Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1992 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 267, No. 10, Issue of April 5, pp. 7048-7052,1992
Printed in U.S.A.
Inhibition of Tissue Kallikrein by Protein C Inhibitor EVIDENCE FOR IDENTITY OF PROTEIN C INHIBITOR WITH THE KALLIKREIN BINDING PROTEIN*
(Received for publication, July 16, 1991)
Sonja Ecke$, Margarethe Geiger$#, Irene Resch$, Ingrid Jerabek$, Laura Stinglll, Manfred Maier$, and Bernd R. Binder$ From the $Laboratory for Clinical Experimental Physwlogy, Department of Medical Physiology, University of Vienna, Schwarzspanierstrasse 17, A-1090 Vienna and YTechnoclone Ltd., Vienna A-1090, Austria
We studied the inhibition of tissue kallikrein by pro- tein C inhibitor (PCI), a relatively unspecific heparin- dependent serine protease inhibitor present in plasma and urine. PC1 inhibited the amidolytic activity (cleav- age of H-D-vdyl-L-leucyl-arginine-p-nitroaniline) of urinary kallikrein with an apparent second order rate constant of 2.3 X lo4 M" s" and formed stable com- plexes (85 kDa) with urinary kallikrein as judged from silver-stained sodium dodecyl sulfate-polyacrylamide gels. Complex formation was time-dependent and was paralleled by a decrease in the intensity of the main PC1 protein band (M. = 57,000) and an increase in the intensity of the lower M, (54,000) PC1 form (cleaved inhibitor). Heparin interfered with the inhibition of tissue kallikrein by PC1 and with the formation of tissue kallikrein-PC1 complexes in a dose-dependent fashion and completely abolished PCI-tissue kallikrein interaction at 300 pg/ml. This is in contrast to findings on the interaction of PC1 with all other target proteases studied so far (i.e. stimulation of inhibition by heparin) but is similar to the reaction pattern of '261-labeled tissue kallikrein with so called kallikrein binding pro- tein described in serum and other systems. To study a possible relationship between PC1 and this kallikrein binding protein we incubated '261-labeled urinary kal- likrein in serum and in PCI-immunodepleted serum in the absence and presence of heparin and analyzed com- plex formation using sodium dodecyl sulfate-poly- acrylamide gel electrophoresis. In normal serum, formed complexes co-migrated with complexes of pu- rified PC1 and '261-kallikrein and were less intense in the presence of heparin. No complex formation at all was seen in PCI-depleted serum. Our data indicate that PC1 may be a physiologically important endogenous inhibitor of tissue kallikrein and provide evidence that PC1 may be identical to the previously described kal- likrein binding protein.
Tissue/glandular kallikreins are trypsin-like serine pro- teases that have been isolated from several tissues, including kidney, salivary glands, and pancreas (1-6). They are also present in the exocrine secretions of the respective organs (2,
* This work was supported in part by Grants P7579-M and P8108- M from the Austrian Funds for the Promotion of Scientific Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
8 To whom correspondence should be addressed Laboratory for Clinical Experimental Physiology, Dept. of Medical Physiology, Uni- versity of Vienna, Schwarzspanierstr. 17, A-1090 Vienna, Austria, Tel.: 43-1-40480 (ext. 251); Fax: 43-1-4087500.
6, 7). The primary physiological function of tissue kallikreins seems to be kinin release from high and low M, kininogens by the cleavage of two peptide bonds within the kininogen molecules (2). Kinins have a variety of species and tissue- specific biological activities, including vasodilation and con- traction of visceral smooth muscles (2). In addition to kinin generation, kallikrein may also be involved in renin release (8) and angiotensin I1 generation (9-11). Therefore, the reg- ulation of kallikrein activity seems to be of major physiological importance. So far, no efficient endogenous inhibitor of tissue kallikrein has been identified. The only proteinase inhibitor exhibiting some inhibitory activity toward tissue kallikrein is al-protease inhibitor (12). However, the interaction of tissue kallikrein with al-protease inhibitor is slow, with a half-life of several hours (12, 13). Recently, a tissue kallikrein binding protein has been described in several biological systems in- cluding serum (13-15). This binding protein interacts with tissue kallikrein by forming SDS'-stable complexes with a M, of 92,000. Complex formation is only observed with enzymat- ically active kallikrein (14) and is, at least in human systems, abolished in the presence of heparin (13-15). The identity of this kallikrein binding protein has not yet been defined. However, its reaction pattern suggests that it may be a serine protease inhibitor with a M, of approximately 50,000 and with affinity for heparin.
Protein C inhibitor (PCI; M , = 57,000) is a relatively nonspecific heparin-dependent serine protease inhibitor (ser- pin) that inhibits a variety of proteases (16-18). The presence of PC1 has been described in plasma (05 pg/ml) (16) and urine (19-21), and the urinary concentration is higher than expected from plasma filtration (0200 ng/ml, 21). So far, PC1 has mainly been studied with respect to its interaction with enzymes involved in coagulation and fibrinolysis (16-23). It has been shown that PC1 inhibits activated protein C, throm- bin, factor Xa, factor XIa, plasma kallikrein, and urokinase by forming SDS-stable complexes (16-18). The reaction of PC1 with its target proteases is stimulated by heparin (16- 18) and other glycosaminoglycans (24). Although activated protein C-PC1 complexes have been shown in plasma samples from patients with disseminated intravascular coagulation (25) and urokinase-PC1 complexes in plasma samples from patients receiving exogenous urokinase therapy (26), the physiological role of PC1 is still unclear. The presence of PC1 in urine combined with the fact that urine is a major source
The abbreviations used are: SDS, sodium dodecyl sulfate; H-D- Val-Leu-Arg-pNA (S-2266), H-o-valyl-L-leucyl-arginine-p-nitroani- line-&hydrochloride; <Glu-Pro-Arg-pNA (S-2366), a-pyroglutamyl- L-prolyl-L-arginine-p-nitroanilide-hydrochloride; IgG, immunoglob- ulin G; PAGE, polyacrylamide gel electrophoresis; PCI, protein C inhibitor.
7048

Protein C InhibitorlKallikrein Binding Protein 7049
of tissue kallikrein prompted us to investigate the possible inhibition of tissue kallikrein by PCI.
EXPERIMENTAL PROCEDURES
Materials-Sepharose 4B, activated CH-Sepharose 4B, and hepa- rin-Sepharose CL-GB (Pharmacia, Sweden); CNBr (Merck, Ger- many); heparin sodium salt ( ~ 1 4 0 units/mg; Fluka AG, Switzerland); acrylamide, bisacrylamide, SDS, and calibration proteins for SDS- PAGE (Bio-Rad); H-D-Val-Leu-Arg-pNA (S-2266) and <Glu-Pro- Arg-pNA (S-2366) (Kabi, Sweden); and activated protein C (Techn- oclone GmbH, Austria) were obtained as indicated. A monoclonal antibody against protein C inhibitor (4PCI) was obtained by immu- nizing Balb/c mice with purified urinary PC1 (24). Immunization of mice, fusion of mouse spleen cells, and isolation of single anti-PC1 IgG secreting clones were performed as described previously (27), except that X63-Ag8.653 mouse myeloma cells were used as fusion partners. One clone producing anti PCI-IgG (4PCI) was selected and injected intraperitoneally into Pristan-primed Balb/c mice. Ascitic fluid was collected and 4PCI-IgG was isolated by ammonium sulfate precipitation (50% saturation) of the ascites fluid. The precipitate was dissolved in and dialyzed against 0.1 M NaHC03 and coupled to CNBr-activated Sepharose 4B according to the manufacturer's in- structions at a concentration of 1 mg/ml Sepharose. Aprotinin was coupled to activated CH-Sepharose 4B as described (7). Urinary kallikrein was either purified as described previously (7) or obtained commercially from Protogen AG, Switzerland. '''1 labeling of urinary kallikrein (Protogen AG) was performed by the Iodogen method (28). After radiolabeling, active lZ5I-kallikrein was purified by affinity chromatography on aprotinin-Sepharose (7). The '251-kallikrein used had a specific activity of 9.6 pCi/nmol. PCI-immunodepleted serum was prepared by passing whole blood serum (4 ml) over a 4PCI IgG- Sepharose 4B column (5 ml) equilibrated in 0.01 M Tris-HC1, 0.1 M NaCl, 0.01% Tween 80, pH 7.4. The first flow-through fraction (protein content 2 mg/ml) was used as PC1 immunodepleted serum. Control serum (protein content 2.5 mg/ml) was prepared in the same way but a Sepharose 4B column was used instead of 4PCI IgG- Sepharose. PCI-immunodepleted serum contained less than 1% of PC1 antigen present in control serum as determined by enzyme- linked immunosorbent assay using 4PCI IgG as catching antibody and peroxidase-labelled immunopurified rabbit anti-PC1 IgG as de- tecting antibody (lower detection limit = 3 ng/ml).'
Purification of Urinary Protein C Inhibitor-The PC1 purification protocol described previously (24) was modified in such a way that affinity chromatography on 4PCI-IgG Sepharose 4B was used as a first step and that affinity chromatography on anti-urokinase-IgG Sepharose and ion exchange chromatography on Q-Sepharose fast flow were omitted. Briefly, freshly voided human urine (21) was collected on benzamidine (10 mM final concentration) and aprotinin (100 KIU/ml final concentration), adjusted to a pH between 7.0 and 7.4 by addition of 1 M NaOH and incubated, and end-over-end rotated overnight with 4PCI-IgG-Sepharose 4B (40 ml settled volume) equil- ibrated in 0.01 M Tris-HC1, 1.0 M NaC1, 0.01% Tween 80, 10 mM benzamidine, 100 KIU/ml aprotinin, pH 7.4. Unbound material was separated from the Sepharose gel by means of a sintered glass filter. The 4PCI-IgG Sepharose was washed extensively with 0.01 M Tris- HC1, 1.0 M NaCl, 0.01% Tween 80, 10 mM benzamidine, 100 KIU/ml aprotinin, pH 9.0, on the same filter. Thereafter the gel suspension was filled into a column and washed with 0.01 M Tris-HC1, 1.0 M NaCl, 0.01% Tween 80, pH 9.0, until the Azso was below 0.01. Elution was performed with 0.05 M triethylamine buffer, pH 11.5, containing 0.5 M NaC1. Fractions (3-4 ml each) were collected into tubes con- taining 1 ml of 0.5 M Tris-HC1, pH 7.5, to neutralize pH immediately. Protein containing fractions (Azs0 2 0.03) eluted at pH 11.5 were pooled and stored at -70 "C until further use. Eluates obtained in this way from three to five 4PCI-IgG-Sepharose steps were pooled, dialyzed extensively against 0.05 M Tris-HC1, 0.2 M NaCl, 0.01% Tween 80, pH 7.4, and subjected to heparin-Sepharose CL-GB chro- matography as described previously (24). Elution of the heparin- Sepharose column was performed with a linear gradient between 0.2 and 0.8 M NaCl in 0.05 M Tris-HC1, 0.01% Tween 80, pH 7.4, and fractions (2.5 ml) were collected. Fractions were screened for protein (Am) and inhibition of activated protein C (24). PC1 activity co- eluted with a protein peak at 0.35 M NaC1. PCI-containing fractions were pooled and stored in aliquots a t -70 "C until further use. PC1
'1. Resch, P. Hufnagl, M. Geiger, B. R. Binder, manuscript in preparation.
purified by this procedure was homogeneous and migrated on silver- stained SDS-PAGE gels as a doublet with a major band corresponding to M , of 57,000 and a minor band ( M , = 54,000). The yield of this purification procedure was =lo0 pg PCI/liter urine as compared with =35 pg PCI/liter urine obtained with the previously described puri- fication protocol (24).
Analytical Methods-Molar concentrations of purified active kal- likrein were evaluated from the cleavage of S-2266 using the change in absorbance at 405 nm/min and cm (AA,05/min/cm) of 2 .75/p~ enzyme as given by the manufacturer. The concentration of purified urinary PC1 was determined from its absorbance at 280 nm using a Ai;$1 of 14.1 (16), M, of 57,000 was used to calculate molar concen- trations; SDS-PAGE (10% acrylamide) was performed according to Laemmli (29) using 1.5-mm slab gels. After electrophoresis gels were stained by the silver stain method (30). Each lane of the silver-stained gels was scanned densitometrically at 523 nm using a Hirschmann Elscript 400 densitometer and peaks corresponding to the stained protein bands were obtained. The relative intensity of each stained band, represented by the size of the area of the corresponding peak, was expressed as percent of the total intensity of all stained bands present in a lane. Gels containing lZ5I-kallikrein were fixed with acetic acid/methanol/water (10:50:40), dried, and exposed to Kodak X- Omat AR films.
Amidolytic Assay for Inhibition of Tissue Kallikrein Actiuity by PCI-Inhibition of the amidolytic activity of purified urinary kalli- krein by purified urinary PC1 was tested on 96-well microtiter plates. Urinary kallikrein (3 nM final concentration) was incubated without or with PC1 (30 nM final concentration) in the absence or presence of different concentrations of heparin (0.03-300 pg/ml final concen- tration) at 37 "c in 50 pl of 0.01 M Tris-HC1, 0.1 M NaCl, 0.01% Tween 80, pH 7.4. After 30 min, 50 p1 of S-2266 (0.4 mM final concentration) dissolved in 0.05 M Tris-HC1, pH 8.3, was added to each well and after additional incubation for 3 h at 37 "C, the A405
was determined in an enzyme-linked immunosorbent assay reader (Antos reader 2001).
Kinetic Analysis of Tissue Kallikrein Inhibition by PCZ-Urinary kallikrein (4 nM final concentration) was incubated without or with different concentrations of PC1 (30, 15, and 7.5 nM final concentra- tion) in 40 p1 of 0.01 M Tris-HC1, 0.1 M NaC1, 0.01% Tween 80, pH 7.4, in wells of a 96-well microtiter plate. After different time periods (0, 5, 10, 20,40, and 80 min) heparin (10 pl, 3 mg/ml final concentra- tion) was added to each well in order to stop the enzyme-inhibitor interaction. After stopping the last wells (80 min) 50 p1 of S-2266 (final concentration 0.4 mM) dissolved in 0.05 M Tris-HC1, pH 8.3, was added to each well and the amidolytic activity was measured as described above. Kallikrein activity [E] was determined for each PC1 concentration and each incubation time period and plotted either as [E,]/[EO] uersus incubation time or as ln[Et]/[Eo] uersus incubation time, [Eo] representing the initial amidolytic activity of kallikrein obtained for each PC1 concentration by adding heparin at time ( t ) = 0, ix. before addition of PCI, [E,] representing the remaining enzy- matic activity after stopping the reaction at time = t. Pseudo first order rate constants were calculated as the negative slopes of the initial linear parts of the ln[Et]/[Eo] uersus time plots. The apparent second order rate constant (k,) was obtained by dividing the pseudo first order rate constants by the molar concentrations of PC1 used.
Bioassay for Kallikrein Actiuity-For control purposes, kinin gen- eration by urinary kallikrein in the absence or presence of PC1 and in the absence or presence of heparin was tested using the rat uterus bioassay system (31). Urinary kallikrein (2.3 nM final concentration) was incubated at 37 "C with PC1 (230 nM final concentration) in the absence or presence of heparin (0.05-500 pg/ml final concentration) in a total volume of 30 pl of 0.01 M Tris-HC1, 0.1 M NaC1, 0.01% Tween 80, pH 7.4. After 2 h a 5-pl aliquot of this reaction mixture was transferred to 100 pl of heat-inactivated human plasma as a source of kininogens (31). After a 5-min incubation at 37 "C, the mixture was applied to the rat uterus bioassay system and kinin generated was quantified using known bradykinin standards.
Complex Formation of Urinary Kallikrein with PCI-Purified uri- nary kallikrein (7.7 nM final concentration) was incubated with purified urinary PC1 (200 nM final concentration) in the absence or presence of heparin (0.03-300 pg/ml final concentration) in a total volume of 70 pl of 0.01 M Tris-HC1,O.l M NaCl, 0.01% Tween 80, pH 7.4. Incubations were stopped at various times (0-4 h) by the addition of 35 p1 of 0.26 M Tris-HC1 buffer, pH 6.8, containing 2.5% SDS and 25% glycerol and heating the samples in a boiling water bath for 10 min. Thereafter the samples were applied to SDS-PAGE gels (10% acrylamide) prepared as described above.
7050 Protein C InhibitorlKallikrein Binding Protein Complex Formation of 1z51-Urino~ Kallikrein in Serum and PCI-
immunodepleted Serum-"z61-Labeled urinary kallikrein (4.5 nM, 6000 cpm) was incubated with control serum (3 pl), with PCI- immunodepleted serum (3 pl), or with purified PC1 (270 nM final concentration) in the absence or presence of heparin (3 and 300 pg/ ml final concentrations) in a total volume of 63 pl 0.01 M Tris-HC1, 0.1 M NaC1, 0.01% Tween 80, pH 7.4. After 1 h the reactions were stopped by the addition of 40 pl 0.26 M Tris-HC1 buffer, pH 6.8, containing 2.5% SDS and 25% glycerol and heating the samples in a boiling water bath for 10 min. SDS-PAGE and autoradiography were done as described above.
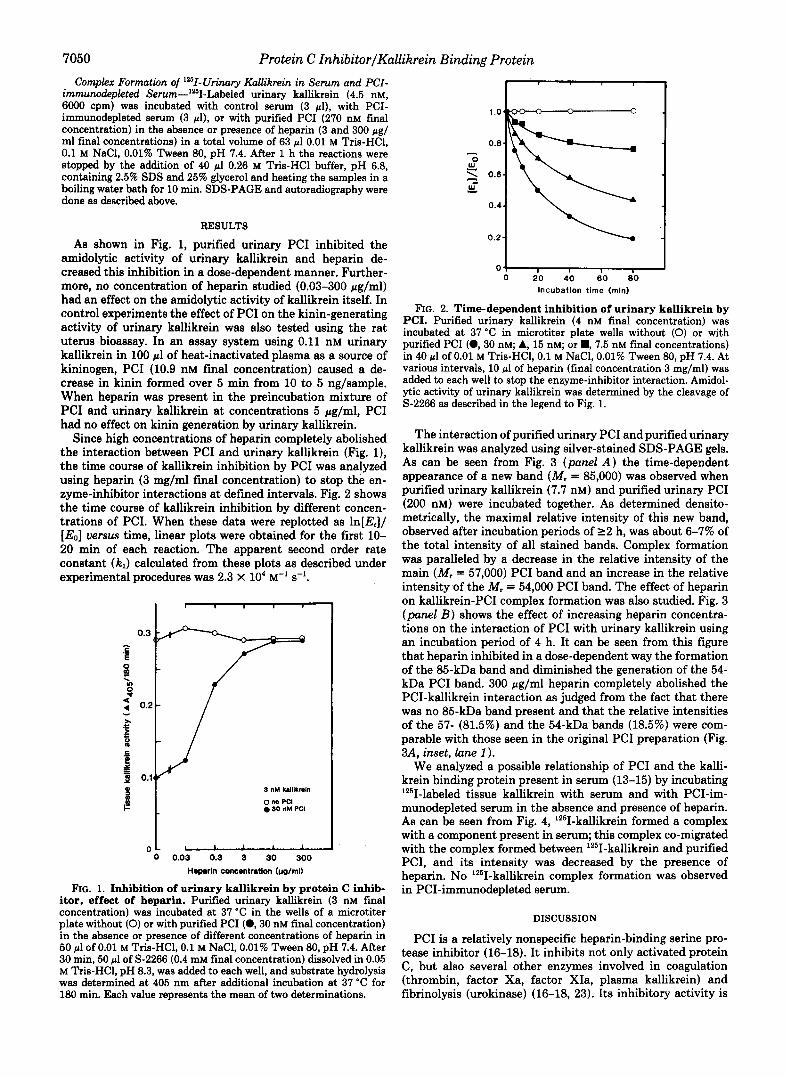
RESULTS As shown in Fig. 1, purified urinary PC1 inhibited the
amidolytic activity of urinary kallikrein and heparin de- creased this inhibition in a dose-dependent manner. Further- more, no concentration of heparin studied (0.03-300 pg/ml) had an effect on the amidolytic activity of kallikrein itself. In control experiments the effect of PC1 on the kinin-generating activity of urinary kallikrein was also tested using the rat uterus bioassay. In an assay system using 0.11 nM urinary kallikrein in 100 pl of heat-inactivated plasma as a source of kininogen, PC1 (10.9 nM final concentration) caused a de- crease in kinin formed over 5 min from 10 to 5 ng/sample. When heparin was present in the preincubation mixture of PC1 and urinary kallikrein at concentrations 5 pg/ml, PC1 had no effect on kinin generation by urinary kallikrein.
Since high concentrations of heparin completely abolished the interaction between PC1 and urinary kallikrein (Fig. l), the time course of kallikrein inhibition by PC1 was.analyzed using heparin (3 mg/ml final concentration) to stop the en- zyme-inhibitor interactions at defined intervals. Fig. 2 shows the time course of kallikrein inhibition by different concen- trations of PCI. When these data were replotted as ln[E,]/ [E0] uersus time, linear plots were obtained for the first 10- 20 min of each reaction. The apparent second order rate constant (kJ calculated from these plots as described under experimental procedures was 2.3 X lo4 M-' s-'.
I I I I
3 nM kalllkmln
o no PCI 0 30 nM PC1
I I I I
0.03 0.3 3 30 300 ~oparh concentratbn bolml)
FIG. 1. Inhibition of urinary kallikrein by protein C inhib- itor, effect of heparin. Purified urinary kallikrein (3 nM final concentration) was incubated at 37 "C in the wells of a microtiter plate without (0) or with purified PC1 (0 ,30 nM final concentration) in the absence or presence of different concentrations of heparin in 50 pl of 0.01 M Tris-HC1,O.l M NaCl, 0.01% Tween 80, pH 7.4. After 30 min, 50 pl of S-2266 (0.4 mM final concentration) dissolved in 0.05 M Tris-HC1, pH 8.3, was added to each well, and substrate hydrolysis was determined at 405 nm after additional incubation at 37 "C for 180 min. Each value represents the mean of two determinations.
I 1.0(
0.8- c
w 0 z 0.6- w- L
0.4.
0.2-
01 I I I 0 20 40 60 80
Incubation time (rnin)
FIG. 2. Time-dependent inhibition of urinary kallikrein by PCI. Purified urinary kallikrein (4 nM final concentration) was incubated at 37 "C in microtiter plate wells without (0) or with purified PC1 ( 0 , 3 0 nM; A, 15 nM; or ., 7.5 nM final concentrations) in 40 pl of 0.01 M Tris-HC1, 0.1 M NaCl, 0.01% Tween 80, pH 7.4. At various intervals, 10 pl of heparin (final concentration 3 mg/ml) was added to each well to stop the enzyme-inhibitor interaction. Amidol- ytic activity of urinary kallikrein was determined by the cleavage of S-2266 as described in the legend to Fig. 1.
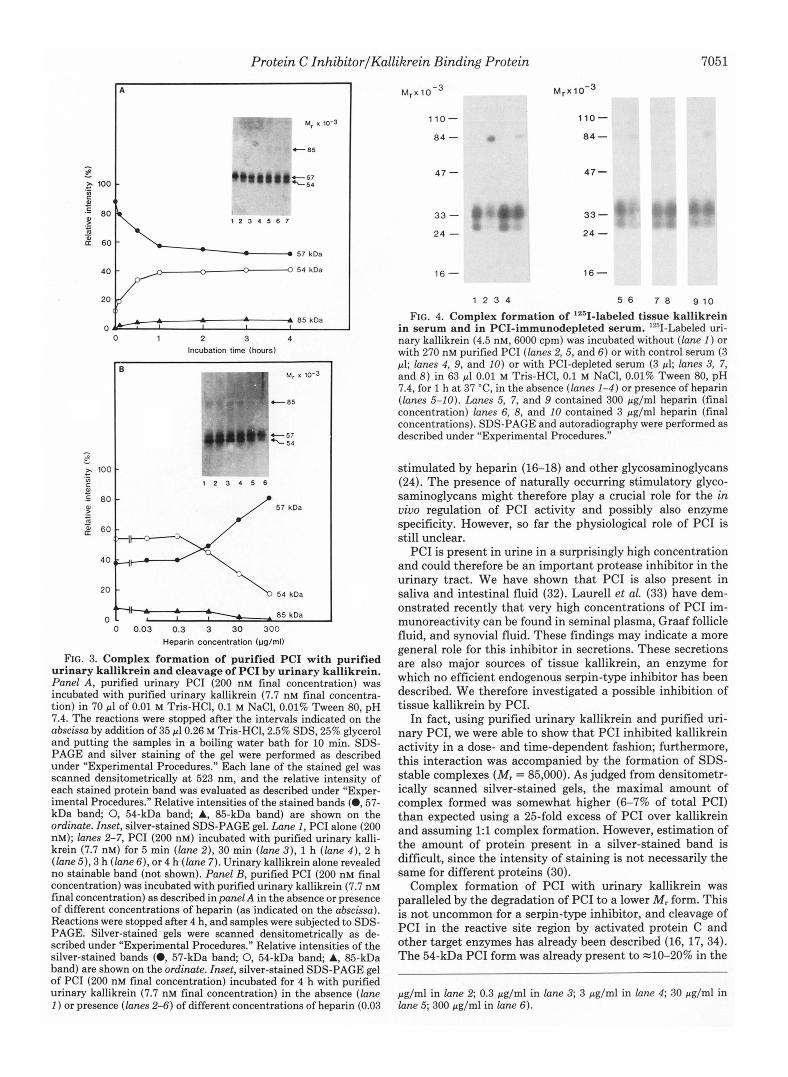
The interaction of purified urinary PC1 and purified urinary kallikrein was analyzed using silver-stained SDS-PAGE gels. As can be seen from Fig. 3 (panel A) the time-dependent appearance of a new band (Mr = 85,000) was observed when purified urinary kallikrein (7.7 nM) and purified urinary PC1 (200 nM) were incubated together. As determined densito- metrically, the maximal relative intensity of this new band, observed after incubation periods of 2 2 h, was about 6-7% of the total intensity of all stained bands. Complex formation was paralleled by a decrease in the relative intensity of the main (MI = 57,000) PC1 band and an increase in the relative intensity,of the MI = 54,000 PC1 band. The effect of heparin on kallikrein-PC1 complex formation was also studied. Fig. 3 (panel B ) shows the effect of increasing heparin concentra- tions on the interaction of PC1 with urinary kallikrein using an incubation period of 4 h. It can be seen from this figure that heparin inhibited in a dose-dependent way the formation of the 85-kDa band and diminished the generation of the 54- kDa PC1 band. 300 pg/ml heparin completely abolished the PCI-kallikrein interaction as judged from the fact that there was no 85-kDa band present and that the relative intensities of the 57- (81.5%) and the 54-kDa bands (18.5%) were com- parable with those seen in the original PC1 preparation (Fig. 3A, inset, lane 1 ).
We analyzed a possible relationship of PC1 and the kalli- krein binding protein present in serum (13-15) by incubating lZ5I-labeled tissue kallikrein with serum and with PCI-im- munodepleted serum in the absence and presence of heparin. As can be seen from Fig. 4, lZ6I-kallikrein formed a complex with a component present in serum; this complex co-migrated with the complex formed between '2SI-kallikrein and purified PCI, and its intensity was decreased by the presence of heparin. No 'z51-kallikrein complex formation was observed in PCI-immunodepleted serum.
DISCUSSION
PC1 is a relatively nonspecific heparin-binding serine pro- tease inhibitor (16-18). It inhibits not only activated protein C, but also several other enzymes involved in coagulation (thrombin, factor Xa, factor XIa, plasma kallikrein) and fibrinolysis (urokinase) (16-18, 23). Its inhibitory activity is
Protein C InhibitorlKallikrein Binding Protein
A
t 85
L 1 2 3 4 5 6 7
57 kDa
A A 85 koa
0 1 2 3 4 Incubation time (hours)
1 “ * ’0-3 ’ C 8 5
57 koa
54 kDa
A 85 kDa A
0 I
0 0.03 0.3 3 30 300 Heparin concentration (ug/ml)
FIG. 3. Complex formation of purified PC1 with purified urinary kallikrein and cleavage of PC1 by urinary kallikrein. Panel A, purified urinary PC1 (200 nM final concentration) was incubated with purified urinary kallikrein (7.7 nM final concentra- tion) in 70 pl of 0.01 M Tris-HC1, 0.1 M NaCI, 0.01% Tween 80, pH 7.4. The reactions were stopped after the intervals indicated on the abscissa by addition of 35 pl0.26 M Tris-HC1,2.5% SDS, 25% glycerol and putting the samples in a boiling water bath for 10 min. SDS- PAGE and silver staining of the gel were performed as described under “Experimental Procedures.” Each lane of the stained gel was scanned densitometrically a t 523 nm, and the relative intensity of each stained protein band was evaluated as described under “Exper- imental Procedures.” Relative intensities of the stained bands (0,57- kDa band; 0, 54-kDa band; A, 85-kDa band) are shown on the ordinate. Inset, silver-stained SDS-PAGE gel. Lane 1, PC1 alone (200 nM); lanes 2-7, PC1 (200 nM) incubated with purified urinary kalli- krein (7.7 nM) for 5 min (lane 2), 30 min (lane 3 ) , 1 h (lane 4 ) , 2 h (lane 5), 3 h (lane 6 ) , or 4 h (lane 7). Urinary kallikrein alone revealed no stainable band (not shown). Panel B, purified PC1 (200 nM final concentration) was incubated with purified urinary kallikrein (7.7 nM final concentration) as described in panel A in the absence or presence of different concentrations of heparin (as indicated on the abscissa). Reactions were stopped after 4 h, and samples were subjected to SDS- PAGE. Silver-stained gels were scanned densitometrically as de- scribed under “Experimental Procedures.” Relative intensities of the silver-stained bands (0, 57-kDa band; 0, 54-kDa band; A, 85-kDa band) are shown on the ordinate. Inset, silver-stained SDS-PAGE gel of PC1 (200 nM final concentration) incubated for 4 h with purified urinary kallikrein (7.7 nM final concentration) in the absence (lane 1 ) or presence (lanes 2-6) of different concentrations of heparin (0.03
M , ~ 1 o - ~
110-
84 - c
47 -
33 - 24 -
16 -
1 2 3 4
7051
M , x ~ o - ~
110-
84 -
47 -
33 - 24 -
16-
5 6 7 8 9 1 0
FIG. 4. Complex formation of 12sI-labeled tissue kallikrein in serum and in PCI-immunodepleted serum. ’2sII-Labeled uri- nary kallikrein (4.5 nM, 6000 cpm) was incubated without (lane 1 ) or with 270 nM purified PC1 (lanes 2,5, and 6 ) or with control serum (3 pl; lanes 4, 9, and 10) or with PCI-depleted semm (3 pl; lanes 3, 7, and 8) in 63 pl 0.01 M Tris-HCI, 0.1 M NaCl, 0.01% Tween 80, pH 7.4, for 1 h at 37 “C, in the absence (lanes 1-4) or presence of heparin (lanes 5-10). Lanes 5, 7, and 9 contained 300 pg/ml heparin (final concentration) lanes 6, 8, and 10 contained 3 pg/ml heparin (final concentrations). SDS-PAGE and autoradiography were performed as described under “Experimental Procedures.”
stimulated by heparin (16-18) and other glycosaminoglycans (24). The presence of naturally occurring stimulatory glyco- saminoglycans might therefore play a crucial role for the in uiuo regulation of PC1 activity and possibly also enzyme specificity. However, so far the physiological role of PC1 is still unclear.
PC1 is present in urine in a surprisingly high concentration and could therefore be an important protease inhibitor in the urinary tract. We have shown that PC1 is also present in saliva and intestinal fluid (32). Laurel1 et al. (33) have dem- onstrated recently that very high concentrations of PC1 im- munoreactivity can be found in seminal plasma, Graaf follicle fluid, and synovial fluid. These findings may indicate a more general role for this inhibitor in secretions. These secretions are also major sources of tissue kallikrein, an enzyme for which no efficient endogenous serpin-type inhibitor has been described. We therefore investigated a possible inhibition of tissue kallikrein by PCI.
In fact, using purified urinary kallikrein and purified uri- nary PCI, we were able to show that PC1 inhibited kallikrein activity in a dose- and time-dependent fashion; furthermore, this interaction was accompanied by the formation of SDS- stable complexes (Mr = 85,000). As judged from densitometr- ically scanned silver-stained gels, the maximal amount of complex formed was somewhat higher (6-7% of total PCI) than expected using a %-fold excess of PC1 over kallikrein and assuming 1:l complex formation. However, estimation of the amount of protein present in a silver-stained band is difficult, since the intensity of staining is not necessarily the same for different proteins (30).
Complex formation of PC1 with urinary kallikrein was paralleled by the degradation of PC1 to a lower M , form. This is not uncommon for a serpin-type inhibitor, and cleavage of PC1 in the reactive site region by activated protein C and other target enzymes has already been described (16, 17,34). The 54-kDa PC1 form was already present to =lo-20% in the
pg/ml in lane 2; 0.3 pglml in lane 3; 3 pg/ml in lane 4; 30 pg/ml in lane 5; 300 pg/ml in lane 6).

7052 Protein C InhibitorIKallikrein Binding Protein
PC1 preparations used as shown in Fig. 3A, indicating that PC1 might have been cleaved either in vivo or during the purification procedure. Kinetics for the inhibition of urinary kallikrein by PC1 were determined using a high heparin concentration to stop the enzyme-inhibitor interaction after defined intervals, assuming that heparin has no effect on the stability of the kallikrein-PC1 complex. Although the appar- ent second order rate constant ( k J calculated under these conditions for the inhibition of urinary kallikrein by PC1 (2.3 X lo4 M-' s-') does not seem to be very high, it is in the same order of magnitude (18) or higher (24) than the kl calculated for the reaction of PC1 with its hitherto known target pro- teases. This indicates that PC1 may, in fact, be a physiologi- cally important inhibitor of tissue kallikrein.
Analysis of the effect of heparin on the PCI-tissue kallikrein interaction revealed an inhibitory effect of heparin. This is in contrast to findings on the interaction of PC1 with all other target proteases studied so far, where heparin enhanced the enzyme-inhibitor interaction. Such a reaction pattern, how- ever, has been described by the group of Chao for the inter- action of tissue kallikrein with the so called kallikrein binding protein (13-15); this peculiar heparin effect even led those authors to exclude PC1 as a possible candidate to be the kallikrein binding protein (13). Observing such a heparin effect during the interaction of PC1 with kallikrein, we, how- ever, were prompted to investigate the possible identity of PC1 with this binding protein. Data obtained show that when '261-kallikrein was incubated either with serum or with puri- fied PCI, the formed complexes displayed identical mobilities on SDS-PAGE. Furthermore, the fact that serum depleted of PC1 by monoclonal antibody affinity chromatography no longer reacted with '251-kallikrein to form detectable com- plexes strongly supports the concept that PC1 is the kallikrein binding protein.
Acknowledgments-The excellent secretarial assistance of Helga Hitschmann and the artwork contribution of Thomas Nardelli are thankfully acknowledged.
1. 2.
3.
4.
5.
REFERENCES Clements, J. A. (1989) Endocr. Reu. 10,393-411 Scicli, A. G., and Carretero, 0. A. (1986) Kidney Znt. 2 9 , 120-
130 Kamada, M., Ikekita, M., Kurakashi, T., Aoki, K., Kizuki, K.,
Moriya, H., Sweeley, C. C., Komo, M., and Tsugita, A. (1990) Chem. Phurm. Bull. (Tokyo) 38,1053-1057
El-Thaher, T. S., Saed, G. M., and Bailey, G. S. (1990) Biochim.
Brady, J. M., and McDonald, R. J. (1990) Arch. Biochem. Biophys. Biophys. Acta 1034 , 157-161
278,342-349
6. Jovov, B., Wills, N. K., Donaldson, P. J., and Lewis, S. A. (1990)
7. Ole-MoiYoi, O., Spragg, J., and Austen, K. F. (1979) Proc. Natl.
8. Hiwada, K., Matsumoto, C., and Kokubu, T. (1983) Hypertension
9. Asakawa, K., and Maruta, J. (1980) Nature 288,705-706
Am. J. Physwl. 2 5 9 , C869-C882
Acad. Sei. ti. S. A. 76,3121-3125
5, 191-197
10. Maruta, H., and Arakawa, K. (1983) Bwchem. J. 213,193-200 11. Ideishi, M., Sasaguri, M., Ikeda, M., and Arakawa, K. (1990)
Nephron 65, (Suppl. 1) 62-64 12. Geiger, R., Stuckstedte U., Clausnitzer, V., and Fritz, H. (1981)
Hoppe-Seyhr's 2. Physiol. Chem. 362,317-325 13. Chen, L. M., Chao, L., Mayfield, R. K., and Chao, J. (1990)
Biochem. J. 2 6 7 , 79-84 14. Chao, J., Tillman, D. M., Wang, M., Margolius, H. S., and Chao,
L. (1986) Biochem. J. 239,325-331 15. Chao, J., and Chao, L. (1987) in Proceedings of the Symposium
on Renal Functbn, Hypertension, and Kallikrein-Kinin System, December 7-9, Sapporo, Japan (Iimura, O., and Margolius, H. S., eds) pp. 67-75, Hokusen-Sha Publishing Co., Japan
16. Suzuki, K., Deyashiki, Y., Nishioka, J., and Toma, K. (1989) Thromb. Haemostasis 61,337-342
17. Geiger, M. (1988) Fibrinolysis 2 , 183-188 18. Espaiia, F., Berrettini, M., and Griffin, J. H. (1989) Thromb. Res.
19. Geiger, M., Heeb M. J., Binder, B. R., and Griffin, J. H. (1988)
20. Heeb, M. J., Espaiia, F., Geiger, M., Collen, D., Stump, D. C.,
21. Stump, D. C., Thienpont, M., andCollen, D. (1986) J. Biol. Chem.
22. Laurell, M., Stenflo, J., and Carlson, T. H. (1990) Blood 7 6 ,
23. Meijers, J. C. M., Kanters, D. H. A. J., Vlooswijk, R. A. A., van Erp, H. E., Hessing, M., and Bouma, B. N. (1988) Biochemistry
24. Geiger, M., Priglinger, U., Griffin, J. H., and Binder, B. R. (1991)
25. Espaiia, F., Vicente, V., Tabernero, D., Scharrer, I., and Griffin,
26. Geiger, M., Huber, K., Wojta, J., Stingl, L., Espaiia, F., Griffin,
27. Wojta, J., Kirchheimer, J. C., Turcu, L., Christ, G., and Binder,
28. Markwel, M. A. K., and Fox, C. F. (1978) J. Am. Chem. SOC. 17 ,
29. Laemmli, U. K. (1970) Nature 227,680-685 30. Morrissey, J. H. (1981) Anal. Biochem. 117,307-310 31. Maier, M., Jerabek, I., Reissert, G., Holtzl, E., and Binder, B. R.
(1988) Clin. Chim. Acta 178 , 127-140 32. Resch, I., Hufnagl, P., Geiger, M., and Binder, B. R. (1991)
Abstract book, Znternational Conference Kinin '91 Munich, Ger- many, p. 199, Abstr. PW-2.3
33. Laurell, M., Christensson, A., Abrahamsson, P.-A., Stenflo, J., and Lilja, H. (1991) Thromb. Haemostasis 6 5 , (Abstr. 570) 854
34. Suzuki, K., Nishioka, J., Kusumoto, H., and Hashimoto, S. (1984) J. Biochem. (Tokyo) 95, 187-195
56,369-384
FASEB J. 2,2263-2267
and Griffin, J. H. (1987) J. Biol. Chem. 262,15813-15816
261,12759-12766
2290-2295
27,4231-4237
J. Biol. Chem. 2 6 6 , 11851-11857
J. H. (1990) Thromb. Res. 69,593-608
J. H., and Binder, B. R. (1989) Blood 74,722-728
B. R. (1986) Thromb. Haemostasis 55, 347-351
4807-4817







![Mechanism of Reaction, Tissue Distribution, and Inhibition of … · [CANCER RESEARCH 34, 1503-1515, June 1974] Mechanism of Reaction, Tissue Distribution, and Inhibition of Arylhydroxamic](https://img.pdfslide.us/doc/110x75/5e58a5ee3cd8ad16721400a4/mechanism-of-reaction-tissue-distribution-and-inhibition-of-cancer-research-34.jpg)








![[Product Monograph Template - Standard]...9.2 Pharmacodynamics . Concentration-dependent inhibition of plasma kallikrein, measured as reduction of cHMWK levels, was demonstrated after](https://img.pdfslide.us/doc/110x75/5f0e37947e708231d43e2ca7/product-monograph-template-standard-92-pharmacodynamics-concentration-dependent.jpg)












