Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1985 by The American Society of Biological Chemists, Inc.
Val. 260, No. 25, Issue of November 5, pp. 13666-13676.1985 Printed in U.S.A.
Characterization of Human Blood Coagulation Factor XI1 cDNA PREDICI"I'ON OF THE PRIMARY STRUCTURE OF FACTOR XI1 AND THE TERTIARY STRUCTURE OF @-FACTOR XIIa*
(Received for publication, May 1, 1985)
Laboratory, Cold SpringHarbor, New York 11 724
A human liver cDNA library was screened by colony hybridization with two mixtures of synthetic oligode- oxyribonucleotides as probes. These oligonucleotides encoded regions of @-factor XIIa as predicted from the amino acid sequence. Four positive clones were isolated that contained DNA coding for most of factor XI1 mRNA. DNA sequence analysis of these overlapping clones showed that they contained DNA coding for part of an amino-terminal extension, the complete amino acid sequence of plasma factor XII, a TGA stop codon, a 3' untranslated region of 150 nucleotides, and a poly(A)+ tail. The cDNA sequence predicts that plasma factor XI1 consists of 596 amino acid residues. Within the predicted amino acid sequence of factor XII, we have identified three peptide bonds that are cleaved by kallikrein during the formation of &factor XIIa. Com- parison of the structure of factor XI1 with other pro- teins revealed extensive sequence identity with regions of tissue-type plasminogen activator (the epidermal growth factor-like region and the kringle region) and fibronectin (type I and type I1 homologies). As the type I1 region of fibronectin contains a collagen-binding site, the homologous region in factor XI1 may be re- sponsible for the binding of factor XI1 to collagen. The carboxyl-terminal region of factor XI1 shares consid- erable amino acid sequence homology with other serine proteases including trypsin and many clotting factors. A preliminary structural model of &factor XIIa is pro- posed based on the known high resolution x-ray dif- fraction structures of trypsin, chymotrypsin, and elas- tase.
Human blood coagulation factor XI1 (Hageman factor) is a glycoprotein that circulates in plasma as an inactive zymogen. The protein has been highly purified from human plasma (Revak et al., 1974; Griffin and Cochrane, 1976; Fujikawa and Davie, 1981) and consists of a single polypeptide chain of M, 80,000. On activation, factor XI1 is converted to a serine protease that is capable of initiating the blood clotting cas- cade, the fibrinolytic system, and the production of kinins (Kaplan, 1978; Ratnoff and Saito, 1979; Heimark et al., 1980).
*This work was supported in part by grants from the Medical Research Council of Canada (to R. T. A. M. and G. D. B.) and the British Columbia Health Care Research Foundation (to R. T. A. M. and G. D. B.) and by National Institutes of Health Grant HL06350 (to C.-J. S. E.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Deborah E. cool$, Cora-Jean S. Edgells, Gordon V. Louie$, Mark J. Zollery, Gary D. Brayer$, and Ross T. A. MacGillivrayl From the $Department of Biochemistry, University of British Columbia, Vancouver, British Columbia V6T 1 w5, the $Department of p a t h o k m University of North Carolina. Chapel Hill, North Carolina 27514, and the %'&ld Spring Harbor
Factor XI1 readily binds to anionic surfaces such as sili- cates, dextran sulfate, and sulfatides in vitro (Ratnoff and Rosenblum, 1958; Fujikawa et al., 1977,1980a). In uiuo, these binding properties may be responsible for the activation of factor XI1 when it comes in contact with collagen or platelet membranes (Wilner et al., 1968; Harpel, 1972). Surface-bound factor XI1 has enzyme activity towards its protein substrates, prekallikrein and factor XI (McMillin et al., 1974; Saito, 1977; Heimark et al., 1980). These two proteins circulate in plasma as inactive zymogens complexed with high molecular weight kininogen (Thompson et al., 1977, 1979; van der Graaf et al., 1982). Upon surface activation of the coagulation system, the high molecular weight kininogen-prekallikrein complex, the high molecular weight kininogen-factor XI complex, and fac- tor XI1 bind to anionic surfaces at the site of injury (Wiggins et al., 1977; Thompson et al., 1979; Kerbiriou and Griffin, 1979). Surface-bound factor XI1 activates prekallikrein by limited proteolysis. The resulting kallikrein cleaves factor XI1 generating factor XIIa. The production of activated factor XI1 results in further reciprocal activation of prekallikrein and factor XII. Factor XIIa then activates surface-bound factor XI and thence the rest of the clotting cascade.
There are multiple sites in factor XI1 that are susceptible to cleavage by a number of proteases including kallikrein, plasmin, and trypsin (Margolis, 1958; Kaplan and Austen, 1971; Meier et al., 1977; Dunn and Kaplan, 1982). Limited proteolysis by kallikrein activates surface-bound factor XI1 and produces two active enzyme forms, a-factor XIIa and p- factor XIIa. A single cleavage of the zymogen generates a- factor XIIa while two more subsequent cleavages by kallikrein yield @-factor XIIa (Revak et al., 1974; Dunn et al., 1982; Fujikawa and McMullen, 1983). Both active enzymes consist of two polypeptide chains held together by a disulfide bond (Revak et al., 1974, 1977). a-Factor XIIa is comprised of two polypeptide chains of MI 52,000 and 28,000 while p-factor XIIa consists of two polypeptide chains of M, 2,000 and 28,000. Amino-terminal sequence analysis has shown that the M, 52,000 fragment in a-factor XIIa is derived from the amino-terminal region of factor XI1 (Fujikawa et al., 1980b) and contains the surface-binding site (Revak and Cochrane, 1976; Revak et al., 1977). The M, 2,000 fragment of p-factor XIIa does not bind to surfaces. Although the complete amino acid sequence of p-factor XIIa has been determined (Fujikawa and McMullen, 1983), the origin of the MI 2,000 fragment in the zymogen was not determined. However, the characteri- zation of a-factor XIIa has since been reported in an abstract by these authors (McMullen and Fujikawa, 1984). The M, 28,000 fragments of both a- and @-factor XIIa are identical and contain the catalytic domain derived from the carboxyl-
13666

Human Factor XII cDNA 13667
terminal region of factor XI1 (Revak et aL, 1974, 1977). The catalytic domain of a- and @-factor XIIa shares extensive amino acid sequence homology with other serine proteases, including many blood clotting factors (see Jackson and Nem- erson, 1980; Fujikawa and McMullen, 1983). The sequence homology is highest at the activation regions and near the active sites of these proteases.
In order to characterize the zymogen form of factor XI1 more thoroughly, we have isolated several cDNA clones coding for human factor XII. Together, these clones code for part of an amino-terminal extension (six amino acid residues), the complete amino acid sequence of plasma factor XII, a TGA stop codon, a 3’ untranslated sequence of 150 bp’ and a poly(A)+ tail. From the. cDNA sequence we predict that plasma factor XI1 consists of a single polypeptide chain of 596 residues. Comparison of the predicted factor XI1 sequence with the sequences of other proteins shows that factor XI1 shares extensive amino acid identity with tissue-type plas- minogen activator (tPA) and fibronectin. Based on the known atomic coordinates of bovine trypsin (Marquart et al., 1983), porcine elastase (Sawyer et al., 1978), and bovine chymotryp- sin (Cohen et al., 1981), a three-dimensional model of @-factor XIIa has been constructed. The model predicts that: (i) there is a close structural relationship between the catalytic regions of these serine proteases and @-factor XIIa; (ii) the disulfide bonding arrangement of the 14 cysteine residues in @-factor XIIa can be assigned; and (iii) the presence of an alanine residue rather than a serine residue at the a-carbon position 190 may explain why some clotting factors cleave preferen- tially at arginine residues rather than at lysine residues.
MATERIALS AND METHODS
Enzymes and Chemicals-Escherichia coli DNA polymerase I and Klenow fragment were purchased from Boehringer Mannheim, and deoxyribonuclease I was purchased from P-L Biochemicals. All other enzymes were purchased from Bethesda Research Laboratories or New England Biolabs. P-L Biochemicals also supplied deoxy- and dideoxyribonucleotide triphosphates, the M13 sequencing primer (heptadecanucleotide), and oligo(dT) cellulose (type 7). [LY-~’P] dNTPs (3000 Ci/mmol; 1 Ci = 3.7 X 10” Bq) and [y3’P]ATP (3000 Ci/mmol) were purchased from Amersham. Cellulose nitrate (BA-85) was purchased from Schleicher & Schuell.
Synthesis of Oligodeozyribonucleotides-Two pools of mixed hep- tadecadeoxyribonucleotides were used as hybridization probes. Pool I (5’-dTCRAAYTGRTGNCCCCA-3’, where R represents both dG and d A , Y represents dT and dC, and N represents dG, dA, dT, and dC) was complementary to the mRNA sequence coding for amino acid residues 133-138 of @-factor XIIa (Fujikawa and McMullen, 1983). Pool I1 (5’-dCCYTGRCANGCYTCNGT-3’) was complemen- tary to the mRNA coding for residues 182-188 of @-factor XIIa. Both pools of oligonucleotides were synthesized by using an Applied Bio- systems 380A DNA Synthesizer, and the heptadecanucleotide frac- tions were purified by polyacrylamide gel electrophoresis in the pres- ence of 8.3 M urea (Atkinson and Smith, 1984). The oligonucleotide mixtures were labeled with [y-3ZP]ATP and T4 polynucleotide kinase (Chaconas and van de Sande, 1980) to a specific activity of 1.1 X lo6 cpm/pmol. Unincorporated [Y-~’P]ATP was removed by chromatog- raphy on a column of Sephadex G-25 (superfine) equilibrated and eluted with 5 mM Na2EDTA (pH 8.0), and the excluded fraction was added directly to the hybridization mixture.
Screening a Human Liver cDNA Library-An adult human liver cDNA library (Prochownik et al., 1983) was generously provided by S. H. Orkin (Children’s Hospital Medical Center, Boston). This library consists of double-stranded cDNA (>500 bp in length) cloned into the PstI site of pKT218 by homopolymeric dGdC tailing. Ini- tially, 2 X lo5 colonies of the cDNA library were plated directly onto 10 cellulose nitrate filters placed on Luria broth,plates containing tetracycline (12.5 pg/mI). The colonies were grown and two sets of
‘The abbreviations used are: bp, base pair(s); tPA, tissue-type plasminogen activator.
replica filters were prepared (Hanahan and Meselson, 1980; Fung et al., 1984). One set of the replica filters was hybridized with the 32P- labeled Pool I oligonucleotides and the other set of filters was hybrid- ized to 32P-labeled Pool 11. Hybridization and washing conditions were as described by Fung et al. (1984). Colonies that hybridized to both pools of oligonucleotides were rescreened at lower colony density. Plasmid DNA was prepared from positive colonies using the alkaline lysis method (Maniatis et al., 1982). The cDNA library was subse- quently rescreened by colony hybridization (Hanahan and Meselson, 1980) using nick-translated factor XI1 cDNA restriction enzyme fragments as hybridization probes (Maniatis et al., 1975). All cloning experiments were performed in compliance with the Medical Re- search Council guidelines for recombinant DNA research.
Restriction Endonuclease Mapping-Plasmid DNA was digested with one or more restriction endonucleases under conditions recom- mended by the manufacturers. The resulting fragments were analyzed by agarose or polyacrylamide gel electrophoresis.
DNA Sequence Analysis-DNA sequence analysis was carried out essentially as described by Deininger (1983). Plasmid DNA was randomly sheared by sonication and the resulting DNA fragments were fractionated on a 5% polyacrylamide gel. Fragments (300-500 bp in size) were recovered by electroelution, and the ends were repaired with T4 DNA polymerase. The blunt-ended fragments were then ligated into the SmaI site of M13mp8 and used to transform E. coli strain JM103 (Messing, 1983). Phage containing cDNA sequences were identified by plaque hybridization (Benton and Davis, 1977) using nick-translated cDNA restriction fragments as probes. Single- stranded DNA was isolated from positive plaques (Messing, 1983) and subjected to DNA sequence analysis using the chain termination method (Sanger et al., 1977). This strategy resulted in the determi- nation of most of the cDNA sequence. To complete the sequence analysis, three restriction enzyme fragments were subcloned into M13 (see “Results”). All DNA sequence data were analyzed by the DBU- TIL computer program of Staden (1982).
Northern Blot Analysis-Poly(A)+ RNA was isolated from a sample of human liver by the guanidine hydrochloride method (Chirgwin et al., 1979). The poly(A)+ RNA (20 pg) was denatured with formalde- hyde/formamide, electrophoresed on a 1% agarose gel containing formaldehyde, and transferred to nitrocellulose as described by Man- iatis et al. (1982). The blot was analyzed using nick-translated pc- HXII-501 (specific activity, 1.4 X 10’ cpm/pg) as a hybridization probe. Hybridization and washing conditions were as described for Southern blots by Kan and Dozy (1978).
Southern Blot Analysis-High molecular weight DNA was isolated from a liver sample of a female with no known hematological disorders using the method of Blin and Stafford (1976). DNA samples (10 pg) were digested with restriction endonucleases (40 units) at 37 “C for 18 h. The DNA fragments were separated by electrophoresis on a 1% agarose gel, denatured, and transferred to nitrocellulose as described by Southern (1975). The blot was analyzed using pcHXII-501, pre- viously labeled by nick translation (specific activity, 9 X 10’’ cpm/pg) as a hybridization probe. Hybridization and washing conditions were as described for the Northern blot analysis.
Sequence Alignment and Structural Modelling-The amino acid sequence alignment of @-factor XIIa to the pancreatic serine proteases trypsin, chymotrypsin, and elastase was based on considerations of both sequence homology and topological equivalence (James et al., 1978). During this procedure the preservation of regions of identical amino acid sequences near the catalytic residues and the homologous positioning of disulfide bridges were given the highest priority. Inser- tions were introduced only when such residues were absent in the amino acid sequences of all three pancreatic enzymes and placed only where inspection of the known tertiary structures of the pancreatic enzymes indicated that sufficient room was available to accommodate them.
The modelling procedure used to predict the tertiary structure of &factor XIIa was based primarily upon the known high resolution structure of bovine pancreatic trypsin (Marquart et al., 1983). An initial model was constructed with coordinates from the polypeptide chain backbone of trypsin and those amino acid side chains conserved in the primary structures of both proteins (84 residues). Changes in the identity of nonhomologous amino acids (139 residues) were ac- complished in the following manner. The original trypsin side chain was replaced by the appropriate @factor XIIa side chain (as obtained from a standard amino acid group dictionary (Sielecki et al., 1979)) utilizing a least squares fitting procedure. In this process, the maximal number of conformational torsional angles from the original trypsin

13668 Human Factor XII cDNA side chain were conserved in the positioning of the new @-factor XIIa side chain.
With the exception of residue 245 at the carboxyl terminus, there are no deletions in the amino acid sequence of @-factor XIIa with respect to trypsin (Table I). Insertions in the @-factor XIIa sequence were handled in one of two ways. The first method was based on the strong tertiary structural homology known to exist among the pan- creatic serine proteases, chymotrypsin, elastase, and trypsin (Bode et al., 1976; James et al., 1978; Sawyer et al., 1978; Cohen et al., 1981). When @-factor XIIa insertions (with respect to the trypsin sequence)
were also present in the sequences of chymotrypsin or elastase, the portion of polypeptide chain from the appropriate protein was used as a guide in the placement of the comparable insertion in the @- factor XIIa model. Amino acid positions modelled in this way included residues 67-68, 126, 131, 170A, 172, 203-205, and 206. Amino acid insertions unique to @-factor XIIa (residues 61 A-C, 109 A-E, and 205 A-C) occurred at the ends of existing @-loops in the structures of the pancreatic enzymes and were handled by a second method. The inserted residues were modelled as extended @-loops and merged to the polypeptide chain backbone at the appropriate positions. All three
TABLE I Amino acid sequence alignment of the catalytic regions of human @-factor XIIa (FXII), bovine trypsin (TRYP),
porcine elastase (ELAS), bovine chymotrypsin (CHYM), and human tissue-type plasminogen activator (tPA) The sequences of trypsin, elastase, chymotrypsin, and tPA are taken from Marquart et al. (1983), Sawyer et al.
(1978), Cohen et al. (1981), Pennica et al. (1983), respectively. The numbering system used is based on that of chymotrypsinogen A (Hartley and Kauffman, 1966), with insertions in the sequences of related enzymes denoted by letters (36A, 36B, etc.). Deletions are indicated by dashed lines. Identical residues in corresponding positions of all five moteins are boxed. The standard single letter code for amino acids is used.
F XI1 TRY P eLlS M Y U tPA
F XI1 TRYP ELAS c w Y U tPA
F X I 1 TRYP ELAS M Y * t P A
TRYP F XI1
ELAS CHYM tPA
F X I 1 TRYP ELAS CHYU tPA
TRYP F X I 1
CHYU ELAS
tPA
F XI1 TRY P m S M Y U tPA
TRYP P X I 1
ELM CWYU tPA
F X11 TRYP ELAS CHY" tPA
F X11 TRYP
CHYU ELAS
tPA
F X11 T R Y P ELAS CHYll tPA
TRYP F X I 1
CHYU ELAS
tPA
-5 -4 -3 -2 -1 1 2 3 4 5 6 7 8 9 10 11. 12 1 3 N G P L S C G Q R - " " " " " " " _ " " " _ _ . . . " """""""" _ " " C G V P A I Q P V L S G L P S C S T C G L R Q Y S Q P Q F R -
A B C D 16 17 18 19 20 21 22 23 24 25 26 21 28 29 30. 31 32 3 3 34 35 36 36 36 36 36 : i i [ Y T C G A N T V L V A L R G A H Y I A A L Y " " "
Y Q V S L N " " " T B A Q R N S W S Q I S L Q Y R S G S -
I V N E E A V P G S W W Q V S L Q D K " " I I G L F A D I A S H W Q A A I F A K H R R S
A B C A 61 61 61 61 62 63 64 65 65 66 67 68 69 70 11 12 13 74 15 76 17 78 79 80 81
D R P A P . .
82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 99 99 100 101 102 103 104 A B
T L A V R S Y R L H E A F S P V S Y " F I S A S K S I V H P S Y N S N T L " N
A B C D E 105 106 101 108 109 109 109 109 109 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124
Q E D A D G S C A L L S
125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142.143 144 145 146 147 148 149 s G A A R e s a T T L r
150 151 152 153 154 155 156 151 158 159 160 161 162 163 164 165 166 167 168 169 110 110 170 171 172 E Y A S S Y P ~ ~ ~ ~ ~ ~ ~ ~ P l L S N ~ ~ ~ ~ S - A Y - P F L S L E S A P D V H
Q L A N T P D R Q Q A S L P L L S N T N K K " Y W
Y L P T V D Y S S S Y Y
F Y S E R K E A H V R L Y P S S R T S Q H L L
173 114 115 G S S P G Q G S T G T K N R T
190 191 192 193 194 195 196 191 198 G D S G G P G D S G G P
182 183 184
C A G
199 200 201
f L V L V
A 184 185 186 181 188 188 188 188 188 188 188 188 189 F L E G G T " " " D Y L E G G K " " " D . G N G " R " " " S - k S G V " " " - S D T R S G C P Q A N L H D
A B C D E F G
l r s c 202 203 204 205 205 205 205 206 207 208 209 210 211 E D Q A A E R R L T L Q S""". L V N G - - - Q : i t Z I K K N G - " A W T L V L N D G - - - R M T L V
A I 212 213 214 215 216 211 217 218 219 220 221 221 222 223 224 225 226 227'228 229 230 231 232 233 234
W G S "
N V T R K S A Y
I I M W G L - - G M G Q X D V M G M Y T K M T N Y
235 236 231 238 239 240 241 242 243 244 245 ; ; n ; ; E H T V S -
O T I A S N

Human Factor XII cDNA 13669
insertions occur at the surface of the enzyme where there is sufficient room for their placement. No disruption of nearby segments of conserved polypeptide chain structure was required during this fitting process.
In order to obtain the best overall structural model, the full set of initial atomic coordinates for &factor XIIa were further subjected to 10 cycles of structural idealization (final root mean square 6 for bond distances, 0.006 A, Konnert and Hendrickson, 1980). It is this final predicted model that is discussed herein.
RESULTS
Isolation of Factor XZZ cDNA Clones-Two hundred thou- sand bacterial colonies of a human liver cDNA library were screened with two mixtures of synthetic oligonucleotides that encoded two regions of ,&factor XIIa as predicted from the known amino acid sequence. Although each of the oligonucle- otide mixtures hybridized to about 60 different colonies, only two colonies hybridized to both. After rescreening at lower colony density, plasmid DNA was isolated from the two positives that were designated pcHXII-11 and pcHXII-14 (Fig. 1). Subsequent sequence analysis revealed that these plasmids contained DNA coding for the known amino acid sequence of p-factor XIIa. Because pcHXII-11 and pcHXII- 14 contained only short cDNA inserts, 2 x lo5 colonies of the cDNA library were rescreened with the 32P-labeled PstI insert of pcHXII-11 as a hybridization probe. Thirty-six positive clones of various lengths were obtained; plasmids pcHXII-17 and pcHXII-501 contained the longest cDNA inserts and were studied further. Restriction endonuclease mapping of the four positive plasmids showed that they contained overlapping DNA (Fig. 1) except that pcHXII-17 appeared to contain a deletion (Fig. 1, open dashed bar). This was confirmed by subsequent DNA sequence analysis (see next section).
Sequence Analysis of Human Factor XZZ cDNA Clones- The strategy used to determine the nucleotide sequence of the four clones, pHXII-11, -14, -17, and -501 is shown in Fig. 1. The thick arrows represent the sequence determined from M13 phage containing randomly sheared DNA. Although
*e+
more than 90% of the nucleotide sequence was determined using this strategy, few random clones were isolated that overlapped the AuaI site of factor XI1 cDNA (Fig. 1). To complete the nucleotide sequence analysis, plasmid pcHXII- 501 (Fig. 1) was digested with PstI and the 1200- and 280-bp fragments were isolated by polyacrylamide gel electrophoresis. The 280-bp fragment was cloned directly into the PstI site of M13mp8. The 1200-bp fragment was digested with both AuaI and SmaI, and the ends were made blunt-ended using the Klenow fragment of E. coli DNA polymerase I (Smith et al., 1979). The resulting PstIIAuaI and AuaIISmaI fragments were isolated by polyacrylamide gel electrophoresis and sub- sequently ligated into the SmaI site of M13mp18. The DNA sequence analysis of these clones is shown in Fig. 1 (thin arrows).
The nucleotide sequences of the four factor XI1 cDNA clones were compiled into a consensus sequence (Fig. 2). Each nucleotide in the sequence was determined an average of 6.6 times, and 84% of the sequence was determined on both strands. In the regions of overlap, the four factor XI1 cDNA clones contained identical nucleotide sequences, with one exception. In agreement with the restriction map of pcHXII- 17 (Fig. l), the DNA sequence analysis showed that this plasmid contains a deletion of 53 bp between nucleotides 1330 and 1383 (Fig. 2). The deleted region is flanked by an inverted repeat having the sequence: CTCGC CGTC (nucleotides 1320-1329 and nucleotides 1383-1392, Fig. 2). This deletion alters the reading frame of factor XI1 mRNA and probably represents a cloning artifact.
Predicted Amino Acid Sequence of Human Factor XZZ- Translation of the cDNA sequence using the standard genetic code results in a single open reading frame coding for human factor XI1 (Fig. 2). The derived amino acid sequence is iden- tical to those regions of human factor XI1 that have been determined by protein chemistry techniques. Nucleotides 19- 78 encode the amino-terminal region of factor XI1 (Fujikawa
G
*e* *e+ *.+ *e+ I I I 1 I I
I I P
1 pcHXII-11 pcHXII-14 pcHXII-17 r I pcHXII-501 1;1 I
- 4 "-" - """ -" - "- """ - - - - -c" - - 4"
"c- - " " -c- - " " C" - -
0 0.2 0.4 0.6 0.8 1.0 t.2 1.4 1.6 1.8 2.0 I I I I I I I I I I I
Nucieotldes (kb)
FIG. 1. Restriction map and sequencing strategy for human factor XI1 cDNA. The bars below the restriction map represent cDNA clones pcHXII-11, pcHXII-14, pcHXII-17, and pcHXII-501 and include regions coding for the signal peptide (dotted bar), the heavy chain of a-factor XIIa (open bar), the light and heavy chains of &factor XIIa (solid bars), and the 3' untranslated region (hatched bar). The open dashed line in pcHXII-17 represents a deletion (see text for details). Arrows below the cDNA clones represent the sequencing strategy used, where each arrow represents an M13 clone. The extent of sequencing is indicated by the length of the arrows. DNA sequence determined on the coding strand is shown by an arrow pointing left; sequence determined on the noncoding strand is shown by an arrow pointing right. Thick arrows represent DNA sequences obtained from randomly sheared cDNA fragments and thin arrows represent sequences obtained from restriction enzyme fragments. The scale at the bottom represents nucleotides in kilobase pairs (kb).

13670 Human Factor XII cDNA
10
A m CCC C U GTG Ca C U GCT Cffi ffiC UG CIT GGC GU uil llCT CIL T M ACT Gff W ATG nC Poly A 1.905 1.920 1.935 - 1,950 1.959
FIG. 2. Nucleotide sequence of human factor XI1 cDNA. The sequence was determined by analysis of the overlapping clones shown in Fig. 1. The predicted amino acid sequence of human factor XI1 is shown above the DNA sequence. The oligonucleotide sequences used for screening the cDNA library are represented by boxes at nucleotides 1474-1490 and 1627-1643. The putative signal peptide is numbered backwards from the putative signal peptidase cleavage site (open arrow). The proteolytic cleavage sites that give rise to p-factor XIIa are shown by the heauy arrows (at amino acid residues 334, 343, and 353). The residues that make up the catalytic triad (His-393, Asp-442, and Ser-544) are underlined. The TGA stop codon is located at nucleotides 1807-1809 and followed by the 3' untranslated region (nucleotides 1810-1959) and a poly(A)+ tail. The poly(A)+ recognition site AATAAA is encoded by nucleotides 1937-1942. A carbohydrate attachment site (Fujikawa and McMullen, 1983) is indicated by the solid diamond.
et al., 1980b; Fujikawa and Davie, 1981). Since the amino- terminal residue of plasma factor XI1 is encoded by nucleo- tides 19-21 (Fig. 2), the factor XI1 cDNA sequence also encodes part of an amino-terminal extension (nucleotides 1- 18, Fig. 2). Nucleotides 1021-1047 and 1078-1806 encode the light and heavy chains of p-factor XIIa as determined by Fujikawa and McMullen (1983). In the presence of an anionic surface, kallikrein cleaves factor XI1 at a position carboxyl- terminal to Arg-353, resulting in the formation of a-factor XIIa (Fujikawa and McMullen, 1983). Further proteolytic cleavages occur carboxyl-terminal to Arg-334 and Arg-343
and give rise to @-factor XIIa. During the formation of @- factor XIIa, residues 1-334 and 345-353 of factor XI1 are removed. The codon for the carboxyl-terminal residue of the heavy chain of p-factor XIIa (Ser-596) is followed by a TGA stop codon (nucleotides 1807-1809, Fig. 2), a 3' untranslated region of 150 nucleotides and a poly(A)+ tail. Nucleotides 1937-1942 contain the sequence AATAAA that is involved in the polyadenylation of mRNA (Proudfoot and Brownlee, 1976; Wickens and Stephenson, 1984). This sequence occurs 17 nucleotides upstream of the poly(A)+ tail.
The predicted amino acid sequence of factor XI1 corre-

Human Factor XII cDNA 13671
sponds to a mature protein of 596 amino acids. The amino acid composition of plasma factor XI1 was determined to be as follows: Alasl, Arg39, Asn13, Asp22, 1/2-Cys40, Gln37, GlU31, Gly4s, His27, Ileg, Lew, Lys19, Met4, Phe15, pro.056, S e s ~ , Thr34, Trp,,, Tyr19, and Vab. From the predicted amino acid se- quence, the molecular weight of plasma factor XI1 is 66,915 in the absence of carbohydrate and 80,427 with the addition of 16.8% carbohydrate (Fujikawa and Davie, 1981). Within experimental error, both the predicted amino acid composi- tion and molecular weight of factor XI1 are in excellent agreement with those determined for the purified protein (Griffin and Cochrane, 1976; Fujikawa and Davie, 1981).
A 2.40-kb mRNA from Liver Codes for Human Factor XII- To determine the size of factor XI1 mRNA, human liver poly(A)+ RNA was electrophoresed on an agarose gel under denaturing conditions and transferred to nitrocellulose. The blot was hybridized to 32P-labeled pcHXII-501, which con- tains 1959 bp of coding sequence. Autoradiography revealed a single band that was 2,400 f 100 nucleotides in size (Fig. 3A). Since eukaryotic mRNAs usually contain poly(A)+ tails of 180-200 nucleotides (Perry, 1976), we conclude that our longest clone is probably missing about 250 nucleotides of the mRNA sequence.
The Human Factor XZZ Gene-As an initial step towards the characterization of the human factor XI1 gene, Southern blots of human liver DNA digested with several restriction endonucleases were hybridized to 32P-labeled factor XI1 cDNA (pcHXII-501). Autoradiography revealed several hybridizing bands in each of the restriction enzyme digests (Fig. 3B). This suggests that the human genome contains either a single factor XI1 gene containing several introns or contains multi- ple factor XII-like genes or pseudogenes.
A
4.72 - 2.90 - 1.87 I
1.54
23.70 - 9.46 - 6.66 -
4.26 -
2.25 - 1.96 -
0.59 4 FIG. 3. Analysis of the mRNA and the gene coding for factor
XII. A , human liver poly(A)+ RNA was electrophoresed in a dena- turing agarose-formaldehyde gel and was transferred to nitrocellulose. The filter was hybridized with 32P-labeled pcXII-501. The filter was exposed for 18 h at -70°C with Lightning Plus intensifying screens (DuPont). The positions of RNA size standards are indicated, includ- ing mammalian 28 S (4.72 kb) and 18 S (1.87 kb) rRNA and E. coli 23 S (2.90 kb) and 16 S (1.54 kb) rRNA (Noller, 1984). B, human liver DNA was digested by various restriction enzymes and electro- phoresed in a 1.0% agarose gel. After denaturation, the DNA was transferred to nitrocellulose and hybridized with 32P-labeled pcHXII- 501. The filter was autoradiographed for 4 days at -70 "C with intensifying screens. Lane M shows the positions of "P-labeled X- HindIII DNA used as molecular weight markers. The human DNA was digested with EcoRI (lane I), HindIII (lane 2), PstI (lane 3), and BamHI (lane 4).
DISCUSSION
Factor XII cDNA Clones-A factor XI1 cDNA of 1959 bp was isolated from a cDNA library prepared from human liver mRNA. This cDNA (pcHXII-501) codes for part of a leader peptide followed by the entire coding region of plasma factor XII, a TGA stop codon, a 150-bp noncoding region at the 3' end, and a poly(A)+ tail of at least 75 bp.
The cDNA sequence data have enabled us to locate the kallikrein cleavage sites (solid arrows, Fig. 2) that produce p- factor XIIa during zymogen activation (Cochrane et al., 1973; Revak et al., 1977; Dunn et al., 1982). The three cleavage sites occur carboxyl-terminal to arginine residues in the sequences: Leu-Thr-Arg, Gly-Gln-Arg, and Met-Thr-Arg. Because kalli- krein does not recognize a unique sequence, however, the locations of other kallikrein cleavage sites in the M, 52,000 fragment of a-factor XIIa cannot be determined from the cDNA sequence data.
Factor XI1 is a secreted protein and would be expected to contain a signal peptide which functions in the transport of the protein across the rough endoplasmic reticulum mem- brane (Blobel et al., 1979). The cDNA sequence predicts that factor XI1 is synthesized as a precursor containing an amino- terminal extension of at least six amino acid residues (encoded by nucleotides 1-18, Fig. 2). Cleavage of a Ser-Ile bond (en- coded by nucleotides 16-21, Fig. 2) in the precursor would give rise to plasma factor XII. This bond cleavage is consistent with the specificity of signal peptidase (Watson, 1984); from the present study, however, it is unclear whether nucleotides 1-18 encode part of a signal peptide or another type of precursor sequence.
Homologies with Other Proteins-Comparison of residues 1-276 of factor XI1 with protein sequences in the National Biomedical Research Foundation (Washington, D. C.) Protein Data Base reveals extensive sequence homology with both fibronectin and tPA. Based on these sequence homologies, a schematic representation of the structure of factor XI1 is shown in Fig. 4. This structure is based on five different regions that share homology with regions of either fibronectin or tPA. In addition, factor XI1 contains a proline-rich region that does not share sequence homology with any known protein sequence. Each of these six regions of factor XI1 are discussed in detail in the following sections.
The amino-terminal region of factor XI1 (region A-B, Fig. 4) shares sequence homology with the type I1 homology re- gions of fibronectin. The type I1 homologies are each com- posed of about 60 residues including four half-cysteine resi- dues (Petersen et al., 1983) and share approximately 54% sequence homology with each other (31/57 residues in corre- sponding positions are identical). Residues 13-69 of factor XI1 share 39% sequence homology (22/57 residues are iden- tical) and 40% sequence homology (23/57 residues are iden- tical) with the two fibronectin sequences, respectively, includ- ing the four half-cysteine residues (Fig. 5A). The type I1 homologies probably comprise the collagen-binding site in fibronectin (see Yamada, 1983); by analogy, the region of factor XI1 that shares sequence homology to the type I1 regions may be responsible for the collagen-binding properties of factor XII.
Two regions of factor XI1 (regions B-C and D-E, Fig. 4) are homologous to an epidermal growth. factor-like sequence that has been found in many proteins including the 19-kDa protein from Vaccina virus, transforming growth factor type 1, tPA, and several clotting factors (Blomquist et al., 1984; Doolittle et al., 1984). In each of these proteins, there is a highly conserved region of 50 amino acids with nine invariant cys- teine and glycine residues. After the insertion of three gaps

13672 Human Factor XII cDNA
n
FIG. 4. Proposed model for human factor XII. The model was based on amino acid sequence homologies with human tPA (Pennica et al., 1983; Ny et al., 1984) and bovine fibronectin (Petersen et al., 1983). Thin arrows demarcate the proposed protein structural do- mains found in human factor XII. Region A-B represents a fibronec- tin type I1 homology; B-C and D-E are growth factor-like regions; C- D region is a fibronectin type I homology; region E-F is a kringle structure; F-G is a proline-rich region whose structure is undefined, and G-COOH represents the catalytic region (see text for details). Disulfide bonds have been inserted according to their arrangement in the homologous domains of other proteins; however, the disulfide bond arrangement of the fibronectin type I1 region has not been determined. The disulfide bonds in the catalytic region were based on the proposed model of @-factor XIIa (see “Discussion”). Kallikrein cleavage sites that give rise to @-factor XIIa are indicated by the heavy arrows.
in the factor XI1 sequences, all cysteine residues align per- fectly (Fig. 5B). The carboxyl-terminal growth factor domain also contains the invariant glycine residues; in the amino- terminal domain, however, one invariant glycine residue has been replaced with a histidine.
Separating the two growth factor-like regions of factor XI1 is a 43-amino acid peptide (regions C-D, Fig. 4) that shares limited sequence homology with the type I regions of fibro- nectin (Fig. 5C). Fibronectin contains at least nine type I regions each of which are characterized by two disulfide bonds giving a two-loop structure that has been named a “finger” domain (Petersen et al., 1983). The function of the finger domains in fibronectin is unclear but they may be involved in fibrin-binding (see Yamada, 1983).
Another type of homology found in factor XI1 is the kringle domain (region E-F, Fig. 4). Kringle domains are typically 80 amino acids in length and form three characteristic disulfide bonds. Kringles have been found in prothrombin (Magnusson et al., 1975), plasminogen (Sottrup-Jensen et al., 1978), tPA (Pennica et al., 1983), and urokinase (Giinzler et al., 1982) and share approximately 40% homology with each other in- cluding six invariant half-cystine residues (see Jackson and Nemerson, 1980). An alignment of one of the kringle regions of tPA with factor XI1 is shown in Fig. 5 0 . If three gaps are inserted into the factor XI1 sequence, the kringles share 41% sequence homology (36 out of 87 residues are identical). The function of the kringle regions is unclear but they may be involved in the binding of factor XI1 to fibrinogen and fibrin (Sottrup-Jensen et al., 1978; Lucas et al., 1983). The presence of both a finger domain and a kringle suggests that the fibrin binding capacity of factor XI1 may be important in uiuo. One consequence of fibrin binding is that on initiation of the clotting cascade, factor XI1 would be trapped in the growing network of fibrin. Consequently, factor XI1 (or a-factor XIIa)
would be available for the subsequent initiation of the fibrin- olytic cascade.
The kringle structure is followed by a region (residues 279- 330, Fig. 2; region F-G, Fig. 4) in which 33% of the residues are proline (17 out of 52). Despite the high proline content, this region does not share any sequence homology with other proline-rich proteins such as those from salivary glands (Zie- mer et al., 1984). However, residues 299-308 of the proline- rich region of factor XI1 share sequence homology with resi- dues 29-38 of calf thymus high-mobility group protein 17 (Walker et al., 1977) in which 8 out of 10 residues in corre- sponding positions are identical. The significance of this homology and the function of the proline-rich region of factor XI1 are unclear.
Downstream of the proline-rich region in factor XI1 is the catalytic region (region G to the COOH terminus, Fig. 4) that shares amino acid sequence homology with other serine pro- teases. An alignment of the amino acid sequences of the catalytic regions of p-factor XIIa, tPA, and the three pan- creatic serine proteases is given in Table I. An analysis of the sequence homology between these enzymes is summarized in Table 11. It is notable that the sequence homology between 8- factor XIIa and the pancreatic enzymes (average value of 35%) is comparable to the homology present between the pancreatic enzymes themselves (average value of 42%). The strong structural homology between @-factor XIIa and the pancreatic proteases has allowed us to develop a structural model of /3-factor XIIa based on the known three-dimensional structures of the pancreatic enzymes (see next section).
From these sequence comparisons, it can be seen that factor XI1 shares remarkable sequence homology with tPA (Ny et al., 1984), a protease that converts plasminogen to plasmin in the fibrinolytic pathway (see Collen, 1980). The complete amino acid sequence of human tPA has been predicted from the cDNA sequence (Pennica et al., 1983) and shows that tPA consists of a finger domain, a growth factor-like domain, two kringle structures, and the catalytic domain. This organiza- tion is similar to regions C to the COOH terminus of factor XI1 except that one of the tPA kringles is replaced by the proline-rich region (Fig. 4).
It has been suggested that the genes coding for multi- domainal proteins may have evolved by joining of exons coding for separate functional regions (Gilbert, 1979). In the case of factor XII, at least seven potential domains can be identified (Fig. 4). We are now characterizing the positions of the introns in the factor XI1 gene, which will allow a compar- ison of this gene to the tPA (Ny et al., 1984) and fibronectin (Hirano et al., 1983) genes. This analysis should shed light on the evolution of the factor XI1 gene.
A Model of the Tertiary Structure of &Factor XIIa-Thus far, it has not been possible to determine the tertiary structure of any of the clotting proteases using x-ray diffraction tech- niques. The major difficulties encountered in the structural determination of these enzymes have been either an inability to isolate sufficient material or to prepare suitable crystalline forms. The absence of direct structural studies was the pri- mary incentive in the development of the present structural model for @-factor XIIa. Although this model should only be considered a preliminary approximation of the structure of p- factor XIIa, it does provide a vehicle for gaining additional insight into the function of this enzyme.
The level of sequence homology observed between the pan- creatic proteases (41%) has subsequently been shown to trans- late into a substantially higher degree of tertiary structure homology (-85%, James et al., 1978). Since the level of sequence homology between these enzymes and p-factor XIIa

Human Factor XII cDNA 13673
A Fibronectin Type I1 Homology:
FXI I
FXI I
B Growth Factor Homology:
tPA
FXII-1 t t t t t t t t t
c Fibronectin Type I Homoloqy:
D Kringle Homology: tPA
B K B S D D G R O L ( S Y 1 L A R U T L ( s G 1 P U Q P i A b E a T Y R N V T A E Q A D T R A T C Y E D Q G I S Y R G T W S T A E S G A E C T N W N S S A L A Q K P Y S G R R
FXI I
tPA FXI I
FIG. 5. Homologies between the amino acid sequences of human factor XII, bovine fibronectin (Petersen et ul., 1983), and human tissue-type plasminogen activator (Pennica et ul., 1983). Identical residues in corresponding positions are bow& gaps have been inserted in the sequences to maximize homology. A, comparison of segment S3, residues 14-73 (type 11-l), and residues 74-130 (type 11-2) of fibronectin with residues 13-69 of factor XI1 (FXII). B, comparison of residues 51-84 of tissue-type plasminogen activator (tPA) and residues 79-111 (FXII-I) and residues 159-190 (FXZZ-2) of factor XII. Arrows denote glycine and cysteine residues that are invariant in this type of homology (see text for details). C, comparison of segment S1, residues 17-57 (type I-I) and segment S1 residues 66-104 ( t y p e I-2) of fibronectin with residues 116-151 of factor XI1 (FXII). D, comparison of residues 87-173 of tissue-type plasminogen activator ( P A ) with residues 193-276 of factor XI1 (FXII).
TABLE I1 Comparative analysis of the amino acid sequence alignment between
the catalytic regions of @-factor XZIa and the pancreatic serine proteases
The total number of residues in each protein is given in parentheses below its name in the heading. For each pair of proteins aligned in Table I, the matrix contains the number of identical residues followed by the overall sequence homology (given as a percentage of the smaller protein) in parentheses.
Chymotrypsin Elastase (243) (240)
Trypsin (223)
8-Factor XIIa 86 (35%) 79 (33%) 84 (38%) Chymotrypsin 94 (41%) 101 (45%) Elastase 87 (39%)
is similar (-36%), one might also anticipate a corresponding level of tertiary structure homology between @-factor XIIa and the pancreatic serine proteases. The modelled structure of @-factor XIIa and the structure of trypsin, determined by x-ray diffraction techniques, are shown in Fig. 6. Beyond indicating tertiary structure homology, our current model of &factor XIIa provides a basis for understanding the impli- cations of amino acid sequence differences between this en- zyme and its pancreatic counterparts. For example, it is known that all 14 cysteine residues of @-factor XIIa are
involved in disulfide bridging (Fujikawa and McMullen, 1983). From the sequence alignment of @-factor XIIa (Table I), it is apparent that four disulfide bridges (involving residues 42- 58,136-201,168-182, and 191-220) are conserved in all three pancreatic enzymes, and a further homologous disulfide bridge is shared with chymotrypsin (residues 1-122). The disulfide bridging pattern of the remaining four cysteine residues of @- factor XIIa cannot be determined from primary structure alignments alone. However, a tentative placement of these disulfide bridges can be made as shown in Fig. 6, Within the tertiary structure model of @-factor XIIa, pairing of the re- maining cysteines is clearly evident, since Cys-50 lies nearby Cys-111, and Cys-77 is near Cys-80. Indeed, in both cases suitable disulfide bonds can be made without disturbing the surrounding polypeptide chain structure and were incorpo- rated into the final @-factor XIIa structure (see Fig. 6).
Also evident in Fig. 6 are the positions of major insertions that occur in the amino acid sequence of /3-factor XIIa (resi- dues 61 A-C, 109 A-E, and 205 A-C). All three of these occur towards the end of existing surface @-loops in the pancreatic enzymes (see Fig. 6B); two insertions are positioned on the forward active site face of the enzyme (residues 61 A-C and 109 A-E), and the other is positioned towards the back surface of the enzyme (residues 205 A-C). Thus, the insertions ob- served for /3-factor XIIa occur at sites easily accommodated
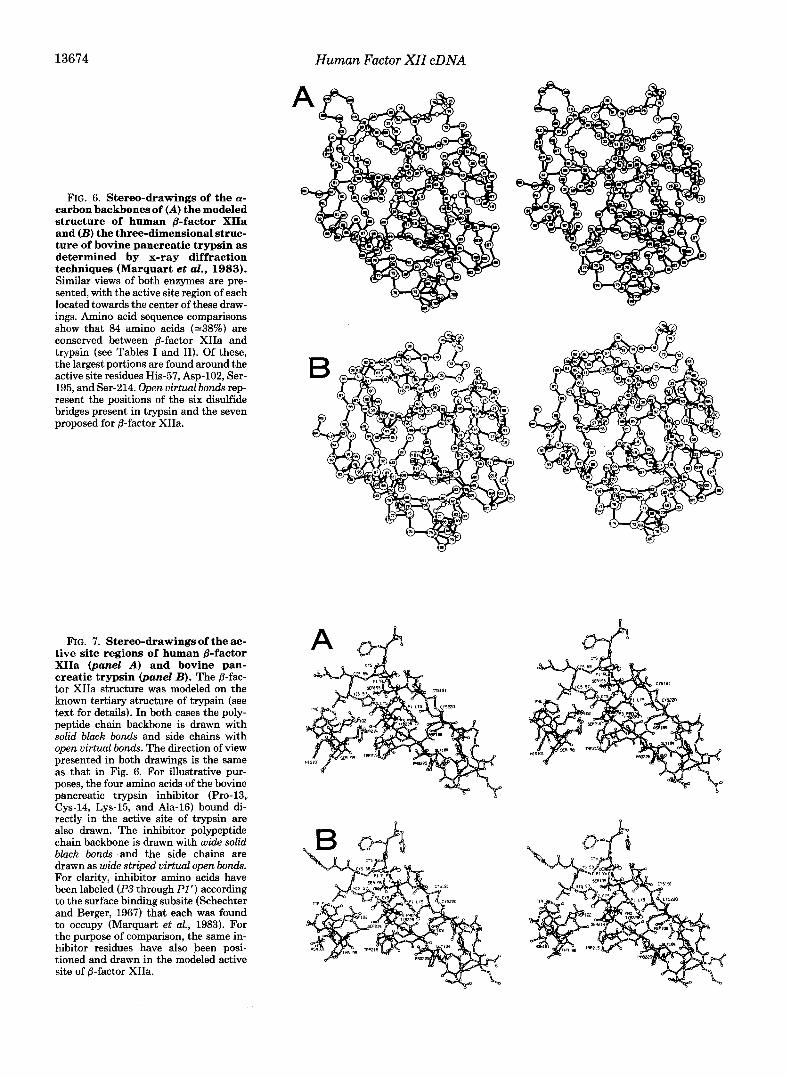
13674 Human Factor XII cDNA
FIG. 6. Stereo-drawings of the a- carbon backbones of (A) the modeled structure of human &factor XIIa and (B) the three-dimensional struc- ture of bovine pancreatic trypsin as determined by x-ray diffraction techniques (Marquart et al., 1983). Similar views of both enzymes are pre- sented, with the active site region of each located towards the center of these draw- ings. Amino acid sequence comparisons show that 84 amino acids (-38%) are conserved between @-factor XIIa and trypsin (see Tables I and 11). Of these, the largest portions are found around the active site residues His-57, Asp-102, Ser- 195, and Ser-214. Open virtual bonds rep- resent the positions of the six disulfide bridges present in trypsin and the seven proposed for @-factor XIIa.
FIG. 7. Stereo-drawings of the ac- tive site regions of human &factor XIIa (panel A) and bovine pan- creatic trypsin (panel B). The p-fac- tor XIIa structure was modeled on the known tertiary structure of trypsin (see text for details). In both cases the poly- peptide chain backbone is drawn with solid black bonds and side chains with open virtual bonds. The direction of view presented in both drawings is the same as that in Fig. 6. For illustrative pur- poses, the four amino acids of the bovine pancreatic trypsin inhibitor (Pro-13, Cys-14, Lys-15, and Ala-16) bound di- rectly in the active site of trypsin are also drawn. The inhibitor polypeptide chain backbone is drawn with wide solid black bonds and the side chains are drawn as wide striped virtual open bonds. For clarity, inhibitor amino acids have been labeled (P3 through PI ') according to the surface binding subsite (Schechter and Berger, 1967) that each was found to occupy (Marquart et al., 1983). For the purpose of comparison, the same in- hibitor residues have also been posi- tioned and drawn in the modeled active site of p-factor XIIa.
"
A

Human Factor XII &DNA 13675
within the overall structural framework of the pancreatic enzymes. Note that, in the present study, the five additional N-terminal amino acids of @-factor XIIa have not been in- cluded in our structural model. These residues have no coun- terparts in the pancreatic enzymes and therefore their place- ment could not be accomplished with an acceptable degree of confidence. However, these additional residues are unlikely to influence the catalytic activity of /3-factor XIIa, being positioned on the extreme rear surface of the enzyme in the orientation given in Fig. 6 (see the placement of residue 1).
The overall pattern of variable and constant regions of sequence homology observed between other blood coagulation and pancreatic proteases is also preserved in comparisons with @-factor XIIa. Thus, the present work, in agreement with other modelling studies (Furie et al., 1982), suggests that the basic core structural framework of the blood coagulation proteins is very similar to that of the pancreatic serine pro- teases. However, the exterior molecular surfaces of the coag- ulation proteins, which are composed in large part of six variable regions of sequence, are unique and are probably related to the individual physiological function of each en- zyme. The insertion of 5 residues at residue 109 in constant region CR3 of @-factor XIIa is the one notable exception to the overall pattern of constant and variable sequence regions proposed by Furie et al., 1982. The sequence alignment of this insertion in &factor XIIa cannot be accommodated elsewhere since it is fixed on either side by the placement of Asp-102 and Cys-122 (involved in a disulfide bridge with Cys-1), a feature conserved in the structure of chymotrypsin. Although absent in thrombin, factor Xa and factor IXa, this insertion is also present in tPA (see Table I) and therefore appears to be more than just a unique structural feature of @-factor XIIa. Thus, we propose that a further variable region be defined to describe insertions occurring at residue 109.
In common with other variable regions of amino acid se- quence, the 5-residue insertion at position 109 occurs on the surface of the enzyme and is most probably related to the ability of @-factor XIIa to recognize the physiological molec- ular surfaces with which it must interact. It is notable that the carbohydrate moiety of @-factor XIIa (bound to Asn-74; Fujikawa and McMullen, 1983) is positioned near this inser- tion and also on the substrate binding face of the enzyme (see Fig. 6A). Specialized surface features such as these and the variable regions of sequence already discussed provide a mech- anism by which complex cascade pathways like those involved in blood coagulation can occur by the specific recognition of substrate molecules (Jackson and Nemerson, 1980; Nemerson and Furie, 1980).
Fig. 7 presents a more detailed structural representation of the active site region in the derived model of ,&factor XIIa and this same region of trypsin. As is evident from Table I, the amino acid sequence homology between @-factor XIIa and trypsin in the active site region is very high, particularly about the catalytic residues His-57 and Ser-195. Thus, not unex- pectedly, the resultant tertiary structure model for ,&factor XIIa in this region in nearly identical to that observed for trypsin.
Also drawn solely for illustrative purposes is that portion of the bovine pancreatic trypsin inhibitor (Pro-13 to Ala-16) bound in the active site of trypsin (Fig. 7B, Marquart et al., 1983). These inhibitor residues serve to demonstrate how substrates bind within the active site of trypsin. Although the identical placement of this inhibitor peptide in the active site of @-factor XIIa is arbitrary, it clearly illustrates that this enzyme could bind peptide substrates in a manner similar to trypsin and other serine proteases (James et al., 1980).
The precise physiological substrates cleaved by @-factor XIIa have yet to be determined definitively. In general, the enzyme shows a trypsin-like cleavage specificity, with a dis- tinct preference for argininyl over lysyl peptide bonds (Rad- cliffe and Nemerson, 1976; Kurachi and Davie, 1977; Gold- smith et al., 1978). One potential factor in this observed preference for argininyl peptide bonds was suggested from our current model building approach. The positively charged amino end of the lysyl side chain in the primary binding pocket of trypsin forms a salt bridge to Asp-189 and hydrogen- bonded interactions to Ser-190. The hydroxyl group of Ser- 190 significantly restricts the positioning of the lysyl side chain at the b$ttom of. the S1 binding subsite (Ser-190 OG- Lys NZ 3.13 A). In our putative model of the @-factor XIIa active site, this interaction is absent since an alanine residue is present at position 190 of this enzyme. This amino acid substitution could reasonably be expected to produce two results (see Fig. 7). First, factor XIIa would have a lower affinity for lysyl side chains due to the absence of a hydrogen- bonded interaction to the hydroxyl of Ser-190. Secondly, the increased volume at the bottom of the primary binding subsite could more easily accommodate the larger and more rigid side chain of arginine. It is worth noting that at least four other serine proteases involved in the blood clotting cascade (throm- bin, factor Xa, tPA, and activated protein C), which have a preference for cleavage at arginines (see Jackson and Nem- erson, 1980), also, have the alanine residue at position 190 in their amino acid sequences. Thus, examination of the @-factor XIIa structural model provides at least a partial explanation for argininyl side chain preference for these enzymes.
Acknourledgments-We would like to thank Dr. Stuart Orkin for providing us with the human liver cDNA library. We would also like to thank Dr. Colin Hay, Marion Fung, and other colleagues for helpful technical suggestions and for critically reviewing this manuscript.
REFERENCES Atkinson, T., and Smith, M. (1984) in Oligonucleotide Synthesis, a
practical approach (Gait, M. J., ed) pp. 35-81, IRL Press Ltd., Oxford
Benton, W. D., and Davis, R. W. (1977) Science 196,180-182 Blin, N., and Stafford, D. W. (1976) Nucleic Acids Res. 3,2303-2308 Blobel, G., Walter, P., Chang, C. N., Goldman, B. M., Erickson, A.
H., and Lingappa, R. (1979) in Secretory Mechanisms (Hopkin, C. R., and Duncan, C. J., eds) Vol. 33, pp. 9-36, Cambridge University Press, London
Blomquist, M. C., Hunt, L. T., and Barker, W. C. (1984) Proc. Natl. Acad. Sci. U. S. A. 81,7363-7367
Bode, W., Schwager, P., and Huber, R. (1976) in Proteolysis and Physiological Regulation (Ribbons, D. W., and Brew, K., eds) Vol. 11, pp. 43-74, Academic Press, New York
Chaconas, G., and van de Sande, J. H. (1980) Methods Enzymol. 6 5 ,
Chirgwin, J. M., Przybyla, A. E., MacDonald, R. J., and Rutter, W.
Cochrane, C. G., Revak, S. D., and Wuepper, K. D. (1973) J. Exp.
Cohen, G. H., Silverton, E. W., and Davies, D. R. (1981) J. Mol. Biol.
Collen, D. (1980) Thromb. Huemostasis 43, 77-89 Deininger, P. L. (1983) Anal. Biochem. 129 , 216-223 Doolittle, R. F., Feng, D. F., and Johnson, M. S. (1984) Nature
Dunn, J. T., and Kaplan, A. P. (1982) J. Clin. Inuest. 70. 627-631
75-85
J. (1979) Biochemistn 18,5294-5299
Med. 138, 1564-1583
148,449-479
(Lo&.) 307,558-560
Dunn, J. T., Silverberg, M., and Kaplan, A. P. (1982) J. Biol. Chem. 257,1779-1784
Fujikawa, K., and Davie, E. W. (1981) Methods Enzymol. 80, 198-
Fujikawa, K., and McMullen, B. A. (1983) J. Biol. Chem. 258,10924-
Fujikawa, K., Kurachi, K., and Davie, E. W. (1977) Biochemistry 16 ,
211
10933
4182-4188

13676 Human Factor XII cDNA Fujikawa, K., Heimark, R. L., Kurachi, K., and Davie, E. W. (1980a)
Biochemistry 19 , 1322-1330 Fujikawa, K., McMullen, B. A., Heimark, R. L., Kurachi, K., and
Davie, E. W. (1980b) Protides Bwl. Fluids Proc. Colloq. 2 8 , 193- 196
Fung, M. R., Campbell, R. M., and MacGillivray, R. T. A. (1984) Nucleic Acids Res. 12,4481-4492
Furie, B., Bing, D. H., Feldmann, R. J., Robison, D. J., Burnier, J. P., and Furie, B. C. (1982) J. Biol. Chem. 2 5 7 , 3875-3882
Gilbert, W. (1979) in Eukaryotic Gene Regulation (Maniatis, T., and Axel, R., eds) pp. 1-12, Academic Press, New York
Goldsmith, G. H., Jr., Saito, H., and Ratnoff, 0. D. (1978) J. Clin. Inuest. 62,' 54-60
Griffin, J. H., and Cochrane, C. G. (1976) Methods Enzymol. 45,56- 65
Giinzler, W. A., Steffens, G. J., Otting, F., Kim, S. M. A., Frankus, E., and Flohe, L. (1982) Hoppe-Seyler's 2. Physiol. Chem. 363 , 1155-1165
Hanahan, D., and Meselson, M. (1980) Gene 10,63-67 Harpel, P. C. (1972) J. Clin. Inuest. 5 1 , 1813-1822 Hartley, B. S., and Kauffman, D. L. (1966) Biochem. J. 101, 229-
231 Heimark, R. L., Kurachi, K., Fujikawa, K., and Davie, E. W. (1980)
Nature (Lond.) 286,456-460 Hirano, H., Yamada, Y., Sullivan, M., de Crombrugghe, B., Pastan,
I., and Yamada, K. M. (1983) Proc. Natl. Acad. Sci. U. S. A. 8 0 ,
Jackson, C. M., and Nemerson, Y. (1980) Annu. Rev. Biochem. 4 9 ,
James, M. N. G., Delbaere, L. T. J., and Brayer, G. D. (1978) Can. J.
James, M. N. G., Sielecki, A. R., Brayer, G. D., Delbaere, L. T. J.,
Kan, Y. W., and Dozy, A. M. (1978) Proc. Natl. Acad. Sci. U. S. A.
Kaplan, A. P. (1978) Prog. Hemostasis Thromb. 4 , 127-175 Kaplan, A. P., and Austen, K. F. (1971) J. Exp. Med. 133,696-712 Kerbiriou, D. M., and Griffin, J. H. (1979) J. Biol. Chem. 254,12020-
Konnert, J. H., and Hendrickson, W. A. (1980) Acta Crystallogr. Sect.
Kurachi, K., and Davie, E. W. (1977) Biochemistry 16,5831-5839 Lucas, M. A., Fretto, L. J., and McKee, P. A. (1983) J. Biol. Chem.
Magnusson, S., Petersen, T. E., Sottrup-Jensen, L., and Claeys, H. (1975) in Proteases and Biological Control (Reich, E., Rifkin, D. B., and Shaw, E., eds) pp. 123-149, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY
Maniatis, T., Jeffrey, A., and Kleid, D. G. (1975) Proc. Natl. Acad. Sei. U. S. A. 72,1184-1188
Maniatis, T., Fritsch, E. F., and Sambrook, J. (1982) Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY
46-50
765-811
Biochem. 56,396-402
and Bauer, C. A. (1980) J. Mol. Biol. 144,43-88
75,5631-5635
12027
A, 36,344-350
258,4249-4256
Margolis, J. (1958) J. Physiol. (Lond.) 1 4 4 , 1-22 Marquart, M., Walter, J., Deisenhofer, J., Bode, W., and Haber, R.
McMillin, C. R., Saito, H., Ratnoff, 0. D., and Walton, A. G. (1974)
McMullen, B., and Fujikawa, K. (1984) Circulation 70,II-351 Meier, H. L., Pierce, J. V., Colman, R. W., and Kaplan, A. P. (1977)
Messing, J. (1983) Methods Enzymol. 101 , 20-78
(1983) Acta Crystallogr. Sect. B, 39,480
J. Clin. Inuest. 5 4 , 1312-1322
J. Clin. Invest. 6 0 , 18-31
Nemerson, Y., and Furie, B. (1980) CRC Crit. Reu. Biochem. 9 , 45-
Noller, H. F. (1984) Annu. Reu. Biochem. 5 3 , 119-162 Ny, T., Elgh, F., and Lund, B. (1984) Proc. Natl. Acad. Sci. U. S. A.
81,5355-5359 Pennica, D., Holmes, W. E., Kohr, W. J., Harkins, R. N., Vehar, G.
A., Ward, C. A., Bennett, W. F., Yelverton, E., Seeburg, P. H., Heyneker, H. L., Goeddel, D. V., and Collen, D. (1983) Nature
85
(Lond.) 301,214-221 Perry, R. P. (1976) Annu. Reu. Biochem. 45,605-629 Petersen, T. E., Thprgersen, H. C., Skorstengaard, K., Vibe-Pedersen,
Natl. Acad. Sci. U. S. A. 8 0 , 137-141 K., Sahl, P., Sottrup-Jensen, L., and Magnusson, S. (1983) Proc.
Prochownik, E. V., Markham, A. F., and Orkin, S. H. (1983) J. Biol. Chem. 258,8389-8394
Proudfoot, J. M., and Brownlee, G. G. (1976) Nature (Lond.) 2 6 3 ,
Radcliffe, R., and Nemerson, Y. (1976) J. Biol. Chem. 2 5 1 , 4797-
Ratnoff, 0. D., and Rosenblum, J. M. (1958) Am. J. Med. 2 5 , 160-
Ratnoff, 0. D., and Saito, H. (1979) Curr. Top. Hematol. 2 , 1-57 Revak, S. D., and Cochrane, C. G. (1976) J. Clin. Inuest. 57,852-860 Revak, S. D., Cochrane, C. G., Johnston, A. R., and Hugli, T. E.
Revak, S. D., Cochrane, C. G., and Griffin, J. H. (1977) J. Clin. Invest.
Saito, H. (1977) J. Clin. Invest. 6 0 , 584-594 Sanger, F., Nicklen, S., and Coulson, A. R. (1977) Proc. Natl. Acad.
Sci. U. 5'. A. 74,5463-5467 Sawyer, L., Shotton, D. M., Campbell, J. W., Wendell, P. L., Muir-
head, H., Watson, H. C., Diamond, R., and Ladner, R. C. (1978) J. Mol. Biol. 1 1 8 , 137-208
Schechter, I., and Berger, A. (1967) Biochem. Biophys. Res. Commun.
Sielecki, A. R., Hendrickson, W. A., Broughton, C. G., Delbaere, L. T. J., Brayer, G. D., and James, M. N. G. (1979) J. Mol. Biol. 134,
Smith, M., Leung, D. W., Gillam, S., Astell, C. R., Montgomery, D. L., and Hall, B. D. (1979) Cell 16 , 753-761
Sottrup-Jensen, L., Claeys, H., Zajdel, M., Petersen, T. E., and Magnusson, S. (1978) in Progress in Chemical Fibrinolysis and Thrombolysis (Davidson, J. F., Rowan, R. M., Samana, M. M., and Desnoyers, P. C., eds) Vol. 3, pp. 191-209, Raven Press, New York
211-214
4802
168
(1974) J. Clin. Inuest. 5 4 , 619-627
5 9 , 1167-1175
27,157-162
781-804
Southern, E. M. (1975) J. Mol. Bwl. 98,503-517 Staden, R. (1982) Nucleic Acids Res. 10,4731-4751 Thompson, R. E., Mandle, R., Jr., and Kaplan, A. P. (1977) J. Clin.
Thompson, R. E., Mandle, R., Jr., and Kaplan, A. P. (1979) Proc.
van der Graaf, F., Tans, G., Bouma, B. N., and Griffin, J. H. (1982)
Walker, J. M., Hastings, J. R. B., and Johns, E. W. (1977) Eur. J.
Watson, M. E. E. (1984) Nucleic Acids Res. 12,5145-5164 Wickens, M., and Stephenson, P. (1984) Science 226,1045-1051 Wiggins, R. C., Bouma, B. N., Cochrane, C. G., and Griffin, J. H.
Wilner, G. D., Nossel, H. L., and LeRoy, E. C. (1968) J. Clin. Invest.
Yamada, K. M. (1983) Annu. Reu. Biochem. 52,761-799 Ziemer, M. A., Swain, W. F., Rutter, W. J., Clements, S., Ann, D. K.,
Invest. 60, 1376-1380
Natl. Acad. Sci. U. S. A. 76,4862-4866
J. Biol. Chem. 257,14300-14305
Biochem. 76,461-468
(1977) Proc. Natl. Acad. Sci. U. S. A. 7 4 , 4636-4640
47,2608-2615
and Carlson, D. M. (1984) J. Biol. Chem. 259,10475-10480





























