Embed Size (px)
Citation preview

The oxidation of iodine to iodate by hydrogen peroxide
Guy Schmitz
des Sciences L ibre de Bruxelles, CP165, Av.F.Roosevelt 50,Faculte� Applique� es, Universite�1050 Bruxelles, Belgium. E-mail : gschmitz=ulb.ac.be
Received 20th July 2001, Accepted 3rd September 2001First published as an Advance Article on the web 3rd October 2001
The kinetics of the oxidation of iodine by hydrogen peroxide, a part of the BrayÈLiebhafsky reaction, has beenstudied between 298 and 323 K using a spectrophotometric method. The rate of reaction passes through amaximum when the concentration of hydrogen peroxide increases. At low hydrogen peroxide concentration,the order of reaction with respect to is roughly one, whereas at high hydrogen peroxide concentration the[I2]apparent order is higher and there is no simple kinetic law. This reaction can be observed only with sufficientacid and iodate concentrations. However, its rate does not change much with perchloric acid concentration inthe range 0.04 to 0.2 mol dm~3 and iodate concentration in the range 0.005 to 0.05 mol dm~3. The maximumÐrst order rate constant is 8 ] 10~3 s~1 at 298 K with an activation energy of 80 kJ mol~1. The results arediscussed considering a model proposed previously for the BrayÈLiebhafsky reaction.
1 IntroductionThe oxidation of iodine to iodate by hydrogen peroxide is apart of the BrayÈLiebhafsky1,2 (BL) reaction, that is thedecomposition (D) of hydrogen peroxide catalysed by iodateand iodine in acidic solutions.
2H2O2 ] 2H2O ] O2 (D)
As already proposed by Bray,1 it is recognised that this cataly-sis results from two reactions, the reduction (R) of iodate toiodine and the oxidation (O) of iodine to iodate.
2IO3~] 2H`] 5H2O2] I2] 5O2 ] 6H2O (R)
I2 ] 5H2O2 ] 2IO3~] 2H`] 4H2O (O)
The sum of reactions (R) and (O) gives reaction (D). Thesereactions are themselves complex and their mechanismsinvolve several intermediate species, especially iodide andhypoiodous acid.
We have shown3,4 that the kinetics of reaction (R) is closelyrelated to the kinetics of the Dushman reaction5
IO3~] 5I~] 6H`] 3I2] 3H2O (1)
Here we study the kinetics of reaction (O), the second maincomponent of the BL reaction. First, it is necessary to con-sider the experimental conditions under which this reactioncan be studied. The reactions between hydrogen peroxide andiodine depend on the composition of the solutions.2 The mainparameter is the acidity. In slightly acidic solutions, hydrogenperoxide does not oxidise iodine but reduces it :
I2 ] H2O2] 2I~] 2H`] O2 (2)
It can also oxidise iodide :
2I~] 2H`] H2O2] I2] 2H2O (3)
When the rates of these two reactions balance each other weget another catalytic pathway to the decomposition (D).Liebhafsky6 has studied these reactions and concluded thathydrogen peroxide does not react directly with iodine but withhypoiodous acid, a product of the hydrolysis (4).
I2 ] H2O H HIO ] I~] H` (4)
Thus, the reactions (2) and (3) should be written in the form
HIO ] H2O2 ] I~] H`] O2 ] H2O (5)
I~] H`] H2O2 ] HIO ] H2O (6)
Reaction (5) is important also during the BL reaction. On theother hand, the concentration [I~] is then so low that thereaction (6) can be neglected.
The oxidation (O) of iodine to iodate cannot take placeunless the acidity is sufficiently high, at least about 0.01 M. Atlower acidities we get only reactions (5) and (6). In order toobserve reaction (O), it is also necessary to have initially someiodate. Its minimal concentration depends on the acidity andthe hydrogen peroxide concentration and can be very small.The reaction can even start without added iodate, but, in thiscase, it is preceded by an induction period7 long enough topermit some iodate formation. A period of slow iodine con-centration decrease8 is also sometimes observed before thestart of reaction (O). These observations can be understoodnoting that the iodide concentration [I~] during reaction (O)is many orders of magnitude lower than during the hydrogenperoxide decomposition following the pathway (5) ] (6). Thus,reaction (O) can be observed only if the acid and iodate con-centrations are such that the iodide is quickly removed by theDushman reaction (1).
Liebhafsky has studied the kinetics of the reaction (O)between 0 and 50 ¡C.7,9 He has proposed a Ðrst order rate law
where the experimental rate constant[d[I2]/dt \ kexp [I2],depends on the hydrogen peroxide, acid and iodate con-kexpcentrations. This dependence cannot be expressed by a simplelaw. It is especially surprising that, although hydrogen peroxi-de is a reactant, when its concentration increases the ratedecreases. Our former results for this reaction10 were in agree-ment with those of Liebhafsky but uncertainties about somee†ects of the concentrations, and their explanation, remained.As understanding of the kinetics of reaction (O) is essential formodelling the BL reaction, we have undertaken a new detailedexamination.
DOI : 10.1039/b106505j Phys. Chem. Chem. Phys., 2001, 3, 4741È4746 4741
This journal is The Owner Societies 2001(
Dow
nloa
ded
by U
nive
rsity
of
Sout
h D
akot
a on
17/
04/2
013
15:0
9:18
. Pu
blis
hed
on 0
3 O
ctob
er 2
001
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B10
6505
JView Article Online / Journal Homepage / Table of Contents for this issue
2. ExperimentalThe iodine concentration was followed by spectrophotometryat 460 nm using either a conventional thermostatic quartz cellor a stopped-Ñow accessory RX.1000 from Applied Photo-physics. The concentration [I~] is so low during reaction (O)that there is no contribution from The temperature wasI3~.controlled to ^0.1 ¡C. One drive syringe contained an acidicsolution of iodine and iodate and the other a solution of
with the same acid concentration. Analytical gradeH2O2 KI and were used without further puri-KIO3 , HClO4 , H2O2Ðcation. The concentration was controlled by per-H2O2manganate titration and the concentration byHClO4carbonate titration. The iodine solutions were fresh preparedbefore each series of experiments. Two di†erent methods ofpreparation were used. Following the Ðrst, solid iodine wassublimed in a nitrogen Ñow and dissolved in deionized water.The concentration of the resulting solution was measured bytitration with standard and controlled spectro-Na2S2O3photometrically before each run. Following the second, theiodine was generated by adding KI to an acidic solution ofiodate and leaving reaction (1) to take place until completion.Its concentration, deduced from the initial concentration ofKI, was controlled spectrophotometrically, as well as theabsence of Both methods gave the same results.I3~.
3. Results and discussion
3.1. Analysis of the data
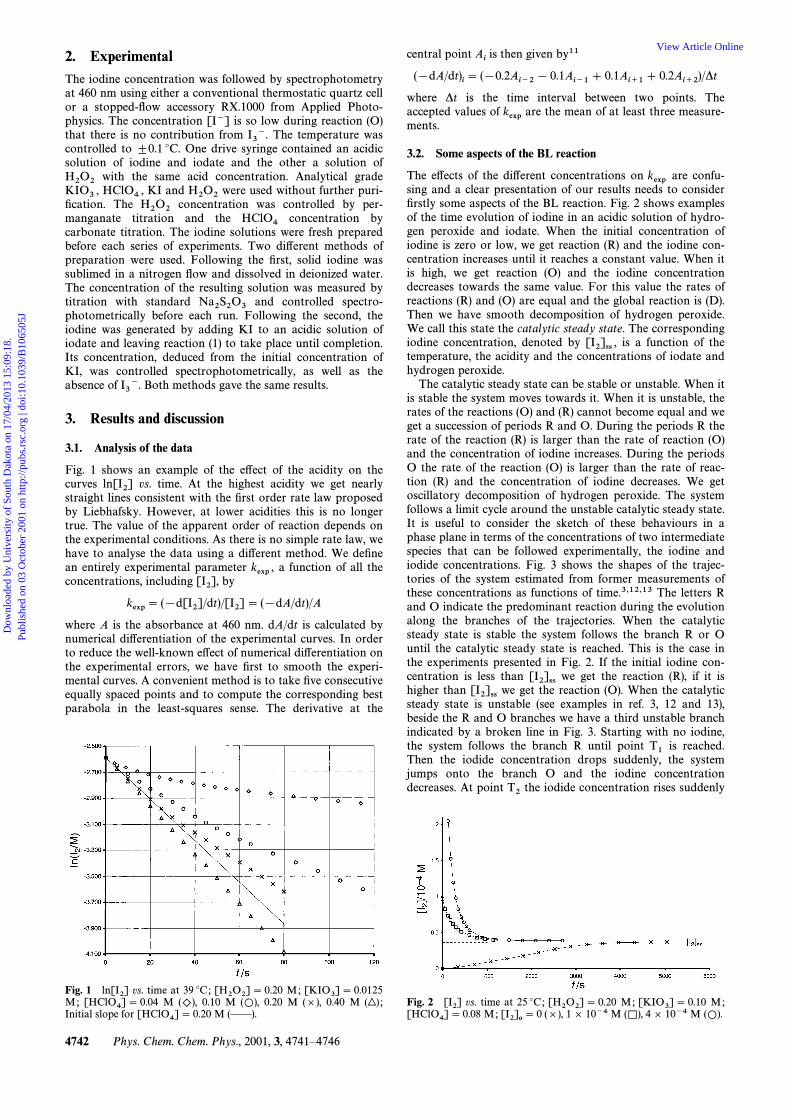
Fig. 1 shows an example of the e†ect of the acidity on thecurves vs. time. At the highest acidity we get nearlyln[I2]straight lines consistent with the Ðrst order rate law proposedby Liebhafsky. However, at lower acidities this is no longertrue. The value of the apparent order of reaction depends onthe experimental conditions. As there is no simple rate law, wehave to analyse the data using a di†erent method. We deÐnean entirely experimental parameter a function of all thekexp ,concentrations, including by[I2],
kexp\ ([d[I2]/dt)/[I2]\ ([dA/dt)/A
where A is the absorbance at 460 nm. dA/dt is calculated bynumerical di†erentiation of the experimental curves. In orderto reduce the well-known e†ect of numerical di†erentiation onthe experimental errors, we have Ðrst to smooth the experi-mental curves. A convenient method is to take Ðve consecutiveequally spaced points and to compute the corresponding bestparabola in the least-squares sense. The derivative at the
vs. time at 39 ¡C; M;Fig. 1 ln[I2] [H2O2]\ 0.20 [KIO3]\ 0.0125M; M 0.10 M 0.20 M (]), 0.40 M[HClO4]\ 0.04 ()), (L), (|) ;Initial slope for M (ÈÈ).[HClO4]\ 0.20
central point is then given by11Ai
([dA/dt)i\ ([0.2A
i~2 [ 0.1Ai~1 ] 0.1A
i`1] 0.2Ai`2)/*t
where *t is the time interval between two points. Theaccepted values of are the mean of at least three measure-kexpments.
3.2. Some aspects of the BL reaction
The e†ects of the di†erent concentrations on are confu-kexpsing and a clear presentation of our results needs to considerÐrstly some aspects of the BL reaction. Fig. 2 shows examplesof the time evolution of iodine in an acidic solution of hydro-gen peroxide and iodate. When the initial concentration ofiodine is zero or low, we get reaction (R) and the iodine con-centration increases until it reaches a constant value. When itis high, we get reaction (O) and the iodine concentrationdecreases towards the same value. For this value the rates ofreactions (R) and (O) are equal and the global reaction is (D).Then we have smooth decomposition of hydrogen peroxide.We call this state the catalytic steady state. The correspondingiodine concentration, denoted by is a function of the[I2]ss ,temperature, the acidity and the concentrations of iodate andhydrogen peroxide.
The catalytic steady state can be stable or unstable. When itis stable the system moves towards it. When it is unstable, therates of the reactions (O) and (R) cannot become equal and weget a succession of periods R and O. During the periods R therate of the reaction (R) is larger than the rate of reaction (O)and the concentration of iodine increases. During the periodsO the rate of the reaction (O) is larger than the rate of reac-tion (R) and the concentration of iodine decreases. We getoscillatory decomposition of hydrogen peroxide. The systemfollows a limit cycle around the unstable catalytic steady state.It is useful to consider the sketch of these behaviours in aphase plane in terms of the concentrations of two intermediatespecies that can be followed experimentally, the iodine andiodide concentrations. Fig. 3 shows the shapes of the trajec-tories of the system estimated from former measurements ofthese concentrations as functions of time.3,12,13 The letters Rand O indicate the predominant reaction during the evolutionalong the branches of the trajectories. When the catalyticsteady state is stable the system follows the branch R or Ountil the catalytic steady state is reached. This is the case inthe experiments presented in Fig. 2. If the initial iodine con-centration is less than we get the reaction (R), if it is[I2]sshigher than we get the reaction (O). When the catalytic[I2]sssteady state is unstable (see examples in ref. 3, 12 and 13),beside the R and O branches we have a third unstable branchindicated by a broken line in Fig. 3. Starting with no iodine,the system follows the branch R until point is reached.T1Then the iodide concentration drops suddenly, the systemjumps onto the branch O and the iodine concentrationdecreases. At point the iodide concentration rises suddenlyT2
vs. time at 25 ¡C; M; M;Fig. 2 [I2] [H2O2]\ 0.20 [KIO3]\ 0.10M; (]), 1] 10~4 M 4] 10~4 M[HClO4]\ 0.08 [I2]o \ 0 (K), (L).
4742 Phys. Chem. Chem. Phys., 2001, 3, 4741È4746
Dow
nloa
ded
by U
nive
rsity
of
Sout
h D
akot
a on
17/
04/2
013
15:0
9:18
. Pu
blis
hed
on 0
3 O
ctob
er 2
001
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B10
6505
J
View Article Online
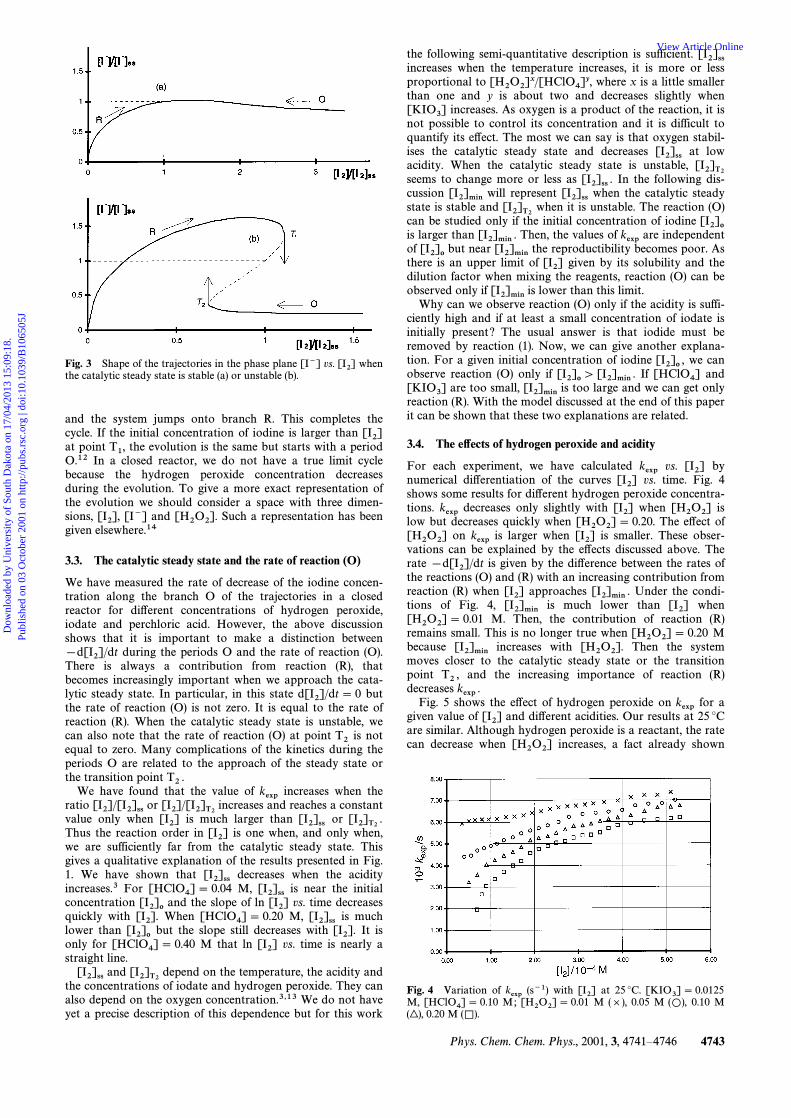
Fig. 3 Shape of the trajectories in the phase plane [I~] vs. when[I2]the catalytic steady state is stable (a) or unstable (b).
and the system jumps onto branch R. This completes thecycle. If the initial concentration of iodine is larger than [I2]at point the evolution is the same but starts with a periodT1,O.12 In a closed reactor, we do not have a true limit cyclebecause the hydrogen peroxide concentration decreasesduring the evolution. To give a more exact representation ofthe evolution we should consider a space with three dimen-sions, [I~] and Such a representation has been[I2], [H2O2].given elsewhere.14
3.3. The catalytic steady state and the rate of reaction (O)
We have measured the rate of decrease of the iodine concen-tration along the branch O of the trajectories in a closedreactor for di†erent concentrations of hydrogen peroxide,iodate and perchloric acid. However, the above discussionshows that it is important to make a distinction between
during the periods O and the rate of reaction (O).[d[I2]/dtThere is always a contribution from reaction (R), thatbecomes increasingly important when we approach the cata-lytic steady state. In particular, in this state butd[I2]/dt \ 0the rate of reaction (O) is not zero. It is equal to the rate ofreaction (R). When the catalytic steady state is unstable, wecan also note that the rate of reaction (O) at point is notT2equal to zero. Many complications of the kinetics during theperiods O are related to the approach of the steady state orthe transition point T2 .
We have found that the value of increases when thekexpratio or increases and reaches a constant[I2]/[I2]ss [I2]/[I2]T2value only when is much larger than or[I2] [I2]ss [I2]T2.
Thus the reaction order in is one when, and only when,[I2]we are sufficiently far from the catalytic steady state. Thisgives a qualitative explanation of the results presented in Fig.1. We have shown that decreases when the acidity[I2]ssincreases.3 For M, is near the initial[HClO4]\ 0.04 [I2]ssconcentration and the slope of ln vs. time decreases[I2]o [I2]quickly with When M, is much[I2]. [HClO4]\ 0.20 [I2]sslower than but the slope still decreases with It is[I2]o [I2].only for M that ln vs. time is nearly a[HClO4]\ 0.40 [I2]straight line.
and depend on the temperature, the acidity and[I2]ss [I2]T2the concentrations of iodate and hydrogen peroxide. They canalso depend on the oxygen concentration.3,13 We do not haveyet a precise description of this dependence but for this work
the following semi-quantitative description is sufficient. [I2]ssincreases when the temperature increases, it is more or lessproportional to where x is a little smaller[H2O2]x/[HClO4]y,than one and y is about two and decreases slightly when
increases. As oxygen is a product of the reaction, it is[KIO3]not possible to control its concentration and it is difficult toquantify its e†ect. The most we can say is that oxygen stabil-ises the catalytic steady state and decreases at low[I2]ssacidity. When the catalytic steady state is unstable, [I2]T2seems to change more or less as In the following dis-[I2]ss .cussion will represent when the catalytic steady[I2]min [I2]ssstate is stable and when it is unstable. The reaction (O)[I2]T2can be studied only if the initial concentration of iodine [I2]ois larger than Then, the values of are independent[I2]min . kexpof but near the reproductibility becomes poor. As[I2]o [I2]minthere is an upper limit of given by its solubility and the[I2]dilution factor when mixing the reagents, reaction (O) can beobserved only if is lower than this limit.[I2]minWhy can we observe reaction (O) only if the acidity is suffi-ciently high and if at least a small concentration of iodate isinitially present? The usual answer is that iodide must beremoved by reaction (1). Now, we can give another explana-tion. For a given initial concentration of iodine we can[I2]o ,observe reaction (O) only if If and[I2]o [ [I2]min . [HClO4]are too small, is too large and we can get only[KIO3] [I2]minreaction (R). With the model discussed at the end of this paperit can be shown that these two explanations are related.
3.4. The e†ects of hydrogen peroxide and acidity
For each experiment, we have calculated vs. bykexp [I2]numerical di†erentiation of the curves vs. time. Fig. 4[I2]shows some results for di†erent hydrogen peroxide concentra-tions. decreases only slightly with when iskexp [I2] [H2O2]low but decreases quickly when The e†ect of[H2O2]\ 0.20.
on is larger when is smaller. These obser-[H2O2] kexp [I2]vations can be explained by the e†ects discussed above. Therate is given by the di†erence between the rates of[d[I2]/dtthe reactions (O) and (R) with an increasing contribution fromreaction (R) when approaches Under the condi-[I2] [I2]min .tions of Fig. 4, is much lower than when[I2]min [I2]M. Then, the contribution of reaction (R)[H2O2]\ 0.01remains small. This is no longer true when M[H2O2]\ 0.20because increases with Then the system[I2]min [H2O2].moves closer to the catalytic steady state or the transitionpoint and the increasing importance of reaction (R)T2 ,decreases kexp .
Fig. 5 shows the e†ect of hydrogen peroxide on for akexpgiven value of and di†erent acidities. Our results at 25 ¡C[I2]are similar. Although hydrogen peroxide is a reactant, the ratecan decrease when increases, a fact already shown[H2O2]
Fig. 4 Variation of (s~1) with at 25 ¡C.kexp [I2] [KIO3]\ 0.0125M, M; M (]), 0.05 M 0.10 M[HClO4]\ 0.10 [H2O2]\ 0.01 (L),
0.20 M(|), (K).
Phys. Chem. Chem. Phys., 2001, 3, 4741È4746 4743
Dow
nloa
ded
by U
nive
rsity
of
Sout
h D
akot
a on
17/
04/2
013
15:0
9:18
. Pu
blis
hed
on 0
3 O
ctob
er 2
001
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B10
6505
J
View Article Online

Fig. 5 E†ect of on (s~1) at 39 ¡C. M;[H2O2] kexp [KIO3]\ 0.0125M and M (]), 0.10 M 0.20 M[I2]\ 4 ] 10~4 [HClO4]\ 0.04 (L),
0.40 M M and M (]) ;(|), ()) ; [I2]\ 2 ] 10~4 [HClO4]\ 0.04or 4 ] 10~4 M gives the same values of when[I2]\ 2 ] 10~4 kexpM.[HClO4]\ 0.40
earlier.7,9,10 Of course, the rate must be zero if [H2O2]\ 0and the real situation is that passes through a maximumkexpwhen increases. If M, the maximum[H2O2] [HClO4]\ 0.40of can be seen when is between 0.02 and 0.05 M.kexp [H2O2]At lower acidities the maximum is obtained for lower [H2O2]values but cannot be located exactly because we cannotmeasure for very low The stoichiometry of reac-kexp [H2O2].tion (O) shows that, in order to have an excess of we[H2O2],must take [H2O2]o [ 5 [I2]o .
Fig. 2 and 3, and the above discussion about the e†ect ofthe approach to the catalytic steady state or the transitionpoint give a qualitative explanation of these results. The e†ectof depends on the acidity because increases[H2O2] [I2]minwhen the acidity decreases. When M,[HClO4]\ 0.40 [I2]minis much lower than The rate of reaction (O) increases[I2].with and seems to reach a limiting maximum value.[H2O2]The slow decrease observed at high can be explained[H2O2]by a small contribution from reaction (R). This e†ect is morepronounced for and 0.10 M. For[HClO4]\ 0.20
M, is still larger and decreases[HClO4]\ 0.04 [I2]min kexpquickly when increases. Near the catalytic steady[H2O2]state, the reproducibility becomes poor and the spectro-photometric measurements can be disturbed by oxygenbubbles. Then, the main stoichiometry is no longer (O) but is(D).
The e†ect of the acidity is di†erent at low or high hydrogenperoxide concentrations. In Fig. 5, we see that at low [H2O2]seems to be independent of except for a smallkexp [HClO4],diminution at the highest acidity, whereas at high [H2O2] kexpincreases with At low we are far from the[HClO4]. [H2O2]catalytic steady state or the transition point and is[d[I2]/dtnear to the rate of the reaction (O), which seems to be inde-pendent of the acidity. At high is higher and[H2O2], [I2]minapproaches when decreases. When the value of[I2] [HClO4]the ratio decreases to nearly one, decreases[I2]/[I2]min kexpquickly.
3.5. Other e†ects
Here we discuss brieÑy the e†ects of iodate, oxygen and tem-perature. Although iodate is a product of the reaction, kexpincreases slightly when increases. According to the[KIO3]results of Fig. 6 it tends to a maximum value. If we assumethat the rate of the isolated reaction (O) is independent of
this observation can be explained, remembering that[KIO3]decreases when increases. The e†ect of reac-[I2]min [KIO3]tion (R) becomes less important at high However, we[KIO3].suspect that things are more complicated and we need toknow more precisely the e†ect of on Some[KIO3] [I2]min .complications come also from the relative weakness of iodic
Fig. 6 E†ect of and on (s~1) at 25 ¡C.[HClO4] [KIO3] kexp [I2]\ 4] 10~4 M; M and M (]), 0.10 M[H2O2]\ 0.01 [HClO4]\ 0.04
0.20 M M and M(L), (|) ; [H2O2]\ 0.02 [HClO4]\ 0.10 (K).
acid and the di†erences between and [H`] and[HClO4]between and[KIO3] [IO3~].15Series of experiments were performed to check the e†ect of
oxygen. In one group of experiments, the solutions were satu-rated with oxygen. In the other group, solutions with the sameconcentrations were prepared under nitrogen. In most cases,they gave identical results. However, oxygen has an e†ect atlow acidities and high Then it increases the rate of[H2O2].reaction and, when the initial concentration of iodine is closeto solutions saturated with oxygen give reaction (O)[I2]min ,while solutions prepared under nitrogen give reaction (R).
As is a complicated function of the concentrations,kexpthere is no real activation energy related to it. However wecan estimate a maximum value of for M.kexp [I2]\ 4 ] 10~4This gives s~1 at 25 ¡C and 0.03 s~1 at(kexp)max \ 8 ] 10~339 ¡C. Liebhafsky found 5 ] 10~4 s~1 at 0 ¡C7 and 0.12 s~1 at50 ¡C.9 A plot of vs. 1/T gives a straight line with aln(kexp)maxslope of 80 kJ mol~1. This value is in agreement with thatobtained in the same range of acidities by a di†erent method(77 kJ mol~1).16
4. A model for the BL reaction
4.1. The simpliÐed model
The model we have proposed3,17 for the BL reaction can beused for simulations with more or less detail. The main formsare the general model, the simpliÐed model and the Braylator.The general model includes three groups of reactions : thereactions of iodine and its compounds constituting the mecha-nism of the Dushman reaction, the reactions of hydrogen per-oxide with these iodine compounds and the reactions ofoxygen. It includes many reactions with unknown rate con-stants and we think it is useless to try to adjust them to agiven set of experimental results. It is well known that a goodÐt is not a good proof when the number of adjustable param-eters is large. This is why we have proposed a simpliÐedmodel, including only the reactions we consider essential tomodel the BL oscillations. It gives a semi-quantitative expla-nation of most experimental facts. Further work shouldimprove the model by adding reactions only when theirimportance is demonstrated. On the basis of a stoichiometricnetwork analysis,18,19 we have also constructed the Braylator.This is the most simple form of the model keeping its essentialfeatures. Although oversimpliÐed, it is of theoretical interestbecause it shows clearly the origin of the BL oscillations.20
The simpliÐed model consists of reactions (M1)È(M6). It isderived from the general model keeping only two reactions ofhydrogen peroxide, one reduction (M5) and one oxidation(M6) and contracting the Ðrst steps of the Dushman reactioninto one pseudo-elementary step (M1). Actually, orH2I2O3 ,its anhydride is an intermediate compound in this reac-I2O2 ,tion.4,21 In order to explain qualitatively the e†ect of oxygen,
4744 Phys. Chem. Chem. Phys., 2001, 3, 4741È4746
Dow
nloa
ded
by U
nive
rsity
of
Sout
h D
akot
a on
17/
04/2
013
15:0
9:18
. Pu
blis
hed
on 0
3 O
ctob
er 2
001
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B10
6505
J
View Article Online

we have added to the simpliÐed model the global reaction(M7)3,13 with an empirical rate law is anr7\ k7 (O2)(I~). k7apparent rate constant, an unknown function of (H`), (IO3~)and (H2O2).
IO3~] I~] 2H`H HIO ] HIO2 (M1)
HIO2 ] I~] H`] I2O ] H2O (M2)
I2O ] H2O H 2HIO (M3)
HIO ] I~] H`H I2] H2O (M4)
HIO ] H2O2 ] I~] H`] O2 ] H2O (M5)
I2O ] H2O2 ] HIO ] HIO2 (M6)
I~] H`] 12O2] HIO (M7)
Numerical simulations have been published showing thatthe model can explain the main features of the BLoscillations3,22h25 and of the reaction (O).10 This reaction isthe result of the combination 2(M [ 1)] 3(M2)] 2(M [ 3)] (M[ 4)] 5(M6). Hydrogen peroxide acts asan oxidant in reaction (M6). As it oxidises produced byI2Oreaction (M [ 3), the rate of (M6) is proportional to [HIO].2On the other hand, hydrogen peroxide acts as a reductant inreaction (M5) with a rate proportional to [HIO]. The key ofthe model is the competition between the oxidation and thereduction of HIO with di†erent orders of reaction.13,20 Theglobal result is an oxidation only if [HIO] is sufficiently large.With the quasi-equilibrium (M4) we see that it is equivalent tosay that [I~] must be sufficiently low, as observed. The directoxidation of I~ by hydrogen peroxide is negligible in the con-ditions of the BL reaction.26,27 I~ can be removed by reaction(M1) at the start of reaction (O) but, during this reaction, wehave another pathway for the iodide removal, the sum of reac-tions (M2) and (M6) giving H2O2] I~] H`] HIO] H2O.
4.2. The catalytic steady state
At the catalytic steady state, the only global reaction is (D)and the rates of production and consumption of all the iodinecompounds are equal. This gives the following relationsbetween the rates of the steps (Mi).r
ir1\ r3\ r4\ 0 r2 \ r6 r5 \ r6 ] r7 .
Introducing the expressions for these rates, we have obtained3the expression
[I2]ss\k4
k~4
C k3 k5k~3 k6
D2C k~1k5[H2O2]k1k2[IO3~][H`]
D1@2 1
a75(7)
where is a parameter function of the oxygen concentration.a7The simpliÐed model explains the increase of when[I2]ssincreases and when or [H`] decrease.[H2O2] [IO3~]
4.3. The limiting rate law
The simplest explanation of the rate law [d[I2]/dt \ kexpwhere is a complex function of the concentrations[I2], kexpbut reaching a maximum value, is based on the rate ofreaction (M[ 4). With [d[I2]/dt \ k~4[I2]this maximum would be reached when[ k4[HIO][I~][H`],
[HIO][I~][H`] with However thek~4[I2]A k4 kexp \ k~4 .value of has been measured directly28,29 and is largerk~4than Thus this explanation cannot be correct. The sim-kexp .pliÐed model gives another explanation and an analyticalanalysis of it is useful. Let us consider the model under condi-tions very far from the catalytic steady state when [I~] is verylow and [HIO] very large. Then, we can neglect the com-ponent from left to right of reaction (M1), neglect the rate ofreaction (M5) compared to (M6) and write the model asfollows.
I2 ] H2O H HIO ] I~] H` (M[ 4)
2HIO H I2O ] H2O (M [ 3)
I2O ] H2O2 ] HIO ] HIO2 (M6)
HIO2 ] I~] H`] I2O ] H2O (M2)
HIO ] HIO2] IO3~] I~] 2H` (M[ 1)
The quasi-steady state approximation for the intermediatespecies gives the relations
12k~3[HIO]2 [ 12k3[I2O]\ 15k6[I2O][H2O2]
\ 13k2[HIO2][I~][H`]
\ 12k~1[HIO][HIO2]Reaction (M4) is at quasi-equilibrium during our experiments :
k~4[I2]\ k4[HIO][I~][H`]
From these relations and [d[I2]/dt \ 12d[IO3~]/dt \ 12r~1,we get
[d[I2]
dt\
k2 k~3 k~43k~1k4
2k6[H2O2]5k3] 2k6[H2O2]
[I2]
This is exactly the form of the rate law observed very far fromthe catalytic steady state, Ðrst order in with a maximum[I2]value when increases. Closer to the catalytic steady[H2O2]state, the rate is lower because the rate of reaction (M5) andthe reversibility of reaction (M [ 1) cannot be neglected.Then, there is no simple rate law.
ConclusionsThe kinetics of the oxidation of iodine by hydrogen peroxideis complex and is the result of two kinds of e†ects : the kineticsof reaction (O) very far from the catalytic steady state and theapproach to this state. Very far from the catalytic steady state,when hydrogen peroxide acts only as an oxidant, the kineticsis relatively simple. The reaction is order one with respect toiodine and its rate increases with the hydrogen peroxide con-centration up to a limiting value. The acidity and the iodateconcentration have no, or a very small, e†ect. The complexitycomes out in conditions where hydrogen peroxide acts also asa reductant, especially when we cannot neglect the well knownreaction In theseHIO] H2O2 ] I~] H`] O2] H2O.conditions, there is no simple relation between the rate andthe ratio but all the factors that decrease this[I2]/[I2]minratio, decrease the rate. The experimental parameter kexpdecreases when decreases and when increases. It[I2] [H2O2]increases slightly with the iodate concentration and increasesor decreases with the acidity, depending on the other concen-trations. Oxygen has no e†ect on the rate of reaction exceptnear the catalytic steady state, where it promotes reaction (O)and increases its rate.
We have discussed these results in terms of our model forthe BrayÈLiebhafsky reaction. It explains the results very farfrom the catalytic steady state. The simpliÐed model (M1)È(M7) gives also a semi-quantative explanation of the otherresults but appears to be an oversimpliÐcation. On the otherhand, the general model includes too many reactions withunknown rate constants. Our aim is to Ðnd a model includingall the important reactions, but no more, based on di†erentkinds of experiments, not only on the shape of the BL oscil-lations. Before improving our earlier numerical simulations,we also need more independent information about the rateconstants. This work gives such information.
AcknowledgementThe author thanks Professor Ljiljana Kolar-Anic for veryuseful discussions during the writing of this paper.
Phys. Chem. Chem. Phys., 2001, 3, 4741È4746 4745
Dow
nloa
ded
by U
nive
rsity
of
Sout
h D
akot
a on
17/
04/2
013
15:0
9:18
. Pu
blis
hed
on 0
3 O
ctob
er 2
001
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B10
6505
J
View Article Online

References1 W. C. Bray, J. Am. Chem. Soc., 1921, 43, 1262.2 W. C. Bray and H. A. Liebhafsky, J. Am. Chem. Soc., 1931, 53, 38.3 G. Schmitz, J. Chim. Phys., 1987, 84, 957.4 G. Schmitz, Phys. Chem. Chem. Phys., 1999, 1, 1909.5 S. Dushman, J. Phys. Chem., 1904, 8, 453.6 H. A. Liebhafsky, J. Am. Chem. Soc., 1932, 54, 1792 ; H. A. Lieb-
hafsky, J. Am. Chem. Soc., 1932, 54, 3499.7 H. A. Liebhafsky, J. Am. Chem. Soc., 1931, 53, 2074.8 L. Cantrel and J. Chopin, Proceedings of the 4th CSNI W orkshop
on Iodine Chemistry in Reactor Safety, Switzerland,Wu� renlingen,1996, pp. 187È211.
9 H. A. Liebhafsky, W. C. McGavock, R. J. Reyers, G. M. Roe andL. S. Wu, J. Am. Chem. Soc., 1978, 100, 87.
10 (a) M. Radenkovic, G. Schmitz and Lj. Kolar-Anic, J. Serb.Chem. Soc., 1997, 62, 367 ; (b) M. Radenkovic, G. Schmitz and L.Kolar-Anic, Phys. ChemÏ98, 4th International Conference on Fun-damental and Applied Aspects of Physical Chemistry, Society ofPhysical Chemists of Serbia, Belgrade, 1998, p. 195 ; (c) M.Radenkovic, Thesis, University of Belgrade, 1996.
11 F. Scheid, Numerical Analysis, Mcgraw-Hill, New York, 1986, ch.21.
12 H. A. Liebhafsky and L. S. Wu, J. Am. Chem. Soc., 1974, 96, 7180.13 G. Schmitz, Phys. Chem. Chem. Phys., 1999, 1, 4605.14 G. Schmitz, Lj. Kolar-Anic, S. Anic and Z. Cupic, J. Chem. Educ.,
2000, 77, 1502.
15 Z. Cupic, S. Anic and Dj. Misljenovic, J. Serb. Chem. Soc., 1996,61, 893.
16 J. Ciric, S. Anic, Z. Cupic and Lj. Kolar-Anic, Sci. Sintering, 2000,32, 187.
17 G. Schmitz, J. Chim. Phys., 1974, 71, 689.18 B. L. Clarke, in Advances in Chemical Physics, ed. I. Prigogine
and S. A. Rice, Wiley, New York, 1980, vol. XLIII, p. 1.19 B. L. Clarke, Cell Biophys., 1988, 12, 237.20 G. Schmitz, J. Chim. Phys., 1991, 88, 15.21 J. A. Agreda, R. J. Field and N. J. Lyons, J. Phys. Chem. A, 2000,
104, 5269.22 Lj. Kolar-Anic and G. Schmitz, J. Chem. Soc., Faraday T rans.,
1992, 88, 2343.23 Lj. Kolar-Anic, Z. Cupic, S. Anic and G. Schmitz, J. Chem. Soc.,
Faraday T rans., 1997, 93, 2147.24 R. Vilcu, T. Danciu and D. Bala, Discrete Dyn. Nat. Soc., 1998, 2,
195 ; R. Vilcu, T. Danciu and D. Bala, Discrete Dyn. Nat. Soc.,2000, 4, 55.
25 K. Kissimonova, I. Valent, L. Adamcikova and P. Sevcik, Chem.Phys. L ett., 2001, 341, 345.
26 H. A. Liebhafsky and A. Mohammad, J. Am. Chem. Soc., 1933,55, 3977.
27 C. Arias, F. Mata and J. F. Perez-Benito, Can. J. Chem., 1990, 68,1499.
28 M. Eigen and K. Kustin, J. Am. Chem. Soc., 1962, 84, 1355.29 I. Lengyel, I. R. Epstein and K. Kustin, Inorg. Chem., 1993, 32,
5880.
4746 Phys. Chem. Chem. Phys., 2001, 3, 4741È4746
Dow
nloa
ded
by U
nive
rsity
of
Sout
h D
akot
a on
17/
04/2
013
15:0
9:18
. Pu
blis
hed
on 0
3 O
ctob
er 2
001
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B10
6505
J
View Article Online





























