Embed Size (px)
Citation preview

The Pathophysiology of the Peritoneal Membrane
Olivier Devuyst,* Peter J. Margetts,† and Nicholas Topley‡
*Division of Nephrology, Universite catholique de Louvain Medical School, Brussels, Belgium; †Division of Nephrology,St. Joseph’s Hospital, Department of Medicine, McMaster University, Hamilton, Ontario, Canada; and ‡Department ofInfection, Immunity and Biochemistry, School of Medicine, Cardiff University, Cardiff, Wales, United Kingdom
Peritoneal dialysis (PD) is a life-sustain-ing therapy used by �100,000 patientswith ESRD worldwide, accounting forapproximately 10 to 15% of the dialysispopulation.1 The major obstacles to suc-cessful long-term PD are infection anddeleterious functional alterations in theperitoneal membrane after exposure todialysis solutions; this loss of dialysis ca-pacity is responsible for increased mor-bidity and mortality. These alterations,involving approximately 50% of all PDpatients, include progressive fibrosis, an-giogenesis, and vascular degeneration as-sociated with increased solute transportand loss of ultrafiltration (UF).2– 4 In asmall percentage of cases, a poorly de-fined but catastrophic fibrogenic re-sponse occurs primarily in the visceralperitoneum, leading to the onset of en-capsulating peritoneal sclerosis (EPS)with an associated high mortality rate.5,6
In the past two decades, clinical im-provements in therapy delivery and pre-scription have been introduced, includ-ing new dialysis solutions, improvedconnections, automated PD, and tailoredantibiotic strategies. Although these ad-vances reduce the incidence of peritoni-tis, infectious complications remain aproblem, as does membrane failure.There is thus a growing need to under-stand the molecular basis of these mem-brane-degenerative events and a need toestablish suitable experimental modelsto define better various aspects of thetherapy. This includes a better under-standing of transport mechanisms acrossthe peritoneal membrane, improved def-inition of the response of the peritoneumto infection and inflammation, and deci-phering the molecular mechanisms thatdrive peritoneal fibrosis and vasculardamage that lead to membrane dysfunc-
tion.4 Many of these investigations usingeither in vivo animal models or in vitrocell-based systems are based on interven-tional studies with pharmacologic agents,with blocking antibodies, or in transientoverexpression systems.7–10 Although thesestudies provide significant insight intoperitoneal pathophysiology, it is widelyacknowledged that they are limited insome cases by a lack of pathway specific-ity, adverse effects, and transient efficacy,so many questions remain to be ad-dressed.11–13 Until recently, the body sizeof a species was considered a major lim-iting factor in performing in vivo studiesrelevant to PD; however, the develop-ment of new approaches for in vivo phe-notyping14 coupled with molecular biol-ogy techniques provides the potential ofusing genetically modified mice for clin-ically relevant mechanistic studies ad-dressing various key aspects of PD patho-physiology.15,16 The purpose of this briefreview is to illustrate how these studies(primarily in rodent models) provide amore complete understanding of basicmechanisms and pave the way for the de-velopment of novel, specifically targeteddiagnostic and therapeutic strategiesaimed at reducing infection and improv-
Published online ahead of print. Publication dateavailable at www.jasn.org.
Correspondence: Dr. Olivier Devuyst, Division ofNephrology, UCL Medical School, 10 Avenue Hip-pocrate, B-1200 Brussels, Belgium. Phone: �32-2-764-54-50; Fax: �32-2-764-54-55; E-mail:[email protected]
Copyright © 2010 by the American Society ofNephrology
ABSTRACTThe development of peritoneal dialysis (PD) as a successful therapy has and stilldepends on experimental models to test and understand critical pieces of patho-physiology. To date, the majority of studies performed in rat and rabbit modelsderive mechanistic insights primarily on the basis of interventional pharmacologicagents, blocking antibodies, or transient expression systems. Because body size nolonger limits the performance of in vivo studies of PD, genetic mouse models areincreasingly available to investigate the molecular and pathophysiologic mecha-nisms of the peritoneal membrane. We illustrate in this review how these inves-tigations are catching up with other areas of biomedical research and providedirect evidence for understanding transport and ultrafiltration, responses toinfection, and structural changes including fibrosis and angiogenesis. Thesestudies are relevant to mechanisms responsible not only for the major compli-cations of PD but also for endothelial biology, host defense, inflammation, andtissue repair processes.
J Am Soc Nephrol 21: 1077–1085, 2010. doi: 10.1681/ASN.2009070694
BRIEF REVIEW www.jasn.org
J Am Soc Nephrol 21: 1077–1085, 2010 ISSN : 1046-6673/2107-1077 1077

ing membrane survival and long-termoutcomes in patients who are on PD.
PERITONEAL TRANSPORT,AQUAPORINS, AND UF
Once technical issues were overcome,mouse models were initially used tocharacterize the general structure of thevisceral and parietal peritoneum that iseffectively undistinguishable from thatdescribed in rats and humans.16,17 Fur-thermore, the specific distribution of thewater channel aquaporin 1 (AQP1) andendothelial nitric oxide synthase (eNOS)in distinct vascular beds have been con-firmed, and exposure of mice to standardglucose dialysis solutions yields equili-bration curves for urea and glucose, so-dium sieving, and a net UF that are re-markably similar to those obtained inrats and humans.16
The capacity for UF across the perito-neal membrane is a major predictor ofoutcome and mortality in PD pa-tients.18,19 According to the three-poremodel, the major transport barrier of themembrane is the capillary endothelium,which contains ultrasmall pores (radius�3Å) that facilitate the osmotic trans-port of water.20 Computer simulationshave predicted these ultrasmall pores ac-count for approximately 50% of the UFand explain the sodium sieving—themarked fall of the dialysate-to-plasmaratio of sodium during the first hour ofPD with hypertonic dialysate.21 Theidentification of AQPs, a family of inte-gral plasma membrane proteins con-served in bacteria, plants, and mammals,provided critical insights in the molecu-lar mechanisms involving water perme-ation across biological membranes.22
The first member of the AQP family to beidentified, AQP1, is abundantly ex-
pressed in endothelial cells lining perito-neal capillaries, consistent with the pre-dicted topology of ultrasmall pores(Figure 1).23,24
This hypothesis has been substantiatedby Yang et al.,25 who showed that osmoti-cally driven water transport across the peri-toneum (estimated by a tracer dilution)was decreased in mice lacking AQP1. Ni etal.26 used an infusion model to demon-strate that, in comparison with control lit-termates, AQP1 knockout mice lack so-dium sieving and have a major decrease inUF, despite unchanged osmotic gradient(Figure 1). The use of AQP1-deficient micethus validates the three-pore model andprovides direct evidence for the role of wa-ter channels in PD. Increasing the expres-sion (or function) of AQP1 in the perito-neum, with, for example, corticosteroidtreatment, might thus be a potential ap-proach to treating UF failure in PD pa-tients.27
m
A
B
C
D
Lumen
0 30 60 90 1200.75
0.85
0.95 *#
*#*#
Dwell time (min)
D/P
sod
ium
+/+ +/– –/–
Initi
al U
F r
ate
(ml/m
in)
Aqp1+/+ (n=6)Aqp1+/– (n=6)Aqp1–/– (n=6)
40
20
0
Aqp1 mice
Figure 1. Distribution and role of AQP1 in the peritoneal membrane. (A) Cross-section of the human parietal peritoneum stained forAQP1. AQP1 is detected in the endothelium lining peritoneal capillaries, venules, and small veins. m, mesothelium. Bar � 40 �m).(B) Immunogold electron microscopy on mouse visceral peritoneum unicryl sections shows a very strong signal for AQP1 in the plasmamembrane and plasma membrane infoldings of capillary endothelial cells. Bar � 500 nm. (C and D) Effect of AQP1 deletion on thetransport of water across the peritoneal membrane. Mice with a targeted deletion of Aqp1 are investigated using a peritonealequilibration test essentially similar to that performed in patients. The dialysate-to-plasma ratio (D/P) of sodium (C) and the initial UFrates calculated from the first derivate of the best fitting curves for each mouse (D) are determined in Aqp1�/� mice (purple symbols),Aqp1�/� mice (blue symbols), and Aqp1�/� mice (red symbols) during a 2-hour exchange with hypertonic dialysate. In comparison withAqp1�/� mice, mice lacking AQP1 show a complete loss of sodium sieving and significantly lower initial UF rates. Intermediate valuesof sodium sieving and initial UF rates are observed in Aqp1�/� mice. Adapted from Ni et al.,26 with permission.
BRIEF REVIEW www.jasn.org
1078 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 1077–1085, 2010
ACUTE PERITONITIS: ROLE OFNOS ISOFORMS
Acute peritonitis is characterized by anincreased endothelial exchange area,with increased transport of small solutesand glucose, loss of proteins into the di-alysate, and dissipation of the osmoticgradient, leading to UF failure.28,29 Vaso-active substances released during the in-flammation reaction, particularly NO,play a role in these changes.30 Indeed, in-hibition of NOS with NG-nitro-L-argi-nine methyl ester improve UF and re-verse permeability changes in rat andmouse models of acute peritonitis.8,31
The three NOS isoforms—neuronalNOS (nNOS, NOS1), inducible NOS(iNOS, NOS2), and eNOS (NOS3)—aredifferentially expressed in the peritonealmembrane.32
The neuronal and endothelial isozymesare constitutive isoforms, their activitybeing controlled by intracellular Ca2�
levels, whereas iNOS is quiescent until itstranscription is activated by LPS and/orcytokines.33 The use of peritonitis mod-els in mice lacking specific NOS isoformsdemonstrates the importance of NO forstructural and transport-related alter-ations induced by acute peritoneal infec-tion or inflammation. The deletion ofeNOS, which has no effect on peritonealstructure or transport at baseline, signif-
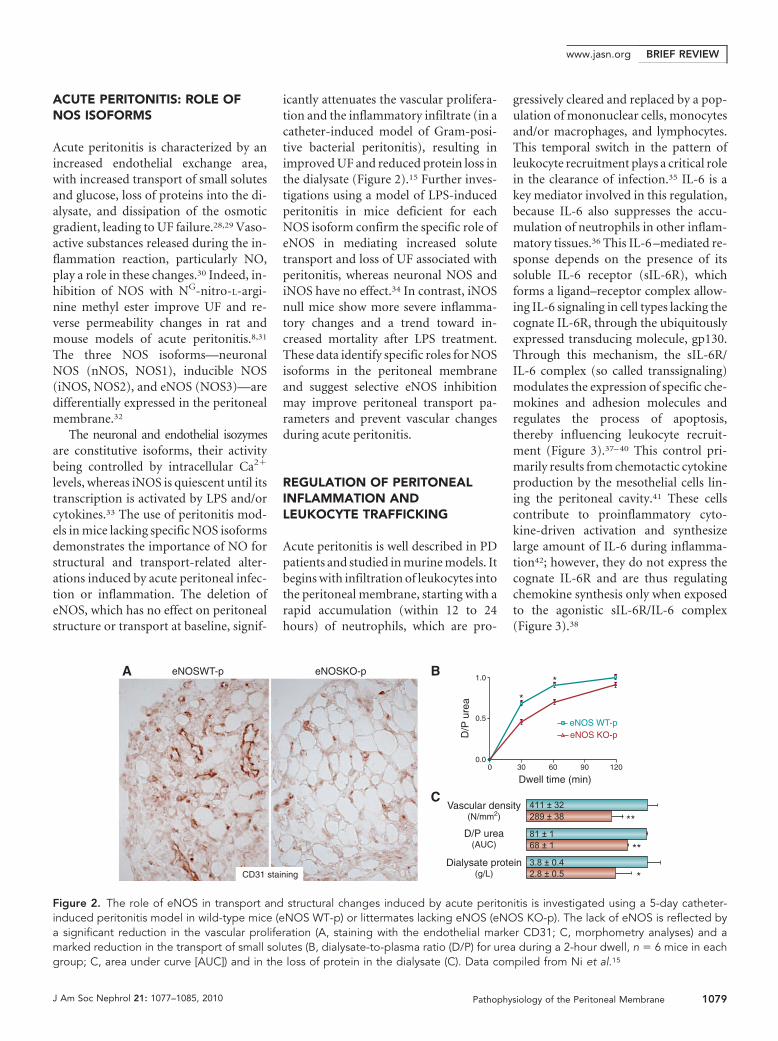
icantly attenuates the vascular prolifera-tion and the inflammatory infiltrate (in acatheter-induced model of Gram-posi-tive bacterial peritonitis), resulting inimproved UF and reduced protein loss inthe dialysate (Figure 2).15 Further inves-tigations using a model of LPS-inducedperitonitis in mice deficient for eachNOS isoform confirm the specific role ofeNOS in mediating increased solutetransport and loss of UF associated withperitonitis, whereas neuronal NOS andiNOS have no effect.34 In contrast, iNOSnull mice show more severe inflamma-tory changes and a trend toward in-creased mortality after LPS treatment.These data identify specific roles for NOSisoforms in the peritoneal membraneand suggest selective eNOS inhibitionmay improve peritoneal transport pa-rameters and prevent vascular changesduring acute peritonitis.
REGULATION OF PERITONEALINFLAMMATION ANDLEUKOCYTE TRAFFICKING
Acute peritonitis is well described in PDpatients and studied in murine models. Itbegins with infiltration of leukocytes intothe peritoneal membrane, starting with arapid accumulation (within 12 to 24hours) of neutrophils, which are pro-
gressively cleared and replaced by a pop-ulation of mononuclear cells, monocytesand/or macrophages, and lymphocytes.This temporal switch in the pattern ofleukocyte recruitment plays a critical rolein the clearance of infection.35 IL-6 is akey mediator involved in this regulation,because IL-6 also suppresses the accu-mulation of neutrophils in other inflam-matory tissues.36 This IL-6 –mediated re-sponse depends on the presence of itssoluble IL-6 receptor (sIL-6R), whichforms a ligand–receptor complex allow-ing IL-6 signaling in cell types lacking thecognate IL-6R, through the ubiquitouslyexpressed transducing molecule, gp130.Through this mechanism, the sIL-6R/IL-6 complex (so called transsignaling)modulates the expression of specific che-mokines and adhesion molecules andregulates the process of apoptosis,thereby influencing leukocyte recruit-ment (Figure 3).37– 40 This control pri-marily results from chemotactic cytokineproduction by the mesothelial cells lin-ing the peritoneal cavity.41 These cellscontribute to proinflammatory cyto-kine-driven activation and synthesizelarge amount of IL-6 during inflamma-tion42; however, they do not express thecognate IL-6R and are thus regulatingchemokine synthesis only when exposedto the agonistic sIL-6R/IL-6 complex(Figure 3).38
eNOSWT-p eNOSKO-p
CD31 staining
Dwell time (min)
D/P
ure
a *
*
eNOS WT-peNOS KO-p
A B
CVascular density
(N/mm2)
D/P urea(AUC)
Dialysate protein(g/L)
**
**
*
1.0
0.5
0.00 30 60 90 120
411 ± 32289 ± 38
81 ± 168 ± 1
3.8 ± 0.42.8 ± 0.5
Figure 2. The role of eNOS in transport and structural changes induced by acute peritonitis is investigated using a 5-day catheter-induced peritonitis model in wild-type mice (eNOS WT-p) or littermates lacking eNOS (eNOS KO-p). The lack of eNOS is reflected bya significant reduction in the vascular proliferation (A, staining with the endothelial marker CD31; C, morphometry analyses) and amarked reduction in the transport of small solutes (B, dialysate-to-plasma ratio (D/P) for urea during a 2-hour dwell, n � 6 mice in eachgroup; C, area under curve [AUC]) and in the loss of protein in the dialysate (C). Data compiled from Ni et al.15
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 21: 1077–1085, 2010 Pathophysiology of the Peritoneal Membrane 1079

For gaining insights into the media-tors controlling the pattern of leukocyterecruitment during peritoneal inflam-mation, a mouse model of acute perito-neal inflammation was established by us-ing a controlled dose of cell-free supernatantof Staphylococcus epidermidis, a majorcause of PD-associated peritonitis.38 Themodel recapitulates the pattern of in-flammatory events encountered duringhuman PD peritonitis, with early activa-tion of proinflammatory cytokines (TNF-�,IL-1, and IFN-�) and subsequentchanges in chemokine expression andthe rapid recruitment of neutrophils andtheir subsequent replacement by mono-cytes.38 By combining clinical and invitro investigations with studies in IL-6null mice, it is believed that the initialattraction of neutrophils by proinflam-matory cytokine-driven expression ofCXC chemokine, MIP-1/KC, is followedby the release of sIL-6R shed from neu-trophils, facilitating the formation ofsIL-6R/IL-6 complexes. In turn, thesecomplexes suppress the release of otherCXC chemokines, ensuring clearance ofneutrophils, and simultaneously pro-moting the secretion of the CC chemo-kines, such as monocyte chemoattrac-tant protein 1 (MCP-1) and RANTES,triggering the recruitment of mononu-clear leukocytes.38 Further studies usingIFN-� null mice establish that IFN-� isalso involved in controlling the initial re-
cruitment of neutrophils, by affecting thelocal activities of IL-6 and IL-1�, but alsoin the promotion of their apoptosis andclearance.39 Using knock-in mice ex-pressing mutant forms of the IL-6 signaltransducer molecule gp130, McLoughlinet al.40 showed that IL-6/sIL-6R signalingalso selectively promotes T cell recruit-ment into the peritoneal membranethrough a gp130-dependent, STAT1/3-dependent activation pathway. Taken to-gether, these studies provide useful in-sight into the actions of IL-6 and itssoluble receptor during acute inflamma-tion and suggest that while the transitionfrom innate immunity to acquired im-munity facilitates the resolution of in-flammation and the clearance of bacte-rial infection in the peritoneum,dysregulation of this pathway as occursin chronic inflammation or after re-peated infections also contributes to in-flammation-induced peritoneal damage.These studies provide clear evidence fortherapeutic intervention to reduce in-flammation43 and to promote the clear-ance of bacterial infections (N.T. andS.A. Jones, unpublished data).
TRANSGENIC MICE USED FORCELLULAR STUDIES
A major interest of transgenic mice is thepossibility of harvesting cells to develop
primary cultures to investigate the role ofspecific molecules in a given cell popula-tion. This approach has been used to in-vestigate the role of Toll-like receptor 4(TLR4) in murine peritoneal mesothelialcells (MPMC) exposed to inflamma-tion.44 Kato et al.44 developed primarycell cultures of MPMC derived from ei-ther C3H/HeN mice (wild-type; LPSsensitive) or C3H/HeJ mice (that lack theresponse to LPS). Using this system, theyobserved the induction of MCP-1 andmacrophage inflammatory protein 2(MIP-2) by MPMC stimulated with lipidA depends on the expression of TLR4.Furthermore, leukocyte recruitment intothe peritoneal cavity and the productionof MCP-1 and MIP-2 in response to LPSare significantly increased in C3H/HeNmice as compared with C3H/HeJ mice.Thus, TLR4 is directly involved in theproduction of chemokines by mesothe-lial cells, suggesting that TLR4-mediatedpathways reduce the detrimental conse-quences of peritoneal inflammation. Re-cent studies45 also showed that treat-ment with the soluble form of TLR2modulates peritoneal inflammationand leukocyte recruitment and doesnot have a negative impact on bacterialclearance in a peritoneal infectionmodel. These data suggest that therapeuticintervention against inflammation can beachieved without compromising peritonealhost defense.
Classical Trans-signaling
IL-6
Signal via STAT3
Signal via STAT3
sIL-6R/IL-6
IL-6R
gp130
Time (days)
Infection
PMN
IL-1IFN-γTNF-α
IL-6
Monocytes
Lymphocytes
IL-6 + sIL-6R
IL-8 MCP-1/RANTES PMN apoptosis
Figure 3. IL-6 and sIL-6R signaling in the regulation of leukocyte trafficking. The regulation of leukocyte trafficking in the peritonealcavity is mediated by proinflammatory cytokine–driven (IL-1, TNF-�, and IFN-�) activation of IL-6/sIL-6R transsignaling mediatedthrough control of STAT3 activation that results in differential control of chemokine secretion (that is responsible for mononuclearleukocyte and T cell recruitment) and polymorphonuclear neutrophils (PMN) apoptosis. The scheme is derived from data in murinemodels of acute inflammation and from measurements in the effluent of patients with episodes of peritonitis.
BRIEF REVIEW www.jasn.org
1080 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 1077–1085, 2010

FIBROSIS PATHWAYS,ANGIOGENESIS, ANDEPITHELIAL-TO-MESENCHYMALTRANSITION
Studies have demonstrated that perito-neal mesothelial cells undergo epithelial-to-mesenchymal transition (EMT) afterexposure to injury46 or associated growthfactors (Figure 4) to form fibroblasts.47
Furthermore, EMT of peritoneal me-sothelial cells is associated with angio-genic stimuli48 and altered solute trans-port.49 Angiogenesis and fibrosis seem tobe intimately linked through commoninitiating growth factors and inflamma-tory cytokines and the EMT process. Un-derstanding the mechanisms of fibrosisand the interaction with angiogenesis istherefore important to developing thera-peutic strategies to preserve the perito-neum as a dialysis membrane.
Epithelial-to-MesenchymalTransitionEMT is an essential process in embryo-genesis,50 is beneficial in normal woundhealing,51 but is pathogenic in malig-nancy52 and fibrosis.53 There is increas-ing evidence to suggest that treatment toprevent EMT may also ameliorate perito-
neal fibrosis and angiogenesis and there-fore preserve the peritoneal mem-brane.54 EMT is a cellular programconsisting of a loss of cell– cell and cell–matrix interaction and cell polarity, cy-toskeletal rearrangement, and basementmembrane degradation with subsequentmigration or invasion.55 Recently, bi-omarkers for EMT have been categorizedand include the loss of the epithelial ad-hesion protein E-cadherin and upregula-tion of mesenchymal markers such as fi-broblast-specific protein 1.56
E-cadherin expression is regulated atmultiple levels, including gene expres-sion and both extracellular and intracel-lular protein cleavage. E-cadherin geneexpression is suppressed by a family ofregulatory proteins, including zinc fingerDNA– binding proteins Snail,57 Slug,58
Twist,59 ZEB1, and ZEB2.60 These pro-teins are, in turn, regulated by growthfactors such as PDGF, TGF-�, and Wntproteins.60
The complex pathways involved inEMT have been investigated in trans-genic mouse models, but these have yetto be studied in the peritoneum to anyextent. Smads are key signal transduc-tion proteins involved in TGF-� signal-ing, and the role of Smad3 in EMT in vivo
is somewhat controversial. In the kidney,Smad3 is essential for EMT,61 but EMT isobserved in Smad3 knockout mice withlocal ocular lens overexpression of TGF-�.62 EMT has been studied in mesothe-lial cell cultures, which reveal mamma-lian target of rapamycin (mTOR) mayhave a role in peritoneal EMT, and themTOR inhibitor, rapamycin, has beenshown to maintain E-cadherin expres-sion in the face of TGF-�–inducedEMT.63 Likewise, bone morphogenicprotein 7 acts as a TGF-� antagonistand in mesothelial cell culture reversesEMT.54
Invasion is essential in the full EMTprocess. Matrix metalloproteinease 2(MMP-2) and MMP-9 have been studiedextensively, because they are gelatinaseswith specificity for basement mem-brane–associated collagen type IV. Asidefrom this collagenase activity, MMPshave a variety of other effects, includingcleavage of growth factor precursors andgrowth factor– binding proteins64 and al-tering activity of other receptors andproteinases.65 MMP-2 is specifically im-plicated in EMT, and inhibition ofMMP-2 inhibits EMT in renal tubularcell culture.66 Metalloproteinase inhibi-tors have been evaluated in a mouse
A
FE
CB
D
Cytokeratin Mergeα-SMA
Figure 4. Peritoneal mesothelial cells undergo EMT. The peritoneum of mice was stained for the epithelial marker cytokeratin in greenand the mesenchymal marker �-smooth muscle actin (�-SMA) in red. (A through C) In animals exposed to TGF-�, cells with epithelialcharacteristics (teal arrows) are intermingled with cells expressing both epithelial and mesenchymal markers (white arrows) and fullydifferentiated myofibroblasts (red arrows). (D through F) Unexposed animals reveal a single mesothelial cell layer positive for cytokeratinwith no �-SMA expression. Nuclei are counterstained with DAPI (blue). Magnification, �200.
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 21: 1077–1085, 2010 Pathophysiology of the Peritoneal Membrane 1081

model of peritoneal fibrosis using re-peated injections of chlorhexidine glu-conate.67 The MMP inhibitor suppressesperitoneal fibrosis and angiogenesis, butthe effect on EMT was not evaluated.
Peritoneal Membrane Fibrosis andAngiogenesisThe most consistent change observed inthe peritoneal tissues of a patient who ison PD is an increase in the submesothe-lial thickness associated with peritonealfibrosis and angiogenesis (Figure 5).3,4 Insome rare cases, peritoneal fibrosis man-ifests as an aggressive encapsulation ofthe bowels (EPS) with significant associ-ated morbidity and mortality.68 Fibrosisand angiogenesis seem to occur togetherin peritoneal tissues,69 and interventionsthat reduce angiogenesis also reduce fi-brosis.70
The cause of peritoneal fibrosis is notclear, but both human biopsy studies andanimal studies suggested that uremiaalone induces fibrotic changes in theperitoneum.3,71 An ongoing focus of re-search concerns the biocompatible na-ture of PD fluids and their possible fibro-genic effects. Aside from a low pH andlactate buffer, standard dialysis fluidshave a high concentration of glucoseand contain glucose degradation prod-ucts (GDPs) as a result of heat steriliza-
tion. High concentration of glucosealone induces fibrogenic growth fac-tors in peritoneal mesothelial cells inculture.72 Both in vivo and in vitro stud-ies find that GDPs induce fibrosis andangiogenesis in the peritoneum. Theuremic milieu, along with nonphysi-ologic PD solutions, leads to the ap-pearance of advanced glycation end-products (AGEs) in the peritonealtissues. These AGEs bind to a cognatereceptor (RAGE), and this direct inter-action induces fibrosis.73 The interac-tion between fibrosis and angiogenesismay occur at the level of inducing cy-tokines; TGF-�69 and inflammatorycytokines74 induce vascular endothelialgrowth factor and angiogenesis. Like-wise, using RAGE null mice, Sch-wenger et al.75 demonstrated thatGDPs induce both fibrosis and angio-genesis.
At the cellular level, the fibroblast is akey mediator of peritoneal fibrosis. Se-lective depletion of fibroblasts using atransgenic mouse with the thymidine ki-nase gene driven by a fibroblast-specificpromoter demonstrated that selectivedepletion of fibroblasts decreases fibrosisand angiogenesis.76
EPS has been identified as a potential im-portantcomplicationofPD;however, the in-cidence remains controversial.77 This clinical
concern has led to a need to develop realisticanimal models of EPS. The standard modelusedtodate includesadaily injectionofchlo-rhexidine gluconate.78 We recently devel-oped a novel model using a helper-depen-dent adenovirus expressing TGF-�1(HDAdTGF�1).79 Unlike the first-genera-tion adenovirus, the helper-dependent ad-enovirus demonstrates prolonged expres-sion. HDAdTGF�1 administered as asingle injection to the peritoneum of miceleads to a progressive fibrosis and bowelencapsulation and suppresses weight gainsimilar to the clinical course in EPS.
CONCLUSIONS ANDPERSPECTIVES
The examples outlined herein reveal howthe use of transgenic mouse and cellularmodels has already made a significantimpact on defining basic mechanismsthat operate in the peritoneal membrane.The development of transgenic mice forpathways and molecules relevant to spe-cific diseases together with the possibilityof investigating minute biologic samplesfor numerous parameters simultaneouslyexplains why the use of such models is setto transform research into practice. Todate, studies in null mice and cells de-rived from these animals provide direct
B
Inflammatory Episodes (cumulative effect of severity?)
A C
Fibrogenesis (vascular and fibrotic changes)
Exposure to solution components
Systemic inflammatory status
Local “membrane”inflammation
EMTMesothelial cell integrity
0 2.5 5
Time on PD(years)
7.5 10+m
m
Figure 5. Deleterious modifications of the peritoneal membrane exposed to PD. (A) Structure at the beginning of therapy. (B) Structuralalterations—loss of mesothelial integrity, submesothelial fibrosis, vasculopathy, and vascular proliferation—after 5 years on PD. Brownstaining indicates immunoreactivity for factor VIII, indicating the presence of blood vessels. m, mesothelium. Bar � 100 �m. (C)Summary of the time course of events and factors held responsible for driving alterations to the structure and function of the peritonealmembrane. EMT, epithelial-to-mesenchymal transition.
BRIEF REVIEW www.jasn.org
1082 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 1077–1085, 2010

mechanistic insights into the transportproperties of the peritoneal membrane,the role of cytokines and chemokines inregulating peritoneal inflammation, bac-terial clearance and leukocyte recruit-ment, and pathways involved in structuraland fibrogenic alterations that contribute totreatment failure (Figure 5).
As more of these models becomeavailable to target other relevant path-ways and with the application of mul-tiplex assay and DNA/RNA array tech-nologies in these models, it will becomepossible to assess the interactive rela-tionships of various physiologic andpathophysiologic pathways in the peri-toneum in relation to systemic param-eters. Mouse models also offer a vitalpreclinical resource in which the test-ing of various therapeutic strategies,arising from the mechanistic ap-proaches mentioned herein, can beevaluated. Limitations of such modelsshould be kept in mind, including thevarious growth and metabolic rates,the effect of the genetic background,and the possibility of adaptive mecha-nisms. Despite these limitations, theynevertheless offer a tremendous re-sources that is poised to transformperitoneal research and lead to targetedinterventions to prolong PD therapy.
ACKNOWLEDGMENTS
O.D. is supported by the Belgian agencies
Fonds National de la Recherche Scientifique
and Fonds de la Recherche Scientifique Medi-
cale, the Action de Recherches Concertees 05/
10-328, an Inter-university Attraction Pole
(IUAP P6/05), the DIANE network, and
grants from Baxter Belgium; N.T. has been
supported by the Wellcome Trust, the Medi-
cal Research Council, Arthritis Research
Council, National Kidney Research Fund, the
Baxter Renal Extramural Grant Program, and
the Welsh Office of Research and Develop-
ment; P.M. is supported by Canadian Insti-
tutes of Health Research.
We are grateful to Eric Goffin, Simon
Jones, Ray Krediet, Norbert Lameire, Bengt
Lindholm, Bengt Rippe, and Jean-Marc Ver-
bavatz for support and discussions and to all
our fellows and technicians for superb assis-
tance in developing and analyzing these
mouse models.
DISCLOSURESNone.
REFERENCES
1. Grassmann A, Gioberge S, Moeller S, BrownG: ESRD patients in 2004: Global overviewof patient numbers, treatment modalitiesand associated trends. Nephrol Dial Trans-plant 20: 2587–2593, 2005
2. Margetts PJ, Churchill DN: Acquired ultrafiltra-tion dysfunction in peritoneal dialysis patients.J Am Soc Nephrol 13: 2787–2794, 2002
3. Williams JD, Craig KJ, Topley N, Von Ruhland C,Fallon M, Newman GR, Mackenzie RK, WilliamsGT, Peritoneal Biopsy Study Group: Morpho-logic changes in the peritoneal membrane ofpatients with renal disease. J Am Soc Nephrol13: 470–479, 2002
4. Devuyst O, van Westrhenen R, Topley N: Long-term peritoneal dialysis patients. In: Nolph andGokal’s Textbook of Peritoneal Dialysis, 3rd Ed.,edited by Khanna R, Krediet RT, New York,Springer, 2009, pp 757–780
5. Garosi G: Different aspects of peritonealdamage: Fibrosis and sclerosis. ContribNephrol 163: 45–53, 2009
6. Augustine T, Brown PW, Davies SD, SummersAM, Wilkie ME: Encapsulating peritoneal scle-rosis: Clinical significance and implications.Nephron Clin Pract 111: c149–c54, 2009
7. Carlsson O, Nielsen S, Zakaria el-R, Rippe B:In vivo inhibition of transcellular water chan-nels (aquaporin-1) during acute peritonealdialysis in rats. Am J Physiol 271: H2254–H2262, 1996
8. Ferrier ML, Combet S, van Landschoot M,Stoenoiu MS, Cnops Y, Lameire N, DevuystO: Inhibition of nitric oxide synthase re-verses changes in peritoneal permeability ina rat model of acute peritonitis. Kidney Int60: 2343–2350, 2001
9. De Vriese AS, Tilton RG, Stephan CC,Lameire NH: Vascular endothelial growthfactor is essential for hyperglycemia-in-duced structural and functional alterationsof the peritoneal membrane. J Am SocNephrol 12: 1734–1741, 2001
10. Margetts PJ, Gyorffy S, Kolb M, Yu L, HoffCM, Holmes CJ, Gauldie J: Antiangiogenicand antifibrotic gene therapy in a chronicinfusion model of peritoneal dialysis in rats.J Am Soc Nephrol 13: 721–728, 2002
11. Miyata T, Devuyst O, Kurokawa K: Towardbetter dialysis compatibility: Advances inthe biochemistry and pathophysiology ofthe peritoneal membranes. Kidney Int 61:375–386, 2002
12. Topley N: Animal models in peritoneal dial-ysis: More questions than answers? Perit DialInt 25: 33–34, 2005
13. Lameire N, Van Biesen W, Mortier S, DeVriese A: What did we learn from animalmodels in peritoneal dialysis? Contrib Neph-rol 150: 70–76, 2006
14. Rao S, Verkman AS: Analysis of organ phys-iology in transgenic mice. Am J Physiol CellPhysiol 279: C1–C18, 2000
15. Ni J, Moulin P, Gianello P, Feron O, BalligandJL, Devuyst O: Mice that lack endothelialnitric oxide synthase are protected againstfunctional and structural modifications in-duced by acute peritonitis. J Am Soc Neph-rol 14: 3205–3216, 2003
16. Ni J, Cnops Y, Debaix H, Boisde I, VerbavatzJM, Devuyst O: Functional and molecularcharacterization of a peritoneal dialysismodel in the C57BL/6J mouse. Kidney Int67: 2021–2031, 2005
17. Goltoib L: Functional structure of the perito-neum as a dialysing membrane. In: Nolphand Gokal’s Textbook of Peritoneal Dialysis,3rd Ed., edited by Khanna R, Krediet RT,New York, Springer, 2009, pp 73–136
18. Brown EA, Davies SJ, Rutherford P, MeeusF, Borras M, Riegel W, Divino Filho JC,Vonesh E, van Bree M, EAPOS Group: Sur-vival of functionally anuric patients on auto-mated peritoneal dialysis: The EuropeanAPD Outcome Study. J Am Soc Nephrol 14:2948–2957, 2003
19. Brimble KS, Walker M, Margetts PJ, KundhalKK, Rabbat CG: Meta-analysis: Peritonealmembrane transport, mortality, and tech-nique failure in peritoneal dialysis. J Am SocNephrol 17: 2591–2598, 2006
20. Rippe B, Stelin G, Haraldsson B: Computersimulations of peritoneal fluid transport inCAPD. Kidney Int 40: 315–325, 1991
21. Rippe B, Venturoli D, Simonsen O, de ArteagaJ: Fluid and electrolyte transport across theperitoneal membrane during CAPD accord-ing to the three-pore model. Perit Dial Int24: 10–27, 2004
22. Agre P: Aquaporin water channels (NobelLecture). Angew Chem Int Ed Engl 43:4278–4290, 2004
23. Nielsen S, Smith BL, Christensen EI, Agre P:Distribution of the aquaporin CHIP in secre-tory and resorptive epithelia and capillaryendothelia. Proc Natl Acad Sci U S A 90:7275–7279, 1993
24. Devuyst O, Nielsen S, Cosyns JP, Smith BL,Agre P, Goffin E: Aquaporin-1 and endothe-lial nitric oxide synthase expression in capil-lary endothelia of human peritoneum. Am JPhysiol 275: H234–H242, 1998
25. Yang B, Folkesson HG, Yang J, Matthay MA,Ma T, Verkman AS: Reduced osmotic waterpermeability of the peritoneal barrier inaquaporin-1 knockout mice. Am J Physiol276: C76–C81, 1999
26. Ni J, Verbavatz JM, Rippe A, Boisde I, MoulinP, Rippe B, Verkman AS, Devuyst O: Aqua-
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 21: 1077–1085, 2010 Pathophysiology of the Peritoneal Membrane 1083

porin-1 plays an essential role in water per-meability and ultrafiltration during perito-neal dialysis. Kidney Int 69: 1518–1525,2006
27. Stoenoiu MS, Ni J, Verkaeren C, Debaix H,Jonas J-C, Lameire N, Verbavatz J-M,Devuyst O: Corticosteroids induce expres-sion of aquaporin-1 and increase transcellu-lar water transport in rat peritoneum. J AmSoc Nephrol 14: 555–565, 2003
28. Krediet RT, Zuyderhoudt FM, Boeschoten EW,Arisz L: Alterations in the peritoneal transportof water and solutes during peritonitis in con-tinuous ambulatory peritoneal dialysis pa-tients. Eur J Clin Invest 17: 43–52, 1987
29. Combet S, van Landschoot M, Moulin P,Piech A, Verbavatz JM, Goffin E, BalligandJL, Lameire N, Devuyst O: Regulation ofaquaporin-1 and nitric oxide synthase iso-forms in a rat model of acute peritonitis.J Am Soc Nephrol 10: 2185–2196, 1999
30. Douma CE, de Waart DR, Struijk DG, KredietRT: Are phospholipase A2 and nitric oxideinvolved in the alterations in peritonealtransport during CAPD peritonitis? J LabClin Med 132: 329–340, 1998
31. Ni J, Cnops Y, McLoughlin RM, Topley N,Devuyst O: Inhibition of nitric oxide synthasereverses permeability changes in a mousemodel of acute peritonitis. Perit Dial Int 25:S11–S14, 2005
32. Combet S, Balligand JL, Lameire N, GoffinE, Devuyst O: A specific method for mea-surement of nitric oxide synthase enzymaticactivity in peritoneal biopsies. Kidney Int 57:332–338, 2000
33. Forstermann U, Kleinert H: Nitric oxide syn-thase: Expression and expressional controlof the three isoforms. Naunyn Schmiede-bergs Arch Pharmacol 352: 351–364, 1995
34. Ni J, McLoughlin RM, Brodovitch A, MoulinP, Brouckaert P, Casadei B, Feron O, TopleyN, Balligand JL, Devuyst O: Nitric oxide syn-thase isoforms play distinct roles duringacute peritonitis. Nephrol Dial Transplant25: 86–96, 2010
35. Bellingan GJ, Caldwell H, Howie SE, DarnsfieldI, Haslett C: In vivo fate of the inflammatorymacrophage during the resolution of inflam-mation. J Immunol 157: 2577–2585, 1996
36. Xing Z, Gauldie J, Cox G, Baumann H, JordanaM, Lei XF, Achong MK: IL-6 is an anti-inflam-matory cytokine required for controlling lo-cal and systemic acute inflammatory re-sponses. J Clin Invest 101: 311–320, 1998
37. Jones SA, Horiuchi S, Topley N, Yamamoto N,Fuller GM: The soluble interleukin-6 receptor:Mechanisms of production and implications indisease. FASEB J 15: 43–58, 2001
38. Hurst SM, Wilkinson TS, McLoughlin RM,Jones S, Horiuchi S, Yamamoto N, Rose-John S, Fuller GM, Topley N, Jones SA: IL-6and its soluble receptor orchestrate a tem-poral switch in the pattern of leukocyte re-cruitment seen during acute inflammation.Immunity 14: 705–714, 2001
39. McLoughlin RM, Witowski J, Robson RL,Wilkinson TS, Hurst SM, Williams AS,Williams JD, Rose-John S, Jones SA, TopleyN: Interplay between IFN-� and IL-6 signal-ing governs neutrophil trafficking and apo-ptosis during acute inflammation. J Clin In-vest 112: 598–607, 2003
40. McLoughlin RM, Jenkins BJ, Grail D,Williams AS, Fielding CA, Parker CR, ErnstM, Topley N, Jones SA: IL-6 trans-signalingvia STAT3 directs T cell infiltration in acuteinflammation. Proc Natl Acad Sci U S A 102:9589–9594, 2005
41. Topley N, Jorres A, Luttmann W, PetersenMM, Lang MJ, Thierauch KH, Muller C,Coles GA, Davies M, Williams JD: Humanperitoneal mesothelial cells synthesize inter-leukin-6: Induction by IL-1 beta and TNFalpha. Kidney Int 43: 226–233, 1993
42. Witowski J, Jorres A, Coles GA, Williams JD,Topley N: Superinduction of IL-6 synthesis inhuman peritoneal mesothelial cells is relatedto the induction and stabilization of IL-6mRNA. Kidney Int 50: 1212–1223, 1996
43. Nowell MA, Williams AS, Carty SA, SchellerJ, Hayes AJ, Jones GW, Richards PJ, Slinn S,Ernst M, Jenkins BJ, Topley N, Rose-John S,Jones SA: Therapeutic targeting of IL-6 transsignaling counteracts STAT3 control of ex-perimental inflammatory arthritis. J Immunol182: 613–622, 2009
44. Kato S, Yuzawa Y, Tsuboi N, Maruyama S,Morita Y, Matsuguchi T, Matsuo S: Endotoxin-induced chemokine expression in murineperitoneal mesothelial cells: The role of toll-like receptor 4. J Am Soc Nephrol 15: 1289–1299, 2004
45. Raby AC, Le Bouder E, Colmont C, Davies J,Richards P, Coles B, George CH, Jones SA,Brennan P, Topley N, Labeta MO: SolubleTLR2 reduces inflammation without compro-mising bacterial clearance by disrupting TLR2triggering. J Immunol 183: 506–517, 2009
46. Yanez-Mo M, Lara-Pezzi E, Selgas R, Ramırez-Huesca M, Domınguez-Jimenez C, Jimenez-Heffernan JA, Aguilera A, Sanchez-Tomero JA,Bajo MA, Alvarez V, Castro MA, del Peso G,Cirujeda A, Gamallo C, Sanchez-Madrid F,Lopez-Cabrera M: Peritoneal dialysis and epithe-lial-to-mesenchymal transition of mesothelialcells. N Engl J Med 348: 403–413, 2003
47. Margetts PJ, Bonniaud P, Liu L, Hoff CM,Holmes CJ, West-Mays JA, Kelly MM: Tran-sient overexpression of TGF-beta1 inducesepithelial mesenchymal transition in the ro-dent peritoneum. J Am Soc Nephrol 16:425–436, 2005
48. Aroeira LS, Aguilera A, Selgas R, Ramırez-Huesca M, Perez-Lozano ML, Cirugeda A, BajoMA, del Peso G, Sanchez-Tomero JA, Jimenez-Heffernan JA, Lopez-Cabrera M: Mesenchymalconversion of mesothelial cells as a mechanismresponsible for high solute transport rate in peri-toneal dialysis: Role of vascular endothelialgrowth factor. Am J Kidney Dis 46: 938–948,2005
49. Del Peso G, Jimenez-Heffernan JA, BajoMA, Aroeira LS, Aguilera A, Fernandez-Perpen A, Cirugeda A, Castro MJ, de GraciaR, Sanchez-Villanueva R, Sanchez-TomeroJA, Lopez-Cabrera M, Selgas R: Epithelial-to-mesenchymal transition of mesothelialcells is an early event during peritoneal di-alysis and is associated with high peritonealtransport. Kidney Int 108: S26-–S33, 2008
50. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA: Epithelial-mesenchy-mal transitions: The importance of changingcell state in development and disease. J ClinInvest 119: 1438–1449, 2009
51. Kalluri R: EMT: When epithelial cells decideto become mesenchymal-like cells. J ClinInvest 119: 1417–1419, 2009
52. Sabbah M, Emami S, Redeuilh G, Julien S,Prevost G, Zimber A, Ouelaa R, Bracke M, DeWever O, Gespach C: Molecular signatureand therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial can-cers. Drug Resist Updat 11: 123–151, 2008
53. Kalluri R, Weinberg RA: The basics of epi-thelial-mesenchymal transition. J Clin Invest119: 1420–1428, 2009
54. Yu MA, Shin KS, Kim JH, Kim YI, Chung SS,Park SH, Kim YL, Kang DH: HGF and BMP-7ameliorate high glucose-induced epithelial-to-mesenchymal transition of peritoneal me-sothelium. J Am Soc Nephrol 20: 567–581,2009
55. Kalluri R, Neilson EG: Epithelial-mesenchy-mal transition and its implications for fibro-sis. J Clin Invest 112: 1776–1784, 2003
56. Zeisberg M, Neilson EG: Biomarkers for epi-thelial-mesenchymal transitions. J Clin In-vest 119: 1429–1437, 2009
57. Cho HJ, Baek KE, Saika S, Jeong MJ, Yoo J:Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal tran-sition by activating PI3 kinase/Akt signalpathway. Biochem Biophys Res Commun353: 337–343, 2007
58. Bolos V, Peinado H, Perez-Moreno MA, FragaMF, Esteller M, Cano A: The transcription fac-tor Slug represses E-cadherin expression andinduces epithelial to mesenchymal transitions:A comparison with Snail and E47 repressors.J Cell Sci 116: 499–511, 2003
59. Yang MH, Wu KJ: TWIST activation by hyp-oxia inducible factor-1 (HIF-1): Implicationsin metastasis and development. Cell Cycle7: 2090–2096, 2008
60. Peinado H, Olmeda D, Cano A: Snail, Zeband bHLH factors in tumour progression: Analliance against the epithelial phenotype?Nat Rev Cancer 7: 415–428, 2007
61. Sato M, Muragaki Y, Saika S, Roberts AB,Ooshima A: Targeted disruption of TGF-be-ta1/Smad3 signaling protects against renaltubulointerstitial fibrosis induced by unilat-eral ureteral obstruction. J Clin Invest 112:1486–1494, 2003
62. Banh A, Deschamps PA, Gauldie J, OverbeekPA, Sivak JG, West-Mays JA: Lens-specific
BRIEF REVIEW www.jasn.org
1084 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 1077–1085, 2010

expression of TGF-beta induces anteriorsubcapsular cataract formation in the ab-sence of Smad3. Invest Ophthalmol Vis Sci47: 3450–3460, 2006
63. Aguilera A, Aroeira LS, Ramirez-Huesca M,Perez-Lozano ML, Cirugeda A, Bajo MA, DelPeso G, Valenzuela-Fernandez A, Sanchez-Tomero JA, Lopez-Cabrera M, Selgas R: Effectsof rapamycin on the epithelial-to-mesenchymaltransition of human peritoneal mesothelial cells.Int J Artif Organs 28: 164–169, 2005
64. Hemers E, Duval C, McCaig C, Handley M,Dockray GJ, Varro A: Insulin-like growth fac-tor binding protein-5 is a target of matrixmetalloproteinase-7: Implications for epi-thelial-mesenchymal signaling. Cancer Res65: 7363–7369, 2005
65. Egeblad M, Werb Z: New functions for thematrix metalloproteinases in cancer pro-gression. Nat Rev Cancer 2: 161–174, 2002
66. Cheng S, Pollock AS, Mahimkar R, Olson JL,Lovett DH: Matrix metalloproteinase 2 andbasement membrane integrity: A unifyingmechanism for progressive renal injury.FASEB J 20: 1898–1900, 2006
67. Ro Y, Hamada C, Inaba M, Kaneko K, TominoY: Inhibitory effects of matrix metalloprotein-ase inhibitor ONO-4817 on morphological al-terations in chlorhexidine gluconate-inducedperitoneal sclerosis rats. Nephrol Dial Trans-plant 22: 2838–2848, 2007
68. Kawaguchi Y, Kawanishi H, Mujais S, TopleyN, Oreopoulos DG: Encapsulating perito-
neal sclerosis: Definition, etiology, diagno-sis, and treatment. International Society forPeritoneal Dialysis Ad Hoc Committee onUltrafiltration Management in Peritoneal Di-alysis. Perit Dial Int 20[Suppl 4]: S43–S55,2000
69. Margetts PJ, Kolb M, Galt T, Hoff CM,Shockley TR, Gauldie J: Gene transfer oftransforming growth factor-beta1 to the ratperitoneum: Effects on membrane function.J Am Soc Nephrol 12: 2029–2039, 2001
70. Yoshio Y, Miyazaki M, Abe K, Nishino T,Furusu A, Mizuta Y, Harada T, Ozono Y,Koji T, Kohno S: TNP-470, an angiogenesisinhibitor, suppresses the progression ofperitoneal fibrosis in mouse experimentalmodel. Kidney Int 66: 1677–1685, 2004
71. Combet S, Ferrier ML, Van Landschoot M,Stoenoiu M, Moulin P, Miyata T, Lameire N,Devuyst O: Chronic uremia induces perme-ability changes, increased nitric oxide syn-thase expression, and structural modifica-tions in the peritoneum. J Am Soc Nephrol12: 2146–2157, 2001
72. Ha H, Lee HB: Effect of high glucose onperitoneal mesothelial cell biology. PeritDial Int 20[Suppl 2]: S15–S18, 2000
73. De Vriese AS, Flyvbjerg A, Mortier S, Til-ton RG, Lameire NH: Inhibition of the in-teraction of AGE-RAGE prevents hypergly-cemia-induced fibrosis of the peritonealmembrane. J Am Soc Nephrol 14: 2109 –2118, 2003
74. Margetts PJ, Kolb M, Yu L, Hoff CM, HolmesCJ, Anthony DC, Gauldie J: Inflammatory cy-tokines, angiogenesis, and fibrosis in the ratperitoneum. Am J Pathol 160: 2285–2294,2002
75. Schwenger V, Morath C, Salava A, Amann K,Seregin Y, Deppisch R, Ritz E, Bierhaus A,Nawroth PP, Zeier M: Damage to the peri-toneal membrane by glucose degradationproducts is mediated by the receptor foradvanced glycation end-products. J Am SocNephrol 17: 199–207, 2006
76. Okada H, Inoue T, Kanno Y, Kobayashi T,Watanabe Y, Ban S, Neilson EG, Suzuki H:Selective depletion of fibroblasts preservesmorphology and the functional integrity ofperitoneum in transgenic mice with perito-neal fibrosing syndrome. Kidney Int 64:1722–1732, 2003
77. Kawanishi H, Moriishi M: Encapsulatingperitoneal sclerosis: Prevention and treat-ment. Perit Dial Int 27[Suppl 2]: S289 –S292, 2007
78. Sawada T, Ishii Y, Nakajima I, Fuchinoue S,Kubota K, Teraoka S: An experimentalmodel of encapsulating peritoneal sclero-sis. Perit Dial Int 29[Suppl 2]: S49 –S50,2009
79. Liu L, Shi CX, Ghayur A, Zhang C, Su JY,Hoff CM, Margetts PJ: Prolonged perito-neal gene expression using a helper-de-pendent adenovirus. Perit Dial Int 29:508 –516, 2009
BRIEF REVIEWwww.jasn.org
J Am Soc Nephrol 21: 1077–1085, 2010 Pathophysiology of the Peritoneal Membrane 1085




![An Update on Pathophysiology and Medical Management of ...peritoneal, ovarian and rectovaginal septum endometriosis (RVS) [8]. Significantly, more nerve fibres are present ... regulates](https://img.pdfslide.us/doc/110x75/5e68089093d4236c347752b2/an-update-on-pathophysiology-and-medical-management-of-peritoneal-ovarian-and.jpg)























