metabolic complications such as diabetes, hypertriglyceridemia and lowlevelsofhighdensitylipoproteincholesterol.AffectedwomenwithFPLDaremorepre-disposedtocoronaryarterydiseaseandothertypesofatherosclerotic vascular disease.
Molecular Basis
FPL Type 1: Kobberling variety: unknown
Kobberlingandco-workersfromGermanyreportedaphenotypeofFPLwhichwasdistinctfromthatreportedbyDunnigan.TheKobberlingvarietyis less common and has been reported in only two small pedigrees and four sporadic cases. The age of onset of lipodystrophy and the mode of inheritance are not clear. On the basis of clinical findings, the loss of adiposetissueintheKobberlingvarietyissaidtoberestrictedtoextremitiesonly.Patientshavenormalamountsoffatinthefaceareaandmayhavenormal,orevenexcess,scfatinthetruncalarea.Thegeneticbasisforthisparticularvarietyisunknown.
FPL Type 2: Dunnigan variety (FPLD): LMNA mutations
Usingagenomewidelinkageanalysisapproachinfivelargeinformativepedigrees,ithasbeenreportedthattheFPLDlocusisonchromosome1q21-22. Subsequently, many missense mutations (alterations) have been

6
identifiedinthelaminA/C(LMNA)geneinpatientswithFPLD.LMNAhas12exonsandbyalternativesplicinginexon10twoproteinsprelaminA(fullform)orC(shortform),areencoded.LaminsAandCarecomponentsof the nuclear lamina which is located between chromatin and the inner nuclearmembrane.Thus,itislikelythatmissensemutationsmayaffectnuclear function and result in premature cell death of adipocytes (fat cells),thuscausinglipodystrophy.Three-fourthsoftheFPLDpatientshave mutations at the codon position 482 where arginine is replaced by glutamine,leucineortryptophan.Somepatientswithmutationsinexon11 have been observed to have less severe form of lipodystrophy than thosewithexon8mutations.RarepatientswithFPLDrevealmutationsinexon1andthesepatientsdevelopcardiomyopathy(diseaseofheartmuscles) which manifests as premature congestive heart failure and cardiacarrhythmias(rhythmdisturbances),suchasheartblocksandatrialfibrillationnecessitatingtheuseofcardiacpacemakers.Someofthesepatients require cardiac transplantation.
FPLType 3: PPARG mutations
Employing a candidate gene approach, we were the first to report a heterozygousmissensemutation,p.Arg397Cys,inthePPARGina64-yearoldwomanwhopresentedwithFPL,diabetes,hypertriglyceridemia,hypertension and hirsutism. She had lipodystrophy of the face and extremitiesthatwasnoticedmuchlaterinlife.Sincethen,approximately30patientswithFPLduetoPPARGmutationshavebeenreported.PPARGisLocatedonthechromosome3p25whichencodesperoxisomeproliferatoractivatorreceptorgamma(PPARγ)akeytranscriptionfactorinvolvedinadipocytedifferentiation.Itishighlyexpressedintheadiposetissue.MissensemutationsinPPARγcauseFPLduetodefectivedifferentiationofadipocytes.PatientswithPPARGmutationsdeveloplessseverelipodystrophythanthosewithFPLDandscfatlossismoreprominentfromthedistalextremities(calfandforearm)thanfromthethighsandarms.
FPL Type 4: PLIN1 mutations
Perilipin(PLIN1)isthemostabundantproteincoatinglipiddropletsinadipocytes.Thegeneislocatedonchromosome15q26.Itisconsideredto be essential for formation and maturation of lipid droplets and storage of triglycerides as well as release of fatty acids from these lipid droplets. OverexpressionofmutantPLIN1in3T3-L1pre-adipocyteshaveresultedinsmallerlipiddropletsascomparedtothewildtypePLIN1.Histopathology

7
reportofscadiposetissuefromfourpatientswithPLIN1mutationshave also shown reduced size of adipocytes and increased macrophage infiltrationandadiposetissuefibrosis.AtotaloffiveFPLpatientshavebeenreportedtohavemutationsinPLIN1andallofthemhadfattyliver,hypertriglyceridemia and hyperinsulinemia. Lipodystrophy was most strikinginthelowerlimbsandgluteo-femoral(buttocks)depots.RecentinformationsuggeststhatmutantformsofPLIN1failtobindtoAB-hydrolase containing 5, which results in constitutive co-activation of adipose triglyceride lipase and increase basal lipolysis.
FPL Type 5: AKT2 mutation
AKT2isaphosphoinositide-dependentserine/threoninekinaseandalsoknownasproteinkinaseB.AKT2ispredominantlyexpressedininsulinsensitivetissuesandisinvolvedinpost-receptorinsulinsignaling.AKT2islocatedonthechromosome19q13.2.Aheterozygousmissensemutation,p.Arg274His,inAKT2wasdiscoveredinfoursubjectsfromafamilywhopresentedwithinsulin resistance and diabetes mellitus. The loss of adipose tissue in patients withheterozygousmutationsinAKT2mayeitherbeduetoreducedadipocytedifferentiation or dysfunctional post insulin receptor signaling.
FPL AUTOSOMAL RECESSIVE: CIDEC MUTATIONRecently,apatientwithautosomalrecessiveFPLhasbeenidentifiedwithhomozygousmutationinCIDEC.Cidec/Fsp27isalipiddropletproteinmosthighlyexpressedinadipocyteswhereitsexpressionisinducedduringadipocytedifferentiation.Adiposetissuebiopsyoftheaffectedpatientshowed multilocular lipid droplets in comparison to normal one large lipid droplet in adipocytes.
Other Types of FPL
ItappearsthatfivelociforFPL,LMNA,PPARγ,AKT2,CIDECandPLIN1maynotbeabletoexplainthegeneticbasisofallthepatientswithFPLandthereislikelihoodofadditionalloci.IndepthcharacterizationoftheclinicalphenotyperelatedtothepatternoflossoffatinFPLpatientswithmutations in different genes may be helpful in identification of different phenotypes without resorting to molecular diagnosis.
Mandibuloacral Dysplasia (MAD) associated Lipodystrophy
Mandibuloacraldysplasia(MAD)isarareautosomalrecessivesyndrome

8
characterized by mandibular hypoplasia, delayed cranial suture closure, dysplastic clavicles, club-shaped terminal phalanges, acro-osteolysis, and atrophyoftheskinofthehandsandfeet,andtypicalfacialchanges.Patientsalsoshow“progeroidfeatures”suchasbird-likefacies,high-pitchedvoice,skinatrophy,pigmentation,alopecia,andnaildysplasia.Thedisorderhasbeenreportedinabout40patientssofar.PatientswithMADeitherhavepartiallossofscfatfromtheextremities(typeA)ormoregeneralizedlossofscfatinvolvingtheface,trunkandextremities(typeB).The syndrome is also associated with lipodystrophy and clinical features of metabolic syndrome such as insulin resistance, impaired glucose tolerance, diabetes mellitus and hyperlipidemia.
Molecular Basis
MAD Type A: LMNA mutations
PatientswithMADfrequentlyhavepartiallipodystrophyandinsulinresistance,andthediseaseiscausedbymutationsintheLMNAgene.ManypatientswithMADandtypeA(partial)lipodystrophyhavehomozygousp.Arg527HismutationinLMNA.Sofar,atotalof30patientswithMADduetovariousLMNAmutationshavebeenreported.Somepatientsmaydevelop severe progeroid manifestations, similar to those seen in progeria patientssuchasalopecia,lossofeyebrows,delayedsexualmaturationandprematurelossofteeth.MostoftheLMNAmutationscausingMADarelocatedintheC-terminalregionaffectingexons8-10.HowthesespecificLMNAmutationscauseresorptionofbonessuchasmandible,claviclesandterminal phalanges remains unclear.
MAD Type B: Zinc metalloproteinase (ZMPSTE24) mutations
CompoundheterozygousmutationsinZMPSTE24inaBelgianwomanwithMADhavebeenreported.Shealsohadprogeroidfeaturesandgeneralizedlipodystrophy. She died at age 24 years as a result of complications of chronic renal failure due to focal segmental glomerulosclerosis. Three other MADpatientswithmutationsinthesamegenehavebeenreported.ItissuggestedthataccumulationofprelaminAand/orlackofmaturelaminAinthecellsmaybetheunderlyingmechanism.Atotalofeightpatientswith this subtype have been reported and most of them have been young children.Therearenoreportsofdiabetesamongthem.PatientswithZMPSTE24mutationsareprematureatbirth,haveearlyonsetofskeletaldefects including acro-osteolysis, have more progeroid appearance and develop subcutaneous calcified nodules on the phalanges.

9
Other Types of MAD
Some patients with mandibuloacral dysplasia have no apparent alterations ineithertheLMNAorZMPSTE24gene,suggestingtheexistenceofotherasyet unmapped loci for this disorder.
Mandibular hypoplasia, Deafness, Progeroid features (MDP) associated lipodystrophy syndrome
This syndrome was recently reported in seven patients and is characterized bymandibularhypoplasia,deafness;progeroidfeatures(MDP)-associatedlipodystrophy.NoneofthemhadanymutationsinLMNAorZMPSTE24.AscomparedtoMADpatients,theyshoweddistinctcharacteristicssuchassensorineural hearing loss, and absence of clavicular hypoplasia and acro-osteolysis.AllmaleswithMDPhadundescendedtestesandhypogonadism.Oneadultfemaleshowedlackofbreastdevelopment.Twoofthesevenpatientshaddiabetesmellitus.ThemolecularbasisofMDPsyndromeremains to be elucidated.
Autoinflammatory Lipodystrophy Syndrome
This autosomal recessive autoinflammatory disorder is characterized by childhood onset of recurrent fever, joint stiffness and severe contractures ofthehandsandfeet,erythematousskinlesionswithsubsequentdevelopment of lipodystrophy. Recently, joint contractures, muscular atrophy,microcyticanemia,andpanniculitis-inducedlipodystrophy(JMP)were reported in two pedigrees where affected patients developed progressive lipodystrophy during childhood. Similar patients had been previouslyreportedfromJapan.Additionalclinicalfeaturesincludemuscleweaknessandatrophyandhepatosplenomegaly.TwogroupsrecentlyhavealsoreportedfivepatientswithChronicAtypicalNeutrophilicDermatosiswithLipodystrophyandElevatedTemperature(CANDLE)syndromewhohave recurrent fever, annular violaceous plaques during infancy which result in loss of sc fat from the face and upper limbs.
Molecular Basis
Ahomozygous,missense,lossoffunction,mutationinproteasomesubunit,beta-type,8(PSMB8)genehasbeenreportedinaffectedpatientswithJMPsyndrome.Subsequently,missensePSMB8mutationswerereportedin Japanese patients with auto inflammatory lipodystrophy and also in thosewithCANDLEsyndrome.ThePSMB8geneislocatedonchromosome6p21.3andencodestheβ5i subunit of the immunoproteasome.

10
Immunoproteasomes are responsible for proteolysis of antigens presented bymajorhistocompatibilitycomplex(MHC)classImoleculesandresultingenerationofimmunogenicepitopes.MutationsinPSMB8maytriggeranautoinflammatory response that results in panniculitis and other clinical manifestations.
Short stature, hyperextensibility of joints and/or inguinal hernia, ocular depression, Reiger anomaly and teething delay (SHORT) Syndrome
Atotalof30patientshavebeenreportedwithSHORTsyndrome.Thepedigrees reveal both autosomal recessive and dominant modes of transmission. Reiger anomaly consists of eye abnormalities such as iris hypoplasia, Schwalbe ring, iridocorneal synechiae, micro- or megalo-cornea and dental anomalies such as hypodontia (missing teeth), microdontia (teeth smaller than normal), incomplete or underdevelopment of enamel and atypical teeth. Other clinical features include intrauterine growth retardation, failure to thrive, delayed speech development, small head circumference, bilateral clinodactyly (fifth finger bending towards adjacent 4th finger) and sensorineural hearing loss. Different patterns of fat loss have been reported. In many patients, lipodystrophy affects the face, upper extremitiesandsometimesthetrunk,withrelativesparingofthelowerextremities.Othershadlipodystrophyaffectingonlytheface,glutealregionand elbows. Diabetes occurs as early as the second and third decade of life.ThemolecularbasisofSHORTsyndromeremainsunknown.
Neonatal progeroid syndrome (Wiedemann-Rautenstrauch syndrome)
Thisisanautosomalrecessivesyndromewithatotalofapproximately25reportedcases.Newbornswiththissyndromehaveatriangular,old-lookingfacewithrelativelylargeskull(progeroidappearance),prominentveinson the scalp, sparse scalp hair, large anterior fontanelle and generalized lipodystrophy. However, sc fat in the sacral and gluteal areas is spared. Approximately50percentofpatientsreportedlydiebeforetheageof6yearsbutpatientssurvivinguptotheageof16yearshavebeenreported.Recently,twopatientswhoalsomanifestedclinicalfeaturesofMarfansyndrome were reported to harbor de novo heterozygous mutations in fibrillin1(FBN1)gene.Nopatienthasbeenreportedtodevelopdiabetes.

11
Acquired LipodystrophiesAcquiredlipodystrophiesarecausedbymedications,autoimmunemechanismsorotherunknownmechanisms.Theseincludehighlyactiveantiretroviraltherapy(HAART)inducedlipodystrophyinHIV-infectedpatients(LD-HIV),acquiredgeneralizedlipodystrophy(AGL),acquiredpartiallipodystrophy(APL)andlocalizedlipodystrophy.Acquiredlipodystrophiesdonot have a direct genetic basis. Rather, many mechanisms may be involved.
Despite recognition of acquired lipodystrophies for more than a century, progress in understanding underlying pathogenetic mechanisms has beenslow.Acquiredpartiallipodystrophy(APL)hasbeenreportedinapproximately250casesofvariousethnicitieswithmaletofemaleratioof1:4andacquiredgeneralizedlipodystrophy(AGL)inapproximately100cases,mostlyCaucasianswithamaletofemaleratioof1:3.LD-HIVisestimated to affect more than 100,000 patients in the United States and many more in other countries.
Acquired Generalized Lipodystrophy (AGL, Lawrence syndrome)
EventhoughtheonsetoflossofscfatinpatientswithAGLoccursduringchildhood,theexactmechanismsoffatlossarenotknown.Thepatternandextentoffatlossisquitevariable.Mostofthepatientshavegeneralized loss of fat, but in a few of them, some areas of the body such as intraabdominal fat and bone marrow fat are spared. However, patientsdevelopextremelyseverehepaticsteatosisandfibrosis,diabetes,and hypertriglyceridemia, which are difficult to manage. The panniculitis–associatedAGLusuallypresentswithlessseverefatlossandmetaboliccomplications than the autoimmune or idiopathic subtypes. Some patients withAGLhavebeenreportedtohavechronichepatitiswithautoimmunefeatures and low serum complement 4 levels, suggesting involvement of the classical complement pathway in the pathogenesis of fat loss.
Acquired Partial Lipodystrophy (APL, Barraquer-Simons syndrome)
APLdevelopsinmostpatientsbeforeage15.Patientslosescfatgraduallyin a symmetric fashion, first affecting the face and then spreading downward.Mostofthempresentwithfatlossfromtheface,neck,upperextremities,andtrunkwithsparingofscabdominalfatandlowerextremities.Metaboliccomplicationsareusuallynotseen.However,approximatelyonefifthofthepatientsdevelopmembranoproliferativeglomerulonephritis, and later on some of them develop drusen (yellow or

12
whiteextracellularmaterialbuildupinthemembraneofeye).Thereisstrongevidence to suggest that fat loss involves autoimmune-mediated destruction of adipocytes because more than 80% of the patients have low serum levels of complement 3 and a circulating autoantibody called complement 3- nephriticfactorthatblocksdegradationoftheenzymeC3convertase..
HAART-induced lipodystrophy in HIV–infected patients (LD-HIV)
LD-HIVusuallyappearsafterreceivingHIV-1proteaseinhibitor-containingHAARTfor2yearsormore.Mostpatientsgraduallylosescfatfromthearms, legs, and face. Some areas of the body are spared, and some patients accumulateexcessfatintheseareasmanifestingasbuffalohump,doublechin, and increased waist circumference. It is also observed that the fat lossprogressivelygetsworsewithongoingHAARTtherapyanddoesnotreverseondiscontinuationofproteaseinhibitors.Manypatientsdevelophypertriglyceridemia, but only a few develop diabetes mellitus.
In many patients, protease inhibitors and nucleoside reverse transcriptase inhibitorsareimplicatedincausinglipodystrophy.ProteaseinhibitorsmaycauselipodystrophybyinhibitingZMPSTE24,resultinginaccumulationofprelaminA.Othermechanismsmayincludeproteaseinhibitor-inducedalterationofexpressionofkeytranscriptionfactorsinvolvedinlipogenesisand adipocyte differentiation such as sterol regulatory element-binding proteinIc,andPPARγ.Proteaseinhibitorsmayalsoinduceinsulinresistancebyinhibitingglucosetransporter4expressions.Sinceproteaseinhibitorsornucleoside reverse transcriptase inhibitors are usually given together as part oftheHAART,theindividualeffectsofthesedrugsonthephenotyperemainunclear.
Localized Lipodystrophy
Localized lipodystrophy can occur due to sc injection of various drugs, panniculitis, pressure, and other mechanisms. It present with sc fat loss from afocalregionresultinginadimpleoracraterwithoverlyingskinusuallyunaffected. In some patients, large contiguous or anatomically distinct areas on any region of the body may be involved.

13
KeyPIs(Proteaseinhibitors)NRTIs (Nucleoside reverse transcriptase inhibitors)ZMPSTE24,(Zincmetalloproteinase)
MGPN,(Membranoproliferativeglomerulonephritis)
DiagnosisLipodystrophies should be suspected in differential diagnosis of “lean or non-obese” patients presenting with early diabetes, severe hypertriglyceridemia, hepatic steatosis, hepatosplenomegaly, acanthosis nigricans, and polycystic ovariansyndrome.Thesepatientsshouldbeexaminedcarefullyforevidenceof loss of sc fat especially from the hips and thighs and muscular prominence. Somepatientsmaypresentwithexcessscfatdepositioninvariousanatomic regions and may resemble patients with Cushing’s syndrome and truncalobesity.Patientswithlipodystrophyshouldalsobedifferentiatedfromthosewithanorexianervosa,cachexia,starvation,diencephalic
Type Subtype Clinical features Pathogenetic basi
Lipodystrophy in HIV infected patients
Acquired partial lipodystrophy
Acquired generalized lipodystrophy
Localized lipodystrophy
Loss of sc fat from the face and extremities and excess fat deposition in the neck and abdomen.
Loss of sc fat from the face, neck, upper limbs, and trunk, sparing the lower abdomen and lower limbs.
Generalized loss of fat associated with tender sc nodules, autoimmune or other diseases.
Loss of sc fat from small areas of the body.
PIs may inhibit ZMPSTE24 and/or cause dysregulation of transcription factors involved in adipogenesis. NRTIs may inhibit mitochondrial polymerase- γ and cause mitochondrial toxicity.
Low serum complement 3 levels and presence of an autoantibody, C3 nephritic factor in most patients suggest autoimmune –mediated loss of adipose tissue.
Panniculitis preceding the loss of sc fat and association of autoimmune diseases suggest immune-mediated loss of adipose tissue.
Multiple mechanisms involved.
•PI-induced•NRTI-induced
•Autoimmune•MPGN-associated•Idiopathic
•Autoimmune•Panniculitis-associated•Idiopathic
•Drug•Panniculitis-induced•Pressure-induced•Centrifugal•Idiopathic
Table 2: Classification, Clinical Features, and Pathogenetic Basis of Acquired Lipodystrophies.

14
syndrome, multiple symmetric lipomatosis and other rare progeroid syndromes and disorders affecting growth and development.
History
If a lipodystrophy phenotype is discovered at or shortly after birth, CGLshouldbeconsidered;otherwisethepatientmayhaveacquiredlipodystrophy. In those suspected of having genetic lipodystrophies, an in-depthpedigreeanalysisshouldbeconducted.Patientsshouldbeaskedabouttheageofonsetandprogressionoflipodystrophyandotherassociatedmanifestations.Takingadetailedfamilyhistory,includingthehistory of consanguinity, is very important to understand the mode of inheritance.DiagnosisofAPL,atypicalprogeroidsyndrome,andAGLcanbedelayedinchildrenforseveralyearsuntiltheyseeaspecialist.Associatedautoimmune diseases, especially juvenile dermatomyositis, should be considered in patients with acquired lipodystrophies. Those with localized lipodystrophiesshouldbeaskedaboutlocalinjections,trauma,pressure,orother insults.
Complications
Some patients with generalized lipodystrophies are predisposed to developingextremehypertriglyceridemiaandchylomicronemia,whichresultinacutepancreatitisandevendeath.ManypatientswithFPLdevelopcoronary heart disease and other atherosclerotic vascular complications. Hepatic steatosis can lead to cirrhosis, necessitating liver transplantation. SomepatientswithMADdieduringchildhoodofunknownreasons,andpatientswithAPLwhodevelopmembranoproliferativeglomerulonephritismay eventually succumb to renal failure. Sudden death has been reported duringchildhoodinCGL,type4,likelyduetoarrhythmias.
Laboratory Tests
The diagnosis of various types of lipodystrophies is mainly clinical. Laboratory test depends upon the type of lipodystrophy and may provideadditionalsupportiveevidence.Exceptforpatientswithlocalizedlipodystrophies, a serum chemistry profile for glucose, lipids, liver enzymes, anduricacidshouldbeobtained.Measurementofserumleptindoesnothelp diagnostically but may predict response to investigational metreleptin replacementtherapy.PatientswithAPLshouldbetestedforserumC3and C3-nephritic factor and urinalysis for proteinuria should be conducted on these patients. Radiographs can show the presence of lytic lesions in appendicularbonesinpatientswithCGLandskeletaldefectsinthose

15
withMAD.Skinbiopsyisusefulforpatientswithlocalizedlipodystrophyor panniculitis-associated varieties. Holter monitoring, echocardiography, and stress test should be conducted for patients suspected of having cardiomyopathy or coronary heart disease.
Distinction between various types of lipodystrophy can be made by physicalexamination.Skinfoldthicknessmeasurement,dual–energyX-ray absorptiometry, and a whole body T-1 weighted magnetic resonance imaging can provide information on the pattern of fat loss. For genetic lipodystrophieswhosemolecularbasisisknown,genetictesting,includingprenataldiagnosis,isavailableforAGPAT2,BSCL2,LMNA,ZMPSTE24,andPPARGinclinicallaboratories.GenotypingforotherlipodystrophygenessuchasCAV1,PTRF,AKT2,CIDEC,PLIN1andPSMB8isavailableonaresearch basis.
TreatmentTreatment of various types of lipodystrophies is quite challenging. Propercounselingofparentsiscriticalforpreventingunwantedstressandpsychologicalsequelaeinchildrenaffectedwithlipodystrophy.Becausereversal of the lost adipose tissue is not possible, cosmetic surgery to improve appearance and management of metabolic complications are the onlytherapeuticoptions.Unwantedexcessadiposetissuecanbesurgicallyexcisedorremovedbyliposuction.Thosewithseverefaciallipodystrophycanundergo reconstructive facial surgery including fascial grafts from thighs, free flaps from anterolateral thigh, anterior abdomen, or temporalis muscle.
Forthelackofclinicaltrialevidence,allpatientsareadvisedtoconsumehigh carbohydrate, low–fat diets. These diets can improve chylomicronemia in patients presenting with acute pancreatitis however, it may also raise very low density lipoprotein triglyceride concentration. Reduction of energy intakeandincreasedphysicalactivityisimportantinpatientswithFPLtoavoidexcessfatdepositioninnonlipodystrophicregions.ManypatientswithFPLhaveincreasedriskofcoronaryheartdiseaseandtheyshouldlimitintakeofsaturatedandtrans-unsaturatedfatsanddietarycholesterol.However, whether such diet will be beneficial in the long term to reduce hepatic steatosis, serum triglycerides and improve glycemic control remains unclear.
Investigative TherapiesNo controlled clinical trials have been conducted to help guide drug

16
therapyformetaboliccomplications.Sincemanypatientshaveextremeinsulin resistance, they may require high doses of insulin. However, some patients can achieve good glycemic control with oral hypoglycemic drugs suchasmetforminandsulfonylureas.Metformincanimproveinsulinsensitivity, reduce appetite and induce ovulation in patients with polycystic ovarian syndrome. Thiazolidinediones can also be used; however, they can induce unwanted growth of adipose tissue in nonlipodystrophic regions in patientswithFPL.AlthoughpatientswithPPARGmutationsandFPLshouldrespond better to thiazolidinediones, there is not much data to support this. For many patients with generalized lipodystrophy, insulin therapy is needed. Subcutaneous metreleptin replacement therapy has been shown to improve diabetes control, hepatic steatosis, and hypertriglyceridemia in markedlyhypoleptinemicpatientswithgeneralizedlipodystrophies,butitseffectsinpatientswithFPLsofarhavebeenmodest.Metreleptintherapyremains investigational and is not yet approved by the U.S. Food and Drug Administration(FDA).
Information on current clinical trials is posted at www.clinicaltrials.gov
AllstudiesreceivingU.S.governmentfunding,andsomesupportedbyprivate industry, are posted on this government web site.
For information about clinical trials sponsored by private sources, contact: www.centerwatch.com
NORD does not endorse or recommend any particular studies.
Biochemistry
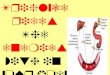
Figure 1

17
Figure Legends:
Fig. 1 The Role of Lipodystrophy Genes in Lipid Droplet formation in Adipocytes. Lipid droplets (LD) are organelles that store triglycerides (TG)intracellularly.Theyformasbuddingvesiclesattheendoplasmicreticulum(ER)whichfuseinadipocytestoformonelargeLD.Manyproteins,suchascelldeath-inducingDNAfragmentationfactora-likeeffectorc(CIDEC,showninbluetriangles),seipin(pinksquares)andperilipin 1 (green circles) are present on the LD membrane. CIDEC and seipin may be involved in fusion of LDs to form a larger LD while perilipin 1 is essential for lipid storage and hormone mediated lipolysis. Caveolae are formed from lipid rafts on the cell surface which include cholesterol (yellowsymbols),glycosphingolipids(greensymbols)andcaveolin-1(blackhairpinlikesymbols).Endocytosisofcaveolaeformscaveolinvesicleswhichmay directly merge with lipid droplets and thus translocating fatty acids to LDs.PolymeraseIandtranscriptreleasefactor(PTRF)controlsexpressionof caveolin 1 and 3 (not shown). The classical and alternative pathways involvedinthebiosynthesisofTGareshowninsidethelipiddroplet.Intheadiposetissue,TGsynthesisrequiresglycerol-3-phosphateastheinitialsubstrate (classical pathway), whereas in the small intestine, synthesis of TGcanoccurviaanalternativepathwayusingmonoacylglycerol(MAG)astheinitialsubstrate.Acylationofglycerol-3-phosphateusingfattyacylcoenzymeA(FA-CoA)atthesn-1positioniscatalyzedbyglycerol-3-phosphateacyltransferases(GPATs)resultingintheformationof1-acylglycerol-3-phosphateorlysophosphatidicacid(LPA).LPAisthenacylated at the sn-2 position by 1-acylglycerol-3-phosphate acyltransferases (AGPATs)toyieldphosphatidicacid(PA).RemovalofphosphategroupfromPAbyPAphosphatases(PAPs)producesdiacylglycerol(DAG).FurtheracylationofDAGatthesn-3positionbydiacylglycerolacyltransferases
Figure 2

18
(DGATs)finallyproducesTG.Inthealternativepathway,MAGisacylatedtoDAGbymonoacylglycerolacyltransferases(MGATs)whichisthenfurtherconvertedtoTG.LaminA/Careintegralcomponentsofnuclearlamina(shown in blue color) and interact with nuclear membrane proteins as wellaschromatin.Zincmetalloproteinase(ZMPSTE24)iscriticalforpost-translationalprocessingofprelaminAtoitsmatureform,laminA.
PublishedwithpermissionfromGargA.Lipodystrophies:Geneticandacquiredbodyfatdisorders.JCEM:2011;96:3313-3325.Copyright2011,The Endocrine Society.
Fig. 2 The Role of Lipodystrophy Genes in Pathways involved in the Development, Differentiation and Death of Adipocytes. The pluripotent mesenchymal stem cells can form preadipocytes, myocytes or osteoblasts depending upon the various cues. In response to various signals from hormones such as insulin and steroids and induction of adipogenic transcription factors, a series of changes are initiated in preadipocytes which lead to their differentiation to adipocytes. The transcription factors, CCAAT(cytidine-cytidine-adenosine-adenosine-thymidine)-enhancer-bindingproteins(C/EBP)b/darethefirsttobeupregulatedandthenstimulateothertranscriptionfactorssuchasPPARg,C/EBPa,andsterolregulatoryelement-bindingprotein(SREBP)1c.Someothergenessuchaspreadipocytefactor1(Pref1),aknownadipogenesisinhibitoraredownregulated.Matureadipocytesareactivatedresultingintheoverexpressionoflipogenicgeneslikefattyacidsynthase(FAS),acetylcoenzymeAcarboxylase(ACC),GPATs,AGPATs,andDGATsforbiosynthesisoftriglyceridesandphospholipids.Thesize of the lipid droplets is reduced upon fasting and increases with increased substrateavailability.AvailabledatasuggestthattheBSCL2-encodedprotein,seipin,andv-AKTmurinethymomaoncogenehomolog2(AKT2)maybeinvolvedinadipocytedifferentiation,whereastheAGPAT2geneaffects triglyceride synthesis. Clinical evidence from lipodystrophy patients harboringLMNAorZMPSTE24mutationssuggeststhatnucleardysfunctionmayaccelerateapoptosis/deathofmatureadipocytes.Interstitialtissuemayalsoplayanimportantroleinadipocytesurvival.MutationsinPSMB8whichencodesγ5i,acatalyticsubunitoftheimmunoproteasomesmayinduceautoinflammatory syndrome resulting in infiltration of lymphocytes in adipose tissue (panniculitis) and death of nearby adipocytes.
PublishedwithpermissionfromGargA.Lipodystrophies:Geneticandacquiredbodyfatdisorders.JCEM:2011;96:3313-3325.Copyright2011,The Endocrine Society.

19
REFERENCES1.GargA2000Lipodystrophies.AmJMed108:143-152.
2.GargA2004Acquiredandinheritedlipodystrophies.NEnglJMed350:1220-1234.
3.BerardinelliW1954Anundiagnosedendocrinometabolicsyndrome:reportof2cases.JClinEndocrinolMetab14:193-204.
4.CarrA,SamarasK,BurtonS,LawM,FreundJ,ChisholmDJ,CooperDA1998Asyndromeofperipherallipodystrophy,hyperlipidaemiaandinsulinresistanceinpatientsreceivingHIVproteaseinhibitors.AIDS12:F51-58.
5.GargA,MisraA2004Lipodystrophies:raredisorderscausingmetabolicsyndrome.EndocrinolMetabClinNorthAm33:305-331.
6.SeipM1959Lipodystrophyandgigantismwithassociatedendocrinemanifestations:anewdiencephalicsyndrome?ActaPaediatrica48:555-574.
7.VanMaldergemL,MagreJ,KhalloufTE,Gedde-DahlT,Jr.,DelepineM,TrygstadO,SeemanovaE,StephensonT,AlbottCS,BonniciF,PanzVR,MedinaJL,BogalhoP,HuetF,SavastaS,VerloesA,RobertJJ,LoretH,DeKerdanetM,Tubiana-RufiN,MegarbaneA,MaassenJ,PolakM,LacombeD,KahnCR,SilveiraEL,D’AbronzoFH,GrigorescuF,LathropM,CapeauJ,O’RahillyS2002Genotype-phenotyperelationshipsinBerardinelli-Seipcongenitallipodystrophy.JMedGenet39:722-733.
8.AgarwalAK,SimhaV,OralEA,MoranSA,GordenP,O’RahillyS,ZaidiZ,GurakanF,ArslanianSA,KlarA,RickerA,WhiteNH,BindlL,HerbstK,KennelK,PatelSB,Al-GazaliL,GargA2003Phenotypicandgeneticheterogeneityincongenitalgeneralizedlipodystrophy.JClinEndocrinolMetab88:4840-4847.
9.PardiniVC,VictoriaI,M.,RochaSM,AndradeDG,RochaAM,PieroniFB,MilagresG,PurischS,VelhoG1998Leptinlevels,beta-cellfunction,andinsulinsensitivityinfamilieswithcongenitalandacquiredgeneralizedlipoatrophicdiabetes.JClinEndocrinolMetab83:503-508.
10.MagreJ,DelepineM,VanMaldergemL,RobertJJ,MaassenJA,MeierM,PanzVR,KimCA,Tubiana-RufiN,CzernichowP,SeemanovaE,BuchananCR,LacombeD,VigourouxC,LascolsO,KahnCR,CapeauJ,LathropM2003PrevalenceofmutationsinAGPAT2amonghuman lipodystrophies. Diabetes 52:1573-1578.
11.GargA,WilsonR,BarnesR,AriogluE,ZaidiZ,GurakanF,KocakN,O’RahillyS,TaylorSI,PatelSB,BowcockAM1999Ageneforcongenitalgeneralizedlipodystrophymapstohumanchromosome9q34.JClinEndocrinolMetab84:3390-3394.
12.AgarwalAK,AriogluE,deAlmeidaS,AkkocN,TaylorSI,BowcockAM,BarnesRI,GargA2002AGPAT2ismutatedincongenitalgeneralizedlipodystrophylinkedtochromosome9q34.NatGenet31:21-23.
13.MagreJ,DelepineM,KhalloufE,Gedde-DahlT,Jr.,VanMaldergemL,SobelE,PappJ,MeierM,MegarbaneA,BachyA,VerloesA,d’AbronzoFH,SeemanovaE,AssanR,BaudicN,BourutC,CzernichowP,HuetF,GrigorescuF,deKerdanetM,LacombeD,LabruneP,LanzaM,LoretH,MatsudaF,NavarroJ,Nivelon-ChevalierA,PolakM,RobertJJ,TricP,Tubiana-RufiN,VigourouxC,WeissenbachJ,SavastaS,MaassenJA,TrygstadO,BogalhoP,FreitasP,MedinaJL,BonnicciF,JoffeBI,LoysonG,PanzVR,RaalFJ,O’RahillyS,StephensonT,KahnCR,LathropM,CapeauJ2001IdentificationofthegenealteredinBerardinelli-Seipcongenitallipodystrophyonchromosome11q13.NatGenet28:365-370.
14.KimCA,DelepineM,BoutetE,ElMourabitH,LeLayS,MeierM,NemaniM,BridelE,LeiteCC,BertolaDR,SempleRK,O’RahillyS,DugailI,CapeauJ,LathropM,MagreJ2008

20
Associationofahomozygousnonsensecaveolin-1mutationwithBerardinelli-Seipcongenitallipodystrophy.JClinEndocrinolMetab93:1129-1134.
15.HayashiYK,MatsudaC,OgawaM,GotoK,TominagaK,MitsuhashiS,ParkYE,NonakaI,Hino-FukuyoN,HaginoyaK,SuganoH,NishinoI2009HumanPTRFmutationscause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy.JClinInvest119:2623-2633.
16.ShastryS,DelgadoMR,DirikE,TurkmenM,AgarwalAK,GargA2010Congenitalgeneralizedlipodystrophy,type4(CGL4)associatedwithmyopathyduetonovelPTRFmutations.AmJMedGenetA152A:2245-2253.
17.RajabA,StraubV,McCannLJ,SeelowD,VaronR,BarresiR,SchulzeA,LuckeB,LutzkendorfS,KarbasiyanM,BachmannS,SpulerS,SchuelkeM2010Fatalcardiacarrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with musclerippling(CGL4)duetoPTRF-CAVINmutations.PLoSGenet6:e1000874.
18.TakeuchiK,ReueK2009Biochemistry,physiology,andgeneticsofGPAT,AGPAT,andlipinenzymesintriglyceridesynthesis.AmJPhysiolEndocrinolMetab296:E1195-1209.
19.AgarwalAK,GargA2010Enzymaticactivityofthehuman1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: upregulated in breast and cervical cancers. J Lipid Res 51:2143-2152.
20.AgarwalAK,GargA2003Congenitalgeneralizedlipodystrophy:significanceoftriglyceridebiosyntheticpathways.TrendsEndocrinolMetab14:214-221.
21.SzymanskiKM,BinnsD,BartzR,GrishinNV,LiWP,AgarwalAK,GargA,AndersonRG,GoodmanJM2007Thelipodystrophyproteinseipinisfoundatendoplasmicreticulumlipiddropletjunctionsandisimportantfordropletmorphology.ProcNatlAcadSciUSA104:20890-20895.
22.FeiW,ShuiG,GaetaB,DuX,KuerschnerL,LiP,BrownAJ,WenkMR,PartonRG,YangH 2008 Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast.JCellBiol180:473-482.
23.PayneVA,GrimseyN,TuthillA,VirtueS,GraySL,DallaNoraE,SempleRK,O’RahillyS,RochfordJJ2008ThehumanlipodystrophygeneBSCL2/seipinmaybeessentialfornormaladipocytedifferentiation.Diabetes57:2055-2060.
24.GargA,AgarwalAK2008Caveolin-1:anewlocusforhumanlipodystrophy.JClinEndocrinolMetab93:1183-1185.
25.SimhaV,GargA2003PhenotypicheterogeneityinbodyfatdistributioninpatientswithcongenitalgeneralizedlipodystrophyduetomutationsintheAGPAT2orSeipingenes.JClinEndocrinolMetab88:5433-5437.
26.SimhaV,AgarwalAK,AroninPA,IannacconeST,GargA2008Novelsubtypeofcongenitalgeneralizedlipodystrophyassociatedwithmuscularweaknessandcervicalspineinstability.AmJMedGenetA146A:2318-2326.
27.SimhaV,AgarwalAK,OralEA,FrynsJP,GargA2003Geneticandphenotypicheterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin EndocrinolMetab88:2821-2824.
28.FreidenbergGR,CutlerDL,JonesMC,HallB,MierRJ,CullerF,JonesKL,LozzioC,KaufmannS1992Severeinsulinresistanceanddiabetesmellitusinmandibuloacraldysplasia.AmJDisChild146:93-99.
29.NovelliG,MuchirA,SangiuoloF,Helbling-LeclercA,D’ApiceMR,MassartC,CaponF,SbracciaP,FedericiM,LauroR,TudiscoC,PallottaR,ScaranoG,DallapiccolaB,MerliniL,

21
BonneG2002MandibuloacraldysplasiaiscausedbyamutationinLMNA-encodinglaminA/C.AmJHumGenet71:426-431.
30.AgarwalAK,FrynsJP,AuchusRJ,GargA2003Zincmetalloproteinase,ZMPSTE24,ismutatedinmandibuloacraldysplasia.HumMolGenet12:1995-2001.
31.AgarwalAK,ZhouXJ,HallRK,NichollsK,BankierA,VanEschH,FrynsJ-P,GargA2006Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia due to zinc metalloproteinasedeficiency.JInvestigMed54:208-213.
32.AhmadZ,ZackaiE,MedneL,GargA2010EarlyonsetmandibuloacraldysplasiaduetocompoundheterozygousmutationsinZMPSTE24.AmJMedGenetA152A:2703-2710.
33.GargA,HernandezMD,SousaAB,SubramanyamL,MartinezdeVillarrealL,dosSantosHG,BarbozaO2010Anautosomalrecessivesyndromeofjointcontractures,muscularatrophy,microcyticanemia,andpanniculitis-associatedlipodystrophy.JClinEndocrinolMetab95:E58-63.
34.HorikoshiA,IwabuchiS,IizukaY,HagiwaraT,AmakiI1980[Acaseofpartiallipodystrophywith erythema, dactylic deformities, calcification of the basal ganglia, immunological disorders andlowIQlevel(author’stransl)].RinshoShinkeigaku–ClinicalNeurology20:173-180.
35.TanakaM,MiyataniN,YamadaS,MiyashitaK,ToyoshimaI,SakumaK,TanakaK,YuasaT,MiyatakeT,TsubakiT1993Hereditarylipo-muscularatrophywithjointcontracture,skineruptionsandhyper-gamma-globulinemia:anewsyndrome.InternMed32:42-45.
36.AgarwalAK,XingC,DeMartinoGN,MizrachiD,HernandezMD,SousaAB,MartinezdeVillarrealL,dosSantosHG,GargA2010PSMB8encodingthebeta5iproteasomesubunitismutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophysyndrome.AmJHumGenet87:866-872.
37.RivettAJ,HearnAR2004Proteasomefunctioninantigenpresentation:immunoproteasomecomplexes,Peptideproduction,andinteractionswithviralproteins.CurrProteinPeptSci5:153-161.
38.TorreloA,PatelS,ColmeneroI,GurbindoD,LendinezF,HernandezA,Lopez-RobledilloJC,DadbanA,RequenaL,PallerAS2010Chronicatypicalneutrophilicdermatosiswithlipodystrophyandelevatedtemperature(CANDLE)syndrome.JAmAcadDermatol62:489-495.
39.RamotY,CzarnowickiT,MalyA,Navon-ElkanP,ZlotogorskiA2010ChronicAtypicalNeutrophilicDermatosiswithLipodystrophyandElevatedTemperatureSyndrome:ACaseReport.PediatrDermatol5:538-41.
40.DunniganMG,CochraneMA,KellyA,ScottJW1974Familiallipoatrophicdiabeteswithdominanttransmission.Anewsyndrome.QJMed43:33-48.
41.KobberlingJ,DunniganMG1986Familialpartiallipodystrophy:twotypesofanXlinkeddominantsyndrome,lethalinthehemizygousstate.JMedGenet23:120-127.
42.GargA,PeshockRM,FleckensteinJL1999Adiposetissuedistributioninpatientswithfamilialpartiallipodystrophy(Dunniganvariety).JClinEndocrinolMetab84:170-174.
43.GargA2000Genderdifferencesintheprevalenceofmetaboliccomplicationsinfamilialpartiallipodystrophy(Dunniganvariety).JClinEndocrinolMetab85:1776-1782.
44.GargA,SpeckmanRA,BowcockAM2002Multisystemdystrophysyndromeduetonovelmissense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/Cgene.AmJMed112:549-555.
45.SubramanyamL,SimhaV,GargA2010Overlappingsyndromewithfamilialpartiallipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous

22
missenselaminA/Cmutations.ClinGenet78:66-73.
46.PetersJM,BarnesR,BennettL,GitomerWM,BowcockAM,GargA1998Localizationofthe gene for familial partial lipodystrophy (Dunnigan variety) to chromosome 1q21-22. Nat Genet18:292-295.
47.CaoH,HegeleRA2000NuclearlaminA/CR482QmutationinCanadiankindredswithDunnigan-typefamilialpartiallipodystrophy.HumMolGenet9:109-112.
48.ShackletonS,LloydDJ,JacksonSN,EvansR,NiermeijerMF,SinghBM,SchmidtH,BrabantG,KumarS,DurringtonPN,GregoryS,O’RahillyS,TrembathRC2000LMNA,encodinglaminA/C,ismutatedinpartiallipodystrophy.NatGenet24:153-156.
49.SpeckmanRA,GargA,DuF,BennettL,VeileR,AriogluE,TaylorSI,LovettM,BowcockAM2000Mutationalandhaplotypeanalysesoffamilieswithfamilialpartiallipodystrophy(Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of laminA/C.AmJHumGenet66:1192-1198.
50.AgarwalAK,GargA2002Anovelheterozygousmutationinperoxisomeproliferator-activated receptor-g gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab87:408-411.
51.HegeleRA,CaoH,FrankowskiC,MathewsST,LeffT2002PPARGF388L,atransactivation-deficientmutant,infamilialpartiallipodystrophy.Diabetes51:3586-3590.
52.SempleRK,ChatterjeeVK,O’RahillyS2006PPARgammaandhumanmetabolicdisease.JClinInvest116:581-589.
53.GeorgeS,RochfordJJ,WolfrumC,GraySL,SchinnerS,WilsonJC,SoosMA,MurgatroydPR,WilliamsRM,AceriniCL,DungerDB,BarfordD,UmplebyAM,WarehamNJ,DaviesHA,SchaferAJ,StoffelM,O’RahillyS,BarrosoI2004AfamilywithsevereinsulinresistanceanddiabetesduetoamutationinAKT2.Science304:1325-1328.
54.Rubio-CabezasO,PuriV,MuranoI,SaudekV,SempleRK,DashS,HydenCS,BottomleyW,VigourouxC,MagreJ,Raymond-BarkerP,MurgatroydPR,ChawlaA,SkepperJN,ChatterjeeVK,SulimanS,PatchAM,AgarwalAK,GargA,BarrosoI,CintiS,CzechMP,ArgenteJ,O’RahillyS,SavageDB2009PartiallipodystrophyandinsulinresistantdiabetesinapatientwithahomozygousnonsensemutationinCIDEC.EMBOMolMed1:280-287.
55.GandotraS,LeDourC,BottomleyW,CerveraP,GiralP,ReznikY,CharpentierG,AuclairM,DelepineM,BarrosoI,SempleRK,LathropM,LascolsO,CapeauJ,O’RahillyS,MagreJ,SavageDB,VigourouxC2011Perilipindeficiencyandautosomaldominantpartiallipodystrophy.NEnglJMed364:740-748.
56.OlofssonSO,BostromP,AnderssonL,RutbergM,LevinM,PermanJ,BorenJ2008Triglyceride containing lipid droplets and lipid droplet-associated proteins. Curr Opin Lipidol 19:441-447.
57.NishinoN,TamoriY,TateyaS,KawaguchiT,ShibakusaT,MizunoyaW,InoueK,KitazawaR,KitazawaS,MatsukiY,HiramatsuR,MasubuchiS,OmachiA,KimuraK,SaitoM,AmoT,OhtaS,YamaguchiT,OsumiT,ChengJ,FujimotoT,NakaoH,NakaoK,AibaA,OkamuraH,FushikiT,KasugaM2008FSP27contributestoefficientenergystorageinmurinewhiteadipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest 118:2808-2821.
58.CaronM,AuclairM,DonadilleB,BereziatV,GuerciB,LavilleM,NarbonneH,BodemerC,LascolsO,CapeauJ,VigourouxC2007HumanlipodystrophieslinkedtomutationsinA-typelaminsandtoHIVproteaseinhibitortherapyarebothassociatedwithprelaminAaccumulation,oxidativestressandprematurecellularsenescence.CellDeathDiffer14:1759-

23
1767.
59.GargA,VinaitheerthanM,WeatherallP,BowcockA2001Phenotypicheterogeneityinpatients with familial partial lipodystrophy (Dunnigan variety) related to the site of mis-sense mutationsinLaminA/C(LMNA)gene.JClinEndocrinolMetab86:59-65.
60.Al-ShaliK,CaoH,KnoersN,HermusAR,TackCJ,HegeleRA2004Asingle-basemutationintheperoxisomeproliferator-activatedreceptorgamma4promoterassociatedwithalteredinvitroexpressionandpartiallipodystrophy.JClinEndocrinolMetab89:5655-5660.
61.GargA,SubramanyamL,AgarwalAK,SimhaV,LevineB,D’ApiceMR,NovelliG,CrowY2009AtypicalprogeroidsyndromeduetoheterozygousmissenseLMNAmutations.JClinEndocrinolMetab94:4971-4983.
62.ShastryS,SimhaV,GodboleK,SbracciaP,MelanconS,YajnikCS,NovelliG,KroissM,GargA2010Anovelsyndromeofmandibularhypoplasia,deafness,andprogeroidfeaturesassociated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab95:E192-197.
63.SensenbrennerJA,HusselsIE,LevinLS1975CC–Alowbirthweightsyndrome,Riegersyndrome.BirthDefects11:423-426.
64.GorlinRJ,CervenkaJ,MollerK,HorrobinM,WitkopJ1975Riegeranomalyandgrowthretardation(TheS-H-O-R-TSyndrome).BirthDefects11:46-48.
65.O’NeillB,SimhaV,KothaV,GargA2007Bodyfatdistributionandmetabolicvariablesinpatientswithneonatalprogeroidsyndrome.AmJMedGenetA143:1421-1430.
66.MisraA,PeethambaramA,GargA2004Clinicalfeaturesandmetabolicandautoimmunederangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine(Baltimore)83:18-34.
67.MisraA,GargA2003Clinicalfeaturesandmetabolicderangementsinacquiredgeneralizedlipodystrophy:casereportsandreviewoftheliterature.Medicine82:129-146;
68.ChenD,MisraA,GargA2002Lipodystrophyinhumanimmunodeficiencyvirus-infectedpatients.JClinEndocrinolMetab87:4845-4856.
69.CarrA2000HIVproteaseinhibitor-relatedlipodystrophysyndrome.ClinicalInfectiousDiseases 30 Suppl 2:S135-142.
70.GrinspoonS,CarrA2005Cardiovascularriskandbody-fatabnormalitiesinHIV-infectedadults.NEnglJMed352:48-62.
71.SavageDB,SempleRK,ClatworthyMR,LyonsPA,MorganBP,CochranEK,GordenP,Raymond-BarkerP,MurgatroydPR,AdamsC,ScobieI,MuftiGJ,AlexanderGJ,ThiruS,MuranoI,CintiS,ChaudhryAN,SmithKG,O’RahillyS2009Complementabnormalitiesin
acquiredlipodystrophyrevisited.JClinEndocrinolMetab94:10-16.
72.HudonSE,CoffinierC,MichaelisS,FongLG,YoungSG,HrycynaCA2008HIV-proteaseinhibitorsblocktheenzymaticactivityofpurifiedSte24p.BiochemBiophysResCommun374:365-368.
73.BastardJP,CaronM,VidalH,JanV,AuclairM,VigourouxC,LuboinskiJ,LavilleM,MalachiM,GirardPM,RozenbaumW,LevanP,CapeauJ2002AssociationbetweenalteredexpressionofadipogenicfactorSREBP1inlipoatrophicadiposetissuefromHIV-1-infectedpatientsandabnormaladipocytedifferentiationandinsulinresistance.Lancet359:1026-1031.
74.MurataH,HruzPW,MuecklerM2000ThemechanismofinsulinresistancecausedbyHIVproteaseinhibitortherapy.JBiolChem275:20251-20254.
75.CarrA,MillerJ,LawM,CooperDA2000Asyndromeoflipoatrophy,lacticacidaemiaand

24
liverdysfunctionassociatedwithHIVnucleosideanaloguetherapy:contributiontoproteaseinhibitor-relatedlipodystrophysyndrome.AIDS14:F25-32.
76.LeeH,HanesJ,JohnsonKA2003ToxicityofnucleosideanaloguesusedtotreatAIDSandtheselectivityofthemitochondrialDNApolymerase.Biochemistry42:14711-14719.
77.KlopstockT,NaumannM,SeibelP,ShalkeB,ReinersK,ReichmannH1997MitochondrialDNAmutationsinmultiplesymmetriclipomatosis.Molecular&CellularBiochemistry174:271-
275.
78.OralEA,SimhaV,RuizE,AndeweltA,PremkumarA,SnellP,WagnerAJ,DePaoliAM,ReitmanML,TaylorSI,GordenP,GargA2002Leptin-replacementtherapyforlipodystrophy.NEnglJMed346:570-578.
79.SimhaV,SzczepaniakLS,WagnerAJ,DePaoliAM,GargA2003Effectofleptinreplacement on intrahepatic and intramyocellular lipid content in patients with generalized
lipodystrophy.DiabetesCare26:30-35.
80.ParkJY,JavorED,CochranEK,DePaoliAM,GordenP2007Long-termefficacyofleptinreplacementinpatientswithDunnigan-typefamilialpartiallipodystrophy.Metabolism56:508-
516.
81.SimhaV,SubramanyamL,SzczpaniakL,QuittnerC,Adams-HuetB,SnellP,Garg,A2012Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin EndocrinolMetab97(3):785-92.
ResourcesUT Southwestern Lipodystrophy website:www.lipodystrophy.info
GeneTestswebsite:http://www.ncbi.nlm.nih.gov/sites/genetests/
Clinical Centers & Medical ExpertsAbhimanyuGarg,M.D.ProfessorofInternalMedicine,Chief,DivisionofNutritionandMetabolicDiseases,Distinguished Chair in Human Nutrition ResearchUTSouthwesternMedicalCenteratDallas5323HarryHinesBoulevard,K5.214Dallas,TX75390-8537

25
PhillipGorden,M.D.
Clinical&CellularBiologySectionNIDDK,NationalInstitutesofHealthBuilding10-CRC,Room6-595210 Center Dr. Bethesda,MD20892-1770
ElifOra,M.D.AssociateProfessor,Endocrinology
UniversityofMichiganInternalMedicineMetabolism,Endocrinology&DiabetesDomino’s Farms24FrankLloydWrightDr,GPS:4000AveMariaDriveAnnArbor,MI48109
Acknowledgements
NORDisgratefultothefollowingmedicalexpertforservingasauthorofthisPhysicianGuide:
AbhimanyuGarg,M.D.ProfessorofInternalMedicine,Chief,DivisionofNutritionandMetabolicDiseases,Distinguished Chair in Human Nutrition ResearchUTSouthwesternMedicalCenteratDallas5323HarryHinesBoulevard,K5.214Dallas,TX75390-8537
ThisNORDPhysicianGuidewasmadepossiblebyaneducationalgrantfromAmylinPharmaceuticals

26
Patient Support and Resources National Organization for Rare Disorders (NORD) 55KenosiaAvenue POBox1968 Danbury,CT06813-1968 Phone:(203)744-0100 Toll free: (800) 999-NORD Fax:(203)798-2291
www.rarediseases.org [email protected]
NORD is grateful to the following medical expert for serving as author of this Physician Guide:
Abhimanyu Garg, M.D.Professor of Internal Medicine,Chief, Division of Nutrition and Metabolic Diseases,Distinguished Chair in Human Nutrition ResearchUT Southwestern Medical Center at Dallas5323 Harry Hines Boulevard, K5.214Dallas, TX 75390-8537
This NORD Physician Guide was made possible by an educational grant from Amylin Pharmaceuticals

NORD Guides for Physicians
#1 The Pediatrician’s Guide to Tyrosinemia Type 1
#2 The Pediatrician’s Guide to Ornithine Transcarbamylase Deficiency...and other Urea Cycle Disorders
#3 The Physician’s Guide to Primary Lateral Sclerosis
#4 The Physician’s Guide to Pompe Disease
#5 The Physician’s Guide to Multiple System Atrophy
#6 The Physician’s Guide to Hereditary Ataxia
#7 The Physician’s Guide to Giant Hypertrophic Gastritis and Menetrier’s Disease
#8 The Physician’s Guide to Amyloidosis
#9 The Physician’s Guide to Medullary Thyroid Cancer
#10 The Physician’s Guide to Hereditary Angioedema (HAE)
#11 The Physician’s Guide to The Homocystinurias
#12 The Physician’s Guide to Treacher Collins Syndrome
#13 The Physician’s Guide to Urea Cycle Disorders
#14 The Physician’s Guide to Myelofibrosis
#15 The Physician’s Guide to Lipodystrophy Disorders
These booklets are available free of charge. To obtain copies, call or write to NORD or download the text from www.NORDPhysicianGuides.org.
For information on rare disorders and the voluntary health organi-zations that help people affected by them, visit NORD’s web site at www.rarediseases.org or call (800) 999-NORD or (203) 744-0100.
NORD helps patients and families affected by rare disorders by providing:
• Physician-reviewedinformation in understandable language
• Referralstosupportgroupsand other sources of help
• Networkingwithotherpatients and families
• Medicationassistanceprograms
• Grantsandfellowshipsto encourage research on rare diseases
• Advocacyforhealth-related causes that affect the rare- disease community
• Publicationsforphysiciansand other medical professionals
Contact NORD at [email protected].
National Organization for Rare Disorders (NORD)POBox1968Danbury,CT06813-1968Phone:(203)744-0100Toll free: (800) 999-NORDFax:(203)798-2291
This booklet was made possible through an educational grant from Amylin Pharmaceuticals.