Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL. CHEMISTRY 0 1990 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 265, No. 27, Issue of September 25, pp. X330-16336,199O Printed in U.S. A.
The Oxidative Inactivation of Mitochondrial Electron Transport Chain Components and ATPase*
(Received for publication, April 10, 1990)
Yin ZhangS, Olivier Marcillat, Cecilia Giulivi, Lars Ernsterg, and Kelvin J. A. Davies!l From the Institute for Toxicoloev & Deuartment of Biochemistry, The University of Southern California, Health Sciences Ca’mpus, Los ~&eltk, k’alifornia 60033
Bovine heart submitochondrial particles (SMP) were exposed to continuous fluxes of hydroxyl radical (‘OH) alone, superoxide anion radical (0;) alone, or mixtures of ‘OH and 02, by y radiolysis in the presence of 100% NzO (‘OH exposure), 100% O2 + formate (02 exposure), or 100% O2 alone (*OH + 0; exposure). Hydrogen peroxide effects were studied by addition of pure HzOz. NADH dehydrogenase, NADH oxidase, succinate de- hydrogenase, succinate oxidase, and ATPase activities (V,,,,,) were rapidly inactivated by ‘OH (10% inacti- vation at 15-40 nmol of ‘OH/mg of SMP protein, 50- 90% inactivation at 600 nmol of ‘OH/mg of SMP pro- tein) and by ‘OH + 0; (10% inactivation at 20-80 nmol of ‘OH + O;/mg of SMP protein, 45-75% inactivation at 600 nmol of ‘OH + O;/mg of SMP protein). Impor- tantly, 0; was a highly efficient inactivator of NADH dehydrogenase, NADH oxidase, and ATPase (10% in- activation at 20-50 nmol of O;/mg of SMP protein, 40% inactivation at 600 nmol of O;/mg of SMP pro- tein), a mildly efficient inactivator of succinate dehy- drogenase (10% inactivation at 150 nmol of Oplmg of SMP protein, 30% inactivation at 600 nmol of O;/mg of SMP protein), and a poor inactivator of succinate oxidase (~10% inactivation at 600 nmol of Oz/mg of SMP protein). H202 partially inactivated NADH de- hydrogenase, NADH oxidase, and cytochrome oxidase, but even 10% loss of these activities required at least 500-600 nmol of HzOz/mg of SMP protein. Cyto- chrome oxidase activity (oxygen consumption sup- ported by ascorbate + N,N,N’,N’-tetramethyl-p-phen- ylenediamine) was remarkably resistant to oxidative inactivation, with less than 20% loss of activity evident even at ‘OH, 02, ‘OH + 02, or HzOz concentrations of 600 nmol/mg of SMP protein. Cytochrome c oxidase activity, however (oxidation of, added, ferrocyto- chrome c), exhibited more than a 40% inactivation at 600 nmol of ‘OH/mg of SMP protein. The ‘OH-depend- ent inactivations reported above were largely inhibit- able by the ‘OH scavenger mannitol. In contrast, the O;-dependent inactivations were inhibited by active
* This research was supported by Grant ES 03598 from the Na- tional Institutes of Health, National Institute of Environmental Health Sciences (to K. J. A. D.). Some aspects of this work have been published in preliminary form (Zhang, Y., Marcillat, O., and Davies, K. J. A. (1990) FASEB J. 2, A488, abstr. 4135). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “adver- tisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Current address: Dept. of Radiation Therapy, University of Texas, Galveston, TX 77550.
§ Distinguished Visiting Professor. Permanent address: Dept. of Biochemistry, University of Stockholm, Stockholm S-10691, Sweden.
n To whom all correspondence and reprint requests should be addressed.
superoxide dismutase, but not by denatured superoxide dismutase or catalase. Membrane lipid peroxidation was evident with ‘OH exposure but could be prevented by various lipid-soluble antioxidants which did not protect enzymatic activities at all. We conclude that both ‘OH and 02 are effective inactivators of specific proteins in the respiratory chain, but that the patterns of inactivation are quite distinct for the two active oxygen species: in contrast, the respiratory chain ap- pears to be far more resistant to HzOz. Generalized lipid peroxidation, although a simple and widely used indicator of oxidative stress, appears to be unrelated to electron transport chain inactivation. The pattern of results presented here may prove useful in probing molecular mechanisms of oxidative damage to mito- chondria, and in the development of effective antioxi- dant strategies.
Mitochondrial electron transport has long been recognized as a major intracellular source of oxygen radicals and hydro- gen peroxide (1). In the presence of various drugs or toxins (e.g. electron transport inhibitors and uncouplers, various quinonoid compounds, etc.) the mitochondrial generation of reactive oxygen species can increase severalfold (2, 3). The superoxide anion radical (0,)’ appears to be the first oxygen reduction product generated under both physiological and pathological conditions (1). Subsequent dismutation of 0; generates hydrogen peroxide (H202), and reaction of 0; with H202 can result in the (metal-catalyzed) production of the hydroxyl radial ( ’ OH) (1, 4, 7).
While some 0; and H202 may diffuse from the mitochon- drion to damage distant cellular components, the reactions of ‘OH are largely diffusion controlled. Furthermore, the effects of all three species should be greatest at the mitochondrial inner membrane where they are generated in the course of respiration, and where their concentrations are probably high- est. In addition, the mitochondrial inner membrane contains several iron and copper enzyme complexes which may catalyze further reactions with 0; and Hz02.
The above considerations have prompted numerous inves- tigators to study the oxidative inactivation of mitochondrial functions under physiological conditions, and after exposure to various drugs and toxins (e.g. Refs. 4-19). Such studies have clearly demonstrated the sensitivity of mitochondrial proteins, lipids, and DNA to oxidative stress. Indeed, such studies indicate that oxidative damage is one of the major
’ The abbreviations used are: O;, superoxide anion radical; SMP, submitochondrial preparations inner mitochondrial membranes; ‘OH, hydroxyl radical; DCPIP, 2,6-dichlorophenolindophenol; TMPD, N,N,N’,N’,-tetramethyl-p-phenylenediamine; BHT, buty- lated hydroxytoluene; DPPD, N,N’-diphenyl-1,4-phenylenediamine.
16330
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Oxidative Inactivation of Mitochondrial Enzymes 16331
determinants of mitochondrial turnover. Recently, we have reported that mitochondria (20), like
bacteria (21-25) and the eucaryotic cytoplasm (21, 26-32), contain a proteolytic system which can selective degrade oxidatively modified proteins. Such proteolytic systems are thought to form part of the intracellular repair systems for oxidative stress (21, 33). In related studies, however, we (26- 29) and others (34-36) have demonstrated that 02, H202, and ‘OH have entirely different effects on a variety of purified proteins.
Studies of mitochondrial oxidative inactivation to date have been seriously hampered by incomplete quantification of re- active oxygen species and by ignorance of the effects of each oxygen species on mitochondrial components. The typical experimental situation involves widespread mitochondrial damage following incubation with substrates, drugs, or toxi- cants; such conditions generate complex mixtures of O,, HzOz, and ‘OH. Despite considerable efforts to determine the im- portance of each oxygen species, via inhibition’studies with various antioxidant enzymes and compounds, it has not al- ways been possible to discriminate damage mechanisms at a molecular level.
The present study was devised with two major goals: 1) to determine the reactivity of individual oxygen species (02, H202, and ‘OH) with various mitochondrial inner membrane components, and 2) to provide the damage profile or pattern of each oxygen species which we and other investigators could utilize in the interpretation of experiments involving mixtures of O;, HzOz, and ‘OH. Our experiments center on the major bioenergetic protein complexes of the inner mitochondrial membrane, following exposure of submitochondrial prepara- tions (SMP) to known fluxes of 0; or ‘OH (generated by y radiolysis) or to H202.
MATERIALS AND METHODS
Exposure of Submitochondrial Preparations to Reactive Oxygen Species-SMP were prepared, as previously described (19) by soni- cation of “washed” mitochondria under an atmosphere of argon. After removal of unbroken mitochondria, SMP were recovered by two centrifugal washes at 100,000 x g (30 min) and suspended (20 mg of biuret protein/ml) in 50 mM pot&sium phosphate buffer (pH 7.4).
SMP were exnosed to ‘OH. 0;. or to mixtures of ‘OH + 0; bv “Co radiation 0; dilute (0.5 rng oiprotein/ml) solutions in 1.25mi potassium phosphate buffer (pH 7.4). Exposure conditions were ex- actly as described previously for purified proteins (26-29). Exposure to ’ OH alone was achieved by radiolysis of SMP solutions under an atmosphere of 100% NzO. Exposure to 0; alone was achieved by radiolysis of SMP solutions in 10 mM sodium formate, under an atmosphere of 100% oxygen. Exposure to (equimolar concentrations of) ‘OH + 0; was achieved by radiolysis of SMP solutions under an atmosphere of 100% oxygen. All radiolytic procedures were performed at 4 “C, at a dose rate of 400 f 5 rads/min (Frick dosimetry (37)), in a “Gammacel 200” 6oCo source (Atomic Energy of Canada, Ltd.). Radiolysis times were varied in order to achieve cumulative oxygen radical exposures of 60-600 nmol/mg of SMP protein (26-29).
Exposure to HZOZ was conducted (at 4 “C) with 0.5 mg of SMP protein/ml suspensions in 1.25 mM potassium phosphate buffer (pH 7.4). In each experiment bolus additions of HZ02 (quantified spectro- photometrically; cZ40 = 40 M-’ cm-‘) were made to the SMP suspen- sions. The length of incubation with 60-600 nmol of HZOZ/mg of SMP protein was varied to match exactly the radiolytic exposures described above. Following exposure, catalase (0.1 mg/ml) was added to remove any potential residual H202.
Enzyme Assays-NADH dehydrogenase activity was assayed spec- trophotometrically by the rate of NADH-dependent ferricyanide re- duction’at 420 nm, as described by Singer (38). Four separate ferri- cyanide concentrations (0.5-1.0 mM) were used to determine each V,,,., or KM data point. In one set of experiments (Table I) NADH dehydrogenase was assayed by the alternate method of 2,6-dichloro- phenolindophenol (DCPIP) reduction (38). Five DCPIP concentra- tions (20-80 pM) were used to determine each V,,,., or KM data point, by reduction of 37 pM DCPIP.
In each NADH dehydrogenase or succinate dehydrogenase assay, enzymatic velocities were extrapolated to infinite ferricyanide (or DCPIP), or phenazine methosulfate concentrations (as appropriate) by least-squares linear regression analysis of Lineweaver-Burk plots. V,,, values for NADH dehydrogenase activities are given as micro- mole of NADH .min-’ .rng-’ (1 mol of NADH reduces 2 mol of ferricyanide or 1 mol of DCPIP). Rates of DCPIP reduction by NADH dehydrogenase (Table I) were not converted to V,,,, values for NADH oxidation because DCPIP does not measure the full activity of the enzyme. V,,,., values for succinate dehydrogenase activities are given as nanomole of succinate 1 min-’ . ma-’ (1 mol of DCPIP is reduced as 1 mol of succinate is oxidized).
NADH oxidase, succinate oxidase, and cytochrome oxidase activi- ties were measured polarographically (6), using a Gilson “Oxygraph” fitted with a Clark-type electrode (Rank Bros., Bottisham, United Kingdom). All measurements were performed at 30 “C in 50 mM potassium phosphate buffer (pH 7.4) at an SMP concentration of 0.1 mg of protein/ml. Substrate concentrations, tested to produce maxi- mal oxidase activities, were: 0.2 mM NADH, 10 mM succinate; or 5 mM ascorbate plus 0.5 mM, N,N,N’,N’-tetramethyl-p-phenylenedi- amine (TMPD). Following incubation of SMP with NADH, succinate oxidase activity was measured in the presence of succinate + 0.2 pM rotenone. Finally, cytochrome oxidase activity was measured in the presence of rotenone, 0.4 jtM antimycin, ascorbate, and TMPD.
Cytochrome c oxidase activity was measured (in separate samples) spectrophotometrically (Shimadzu UV-3000, in the “dual wavelength mode”) by the oxidation of (added) 1.0 pM ferrocytochrome c at 550 and 540 nm (6 = 21 mM-’ cm-‘). Ferrocytochrome c was prepared by reduction of the oxidized protein with dithionite in deaerated buffer, followed by Sephadex G-25 chromatography.
ATPase activity was assayed by coupling the reaction to the pyruvate kinase and lactate dehydrogenase systems and measuring NADH oxidation at 340 nm (39). The assay system for ATPase activity contained 50 mM Tris acetate buffer (pH 7.4), 1 mM magne- sium chloride, 2 mM ATP, 2 mM phosphoenolpyruvate, 0.2 mM NADH, 15 pg/ml pyruvate kinase, and 5 fig/ml lactate dehydrogenase.
RESULTS
NADH Dehydrogenase Activity-NADH dehydrogenase ac- tivity, as measured by ferricyanide reduction, declined expo- nentially during exposure to ‘OH or ‘OH + 0; (Fig. lA). Maximal loss of activity (approximately 60%) was observed at 600 nmol of * OH/mg of SMP protein. A more modest decline in activity (approximately 40%) was observed with 0; exposure. At 600 nmol of radicals/mg of SMP protein, ‘OH, 02, and ‘OH + 0; all produced 50% decreases in the KM for ferricyanide, although the shapes of the three KM modification curves were quite different (Fig. 1B).
Our SMP exhibited relatively low peroxisomal contamina- tion, nevertheless a contaminating catalase activity (azide- inhibitable HzOz consumption) of 16.2 -C 3.3 nmol. min-’ . mg
5 > “---c-z? 0 200 4 o”o- 3000 nmol radicals or H202/ mg SMP protem
FIG. 1. Effect of oxygen radicals and Hz02 on NADH dehy- drogenase activity. SMP were exposed to the following oxygen radicals or H202: ‘OH (0); ‘OH + 0; (A, 50% ‘OH and 50% 0;); 0; (0); H202 (Cl). V,,,,, (Panel A) and KM (Panel B) values for NADH dehydrogenase were determined (38) by ferricyanide reduction (see “Materials and Methods”). All values are means & S.E. of three independent determinations.
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from
16332 Oxidative Inactivation of Mitochondrial Enzymes
protein-’ was measured. For this reason, the results in Fig. 1 (and all subsequent figures) have been extended to 3000 nmol of H202.mg of protein-‘, which completely accounts for the maximal Hz02 consumption by contaminating catalase. Ex- posure to H,O, caused only a 15% loss of NADH dehydrogen- ase activity at 3000 nmol/mg protein (Fig. L4). A 29% de- crease in KIM was observed at low H202 exposures, but higher H202 concentrations produced no further change (Fig. 1B).
The ferricyanide reduction assay is thought to measure the full activity of the NADH dehydrogenase complex (38). In contrast, the assay using DCPIP reduction measures a lower activity, which may reflect an alternate site of reduction in the complex (38). Nevertheless, assays of V,,, and KM for DCPIP (Table I) revealed essentially identical results to those seen with ferricyanide (Fig. l), except that no change in the KM for DCPIP was observed with 0: exposure, and Hz02 did not appear to affect DCPIP reduction at all (Table I).
NASH Ox&se Actiuity-NADH oxidase activity declined exponentially with ‘OH exposure, with a decrease of approx- imately 90% at 600 nmol of ‘OH/mg of SMP protein (Fig. 2). In contrast, 0; exposure produced an initial decline of some 40% (at 100 nmol of O;/mg of SMP) and no further decrease. Results with ‘OH + 0; exposure were intermediate between those of ‘OH alone and 0, alone. In contrast, Hz02 produced a 16% decrease in NADH oxidase activity at 600 nmol/mg of protein, and a 26% loss of activity at 3000 nmol/mg of protein (Fig. 2).
TABLE I NADH dehydrogenase activity measured
by the DCPIP reduction assay SMP were exposed to 600 nmol of oxygen radicals or HzOZ/mg of
protein exactly as described in the legend to Fig. 1. NADH dehydro- genase activity (V,., and KM) was measured by the reduction of DCPIP (38), using least-squares linear regression analysis of Line- weaver-Burke plots, as described under “Materials and Methods.”
Treatment v 0 “Iax K.Wb
None 955 * 19 88 + 5 ‘OH 393 k 13 48 f 6 02 689 f 80 80 f 6 ‘OH+O; 447 + 3 63 f 2 HAA 950 * 20 85 f 4
y V,,,., values (means + SE. of three independent determinations) are expressed as nanomole of NADH oxidized. min-’ . mg SMP pro- tein-‘.
*KM values (means f S.E. of three independent determinations) are expressed as micromolar of DCPIP necessary for one-half V,.. activity.
nmol radicals or H20Z/mg protem
FIG. 2. Effect of oxygen radicals and Hz02 on NADH oxi- dase activity. SMP were exposed to the following active oxygen species: ‘OH (0); ‘OH + 0; (A, 50% ‘OH and 50% 02); 0, (0); H20, (Cl). NADH oxidase was measured polarographically (6) after addition of 0.2 mM NADH (see “Materials and Methods”). All values are means rt SE. of three independent determinations.
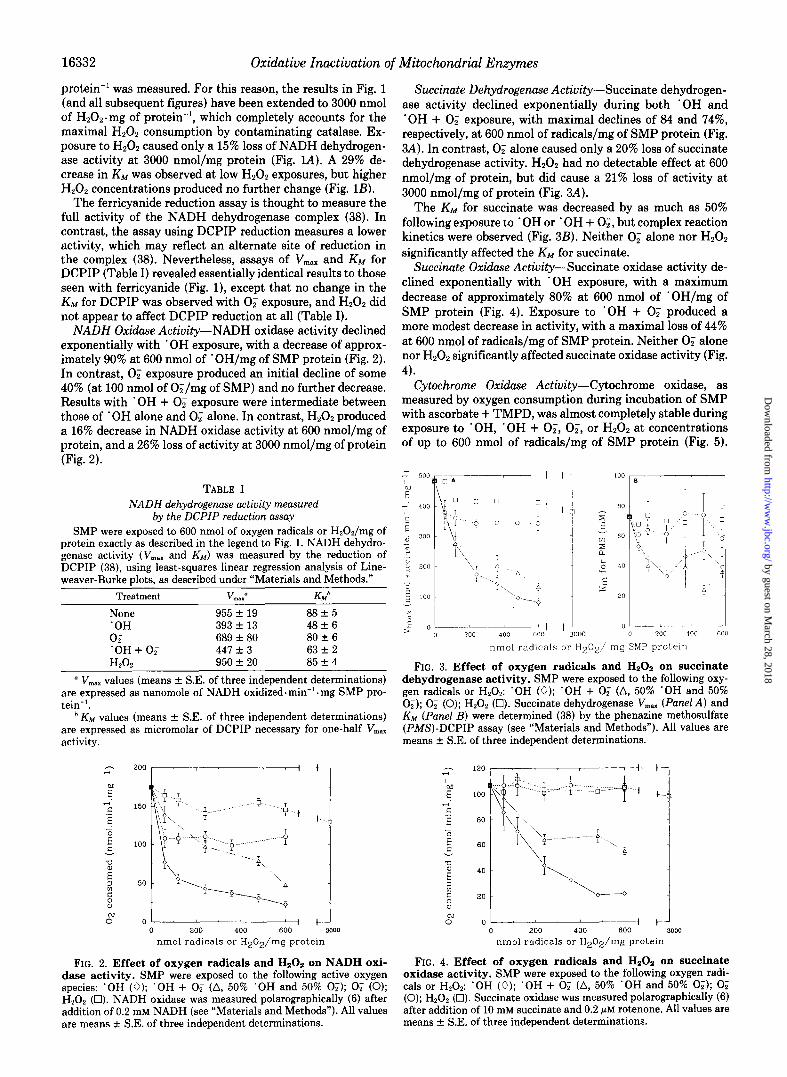
Succinnte Dehydrogenase Activity-Succinate dehydrogen- ase activity declined exponentially during both ‘OH and ‘OH + 0; exposure, with maximal declines of 84 and 74%, respectively, at 600 nmol of radicals/mg of SMP protein (Fig. 3A). In contrast, 0; alone caused only a 20% loss of succinate dehydrogenase activity. H,O, had no detectable effect at 600 nmol/mg of protein, but did cause a 21% loss of activity at 3000 nmol/mg of protein (Fig. 3A).
The Knn for succinate was decreased by as much as 50% following exposure to ‘OH or ‘OH + O;, but complex reaction kinetics were observed (Fig. 3B). Neither 0; alone nor H,O, significantly affected the KM for succinate.
Succinute Oxidase Activity-Succinate oxidase activity de- clined exponentially with ‘OH exposure, with a maximum decrease of approximately 80% at 600 nmol of ’ OH/mg of SMP protein (Fig. 4). Exposure to ‘OH + 0; produced a more modest decrease in activity, with a maximal loss of 44% at 600 nmol of radicals/mg of SMP protein. Neither 0; alone nor H202 significantly affected succinate oxidase activity (Fig. 4).
Cytochrome Oxidase Activity-Cytochrome oxidase, as measured by oxygen consumption during incubation of SMP with ascorbate + TMPD, was almost completely stable during exposure to ‘OH, ‘OH + O;, O;, or Hz02 at concentrations of up to 600 nmol of radicals/mg of SMP protein (Fig. 5).
1 l E 0 -----I--
> 0 200 JO0 6001 I 3000 II PO0 ‘100 600 nmol radkcnls or H202/ mg SMP protem
FIG. 3. Effect of oxygen radicals and HzOz on succinate dehydrogenase activity. SMP were exposed to the following oxy- gen radicals or H,O,: ‘OH (0); ‘OH + 0; (A, 50% ‘OH and 50% 0;); 0; (0); H202 (0). Succinate dehydrogenase V,., (Panel A) and KM (Panel B) were determined (38) by the phenazine methosulfate (PMS)-DCPIP assay (see “Materials and Methods”). All values are means rt S.E. of three independent determinations.
nmol radicals or HZOZ/mg protein
FIG. 4. Effect of oxygen radicals and Hz02 on succinate oxidase activity. SMP were exposed to the following oxygen radi- cals or H,O,: ‘OH (0); ‘OH + 0; (A, 50% ‘OH and 50% 0;); 0; (0); H,Oz (Cl). Succinate oxidase was measured polarographically (6) after addition of 10 mM succinate and 0.2 +M rotenone. All values are means f S.E. of three independent determinations.
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Oxidative Inactivation of Mitochondrial Enzymes
0” nmol radicals or H20Z/mg protem
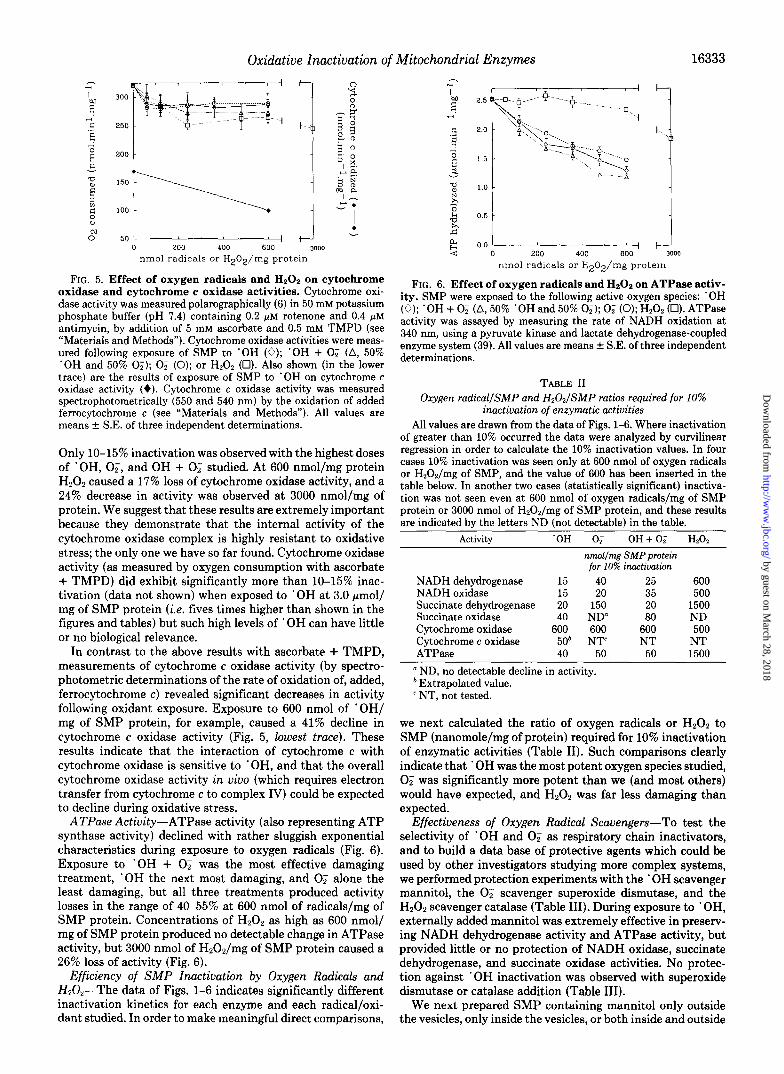
FIG. 5. Effect of oxygen radicals and Hz02 on cytochrome oxidase and cytochrome c oxidase activities. Cytochrome oxi- dase activity was measured polarographically (6) in 50 mM potassium phosphate buffer (pH 7.4) containing 0.2 pM rotenone and 0.4 pM antimycin, by addition of 5 mM ascorbate and 0.5 mM TMPD (see “Materials and Methods”). Cytochrome oxidase activities were meas- ured following exposure of SMP to ‘OH (0); ‘OH + 0; (A, 50% ‘OH and 50% 0;); 0; (0); or H202 (Cl). Also shown (in the lower trace) are the results of exposure of SMP to ‘OH on cytochrome c oxidase activity (+). Cytochrome c oxidase activity was measured spectrophotometrically (550 and 540 nm) by the oxidation of added ferrocytochrome c (see “Materials and Methods”). All values are means f SE. of three independent determinations.
Only IO-15% inactivation was observed with the highest doses of ‘OH, 02, and OH + 0; studied. At 600 nmol/mg protein H202 caused a 17% loss of cytochrome oxidase activity, and a 24% decrease in activity was observed at 3000 nmol/mg of protein. We suggest that these results are extremely important because they demonstrate that the internal activity of the cytochrome oxidase complex is highly resistant to oxidative stress; the only one we have so far found. Cytochrome oxidase activity (as measured by oxygen consumption with ascorbate + TMPD) did exhibit significantly more than lo-15% inac- tivation (data not shown) when exposed to ‘OH at 3.0 pmol/ mg of SMP protein (i.e. fives times higher than shown in the figures and tables) but such high levels of ’ OH can have little or no biological relevance.
In contrast to the above results with ascorbate + TMPD, measurements of cytochrome c oxidase activity (by spectro- photometric determinations of the rate of oxidation of, added, ferrocytochrome c) revealed significant decreases in activity following oxidant exposure. Exposure to 600 nmol of ‘OH/ mg of SMP protein, for example, caused a 41% decline in cytochrome c oxidase activity (Fig. 5, lowest truce). These results indicate that the interaction of cytochrome c with cytochrome oxidase is sensitive to *OH, and that the overall cytochrome oxidase activity in viva (which requires electron transfer from cytochrome c to complex IV) could be expected to decline during oxidative stress.
ATPase Actiuity-ATPase activity (also representing ATP synthase activity) declined with rather sluggish exponential characteristics during exposure to oxygen radicals (Fig. 6). Exposure to ‘OH + 0; was the most effective damaging treatment, ‘OH the next most damaging, and 0; alone the least damaging, but all three treatments produced activity losses in the range of 40-55% at 600 nmol of radicals/mg of SMP protein. Concentrations of H202 as high as 600 nmol/ mg of SMP protein produced no detectable change in ATPase activity, but 3000 nmol of H202/mg of SMP protein caused a 26% loss of activity (Fig. 6).
Efficiency of SMP Inactivation by Oxygen Radicals and I&O,--The data of Figs. 1-6 indicates significantly different inactivation kinetics for each enzyme and each radical/oxi- dant studied. In order to make meaningful direct comparisons,
4 1.0 } ,h E -d 0.5
2 t
-4 0 200 400 600 3000
nmol radicals or H202/mg protein
FIG. 6. Effect of oxygen radicals and Hz02 on ATPase activ- ity. SMP were exposed to the following active oxygen species: ‘OH (0); ‘OH + 0; (A, 50% ‘OH and 50% 0;); 0; (0); H202 (0). ATPase activity was assayed by measuring the rate of NADH oxidation at 340 nm, using a pyruvate kinase and lactate dehydrogenase-coupled enzyme system (39). All values are means f S.E. of three independent determinations.
16333
I
TABLE II Oxygen radical/SMP and H2021SMP ratios required for 10%
inactivation of enzymatic activities All values are drawn from the data of Figs. l-6. Where inactivation
of greater than 10% occurred the data were analyzed by curvilinear regression in order to calculate the 10% inactivation values. In four cases 10% inactivation was seen only at 600 nmol of oxygen radicals or H202/mg of SMP, and the value of 600 has been inserted in the table below. In another two cases (statistically significant) inactiva- tion was not seen even at 600 nmol of oxygen radicals/mg of SMP protein or 3000 nmol of H20,/mg of SMP protein, and these results are indicated by the letters ND (not detectable) in the table.
Activity ‘OH 0; OH+O; HZOZ
nmol/mg SMP protein for 10% inactivation
NADH dehydrogenase 15 40 25 600 NADH oxidase 15 20 35 500 Succinate dehydrogenase 20 150 20 1500 Succinate oxidase 40 ND 80 ND Cytochrome oxidase 600 600 600 500 Cytochrome c oxidase 50’ NT= NT NT ATPase 40 50 50 1500
a ND, no detectable decline in activity. * Extrapolated value. ’ NT, not tested.
we next calculated the ratio of oxygen radicals or HZ02 to SMP (nanomole/mg of protein) required for 10% inactivation of enzymatic activities (Table II). Such comparisons clearly indicate that ’ OH was the most potent oxygen species studied, 0; was significantly more potent than we (and most others) would have expected, and H202 was far less damaging than expected.
Effectiveness of Oxygen Radical Scavengers-To test the selectivity of ‘OH and 0; as respiratory chain inactivators, and to build a data base of protective agents which could be used by other investigators studying more complex systems, we performed protection experiments with the ‘OH scavenger mannitol, the 0; scavenger superoxide dismutase, and the H202 scavenger catalase (Table III). During exposure to ‘OH, externally added mannitol was extremely effective in preserv- ing NADH dehydrogenase activity and ATPase activity, but provided little or no protection of NADH oxidase, succinate dehydrogenase, and succinate oxidase activities. No protec- tion against ‘OH inactivation was observed with superoxide dismutase or catalase addition (Table III).
We next prepared SMP containing mannitol only outside the vesicles, only inside the vesicles, or both inside and outside
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16334 Oxidative Inactivation of Mitochondrial Enzymes
TABLE III Effects of mannitol, superoxide dismutase, and catalase on enzyme
inactivation by the hydroxyl radical SMP were exposed to 600 nmol of ‘OH/mg of protein in the
presence or absence of 10 mM mannitol, 100 mM mannitol, 100 rg/ ml superoxide dismutase (SOD), or 100 rg/ml catalase. NADH de- hydrogenase (V,,,,, with ferricyanide), succinate dehydrogenase (V,,,., with phenazine methosulfate), NADH oxidase, succinate oxidase, and ATPase activities were measured as described under “Materials and Methods.” Percent protection values were determined as follows: 1 - [ % inactivation with potential protector/% inactivation without po-
tential protector] [ lOO]. Inactivation data (in the absence of protective agents) are given in Figs. l-6. All values are means of at least three independent determinations for which S.E. was less than 10%.
Enzyme 10 rnM 100 activity Mannitol Matritol SOD Cat&se
% protection against ‘OH damage NADH dehydrogenase 64 87 0 0 NADH oxidase 0 12 0 0 Succinate dehydrogenase 13 8 14 4 Succinate oxidase 21 10 0 0 ATPase 81 93 8 6
TABLE IV Effects of external and internal mannitol on enzyme
inactivation by the hydroxyl radical SMP were exposed to 600 nmol of ‘OH/mg of protein, and assayed
for enzymatic activities as described in the legend to Table II. Expo- sure to ‘OH was conducted without mannitol, with externally added 100 mM mannitol, with SMP which had been prepared by sonication in the presence of 100 mM mannitol (Internal mannitol), or with both external and internal mannitol. Percent protection was calculated as described in the legend to Table III. Percent protection values are means of at least three independent determinations, for which S.E. was less than 10%.
Enzyme EXtHIA Internal External activitv mannitol mannitol + internal
NADH dehydrogenase NADH oxidase Succinate dehydrogenase Succinate oxidase ATPase
% protection against ‘OH damage 87 48 88 12 12 16
8 38 36 13 43 44 93 15 93
(Table IV). During exposure to ‘OH, mannitol provided much greater protection of NADH dehydrogenase activity when present outside than inside the SMP. NADH oxidase protec- tion was equally poor whether mannitol was added to the inside or to the outside of the SMP. Significant protection of succinate dehydrogenase and succinate oxidase activities was observed with mannitol on the inside, or on both sides of the membrane (Table IV). ATPase activity, however, was equally preserved by external mannitol and by mannitol on both sides of the membrane, but internal mannitol was ineffective.
The results of Table IV are at least fairly consistent with the known membrane topology of the SMP, and the reactivity of ‘OH in an aqueous phase, however, some rather surprising results were obtained. The NADH dehydrogenase active site (NADH oxidation) is on the external surface of SMP (i.e. the matrix side of the inner membrane) where mannitol provides greatest protection. The F1 portion of the ATPase complex is also on the external surface of SMP (i.e. the matrix side of the inner membrane) where it protrudes quite significantly into the aqueous phase, and where mannitol provided protec- tion. Unexplained, however, is the lack of protection of NADH oxidase by either internal or external mannitol, and the protection of succinate dehydrogenase and succinate oxidase afforded by internal mannitol (the succinate dehydrogenase active site is on the external surface of SMP).
We next tested the ability of (external) superoxide dismu- tase, catalase, and mannitol to protect against the respiratory chain inactivation exerted by 0; (Table V). Since NADH dehydrogenase, NADH oxidase, and ATPase activities were most affected by 0; (see Figs. l-6), the protection of only these three activities was tested in Table V. Active superoxide dismutase provided essentially complete protection against 0: damage, but heat-denatured superoxide dismutase was ineffective. Catalase provided minor protection in Table V, but this result may be explained by a combination of nonspe- cific protein scavenging (e.g. see results with denatured cata- lase and superoxide dismutase) and the fact that the heme group of catalase reacts with 0; (40). Mannitol, which does not scavenge 02, provided little or no protection of SMP activities (Table V). Thus, 0; inactivation appears to be both specific and preventable.
Dissociation of Enzyme Inactivation and Lipid Peroxida- tion-Mannitol failed to protect NADH oxidase activity, par- tially protected succinate oxidase activity, and completely protected ATPase activity during exposure to ‘OH (Tables III and IV). These results might indicate that lipid peroxida- tion, in the region of the respiratory chain between the dehydrogenases and cytochrome oxidase, was partially re- sponsible for the loss of oxidase activity. To test this possi- bility, we measured oxidase activities, ATPase activity, and lipid peroxidation in the presence and absence of the lipid- soluble antioxidants n-propyl gallate, trolox, vitamin E, BHT, and DPPD (Table VI).
Significant lipid peroxidation was, indeed, observed in un- protected samples and n-propyl gallate, trolox, vitamin E, BHT, and DPPD provided significant protection (Table VI). Despite inhibiting lipid peroxidation, however, these lipid- soluble antioxidants provided little or no protection of enzy- matic activities. In contrast, the water-soluble protein cyto- chrome c, at one-fifth the concentration of SMP protein (0.1 mg of cytochrome c/ml), protected enzyme activities without inhibiting lipid peroxidation. At twice the SMP protein con- centration (1.0 mg/ml cytochrome c) cytochrome c protected both enzyme activities and membrane lipids, presumably by scavenging essentially all the ‘OH produced (Table VI). Thus, lipid peroxidation, although significant during ‘OH exposure, does not appear to account for loss of enzyme activities.
DISCUSSION
Exposure to ’ OH produced significant inactivation (50% or more at 600 nmol of ’ OH/mg of SMP) of all SMP functions
TABLE V Effects of superoxide dismutase (SOD), catalase, and mannitol on
enzyme inactivation by the superoride anion radical SMP were exposed to 600 nmol of O;/mg of protein in the presence
or absence of active SOD (100 pg/ml), heat-denatured superoxide dismutase (100 rg/ml), active catalase (100 rg/ml), heat-denatured catalase (100 rg/ml), or externally added mannitol (10 or 100 mM). NADH dehydrogenase (V,,,., with ferricyanide), NADH oxidase, and ATPase activities were measured as described under “Materials and Methods.” Inactivation data (in the absence of protective agents) are reported in Figs. 1, 2, and 6. Percent protection values were deter- mined as follows: 1 - [% inactivation with potential protector/% protection without potential protector] [loo]. All values are means of at least three independent determinations for which SE. was less than 10%.
Enzyme SOD Denatured Catalase Denat”red Mannitol
activity SOD cata1ase 10 mM 100 mM
% protection against 0; damage NADH dehy- 100 17 25 20 7 0
drogenase NADH oxidase 101 10 25 19 16 13 ATPase 95 9 35 26 6 0
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Oxidative Inactivation of Mitochondrial Enzymes 16335
TABLE VI Dissociation of enzyme inactiuation and lipid peroxidation during
exposure to the hydroxyl radical SMP were untreated or were exposed to 600 nmol of ‘OH/mg of
protein in the presence or absence of various potential antioxidants. NADH oxidase activity (nanomole of 0, consumed. min-’ . mg of SMP protein-‘), succinate oiidase activity (nanomole 02 consumed. min-’ rnz of SMP nrotein-I). and ATPase activity (micromole of ATP h&olyzed . An-’ . mg of SMP protein-‘) were-measured as described under “Materials and Methods.” Lipid peroxidation (nanomole of malonyldialdehyde/mg of SMP protein) was measured by the thio- barhituric acid assay (with BHT added during the boiling step) described by Buege and Aust (41). Where used, n-propyl gallate and trolox were dissolved in distilled water (by sonication for trolox) and added to 5 mM final concentrations. Vitamin E, butylated hydroxy- toluene (BHT). and N.N’-dinhenvl-1.4-nhenvlenediamine (DPPD) _“.^_ were dissolved in dimethyl sulfoxide and added to 10 PM final con- centrations. Cytochrome c was added (in distilled water) to the final concentrations indicated. Appropriate solvent blanks were measured where necessary. All values are means of at least three independent determinations for which S.E. was less than 10%.
Treatments/additions NADH Succinate ATPase Lipid
oxidase oxidase peroxi- datinn
No ‘OH/no additions . OH treated/no additions ’ OH treated/n-propyl gallate ‘OH treated/trolox . OH treated/vitamin E
OH treated/BHT ‘OH treated/DPPD ’ OH treated/O.1 mg/ml cyto-
chrome c ‘OH treated/l.0 mg/ml cyto-
chrome c
178 110 2.6 0.4 19 20 1.1 6.8 18 24 1.4 0.4 59 19 1.2 0.7 18 19 1.0 1.5 17 21 0.9 1.2 20 21 1.2 1.1
108 70 2.1 6.4
115 82 2.3 1.8
studied, with the exception of cytochrome oxidase measured by the ascorbate + TMPD assay. Cytochrome c oxidase activ- ity (oxidation of added ferrocytochrome c) was inhibited 41% by exposure to 600 nmol of ’ OH/mg of SMP protein. Expo- sure to 0; alone caused 40% inactivations (at 600 nmol of OJmg of SMP) of NADH dehydrogenase, NADH oxidase, and ATPase activities, a 30% loss of succinate dehydrogenase activity, a 10% inactivation of cytchrome oxidase activity, and no significant, decrease in succinate oxidase activity. Exposure to ‘OH + 0; produced results which were inter- mediate between those of ’ OH alone and 0; alone, but which resembled ‘OH more than 0;. Hydrogen peroxide exerted relatively minor effects at concentrations up to 600 nmol of H202/mg of SMP; including a 13% loss of NADH dehydro- genase activity, a 16% loss of NADH oxidase activity, and a 17% loss of (ascorbate + TMPD dependent) cytochrome oxidase activity. At 3000 nmol of HPOn/mg of SMP protein we observed a 15% loss of NADH dehydrogenase, a 26% loss of NADH oxidase, a 21% loss of succinate dehydrogenase, a 24% loss of (ascorbate + TMPD dependent) cytochrome oxidase, and a 26% loss of ATPase activities. If we exclude (ascorbate + TMPD dependent) cytochrome oxidase from consideration, the ratio of radicals or Hz02 to SMP (nano- mole/mg protein) required for 10% inactivation of enzymatic activities was 15-40 for ‘OH, 20-150 for O;, 20-80 for ‘OH + O;, and 500 or more for H202 (Table II).
The aqueous ‘OH scavenger mannitol provided (statisti- cally) significant protection against ’ OH-induced inactivation of most SMP functions but did not protect against inactiva- tion during 0; exposure. In contrast, superoxide dismutase protected against inactivation by 0, but not ‘OH. Generalized lipid peroxidation, although evident with ‘OH exposure, did not appear to be responsible for enzyme inactivation since lipid-soluble antioxidants (n-propyl gallate, trolox, vitamin E,
BHT, and DPPD) inhibited malonyldialdehyde production without protecting enzymatic activities. Furthermore, the presence of low concentrations of cytochrome c (0.1 mg/ml) during ‘OH exposure protected enzyme activities without inhibiting lipid peroxidation, whereas higher concentrations of cytochrome c protected both enzymes and lipids.
The summary of inactivation kinetics provided in Table II, and the inhibitor profiles summarized above, allow for several useful conclusions. Although ’ OH was clearly the most potent oxygen species studied, 0; clearly exerts far more damaging effects on NADH dehydrogenase, NADH oxidase, and ATP- ase activities than was previously suspected. Such 02 me- diated damage is consistent with the proposal that superoxide dismutase protects respiratory chain components (from 0;) during the oxidation of NADH (42). In contrast, H202 alone was much less damaging than might have been expected (particularly when the high concentrations of potentially re- active heme groups and iron-sulfur proteins in SMP are considered).
The inability of mannitol to protect NADH oxidase activity during ’ OH exposure, despite significant protection of NADH dehydrogenase activity by mannitol (Tables III and IV), de- serves further mention. The active site of the NADH dehy- drogenase region of complex I lies on the external surface of SMP. Thus, it appears reasonable that mannitol provided greatest protection of NADH dehydrogenase (during ‘OH exposure) when added outside the SMP. Since NADH oxidase activity was essentially unaffected by mannitol addition, how- ever, it would appear that the oxidase activity was limited by another (oxidatively damaged) component of the respiratory chain. This (unknown) component could simply involve the transfer of electrons from complex I to ubiquinone, which is not measured by the ferricyanide reduction assay. The alter- native explanation that a more distal region of the electron transport chain may limit NADH oxidase activity would appear less likely, since succinate oxidase was (at, least par- tially) protected by internal mannitol (Table IV). Both NADH oxidase and succinate oxidase activities share the region of the respiratory chain between ubiquinone and cytochrome oxidase, and if this region were damaged, one would expect that the loss of both activities and the protection of both activities might be equivalent. Thus, a diminished ability to transfer electrons from complex I to ubiquinone (following ‘OH exposure) would appear to be a more reasonable expla- nation for the loss of NADH oxidase activity. In this regard it should be noted that reduced ubiquinone (ubiquinol) can provide protection against peroxidation of mitochondrial membranes (43). Indeed, ubiquinol has been suggested as an antioxidant of wide biological significance (44).
It should also be noted that the (partial) effectiveness of internal mannitol, against succinate dehydrogenase/oxidase inactivation, discussed above was a surprising result. The active site of succinate dehydrogenase (succinate binding) lies on the external surface of SMP. Thus, we had expected that external mannitol would have been more effective than inter- nal mannitol. The explanation of this phenomenon is beyond the scope of the present investigation, but may be related to the report that cytochrome bs6,, of complex II is a transmem- brane protein which may be required for electron transfer from complex II to ubiquinone (45); further study of this phenomenon is clearly indicated.
Generalized lipid peroxidation (malonyldialdehyde forma- tion) accompanied exposure of SMP to ‘OH, but inhibition of lipid peroxidation did not protect against enzyme inacti- vation. Thus, the inactivation of mitochondrial inner mem- brane enzymes by ‘OH does not appear to depend upon a
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Oxidative Inactivation of Mitochondrial Enzymes
generalized peroxidation of membrane lipid, in good agree- ment with the in uiuo results of Hillered and Ernster (8). A similar conclusion was also recently reached by Guilivi et al. (47) following exposure of SMP to singlet oxygen. We would, naturally, expect that extensive lipid peroxidation would eventually interfere with enzymatic activities, but such high levels of peroxidation often appear to follow, rather than to precede, the initial oxidative modification of membrane pro- teins (30). Thus, the measurement of lipid peroxidation (a simple and widely used tool in studies of oxidative stress) would appear to be of limited value in interpreting the oxi- dative inactivation of mitochondrial enzymes. The possibility of a specific peroxidation of cardiolipin in the membrane is not excluded by our studies. Indeed, the observation that (ascorbate + TMPD dependent) cytochrome oxidase activity was only mildly affected by ‘OH, whereas cytochrome c oxidase activity (oxidation of ferrocytochrome c) was 41% inactivated, may indicate that cardiolipin (which is required for the correct interaction of cytochrome c with cytochrome oxidase) was specifically oxidized. This interpretation pro- vides further support for the conclusions of a recent study by Soussi et al. (46) where it was proposed that cytochrome c oxidase activity is specifically inhibited by peroxidation of cardiolipin during (skeletal muscle) ischemia/reperfusion in- jury.
It is clear that the patterns of SMP enzyme inactivation by ‘OH and 0; are quite different, and *OH scavengers and superoxide dismutase allow for further discrimination of the effects of each oxygen radical. The use of such inactivation patterns and differential ‘OH/OF scavengers in more complex systems, such as intact mitochondria or even intact organs, now seems to be an entirely appropriate manner in which to identify mechanisms of oxidative mitochondrial inactivation.
Acknowledgment-We wish to thank Jane V. Radaza for prepara- tion of the manuscript.
1.
2. 3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
REFERENCES
Chance, B., Sies, H., and Boveris, A. (1979) Physiol. Reu. 59, 527-605
Boveris, A., and Chance, B. (1973) Biochem. J. 34,707-716 Boveris, A., Cadenas, E., and Stoppani, A. 0. M. (1976) Biochem.
J. 156,435-444 Davies, K. J. A., Doroshow, J. H., and Hochstein, P. (1983) FEBS
Z&t. 153,227-230 Doroshow, J. H., and Davies, K. J. A. (1987) Biochem. Pharmacol.
32,2935-2939 Davies, K. J. A., and Doroshow, J. H. (1986) J. Biol. Chem. 261,
3060-3067 Doroshow, J. H., and Davies, K. J. A. (1986) J. Biol. Chem. 261,
3068-3074 Hillered, L., and Ernster, L. (1983) J. Cereb. Blood Flow Metab.
3,207-214 Hillered, L., Ernster, L., and Arfors, K. E. (1984) in Cerebral
Ischemia (Bes, A., Braquet, P., Paoletti, R., and Siesjo, K., eds) pp. 283-299, Elsevier Science Publishing Co., New York
Erickson, G. A., and Koppenol, W. H. (1987) Znt. J. Radiat. Biol. 51,147-155
Ytrehus, K., Myklebust, R., Olsen, R., and Mjos, 0. D. (1987) J. Mol. Cell Cardiol. 19, 379-389
Quintanilha, A. T., Paker, L., Davies, J. M. S., Racanelli, T. L., and Davies, K. J. A. (1982) Ann. N. Y. Acad. Sci. 393, 32-47
13. Vladimirov, Y. A., Olenev, V. I., Suslova, T. B., and Cheremisina, Z. P. (1980) Adu. Lipid Res. 17, 173-249
14. Narabayashi, H., Takeshige, K., and Minakami, S. (1982) Biochem. J. 202.97-105
15. Aono, K., Shiraishi, N., Arita, T., Inouye, B., Nakazawa, T., and Utsumi, K. (1981) Physiol. Chem. Phys. 13, 137-144
16. Davies, K. J. A., Quintanilha, A. T., Brooks,.G. A., and Packer, L. (1982) Biochem. Biophvs. Res. Commun. 107. 1198-1205
17.
18.
19.
20.
21. 22.
23.
24.
25.
26. 27.
28.
29.
30.
31.
32. 33.
34.
35. 36.
37.
38.
39.
40. 41.
Rouach, H., Clement, M., Orfanelli, M. T., Janvier, B., Nord- mann, J., and Nordmann, R. (1983) Biochim. Biophys. Acta 753,439-444
Klimek, J., Schaap, A. P., and Kimura, T. (1983) Biochem. Biophys. Res. Commun. 110, 559-566
Marcillat, O., Zhang, Y., and Davies, K. J. A. (1989) Biochem. J. 259,181-189
Marcillat, O., Zhang, Y., Lin, S. W., and Davies, K. J. A. (1988) Biochem. J. 254, 677-683
Davies, K. J. A. (1986) J. Free Radicals Biol. & Med. 2, 155-173 Davies, K. J. A., and Lin, S. W. (1988) Free Radical Biol. & Med.
5,215-223 Davies, K. J. A., and Lin, S. W. (1988) Free Radical Biol. & Med.
5,225-236 Levine, R. L., Oliver, C. N., Fulks, R. M., and Stadtman, E. R.
(1981) Proc. N&l. Acad. Sci. U. S. A. 78. 2120-2124 Stadtman, E. R., and Wittenberger, M. E. (1985) Arch. Biochem.
Biophys. 239,379-387 Davies, K. J. A. (1987) J. Biol. Chem. 262,9895-9901 Davies, K. J. A., Delsignore, M. E., and Lin, S. W. (1987) J. Biol.
Chem. 262,9902-9907 Davies, K. J. A., and Delsignore, M. E. (1987) J. Biol. Chem.
262,9908-9913 Davies, K. J. A., Lin, S. W., and Pacitici, R. E. (1987) J. Biol.
Chem. 262,9914-9920 Davies, K. J. A., and Goldberg, A. L. (1987) J. Biol. Chem. 262,
8220-8266 Davies, K. J. A., and Goldberg, A. L. (1987) J. Biol. Chem. 262,
8227-8234 Rivett, A. J. (1985) Arch. Biochem. Biophys. 243,624-632 Davies, K. J. A., Wiese, A. G., Sevanian, A., and Kim, E. (1990)
in Molecular Biology of Aging (Finch, C. E., and Johnson, T. E., eds) pp. 123-141, Wiley-Liss, New York
Wolff, S. P.: Garner, A., andDean; R. T. (1986) Trends Biochem. Sci. 11,27-31
Wolff, S. P., and Dean, R. T. (1986) Biochem. J. 234,399-403 Dean, R. T., Thomas, S. M., and Garner, A. (1986) Biochem. J.
240,489-494 Frick, H., and Hart, E. J. (1966) in Radiation Dosimetry (Attix,
F. H., and Roesch, W. C., eds) p. 167, Academic Press, Orlando, FI,
Singer, T. P. (1974) in Methods of Biochemical Analysis (Glick, D., ed) pp. 123-175, John Wiley & Sons, New York
Pullman, M. E., Penefsky, H. S., Datta, A., and Racker, E. (1960) J. Biol. Chem. 235,3322-3329
Kono, Y., and Fridovich, I. (1982) J. Biol. Chem. 257,5751-5754 Buege, J. A., and Aust, S. D. (1978) Methods Enzymol. 52, 302-
310 42. Tyler, D. D. (1975) Biochim. Biophys. Acta 396, 335-346 43. Takayanagi, R., Takeshige, K., and Minakami, S. (1980) Biochem.
J. 192,853-860 44. Beyer, R. E., and Ernster, L. (1990) in Biochemistry, Bioenergetics
and Clinical Aspects of Ubiquinone (Lenaz, G., Barnabei, O., Rabbi, A., and Battino, M., eds) pp. 191-213, Taylor & Francis Ltd., London
45. Hatefi, Y.. and Galante, Y. M. (1980) J. Biol. Chem. 255. 5530- 5537
46. Soussi, B., Idstrom, J.-P., Schersten, T., and Bylund-Fellenius, A.-C. (1990) Acta Physiol. Stand. 138, 107-114
47. Giulivi, C., Sarcansky, M., Rosenfeld, E., and Boveris, A. (1990) Photochem. Photobiol. 52, 745-751
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Y Zhang, O Marcillat, C Giulivi, L Ernster and K J Daviesand ATPase.
The oxidative inactivation of mitochondrial electron transport chain components
1990, 265:16330-16336.J. Biol. Chem.
http://www.jbc.org/content/265/27/16330Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/265/27/16330.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from





























